 Lars Tönges
Lars Tönges Eun Hae Kwon1
Eun Hae Kwon1 Stephan Klebe
Stephan Klebe
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 03 March 2022
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.822949
This article is part of the Research Topic New Challenges and Future Perspectives in Parkinson's disease and age-related movement disorders View all 11 articles
The therapy of neurodegenerative diseases such as Parkinson’s disease (PD) is still limited to the treatment of symptoms and primarily aimed at compensating for dopaminergic hypofunction. Numerous disease-modifying therapies currently in the pipeline attempt to modify the underlying pathomechanisms. In recent decades, the results of molecular genetics and biomarker research have raised hopes of earlier diagnosis and new neuroprotective therapeutic approaches. As the disease-causing processes in monogenetic forms of PD are better understood than in sporadic PD, these disease subsets are likely to benefit first from disease-modifying therapies. Recent studies have suggested that disease-relevant changes found in genetically linked forms of PD (i.e., PARK-LRRK2, PARK-GBA) can also be reproduced in patients in whom no genetic cause can be found, i.e., those with sporadic PD. It can, therefore, be assumed that as soon as the first causal therapy for genetic forms of PD is approved, more patients with PD will undergo genetic testing and counseling. Regarding future neuroprotective trials in neurodegenerative diseases and objective parameters such as biomarkers with high sensitivity and specificity for the diagnosis and course of the disease are needed. These biomarkers will also serve to monitor treatment success in clinical trials. Promising examples in PD, such as alpha-synuclein species, lysosomal enzymes, markers of amyloid and tau pathology, and neurofilament light chain, are under investigation in blood and CSF. This paper provides an overview of the opportunities and current limitations of monogenetic diagnostic and biomarker research in PD and aims to build a bridge between current knowledge and association with PD genetics and biomarkers.
Parkinson’s disease (PD) [sporadic (sPD) and (mono)genetic forms of PD (gPD)] represents a heterogeneous group of disorders with the pathophysiological shared end of a dopaminergic deficit. Therapy of PD is still limited to the treatment of symptoms and is primarily aimed at compensating for dopaminergic hypofunction. Accurate diagnosis of PD according to the underlying pathophysiology is important because of current and future treatment options. In recent years, many efforts have been made to identify specific and sensitive biomarkers (Parnetti et al., 2019; Lawton et al., 2020). Because of its proximity to neuronal structures, cerebrospinal fluid (CSF) is, of course, the most promising liquid biomarker for neurodegenerative diseases. Nevertheless, blood, saliva, skin biopsies, and urine also appear to be good candidates as they are easily accessible and less invasive. Biomarkers from body fluids, skin biopsies, or imaging studies have the advantage of identifying the risk for future PD, diagnosing PD early, and monitoring disease progression, and can serve to monitor possible treatment success in clinical trials. Hence, biomarkers should also be able to distinguish between different entities of neurodegenerative diseases [e.g., sPD vs. gPD vs. atypical Parkinson’s syndromes (aPD)] to decrease the number of false-positive diagnosed patients. An unmet need for future neuroprotective trials in neurodegenerative diseases is an objective parameter such as biomarkers with high specificity and sensibility for the diagnosis and course of the disease. Promising examples in PD, such as alpha-synuclein species, lysosomal enzymes, markers of amyloid and tau pathology, and neurofilament light chain, are under investigation in the blood and CSF (Parnetti et al., 2019). Unfortunately, it has not been possible to date to implement a robust biomarker in the early diagnosis or follow-up of PD. One reason is that biomarker profiles vary from patient to patient due to the great heterogeneity of the disease.
In contrast to sPD, in which the etiology remains elusive for most cases, gPD offer the possibility of better understanding the cause of the disease. Many autosomal dominant and recessive genes have been found since the first description of SNCA as a genetic cause of PD in 1997 (Polymeropoulos et al., 1997). The clinical symptoms between sPD and a monogenetic form are similar in most cases. However, since the course of PD can differ greatly from individual to individual, the question arises whether the etiological heterogeneity might have different causes. Examples include the tremor-dominant or akinetic rigid subtypes of sPD or the early cognitive involvement in some patients with PD. The results of molecular genetics and biomarker research have raised hopes of resolving these unanswered questions in PD and finding new neuroprotective therapeutic approaches. As the disease-causing processes in gPD are better understood than in sPD, patients with these subsets of the disease are likely to benefit first from disease-modifying therapies. A synergistic effect between research on monogenetic forms of the disease and sPD is also likely. Recent studies have suggested that disease-relevant changes found in genetically linked forms of PD (i.e., PARK-LRRK2, PARK-GBA) can also be reproduced in patients in whom no genetic cause can be found (Di Maio et al., 2018). Even if these patients with gPD currently represent only a small group (approximately 10%) (Cook et al., 2021), it is expected that availability of a causal therapy for genetic forms of PD will substantially increase demand for genetic testing and counseling. Therefore, we aim to provide an overview of the most common genetically linked forms of PD and the associated biomarker studies. Table 1 summarizes the genetic causes of PD with regard to the corresponding biomarker.
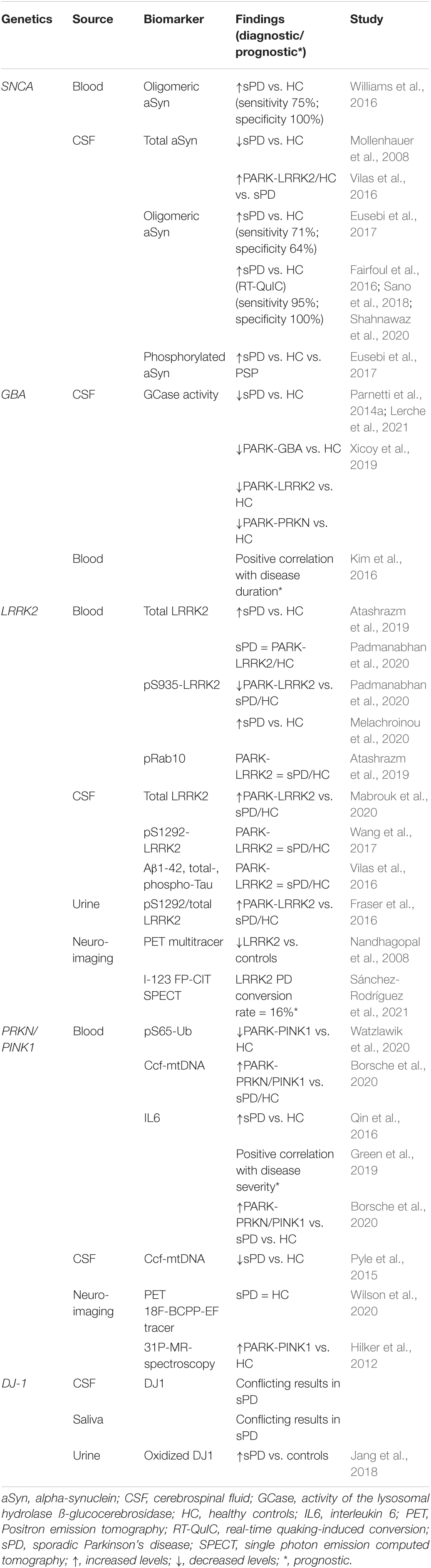
Table 1. An overview of the genetic causes of Parkinson’s disease (PD), the corresponding biomarker, and the diagnostic and/or prognostic value.
Alpha-synuclein (aSyn) is a rather small protein (14 kDa) that is widely expressed in the brain (Braak et al., 2003). It seems to play a role in the stability of neuronal membranes, influencing presynaptic signaling and membrane trafficking through vesicular transport (Henderson et al., 2019). Several papers show that aSyn can be released from neurons and taken up by surrounding neurons or other cell types (Henderson et al., 2019). Therefore, it is not surprising that aSyn, despite constituting an intracellular protein, can be found in CSF, blood, and plasma (Atik et al., 2016; Maass et al., 2019). aSyn aggregates represent the main component of Lewy bodies (LB) (Spillantini et al., 1997). Therefore, PD, together with dementia with Lewy bodies (DLB) and multiple system atrophy (MSA), is referred to as a-synucleinopathy. LB are found in various regions of the CNS, depending on the disease stage (for example, substantia nigra, the dorsal motor nucleus of the vagus, nucleus basalis of Meynert, the locus coeruleus, and diffusely in late stages of the disease) (Forno, 1996). However, LB are also found in the peripheral nervous system in early or prodromal stages of the disease (e.g., sympathetic ganglia, sympathetic cardiac nerves, cholinergic nerve endings of salivary glands, autonomic nerve fibers of the skin).
First described with point mutations, the gene for aSyn (SNCA) was the first gene to be associated with the development of PD with an autosomal-dominant transmission (Polymeropoulos et al., 1997). Over time, duplications and triplications of SNCA have also been shown to play a causal role in hereditary forms of PD. Regarding symptoms, SNCA point mutations are associated with an earlier age at the onset (Ibáñez et al., 2009). SNCA gene duplications are associated with a mean age of the onset and clinical phenotype that overlaps with sPD (Ibáñez et al., 2009). SNCA triplications are associated with the early age of the onset, more rapid progression, and memory impairment (Ibáñez et al., 2009). Nonetheless, mutations in SNCA as a disease cause in PD remain rare.
