 Yi Zhu
Yi Zhu Amanda G. Lobato
Amanda G. Lobato R. Grace Zhai
R. Grace Zhai Milena Pinto
Milena Pinto
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 24 March 2022
Sec. Alzheimer's Disease and Related Dementias
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.852972
This article is part of the Research Topic Clearance of Neurodegenerative Disease-associated Misfolded Proteins View all 4 articles
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by irreversible cognitive decline with limited therapeutic approaches. We characterized a Drosophila model of amyloid pathology that expresses human amyloid-beta precursor protein (APP695) and β-site APP cleaving enzyme (BACE) in the nervous system. Our model recapitulates in vivo the age-dependent accumulation of BACE-derived C-terminal fragment (CTF) and amyloid plaques in the brain, one of the key pathological hallmarks of AD. Using this model, we assessed the effects on plaque formation of Nicotinamide mononucleotide adenylyltransferase (Nmnat), an evolutionarily conserved nicotinamide adenine dinucleotide (NAD+) synthase involved in cellular metabolism and neuroprotection. We compared the effects of overexpression of Drosophila Nmnat (dNmnat), human Nmnat1 (hNmnat1), human Nmnat2 (hNmnat2), and human Nmnat3 (hNmnat3), and observed that hNmnat1 has the highest efficacy in reducing amyloid aggregation and APP-CTF accumulation. Interestingly, we demonstrated that overexpression of hNmnat1 reduces amyloid plaques by promoting autophagic clearance. Our findings uncover a role of hNmnat1 in amyloid clearance and suggest an exciting neuroprotective potential of hNmnat1 in amyloid pathology.
Alzheimer’s disease (AD) is a neurodegenerative disorder that leads to progressive memory loss and cognitive decline and represents the most common form of late-onset dementia (LaFerla et al., 2007; Masters et al., 2015). Two of the main pathological features of AD are amyloid plaques, extracellular insoluble aggregates composed of amyloid-beta fragments (Aβ), and neurofibrillary tangles, intracellular accumulation of hyperphosphorylated Tau (Ingelsson et al., 2004; LaFerla et al., 2007). Together, amyloid pathology and Tau aggregates drive the neuronal impairment that widely affects the cerebral cortex and hippocampus of AD patients (Masters et al., 2015). The effects of these plaques and tangles on cellular functions include mitochondrial dysfunction, synaptic degeneration, and autophagy dysfunction (Ingelsson et al., 2004).
Autophagy is a cellular pathway that controls proteostasis by sequestering and delivering protein aggregates and cellular organelles to lysosomes for degradation. Multiple stages of autophagy are disrupted in AD. Postmortem brain samples of AD patients show decreased levels of Beclin 1, which is essential for autophagy initiation by recruiting membranes to form autophagosomes before fusion to lysosome. Heterozygous deletion of Beclin 1 in transgenic mice expressing human amyloid precursor protein (hAPP) shows compromised neuronal autophagy and accelerated neurodegeneration due to Aβ accumulation (Pickford et al., 2008). Enhancing autophagy through overexpression of Atg8a, a homolog of mammalian LC3, in a Drosophila model of AD increases stress resistance and extends lifespan (Tsakiri et al., 2021). Targeting autophagy and improving protein quality control shows promising therapeutic potential in various neurodegenerative disorders including AD, Huntington’s disease (HD), and Parkinson’s disease (PD) (Nixon, 2013).
Activation of autophagy is involved in the neuroprotection conferred by overexpression of Nmnat (Nicotinamide mononucleotide adenylyltransferase), an evolutionarily conserved rate-limiting enzyme involved in the synthesis of NAD+. In a hypertensive glaucoma model, Nmnat protects against optic nerve degeneration through increasing autophagic flux in retinal ganglion cells (Kitaoka et al., 2013). Under hypoxic stress, Nmnat functions upstream of autophagy to mitigate the damage incurred by dendrites in neurons (Wen et al., 2013). We have previously shown that in a Drosophila model of HD, mutant Huntingtin (Htt) aggregation impairs autophagic pathway, while overexpressing Nmnat promotes autophagic clearance of Htt aggregates and protects against neurodegeneration (Zhu et al., 2019). Drosophila has a single Nmnat gene (dNmnat), and its loss-of-function causes post-development photoreceptor neurodegeneration (Zhai et al., 2006). Overexpression of dNmnat rescues neurodegeneration caused by aggregation of toxic proteins including Tau (Ali et al., 2012; Ma et al., 2020), Ataxin (Zhai et al., 2008; Ruan et al., 2015), and Htt (Zhu et al., 2019).
In mammals, three Nmnat genes produce three different Nmnat protein isoforms with distinct subcellular localizations: Nmnat1 in nuclear, Nmnat2 is present in the Golgi and in the cytoplasm, and Nmnat3 is mainly mitochondrial (Brazill et al., 2017). Nmnat1 is ubiquitously expressed and is one of the most studied isoforms of Nmnat. Its expression is protective against axon degeneration caused by mechanical or toxic insults. Mutations in this gene cause a recessive, early form of blindness genetically defined as Leber Congenital Amaurosis 9 (LCA9) (Falk et al., 2012). Nmnat1 knockout mice do not survive birth, while heterozygous mice develop normally without detectable neurodegeneration or axonal pathology (Conforti et al., 2011). On the other side, Nmnat1 overexpression reduces early behavioral impairment in a mouse model of tauopathy (Rossi et al., 2018) and reverses the loss of tyrosine hydroxylase (TH) neurons in the nigrostriatal pathway of the 3xTgAD mice (Jiang et al., 2021). Nmnat2 is predominantly neuronal and has the most influence over axon survival under physiological conditions: depletion of Nmnat2 causes a primary axonal phenotype (Gilley and Coleman, 2010) and mice lacking Nmnat2 have a severe axon outgrowth defect resulting in axon truncation in the peripheral and central nervous system that is incompatible with postnatal survival (Gilley et al., 2019). Reduced Nmnat2 mRNA levels are seen in AD, PD, and HD patients (Ali et al., 2016), and in a mouse model of tauopathy (Ljungberg et al., 2012). Nmnat3 is also ubiquitously expressed but predominantly present in the liver, heart, red blood cells, and skeletal muscle (Brazill et al., 2017). Nmnat3 is upregulated upon hypoxia-ischemia insult (Galindo et al., 2017). Overexpression of Nmnat3 prevents cortical and hippocampal tissue loss, while Nmnat3 knockdown causes neurodegeneration and increases excitotoxic cell death (Galindo et al., 2017).
