
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 23 March 2022
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.851135
This article is part of the Research Topic Mitochondrial and Lysosomal Dysfunction in Neurodegenerative Diseases: Molecular Mechanisms and Therapeutic Strategies View all 7 articles
 Wei Zheng1,2,3
Wei Zheng1,2,3 Dongsheng Fan1,2,3*
Dongsheng Fan1,2,3*
Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized by multiple motor and non-motor symptoms. Mutations in the glucocerebrosidase (GBA) gene, which encodes the lysosomal enzyme glucocerebrosidase (GCase), which hydrolyzes glucosylceramide (GlcCer) to glucose and ceramide, are the most important and common genetic PD risk factors discovered to date. Homozygous GBA mutations result in the most common lysosomal storage disorder, Gaucher’s disease (GD), which is classified according to the presence (neuronopathic types, type 2 and 3 GD) or absence (non-neuronopathic type, type 1 GD) of neurological symptoms. The clinical manifestations of PD in patients with GBA mutations are indistinguishable from those of sporadic PD at the individual level. However, accumulating data have indicated that GBA-associated PD patients exhibit a younger age of onset and a greater risk for cognitive impairment and psychiatric symptoms. The mechanisms underlying the increased risk of developing PD in GBA mutant carriers are currently unclear. Contributors to GBA-PD pathogenesis may include mitochondrial dysfunction, autophagy-lysosomal dysfunction, altered lipid homeostasis and enhanced α-synuclein aggregation. Therapeutic strategies for PD and GD targeting mutant GCase mainly include enzyme replacement, substrate reduction, gene and pharmacological small-molecule chaperones. Emerging clinical, genetic and pathogenic studies on GBA mutations and PD are making significant contributions to our understanding of PD-associated pathogenetic pathways, and further elucidating the interactions between GCase activity and neurodegeneration may improve therapeutic approaches for slowing PD progression.
Parkinson’s disease (PD) is a heterogeneous neurodegenerative disorder characterized by the motor manifestations of resting tremor, rigidity, bradykinesia and postural instability, as well as some non-motor symptoms. Degeneration of dopaminergic neurons in the substantia nigra pars compacta and the presence of α-synuclein aggregates, that is, Lewy bodies, are the two main pathological hallmarks of PD. Many etiological factors, including multiple genetic causes and environmental and aging factors, have been demonstrated to underlie dopaminergic neurodegeneration (Obeso et al., 2017). More than 20 genetic loci that contribute to familial PD, which accounts for 5–10% of PD cases, have been discovered to date (Balestrino and Schapira, 2018). Glucocerebrosidase (GBA) gene mutations are currently recognized as the most important and common risk factors for developing PD (Sidransky and Lopez, 2012). The GBA gene mainly encodes the lysosomal enzyme glucocerebrosidase (GCase), which hydrolyzes glucosylceramide (GlcCer) to glucose and ceramide. In addition, GCase catalyzes the transfer of glucose from GlcCer to cholesterol to contribute to in the synthesis of β-cholesteryl glucoside (Franco et al., 2018). The GBA gene is located on chromosome 1q21 and is composed of 11 exons and 10 introns spanning a 7.6 kb sequence (Gan-Or et al., 2018). More than 300 known GBA mutations, among which L444P and N370S are the most common, have been found (O’Regan et al., 2017). The N370S mutation is most common in the Ashkenazi Jewish population and is often regarded as a mild mutation (Sidransky and Lopez, 2012), whereas the L444P mutation is found worldwide and is often regarded as a severe mutation (Gan-Or et al., 2008; Do et al., 2019). Homozygous mutations in GBA result in the most common lysosomal storage disorder, Gaucher’s disease (GD) (Grabowski, 2008). Approximately, 7–12% of PD patients carry GBA mutations (García-Sanz et al., 2021), and 25% of GD patients have a first- or second-degree relative with PD (Aflaki et al., 2017). A large multicenter study estimated that the odds ratio of PD patients harboring a GBA mutation was 5.43 compared with that of controls, confirming that GBA mutation is the single largest risk factor for developing PD identified to date (Sidransky et al., 2009). The proportion of GBA mutation carriers among PD patients ranges depending on the population studied and whether the whole exome is sequenced, with highest frequencies found among the Ashkenazi Jewish population (Schapira et al., 2016).
To date, the mechanisms that underlie GBA mutations that increase the risk of developing PD have not been fully elucidated. Several perspectives have been provided, including enhanced SNCA aggregation, lysosomal dysfunction, impaired autophagy, altered lipid homeostasis and mitochondrial dysfunction. In this review, we discuss GBA mutations and PD and focus on the current advances in understanding the pathogenesis by which GBA mutations increase the risk for developing PD, with the goal of improving our understanding of the role of GCase deficiency in the neurodegeneration of PD and gaining further insights into the pathogenetic pathways in PD. Finally, we discuss the implications for PD therapy based on an investigation of the relationship between GBA and PD, with the goal of finding new therapeutic targets that slow PD progression.
Gaucher’s disease is a rare autosomal recessive disorder caused by insufficient activity of the lysosomal enzyme GCase (Stirnemann et al., 2017). A deficient GCase level leads to the accumulation of its substrate GlcCer in lysosomes of reticuloendothelial lineage, causing the acquisition of a variety of clinical phenotypes, including hepatosplenomegaly, anemia, thrombocytopenia, and bone disease (Grabowski et al., 2015). GD is a panethnic disease but is most common in the Ashkenazi Jewish population, which has an incidence of 1/800, significantly higher than the 1/40,000 to 1/60,000 incidence in the general population but rises to Grabowski (2008). GD is classified by the involvement neurological symptoms (neuronopathic types, type 2 and 3 GD) or not (non-neuronopathic type, type 1 GD) (Sidransky, 2004). Type 1 GD, the most common phenotype of GD, is considered to indicate the absence of neurological involvement. The symptoms of type 1 GD are variable and can include hepatosplenomegaly, fatigue, anemia, thrombocytopenia, pulmonary hypertension, and osteopenia/osteoporosis, and type 1 GD symptoms can appear at any age (Stirnemann et al., 2017). Type 2 or acute neuronopathic GD is the most severe form, often appearing in infancy, with organomegaly, pancytopenia, skin abnormalities and severe central nervous system (CNS) impairment, followed by death in the first years of life (Weiss et al., 2015). Type 3 or subacute neuronopathic GD primarily involves visceral signs and impaired horizontal saccadic eye movement and usually presents in adolescence (Stirnemann et al., 2017).
The association between GBA mutations and PD was first described in the 1990s, and GD patients also show concomitant parkinsonism (Neudorfer et al., 1996; Tayebi et al., 2003). A clinical study screened 99 Ashkenazi Jewish patients with idiopathic PD and 1543 healthy Ashkenazi Jewish individuals for six GBA mutations and found that 31.3% of the PD patients expressed one or two GBA mutant alleles, compared with 6.2% of the controls (Aharon-Peretz et al., 2004). Another study reported the genotyping of 57 subjects with PD using brain bank samples found that 12 samples (21%) obtained from PD patients showed alterations in the GBA gene (Lwin et al., 2004). Since these initial studies were reported, multiple studies with PD cohorts characterized by different regions and ethnic origins have been performed to determine the frequency of GBA mutations. The frequency of GBA mutations ranged from 10.7 to 31.3% in the Ashkenazi Jewish population with PD (Aharon-Peretz et al., 2004; Clark et al., 2005) but ranged from 2.3%, the lowest frequency and reported in the Norwegian population with PD, to 9.8% for individuals of other ethnic origins (Toft et al., 2006; Seto-Salvia et al., 2012). In fact, on the one hand, ethnic origin certainly contributes to different frequencies of GBA mutations in PD; on the other hand, the methods used for genotyping affect the identification of mutants, with some studies detecting specific common GBA mutations, for instance, N370S and L444P, and others sequencing all exons of the GBA gene.