Total aSyn and isoforms of aSyn appear to play a major role in PD pathogenicity and, accordingly, are also available as a candidate biomarker. The concentration of total aSyn in CSF may discriminate between patients with PD and healthy controls (Wang et al., 2012), and has been found at a lower concentration in patients with PD (Mollenhauer et al., 2008). This was confirmed in early-diagnosed, treatment-naïve patients (Stewart et al., 2014).
However, no uniform picture emerges regarding total aSyn. In patients with LRRK2 mutations, total aSyn CSF levels were increased in LRRK2-associated PD, non-manifesting carriers, and healthy controls compared to sPD (Vilas et al., 2016). In another study, lower but not statistically significant total aSyn CSF levels were recognized in LRRK2-associated PD, non-manifesting carriers, and sPD compared to healthy controls (Aasly et al., 2014). Follow-up data in patients with PD from the DATATOP study revealed an increase of total aSyn after 2 years (Majbour et al., 2016). An increase in total aSyn has also been reported over 2 years in another study with 63 patients with PD and 21 controls (Hall et al., 2016). In the latter study, higher total aSyn values were associated with longer disease duration (Hall et al., 2016). The authors hypothesized that total aSyn levels were bimodal, with a decrease in early disease stages followed by an increase with advanced neurodegeneration.
Unfortunately, unlike MSA or progressive supranuclear palsy (PSP), it is not possible to distinguish between sPD and aPD based on the aSyn concentration. Therefore, total aSyn is unable to distinguish between alpha-synucleinopathies (sPD, MSA) or between alpha-synucleinopathies and aPD such as PSP or corticobasal degeneration. Because of the cognitive impairment in SNCA triplication carriers, an association between total aSyn and patients with PD with dementia (PDD) could be considered. However, similar levels of total aSyn have been reported for patients with PD and PDD (Farotti et al., 2017).
The reason for the large heterogeneity of the results with respect to total aSyn is likely due to clinical heterogeneity, the number of included patients, and methodological differences that could compromise diagnostic accuracy. Another important confounding variable is hemolysis during the specimen collection, where contamination of blood or CSF leads to increased total aSyn levels (Barkovits et al., 2020).
Before aSyn aggregates into mature amyloid fibrils in LB, aSyn undergoes oligomerization. There is strong evidence that oligomeric aSyn is a key player in PD pathophysiology (Conway et al., 2000). It has been found at higher concentration in patients with PD compared to controls, but with poor diagnostic sensitivity and specificity (Parnetti et al., 2014b; Eusebi et al., 2017). However, there are conflicting results with respect to oligomeric aSyn reported over recent years. One example is the results in asymptomatic and symptomatic PARK-LRRK2 mutation carriers. Asymptomatic PARK-LRRK2 mutation carriers showed elevated oligomeric aSyn levels compared to healthy controls (Aasly et al., 2014); that oligomeric aSyn reflects a presymptomatic precursor of the disease cannot be concluded with certainty because symptomatic PARK-LRRK2 mutation carriers in the same study showed no difference to healthy controls (Aasly et al., 2014). Again, there is no evidence that oligomeric aSyn in the CSF might distinguish between sPD, gPD, and/or aPD. Among the isoforms of aSyn, phosphorylated 129 aSyn has been reported to enhance synuclein toxicity both in vivo and in vitro due to increasing formation of aSyn aggregates (Fujiwara et al., 2002; Smith et al., 2005), whereas a protective effect of phosphorylated 129 aSyn has also been described (Gorbatyuk et al., 2008; Da Azeredo Silveira et al., 2009). Another biomarker could be the 129 aSyn/total aSyn ratio, which seems to be elevated in PD (Wang et al., 2012). Regarding aSyn aggregates, in vitro conversion methods such as real-time quaking-induced conversion (RT-QuIC) have been developed as promising new methods for measuring aggregated aSyn in the CSF of patients with PD. RT-QuIC was originally developed for the detection of the abnormal form of prion protein, e.g., in Creutzfeldt-Jakob disease. In alpha-synucleinopathies, RT-QuIC has been reported with a high sensitivity and specificity of 95-100% for alpha-synucleinopathies (Fairfoul et al., 2016; Sano et al., 2018; Shahnawaz et al., 2020) and a positive predictive value of > 90% (van Rumund et al., 2019), but only 75% sensitivity in patients with an unclear diagnosis of parkinsonism (van Rumund et al., 2019). In addition, RT-QuIC does not appear to be suitable for monitoring the disease course (Nakagaki et al., 2021). The method has also been used to distinguish between alpha-synucleinopathies and other neurodegenerative diseases (Groveman et al., 2018). Due to an improved aSyn RT-QuIC test, a diagnostic sensitivity of 93% and 100% specificity has been achieved when comparing nine patients with synucleinopathy (12 PD; 17 DLB) and 31 non-synucleinopathy controls (including 16 patients with Alzheimer’s diseases). A recent study has presumed that RT-QuIC of aSyn in the CSF could be a promising candidate for prodromal stages for PD and DLB. In patients with isolated REM sleep behavior disorder (RBD), a known risk factor in the development of PD and DLB, a sensitivity and specificity of 90% have been shown (Iranzo et al., 2021). In addition, these patients had a subsequent higher risk of developing PD or DLB. Nearly two-thirds of the patients with RBD were diagnosed with PD or DLB, a mean 3.4 years after the aSyn measurement, of whom 97% were aSyn positive at the baseline (Iranzo et al., 2021). This finding of aSyn in the CSF by RT-QuIC and its value in presymptomatic patients is important, but must be confirmed by further studies.
The different forms of aSyn (total aSyn, oligomeric aSyn, phosphorylated aSyn) also reveal different results in blood serum and plasma. For total aSyn, conflicting results of increased, decreased, or unchanged total aSyn levels have been reported (Parnetti et al., 2019). Oligomeric aSyn in blood serum or red blood cells seems valuable candidates, with a sensitivity of 75% and specificity of up to 100% (Williams et al., 2016). However, these results must be confirmed in larger studies.
Lysosomes are one of the key players for protein degradation in human cells, including a-Syn (Webb et al., 2003). It is obvious that lysosome malfunction leads to an accumulation of dysfunctional proteins and cell organelles. This pathway is shared by several monogenetic forms of PD, including SNCA, ATP13A2, VPS35, DNAJC6, SYNJ1, LRRK2, RAB39B9) (Abeliovich and Gitler, 2016). An association between aSyn and lysosomes has long been hypothesized. It is assumed that a disturbed autophagy-lysosomal pathway is directly related to aSyn aggregates (Sardi et al., 2011; Bae et al., 2015; Suzuki et al., 2015).
Mutations in the corresponding gene for ß-glucocerebrosidase (GBA) are considered the most common genetic risk factor in PD, meaning that not every carrier of a GBA mutation will develop PD. Historically, the observation of an increased risk of PD in type I Gaucher disease (GD) and its families was first surmised from clinical findings (Neudorfer et al., 1996). GD type I represents an autosomal-recessive systemic disorder that can present with various degrees of systemic and neurological manifestations, usually manifesting in childhood. Since the first description, more than 300 different genetic alterations, such as point mutations, insertion, deletion, missense mutations, and splice junctions, have been described in the literature (Beutler et al., 2005; Hruska et al., 2008; Smith et al., 2017). Among GBA mutation carriers, an increasing penetrance of PD (PARK-GBA) with age has been reported (Anheim et al., 2012). Penetrance ranges from 7.6% at age 50 years to 30% at age 80 (Anheim et al., 2012). If patients have two homozygous mutations and thus affected with GD, the risk of PD is higher and the age at the onset (AAO) is earlier (Thaler et al., 2017). Depending on the origin of the patients, the frequency of GBA mutations varies. For example, GBA mutations can be found in 10–31% of patients with Ashkenazi background but only in 2.3% of Norwegian patients (Neumann et al., 2009). For the European non-Ashkenazi patients, the range is between 3 and 12% (Neumann et al., 2009).
Clinical differentiation between sPD and gPD caused by a heterozygous GBA mutation (PARK-GBA) is not possible. However, the AAO of PARK-GBA is 3–6 years earlier than in sPD (Riboldi and Di Fonzo, 2019). The progression of PARK-GBA is more often characterized by prominent cognitive impairment, hallucination, and RBD. Due to the phenotypic description, it is not surprising that GBA mutations have also been found in cases of DLB (Nalls et al., 2013). The frequency of patients with DLB with GBA mutations was up to 13% in pathologically proven patients with DLB (Gámez-Valero et al., 2016).