In this report, we analyzed the effect of expressing dNmnat and human Nmnat1, 2, and 3 in suppressing the proteotoxic phenotype in a Drosophila model of Aβ pathology. We focused on the APP processing and Aβ plaque deposition in the brain and described the regulation of autophagy in Nmnat-facilitated clearance of amyloid aggregation.
The age-dependent accumulation of Aβ plaques and the ensuing oxidative and inflammatory responses are some of the key pathogenic factors of Alzheimer’s disease (LaFerla et al., 2007; Masters et al., 2015). The plaque-forming amyloid protein is produced by an initial cleavage of the APP by β-secretase (β-APP-cleaving enzyme-1 or BACE1) to generate a membrane-bound C-terminal fragment (CTF99), and a subsequent cleavage by γ-secretase to generate amyloidogenic peptides Aβ1–40 and Aβ1–42 (LaFerla et al., 2007). To model amyloid pathology in Drosophila, we used a pan-neuronal constitutive driver elav-GAL4 to express a Myc-tagged APP695 isoform of the human APP (UAS-APP695-N-myc), which is the most highly expressed isoform in neurons (Belyaev et al., 2010), together with BACE1 (UAS-BACE1) in the Drosophila nervous system. To determine if APP and BACE1 were expressed and if the secretase was functional, we performed a western blot analysis using an antibody that recognizes the C-terminal residues of APP (amino acid 676–695). The antibody recognizes the full-length (FL) APP (110 kDa) and the CTF (15 kDa) that remain membrane-bound after the first APP cleavage by β-secretase. In the homogenates of Drosophila brains at 10, 30, and 60 days after eclosion (DAE), both full-length APP and the CTF can be detected (Figure 1A), indicating that the APP was expressed, and that the beta-secretase was functional. Notably, we observed an age-dependent increase of the CTF/FL ratio (Figures 1A,B) and a significant increase of more than double by 60 DAE, indicating continuous APP processing and CTF accumulation in the fly brain over time.

Figure 1. Neuronal expression of APP and BACE1 leads to an age-dependent amyloid aggregation in the brain. (A) Western blot analysis showing full-length amyloid precursor protein (APP-FL) and C-terminal fragment (APP-CTF) of 10, 30, and 60 DAE flies expressing APP. Stain-Free imaging of a representative portion of the membrane is presented to show the protein loading. (B) Quantification of CTF/FL ratio in each group. Data are expressed as mean ± SD. n = 5/group. One-way ANOVA with Bonferroni’s post hoc test. *P < 0.05, **P < 0.01. (C) Top row: fly brains at 10, 30, and 60 DAE stained with antibody anti APP (heatmap). Bottom row: higher magnification images of the boxed areas in the top row. Scale bars = 30 μm. (D) Scatter plot showing quantification of the number of amyloid plaques in panel (C). The whiskers represent the minimum and maximum values of the dataset. One-way ANOVA with Bonferroni’s post hoc test. n = 7–8/group, ****P < 0.0001.
To determine if the CTF accumulation was further processed and able to form amyloid-like plaques in the fly brain, we performed an immunohistochemistry assay using the 6E10 antibody that recognizes amino acids 1–16 of APP and reacts to the abnormally processed isoforms as well as the precursor form. At an early stage (10 DAE), the APP was diffusely expressed in the brain, with a few high-intensity amyloid-like plaques detected (Figure 1C). With aging, at 30 and 60 DAE, we observed a significant increase in plaque number (Figure 1D), consistent with the CTF accumulation over time (Figures 1A,B). Of note, although APP was expressed in all neurons, the plaques were mainly located in the cortex layer of the fly brain, recapitulating the neuroanatomical pattern of APP accumulation in vulnerable regions such as cortical and subcortical layers in postmortem human AD brains (Ingelsson et al., 2004). Taken together, our Drosophila model of AD demonstrated two key pathological signatures in vivo: the biochemical feature of age-dependent APP cleavage and accumulation, and the morphological feature of amyloid plaques deposition in the brain.
We have previously shown that dNmnat and mammalian Nmnat3 protect against neurodegeneration in AD models by chaperoning hyperphosphorylated Tau (pTau) species and ameliorating pathological pTau aggregation (Ma et al., 2020). Moreover, the level of human Nmnat2 has been reported to negatively correlate with the burden of neuritic plaques and neurofibrillary tangles in postmortem human AD brains (Ali et al., 2016). To investigate the effect of different Nmnat isoforms on APP aggregation, we expressed APP together with dNmnat, hNmnat1, hNmnat2, hNmnat3, or mGFP (mitochondrial GFP as control) in the Drosophila nervous system and stained for APP at 60 DAE. We found that only the expression of hNmnat1, the nuclear isoform, significantly reduced amyloid plaques deposition in the brain (Figure 2A). Further quantification showed a significant reduction of amyloid plaque number and size in the hNmnat1 overexpression group when compared to those in the mGFP group (Figures 2B,C). We did not observe a significant change of amyloid plaque burden when overexpressing dNmnat, hNmnat2, or hNmnat3. All the overexpressed Nmnat isoforms can be detected in the fly brains (Figure 2D).