Since most previous single-center PD cohort studies based on GBA mutations were limited in the number of ethnic populations included, GBA genotyping methods and sample size, an international multicenter collaborative study including 5691 PD patients (780 were Ashkenazi Jewish patients) and 4898 controls (387 were Ashkenazi Jewish patients) from 16 centers was performed in 2009 (Sidransky et al., 2009). All the centers screened at least two mutations, N370S and L444P, and found that 15% of the PD patients carried GBA mutations compared with 3% of the controls among the Ashkenazi Jewish patients; 3% of the PD patients carried GBA mutations compared with 1% of the controls among non-Ashkenazi patients (Sidransky et al., 2009). Importantly, when GBA was fully screened in 1883 non-Ashkenazi Jewish patients and 1611 non-Ashkenazi Jewish controls, 7% of the patients were found to have GBA mutations, indicating that approximately 45% of the mutant loci could be missed when only N370S and L444P are sequenced (Sidransky et al., 2009). This study confirms that an adequate sample size and accurate genotyping are imperative to ascertain the frequency of GBA mutations among populations. In a large study with a European cohort, all exons of GBA were screened in 786 PD patients who had a familial PD history, 605 sporadic PD patients and 391 controls, and an overall GBA carrier frequency of 6.7% was found for the PD patients, including sporadic and familial subjects, compared with 1% of the control individuals (Lesage et al., 2011). Furthermore, GBA mutations were found more frequently in patients with a family history of PD (8.4%) than in isolated cases (5.3%) (Lesage et al., 2011).
The clinical manifestations of PD patients with GBA mutations are indistinguishable from those of sporadic PD patients on an individual level. However, when analyzing PD associated with GBA mutations in a group, patients with PD and GBA mutations exhibited a 1.7– 6-year earlier age of onset than those with idiopathic PD (Aharon-Peretz et al., 2004; Clark et al., 2007; Neumann et al., 2009; Nichols et al., 2009). PD patients with GBA mutations who had two mutant GBA alleles developed PD at an earlier age (54.2 versus 65.2 years) than patients who heterozygous, carrying one mutant allele (Alcalay et al., 2014). However, the age-specific risk of developing PD at age 60 and 80 years was higher in GD patients (4.4 and 9.1%) than in heterozygous individuals (1.5 and 7.7%) but this difference was not significant (Alcalay et al., 2014). These data suggested that a second GBA mutant allele contributes to a younger age of PD onset but does not increase the overall risk of developing PD. In addition, patients with PD and GBA mutations had a higher frequency of cognitive impairment or dementia (Neumann et al., 2009; Winder-Rhodes et al., 2013; Creese et al., 2018). Compared with idiopathic PD patients, GBA carriers with a severe mutation, such as L444P, presented with a fivefold greater risk for developing dementia, while those with a mild mutation, such as N370S, exhibited a twofold greater risk for developing dementia (Cilia et al., 2016), suggesting that the extent of cognitive impairment differs on the basis of the severity of mutations. PD patients with GBA mutations were also found to be susceptible to psychiatric symptoms, including depression, hallucinations, sleep disturbances and anxiety (Brockmann et al., 2011). However, the clinical features of GBA-associated PD resemble those of idiopathic PD in terms of tremor, rigidity, and bradykinesia (Clark et al., 2007; Westbroek et al., 2011). In other studies, PD patients carrying GBA mutations were reported to be less likely to have tremor at onset but more likely to present bradykinesia at onset (Gan-Or et al., 2010; Lesage et al., 2011; Swan and Saunders-Pullman, 2013), which may suggest the need to expand the sample size and extend the follow-up times. In addition to an earlier age of onset, more severe symptoms and more rapid progression have been observed in PD patients carrying GBA mutations than in idiopathic PD patients in most studies (Mata et al., 2016).
Recent studies have been exploring the contribution of mutant GBA to PD pathogenesis. To date, the mechanisms that underlie the increased risk of developing PD among GBA mutation carriers have not been fully elucidated. In general, in autosomal dominant forms of PD, such as those involving LRRK2 and α-synuclein, gain-of-function mutations are usually involved in PD pathogenesis. In contrast, loss-of-function mutations, such as those in such as parkin, DJ-1 and PINK1, are implicated in most autosomal recessive forms of PD.
Notably, the inheritance of GBA-associated PD does not follow strict Mendelian law, although GD is an autosomal recessive disease. Therefore, both gain-of-function and loss-of-function have been proposed as explanations for GBA mutation increasing the risk of PD development (Figure 1).
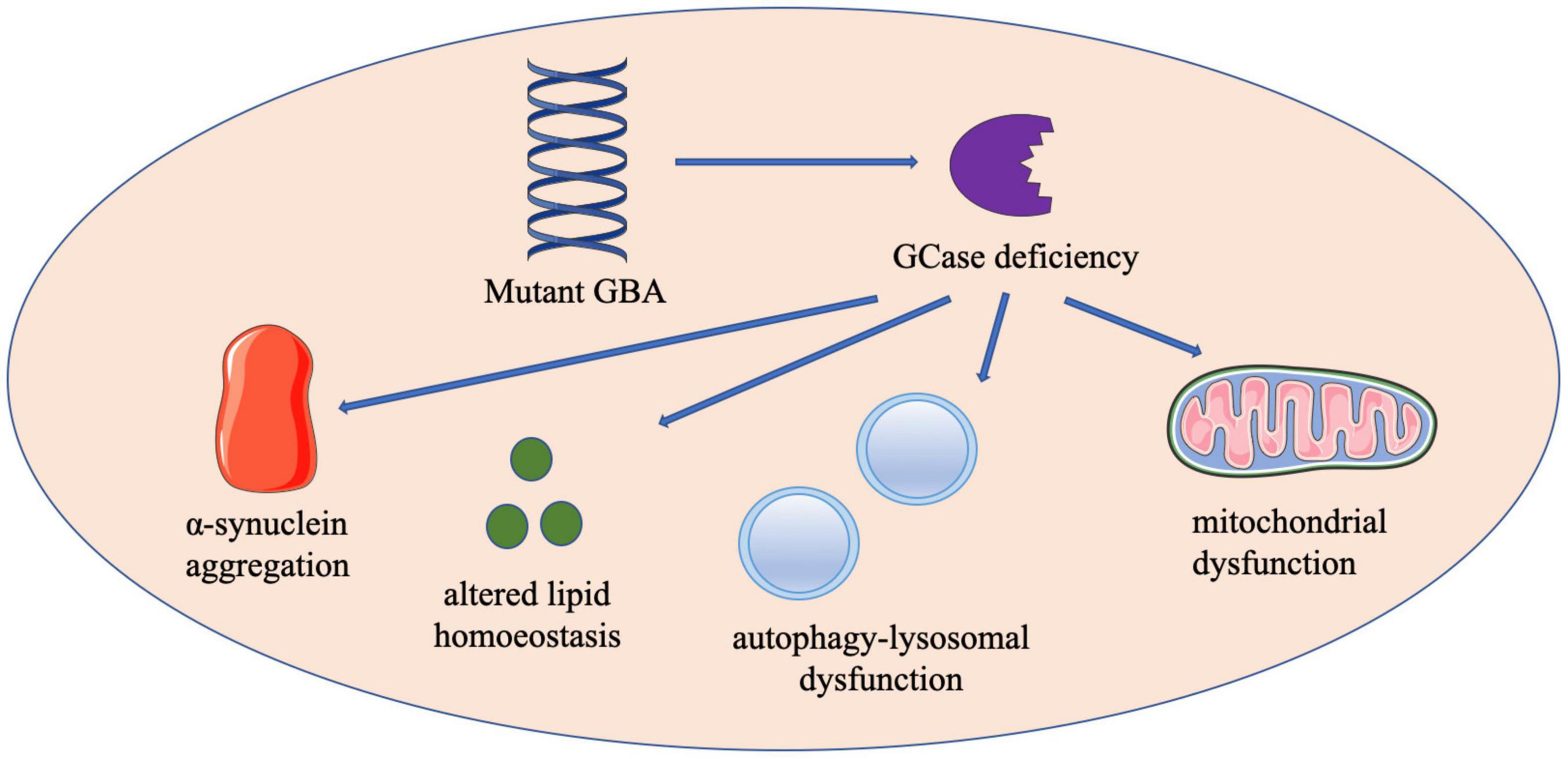
Figure 1. The schematic diagram of proposed mechanisms by which GBA mutations contribute to the development of PD. Contributions to GBA-PD pathogenesis may include enhanced α-synuclein aggregation, altered lipid homeostasis, autophagy-lysosomal dysfunction and mitochondrial dysfunction. GBA, glucocerebrosidase; GCase, glucocerebrosidase.
