 Yu-Chao Li1
Yu-Chao Li1 Wei Zou
Wei Zou- 1Heilongjiang University of Chinese Medicine, Harbin, China
- 2School of Traditional Chinese Medicine, Ningxia Medical University, Yinchuan, China
- 3First Affiliated Hospital, Heilongjiang University of Chinese Medicine, Harbin, China
Stroke’s secondary damage, such as inflammation, oxidative stress, and mitochondrial dysfunction, are thought to be crucial factors in the disease’s progression. Despite the fact that there are numerous treatments for secondary damage following stroke, such as antiplatelet therapy, anticoagulant therapy, surgery, and so on, the results are disappointing and the side effects are numerous. It is critical to develop novel and effective strategies for improving patient prognosis. The ubiquitin proteasome system (UPS) is the hub for the processing and metabolism of a wide range of functional regulatory proteins in cells. It is critical for the maintenance of cell homeostasis. With the advancement of UPS research in recent years, it has been discovered that UPS is engaged in a variety of physiological and pathological processes in the human body. UPS is expected to play a role in the onset and progression of stroke via multiple targets and pathways. This paper explores the method by which UPS participates in the linked pathogenic process following stroke, in order to give a theoretical foundation for further research into UPS and stroke treatment.
Introduction
Stroke is a potentially fatal cerebrovascular event defined by brain tissue damage produced by a sudden rupture of cerebral vessels or cessation of cerebral blood supply, resulting in neurological dysfunction, including ischemic and hemorrhagic stroke (Landowski et al., 2020). Stroke has overtaken ischemic heart disease as the second largest cause of death worldwide (Benjamin et al., 2017; Li et al., 2021). According to a comprehensive analysis of Chinese population’s health, stroke has become the leading cause of mortality in the country (Zhou et al., 2019). According to current research, the ubiquitin proteasome system (UPS) plays a role in the molecular processes that lead to the occurrence and progression of stroke. UPS is also linked to a number of signaling pathways that cause injuries after stroke.
Ubiquitin Proteasome System
In cells, UPS is the primary non-lysosomal route for protein breakdown. This system destroys proteins with basic functions in addition to misfolded or oxidized proteins. Under physiological and pathological situations, it is a critical mechanism for maintaining protein homeostasis (Kramer et al., 2021). In addition, the system is participated in a variety of cellular functions, including DNA repair (Uckelmann and Sixma, 2017), endocytic trafficking (Hicke, 2001), and immunological response (Bednash and Mallampalli, 2016). UPS is vital in central nervous system disorders because it can clean up aberrant proteins in neurodegenerative diseases including Alzheimer’s and Parkinson’s disease (Graham and Liu, 2017). At the same time, the system controls the primary risk factors for cerebrovascular disease, such as atherosclerosis (Wilck and Ludwig, 2014), hypertension (Li et al., 2013), hyperlipidemia (Sharpe et al., 2020), type 2 diabetes (Sun-Wang et al., 2021), and so on.
Ubiquitination Process
Ubiquitination is usually mediated by UPS. Ubiquitin, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), ubiquitin protein ligase (E3), deubiquitinase (DUB), and the proteasome are all important components of the UPS (Kane et al., 2021). Ubiquitin is a tiny peptide with a molecular weight of about 8.5 kDa that consists of 76 amino acids. It has a highly conserved sequence and is found in eukaryotic cells in large quantities. Ubiquitin molecule contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine residue (M1), which allow ubiquitin molecules to connect to one another (Kwon and Ciechanover, 2017). Homotypic polyubiquitination modification and heterotypic polyubiquitination modification are two types of ubiquitin-ubiquitination modification (Komander and Rape, 2012). Furthermore, ubiquitin can undergo post-translational modifications such as phosphorylation or acetylation (Yau and Rape, 2016; Ohtake and Tsuchiya, 2017). This complicates the ubiquitination process. Generally, E1 forms a high-energy thiohydroxyester bond between the carboxyl group of ubiquitin’s C-terminal glycine and the sulfhydryl side chain of E1’s active cysteine under the action of ATP (Ciechanover, 2015). E2 is an intermediate enzyme in ubiquitination reaction, which can bind E1 and E3. E2 enzyme has a highly conserved ubiquitin-conjugating enzyme domain (UBC). On the UBC sequence, there is a cysteine site with symbolic activity, which can accept ubiquitin molecules activated by E1 and form a thioester bond with ubiquitin (Zheng and Shabek, 2017). Subsequently, E2 with activated ubiquitin binds to E3; Finally, E2 transfers the ubiquitin molecule to the substrate by inducing the formation of an isopeptide bond between the C-terminus of ubiquitin and a target lysine of the substrate. E3 specifically recognizes target proteins during ubiquitination (Buetow and Huang, 2016).
E3’s function reflects selectivity and efficiency of ubiquitination. Single subunit proteins and multisubunit complexes are two types of E3. There are four families of E3 single subunit proteins that have been identified: the HECT domain family, the RING domain family, the U-box domain family, and the N-recognition family. Cullin-RING and APC/C are the most common multisubunit complexes found in E3 (Zheng and Shabek, 2017). The human genome encodes approximately more than 600 ubiquitin ligases E3. Different E3 enzymes can specifically mediate the formation of different ubiquitin chain types. According to the different types of ubiquitin chain modification, it shows different functions. Polyubiquitination modification mediated by K48 or K11 ubiquitin chain is involved in proteasome degradation. Many E3 enzymes, such as SCF, gp78 and E6AP, can form K48 ubiquitin chains on substrate proteins, thus mediating the degradation of substrate proteins through proteasome (Wang and Pickart, 2005; Gao et al., 2016; Thacker et al., 2020). K11 ubiquitin chain was first considered as a regulator of cell fission, and its abundance increased with the increase of APC/C activity. APC/C, as an E3 enzyme, mediates the substrate Cyclin B1 to form K11 ubiquitin chain, which is finally degraded by proteasome, so as to promote the normal progress of cell mitosis (Song and Rape, 2010). In addition, studies have shown that K11 ubiquitin chain is often difficult to bind to 26S proteasome, but its degradation efficiency will be significantly enhanced when it forms a composite chain with K48 (Grice et al., 2015). Previous studies have shown that linear ubiquitination assembly complex (LUBAC) can modify NEMO by M1 ubiquitin chain to activate NF-κB pathway (Rahighi et al., 2009). This indicates that M1 ubiquitin chain is involved in activation of NF-κB pathway. ITCH and SMURF1 of the HECT family are members of the K29 ubiquitin chain’s ubiquitin ligases. SMURF1 has been found to add K29 ubiquitin chain to AXIN in WNT pathway. AXIN modified by K29 ubiquitin chain will not be degraded, but will lose the ability to interact with receptor LRP5/6 of WNT pathway (Fei et al., 2013). ITCH has been reported to catalyze Deltex in the NOTCH pathway to form K29 ubiquitin chain, which enables Deltex to enter the lysosomal pathway (Chastagner et al., 2006). In the process of DNA damage repair, RNF168 will form K27 polyubiquitin chain on histone, and then recruit 53BP1 and BRCA1 to DNA damage site to start the DNA damage repair (Brown and Jackson, 2015; Gatti et al., 2015).
Proteasome
The proteasome is a multi-subunit protease complex that is found in both the cytoplasm and the nucleus. In mammalian cells, it is the primary neutral protein hydrolase. It is ubiquitin-dependent and possesses a variety of protein hydrolase functions. In UPS, the term “proteasome” usually refers to the 26S proteasome. The 20S proteasome and the 19S subcomplex make up the 26S proteasome (Gillette et al., 2008). Before the target proteins enter the 20S proteasome, each 19S sub complex comprises several ATPase active sites and ubiquitin binding sites that can recognize ubiquitinated target proteins, deubiquitinate the target proteins, and open the structural folding of the target proteins (Matyskiela et al., 2013). The 20S proteasome of eukaryotic cells contains two α rings and two β rings, these α rings and β rings together form a cylindrical structure, both α rings or β rings are composed of seven subunits. α rings are found in the outer layer of the 20S protease and are mostly employed for substrate identification; β rings are found in the inner layer and are primarily responsible for substrate degradation (Fricker, 2020). The β1, β2, and β5 of the seven β subunits have proteolytic activities, which are referred to as caspase-like activity, trypsin-like activity, and chymotrypsin-like activity, respectively, enzymolysis of acidic, alkaline, hydrophobic, or aromatic amino acid residues at the carboxyl end (Unno et al., 2002; Figure 1).