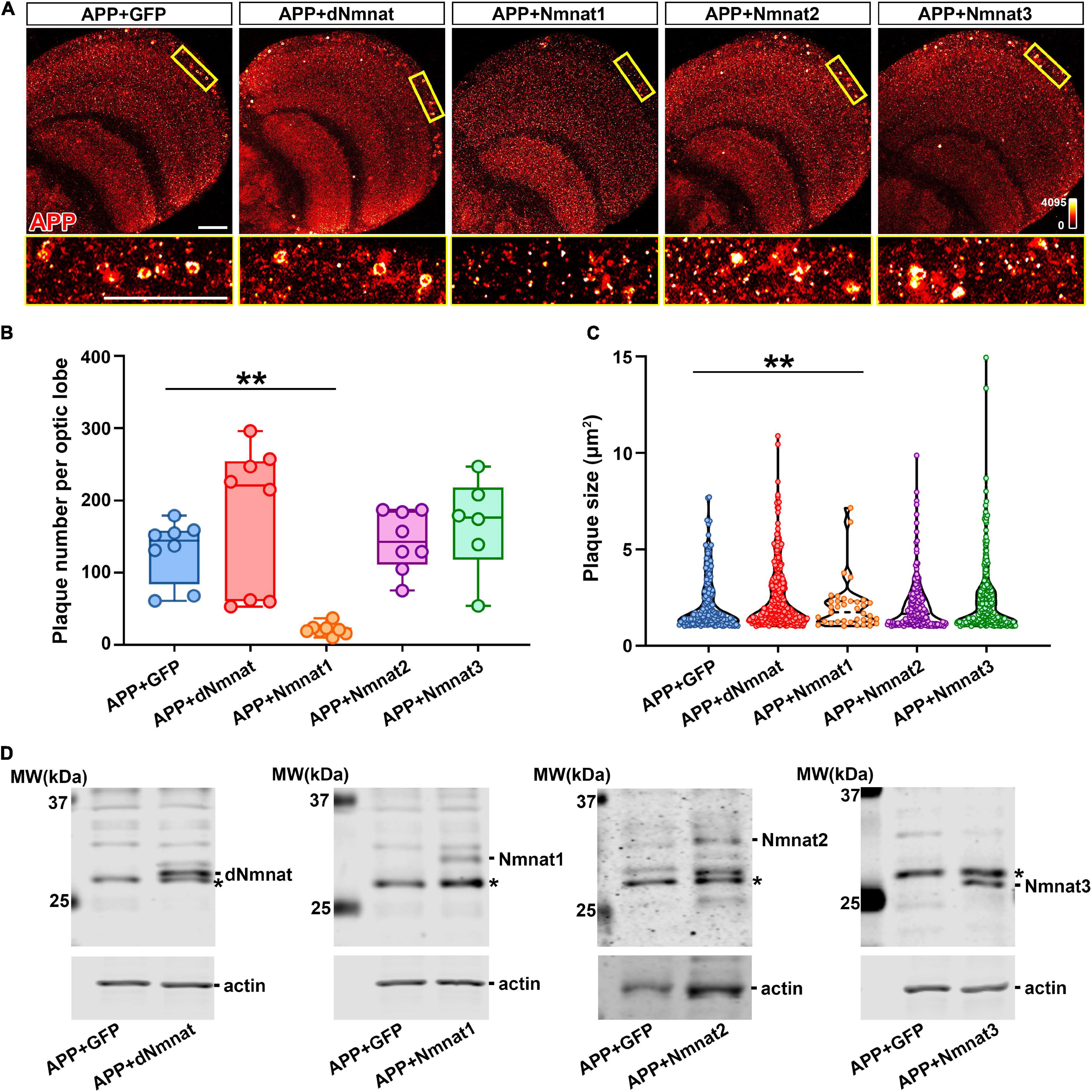
Figure 2. hNmnat1 reduces the accumulation of amyloid plaques in the brain. (A) Top row: APP staining (heatmap) of brains of flies expressing APP + GFP, APP + dNmnat, APP + hNmnat1, APP + hNmnat2, or APP + hNmnat3 at 60 DAE. Bottom row: higher magnification images of the boxed areas in the top row. Scale bars = 30 μm. (B) Scatter plot showing quantification of the number of amyloid plaques in panel (A). The whiskers represent the minimum and maximum values of the dataset. One-way ANOVA with Bonferroni’s post hoc test. n = 6–8/group, **P < 0.01. (C) Violin plot showing quantification of the size of amyloid plaques in panel (A). One-way ANOVA with Bonferroni’s post hoc test. **P < 0.01. (D) Western blot analysis showing expression of Nmnat isoforms in fly brains at 60 DAE. Actin is used as an internal control. * Indicates a non-specific band.
Next, we performed biochemical analysis to investigate how different Nmnat isoforms affect APP processing. As we have shown in Figure 1A, neuronal expression of APP led to an age-dependent accumulation of CTF in the brain. When APP was co-expressed with dNmnat, Nmnat2, or Nmnat3, we still observed a significant increase of CTF level with age, while Nmnat1 overexpression remarkably inhibited CTF accumulation (Figures 3A,B). Collectively, our data identified Nmnat1 as a potent inhibitor of amyloid plaque deposition and APP-CTF accumulation.

Figure 3. hNmnat1 reduces APP cleavage and production of APP-CTF in the brain. (A) Western-blot analysis of full-length amyloid precursor protein (APP-FL) and C-terminal fragment (APP-CTF) of 10, 30, and 60 DAE flies expressing APP + dNmnat, APP + hNmnat1, APP + hNmnat2, or APP + hNmnat3. Stain-Free imaging of a representative portion of the membrane is presented to show the protein loading. (B) Quantification of CTF/FL ratio in each group. Data are expressed as mean ± SD. n = 4–6/group. One-way ANOVA with Bonferroni’s post hoc test. *P < 0.05, **P < 0.01).