Multiple studies point to the vital role of α-synuclein in the pathogenesis of GBA-associated PD, supporting a gain-of-function mechanism in which mutant GCase may directly lead to α-synuclein aggregation. In the brains of PD patients with GBA mutations, GCase is present in 32–90% (mean 75%) of Lewy bodies (Goker-Alpan et al., 2010). In contrast, GCase is positive in <10% of Lewy bodies of subjects without GBA mutations (Goker-Alpan et al., 2010), suggesting that mutant GCase may lead to the aberrant aggregation of α-synuclein, the major element in Lewy bodies. Several independent studies have detected α-synuclein accumulation in transgenic GBA mouse models (Migdalska-Richards et al., 2017b), inhibited GCase activity in conduritol-β-epoxide-treated SH-SY5Y cell cultures and mice (Manning-Bog et al., 2009; Cleeter et al., 2013), and induced pluripotent stem cell (iPSC)-derived neurons obtained from GBA-associated PD patients and GD patients (Schondorf et al., 2014; Woodard et al., 2014; Fernandes et al., 2016). Mazzulli et al. (2011) proposed that a reciprocal relationship between GCase and α-synuclein in synucleinopathies. GlcCer accumulation due to decreased activity of GCase may lead to aggregation of α-synuclein by stabilizing soluble toxic α-synuclein oligomeric intermediates, which tend to transition into insoluble deposits such as Lewy bodies; at the same time, elevation of α-synuclein levels inhibits lysosomal GCase maturation and activity, in turn augmenting α-synuclein aggregation, forming a self-propagating feedback loop in PD and other synucleinopathies that eventually leads to neurodegeneration (Mazzulli et al., 2011). Furthermore, in another study, by Kim et al. (2018) demonstrated that GCase deficiency due to GBA loss-of-function mutations influences the phase transition of α-synuclein. Accumulation of GlcCer resulting from GBA mutations increases α-synuclein monomers that tend to aggregate and convert into oligomers and destabilize α-synuclein tetramers and related multimers, which are resistant to aggregation (Kim et al., 2018). α-Synuclein, as the main pathological factor in PD, has been demonstrated to spread between neurons in a prion-like manner (Gomez-Benito et al., 2020). In addition to influencing the phase transition of α-synuclein, loss of GBA function was found to promote perpetual cell-to-cell transmission of α-synuclein aggregates, including both exogenous and endogenous α-synuclein aggregates (Bae et al., 2014).
Accumulation of α-synuclein not only inhibits lysosomal GCase maturation and activity but also affects the transport of GCase from the endoplasmic reticulum (ER) to lysosomes through the Golgi apparatus. In normal GCase-functioning cells, wild-type GCase is synthesized within polyribosomes on the ER and is translocated into the ER (Ron and Horowitz, 2005). Then, GCase is transported to the Golgi, from where to be transported to lysosomes (Ron and Horowitz, 2005). Lysosomes are the key organelles critical for the degradation of proteins, such as α-synuclein (Lawrence and Zoncu, 2019). In GBA-mutated cells, GlcCer accumulation due to lysosomal GCase deficiency causes lysosomal dysfunction, eventually leading to α-synuclein aggregation. The accumulation of α-synuclein can impair the transport of GCase, in turn reducing α-synuclein turnover (Schapira and Gegg, 2013). However, the mechanism by which α-synuclein affects GCase transport remains unclear. Lysosomal integral membrane protein type-2 (LIMP-2), the lysosomal integral membrane protein critical for transporting GCase from the ER to lysosomes, does not bind α-synuclein (Gegg et al., 2012). The level of LIMP-2 is unaffected in the substantia nigra of PD patients with GBA mutations (Gegg et al., 2012). These data indicate that we should consider whether the degree of GCase decrease depends on decreased transcription or increased degradation of mRNA and the role of α-synuclein in this process.
Lysosomes are acidic dynamic organelles involved in the degradation of harmful or unnecessary cellular contents, such as long-lived proteins and damaged organelles, via enzymatic degradation, and autophagic pathways, including macroautophagy, microautophagy and chaperone-mediated autophagy (Dehay et al., 2013; Chun and Kim, 2018). Disruption of the autophagy-lysosomal system has been identified in studies of PD pathogenesis in recent years. Given the lysosomal localization enzyme GCase, the link between GCase and autophagy-lysosomal pathways has attracted attention. Schondorf et al. (2014) found that iPSC-derived neurons from GBA-associated PD patients and GD patients exhibited autophagic defects due to impaired autophagosome-lysosome fusion. GCase influenced α-synuclein accumulation not only by enhancing the production of α-synuclein but also by decreasing the degradation of α-synuclein via autophagy-lysosomal dysfunction. Autophagy-lysosomal disruptions result in enlargement of the lysosomal compartment in iPSC-derived dopamine neurons of PD patients with GBA-N370S mutations, subsequently increasing the extracellular α-synuclein level (Fernandes et al., 2016). Consistent with iPSC-derived neurons with GBA mutations, impaired autophagy and dysfunctional lysosomes have been found in fibroblasts obtained from PD patients with GBA mutations (Garcia-Sanz et al., 2017). Additionally, GBA deficiency leads to α-synuclein accumulation by inhibiting autophagy in neuroblastoma cells (Du et al., 2015). Protein phosphatase 2A inactivation is involved in the suppression of autophagy induced by GBA mutants (Du et al., 2015). Importantly, the autophagy inducer rapamycin reversed α-synuclein accumulation in GBA-knockdown cells (Du et al., 2015). Defective autophagic clearance due to lysosomal dysfunction has also been detected in GD iPSC-derived neural cells (Awad et al., 2015). Further analysis revealed that decreased level of the transcription factor EB, the main regulator of lysosomal genes, impaired lysosomal biogenesis in GD iPSC-derived neural cells (Awad et al., 2015). Lysosomal function was rescued by recombinant GCase and the overexpressed transcription factor EB, but overexpression of the transcription factor EB alone did not restore lysosomal function, suggesting that GCase leads to autophagy-lysosomal dysfunction in part via transcription factor EB (Awad et al., 2015). Further studies are needed to investigate the role of autophagy-lysosomal dysfunction in GBA-related pathogenesis.
Lipids, as the main component of the cell membrane, are central to maintaining membrane structure and cellular processes, including synaptic transmission and molecular trafficking. Accumulating evidence has demonstrated that lipid homeostasis and its interaction with GCase are involved in PD pathogenesis. GCase normally cleaves GlcCer into glucose and ceramide. Based on a loss-of-function mechanism, the accumulation of GlcCer substrates caused by a reduced GCase level promotes the formation of toxic α-synuclein aggregation by stabilizing α-synuclein oligomers, subsequently resulting in neurodegeneration (Mazzulli et al., 2011). Dysfunction of cholesterol metabolism could result in changes in lipid rafts, which play a vital role in synaptic function (García-Sanz et al., 2021). Utilizing iPSC-derived neuronal models, Zunke et al. (2018) revealed equilibrium between two α-synuclein forms, a 100 Å-sized high molecular weight conformer and a low-molecular-weight 35 Å-sized monomeric species. Glycosphingolipids preferentially convert high-molecular-weight α-synuclein conformers into compact, assembled toxic oligomers and do not convert high-molecular-weight species into monomers, as indicated by fewer interactions with low-molecular-weight monomeric species (Zunke et al., 2018). In addition, cholesterol accumulation was detected within the lysosomes of fibroblasts from patients with GBA mutant-associated PD (Garcia-Sanz et al., 2017). However, it is still unclear whether sphingolipids accumulate in other organelles and the exact location of sphingolipid accumulation. Mitochondria purified from GBA-knockout iPSC-derived neurons were demonstrated to have a significant accumulation of GlcCer and deacylated GlcCer glucosylsphingosine (Schondorf et al., 2018). Notably, only certain species of GlcCer were increased, with 65% increases in C16:0 and C24:0 species and a 30% reduction in C20:0 species (Fernandes et al., 2016). These data suggest that mutant GCase may promote the accumulation of certain GlcCer species and hydrolyze certain GlcCer species at the same time. However, the association between GBA mutations and lipid homeostasis remains controversial. Gegg et al. (2015) reported no changes in total GlcCer levels in the putamen or cerebellum in PD patients carrying GBA mutations compared with control individuals but observed loss of GCase activity. The putamen and cerebellum in PD patients with GBA mutations have previously been shown to exhibit decreased activity of GCase by 48 and 47%, respectively (Gegg et al., 2012). No GlcCer accumulation has been found in the brains of patients with sporadic PD, as GCase activity was decreased by 33% in the substantia nigra of patients with sporadic disease but decreased by 58% in the substantia nigra of GBA-PD patients (Gegg et al., 2012, 2015; Boutin et al., 2016). As lipidomic analyses were performed on postmortem brain tissue, the samples may have been contaminated with glia, which would discredit findings of GCase accumulation in neurons. Another explanation for the lack of lipid substrate accumulation upon loss of GCase activity is that the residual enzyme could prevent lipid accumulation. Further studies are needed to clarify whether GCase deficiency leads to GlcCer accumulation and whether functional compensatory pathways exist in mutant GCase cells.