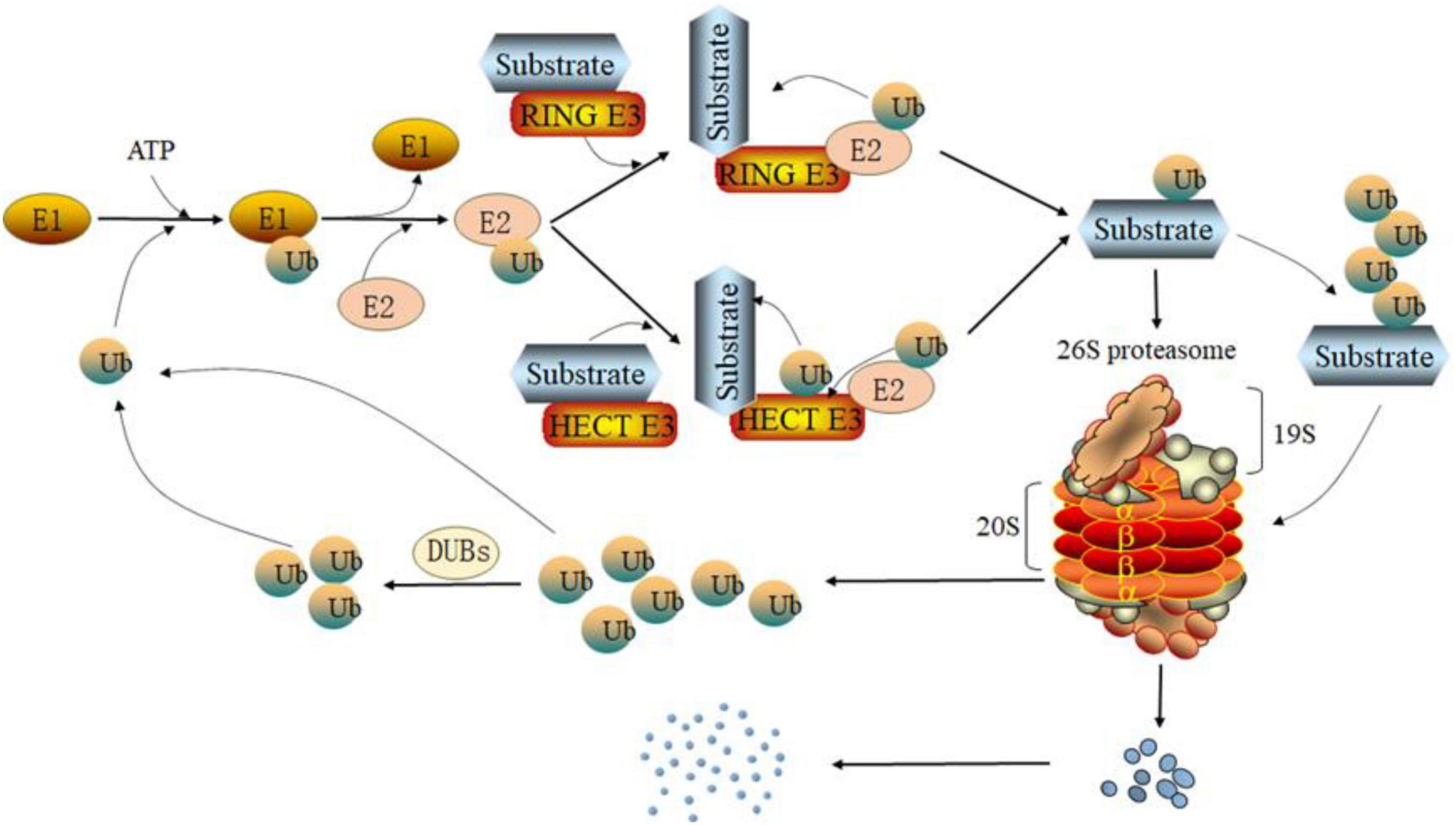
Figure 1. Degradation pathway of the ubiquitin proteasome system (UPS). Under the action of ATP, the ubiquitin activating enzyme E1 forms a high-energy thiohydroxyester bond between the carboxyl group of ubiquitin’s C-terminal glycine and the sulfhydryl side chain of E1’s active cysteine. Then, ubiquitin-conjugating enzyme E2 receives ubiquitin activated by E1 through a cystine site on its ubiquitin-conjugating enzyme domain (UBC) and forms a thioester bond. Depending on the type of ubiquitin protein ligase E3, E2 can transfer ubiquitin to the substrate directly (RING E3s) or indirectly (HECT E3s) and form an isopeptide between the substrate and ubiquitin. In addition, ubiquitin can itself be the target of the ubiquitination cascade. Finally, the ubiquitinated substrate is degraded by the 26S proteasome. The ubiquitin separated from the substrate will re-enter the ubiquitination cycle.
Occurrence and Development of Stroke
According to the causes, stroke can be classified as hemorrhagic or ischemic, with ischemic stroke accounting for around 87% of all cases (Virani et al., 2021). Due to cerebral ischemia or hemorrhage, the normal blood supply of neurons is destroyed, which leads to a series of pathophysiological reactions and finally to the death of nerve cells. The mechanisms involved in nerve injury include excitotoxicity, mitochondrial dysfunction, free radical disorder, inflammation, apoptosis, necrosis, autophagy and so on. According to analyses based on data from the Global Burden of Disease (GBD) study, modifiable risk factors such as hypertension, obesity, hyperglycemia, hyperlipidemia, atrial fibrillation and renal dysfunction account for 87% of stroke risk, while behavioral risk factors such as smoking, sedentary behavior, and an unhealthy diet account for 47%. Air pollution was found to be responsible for 30% of the global risk of stroke (Collaborators, 2020). In addition, age, gender and race are also associated with stroke (Diseases and Injuries, 2020). The combined action of multiple risk factors leads to pathological changes of cardiovascular and cerebrovascular system, including but not limited to atherosclerosis, arteriolar fat hyalinization and fibrin like necrosis, coronary artery disease, and myocardial injury. These pathological changes provide clues for the occurrence, recurrence, and secondary prevention of stroke.
Although brain tissue makes up only 2% of total body weight, cerebral blood flow accounts for 15% of cardiac output. The aerobic oxidation of glucose provides the majority of the energy required by brain tissue, although the glucose reserve is limited. Though brain oxygen use contributes for 23% of total body oxygen consumption, the oxygen reserve is small (Hyder et al., 2013). For these factors, the brain is extremely vulnerable to ischemia and hypoxia. Following a stroke, brain ischemia, and hypoxia are major concerns. In the ischemic core the major mechanism of cell death is energy failure. Neurons cannot create the ATP needed to supply the ionic pumps that maintain the ionic gradient across the neuronal membrane, primarily the Na+/K+ ATPase, without oxygen or glucose. As a result, a substantial amount of Na+ and Ca2+ accumulate in the cytoplasm, causing organelles swelling and degeneration, loss of cell membrane integrity, and ultimately cell necrosis (Lipton, 1999). In the ischemic penumbra, the decrease in blood flow due to collateral blood supply is not enough to lead to rapid energy failure, and neurons still survive for a long time after ischemia and hypoxia. The excessive accumulation of extracellular glutamate is an important factor leading to ischemic penumbra injury (Mazzocchetti et al., 2020). Due to the excessive accumulation of glutamate, the overactivation of subtypes of N-methyl-D-aspartate receptors (NMDARs) leads to cellular calcium overload. Therefore, calcium-dependent proteases are activated, such as calpains, resulting in nerve cell damage (Rami et al., 2000). In addition, some studies have shown that calpains are involved in the activation of Caspase-3, which may be an important mechanism of neuronal apoptosis after stroke (Blomgren et al., 2001). Meanwhile, mitochondrial dysfunction is caused by Ca2+ overload which leads to mitochondrial permeability transition pore (mPTP) opening (Zhu et al., 2018). Of note, mitochondrial dysfunction leads to insufficient ATP production, which will further lead to Ca2+ accumulation and form a vicious circle. Nerve cell survival is aided by the removal of defective mitochondria. Mitochondrial autophagy is an important regulatory process for the quality and quantity of mitochondria. According to a report, UPS plays a key function in the regulation of mitochondrial autophagy (Alsayyah et al., 2020). Mitochondria are organelles that operate as oxidative energy centers and are required for cell survival, but aging or damaged mitochondria produce deadly reactive oxygen species (ROS; Liu et al., 2018). ROS are important signaling molecules in oxidative stress. In the pathological manifestations of stroke, relatively excessive ROS will destroy the homeostasis of intracellular environment, resulting in oxidative stress and mitochondrial damage (Sims and Muyderman, 2010). The main defensive response to oxidative and electrophilic stressors is the Keap1-Nrf2 pathway. UPS can strictly regulate the transcription of nuclear factor erythroid2-related factor (Nrf2) through this pathway, thereby affecting the antioxidant process (Baird and Yamamoto, 2020). Whether it is mitochondrial dysfunction or oxidative stress, it will eventually lead to neuronal damage. Inflammation is one of the most prevalent pathological signs following a stroke, and it is induced by a range of factors, including microglia activation (Koh et al., 2018) and cytokine involvement (Xu et al., 2018). Inflammation has the potential to be neurotoxic., which can lead to neuronal death (Liddelow et al., 2017). Hypoxia-inducible factor-1 (HIF-1) comprised by α and β subunits is a protein that regulates the expression of genes that code for erythropoietin (EPO) and vascular endothelial growth factor (VEGF), as well as genes involved in glucose transport and glycolysis, such as glucose transporter-1 (GLUT1), pyruvate dehydrogenase kinase 1 (PDK1), and lactate dehydrogenase A (LDHA; Leu et al., 2019). In the hypoxic state caused by stroke, the oxygen-dependent HIF prolyl hydroxylase domain (PHD) is inactivated, leading to the stabilization of HIF-1α, followed by its translocation into the nucleus, where it forms a heterodimeric complex with HIF-1β. This complex, called HIF-1, interacts with DNA and activates the expression of multiple target genes encoding proteins that help increase the tissue’s oxygen supply by boosting erythropoiesis and angiogenesis (Semenza, 2004, 2009). UPS has been implicated in the control of HIF in recent research (Delpeso et al., 2003). Therefore, regulating related pathways and their key proteins through UPS may be an effective solution to protect neurons and prevent cell death during stroke.