Autophagy has been reported to play a key role in the degradation of Aβ plaques and protects against Aβ-induced neurotoxicity (Hung et al., 2009). To delineate how hNmnat1 inhibits APP-CTF accumulation and amyloid plaques deposition, we applied an immunofluorescence approach to analyze endogenous autophagy genes (Atg)8a, the Drosophila homolog of mammalian LC3 that is recruited on autophagosomal membranes (Simonsen et al., 2008). We observed that at 60 DAE, hNmnat1-overexpressing flies exhibited a significant reduction of Atg8a level in the cortex layer of the brain where most APP plaques accumulated (Figures 4A,B). Atg8a in Drosophila exists in two forms: an unprocessed cytosolic form (Atg8a-I) and a processed phosphatidylethanolamine-modified form that associates with autophagosomal membranes (Atg8a-II) (Mauvezin et al., 2014). During autophagosome formation, the soluble Atg8a-I is converted into Atg8a-II through covalent binding to phagophore membranes, and Atg8a-II is eventually degraded inside mature autolysosomes (Manzéger et al., 2021). To assess the levels of Atg8a-I and Atg8a-II and the conversion of Atg8a-I to Atg8a-II, we performed biochemical analysis using an anti-GABARAP (γ-aminobutyric acid receptor-associated protein) antibody (Jacomin et al., 2020). GABARAP is a subfamily of Atg8 and the anti-GABARAP antibody has been verified for recognizing Drosophila Atg8 (Jipa et al., 2021). Consistent with imaging, we observed an overall reduction of total Atg8a in the fly brains with hNmnat1 overexpression at 60 DAE (Figures 4D,E), as well as a significant increase of Atg8a-II/Atg8a-I ratio, indicating promotion of autophagic flux (Figure 4F; Ratliff et al., 2016). Next, we analyzed Ref(2)P, the Drosophila homolog of human p62/SQSTM1, an adaptor protein that tethers ubiquitinated protein, binds to Atg8a, and is degraded in autolysosomes (Bjørkøy et al., 2005; Nezis et al., 2008). In brain imaging, we found a significant decrease of Ref(2)P level in the brain cortex layer at 60 DAE when hNmnat1 was overexpressed (Figure 4C), consistent with western blot analysis (Figures 4G,H). Notably, in the APP + mGFP group, we observed increased levels of total Atg8a and Ref(2)P at 60 DAE when compared to those at 30 DAE (Figures 4D–H), indicating an autophagy defect in older flies as previously reported (Bartlett et al., 2011). Taken together, our data indicated that hNmnat1 reduced the load of amyloid plaques in the brain through the promotion of autophagic clearance.
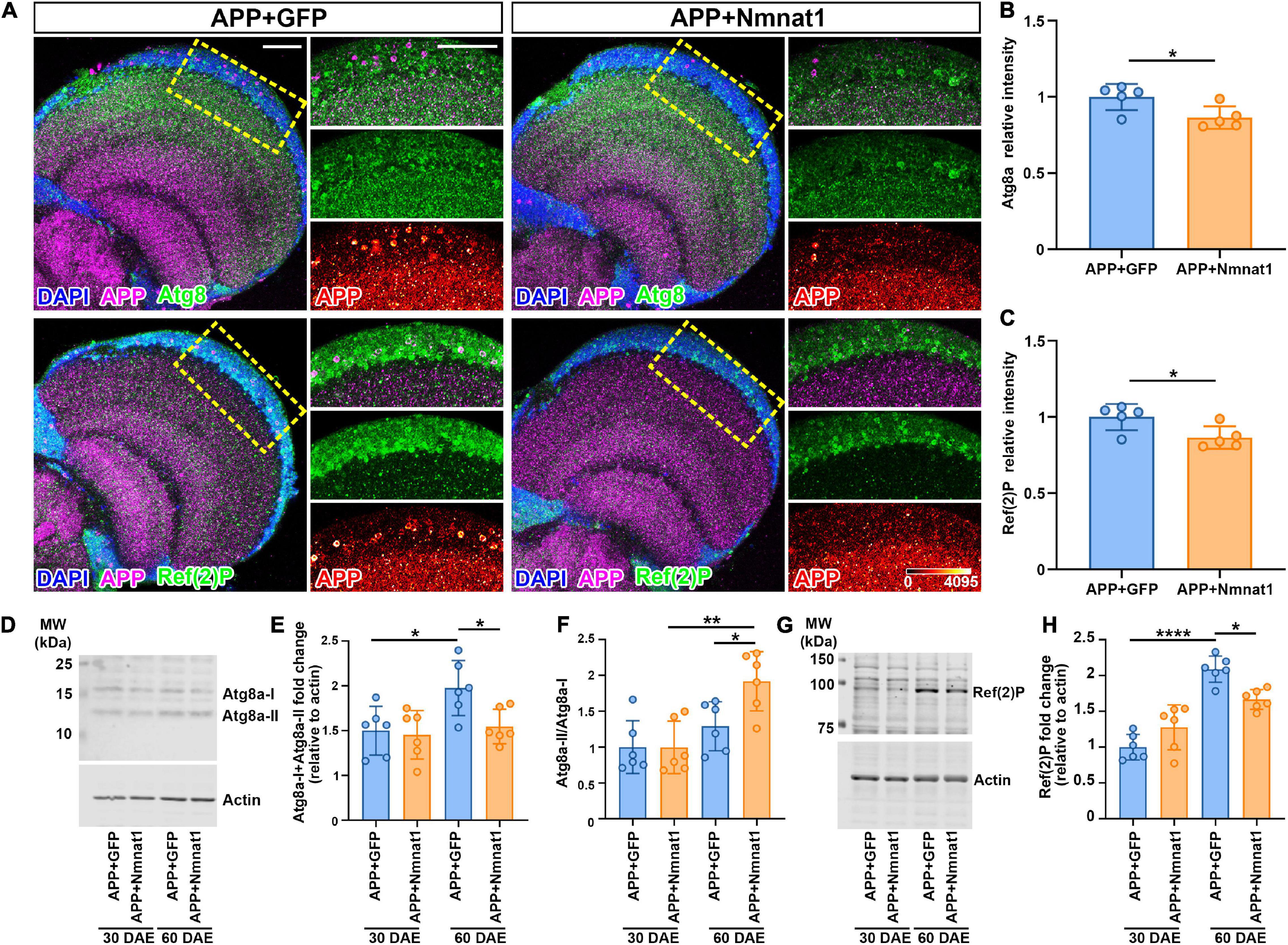
Figure 4. hNmnat1 promotes amyloid aggregates clearance through autophagy. (A) Fly brains at 60 DAE were stained with DAPI (blue), APP (magenta), and Atg8 or Ref(2)P (green). Images in the right column are high magnification of the boxed areas (APP is shown in heatmap). Scale bars = 30 μm. (B,C) Quantification of relative Atg8a and Ref(2)P intensity shown in panel (A). Data are expressed as mean ± SD. n = 5/group. Independent sample t-test. *P < 0.05. (D) Western blot analysis showing levels of endogenous Atg8a-I and Atg8a-II in fly brains at 30 and 60 DAE. Actin is used as an internal control. (E,F) Quantification of total Atg8a level (Atg8a-I + Atg8a-II) (E) and the ratio of Atg8a-II to Atg8a-I (F). Data are expressed as mean ± SD. n = 6/group. One-way ANOVA with Bonferroni’s post hoc test. *P < 0.05, **P < 0.01. (G) Western blot analysis showing levels of endogenous Ref(2)P in fly brains at 30 and 60 DAE. Actin is used as an internal control. (H) Quantification of Ref(2)P level. Data are expressed as mean ± SD. n = 6/group. One-way ANOVA with Bonferroni’s post hoc test. *P < 0.05, ****P < 0.0001.