Mitochondrial dysfunction has long been thought to participate in PD pathogenesis. Accumulating evidence has shown that mutant GCase is associated with mitochondrial defects in GD and PD. In GD mouse models, mitochondria showed impaired function and morphology, exhibiting respiratory chain defects, a decreased mitochondrial membrane potential due to reversal of ATPase, and fragmented mitochondrial morphology (Osellame et al., 2013). Like mice with GD, N370S fibroblasts exhibit fragmented mitochondria and increased reactive oxygen production (Garcia-Sanz et al., 2017). GCase inhibition in a human dopaminergic cell line resulted in increased free radical formation and mitochondrial dysfunction, including reduced mitochondrial membrane potential and decreased adenosine diphosphate phosphorylation (Cleeter et al., 2013). Consistent with these findings, iPSC-derived neurons from GBA-associated PD patients showed reduced respiration, increased morphological changes, higher levels of reactive oxygen species and defects in mitochondrial dynamics (Schondorf et al., 2018). Interestingly, a gene dosage effect was not observed between heterozygous GBA-PD and GBA-knockout neurons, suggesting that different pathways may contribute to mitochondrial dysfunction in heterozygous and homozygous GBA carriers. One such pathway may be involved in the activation of the unfolded protein response and increased ER stress in GBA-PD neurons, as the complete loss of GCase in GBA-knockout cells was incapable of triggering ER stress, supporting gain-of-function mechanisms (Fernandes et al., 2016; Schondorf et al., 2018). Similarly, Yun et al. (2018) found reduced mitochondrial size, increased reactive oxygen species production and decreased complex I activity in heterozygous L444P GBA-knock-in mice. The L444P GBA heterozygous mutation increased the susceptibility of mice to loss of nigrostriatal dopaminergic neurons and mitochondrial damage following MPTP administration (Yun et al., 2018). Under normal conditions, damaged and fragmented mitochondria are degraded via mitophagy, a specific form of autophagy, with serine/threonine kinase PTEN-induced putative kinase protein 1 (PINK1) and the ubiquitination E3 ligase parkin playing pivotal roles (Youle and Narendra, 2011). When mitochondria are damaged, PINK1 accumulates on the outer membrane in response to decreased membrane potential, thus triggering recruitment of parkin for the elimination of damaged mitochondria (Narendra et al., 2008; Twig et al., 2008). However, mutant GCase has been observed to impair mitophagy in GBA-mutant cells, leading to the accumulation of damaged mitochondria and the production of reactive oxygen species, subsequently leading to neurodegeneration and, eventually, to cell death. Osellame et al. (2013) found defective mitophagy in GBA-knockout neurons and astrocytes because the mitochondrial membrane potential in the GBA-knockout cells was not low enough for parkin recruitment, which is recruited to damaged mitochondria when the mitochondrial membrane potential is dissipated. Furthermore, Li et al. (2019) reported that the L444P GBA mutation impaired mitophagy via a dual mechanism, impairing autophagy induction and abrogating damaged mitochondria priming, processes involved in triggering autophagy machinery recruitment to damaged mitochondria. The L444P GBA mutation was impeded both PINK1-parkin-dependent and PINK1-parkin-independent pathways (Li et al., 2019).
Enzyme-replacement therapy (ERT) and substrate reduction therapy (SRT) are two Food and Drug Administration (FDA)-approved therapies for type I GD. ERT is now accepted as the first-line treatment of type I GD, and it is aimed at complementing deficient GCase enzyme activity upon intravenous administration (Shemesh et al., 2015). While ERT treatments significantly improve the visceral and hematologic symptoms of GD, current ERT treatments are ineffective against the neuropathic manifestations of GD since the enzyme does not cross the blood–brain barrier (BBB) (Boer et al., 2020). Thus, there are no data showing ERT treatments prevent the development of PD. To enhance ERT delivery to the CNS, Gramlich et al. (2016) tagged GCase with BBB-crossing peptides, and they effectively bound neurons, as expected. Another alternative is delivering therapeutic proteins through extracellular vesicles, such as exosomes, which are capable of transporting therapeutic proteins across the BBB (Hall et al., 2016). The system of delivering GCase across the BBB may increase the options for using ERT treatment to attenuate the neurological symptoms of GD or PD.
Substrate reduction therapy is accepted as the second-line treatment for GD with patients who are unsuitable for or unwilling to receive ERT; however, SRT leads to more side effects than ERT, the prominent of which are gastrointestinal and neurological symptoms such as onset of tremor or peripheral neuropathy (Cox et al., 2003; Hollak et al., 2009). SRT is administered to inhibit GlcCer synthase, reducing the level of lipid substrates, which effectively improves the hematological and visceral manifestations (Sardi et al., 2018). Miglustat and eliglustat are two SRT-approved drugs currently prescribed that can be administered orally (Shemesh et al., 2015; Mistry et al., 2017). Miglustat can partially cross the BBB and has been tested in an open-label randomized clinical trial for the treatment of GD type III; however, it did not significantly improve the neurological symptoms of GD type III (Schiffmann et al., 2008). SRT may be less effective than ERT in some aspects GD treatment; for example, hematological responses of miglustat are slower and of lesser magnitude compared to ERT, while the visceral symptom improvement obtained with miglustat is similar to that realized with ERT (Mistry et al., 2017). Furthermore, the novel GlcCer synthase inhibitor Genz-682452 has shown good CNS penetration and attenuation of several neuropathic and behavioral symptoms in GD mouse models (Marshall et al., 2016). Another brain-penetrating GlcCer synthase inhibitor, GZ667161, was found to reduce the levels of GlcCer and glucosylsphingosine in the CNS. Remarkably, prolonged administration of GZ667161 decreased α-synuclein accumulation and improved behavioral outcomes in synucleinopathy models (Sardi et al., 2017). Based on these studies, a multicenter, randomized, double-blind clinical study was launched to determine the safety and efficacy of the GlcCer synthase inhibitor GZ/SAR402671 in PD patients with GBA mutations (clinicalTrials.gov identifier: NCT02906020). These studies have provided promising therapeutic strategies for GBA-associated PD patients. However, SRT, which aims to reduce the levels of lipid substrates, does not prevent the other mechanisms that may play a role in the development of GBA-associated PD.
In recent years, gene therapy, which aims to deliver corrected genetic material into human cells, has emerged as a novel therapeutic strategy for PD (Hitti et al., 2019). Adeno-associated virus (AAV) vector-mediated delivery of GBA (using AAV-GBA) has been reported to be effective in augmenting GCase expression and protecting neurons against α-synuclein-mediated neuronal damage in mice and macaques (Rocha et al., 2015; Sucunza et al., 2021). AAVs are popular due to their strong neuronal tropism and good safety profiles, as they are not known to integrate into the host genome (Hudry and Vandenberghe, 2019). Research reported by Sardi et al. (2013) showed that augmenting GCase activity with an AAV vector encoding human GCase in GD mice reduced the levels of lipid glucosylsphingosine and aggregated proteins, such as α-synuclein, tau, and ubiquitin. Hippocampal administration of AAV-mediated GCase reversed the memory deficit of GD mice (Sardi et al., 2013). Massaro et al. (2018) demonstrated that fetal intracranial injection of AAV-GBA into GD mice elevated the level of GCase and ameliorated neuroinflammation and neurodegeneration. AAV-GBA gene therapy also ameliorated motor coordination and microglial and astrocyte activation, which correlated with neuronal loss, in fetal mice and macaques (Massaro et al., 2018). However, GCase normalization did not completely abolish GD pathology; that is, it did not completely normalize brain glycosphingolipid levels or prevent long-term microglial and astrocyte activation (Massaro et al., 2018). Based on these findings, different transduction efficiencies of individual neurons and peripheral macrophages in a CNS pathology model should be taken into consideration. In addition, intravenous injection of AAV-PHP. B, encoding GBA, a novel engineered AAV-GBA vector, restored the level of GCase, prevented α-synuclein inclusion formation, and recovered the loss of lifespan and cognitive performance in A53T-SNCA mice (Morabito et al., 2017). Remarkably, AAV-PHP. B targeting the BBB did not alter BBB integrity or selectivity (Morabito et al., 2017). Altogether, AAV-GBA gene therapy enables non-invasive and effective expression of GCase, possessing great potential as a therapeutic for GD and PD.