A few studies have focused on the activity of the lysosomal hydrolase ß-glucocerebrosidase (GCase) in alpha-synucleinopathies, with a significant decrease of GCase activity in the CSF of patients with PD compared to controls (Parnetti et al., 2014a; Lerche et al., 2021). As a biomarker decreased, GCase activity was consistently found in PARK-GBA (see below) but also in other gPD (PARK-LRRK2, PARK-SNCA, PARK-RKN) (Xicoy et al., 2019). The overall utility of GBA as biomarkers in peripheral biosamples of sPD is highly controversial because, as with the results in CSF, dried blood spots are conflicting (Balducci et al., 2007; Parnetti et al., 2017; Pchelina et al., 2017). In a very recently published work, the authors have hypothesized that low GCase activity was not responsible for the phenotype because low GCase activity has been found in patients with PARK-GBA and non-manifesting GBA carriers (Omer et al., 2022). However, patients with PARK-GBA were nearly 10 years older than non-manifesting GBA carriers (64.9 vs. 53.4 years, respectively). An additional role could be the respective GBA mutation, as there are indications that this causes different changes in GCase activity (Lerche et al., 2021). GCase activity could also be promising as a progression parameter in leukocytes, as a positive correlation between GCase levels in leukocytes and disease duration has been reported in sPD (Kim et al., 2016). Unfortunately, the treatments currently available for patients with GD do not reach the CNS. Hence, these treatment options do not play a role in PARK-GBA. However, there are several approaches to therapy development. One example is the capacity to increase GCase levels, currently being tested using the chaperone ambroxol in patients with PARK-GBA in an ongoing study (NCT02914366) (Mullin et al., 2020). The rationale behind this is that mutant GBA is unable to fold the endoplasmic reticulum correctly and thus promotes protein aggregation. Chaperone proteins able to facilitate the refolding of their substrates were tested. Another approach is gene replacement therapy via adeno-associated virus 9 to deliver a functional copy of the GBA gene to the CNS (Du et al., 2019; Abeliovich et al., 2021). In several animal models of PD or GBA-associated PD, direct application to the CNS was successful (Rocha et al., 2015; Jackson et al., 2019). Accordingly, a phase 1/2a study using these techniques has been recently started (NCT04127578).
Mutations in the leucine-rich repeat kinase (LRRK2) gene are the most common genetic cause of autosomal-dominant late-onset PD (Paisán-Ruíz et al., 2004; Zimprich et al., 2004). LRRK2 variants are also considered significant risk factors in sPD cases (Kluss et al., 2019). G2019S is the most frequent variant, accounting for 4% of gPD and 1% of sPD, with variable distribution among different ethnic populations and incomplete, age-related penetrance (26–84%) (Healy et al., 2008; Lee et al., 2017). LRRK2 encodes a multifunctional protein with a catalytic core of kinase and GTPase activity. Physiologically, LRRK2 is involved in various cellular processes, encompassing cytoskeletal maintenance, vesicular trafficking, mitochondrial function, autophagy, lysosomal degradation, and the inflammatory response (Jeong and Lee, 2020; Mancini et al., 2020). Most LRRK2 mutations cause a toxic gain of function with increased kinase activity. In viral and transgenic rodent models, overexpression of LRRK2 G2019S induced striatal neurodegeneration in a kinase-dependent manner (Lee et al., 2010; Tsika et al., 2015). There is also evidence that LRRK2 interacts with aSyn, mediating its aggregation and propagation (O’Hara et al., 2020).
The phenotype of PARK-LRRK2 resembles that of sPD in terms of cardinal motor features and good response to levodopa (Healy et al., 2008; Marras et al., 2011). PD symptoms manifest at a slightly younger age (57 years). Several studies have observed a more benign disease course in G2019S mutation carriers with a slower decline in motor scores, albeit a similar risk for motor complications (Healy et al., 2008; Marras et al., 2016; Ben Romdhan et al., 2018; Saunders-Pullman et al., 2018) that could not be confirmed by other studies that report no difference (Nabli et al., 2015) or even worse motor symptoms and a higher rate of dyskinesia (Nishioka et al., 2010; Shu et al., 2018). Various studies have indicated that PARK-LRRK2 is associated with less non-motor involvement. Hyposmia and RBD are less prevalent compared to sPD (Saunders-Pullman et al., 2015; Marras et al., 2016), and cognitive function seems to be more mildly impaired, even after many years of disease duration (Aasly et al., 2005; Alcalay et al., 2015).
Given the therapeutic advances of small molecule kinase inhibitors and antisense oligonucleotides to diminish LRRK2 activity, reliable and feasible biomarkers that reflect LRRK2-related pathways are mandatory. LRRK2 is highly expressed in peripheral tissues, such as lung, kidney, and blood cells (Fuji et al., 2015), and the latter serves as an easily accessible source [i.e., peripheral blood mononuclear cells (PBMCs)]. Indeed, by using flow cytometry, significantly elevated LRRK2 levels have been detected in B cells, T cells, CD16 + monocytes (Cook et al., 2017), and neutrophils of subjects with sPD compared to controls (Atashrazm et al., 2019), supporting the link to immune regulation. However, no difference in total LRRK2 levels could be detected in PBMCs among various sample groups, irrespective of PD or LRRK2 mutation status (Melachroinou et al., 2020; Padmanabhan et al., 2020). Alterations of total LRRK2 levels in PBMCs could be obscured by cellular heterogeneity. Considering cell-specific expression patterns of LRRK2, it seems important to purify and analyze different subcellular types separately in future investigations. A prominent site for heterologous LRRK2 phosphorylation is at serine 935 (pS935) that has been shown to be significantly reduced in PBMCs of affected G2019S carriers, distinguishing them from sPD, healthy G2019S carriers, and controls (Padmanabhan et al., 2020). Another study reported significantly elevated in vitro kinase activity of LRRK2 in PBMCs from LRRK2 G2019S mutation carriers in comparison to non-carriers, while the pS935-LRRK2 level was increased in sPD compared to controls (Melachroinou et al., 2020). The molecular mechanisms that alter the regulation of S935 phosphorylation are not fully understood and need further clarification. Moreover, LRRK2 is sensitized to dephosphorylation by LRRK2 kinase inhibitors, with reduced pS935-LRRK2 that could be used as a marker of pharmacological LRRK2 inhibition (Dzamko et al., 2010). Whether the GTPase Rab10, the key substrate of LRRK2 that can be pharmacologically modified, could serve as a marker for target engagement is still inconclusive and needs further assessment (Steger et al., 2016; Thirstrup et al., 2017). At least, for discriminative purposes, Rab10 cannot be suggested since no difference could be found in blood cells between patients with PD and controls (Fan et al., 2018; Atashrazm et al., 2019). Although it has been challenging to directly measure LRRK2 in CSF, LRRK2 quantification in CSF could be obtained by an improved monoclonal antibody technique that has revealed significantly higher CSF LRRK2 levels in G2019S-PD compared to sPD, healthy controls, and non-manifesting G2019 carriers (Mabrouk et al., 2020). Auto-phosphorylation of LRRK2 at serine 1292 (pS1292) reflects kinase activity that is measurable in CSF, but possibly masked by saturation effects no changes in pS1292 levels could be detected in the CSF of G2019S carriers (Wang et al., 2017). As mentioned in the previous section, CSF protein analysis revealed the differential pha-synuclein profiles of individuals with LRRK2 mutation (Aasly et al., 2014; Vilas et al., 2016) that could result from the variable presence of LB pathology in PARK-LRRK2. No differences were found in terms of the AD-related proteins amyloid-β1-42, total-tau, or phospho-tau (Vilas et al., 2016). Fraser et al. (2013) demonstrated the presence of LRRK2 in urinary exosomes, small endosomal-derived vesicles released from cells to the periphery. Intriguingly, G2019S carriers with PD displayed higher pS1292 to the total LRRK2 ratio in urinary exosomes compared to non-carriers and even asymptomatic G2019S carriers, indicating the potential for risk prediction (Fraser et al., 2016). Further studies on LRRK2 exosomes will explore their biomarker potential in PD.
Regarding imaging biomarkers, positron emission tomography (PET) detected reduced tracer binding in presymptomatic LRRK2 mutation carriers compared to controls, with greater progression of dopaminergic dysfunction (Nandhagopal et al., 2008). In this PET study, multiple tracers were applied, including multitracer PET using 18F-6-fluoro-l-dopa, 11C-(±)-α-dihydrotetrabenazine and 11C-d-threo-methylphenidate. Dopamine transporter imaging with I-123 FP-CIT showed potential to monitor progression, although a certain degree of intraindividual variability limited its ability to predict phenoconversion (Sánchez-Rodríguez et al., 2021). Studies that investigate markers to discriminate LRRK2-PD from aPD are lacking.
Given the common biological pathways affecting mitochondrial function and biomarker studies, including both PRKN and PINK1 mutations, these genes are discussed together.
The majority of early-onset parkinsonism with autosomal-recessive inheritance is linked to mutations of the PRKN/PARK2 gene (Kitada et al., 1998; Lücking et al., 2000). More than 130 disease-causing mutations have been identified throughout all 12 exons, including point mutations, exon rearrangements, and small deletions (Kasten et al., 2018). Whereas biallelic PRKN mutation results in overt disease, the question whether heterozygous mutations predispose to disease risk still remains an issue of debate (Huttenlocher et al., 2015; Lubbe et al., 2021; Yu et al., 2021). The encoded Parkin protein is an E3-ubiquitin ligase that transfers ubiquitin to protein substrates, thus targeting them to proteasomal degradation (Shimura et al., 2000; Moore, 2006). Moreover, Parkin regulates mitochondrial quality control and clearance of damaged mitochondria through mitophagy in concert with the other PD-linked gene PINK1 (Narendra et al., 2012). In the Drosophila model, mutant PRKN caused a degenerative phenotype with flight muscle defects, mitochondrial alterations, and abnormalities of dopaminergic neurons (Greene et al., 2003; Pickrell and Youle, 2015).