In this study, we investigated the effect of different Nmnat isoforms on amyloid clearance in a Drosophila model of amyloid aggregation. Our model recapitulated the age-dependent processing of APP with CTF accumulation, and deposition of amyloid plaques in the brain cortex. Among different Nmnat isoforms, we identified hNmnat1 as a potent inhibitor for APP aggregation as evidenced by a remarkable reduction of the processed APP-CTF and decreased number and size of amyloid plaques in the brain. Finally, we showed an increased conversion of Atg8a-I to Atg8a-II as well as a reduction of total Atg8a and Ref(2)P in hNmnat1 overexpressing brains, indicating that hNmnat1 reduces amyloid plaque load by promoting autophagy.
Nmnat has been identified as a neuronal maintenance factor (Zhai et al., 2006). Cytoplasmic Nmnat isoforms like dNmnat in Drosophila used in this study, hNmnat2, and hNmnat3 are essential for maintaining neuronal integrity in a physiological environment and protecting against neurodegeneration in various neurodegenerative models (Brazill et al., 2017). These Nmnat isoforms can act as chaperones to interact with client proteins, facilitate protein folding and clearance, and thereby maintain proteostasis in neurons (Ali et al., 2016; Zhu et al., 2019; Ma et al., 2020). For example, Nmnat2 forms a complex with HSP90 chaperone to promote refolding of toxic tau (Ali et al., 2016) and Nmnat3 uses its NAD+ substrate-binding site to bind and chaperone pTau (Ma et al., 2020).
Aggregates formed from APP processing differ from other protein aggregates in that APP is processed on the plasma membrane and deposited extracellularly (LaFerla et al., 2007). As Nmnat1 is a predominantly nuclear-localized enzyme, it is unlikely for Nmnat1 to directly interact with APP or amyloid plaques. Our findings indicate that hNmnat1 promotes the autophagic clearance of unfolded or misfolded amyloid proteins, likely due to NAD+-dependent transcriptional or post-translational regulations. NAD+ is a coenzyme involved in hundreds of metabolic redox reactions, ADP-ribosylation, histone deacetylation, and calcium signaling pathways (Brazill et al., 2017). NAD+ can be synthesized by a salvage pathway or by a de novo pathway and Nmnat plays an essential role in both: it synthesizes NAD+ from nicotinamide mononucleotide (NMN) in the salvage pathway or it converts nicotinic acid mononucleotide (NaMN) to nicotinic acid adenine dinucleotide (NaAD) in the de novo pathway (Lau et al., 2009). Nmnat1 is essential for the supply of nuclear NAD+, a substrate for poly (ADP-ribose) polymerase (PARP)-mediated ADP-ribosylation and sirtuin (SIRT)-mediated deacetylation, both of which are critical regulators of autophagy. For example, PARP1, a ubiquitous nuclear enzyme, catalyzes PARylation of nuclear AMP-activated protein kinase (AMPK), inducing AMPK nuclear-cytosolic export for autophagosome formation and autophagy initiation (Rodríguez-Vargas et al., 2016). Upon DNA damage, PARP1 is required for autophagy induction by increasing the expression of Bnip-3, Cathepsin b and l, and Belin-1 (Muñoz-Gámez et al., 2009). We also recently demonstrated that Nmnat1 directly forms a complex with PARP1 and increases local NAD+ availability to promote PARylation and regulate cell survival under stress (Liu et al., 2021). In addition, nuclear SIRTs carry out multifaceted functions in autophagy. For example, SIRT1 deacetylates essential components of the autophagy machineries, including Atg5, Atg7, and Atg8, to promote autophagy initiation (Lee et al., 2008). SIRT1 also deacetylates and activates FOXO1 and FOXO3, two essential transcriptional regulators for autophagy induction (Brunet et al., 2004; Zhou et al., 2012). We recently showed that Nmnat1 upregulates SIRT1 and inhibits the activity of p53 (Liu et al., 2021), a negative regulator of autophagy (Tasdemir et al., 2008).
In addition to promoting autophagy, the nuclear NAD+ pool regulates the transcription of the major cleaving enzymes involved in APP processing. For example, BACE1 is tightly regulated by peroxisome proliferator-activated receptor-γ coactivator 1 (PGC)-1α, a transcriptional coactivator that enhances BACE1 ubiquitination and proteasomal degradation and ameliorates Aβ production (Gong et al., 2013; Wang et al., 2013). A decreased level of PGC-1α has been reported in the cortex of AD patients, while exogenous expression of PGC-1α can significantly inhibit Aβ plaque formation by suppressing BACE1 transcription (Katsouri et al., 2011). The transcriptional activity of PGC-1α depends on its subcellular distribution and is promoted by SIRT1-dependent nuclear translocation (Anderson et al., 2008). A previous study showed that nicotinamide riboside (NR), an NAD+ precursor, upregulates the expression of PGC-1α, enhances BACE1 ubiquitination and proteasomal degradation, and thereby ameliorates Aβ production (Gong et al., 2013).