However, the route of delivery, optimal serotype, different transduction efficiencies of individual neurons, accessibility to widespread neuronal circuits and potential side effects of long-term treatment with GCase need be investigated before AAV-GBA gene therapy is translated into the clinic.
Under normal conditions, GCase is subject to ER repair machinery that corrects folding or transported to lysosomes. When GBA is mutated, the number of misfolded proteins exceeds the ER capacity, which triggers the unfolded protein response and ER-associated degradation, through which misfolded enzymes are broken down, leading to a reduction in GCase protein resident in the lysosome (Meusser et al., 2005; Bendikov-Bar et al., 2011; Schapira and Gegg, 2013). Pharmacological small-molecule chaperones may selectively bind to mutant GCase and stabilize and correct misfolded GCase to facilitate protein tracking through the ER to lysosomes (Chen et al., 2020). Small-molecule chaperones also have the ability to prevent ER stress, subsequently attenuating apoptosis and mitochondrial dysfunction (de la Mata et al., 2015). Small-molecule chaperones can be divided into inhibitory chaperones, which interact with the active site of an enzyme, and non-inhibitory chaperones, which primarily enhance enzymatic activity.
Most small-molecule chaperones developed in recent studies are inhibitory chaperones that bind to the active site of misfolded GCase and facilitate its correct folding or translocation to lysosomes (Jung et al., 2016). However, when mutant GCase and its bound inhibitor reach lysosomes, the substrate GlcCer out-competes the inhibitor to optimize substrate degradation (Aflaki et al., 2017). This competition between inhibitors and substrates makes it a challenge to optimize drug dosing to balance the chaperoning and inhibitory capacity of inhibitory small-molecule chaperones for clinical applications. From this drug collection, ambroxol and isofagomine have shown their potential and have been investigated in preclinical and early-stage clinical studies. Ambroxol is a potential inhibitory chaperone candidate that has been selected via high-throughput screening of a library of FDA-approved drugs (Maegawa et al., 2009). Ambroxol, a cough medicine widely used to treat airway mucus hypersecretion and hyaline membrane disease in newborns, was identified as a mixed inhibitor of GCase activity. Ambroxol exerts its maximal activity in the ER where the pH is neutral and loses its binding affinity in lysosomes where the pH is acidic (Maegawa et al., 2009). This pH dependence of ambroxol prevents degradation blockades of the substrate, making ambroxol a desirable small molecule. Ambroxol has been demonstrated to increase GCase activity and lysosomal localization of mutant GCase significantly in various in vitro and in vivo models, such as patient cells (Bendikov-Bar et al., 2013; Luan et al., 2013; McNeill et al., 2014; Yang et al., 2017), mice (Sanders et al., 2013; Migdalska-Richards et al., 2016), and non-human primates (Migdalska-Richards et al., 2017a), in independent studies. In an open-label, non-randomized, non-controlled clinical trial with 17 PD patients, ambroxol crossed the BBB and bound to GCase and increased GCase protein levels and α-synuclein concentrations in the cerebrospinal fluid in patients both with and without GBA mutations, and it induced no serious adverse effects (Mullin et al., 2020). In addition, another clinical trial on ambroxol as a treatment for PD dementia is currently underway (clinicalTrials.gov identifier: NCT02914366). Another inhibitory chaperone is the iminosugar isofagomine, which binds and stabilizes GCase and increases cellular and lysosomal GCase levels (Lieberman et al., 2007). Treatment of patient cells and mice with isofagomine increased GCase activity in brain and visceral tissue, reduced GlcCer levels, attenuated proinflammatory responses, delayed the onset of neurological disease and extended the lifespan (Khanna et al., 2010; Sun et al., 2012). However, isofagomine did not lead to significant clinical improvement in 18 patients of a GD clinical study, and therefore, further development was discontinued, suggesting that more elaborate early-stage clinical trials are needed to ensure the safety and efficiency of isofagomine in GD or PD patients.
A major limitation of inhibitory chaperones is the optimization of drug dosing to balance their chaperoning and inhibitory capacities, as discussed in the previous section. Non-inhibitory chaperones promote the folding of mutant GCase in the ER and translocate it to lysosomes by binding to a site that differs from the active site, thus directly inducing the residual activity of mutant GCase (Baden et al., 2019), making non-inhibitory chaperones promising candidate pharmacological targets. A novel non-inhibitory GCase chaperone, NCGC607, which was identified by high-throughput screening, was demonstrated to restore GCase activity and reduce glycolipid storage in dopaminergic neurons of patients with GD, highlighting the potential of NCGC607 treatment for GD (Aflaki et al., 2016). Additionally, NCGC607 was capable of reducing α-synuclein levels in dopaminergic neurons of patients with parkinsonism, indicating its potential as a therapy for PD (Aflaki et al., 2016). Another non-inhibitory small-molecule chaperone, NCGC758, was reported to enhance GCase activity and reduce the level of GlcCer, ultimately enhancing the clearance of α-synuclein in iPSC-derived dopaminergic neurons of PD and GD patients (Mazzulli et al., 2016). Overall, pharmacological small-molecule chaperones have exhibited some advantages over standard therapy, including ERT and SRT. Small-molecule chaperones not only exhibit the ability to cross the BBB but can also be orally administered and are inexpensive to manufacture.
GBA mutations are the most significant and common genetic risk factors associated with PD and are especially common in the Ashkenazi Jewish population. The mechanism by which GBA mutations result in an increased risk of developing PD remains unclear. Several mechanisms have been suggested to contribute to the pathogenesis of GBA-associated PD, including enhanced α-synuclein aggregation, autophagy-lysosomal dysfunction, altered lipid homeostasis and mitochondrial dysfunction. Both gain-of-function and loss-of-function explanations have been proposed to describe the pathogenesis of GBA-associated PD. The gain-of-function theory suggests that mutant GCase increases α-synuclein aggregation, leading to neurodegeneration. Alternatively, enhanced α-synuclein aggregation results in lysosomal dysfunction or impairment of autophagy, subsequently contributing to the development of parkinsonism. The loss-of-function theory posits that parkinsonism arises as a result of GCase deficiency, which influences lysosomal degradation function, negatively affecting α-synuclein turnover and substrate degradation. However, each of the models and potential pathways attributed to these theories has limitations. First, neither theory adequately explains the reason that only a fraction of GD patients and GBA carriers develop PD. Second, some patients with PD express GBA-null alleles; for example, those with c.84dupG also develop PD, in conflict with the gain-of-function theory. GBA carriers with null alleles may exhibit a higher risk of developing PD (Gan-Or et al., 2015). The current mechanisms can only be supported when GBA is mutated and has an enhanced effect on neurodegeneration but is not the initiator of pathogenesis. Other factors may contribute and may help determine whether mutant GBA can disrupt the cellular homeostatic system and subsequent α-synuclein pathology and PD, to an extent. Alternatively, GBA may act as a ‘second hit’ in some carriers and patients who are genetically predisposed to developing PD in the presence of other factors, such as aging and environmental factors.