PARK-PRKN is characterized by a slowly progressive disease course at an early age of the onset (mean, 31 years) (Khan et al., 2003; Kasten et al., 2018). Beyond the typical clinical triad, hyperreflexia can be present, and dystonia, often affecting the lower limb, can be an initial sign (Khan et al., 2003; Grünewald et al., 2013). The marked and sustained response to levodopa is complicated by frequently associated levodopa-induced motor fluctuations and dyskinesias (Clarimon et al., 2005). With regard to non-motor aspects, olfactory function and cognition are mostly well preserved (Khan et al., 2004; Alcalay et al., 2015; Abeliovich and Gitler, 2016). However, a multicenter, case-control study reported an earlier occurrence and a higher frequency and severity of impulsive-compulsive behaviors in PARK-PRKN (Morgante et al., 2016).
Mutations in the phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) gene represent the second most frequent cause of autosomal-recessive inherited early-onset parkinsonism (Valente et al., 2004). The frequency of PINK1 mutations ranges between 1 and 9%, with variations across different ethnic populations (Schulte and Gasser, 2011; Klein and Westenberger, 2012). Mutations are highly diverse and include missense, non-sense, frameshift mutations, and deletions. As with PRKN, the role of heterozygous PINK1 mutation as a risk factor in PD has not yet been clarified (Krohn et al., 2020). PINK1 encodes a serine/threonine kinase involved in the regulation of the mitophagy pathway by activating ubiquitin and Parkin through phosphorylation (Valente et al., 2004; Narendra et al., 2010; Kane et al., 2014). Drosophila-lacking PINK1 exhibited mitochondrial abnormalities and muscle and neuronal degeneration that could be rescued by PRKN overexpression but not vice versa, suggesting that PINK1 acts upstream of Parkin in the shared pathway (Clark et al., 2006; Park et al., 2006; Yang et al., 2006).
In general, PARK-PINK1 shares similar features as PARK-PRKN regarding early AAO (mean, 32 years), slow disease progression, and excellent levodopa response, although associated with motor fluctuations and dyskinesias (Bonifati et al., 2005; Ibáñez et al., 2006; Kasten et al., 2018). Dystonia at the onset and hyperreflexia are also described in patients with PINK1 mutation. However, non-motor symptoms are more pronounced. Hyposmia is commonly reported (Ferraris et al., 2009). Neuropsychiatric features are multifaceted, including mostly depression and anxiety but also traits of the schizophrenic spectrum and obsessive-compulsive personality disorder (Ephraty et al., 2007; Steinlechner et al., 2007; Ricciardi et al., 2014). Although cognitive function was thought to be less affected (Bonifati et al., 2005; Kasten et al., 2018), a recent study has observed a higher rate of cognitive dysfunction in PINK1 mutations (Piredda et al., 2020).
Biomarkers for mitochondrial integrity are not only required to prove target engagement and drug-induced effects but also to identify patients who will most likely benefit from mitophagy-targeted treatments. PINK1 phosphorylates ubiquitin at a serine residue (pS65-Ub), which could reflect PINK1 activity. Autopsy studies have revealed elevated pS65-Ub levels in brain tissues of patients with sPD, DLB, and normal elderly controls, suggesting that pS65-Ub accumulates upon mitochondrial stress during disease or aging (Fiesel et al., 2015; Hou et al., 2018). In contrast, PINK1/PRKN mutants lacked pS65-Ub signals, consistent with their loss of function to label damaged mitochondria. More intriguingly, pS65-Ub signals detected by sensitive enzyme-linked immunosorbent assay were significantly reduced in blood plasma of patients with homozygous PINK1-mutation carriers compared to controls with heterozygous mutation and non-carriers (Watzlawik et al., 2020). Whether pS65-Ub levels can be used as a diagnostic or prognostic disease marker in blood needs to be confirmed in further investigations. Mitochondrial DNA (mtDNA) is closely related to mitochondrial function. Whereas reduced mtDNA copy numbers were reported in the substantia nigra of PD postmortem brains (Pyle et al., 2016), the findings in peripheral blood are inconclusive (Gui et al., 2015; Pyle et al., 2016; Davis et al., 2020). Circulating cell-free mtDNA (ccf-mtDNA) is a fragment of mtDNA released from cells in response to stress. Reduced ccf-mtDNA levels were reported in the CSF of patients with sPD (Pyle et al., 2015), showing an inverse correlation with treatment (Lowes et al., 2020). Notably, a recent study has observed higher ccf-mtDNA levels in the sera of PRKN and PINK1 biallelic mutation carriers and affected heterozygotes compared to patients with sPD, possibly resulting from impaired mitophagy in PRKN/PINK1-associated PD (Borsche et al., 2020). As inflammation evidently contributes to PD pathogenesis and progression, peripheral inflammatory cytokines have been attributed biomarker potential. Among these, interleukin-6 (IL6) levels were elevated in blood samples from patients with PD (Qin et al., 2016), correlating with motor severity. A recent study has reported higher serum IL6 levels in patients with biallelic PRKN/PINK1 mutations compared to healthy controls, whereas heterozygous PRKN/PINK1 mutation carriers and sPD showed only a trend toward elevated IL6 levels, indicating a gene dosage effect (Borsche et al., 2020). Moreover, IL6 levels correlated only in affected PRKN/PINK1 mutation carriers with disease duration. Mitochondrial dysfunction is also implied in the pathophysiology of a PD such as MSA and PSP (Nicoletti et al., 2021). Comparative studies exploring discriminative biomarkers of PRKN/PINK1-associated PD vs. aPD have not yet been conducted.
Imaging methods can be useful to monitor mitochondrial energy metabolism. Mitochondrial complex I activity can be assessed in vivo by the PET radiotracer 18F-BCPP-EF, which is reduced in a non-human primate model for PD (Tsukada et al., 2016) but could not be translated to patients with PD (Wilson et al., 2020). Alternatively, phosphorus magnetic resonance spectroscopy (31P-MRS) can visualize cerebral mitochondrial metabolism by measuring phosphorous-containing energy metabolites (Henchcliffe et al., 2008). Increased putaminal levels of high-energy phosphates distinguished homozygous PINK1 mutation carriers from heterozygous mutation carriers and controls (Hilker et al., 2012). This finding was interpreted as compensatory mechanisms with respect to the impaired cellular stress resistance.
Two current trials testing vitamin K2 (Prasuhn et al., 2020) and coenzyme Q10 (Prasuhn et al., 2019) include Parkin/PINK1 mutation carriers and use 31P-MRS for patient stratification and as a secondary end point.
DJ-1 is a widely expressed protein that participates in antioxidative stress mechanisms and mitochondrial regulation (Dolgacheva et al., 2019). Mutations in the corresponding gene DJ-1 are responsible for an autosomal-recessive form of gPD (PARK-DJ-1). The phenotype of patients with PARK-DJ-1 consists of an early onset and slow progression of the disease and is, therefore, comparable with other autosomal-recessive forms like PARK-PRKN or PARK-PINK1. However, mutations in DJ-1 are less frequent than PARK-PRKN or PARK-PINK1.
There are conflicting results regarding DJ-1 as a biomarker in the CSF and peripheral tissues, with increased and decreased DJ-1 levels in patients with sPD (Saito, 2014; Farotti et al., 2017). The same conflicting results were obtained when DJ-1 in the CSF was examined in the differential diagnosis of sPD vs. aPD (Farotti et al., 2017). A few studies with a low number of patients have investigated the concentration of DJ-1 in saliva in sPD with conflicting results. Therefore, DJ-1 in saliva does not play a role in daily routine (Farah et al., 2018). Another option is to determine the concentration of oxidized DJ-1 in blood or urine. Oxidized DJ-1 is induced by oxidative stress, which plays a major role in the pathophysiology of neurodegenerative diseases. Increased levels of oxidized DJ-1 have been described in patients with sPD compared to controls (Jang et al., 2018).
A biomarker with a high sensitivity and specificity that could be introduced into clinical routine is currently not available. Biomarker research for PD has made great progress in recent years. Nevertheless, there is still a lack of robust biomarkers with high sensitivity and specificity that would satisfy diagnostic requirements. This is also true for monitoring disease progression and differential diagnosis of sPD, gPD, and aPD. Even for the most common monogenetic forms described here, this has not yet been achieved. This is true for the different forms of PD, including sPD, gPD, and aPD. Further research activity will be necessary to improve or find new techniques (e.g., RT-QuIC) and find the most suitable liquid biomaterial. The key to the diagnosis of PD, especially for future disease-modifying therapies, is likely to be a combination of different biomarkers with different modalities. In addition to peripheric biomarkers and CSF, imaging techniques (nuclear medicine, MRI, etc.) will play a crucial role.