Taken together, by using a Drosophila model of AD, our study demonstrated that overexpressing hNmnat1 inhibits amyloidogenic processing of APP and reduces amyloid plaque accumulation. Recently, high-throughput screens have identified small molecules to boost Nmnat expression in neurons or enhance NAD+ production (Ali et al., 2017; Gardell et al., 2019), highlighting the therapeutic potential of targeting Nmnat/NAD+ in treating AD and other proteinopathies.
Flies were maintained on cornmeal-molasses-yeast medium at room temperature (RT) with 65% humidity, 12 h light/12 h dark cycles. The following fly strains were obtained from Bloomington Drosophila Stock Center: UAS-APP695, UAS-BACE1; UAS-GFP; elav-GAL4. The following fly strains were generated in the lab (Zhai et al., 2006, 2008): UAS-dNmnat; UAS-Nmnat1; UAS-Nmnat2; UAS-Nmnat3. Only female flies were used in the experiments.
For analyzing Nmnat expression and autophagy (Figures 2, 4), 10 heads of each genotype were homogenized using radioimmunoprecipitation assay (RIPA) buffer. Extracted samples were mixed with Laemmli sample buffer (2% SDS, 10% glycerol, 62.5 mM Tris–HCl, 0.001% bromophenol blue, and 5% β-mercaptoethanol), and denatured at 95°C for 10 min. Proteins were separated by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and transferred to a nitrocellulose membrane. After blocking at RT for 1 h, the membrane was incubated with primary antibody at 4°C overnight, followed by secondary antibody incubation for 1 h at RT. The membrane was scanned by an Odyssey Infrared Imaging system (LI-COR Biosciences) and images were analyzed using Image Studio (version 4.0). The following primary antibody dilutions were used: anti-dNmnat [1:1,000, generated in the lab (Zhai et al., 2006, 2008)], anti-Nmnat1 (sc-271557, 1:1,000, Santa Cruz, Dallas, TX, United States), anti-Nmnat2 (ab56980, 1:1,000, Abcam, Cambridge, MA, United States), anti-Nmnat3 (ab71904, 1:1,000, Abcam), anti-Ref(2)P (1:500, Abcam), anti-GABARAP (1:1,000, MBL International Corporation), anti-β-actin (1:10,000, Sigma-Aldrich, St. Louis, MO, United States). The following secondary antibody dilutions were used: IRDye 700DX conjugated anti-Guinea pig (1:10,000, Rockland, PA, United States), DyLight 680 conjugated anti-Rabbit IgG (1:10,000, Rockland), DyLight 800 conjugated anti-Mouse IgG (1:10,000, Rockland).
For analyzing APP processing (Figures 1, 3), heads homogenates were run on a 4–20% gradient Tris–HCl gel (BioRad) and transferred to a PVDF membrane (BioRad). Membranes were blocked in 4% milk in PBST (PBS + 0.1% Tween 20) for 1 h at room temperature. Anti APP C-terminal Fragment C1/6 (Covance SIG-39152) was diluted 1:1,000 in 1% milk in PBST and incubated overnight at 4°C. Goat anti-mouse HRP-conjugated secondary antibody (Cell signaling) was used, and the reaction was developed by chemiluminescence using SuperSignal West reagent (Rockford, IL, United States). Blots were visualized with Chemidoc Imaging System (BioRad). Optical density measurements were taken by software supplied by BioRad. Bands were normalized for the total protein loading (visualized by stain-free technology, in the Chemidoc system, Biorad).
Dissection of the Drosophila brain was performed in dissection dishes with an elastomer bottom. Dissection was performed in phosphate-buffered saline (PBS). Dissected fly brains were immediately fixed with freshly made 4% formaldehyde for 15 min, washed in PBS containing 0.4% (v/v) Triton X-100 (PBTX) 3 times (10 min each), and incubated with primary antibodies diluted in 0.4% PBTX with 5% normal goat serum at 4°C overnight. Brains were incubated with conjugated secondary antibodies at RT for 1 h, followed by 4′,6-diamidino-2-phenylindole (DAPI, 1:300, Invitrogen, Carlsbad, CA, United States) staining for 15 min. Brains were mounted on glass slides with VECTASHIELD Antifade Mounting Medium (Vector Laboratories Inc., Burlingame, CA, United States). The following primary antibodies were used: anti-APP (6E10, 1:250, BioLegend), anti-Ref(2)P (1:250, Abcam), anti-GABARAP (1:250, MBL International Corporation). The following secondary antibodies were used: Alexa Fluor 555 Goat anti-Mouse IgG (1:250, Thermo Fisher Scientific, MA, United States), Cy5 conjugated anti-Rabbit IgG (1:250, Rockland). Fly brains were imaged using an Olympus IX81 confocal microscope coupled with a 60 × oil immersion objective lens, 1.30 numerical aperture (NA), with a scan speed of 8.0 μs per pixel and spatial resolution of 1,024 by 1,024 pixels (12 bits per pixel). Images were processed using FluoView 10-ASW (Olympus) and Adobe Photoshop CS6 (Adobe Systems). Amyloid plaques were quantified in Fiji/Image J (v1.52).
Details regarding each statistical test, sample size (n), and P value are indicated in the figure legends. P < 0.05 was considered statistically significant. All statistical analyses were carried out in GraphPad (v8.0).
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
MP and RZ conceived the project. YZ, AL, and MP carried out the experiments. YZ, AL, RZ, and MP analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Research reported in this publication was supported by the National Institutes of Health (NIH) award K01AG057815 (to MP), and R61AT010408 (to RZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Zoraida Diaz-Perez for technical support.