The activity of GCase is diminished in the substantia nigra of brains with GBA mutations; however, it is also decreased in sporadic PD brains (Gegg et al., 2012). These findings suggest that therapies modulating GCase may not only be applied for the treatment of GBA-associated PD but may also provide a novel therapeutic target for sporadic PD. Importantly, AAV-mediated GCase expression reduced the accumulation of substrate and α-synuclein in both early and late symptomatic GD mice and was effective in reversing cognitive impairment when added before or after the protein aggregate (Sardi et al., 2013). These data demonstrate that modulating GCase activity may be beneficial before or after the diagnosis of PD to prevent the onset of PD or impede the progression of some aspects of GD-associated parkinsonism and PD. In general, the understanding of the function of GCase and the link between GBA mutations, α-synuclein accumulation and PD have made a significant contribution to the thinking on the pathogenesis of PD, and modulation of GCase may be beneficial to all PD patients, GD patients and those with other synucleinopathies. Although many studies have focused on the association between GBA mutations and PD, the GCase story may remain incomplete. Further exploration into the role of this enzyme in PD pathogenesis, manipulation of the GCase pathway, and potential genetic modifications may enable us to better understand the pathogenetic factors in the etiology of PD.
WZ designed and prepared the manuscript. DF supervised the manuscript. Both authors contributed to the article and approved the submitted version.
This work was funded by the National Natural Science Foundation of China (81873784 and 82071426) and the Clinical Cohort Construction Program of Peking University Third Hospital (BYSYDL 2019002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aflaki, E., Borger, D. K., Moaven, N., Stubblefield, B. K., Rogers, S. A., Patnaik, S., et al. (2016). A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-Derived dopaminergic neurons from patients with gaucher disease and parkinsonism. J. Neurosci. 36, 7441–7452. doi: 10.1523/JNEUROSCI.0636-16.2016
Aflaki, E., Westbroek, W., and Sidransky, E. (2017). The complicated relationship between gaucher disease and parkinsonism: insights from a rare disease. Neuron 93, 737–746. doi: 10.1016/j.neuron.2017.01.018
Aharon-Peretz, J., Rosenbaum, H., and Gershoni-Baruch, R. (2004). Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 351, 1972–1977. doi: 10.1056/NEJMoa033277
Alcalay, R. N., Dinur, T., Quinn, T., Sakanaka, K., Levy, O., Waters, C., et al. (2014). Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 71, 752–757. doi: 10.1001/jamaneurol.2014.313
Awad, O., Sarkar, C., Panicker, L. M., Miller, D., Zeng, X., Sgambato, J. A., et al. (2015). Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 24, 5775–5788. doi: 10.1093/hmg/ddv297
Baden, P., Yu, C., and Deleidi, M. (2019). Insights into GBA Parkinson’s disease pathology and therapy with induced pluripotent stem cell model systems. Neurobiol. Dis. 127, 1–12. doi: 10.1016/j.nbd.2019.01.023
Bae, E. J., Yang, N. Y., Song, M., Lee, C. S., Lee, J. S., Jung, B. C., et al. (2014). Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat. Commun. 5:4755. doi: 10.1038/ncomms5755
Balestrino, R., and Schapira, A. H. V. (2018). Glucocerebrosidase and Parkinson Disease: Molecular, Clinical, and Therapeutic Implications. Neuroscientist 24, 540–559. doi: 10.1177/1073858417748875
Bendikov-Bar, I., Maor, G., Filocamo, M., and Horowitz, M. (2013). Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 50, 141–145. doi: 10.1016/j.bcmd.2012.10.007
Bendikov-Bar, I., Ron, I., Filocamo, M., and Horowitz, M. (2011). Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 46, 4–10. doi: 10.1016/j.bcmd.2010.10.012
Boer, D. E. C., van Smeden, J., Bouwstra, J. A., and Aerts, J. (2020). Glucocerebrosidase: Functions in and Beyond the Lysosome. J. Clin. Med. 9:736. doi: 10.3390/jcm9030736
Boutin, M., Sun, Y., Shacka, J. J., and Auray-Blais, C. (2016). Tandem mass spectrometry multiplex analysis of glucosylceramide and galactosylceramide isoforms in brain tissues at different stages of parkinson disease. Anal. Chem. 88, 1856–1863. doi: 10.1021/acs.analchem.5b04227
Brockmann, K., Srulijes, K., Hauser, A. K., Schulte, C., Csoti, I., Gasser, T., et al. (2011). GBA-associated PD presents with nonmotor characteristics. Neurology 77, 276–280. doi: 10.1212/WNL.0b013e318225ab77
Chen, Y., Sam, R., Sharma, P., Chen, L., Do, J., and Sidransky, E. (2020). Glucocerebrosidase as a therapeutic target for Parkinson’s disease. Expert. Opin. Ther. Targets 24, 287–294. doi: 10.1080/14728222.2020.1733970
Chun, Y., and Kim, J. (2018). Autophagy: an essential degradation program for cellular homeostasis and life. Cells 7:278. doi: 10.3390/cells7120278
Cilia, R., Tunesi, S., Marotta, G., Cereda, E., Siri, C., Tesei, S., et al. (2016). Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 80, 662–673. doi: 10.1002/ana.24777
Clark, L. N., Nicolai, A., Afridi, S., Harris, J., Mejia-Santana, H., Strug, L., et al. (2005). Pilot association study of the beta-glucocerebrosidase N370S allele and Parkinson’s disease in subjects of Jewish ethnicity. Mov. Disord. 20, 100–103. doi: 10.1002/mds.20320
Clark, L. N., Ross, B. M., Wang, Y., Mejia-Santana, H., Harris, J., Louis, E. D., et al. (2007). Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69, 1270–1277. doi: 10.1212/01.wnl.0000276989.17578.02
Cleeter, M. W., Chau, K. Y., Gluck, C., Mehta, A., Hughes, D. A., Duchen, M., et al. (2013). Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 62, 1–7. doi: 10.1016/j.neuint.2012.10.010
Cox, T. M., Aerts, J. M., Andria, G., Beck, M., Belmatoug, N., Bembi, B., et al. (2003). The role of the iminosugar N-butyldeoxynojirimycin (miglustat) in the management of type I (non-neuronopathic) Gaucher disease: a position statement. J. Inherit. Metab. Dis. 26, 513–526. doi: 10.1023/a:1025902113005
Creese, B., Bell, E., Johar, I., Francis, P., Ballard, C., and Aarsland, D. (2018). Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson’s disease and Lewy body dementias: review and meta-analyses. Am. J. Med. Genet. B Neuropsychiatr. Genet. 177, 232–241. doi: 10.1002/ajmg.b.32549
de la Mata, M., Cotan, D., Oropesa-Avila, M., Garrido-Maraver, J., Cordero, M. D., Villanueva Paz, M., et al. (2015). Pharmacological Chaperones and Coenzyme Q10 treatment improves mutant beta-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of gaucher disease. Sci. Rep. 5:10903. doi: 10.1038/srep10903
Dehay, B., Martinez-Vicente, M., Caldwell, G. A., Caldwell, K. A., Yue, Z., Cookson, M. R., et al. (2013). Lysosomal impairment in Parkinson’s disease. Mov. Disord. 28, 725–732. doi: 10.1002/mds.25462
Do, J., McKinney, C., Sharma, P., and Sidransky, E. (2019). Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 14:36. doi: 10.1186/s13024-019-0336-2
Du, T. T., Wang, L., Duan, C. L., Lu, L. L., Zhang, J. L., Gao, G., et al. (2015). GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 11, 1803–1820. doi: 10.1080/15548627.2015.1086055
Fernandes, H. J., Hartfield, E. M., Christian, H. C., Emmanoulidou, E., Zheng, Y., Booth, H., et al. (2016). ER stress and autophagic perturbations lead to elevated extracellular alpha-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 6, 342–356. doi: 10.1016/j.stemcr.2016.01.013
Franco, R., Sanchez-Arias, J. A., Navarro, G., and Lanciego, J. L. (2018). Glucocerebrosidase mutations and synucleinopathies. potential role of sterylglucosides and relevance of studying both GBA1 and GBA2 Genes. Front. Neuroanat. 12:52. doi: 10.3389/fnana.2018.00052