LT and SK contributed to conception and design of the study. SK and EK wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
We acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen. This work was further supported by the Bundesministerium für Forschung und Bildung (BMBF) through funding for the TreatHSP network (N°01GM1905 to SK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aasly, J. O., Johansen, K. K., Brønstad, G., Warø, B. J., Majbour, N. K., Varghese, S., et al. (2014). Elevated levels of cerebrospinal fluid α-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front. Aging Neurosci. 6:248. doi: 10.3389/fnagi.2014.00248
Aasly, J. O., Toft, M., Fernandez-Mata, I., Kachergus, J., Hulihan, M., White, L. R., et al. (2005). Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann. Neurol. 57, 762–765. doi: 10.1002/ana.20456
Abeliovich, A., and Gitler, A. D. (2016). Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539, 207–216. doi: 10.1038/nature20414
Abeliovich, A., Hefti, F., and Sevigny, J. (2021). Gene therapy for Parkinson’s disease associated with GBA1 mutations. J. Parkinsons Dis. 11, S183–S188. doi: 10.3233/JPD-212739
Alcalay, R. N., Mejia-Santana, H., Mirelman, A., Saunders-Pullman, R., Raymond, D., Palmese, C., et al. (2015). Neuropsychological performance in LRRK2 G2019S carriers with Parkinson’s disease. Parkinsonism Relat. Disord. 21, 106–110. doi: 10.1016/j.parkreldis.2014.09.033
Anheim, M., Elbaz, A., Lesage, S., Durr, A., Condroyer, C., Viallet, F., et al. (2012). Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78, 417–420. doi: 10.1212/WNL.0b013e318245f476
Atashrazm, F., Hammond, D., Perera, G., Bolliger, M. F., Matar, E., Halliday, G. M., et al. (2019). LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson’s disease patients. Mov. Disord. 34, 406–415. doi: 10.1002/mds.27601
Atik, A., Stewart, T., and Zhang, J. (2016). Alpha-synuclein as a biomarker for Parkinson’s disease. Brain Pathol. 26, 410–418. doi: 10.1111/bpa.12370
Bae, E.-J., Yang, N. Y., Lee, C., Lee, H.-J., Kim, S., Sardi, S. P., et al. (2015). Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 47:e153. doi: 10.1038/emm.2014.128
Balducci, C., Pierguidi, L., Persichetti, E., Parnetti, L., Sbaragli, M., Tassi, C., et al. (2007). Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov. Disord. 22, 1481–1484. doi: 10.1002/mds.21399
Barkovits, K., Kruse, N., Linden, A., Tönges, L., Pfeiffer, K., Mollenhauer, B., et al. (2020). Blood contamination in CSF and its impact on quantitative analysis of alpha-synuclein. Cells 9:370. doi: 10.3390/cells9020370
Ben Romdhan, S., Farhat, N., Nasri, A., Lesage, S., Hdiji, O., Ben Djebara, M., et al. (2018). LRRK2 G2019S Parkinson’s disease with more benign phenotype than idiopathic. Acta Neurol. Scand. 138, 425–431. doi: 10.1111/ane.12996
Beutler, E., Gelbart, T., and Scott, C. R. (2005). Hematologically important mutations: gaucher disease. Blood Cells Mol. Dis. 35, 355–364. doi: 10.1016/j.bcmd.2005.07.005
Bonifati, V., Rohé, C. F., Breedveld, G. J., Fabrizio, E., Mari, M., de, et al. (2005). Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65, 87–95. doi: 10.1212/01.wnl.0000167546.39375.82
Borsche, M., König, I. R., Delcambre, S., Petrucci, S., Balck, A., Brüggemann, N., et al. (2020). Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 143, 3041–3051. doi: 10.1093/brain/awaa246
Braak, H., Del Tredici, K., Rüb, U., Vos, R. A. I., de, Jansen Steur, E. N. H., et al. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Clarimon, J., Johnson, J., Djaldetti, R., Hernandez, D., Hattori, N., Sroka, H., et al. (2005). Mutation of the Parkin gene in a Persian family: clinical progression over a 40-year period. Mov. Disord. 20, 887–890. doi: 10.1002/mds.20495
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Conway, K. A., Lee, S. J., Rochet, J. C., Ding, T. T., Williamson, R. E., and Lansbury, P. T. (2000). Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 571–576. doi: 10.1073/pnas.97.2.571
Cook, D. A., Kannarkat, G. T., Cintron, A. F., Butkovich, L. M., Fraser, K. B., Chang, J., et al. (2017). LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis. 3:11. doi: 10.1038/s41531-017-0010-8
Cook, L., Schulze, J., Naito, A., and Alcalay, R. N. (2021). The role of genetic testing for Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 21:17.
Da Azeredo, Silveira, S., Schneider, B. L., Cifuentes-Diaz, C., Sage, D., Abbas-Terki, T., et al. (2009). Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum. Mol. Genet. 18, 872–887. doi: 10.1093/hmg/ddn417
Davis, R. L., Wong, S. L., Carling, P. J., Payne, T., Sue, C. M., and Bandmann, O. (2020). Serum FGF-21, GDF-15, and blood mtDNA copy number are not biomarkers of Parkinson disease. Neurol. Clin. Pract. 10, 40–46. doi: 10.1212/CPJ.0000000000000702
Di Maio, R., Hoffman, E. K., Rocha, E. M., Keeney, M. T., Sanders, L. H., Miranda, B. R., et al. (2018). LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 10:eaar5429. doi: 10.1126/scitranslmed.aar5429
Dolgacheva, L. P., Berezhnov, A. V., Fedotova, E. I., Zinchenko, V. P., and Abramov, A. Y. (2019). Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 51, 175–188. doi: 10.1007/s10863-019-09798-4
Du, S., Ou, H., Cui, R., Jiang, N., Zhang, M., Li, X., et al. (2019). Delivery of glucosylceramidase beta gene using AAV9 vector therapy as a treatment strategy in mouse models of gaucher disease. Hum. Gene Ther. 30, 155–167. doi: 10.1089/hum.2018.072
Dzamko, N., Deak, M., Hentati, F., Reith, A. D., Prescott, A. R., Alessi, D. R., et al. (2010). Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem. J. 430, 405–413. doi: 10.1042/BJ20100784
Ephraty, L., Porat, O., Israeli, D., Cohen, O. S., Tunkel, O., Yael, S., et al. (2007). Neuropsychiatric and cognitive features in autosomal-recessive early parkinsonism due to PINK1 mutations. Mov. Disord. 22, 566–569. doi: 10.1002/mds.21319
Eusebi, P., Giannandrea, D., Biscetti, L., Abraha, I., Chiasserini, D., Orso, M., et al. (2017). Diagnostic utility of cerebrospinal fluid α-synuclein in Parkinson’s disease: a systematic review and meta-analysis. Mov. Disord. 32, 1389–1400. doi: 10.1002/mds.27110
Fairfoul, G., McGuire, L. I., Pal, S., Ironside, J. W., Neumann, J., Christie, S., et al. (2016). Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 3, 812–818. doi: 10.1002/acn3.338
Fan, Y., Howden, A. J., Sarhan, A. R., Lis, P., Ito, G., Martinez, T. N., et al. (2018). Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. Biochem. J. 475, 23–44. doi: 10.1042/BCJ20170803
Farah, R., Haraty, H., Salame, Z., Fares, Y., Ojcius, D. M., and Said Sadier, N. (2018). Salivary biomarkers for the diagnosis and monitoring of neurological diseases. Biomed. J. 41, 63–87. doi: 10.1016/j.bj.2018.03.004
Farotti, L., Paciotti, S., Tasegian, A., Eusebi, P., and Parnetti, L. (2017). Discovery, validation and optimization of cerebrospinal fluid biomarkers for use in Parkinson’s disease. Expert Rev. Mol. Diagn. 17, 771–780. doi: 10.1080/14737159.2017.1341312
Ferraris, A., Ialongo, T., Passali, G. C., Pellecchia, M. T., Brusa, L., Laruffa, M., et al. (2009). Olfactory dysfunction in Parkinsonism caused by PINK1 mutations. Mov. Disord. 24, 2350–2357. doi: 10.1002/mds.22816
Fiesel, F. C., Ando, M., Hudec, R., Hill, A. R., Castanedes-Casey, M., Caulfield, T. R., et al. (2015). (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 16, 1114–1130. doi: 10.15252/embr.201540514
Forno, L. S. (1996). Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 55, 259–272. doi: 10.1097/00005072-199603000-00001
Fraser, K. B., Moehle, M. S., Alcalay, R. N., West, A. B., Bressman, S., Giladi, N., et al. (2016). Urinary LRRK2 phosphorylation predicts parkinsonian phenotypes in G2019S LRRK2 carriers. Neurology 86, 994–999. doi: 10.1212/WNL.0000000000002436
Fraser, K. B., Moehle, M. S., Daher, J. P. L., Webber, P. J., Williams, J. Y., Stewart, C. A., et al. (2013). LRRK2 secretion in exosomes is regulated by 14-3-3. Hum. Mol. Genet. 22, 4988–5000. doi: 10.1093/hmg/ddt346