Regulatory Mechanism of Ubiquitin Proteasome System
NF-κB Pathway and Ubiquitin Proteasome System
NF-κB is a transcription factor family that includes NF-κB1 (P50/p105), NF-κB2 (p52/P100), and three RelA (p65), RelB, and c-Rel (REL) proteins (Caamano and Hunter, 2002). As a hub in signal transduction pathway, NF-κB can regulate the expression of many genes involved in cell inflammation, immune response, cell growth and development (DiDonato et al., 2012). IκB protein is a family of constitutive inhibitors of NF-κB, including IκBα, IκBβ, IκBγ, IκBζ, IκBε, Bcl-3, p100 and p105 (Yamazaki et al., 2001; Chiba et al., 2013). IKK complex consists of IKKα, IKKβ and Nemo, which is the kinase of IκB (Durand and Baldwin, 2017). Under normal conditions, NF-κB and inhibitor IκB binding exists in the cytoplasm in an inactive potential state (Wang et al., 2019).
NF-κB signaling pathway mainly includes classical and non-classical activation pathways (Figure 2). In the classical one, IKK complex is activated when cells are stimulated by proinflammatory factors, growth factors, immune receptor ligands and stress response (Fricker, 2020), in which IKKβ (ser177 and ser181) and IKKα (ser176 and ser180) sites in the amino terminal kinase domain are phosphorylated. Due to their activation, serine residues of IκB are phosphorylated (Ling et al., 1998; Delhase et al., 1999). The polyubiquitination mode of IκBα depends on the phosphorylation of two serine residues (ser32 and ser36), while the phosphorylation sites of IκBβ are ser19 and ser23 (Chen, 2012). The phosphorylation of IκB leads to the covalent binding of lysine residues at positions 21 and 22 of its amino terminal to multiple ubiquitin molecules through SCF type E3 ubiquitin ligase complex. This binding changes the spatial conformation of IκB, resulting in its recognition and degradation by ATP dependent 26S proteasome (Chen et al., 1995; Karin and Ben-Neriah, 2000; Caamano and Hunter, 2002). Following depolymerization, the liberated NF-κB dimer is activated by several post-translational modifications before being transported to the nucleus and combining with particular DNA sequences to increase target gene transcription (Zhang et al., 2017). Inflammatory cytokines like TNF-α, IL-2, IL-6, and INF-γ are then produced, reactivating NF-κB and causing inflammatory damage to cell tissues (Mitchell and Carmody, 2018). The activation of non-classical NF-κB pathway depends on receptor activated kinase NIK, which can activate IKKα (Cildir et al., 2016). The phosphorylation of NF-κB2 p100 mediated by activated IKKα leads to its own ubiquitination, which is recognized by proteasome and partially degraded to p52. Finally, p52-RelB heterodimer enters the nucleus to start transcription (Xiao et al., 2001; Sun, 2011). Therefore, NF-κB plays a crucial role in acute and chronic inflammatory illnesses as the center regulating the expression of pro-inflammatory genes in the inflammatory process (Baker et al., 2011).
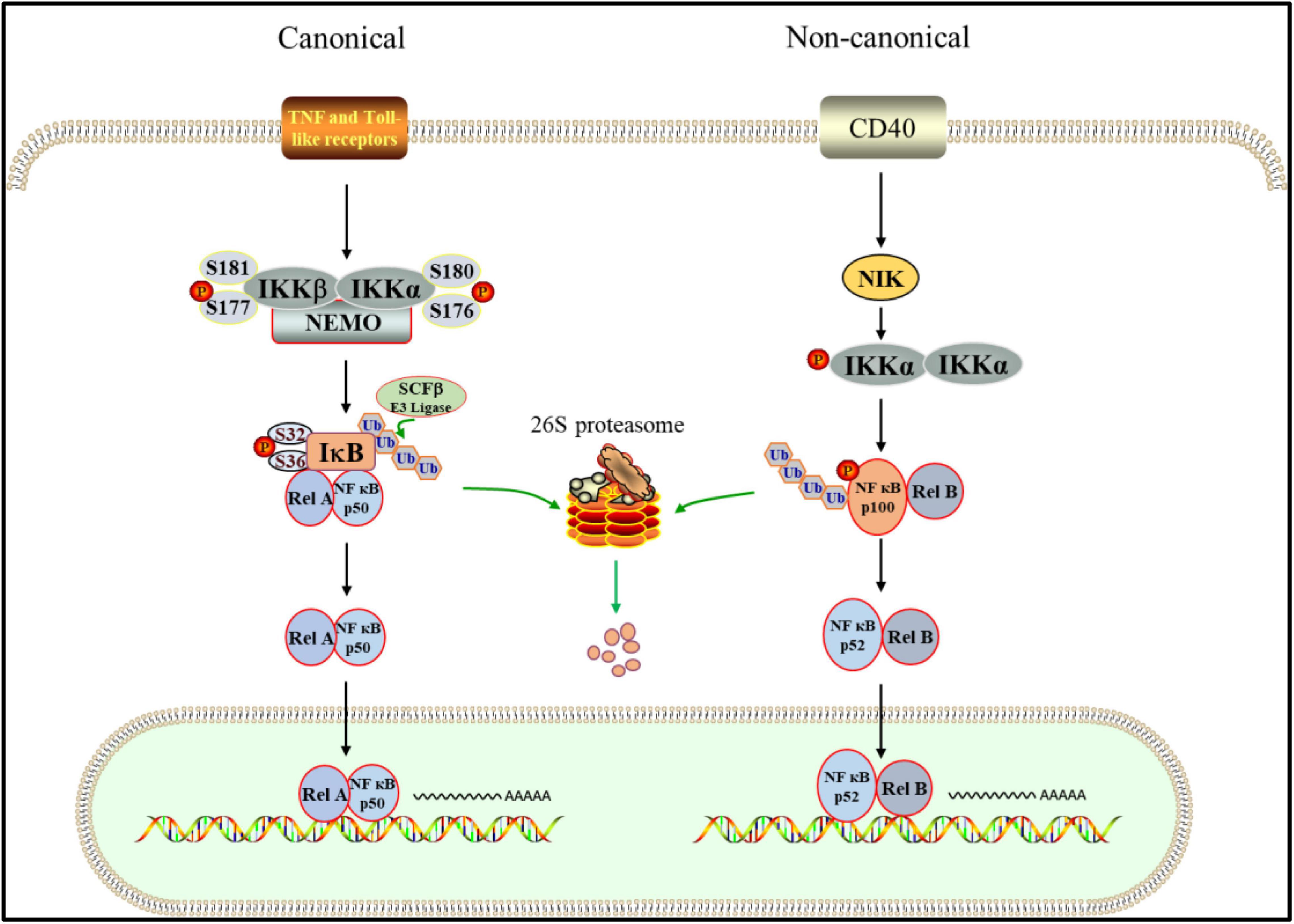
Figure 2. UPS is involved in canonical and non-canonical NF-κB pathway. In the canonical way. After the inflammatory receptor is activated, the activated IKK complex phosphorylates ser32 and ser36 of IκB, which promotes the ubiquitination of IκB mediated by SCFβ. Subsequently, IκB is targeted to the 26S proteasome for proteolysis. NF- κB dimer was released and transferred into the nucleus to start the transcription of target genes. In the non-canonical pattern. IKK is activated by NIK, which leads to phosphorylation of p100. Then, Phosphorylated p100 is degraded by proteasome after ubiquitination, in which p52-RelB heterodimer is produced. Finally, the p52-RelB heterodimer enters the nucleus to start transcription.