Ali, Y. O., Allen, H. M., Yu, L., Li-Kroeger, D., Bakhshizadehmahmoudi, D., and Hatcher, A. (2016). NMNAT2: HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 14:e1002472. doi: 10.1371/journal.pbio.1002472
Ali, Y. O., Bradley, G., and Lu, H.-C. (2017). Screening with an NMNAT2-MSD platform identifies small molecules that modulate NMNAT2 levels in cortical neurons. Sci. Rep. 7:43846.
Ali, Y. O., Ruan, K., and Zhai, R. G. (2012). NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum. Mol. Genet. 21, 237–250. doi: 10.1093/hmg/ddr449
Anderson, R. M., Barger, J. L., Edwards, M. G., Braun, K. H., O’Connor, C. E., and Prolla, T. A. (2008). Dynamic regulation of PGC-1α localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 7, 101–111. doi: 10.1111/j.1474-9726.2007.00357.x
Bartlett, B. J., Isakson, P., Lewerenz, J., Sanchez, H., Kotzebue, R. W., and Cumming, R. C. (2011). p62, Ref (2) P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 7, 572–583. doi: 10.4161/auto.7.6.14943
Belyaev, N. D., Kellett, K. A., Beckett, C., Makova, N. Z., Revett, T. J., and Nalivaeva, N. N. (2010). The Transcriptionally Active Amyloid Precursor Protein (APP) Intracellular Domain Is Preferentially Produced from the 695 Isoform of APP in a β-Secretase-dependent Pathway. J. Biol. Chem. 285, 41443–41454. doi: 10.1074/jbc.M110.141390
Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., and Øvervatn, A. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614. doi: 10.1083/jcb.200507002
Brazill, J. M., Li, C., Zhu, Y., and Zhai, R. G. (2017). NMNAT: It’s an NAD+ synthase…It’sa chaperone…It’sa neuroprotector. Curr. Opin. Genet. Dev. 44, 156–162. doi: 10.1016/j.gde.2017.03.014
Brunet, A., Sweeney, L. B., Sturgill, J. F., Chua, K. F., Greer, P. L., and Lin, Y. (2004). Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015. doi: 10.1126/science.1094637
Conforti, L., Janeckova, L., Wagner, D., Mazzola, F., Cialabrini, L., and Stefano, M. Di (2011). Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J. 278, 2666–2679. doi: 10.1111/j.1742-4658.2011.08193.x
Falk, M. J., Zhang, Q., Nakamaru-Ogiso, E., Kannabiran, C., Fonseca-Kelly, Z., and Chakarova, C. (2012). NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 44, 1040–1045.
Galindo, R., Banks Greenberg, M., Araki, T., Sasaki, Y., Mehta, N., and Milbrandt, J. (2017). NMNAT3 is protective against the effects of neonatal cerebral hypoxia-ischemia. Ann. Clin. Transl. Neurol. 4, 722–738. doi: 10.1002/acn3.450
Gardell, S. J., Hopf, M., Khan, A., Dispagna, M., Sessions, E. H., and Falter, R. (2019). Boosting NAD+ with a small molecule that activates NAMPT. Nat. Commun. 10, 1–12.
Gilley, J., and Coleman, M. P. (2010). Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8:e1000300. doi: 10.1371/journal.pbio.1000300
Gilley, J., Mayer, P. R., Yu, G., and Coleman, M. P. (2019). Low levels of NMNAT2 compromise axon development and survival. Hum. Mol. Genet. 28, 448–458. doi: 10.1093/hmg/ddy356
Gong, B., Pan, Y., Vempati, P., Zhao, W., Knable, L., and Ho, L. (2013). Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 34, 1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005
Hung, S.-Y., Huang, W.-P., Liou, H.-C., and Fu, W.-M. (2009). Autophagy protects neuron from Aβ-induced cytotoxicity. Autophagy 5, 502–510. doi: 10.4161/auto.5.4.8096
Ingelsson, M., Fukumoto, H., Newell, K., Growdon, J., Hedley–Whyte, E., and Frosch, M. (2004). Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62, 925–931. doi: 10.1212/01.wnl.0000115115.98960.37
Jacomin, A.-C., Petridi, S., Monaco, M. Di, Bhujabal, Z., Jain, A., and Mulakkal, N. C. (2020). Regulation of expression of autophagy genes by Atg8a-interacting partners sequoia, YL-1, and Sir2 in Drosophila. Cell Rep. 31:107695. doi: 10.1016/j.celrep.2020.107695
Jiang, H., Wan, Z., Ding, Y., and Yao, Z. (2021). Nmnat1 Modulates Mitochondrial Oxidative Stress by Inhibiting Caspase-3 Signaling in Alzheimer’s Disease. J. Mol. Neurosci. 71, 1467–1472. doi: 10.1007/s12031-020-01781-8
Jipa, A., Vedelek, V., Merényi, Z., Ürmösi, A., Takáts, S., and Kovács, A. L. (2021). Analysis of Drosophila Atg8 proteins reveals multiple lipidation-independent roles. Autophagy 17, 2565–2575. doi: 10.1080/15548627.2020.1856494
Katsouri, L., Parr, C., Bogdanovic, N., Willem, M., and Sastre, M. (2011). PPARγ co-activator-1α (PGC-1α) reduces amyloid-β generation through a PPARγ-dependent mechanism. J. Alzheimers. Dis. 25, 151–162. doi: 10.3233/jad-2011-101356
Kitaoka, Y., Munemasa, Y., Kojima, K., Hirano, A., Ueno, S., and Takagi, H. (2013). Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis. 4, e860–e860. doi: 10.1038/cddis.2013.391
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509.