Fuji, R. N., Flagella, M., Baca, M., Baptista, M. A. S., Brodbeck, J., Chan, B. K., et al. (2015). Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 7:273ra15. doi: 10.1126/scitranslmed.aaa3634
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Gámez-Valero, A., Prada-Dacasa, P., Santos, C., Adame-Castillo, C., Campdelacreu, J., Reñé, R., et al. (2016). GBA mutations are associated with earlier onset and male sex in dementia with lewy bodies. Mov. Disord. 31, 1066–1070. doi: 10.1002/mds.26593
Gorbatyuk, O. S., Li, S., Sullivan, L. F., Chen, W., Kondrikova, G., Manfredsson, F. P., et al. (2008). The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 105, 763–768. doi: 10.1073/pnas.0711053105
Green, H. F., Khosousi, S., and Svenningsson, P. (2019). Plasma IL-6 and IL-17A correlate with severity of motor and non-motor symptoms in Parkinson’s disease. J. Parkinsons Dis. 9, 705–709. doi: 10.3233/JPD-191699
Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B., and Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U.S.A. 100, 4078–4083. doi: 10.1073/pnas.0737556100
Groveman, B. R., Orrù, C. D., Hughson, A. G., Raymond, L. D., Zanusso, G., Ghetti, B., et al. (2018). Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol. Commun. 6:7. doi: 10.1186/s40478-018-0508-2
Grünewald, A., Kasten, M., Ziegler, A., and Klein, C. (2013). Next-generation phenotyping using the parkin example: time to catch up with genetics. JAMA Neurol. 70, 1186–1191. doi: 10.1001/jamaneurol.2013.488
Gui, Y.-X., Xu, Z.-P., Lv, W., Zhao, J.-J., and Hu, X.-Y. (2015). Evidence for polymerase gamma, POLG1 variation in reduced mitochondrial DNA copy number in Parkinson’s disease. Parkinson. Relat. Disord. 21, 282–286. doi: 10.1016/j.parkreldis.2014.12.030
Hall, S., Surova, Y., Öhrfelt, A., Blennow, K., Zetterberg, H., and Hansson, O. (2016). Longitudinal measurements of cerebrospinal fluid biomarkers in Parkinson’s disease. Mov. Disord. 31, 898–905. doi: 10.1002/mds.26578
Healy, D. G., Falchi, M., O’Sullivan, S. S., Bonifati, V., Durr, A., Bressman, S., et al. (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590. doi: 10.1016/S1474-4422(08)70117-0
Henchcliffe, C., Shungu, D. C., Mao, X., Huang, C., Nirenberg, M. J., Jenkins, B. G., et al. (2008). Multinuclear magnetic resonance spectroscopy for in vivo assessment of mitochondrial dysfunction in Parkinson’s disease. Ann. N.Y. Acad. Sci. 1147, 206–220. doi: 10.1196/annals.1427.037
Henderson, M. X., Trojanowski, J. Q., and Lee, V. M.-Y. (2019). α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci. Lett. 709, 134316. doi: 10.1016/j.neulet.2019.134316
Hilker, R., Pilatus, U., Eggers, C., Hagenah, J., Roggendorf, J., Baudrexel, S., et al. (2012). The bioenergetic status relates to dopamine neuron loss in familial PD with PINK1 mutations. PLoS One 7:e51308. doi: 10.1371/journal.pone.0051308
Hou, X., Fiesel, F. C., Truban, D., Castanedes Casey, M., Lin, W., Soto, A. I., et al. (2018). Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 14, 1404–1418. doi: 10.1080/15548627.2018.1461294
Hruska, K. S., LaMarca, M. E., Scott, C. R., and Sidransky, E. (2008). Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 29, 567–583. doi: 10.1002/humu.20676
Huttenlocher, J., Stefansson, H., Steinberg, S., Helgadottir, H. T., Sveinbjörnsdóttir, S., Riess, O., et al. (2015). Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. Hum. Mol. Genet. 24, 5637–5643. doi: 10.1093/hmg/ddv277
Ibáñez, P., Lesage, S., Janin, S., Lohmann, E., Durif, F., Destée, A., et al. (2009). Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch. Neurol. 66, 102–108. doi: 10.1001/archneurol.2008.555
Ibáñez, P., Lesage, S., Lohmann, E., Thobois, S., Michele, G., de, et al. (2006). Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain 129, 686–694. doi: 10.1093/brain/awl005
Iranzo, A., Fairfoul, G., Ayudhaya, A. C. N., Serradell, M., Gelpi, E., Vilaseca, I., et al. (2021). Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 20, 203–212. doi: 10.1016/S1474-4422(20)30449-X
Jackson, K. L., Viel, C., Clarke, J., Bu, J., Chan, M., Wang, B., et al. (2019). Viral delivery of a microRNA to Gba to the mouse central nervous system models neuronopathic Gaucher disease. Neurobiol. Dis. 130:104513. doi: 10.1016/j.nbd.2019.104513
Jang, J., Jeong, S., Lee, S. I., Seol, W., Seo, H., Son, I., et al. (2018). Oxidized DJ-1 levels in urine samples as a putative biomarker for Parkinson’s disease. Parkinsons Dis. 2018:1241757. doi: 10.1155/2018/1241757
Jeong, G. R., and Lee, B. D. (2020). Pathological functions of LRRK2 in Parkinson’s disease. Cells 9:2565. doi: 10.3390/cells9122565
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kasten, M., Hartmann, C., Hampf, J., Schaake, S., Westenberger, A., Vollstedt, E.-J., et al. (2018). Genotype-phenotype relations for the Parkinson’s disease genes parkin, PINK1, DJ1: MDSGene systematic review. Mov. Disord. 33, 730–741. doi: 10.1002/mds.27352
Khan, N. L., Graham, E., Critchley, P., Schrag, A. E., Wood, N. W., Lees, A. J., et al. (2003). Parkin disease: a phenotypic study of a large case series. Brain 126, 1279–1292. doi: 10.1093/brain/awg142
Khan, N. L., Katzenschlager, R., Watt, H., Bhatia, K. P., Wood, N. W., Quinn, N., et al. (2004). Olfaction differentiates parkin disease from early-onset parkinsonism and Parkinson disease. Neurology 62, 1224–1226. doi: 10.1212/01.WNL.0000118281.66802.81
Kim, H.-J., Jeon, B., Song, J., Lee, W.-W., Park, H., and Shin, C.-W. (2016). Leukocyte glucocerebrosidase and β-hexosaminidase activity in sporadic and genetic Parkinson disease. Parkinsonism Relat. Disord. 23, 99–101. doi: 10.1016/j.parkreldis.2015.12.002
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888. doi: 10.1101/cshperspect.a008888
Kluss, J. H., Mamais, A., and Cookson, M. R. (2019). LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 47, 651–661. doi: 10.1042/BST20180462
Krohn, L., Grenn, F. P., Makarious, M. B., Kim, J. J., Bandres-Ciga, S., Roosen, D. A., et al. (2020). Comprehensive assessment of PINK1 variants in Parkinson’s disease. Neurobiol. Aging 91, 168.e1–168.e5. doi: 10.1016/j.neurobiolaging.2020.03.003
Lawton, M., Baig, F., Toulson, G., Morovat, A., Evetts, S. G., Ben-Shlomo, Y., et al. (2020). Blood biomarkers with Parkinson’s disease clusters and prognosis: the oxford discovery cohort. Mov. Disord. 35, 279–287. doi: 10.1002/mds.27888
Lee, A. J., Wang, Y., Alcalay, R. N., Mejia-Santana, H., Saunders-Pullman, R., Bressman, S., et al. (2017). Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov. Disord. 32, 1432–1438. doi: 10.1002/mds.27059
Lee, B. D., Shin, J.-H., VanKampen, J., Petrucelli, L., West, A. B., Ko, H. S., et al. (2010). Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 16, 998–1000. doi: 10.1038/nm.2199
Lerche, S., Schulte, C., Wurster, I., Machetanz, G., Roeben, B., Zimmermann, M., et al. (2021). The Mutation Matters: CSF Profiles of GCase, Sphingolipids, α-Synuclein in PDGBA. Mov. Disord. 36, 1216–1228. doi: 10.1002/mds.28472
Lowes, H., Pyle, A., Santibanez-Koref, M., and Hudson, G. (2020). Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol. Neurodegener. 15:10. doi: 10.1186/s13024-020-00362-y
Lubbe, S. J., Bustos, B. I., Hu, J., Krainc, D., Joseph, T., Hehir, J., et al. (2021). Assessing the relationship between monoallelic PRKN mutations and Parkinson’s risk. Hum. Mol. Genet. 30, 78–86. doi: 10.1093/hmg/ddaa273
Lücking, C. B., Dürr, A., Bonifati, V., Vaughan, J., Michele, G., de, et al. (2000). Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Maass, F., Schulz, I., Lingor, P., Mollenhauer, B., and Bähr, M. (2019). Cerebrospinal fluid biomarker for Parkinson’s disease: an overview. Mol. Cell Neurosci. 97, 60–66. doi: 10.1016/j.mcn.2018.12.005