PINK1/Parkin Pathway and Ubiquitin Proteasome System
The PTEN-induced putative kinase 1(PINK1)/Parkin pathway is one of the most prevalent mechanisms for controlling mitochondrial quality and abundance (Shen et al., 2021). PINK1 is a mitochondrial serine (Ser)/threonine (Thr) kinase with a mitochondrial targeting sequence (MTS), a transmembrane (TM) segment, and a Ser/Thr kinase domain that is nuclear encoded (Eiyama and Okamoto, 2015). PINK1, as a Parkin upstream component, is required for Parkin activation and recruitment to depolarized mitochondria (Miller and Muqit, 2019). Parkin has an N-terminal ubiquitin-like (UBL) domain, three RING domains (RING0, RING1, and RING2), and an in-between RING(IBR) domain that separates RING1 and RING2 as an E3 ligase (Biswas et al., 2020). When the MTS and TM of PINK1 reach the inner mitochondrial membrane under healthy conditions, the transmembrane segment is cleaved in the form of protein hydrolysis by the presenilins-associated rhomboid-like protein (PARL) found in the inner membrane. Cleaved PINK1 is released into the cytoplasm, exposing destabilizing amino acid residues at its N-terminus. E3 ubiquitin ligases (UBR1, UBR2, and UBR4) ubiquitinate them with N-terminal rules and degrade fast by proteasome (Yamano and Youle, 2013; Figure 3A). Therefore, the content of PINK1 in cytoplasm is very low and Parkin is not activated (Pickrell and Youle, 2015). When the mitochondrial membrane potential is abnormal, PINK1 avoids PARL-mediated processing and N-end rule-dependent degradation by forming a stable association with the translocase of the outer membrane (TOM) and accumulating on the outer mitochondrial membrane (OMM; Lazarou et al., 2012). PINK1 accumulates on the OMM can activate parkin in two ways. On the one hand, Parkin is activated by PINK1 phosphorylating ser65 in the Parkin UBL domain (Bingol and Sheng, 2016). On the other hand, PINK1 phosphorylates ser65 in ubiquitin, which is coupled with OMM proteins at basic levels. Parkin’s affinity for phosphorylated ubiquitin is what causes it to be found in mitochondria. The activated Parkin further binds ubiquitin to the OMM protein, and then ubiquitin is phosphorylated by PINK1 (Bragoszewski et al., 2017; Figure 3B). On mitochondria, phospho-ubiquitin produced by PINK1 serves as an autophagy signal, which Parkin amplifies (Lazarou et al., 2015). Subsequently, ubiquitinated OMM protein recruits autophagic adaptor protein SQSTM1/p62 to damaged mitochondria (Geisler et al., 2010), and promotes its degradation through autophagy. There are some negative regulatory mechanisms related to UPS in PINK1/Parkin pathway, which are very important for the stability of mitochondrial autophagy. Ubquitin-specific protease30 (USP30) is an OMM localized DUB, which antagonizes PINK1/Parkin mediated mitochondrial autophagy by deubiquitination of OMM proteins. The presence of USP30 can maintain the steady state of ubiquitination of OMM protein and prevent excessive mitochondrial autophagy (Tanaka, 2020). Furthermore, UBL exhibits a strong affinity for the Rpn13 subunit of the 26S proteasome regulatory granules. The proteasome is attracted to mitochondria by this affinity, which increases proteasome degradation of ubiquitinated OMM protein and Parkin (Aguileta et al., 2015).
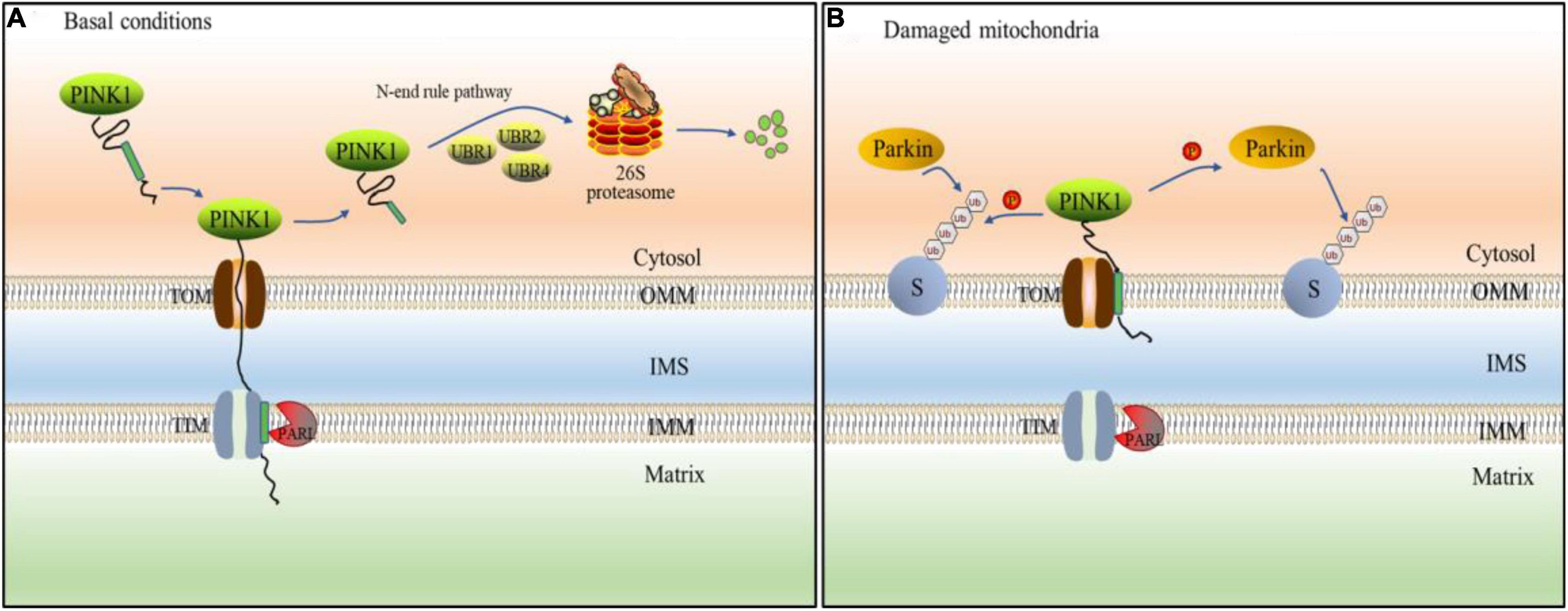
Figure 3. PINK1/Parkin pathway mediates ubiquitination of mitochondria. (A) Under basal condition, PINK1 passes through the outer mitochondrial membrane (OMM) to the inner membrane (IMM), and its transmembrane segment is hydrolyzed by presenilins-associated rhomboid-like protein (PARL) protein. The treated PINK1 is released into the cell matrix and mediated proteasome degradation by E3 ubiquitin ligases (UBR1, UBR2, and UBR4). (B) When mitochondrial disorder, PINK1 cannot be transported across membranes, so that it cannot be destroyed by PARL. Instead, PINK1 stably binds to the OMM and accumulates on it. Therefore, PINK1 can directly phosphorylate ubiquitin or Parkin. Parkin is recruited by phosphorylated ubiquitin to various mitochondrial substrate proteins(S) in which mitochondrion is ubiquitinated.