Gan-Or, Z., Amshalom, I., Kilarski, L. L., Bar-Shira, A., Gana-Weisz, M., Mirelman, A., et al. (2015). Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84, 880–887. doi: 10.1212/WNL.0000000000001315
Gan-Or, Z., Bar-Shira, A., Mirelman, A., Gurevich, T., Kedmi, M., Giladi, N., et al. (2010). LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson disease. Neurogenetics 11, 121–125. doi: 10.1007/s10048-009-0198-9
Gan-Or, Z., Giladi, N., Rozovski, U., Shifrin, C., Rosner, S., Gurevich, T., et al. (2008). Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70, 2277–2283. doi: 10.1212/01.wnl.0000304039.11891.29
Gan-Or, Z., Liong, C., and Alcalay, R. N. (2018). GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 18:44. doi: 10.1007/s11910-018-0860-4
García-Sanz, P., Aerts, M. F. G., and Moratalla, R. (2021). The role of cholesterol in alpha-synuclein and lewy body pathology in GBA1 Parkinson’s Disease. Mov. Disord. 36, 1070–1085. doi: 10.1002/mds.28396
Garcia-Sanz, P., Orgaz, L., Bueno-Gil, G., Espadas, I., Rodriguez-Traver, E., Kulisevsky, J., et al. (2017). N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov. Disord. 32, 1409–1422. doi: 10.1002/mds.27119
Gegg, M. E., Burke, D., Heales, S. J., Cooper, J. M., Hardy, J., Wood, N. W., et al. (2012). Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 72, 455–463. doi: 10.1002/ana.23614
Gegg, M. E., Sweet, L., Wang, B. H., Shihabuddin, L. S., Sardi, S. P., and Schapira, A. H. (2015). No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 30, 1085–1089. doi: 10.1002/mds.26278
Goker-Alpan, O., Stubblefield, B. K., Giasson, B. I., and Sidransky, E. (2010). Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 120, 641–649. doi: 10.1007/s00401-010-0741-7
Gomez-Benito, M., Granado, N., Garcia-Sanz, P., Michel, A., Dumoulin, M., and Moratalla, R. (2020). Modeling parkinson’s disease with the alpha-synuclein protein. Front. Pharmacol. 11:356. doi: 10.3389/fphar.2020.00356
Grabowski, G. A. (2008). Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 372, 1263–1271. doi: 10.1016/S0140-6736(08)61522-6
Grabowski, G. A., Zimran, A., and Ida, H. (2015). Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Hematol. 90, (Suppl. 1), S12–S18. doi: 10.1002/ajh.24063
Gramlich, P. A., Westbroek, W., Feldman, R. A., Awad, O., Mello, N., Remington, M. P., et al. (2016). A peptide-linked recombinant glucocerebrosidase for targeted neuronal delivery: Design, production, and assessment. J. Biotechnol. 221, 1–12. doi: 10.1016/j.jbiotec.2016.01.015
Hall, J., Prabhakar, S., Balaj, L., Lai, C. P., Cerione, R. A., and Breakefield, X. O. (2016). Delivery of therapeutic proteins via extracellular vesicles: review and potential treatments for parkinson’s disease, glioma, and schwannoma. Cell Mol. Neurobiol. 36, 417–427. doi: 10.1007/s10571-015-0309-0
Hitti, F. L., Yang, A. I., Gonzalez-Alegre, P., and Baltuch, G. H. (2019). Human gene therapy approaches for the treatment of Parkinson’s disease: An overview of current and completed clinical trials. Parkinsonism Relat. Disord. 66, 16–24. doi: 10.1016/j.parkreldis.2019.07.018
Hollak, C. E., Hughes, D., van Schaik, I. N., Schwierin, B., and Bembi, B. (2009). Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol. Drug Saf. 18, 770–777. doi: 10.1002/pds.1779
Hudry, E., and Vandenberghe, L. H. (2019). Therapeutic AAV Gene transfer to the nervous system: a clinical reality. Neuron 101, 839–862. doi: 10.1016/j.neuron.2019.02.017
Jung, O., Patnaik, S., Marugan, J., Sidransky, E., and Westbroek, W. (2016). Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert. Rev. Proteomics 13, 471–479. doi: 10.1080/14789450.2016.1174583
Khanna, R., Benjamin, E. R., Pellegrino, L., Schilling, A., Rigat, B. A., Soska, R., et al. (2010). The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. FEBS J. 277, 1618–1638. doi: 10.1111/j.1742-4658.2010.07588.x
Kim, S., Yun, S. P., Lee, S., Umanah, G. E., Bandaru, V. V. R., Yin, X., et al. (2018). GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 115, 798–803. doi: 10.1073/pnas.1700465115
Lawrence, R. E., and Zoncu, R. (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142. doi: 10.1038/s41556-018-0244-7
Lesage, S., Anheim, M., Condroyer, C., Pollak, P., Durif, F., Dupuits, C., et al. (2011). Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 20, 202–210. doi: 10.1093/hmg/ddq454
Li, H., Ham, A., Ma, T. C., Kuo, S. H., Kanter, E., Kim, D., et al. (2019). Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 15, 113–130. doi: 10.1080/15548627.2018.1509818
Lieberman, R. L., Wustman, B. A., Huertas, P., Powe, A. C. Jr., Pine, C. W., Khanna, R., et al. (2007). Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 3, 101–107. doi: 10.1038/nchembio850
Luan, Z., Li, L., Higaki, K., Nanba, E., Suzuki, Y., and Ohno, K. (2013). The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev. 35, 317–322. doi: 10.1016/j.braindev.2012.05.008
Lwin, A., Orvisky, E., Goker-Alpan, O., LaMarca, M. E., and Sidransky, E. (2004). Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 81, 70–73. doi: 10.1016/j.ymgme.2003.11.004
Maegawa, G. H., Tropak, M. B., Buttner, J. D., Rigat, B. A., Fuller, M., Pandit, D., et al. (2009). Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 284, 23502–23516. doi: 10.1074/jbc.M109.012393
Manning-Bog, A. B., Schule, B., and Langston, J. W. (2009). Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 30, 1127–1132. doi: 10.1016/j.neuro.2009.06.009
Marshall, J., Sun, Y., Bangari, D. S., Budman, E., Park, H., Nietupski, J. B., et al. (2016). CNS-accessible inhibitor of glucosylceramide synthase for substrate reduction therapy of neuronopathic gaucher disease. Mol. Ther. 24, 1019–1029. doi: 10.1038/mt.2016.53
Massaro, G., Mattar, C. N. Z., Wong, A. M. S., Sirka, E., Buckley, S. M. K., Herbert, B. R., et al. (2018). Fetal gene therapy for neurodegenerative disease of infants. Nat. Med. 24, 1317–1323. doi: 10.1038/s41591-018-0106-7
Mata, I. F., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., Van Deerlin, V. M., et al. (2016). GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 31, 95–102. doi: 10.1002/mds.26359
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Mazzulli, J. R., Zunke, F., Tsunemi, T., Toker, N. J., Jeon, S., Burbulla, L. F., et al. (2016). Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in parkinson’s patient midbrain neurons. J. Neurosci. 36, 7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016
McNeill, A., Magalhaes, J., Shen, C., Chau, K. Y., Hughes, D., Mehta, A., et al. (2014). Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137, 1481–1495. doi: 10.1093/brain/awu020
Meusser, B., Hirsch, C., Jarosch, E., and Sommer, T. (2005). ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772. doi: 10.1038/ncb0805-766
Migdalska-Richards, A., Daly, L., Bezard, E., and Schapira, A. H. (2016). Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann. Neurol. 80, 766–775. doi: 10.1002/ana.24790
Migdalska-Richards, A., Ko, W. K. D., Li, Q., Bezard, E., and Schapira, A. H. V. (2017a). Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 71:21967. doi: 10.1002/syn.21967
Migdalska-Richards, A., Wegrzynowicz, M., Rusconi, R., Deangeli, G., Di Monte, D. A., Spillantini, M. G., et al. (2017b). The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain 140, 2706–2721. doi: 10.1093/brain/awx221
Mistry, P. K., Lopez, G., Schiffmann, R., Barton, N. W., Weinreb, N. J., and Sidransky, E. (2017). Gaucher disease: progress and ongoing challenges. Mol. Genet. Metab. 120, 8–21. doi: 10.1016/j.ymgme.2016.11.006
Morabito, G., Giannelli, S. G., Ordazzo, G., Bido, S., Castoldi, V., Indrigo, M., et al. (2017). AAV-PHP.B-Mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol. Ther. 25, 2727–2742. doi: 10.1016/j.ymthe.2017.08.004