Mabrouk, O. S., Chen, S., Edwards, A. L., Yang, M., Hirst, W. D., and Graham, D. L. (2020). Quantitative measurements of LRRK2 in human cerebrospinal fluid demonstrates increased levels in G2019S patients. Front. Neurosci. 14:526. doi: 10.3389/fnins.2020.00526
Majbour, N. K., Vaikath, N. N., Eusebi, P., Chiasserini, D., Ardah, M., Varghese, S., et al. (2016). Longitudinal changes in CSF alpha-synuclein species reflect Parkinson’s disease progression. Mov. Disord. 31, 1535–1542. doi: 10.1002/mds.26754
Mancini, A., Mazzocchetti, P., Sciaccaluga, M., Megaro, A., Bellingacci, L., Beccano-Kelly, D. A., et al. (2020). From synaptic dysfunction to neuroprotective strategies in genetic Parkinson’s disease: lessons from LRRK2. Front. Cell Neurosci. 14:158. doi: 10.3389/fncel.2020.00158
Marras, C., Alcalay, R. N., Caspell-Garcia, C., Coffey, C., Chan, P., Duda, J. E., et al. (2016). Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov. Disord. 31, 1192–1202. doi: 10.1002/mds.26614
Marras, C., Schüle, B., Schuele, B., Munhoz, R. P., Rogaeva, E., Langston, J. W., et al. (2011). Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology 77, 325–333. doi: 10.1212/WNL.0b013e318227042d
Melachroinou, K., Kang, M. S., Liong, C., Narayan, S., Levers, N., Joshi, N., et al. (2020). Elevated in vitro kinase activity in peripheral blood mononuclear cells of leucine-rich repeat kinase 2 G2019S carriers: a novel enzyme-linked immunosorbent assay–based method. Mov. Disord. 35, 2095–2100. doi: 10.1002/mds.28175
Mollenhauer, B., Cullen, V., Kahn, I., Krastins, B., Outeiro, T. F., Pepivani, I., et al. (2008). Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp. Neurol. 213, 315–325. doi: 10.1016/j.expneurol.2008.06.004
Moore, D. J. (2006). Parkin: a multifaceted ubiquitin ligase. Biochem. Soc. Trans. 34, 749–753. doi: 10.1042/BST0340749
Morgante, F., Fasano, A., Ginevrino, M., Petrucci, S., Ricciardi, L., Bove, F., et al. (2016). Impulsive-compulsive behaviors in parkin-associated Parkinson disease. Neurology 87, 1436–1441. doi: 10.1212/WNL.0000000000003177
Mullin, S., Smith, L., Lee, K., D’Souza, G., Woodgate, P., Elflein, J., et al. (2020). Ambroxol for the treatment of patients with parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized. Noncontrol. Trial. JAMA Neurol. 77, 427–434. doi: 10.1001/jamaneurol.2019.4611
Nabli, F., Ben Sassi, S., Amouri, R., Duda, J. E., Farrer, M. J., and Hentati, F. (2015). Motor phenotype of LRRK2-associated Parkinson’s disease: a Tunisian longitudinal study. Mov. Disord. 30, 253–258. doi: 10.1002/mds.26097
Nakagaki, T., Nishida, N., and Satoh, K. (2021). Development of α-synuclein real-time quaking-induced conversion as a diagnostic method for α-synucleinopathies. Front. Aging Neurosci. 13:703984. doi: 10.3389/fnagi.2021.703984
Nalls, M. A., Duran, R., Lopez, G., Kurzawa-Akanbi, M., McKeith, I. G., Chinnery, P. F., et al. (2013). A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 70, 727–735. doi: 10.1001/jamaneurol.2013.1925
Nandhagopal, R., Mak, E., Schulzer, M., McKenzie, J., McCormick, S., Sossi, V., et al. (2008). Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology 71, 1790–1795. doi: 10.1212/01.wnl.0000335973.66333.58
Narendra, D., Walker, J. E., and Youle, R. (2012). Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb. Perspect. Biol. 4:a011338. doi: 10.1101/cshperspect.a011338
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D.-F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Neudorfer, O., Giladi, N., Elstein, D., Abrahamov, A., Turezkite, T., Aghai, E., et al. (1996). Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 89, 691–694. doi: 10.1093/qjmed/89.9.691
Neumann, J., Bras, J., Deas, E., O’Sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794. doi: 10.1093/brain/awp044
Nicoletti, V., Palermo, G., Del Prete, E., Mancuso, M., and Ceravolo, R. (2021). Understanding the multiple role of mitochondria in Parkinson’s disease and related disorders: lesson from genetics and protein-interaction network. Front. Cell Dev. Biol. 9:636506. doi: 10.3389/fcell.2021.636506
Nishioka, K., Kefi, M., Jasinska-Myga, B., Wider, C., Vilariño-Güell, C., Ross, O. A., et al. (2010). A comparative study of LRRK2, PINK1 and genetically undefined familial Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 81, 391–395. doi: 10.1136/jnnp.2009.185231
O’Hara, D. M., Pawar, G., Kalia, S. K., and Kalia, L. V. (2020). LRRK2 and α-synuclein: distinct or synergistic players in Parkinson’s disease? Front. Neurosci. 14:577. doi: 10.3389/fnins.2020.00577
Omer, N., Giladi, N., Gurevich, T., Bar-Shira, A., Gana-Weisz, M., Glinka, T., et al. (2022). Glucocerebrosidase activity is not associated with Parkinson’s disease risk or severity. Mov. Disord. 37, 190–195. doi: 10.1002/mds.28792
Padmanabhan, S., Lanz, T. A., Gorman, D., Wolfe, M., Joyce, A., Cabrera, C., et al. (2020). An assessment of LRRK2 serine 935 phosphorylation in human peripheral blood mononuclear cells in idiopathic Parkinson’s disease and G2019S LRRK2 cohorts. J. Parkinsons Dis. 10, 623–629. doi: 10.3233/JPD-191786
Paisán-Ruíz, C., Jain, S., Evans, E. W., Gilks, W. P. Simón, J. van der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. doi: 10.1016/j.neuron.2004.10.023
Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. doi: 10.1038/nature04788
Parnetti, L., Farotti, L., Eusebi, P., Chiasserini, D., Carlo, C., de, et al. (2014b). Differential role of CSF alpha-synuclein species, tau, and Aβ42 in Parkinson’s disease. Front. Aging Neurosci. 6:53. doi: 10.3389/fnagi.2014.00053
Parnetti, L., Chiasserini, D., Persichetti, E., Eusebi, P., Varghese, S., Qureshi, M. M., et al. (2014a). Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 29, 1019–1027. doi: 10.1002/mds.25772
Parnetti, L., Gaetani, L., Eusebi, P., Paciotti, S., Hansson, O., El-Agnaf, O., et al. (2019). CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 18, 573–586. doi: 10.1016/S1474-4422(19)30024-9
Parnetti, L., Paciotti, S., Eusebi, P., Dardis, A., Zampieri, S., Chiasserini, D., et al. (2017). Cerebrospinal fluid β-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 32, 1423–1431. doi: 10.1002/mds.27136
Pchelina, S., Emelyanov, A., Baydakova, G., Andoskin, P., Senkevich, K., Nikolaev, M., et al. (2017). Oligomeric α-synuclein and glucocerebrosidase activity levels in GBA-associated Parkinson’s disease. Neurosci. Lett. 636, 70–76. doi: 10.1016/j.neulet.2016.10.039
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Piredda, R., Desmarais, P., Masellis, M., and Gasca-Salas, C. (2020). Cognitive and psychiatric symptoms in genetically determined Parkinson’s disease: a systematic review. Eur. J. Neurol. 27, 229–234. doi: 10.1111/ene.14115
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Prasuhn, J., Brüggemann, N., Hessler, N., Berg, D., Gasser, T., Brockmann, K., et al. (2019). An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol. Res. Pract. 1:31. doi: 10.1186/s42466-019-0033-1
Prasuhn, J., Kasten, M., Vos, M., König, I. R., Schmid, S. M., Wilms, B., et al. (2020). The use of vitamin K2 in patients with parkinson’s disease and mitochondrial dysfunction (PD-K2): a theranostic pilot study in a placebo-controlled parallel group design. Front. Neurol. 11:592104. doi: 10.3389/fneur.2020.592104
Pyle, A., Anugrha, H., Kurzawa-Akanbi, M., Yarnall, A., Burn, D., and Hudson, G. (2016). Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 38, 216.e7–216.e10. doi: 10.1016/j.neurobiolaging.2015.10.033
Pyle, A., Brennan, R., Kurzawa-Akanbi, M., Yarnall, A., Thouin, A., Mollenhauer, B., et al. (2015). Reduced CSF mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 78, 1000–1004. doi: 10.1002/ana.24515
Qin, X.-Y., Zhang, S.-P., Cao, C., Loh, Y. P., and Cheng, Y. (2016). Aberrations in peripheral inflammatory cytokine levels in parkinson disease: a systematic review and meta-analysis. JAMA Neurol. 73, 1316–1324. doi: 10.1001/jamaneurol.2016.2742
Riboldi, G. M., and Di Fonzo, A. B. (2019). GBA, gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells 8:364. doi: 10.3390/cells8040364