Keap1-Nrf2 Pathway and Ubiquitin Proteasome System
Nrf2 is the main regulator of redox and metabolic homeostasis. It has seven Nrf2 ECH homologous domains (Neh1-Neh7), and each domain has different functions (Hayes and Dinkova-Kostova, 2014). As the most important regulatory domain of Nrf2, Neh2 includes two motifs, DLG and ETGE, which can regulate the stability and ubiquitination of Nrf2 by binding to other proteins such as Keap1 (Tong et al., 2006). Keap1 is a substrate adaptor protein of Cullin3 (Cul3)-dependent E3 ubiquitin ligase complex, which can be assembled with Cul3 and Rbx1 (ring-box1). Keap1 serves as a substrate adapter, whereas Rbx1 binds to ubiquitin-loaded E2 and Cul3 serves as a scaffold between Keap1 and Rbx1, which can regulate Nrf2 (Tonelli et al., 2018). Keap1 contains three functional domains, including a BTB domain, an IVR and a Kelch or DGR domain (Sajadimajd and Khazaei, 2018). The N-terminal BTB domain of Keap1 can bind Cul3, which is necessary for Keap1 dimerization (Suzuki and Yamamoto, 2015). Under normal conditions, the Neh2 domain of Nrf2 interacts with the Kelch/DGR domain in Keap1 through the mediation of DLG and ETGE motifs. Keap1-Cul3-E3 ubiquitin ligase targets multiple lysine residues located in the Neh2 domain at the N-terminal of Nrf2 and promotes the ubiquitination of Nrf2. The ubiquitinated Nrf2 is transported to the 26S proteasome, where it is degraded (Canning et al., 2015; Karunatilleke et al., 2021; Figure 4A). Critical cysteine residues in Keap1, particularly Cys151, operate as sensors of these cellular damages under oxidative stress conditions and become covalently changed by electrophilic molecules or ROS (Zhang and Hannink, 2003). Such changes cause Keap1 to shift conformation, most likely by disrupting the low-affinity link between the Kelch domain and the DLG-motif, resulting in decreased ubiquitylation of Nrf2, inhibiting UPS-mediated degradation and thereby boosting Nrf2 protein levels (Baird et al., 2013). Then Nrf2 translocases to the nucleus and binds to the ARE/EpRE of the target gene through heterodimerization with sMAF protein to induce the expression of a series of cell protective genes, such as NQO1, GST, HMOX1, GCL, GSH, etc. (Kansanen et al., 2013; Fao et al., 2019; Figure 4B), so as to reduce or eliminate oxygen free radicals and improve the antioxidant capacity of cells and tissues.
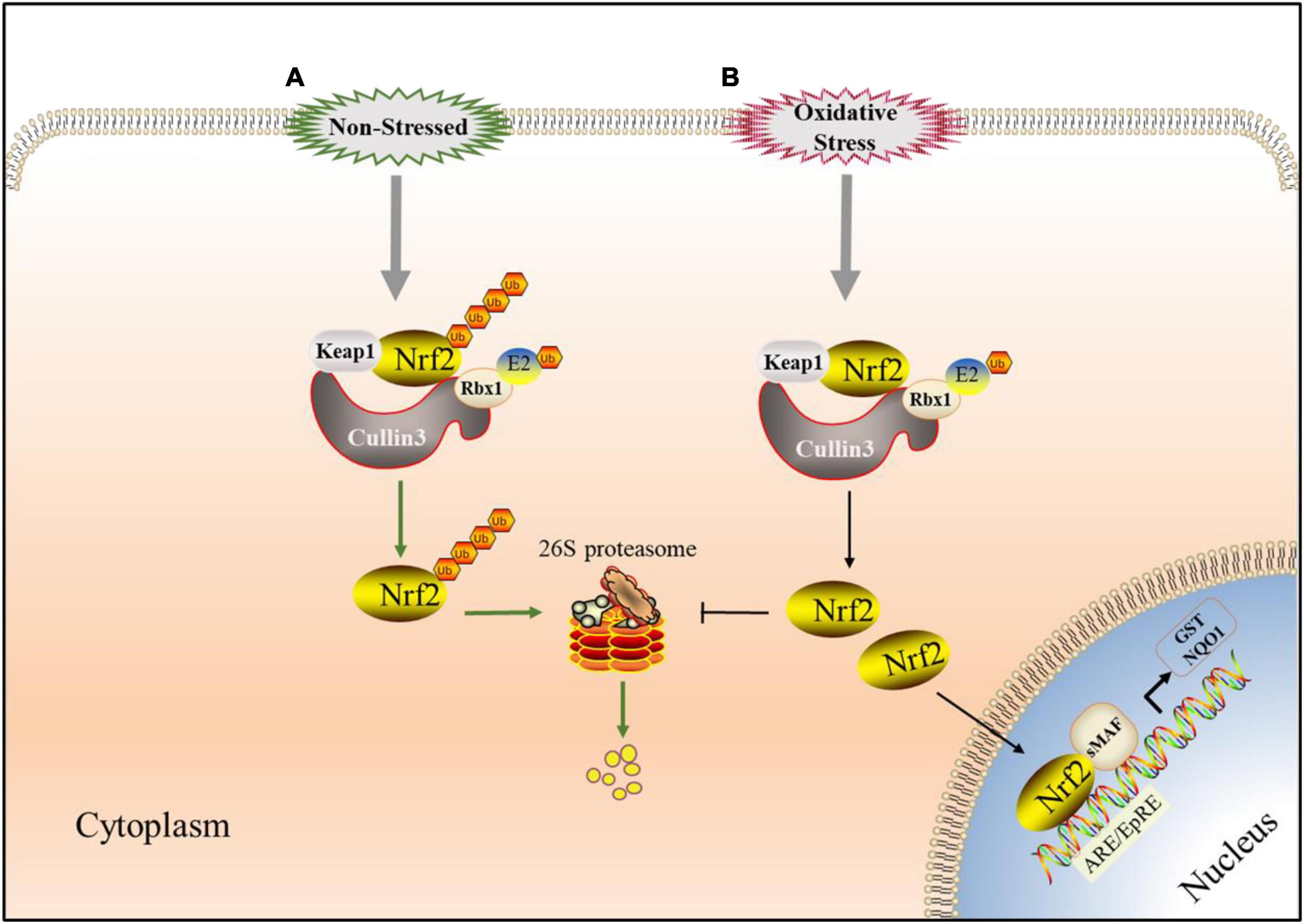
Figure 4. Keap1-Nrf2 signaling pathway involved in UPS. (A) Under non-stressed conditions, Nrf2 binds to Keap1 complex which has E3 ligase activity in the cytoplasm. When Nrf2 is ubiquitinated, it is targeted to the 26S proteasome for proteolysis which keep it low in cytoplasm. (B) Under oxidative stress conditions, Critical cysteine residues in Keap1 is covalently-modified by electrophilic species or reactive oxygen species (ROS) and Nrf2 avoides Keap1-mediated ubiquitination modification. Then Nrf2 translocated to the nucleus, starts ARE/EpRE transcription through heterodimerization with SMAF protein, and induced the expression of a series of cytoprotective genes.
HIF-1 Pathway and Ubiquitin Proteasome System
Hypoxia inducible factor (HIF) is an important transcription factor regulated by the change of oxygen concentration (Wang et al., 1995). It is composed of a α subunit regulated by oxygen concentration and a constitutively expressed β subunit, in which α subunit has three functional forms: HIF-1α, HIF-2α and HIF-3α. HIF is divided into HIF-1, HIF-2 and HIF-3 according to different α subunits. α subunit and β subunit have two important domains: the basic helix-loop-helix (bHLH) and Per-Arnt-Sim (PAS). The bHLH and PAS are required for dimerization between HIF-1α and HIF-1β (Zhang et al., 2011; Shu et al., 2019). HIF-1α is generally expressed in all cells, while HIF-2α and HIF-3α are selectively expressed in some tissues (Majmundar et al., 2010). HIF-1α which has two transactivation domains, N-terminal transactivation domains (N-TAD) and C-terminal transactivation domains (C-TAD), as well as an oxygen-dependent degradation domain (ODDD) that mediates oxygen-regulated stability, is not only the regulatory subunit of HIF-1, but also its active subunit (Ruas et al., 2002). The stability and activity of α subunit are regulated by post-translational modifications such as hydroxylation, ubiquitination, acetylation and phosphorylation. Among them, HIF-1α is mainly regulated by the PHD (Ke and Costa, 2006). Under the normoxic state, the prolyl residues at sites 402 and 564 of HIF-1α are hydroxylated by PHD. Hydroxylated HIF-1α binds to Von Hippel-Lindau (VHL), which together with Elongin C, Elongin B, Cullin-2, and Rbx1, forms the VCB-Cul2 E3 ligase complex. Subsequently, it is ubiquitinated, then recognized by 26S proteasome and degraded (Ohh et al., 2000). Since PHD activity is suppressed during hypoxia, VHL is unable to detect HIF-1α, and hence HIF-1α is not degraded by UPS. Subsequently, accumulated HIF-1α enters the nucleus from the cytoplasm, where it is joined to create a dimer with HIF-1β (Tarhonskaya et al., 2015). In the nucleus, p300/CBP associated factor (PCAF) combines with the C-TAD of HIF-1α to form a complex. The complex combines with the hypoxia response element (HRE) in the promoter region of hypoxia response genes to promote the transcription of hypoxia response gene and cause a series of adaptive responses of cells to hypoxia (Dengler et al., 2014). In addition, HIF-1α is also regulated by the factor inhibiting HIF (FIH; Kaelin and Ratcliffe, 2008). In normoxic environment, FIH hydroxylates the asparagine residue (N803) of HIF-1α to prevent the connection between p300/CBP and C-TAD, thereby reducing the transcriptional activity of HIF-1α (Lando et al., 2002b). Under hypoxia, the hydroxylation of FIH is inhibited, which promotes the interaction between HIF-1α and p300/CBP and leads to the transcription of target genes (Lando et al., 2002a; Figure 5).