Mullin, S., Smith, L., Lee, K., D’Souza, G., Woodgate, P., Elflein, J., et al. (2020). Ambroxol for the treatment of patients with parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. 77, 427–434. doi: 10.1001/jamaneurol.2019.4611
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Neudorfer, O., Giladi, N., Elstein, D., Abrahamov, A., Turezkite, T., Aghai, E., et al. (1996). Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 89, 691–694. doi: 10.1093/qjmed/89.9.691
Neumann, J., Bras, J., Deas, E., O’Sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132(Pt 7), 1783–1794. doi: 10.1093/brain/awp044
Nichols, W. C., Pankratz, N., Marek, D. K., Pauciulo, M. W., Elsaesser, V. E., Halter, C. A., et al. (2009). Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72, 310–316. doi: 10.1212/01.wnl.0000327823.81237.d1
Obeso, J. A., Stamelou, M., Goetz, C. G., Poewe, W., Lang, A. E., Weintraub, D., et al. (2017). Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 32, 1264–1310. doi: 10.1002/mds.27115
O’Regan, G., deSouza, R. M., Balestrino, R., and Schapira, A. H. (2017). Glucocerebrosidase Mutations in Parkinson Disease. J. Parkinsons Dis. 7, 411–422. doi: 10.3233/JPD-171092
Osellame, L. D., Rahim, A. A., Hargreaves, I. P., Gegg, M. E., Richard-Londt, A., Brandner, S., et al. (2013). Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson’s disease. Cell Metab. 17, 941–953. doi: 10.1016/j.cmet.2013.04.014
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Brown, E., Hayes, M. A., et al. (2015). Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 82, 495–503. doi: 10.1016/j.nbd.2015.09.009
Ron, I., and Horowitz, M. (2005). ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 14, 2387–2398. doi: 10.1093/hmg/ddi240
Sanders, A., Hemmelgarn, H., Melrose, H. L., Hein, L., Fuller, M., and Clarke, L. A. (2013). Transgenic mice expressing human glucocerebrosidase variants: utility for the study of Gaucher disease. Blood Cells Mol. Dis. 51, 109–115. doi: 10.1016/j.bcmd.2013.03.006
Sardi, S. P., Cedarbaum, J. M., and Brundin, P. (2018). Targeted therapies for parkinson’s disease: from genetics to the clinic. Mov. Disord. 33, 684–696. doi: 10.1002/mds.27414
Sardi, S. P., Clarke, J., Viel, C., Chan, M., Tamsett, T. J., Treleaven, C. M., et al. (2013). Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 110, 3537–3542. doi: 10.1073/pnas.1220464110
Sardi, S. P., Viel, C., Clarke, J., Treleaven, C. M., Richards, A. M., Park, H., et al. (2017). Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 114, 2699–2704. doi: 10.1073/pnas.1616152114
Schapira, A. H., Chiasserini, D., Beccari, T., and Parnetti, L. (2016). Glucocerebrosidase in Parkinson’s disease: Insights into pathogenesis and prospects for treatment. Mov. Disord. 31, 830–835. doi: 10.1002/mds.26616
Schapira, A. H., and Gegg, M. E. (2013). Glucocerebrosidase in the pathogenesis and treatment of Parkinson disease. Proc. Natl. Acad. Sci. USA 110, 3214–3215. doi: 10.1073/pnas.1300822110
Schiffmann, R., Fitzgibbon, E. J., Harris, C., DeVile, C., Davies, E. H., Abel, L., et al. (2008). Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann. Neurol. 64, 514–522. doi: 10.1002/ana.21491
Schondorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5:4028. doi: 10.1038/ncomms5028
Schondorf, D. C., Ivanyuk, D., Baden, P., Sanchez-Martinez, A., De Cicco, S., Yu, C., et al. (2018). The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 23, 2976–2988. doi: 10.1016/j.celrep.2018.05.009
Seto-Salvia, N., Pagonabarraga, J., Houlden, H., Pascual-Sedano, B., Dols-Icardo, O., Tucci, A., et al. (2012). Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov. Disord. 27, 393–399. doi: 10.1002/mds.24045
Shemesh, E., Deroma, L., Bembi, B., Deegan, P., Hollak, C., Weinreb, N. J., et al. (2015). Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst. Rev. 3:324. doi: 10.1002/14651858.CD010324
Sidransky, E. (2004). Gaucher disease: complexity in a “simple” disorder. Mol. Genet. Metab. 83, 6–15. doi: 10.1016/j.ymgme.2004.08.015
Sidransky, E., and Lopez, G. (2012). The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998. doi: 10.1016/S1474-4422(12)70190-4
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Stirnemann, J., Belmatoug, N., Camou, F., Serratrice, C., Froissart, R., Caillaud, C., et al. (2017). A review of gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 18:441. doi: 10.3390/ijms18020441
Sucunza, D., Rico, A. J., Roda, E., Collantes, M., Gonzalez-Aseguinolaza, G., Rodriguez-Perez, A. I., et al. (2021). Glucocerebrosidase gene therapy induces alpha-synuclein clearance and neuroprotection of midbrain dopaminergic neurons in mice and macaques. Int. J. Mol. Sci. 22:94825. doi: 10.3390/ijms22094825
Sun, Y., Liou, B., Xu, Y. H., Quinn, B., Zhang, W., Hamler, R., et al. (2012). Ex vivo and in vivo effects of isofagomine on acid beta-glucosidase variants and substrate levels in Gaucher disease. J. Biol. Chem. 287, 4275–4287. doi: 10.1074/jbc.M111.280016
Swan, M., and Saunders-Pullman, R. (2013). The association between ss-glucocerebrosidase mutations and parkinsonism. Curr. Neurol. Neurosci. Rep. 13:368. doi: 10.1007/s11910-013-0368-x
Tayebi, N., Walker, J., Stubblefield, B., Orvisky, E., LaMarca, M. E., Wong, K., et al. (2003). Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 79, 104–109. doi: 10.1016/s1096-7192(03)00071-4
Toft, M., Pielsticker, L., Ross, O. A., Aasly, J. O., and Farrer, M. J. (2006). Glucocerebrosidase gene mutations and Parkinson disease in the Norwegian population. Neurology 66, 415–417. doi: 10.1212/01.wnl.0000196492.80676.7c
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Weiss, K., Gonzalez, A., Lopez, G., Pedoeim, L., Groden, C., and Sidransky, E. (2015). The clinical management of Type 2 Gaucher disease. Mol. Genet. Metab. 114, 110–122. doi: 10.1016/j.ymgme.2014.11.008
Westbroek, W., Gustafson, A. M., and Sidransky, E. (2011). Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 17, 485–493. doi: 10.1016/j.molmed.2011.05.003
Winder-Rhodes, S. E., Evans, J. R., Ban, M., Mason, S. L., Williams-Gray, C. H., Foltynie, T., et al. (2013). Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 136(Pt 2), 392–399. doi: 10.1093/brain/aws318
Woodard, C. M., Campos, B. A., Kuo, S. H., Nirenberg, M. J., Nestor, M. W., Zimmer, M., et al. (2014). iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease. Cell Rep. 9, 1173–1182. doi: 10.1016/j.celrep.2014.10.023
Yang, S. Y., Beavan, M., Chau, K. Y., Taanman, J. W., and Schapira, A. H. V. (2017). A Human Neural Crest Stem Cell-Derived dopaminergic neuronal model recapitulates biochemical abnormalities in GBA1 Mutation Carriers. Stem Cell Rep. 8, 728–742. doi: 10.1016/j.stemcr.2017.01.011
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Yun, S. P., Kim, D., Kim, S., Kim, S., Karuppagounder, S. S., Kwon, S. H., et al. (2018). alpha-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 13:1. doi: 10.1186/s13024-017-0233-5
Keywords: glucocerebrosidase (GBA), mitochondrial, lysosomal, Parkinson’s disease (PD), Gaucher’s disease (GD)
Citation: Zheng W and Fan D (2022) Glucocerebrosidase Mutations Cause Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease: Pathogenesis and Therapeutic Implications. Front. Aging Neurosci. 14:851135. doi: 10.3389/fnagi.2022.851135
Received: 09 January 2022; Accepted: 14 February 2022;
Published: 23 March 2022.
Edited by:
Xiaolei Zhu, Nanjing Drum Tower Hospital, ChinaReviewed by:
Rosario Moratalla, Consejo Superior de Investigaciones Científicas (CSIC), SpainCopyright © 2022 Zheng and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongsheng Fan, ZHNmYW4yMDEwQGFsaXl1bi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.