Ricciardi, L., Petrucci, S., Guidubaldi, A., Ialongo, T., Serra, L., Ferraris, A., et al. (2014). Phenotypic variability of PINK1 expression: 12 Years’ clinical follow-up of two Italian families. Mov. Disord. 29, 1561–1566. doi: 10.1002/mds.25994
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Brown, E., Hayes, M. A., et al. (2015). Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 82, 495–503. doi: 10.1016/j.nbd.2015.09.009
Saito, Y. (2014). Oxidized DJ-1 as a possible biomarker of Parkinson’s disease. J. Clin. Biochem. Nutr. 54, 138–144. doi: 10.3164/jcbn.13-108
Sánchez-Rodríguez, A., Martínez-Rodríguez, I., Sánchez-Juan, P., Sierra, M., González-Aramburu, I., Rivera-Sánchez, M., et al. (2021). Serial DaT-SPECT imaging in asymptomatic carriers of LRRK2 G2019S mutation: 8 years’ follow-up. Eur. J. Neurol. 28, 4204–4208. doi: 10.1111/ene.15070
Sano, K., Atarashi, R., Satoh, K., Ishibashi, D., Nakagaki, T., Iwasaki, Y., et al. (2018). Prion-like seeding of misfolded α-synuclein in the brains of dementia with lewy body patients in RT-QUIC. Mol. Neurobiol. 55, 3916–3930. doi: 10.1007/s12035-017-0624-1
Sardi, S. P., Clarke, J., Kinnecom, C., Tamsett, T. J., Li, L., Stanek, L. M., et al. (2011). CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. U.S.A. 108, 12101–12106. doi: 10.1073/pnas.1108197108
Saunders-Pullman, R., Alcalay, R. N., Mirelman, A., Wang, C., Luciano, M. S., Ortega, R. A., et al. (2015). REM sleep behavior disorder, as assessed by questionnaire, in G2019S LRRK2 mutation PD and carriers. Mov. Disord. 30, 1834–1839. doi: 10.1002/mds.26413
Saunders-Pullman, R., Mirelman, A., Alcalay, R. N., Wang, C., Ortega, R. A., Raymond, D., et al. (2018). Progression in the LRRK2-asssociated parkinson disease population. JAMA Neurol. 75, 312–319. doi: 10.1001/jamaneurol.2017.4019
Schulte, C., and Gasser, T. (2011). Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. Appl. Clin. Genet. 4, 67–80. doi: 10.2147/TACG.S11639
Shahnawaz, M., Mukherjee, A., Pritzkow, S., Mendez, N., Rabadia, P., Liu, X., et al. (2020). Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578, 273–277. doi: 10.1038/s41586-020-1984-7
Shimura, H., Hattori, N., Kubo, S. I, Mizuno, Y., Asakawa, S., Minoshima, S., et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305. doi: 10.1038/77060
Shu, L., Zhang, Y., Pan, H., Xu, Q., Guo, J., Tang, B., et al. (2018). Clinical heterogeneity among LRRK2 variants in Parkinson’s disease: a meta-analysis. Front. Aging Neurosci. 10:283. doi: 10.3389/fnagi.2018.00283
Smith, L., Mullin, S., and Schapira, A. H. V. (2017). Insights into the structural biology of Gaucher disease. Exp. Neurol. 298, 180–190. doi: 10.1016/j.expneurol.2017.09.010
Smith, W. W., Margolis, R. L., Li, X., Troncoso, J. C., Lee, M. K., Dawson, V. L., et al. (2005). Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 25, 5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 5:12813. doi: 10.7554/eLife.12813
Steinlechner, S., Stahlberg, J., Völkel, B., Djarmati, A., Hagenah, J., Hiller, A., et al. (2007). Co-occurrence of affective and schizophrenia spectrum disorders with PINK1 mutations. J. Neurol. Neurosurg. Psychiatry 78, 532–535. doi: 10.1136/jnnp.2006.105676
Stewart, T., Liu, C., Ginghina, C., Cain, K. C., Auinger, P., Cholerton, B., et al. (2014). Cerebrospinal fluid α-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am. J. Pathol. 184, 966–975. doi: 10.1016/j.ajpath.2013.12.007
Suzuki, M., Fujikake, N., Takeuchi, T., Kohyama-Koganeya, A., Nakajima, K., Hirabayashi, Y., et al. (2015). Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant α-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 24, 6675–6686. doi: 10.1093/hmg/ddv372
Thaler, A., Gurevich, T., Bar Shira, A., Gana Weisz, M., Ash, E., Shiner, T., et al. (2017). A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat. Disord. 36, 47–51. doi: 10.1016/j.parkreldis.2016.12.014
Thirstrup, K., Dächsel, J. C., Oppermann, F. S., Williamson, D. S., Smith, G. P., Fog, K., et al. (2017). Selective LRRK2 kinase inhibition reduces phosphorylation of endogenous Rab10 and Rab12 in human peripheral mononuclear blood cells. Sci. Rep. 7:10300. doi: 10.1038/s41598-017-10501-z
Tsika, E., Nguyen, A. P. T., Dusonchet, J., Colin, P., Schneider, B. L., and Moore, D. J. (2015). Adenoviral-mediated expression of G2019S LRRK2 induces striatal pathology in a kinase-dependent manner in a rat model of Parkinson’s disease. Neurobiol. Dis. 77, 49–61. doi: 10.1016/j.nbd.2015.02.019
Tsukada, H., Kanazawa, M., Ohba, H., Nishiyama, S., Harada, N., and Kakiuchi, T. (2016). PET imaging of mitochondrial complex I with 18F-BCPP-EF in the brains of MPTP-treated monkeys. J. Nucl. Med. 57, 950–953. doi: 10.2967/jnumed.115.169615
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M. K., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
van Rumund, A., Green, A. J. E., Fairfoul, G., Esselink, R. A. J., Bloem, B. R., and Verbeek, M. M. (2019). α-Synuclein real-time quaking-induced conversion in the cerebrospinal fluid of uncertain cases of parkinsonism. Ann. Neurol. 85, 777–781. doi: 10.1002/ana.25447
Vilas, D., Shaw, L. M., Taylor, P., Berg, D., Brockmann, K., Aasly, J., et al. (2016). Cerebrospinal fluid biomarkers and clinical features in leucine-rich repeat kinase 2 (LRRK2) mutation carriers. Mov. Disord. 31, 906–914. doi: 10.1002/mds.26591
Wang, S., Liu, Z., Ye, T., Mabrouk, O. S., Maltbie, T., Aasly, J., et al. (2017). Elevated LRRK2 autophosphorylation in brain-derived and peripheral exosomes in LRRK2 mutation carriers. Acta Neuropathol. Commun. 5:86. doi: 10.1186/s40478-017-0492-y
Wang, Y., Shi, M., Chung, K. A., Zabetian, C. P., Leverenz, J. B., Berg, D., et al. (2012). Phosphorylated α-synuclein in Parkinson’s disease. Sci. Transl. Med. 4:121ra20. doi: 10.1126/scitranslmed.3002566
Watzlawik, J. O., Hou, X., Fricova, D., Ramnarine, C., Barodia, S. K., Gendron, T. F., et al. (2020). Sensitive ELISA-based detection method for the mitophagy marker p-S65-Ub in human cells, autopsy brain, and blood samples. Autophagy 17, 2613–2628. doi: 10.1080/15548627.2020.1834712
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.M300227200
Williams, S. M., Schulz, P., and Sierks, M. R. (2016). Oligomeric α-synuclein and β-amyloid variants as potential biomarkers for Parkinson’s and Alzheimer’s diseases. Eur. J. Neurosci. 43, 3–16. doi: 10.1111/ejn.13056
Wilson, H., Pagano, G., Natale, E. R., de, Mansur, A., Caminiti, S. P., et al. (2020). Mitochondrial complex 1, sigma 1, and synaptic vesicle 2A in early drug-naive Parkinson’s disease. Mov. Disord. 35, 1416–1427. doi: 10.1002/mds.28064
Xicoy, H., Peñuelas, N., Vila, M., and Laguna, A. (2019). Autophagic- and Lysosomal-Related Biomarkers for Parkinson’s disease: lights and shadows. Cells 8:1317. doi: 10.3390/cells8111317
Yang, Y., Gehrke, S., Imai, Y., Huang, Z., Ouyang, Y., Wang, J.-W., et al. (2006). Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U.S.A. 103, 10793–10798. doi: 10.1073/pnas.0602493103
Yu, E., Rudakou, U., Krohn, L., Mufti, K., Ruskey, J. A., Asayesh, F., et al. (2021). Analysis of heterozygous PRKN variants and copy-number variations in Parkinson’s disease. Mov. Disord. 36, 178–187. doi: 10.1002/mds.28299
Keywords: genetic, biomarkers, Parkinson’s disease, alpha-synuclein, LRRK2
Citation: Tönges L, Kwon EH and Klebe S (2022) Monogenetic Forms of Parkinson’s Disease – Bridging the Gap Between Genetics and Biomarkers. Front. Aging Neurosci. 14:822949. doi: 10.3389/fnagi.2022.822949
Received: 26 November 2021; Accepted: 08 February 2022;
Published: 03 March 2022.
Edited by:
Danling Wang, University of South China, ChinaReviewed by:
Jieqiong Tan, Central South University, ChinaCopyright © 2022 Tönges, Kwon and Klebe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Klebe, c3RlcGhhbi5rbGViZUB1ay1lc3Nlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.