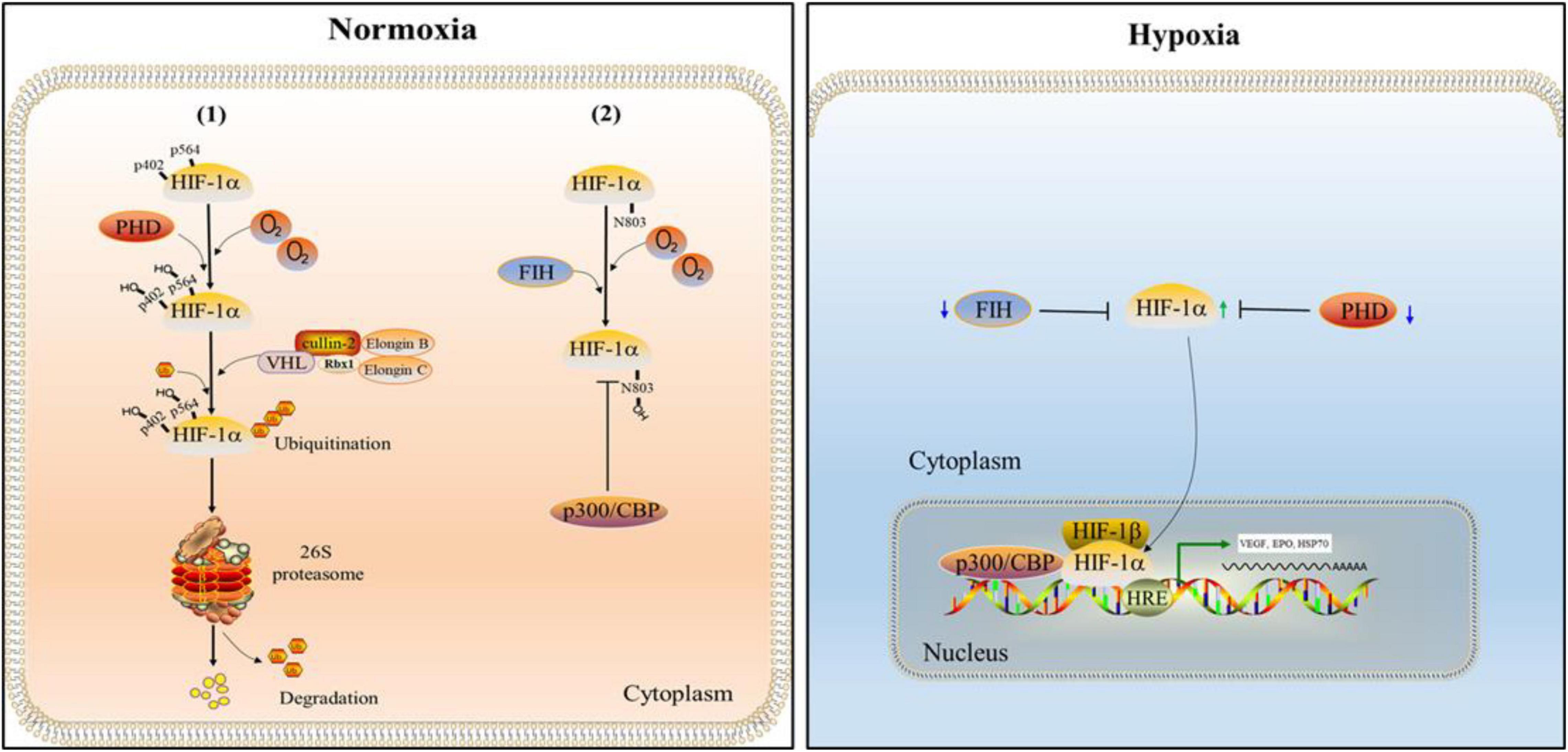
Figure 5. The regulation of HIF-1. Under the normal oxygen condition, the prolyl residues at sites 402 and 564 of HIF-1α are hydroxylated by the prolyl hydroxylase domain (PHD). Subsequently, hydroxylated HIF-1α is ubiquitinated by Von Hippel-Lindau (VHL) and degraded by 26S proteasome. In addition, the asparagine residue (N803) of HIF-1α is hydroxylated by the factor inhibiting HIF (FIH) which inhibits the connection between p300/CBP and C-TAD, thereby reducing the transcriptional activity of HIF-1α. When hypoxia occurs, the activities of FIH and PHD are inhibited. Therefore, HIF-1α can enter the nucleus to form complexes with HIF-1β and p300/CBP, and promote the transcription of hypoxia response genes and cause a series of adaptive responses of cells to hypoxia.
The Role of UPS in Signal Pathway After Stroke
UPS Participation in NF-κB Pathway and Stroke
UPS is essential for maintaining protein homeostasis and preventing damaging protein aggregation in cells (Budenholzer et al., 2017). Inflammation is the result of a complicated interplay between soluble substances and cells (Medzhitov, 2008). Inflammatory cell infiltration and activity frequently result in long-term tissue damage (Feehan and Gilroy, 2019). UPS can play an important role in the inflammatory process by regulating a variety of inflammatory regulatory proteins (Goetzke et al., 2021). It was found that inflammatory response widely exists after cerebral ischemia and acute ischemic stroke (Chamorro et al., 2016). It can be seen that inhibiting the activity of NF-κB can reduce the nerve injury after intracerebral hemorrhage or cerebral ischemia (Liu et al., 2019; Shang et al., 2019). In recent years, many studies have reported that the effect of UPS is related to the activation of NF-κB. Intervening UPS can prevent the degradation of IκB and the activity of NF-κB and combine them in cell solute. Ginsenoside Rd treatment can restore IκB expression in the cytoplast after ischemia injury by decreasing proteasome activity, therefore suppressing NF-κB activity and protecting neurons, according to a study (Zhang et al., 2020). Phillips et al. applied proteasome inhibitor PS519 to rats with focal cerebral ischemia and found that PS519 can reduce the inflammatory response by restraining NF-κB and improve the recovery of neurological function in rats with brain injury (Phillips et al., 2000). According to another study, the neuroprotective effect of PS519 may be related to its involvement in the regulation of cell adhesion molecules ICAM-1 and E-selectin, because these two adhesion molecules play a key role in the adhesion and exudation of neutrophils and macrophages across the blood-brain barrier (Berti et al., 2003). These suggest that the effect of proteasome inhibitors on inflammatory response may be multi-channel. Moreover, although early intervention of UPS has a neuroprotective effect on inhibiting NF-κB overexpression at the transcriptional level, the decrease of long-term proteasome activity is related to intracellular protein aggregation, delayed neuronal degeneration, and death (Meller, 2009). It suggested that UPS may played a dual role in neurons after ischemia. Although proteasome inhibitors have been shown to provide protection in cerebral ischemia, other nerve injury problems caused by decreased proteasome activity cannot be ignored (Caldeira et al., 2014). Due to the non-selectivity of most proteasome inhibitors, the application of proteasome inhibitors is limited (Wojcik and Di Napoli, 2004). Chen et al. suggested that selective immunoproteasome inhibitors may be a promising strategy for stroke treatment. They discovered that inhibiting the expression of p65 and reducing infarction volumes in rats may be accomplished by inhibiting the low molecular weight protein 2 (LMP2), a significant catalytic subunit of immunoproteasome (Chen et al., 2015).