Lau, C., Niere, M., and Ziegler, M. (2009). The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front. Biosci. 14, 410–431. doi: 10.2741/3252
Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., and Bruns, N. E. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. 105, 3374–3379. doi: 10.1073/pnas.0712145105
Liu, J., Tao, X., Zhu, Y., Li, C., Ruan, K., Diaz-Perez, Z., et al. (2021). NMNAT promotes glioma growth through regulating post-translational modifications of P53 to inhibit apoptosis. Elife 10:e70046. doi: 10.7554/eLife.70046
Ljungberg, M. C., Ali, Y. O., Zhu, J., Wu, C.-S., Oka, K., and Zhai, R. G. (2012). CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum. Mol. Genet. 21, 251–267. doi: 10.1093/hmg/ddr492
Ma, X., Zhu, Y., Lu, J., Xie, J., Li, C., and Shin, W. S. (2020). Nicotinamide mononucleotide adenylyltransferase uses its NAD+ substrate-binding site to chaperone phosphorylated Tau. Elife 9:e51859. doi: 10.7554/eLife.51859
Manzéger, A., Tagscherer, K., Lõrincz, P., Szaker, H., Lukácsovich, T., Pilz, P., et al. (2021). Condition-dependent functional shift of two Drosophila Mtmr lipid phosphatases in autophagy control. Autophagy 17, 4010–4028. doi: 10.1080/15548627.2021.1899681
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Primers 1:15056.
Mauvezin, C., Ayala, C., Braden, C. R., Kim, J., and Neufeld, T. P. (2014). Assays to monitor autophagy in Drosophila. Methods 68, 134–139. doi: 10.1016/j.ymeth.2014.03.014
Muñoz-Gámez, J. A., Rodríguez-Vargas, J. M., Quiles-Pérez, R., Aguilar-Quesada, R., Martín-Oliva, D., and de Murcia, G. (2009). PARP-1 is involved in autophagy induced by DNA damage. Autophagy 5, 61–74. doi: 10.4161/auto.5.1.7272
Nezis, I. P., Simonsen, A., Sagona, A. P., Finley, K., Gaumer, S., and Contamine, D. (2008). Ref (2) P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J. Cell Biol. 180, 1065–1071. doi: 10.1083/jcb.200711108
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., and Jaeger, P. A. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 118, 2190–2199. doi: 10.1172/JCI33585
Ratliff, E. P., Kotzebue, R. W., Molina, B., Mauntz, R. E., Gonzalez, A., and Barekat, A. (2016). Assessing basal and acute autophagic responses in the adult Drosophila nervous system: The impact of gender, genetics and diet on endogenous pathway profiles. PLoS One 11:e0164239. doi: 10.1371/journal.pone.0164239
Rodríguez-Vargas, J. M., Rodríguez, M. I., Majuelos-Melguizo, J., García-Diaz, Á, González-Flores, A., and López-Rivas, A. (2016). Autophagy requires poly (adp-ribosyl) ation-dependent AMPK nuclear export. Cell Death Diff. 23, 2007–2018. doi: 10.1038/cdd.2016.80
Rossi, F., Geiszler, P. C., Meng, W., Barron, M. R., Prior, M., and Herd-Smith, A. (2018). NAD-biosynthetic enzyme NMNAT1 reduces early behavioral impairment in the htau mouse model of tauopathy. Behav. Brain Res. 339, 140–152. doi: 10.1016/j.bbr.2017.11.030
Ruan, K., Zhu, Y., Li, C., Brazill, J. M., and Zhai, R. G. (2015). Alternative splicing of Drosophila Nmnat functions as a switch to enhance neuroprotection under stress. Nat. Commun. 6, 1–14.
Simonsen, A., Cumming, R. C., Brech, A., Isakson, P., Schubert, D. R., and Finley, K. D. (2008). Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184. doi: 10.4161/auto.5269
Tasdemir, E., Maiuri, M. C., Galluzzi, L., Vitale, I., Djavaheri-Mergny, M., and D’amelio, M. (2008). Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 10, 676–687. doi: 10.1038/ncb1730
Tsakiri, E. N., Gumeni, S., Manola, M. S., and Trougakos, I. P. (2021). Amyloid toxicity in a Drosophila Alzheimer’s model is ameliorated by autophagy activation. Neurobiol. Aging 105, 137–147. doi: 10.1016/j.neurobiolaging.2021.04.017
Wang, R., Li, J. J., Diao, S., Kwak, Y.-D., Liu, L., and Zhi, L. (2013). Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 17, 685–694. doi: 10.1016/j.cmet.2013.03.016
Wen, Y., Zhai, R. G., and Kim, M. D. (2013). The role of autophagy in Nmnat-mediated protection against hypoxia-induced dendrite degeneration. Mol. Cell. Neurosci. 52, 140–151. doi: 10.1016/j.mcn.2012.11.008
Zhai, R. G., Cao, Y., Hiesinger, P. R., Zhou, Y., Mehta, S. Q., and Schulze, K. L. (2006). Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 4:e416. doi: 10.1371/journal.pbio.0040416
Zhai, R. G., Zhang, F., Hiesinger, P. R., Cao, Y., Haueter, C. M., and Bellen, H. J. (2008). NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452, 887–891. doi: 10.1038/nature06721
Zhou, J., Liao, W., Yang, J., Ma, K., Li, X., and Wang, Y. (2012). FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 8, 1712–1723. doi: 10.4161/auto.21830
Keywords: APP, NAD, Drosophila, autophagy, aggregates
Citation: Zhu Y, Lobato AG, Zhai RG and Pinto M (2022) Human Nmnat1 Promotes Autophagic Clearance of Amyloid Plaques in a Drosophila Model of Alzheimer’s Disease. Front. Aging Neurosci. 14:852972. doi: 10.3389/fnagi.2022.852972
Received: 11 January 2022; Accepted: 24 February 2022;
Published: 24 March 2022.
Edited by:
Zhexing Wen, Emory University, United StatesReviewed by:
Sujyoti Chandra, University of California, Los Angeles, United StatesCopyright © 2022 Zhu, Lobato, Zhai and Pinto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milena Pinto, bXBpbnRvQG1lZC5taWFtaS5lZHU=; R. Grace Zhai, Z3poYWlAbWVkLm1pYW1pLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.