PINK1/Parkin and Cerebral Stroke Regulated by UPS
PINK1/Parkin pathway is one of the classical pathways of mitochondrial autophagy. Under normal conditions, UPS forms a strict PINK1 and Parkin regulatory mechanism to maintain the balance of mitochondrial autophagy and prevent mitochondrial damage caused by excessive mitochondrial autophagy (Oshima et al., 2021). In the stroke induced by mitochondrial dysfunction (Youn et al., 2021; Zhou et al., 2021), PINK1/Parkin pathway mediates ubiquitination of dysfunctional mitochondrial OMM protein, and then clears abnormal mitochondria through crosstalk with autophagy (Geisler et al., 2010). In recent years, there has been more and more studies on this mechanism, which to some extent shows that UPS plays an important role in PINK1/Parkin pathway in the process of brain injury in stroke. At present, the researches on the pathway in stroke mainly focus on its relationship with autophagy, but ignore the role of UPS. In fact, mitochondrial ubiquitination mediated by PINK1/Parkin pathway is the basis of mitochondrial autophagy. Inactive PINK1 cannot activate or recruit Parkin to mitochondria (Narendra et al., 2008; Matsuda et al., 2010). The UPS mechanism can be inhibited to prevent PINK1/Parkin-mediated ubiquitination of OMM proteins (Chan et al., 2011). Furthermore, inhibiting Parkin mitochondrial translocation, lowering Parkin phosphorylation, and lowering the quantity of phosphorylated ubiquitin (pser65 Ub) can all be used to prevent mitochondrial autophagy triggered by various mitochondrial damage causes (Wang et al., 2018a). OMM proteins could be destroyed through an autophagy-independent UPS route, according to Rakovic et al. The bigger OMM proteins MFN2 and TOM70 were only partially ubiquitinated and primarily destroyed by the lysosomal system, whereas the smaller OMM proteins TOM40 and TOM20 were only slightly ubiquitinated and mostly degraded by UPS (Rakovic et al., 2019). It showed that depolarized mitochondrial membrane proteins could be degraded by two different mechanisms: UPS or lysosomal mediated protein hydrolysis. At the same time, the degradation of larger OMM protein affected the stability of OMM, led to its rupture, exposed mitochondrial inner membrane (IMM) to the cytoplasmic environment, and caused drastic changes in IMM structure and morphology, which might eventually lead to the secondary degradation of IMM and matrix protein (Yoshii et al., 2011). In the oxygen-glucose deprivation (OGD) neuronal model, PINK1 knockout mice (PINK1–/–) were more sensitive to ischemic injury than control group (Imbriani et al., 2020). Moreover, in traumatic brain injury (TBI), the loss of Parkin would increase the production of ROS, promote oxidative stress and further lead to neuronal death (Mukhida et al., 2005). In a rat model of ischemia, PINK1/Parkin-mediated mitochondrial autophagy may perform a neuroprotective role in hippocampus neurons (Wu et al., 2018). The deletion of either the PINK1 or Parkin genes has been shown to cause aggregated neuronal damage. Increasing the expression of PINK1 and Parkin, on the other hand, could prevent a huge number of nerve cells from dying. Therefore, it is necessary to pay attention to the role of PINK1/Parkin/UPS mechanism in mitochondrial damage after stroke, rather than autophagy.
UPS Control in Stroke and Keap1-Nrf2
Oxidative stress refers to the imbalance of redox balance caused by the production of excessive ROS after the body is stimulated by the outside world, resulting in the damage of cell tissue (Hybertson et al., 2011). Because of its high oxygen consumption and fat content, the brain is prone to oxidative injury. The role of oxidative stress in ischemia-reperfusion brain injury has long been recognized (Chamorro et al., 2016). Nrf2 is the main regulator of endogenous and exogenous stress defense mechanisms in cells and tissues. Its primary role is to activate the antioxidant response and trigger the transcription of a number of genes in order to protect cells from oxidative stress and restore intracellular homeostasis (Villeneuve et al., 2010; Chen and Maltagliati, 2018). That Nrf2 escape from Keap1 repression is the crucial event for Nrf2-mediated activation (Kaspar et al., 2009). Oxidative stress caused by cerebral ischemia or intracerebral hemorrhage affects the conformation of Keap1. Then, Nrf2 dissociates and transfers to the nucleus, binds to ARE, and stimulates the target gene expression of downstream antioxidant enzymes and other cytoprotective proteins (Hu et al., 2016; Jiang et al., 2017). The expression of Nrf2 is highly up-regulated in ischemic brain tissue, according to studies, and a range of Nrf2 inducers exhibit neuroprotective effects following cerebral ischemia (Wang et al., 2018b). Monomethylfumarate, the immediate metabolite of dimethylfumarate (DMF), causes direct alteration of the cysteine 151 of Keap1, which increases the dissociation of Nrf2 and has neuroprotective properties (Linker et al., 2011; Bomprezzi, 2015). Furthermore, in the human neuroblastoma cell line SH-SY5Y, miR-7 can target to limit Keap1 mRNA translation, preventing the degradation of Nrf2 protein, resulting in Nrf2 activation and cytoprotection (Kabaria et al., 2015). Although these reagents need to be further studied in the stroke model, it is undeniable that preventing Nrf2 from being degraded by proteasome through modification or inhibition of Keap1 is a promising measure in the protection of neurons after stroke. According to a report, p62 expression reduced Nrf2 degradation and increased subsequent Nrf2 nuclear accumulation by inactivating Keap1 (Sun et al., 2016). Li et al. found that sestrin2 stimulated angiogenesis to ameliorate brain injury by activating Nrf2 and modulating the interaction between p62 and Keap1, following photothrombotic stroke in rats (Li et al., 2020). In addition, it has been reported that PINK1/Parkin pathway can promote the release of Nrf2 by inhibiting Keap1 (Xiao et al., 2017). Zhang et al. found that mitophagy reduced oxidative stress via Keap1/Nrf2/PHB2 (Prohibitin 2) pathway after subarachnoid hemorrhage in rats (Zhang et al., 2019). A study has shown that Britanin leading to the induction of the Nrf2 pathway ameliorated cerebral ischemia–reperfusion injury by selectively binding to cysteine 151 of Keap1 and inhibiting Keap1-mediated ubiquitylation of Nrf2 (Wu et al., 2017). Obviously, preventing the proteasome degradation of Nrf2 through the regulation of Keap1 is very important for neuroprotection after stroke.
HIF-1 Pathway and Stroke Regulated by UPS
Tissue oxygen content plays crucial roles in maintaining the normal functioning of cells and regulation of their development (Mohyeldin et al., 2010). HIF-1 is an important transcriptional regulator of hypoxia and plays an important role in cerebrovascular diseases (Correia and Moreira, 2010). Accumulating evidence indicates that the induction of HIF-1 provides protection against cerebral ischemic damage (Zhang et al., 2011). HIF-1α is widely expressed in the hypoxic/ischemic brain (Chavez et al., 2000). Mice with neuron-specific reduction of HIF-1α that were subjected to temporary focal cerebral ischemia showed higher tissue damage and a lower survival rate, indicating that HIF-1-mediated responses have an overall positive impact in the ischemic brain (Baranova et al., 2007). HIF-1α has complex effects on the brain, which largely depends on the time-point after hypoxic damage. At the earliest post-ischemic stage (i.e., within 24 h), HIF-1α accumulation promotes cell death. In contrast, during the later stage (i.e., >4 days), HIF-1α signaling has a pro-survival effect through limitation of the infarct size (Mitroshina et al., 2021). HIF-1α which is an important regulator of hypoxia being regulated by proteasomal degradation (Shi, 2009). There is now overwhelming data suggesting that the UPS contributes to cerebral ischemic injury and proteasome inhibition is a potential treatment option for stroke (Wojcik and Di Napoli, 2004). Growing evidence shows that proteasome inhibitors enhance angioneurogenesis and induces a long-term neuroprotection after cerebral ischemia. Inhibition of immunoproteasome LMP2 was able to enhance angiogenesis and facilitate neurological functional recovery in rats after focal cerebral ischemia/reperfusion. A study highlights an important role for inhibition of LMP2 in promoting angiogenesis events in ischemic stroke, and point to HIF-1α as a key mediator of this response (Chen et al., 2018). The novel proteasome inhibitor BSc2118 protects against cerebral ischemia through HIF1A accumulation and enhanced angioneurogenesis (Doeppner et al., 2012). Although the role of HIF-1α in cerebral ischemia remains complex, the role of HIF-1α as mediator of BSc2118-induced neuroprotection is appealing based on the data present (Yan et al., 2011). Furthermore, a result indicates that 20S proteasomes are involved in HIF-1α degradation in ischemic neurons and that proteasomal inhibition provides more HIF-1α stabilization and neuroprotection than PHD inhibition in cerebral ischemia (Badawi and Shi, 2017).
Conclusion and Future Directions
UPS is the main pathway for the degradation of cytosolic, nuclear and transmembrane proteins, and also the main regulator for maintaining neural development, brain structure and function (Park et al., 2020). Neuron is a highly differentiated terminal cell. Various components of UPS widely exist in synapses and participate in the regulation of synaptic function (Tsakiri and Trougakos, 2015). After stroke, due to the destruction of the internal environment of neuron survival, a series of neuron injury events are caused, which eventually leads to the death of nerve cells and the loss of nerve function. In recent years, there have been more and more researches on UPS. It is found that UPS mediated protein degradation is an important mechanism for the body to regulate the level and function of intracellular proteins. The components involved in this biological process mainly include ubiquitin and its related starting enzymes. UPS plays a very important role in maintaining cell homeostasis (Shang and Taylor, 2011). At the same time, UPS is also involved in the pathological process related to nerve injury after stroke (Ge et al., 2007). At present, there are many research results on the physiological and pathological mechanism of stroke. However, the discussion on UPS and stroke is insufficient and there is a lack of literature for reference. Nevertheless, it may still become a new hotspot in basic research and potential clinical application. It should be noted that the aggregation of ubiquitinated proteins is one of the important features after stroke (Luo et al., 2013). UPS is closely related to the pathways of post-stroke related pathological changes such as mitochondrial autophagy, oxidative stress, hypoxia and inflammatory response (Ahmad et al., 2014). To study the role of UPS in PINK1/Parkin, NF-κB, HIF-1α and the regulatory mechanism of Keap1-Nrf2 pathway is of great significance for the clinical treatment and prognosis of stroke patients. At present, there are still many problems that need more experiments to study. From the perspective of maintaining homeostasis, it is necessary to clarify how to moderately activate or inhibit UPS to play a cytoprotective role. The pathological process of stroke is a dynamic process. It is necessary to observe the changes of UPS by stages. Due to the lack of research on the side effects of drugs in experimental animals and the results of clinical trials, the conclusion whether UPS regulating drugs mediate cell protection or cytotoxicity after stroke is still controversial. To explore the relationship between UPS and stroke and its mechanism has great potential to improve the prognosis of stroke patients.
Author Contributions
Y-CL wrote the manuscript. WZ and YW proofread the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81774416).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguileta, M. A., Korac, J., Durcan, T. M., Trempe, J.-F., Haber, M., Gehring, K., et al. (2015). The E3 ubiquitin ligase parkin is recruited to the 26 S proteasome via the proteasomal ubiquitin receptor Rpn13. J. Biol. Chem. 290, 7492–7505. doi: 10.1074/jbc.M114.614925
Ahmad, M., Dar, N. J., Bhat, Z. S., Hussain, A., Shah, A., Liu, H., et al. (2014). Inflammation in ischemic stroke: mechanisms, consequences and possible drug targets. CNS Neurol. Disord. Drug Targets 13, 1378–1396. doi: 10.2174/1871527313666141023094720
Alsayyah, C., Ozturk, O., Cavellini, L., Belgareh-Touze, N., and Cohen, M. M. (2020). The regulation of mitochondrial homeostasis by the ubiquitin proteasome system. Biochim. Biophys. Acta Bioenerg. 1861:148302. doi: 10.1016/j.bbabio.2020.148302
Badawi, Y., and Shi, H. (2017). Relative contribution of prolyl hydroxylase-dependent and -independent degradation of HIF-1alpha by proteasomal pathways in cerebral ischemia. Front. Neurosci. 11:239. doi: 10.3389/fnins.2017.00239
Baird, L., Llères, D., Swift, S., and Dinkova-Kostova, A. T. (2013). Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. U.S.A 110, 15259–15264. doi: 10.1073/pnas.1305687110
Baird, L., and Yamamoto, M. (2020). The Molecular mechanisms regulating the KEAP1-NRF2 Pathway. Mol. Cell Biol. 40, e99–e20. doi: 10.1128/MCB.00099-20
Baker, R. G., Hayden, M. S., and Ghosh, S. (2011). NF-κB, inflammation, and metabolic disease. Cell Metab. 13, 11–22. doi: 10.1016/j.cmet.2010.12.008
Baranova, O., Miranda, L. F., Pichiule, P., Dragatsis, I., Johnson, R. S., and Chavez, J. C. (2007). Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J. Neurosci. 27, 6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007
Bednash, J. S., and Mallampalli, R. K. (2016). Regulation of inflammasomes by ubiquitination. Cell Mol. Immunol. 13, 722–728. doi: 10.1038/cmi.2016.15
Benjamin, E. J., Blaha, M. J., Chiuve, S. E., Cushman, M., Das, S. R., Deo, R., et al. (2017). Heart disease and stroke statistics-2017 update: a report from the american heart association. Circulation 135, e146–e603. doi: 10.1161/CIR.0000000000000485
Berti, R., Williams, A. J., Velarde, L. C., Moffett, J. R., Elliott, P. J., Adams, J., et al. (2003). Effect of the proteasome inhibitor MLN519 on the expression of inflammatory molecules following middle cerebral artery occlusion and reperfusion in the rat. Neurotox. Res. 5, 505–514. doi: 10.1007/BF03033160
Bingol, B., and Sheng, M. (2016). Mechanisms of mitophagy: pink1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 100, 210–222. doi: 10.1016/j.freeradbiomed.2016.04.015
Biswas, S., Roy, R., Biswas, R., and Bagchi, A. (2020). Structural analysis of the effects of mutations in Ubl domain of parkin leading to parkinson’s disease. Gene 726:144186. doi: 10.1016/j.gene.2019.144186
Bomprezzi, R. (2015). Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: an overview. Ther. Adv. Neurol. Disord. 8, 20–30. doi: 10.1177/1756285614564152
Blomgren, K., Zhu, C., Wang, X., Karlsson, J. O., Leverin, A. L., Bahr, B. A., et al. (2001). Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J. Biol. Chem. 276, 10191–10198. doi: 10.1074/jbc.M007807200
Bragoszewski, P., Turek, M., and Chacinska, A. (2017). Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol. 7:170007. doi: 10.1098/rsob.170007
Brown, J. S., and Jackson, S. P. (2015). Ubiquitylation, neddylation and the DNA damage response. Open Biol. 5:150018. doi: 10.1098/rsob.150018
Budenholzer, L., Cheng, C. L., Li, Y., and Hochstrasser, M. (2017). Proteasome structure and assembly. J. Mol. Biol. 429, 3500–3524. doi: 10.1016/j.jmb.2017.05.027
Buetow, L., and Huang, D. T. (2016). Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 17, 626–642. doi: 10.1038/nrm.2016.91
Caamano, J., and Hunter, C. A. (2002). NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 15, 414–429. doi: 10.1128/CMR.15.3.414-429.2002
Caldeira, M. V., Salazar, I. L., Curcio, M., Canzoniero, L. M., and Duarte, C. B. (2014). Role of the ubiquitin-proteasome system in brain ischemia: friend or foe? Prog. Neurobiol. 112, 50–69. doi: 10.1016/j.pneurobio.2013.10.003
Canning, P., Sorrell, F. J., and Bullock, A. N. (2015). Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 88, 101–107. doi: 10.1016/j.freeradbiomed.2015.05.034
Chamorro, A., Dirnagl, U., Urra, X., and Planas, A. M. (2016). Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 15, 869–881. doi: 10.1016/S1474-4422(16)00114-9
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L., et al. (2011). Broad activation of the ubiquitin–proteasome system by parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Chastagner, P., Israel, A., and Brou, C. (2006). Itch/AIP4 mediates deltex degradation through the formation of K29-linked polyubiquitin chains. EMBO Rep. 7, 1147–1153. doi: 10.1038/sj.embor.7400822
Chavez, J. C., Agani, F., Pichiule, P., and LaMAnnua, J. C. (2000). Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J. Appl. Physiol. 89, 1937–1942. doi: 10.1152/jappl.2000.89.5.1937
Chen, Q. M., and Maltagliati, A. J. J. P. G. (2018). Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 50, 77–97. doi: 10.1152/physiolgenomics.00041.2017
Chen, X., Zhang, X., Chen, T., Jiang, X., Wang, X., Lei, H., et al. (2018). Inhibition of immunoproteasome promotes angiogenesis via enhancing hypoxia-inducible factor-1alpha abundance in rats following focal cerebral ischaemia. Brain Behav. Immun. 73, 167–179. doi: 10.1016/j.bbi.2018.04.009
Chen, X., Zhang, X., Wang, Y., Lei, H., Su, H., Zeng, J., et al. (2015). Inhibition of immunoproteasome reduces infarction volume and attenuates inflammatory reaction in a rat model of ischemic stroke. Cell Death Dis. 6:e1626. doi: 10.1038/cddis.2014.586
Chen, Z., Hagler, J., Palombella, V. J., Melandri, F., Scherer, D., Ballard, D., et al. (1995). Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 9, 1586–1597. doi: 10.1101/gad.9.13.1586
Chen, Z. J. (2012). Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 246, 95–106. doi: 10.1111/j.1600-065X.2012.01108.x
Chiba, T., Inoko, H., Kimura, M., and Sato, T. (2013). Role of nuclear IκBs in inflammation regulation. Biomol. Concepts 4, 187–196. doi: 10.1515/bmc-2012-0039
Ciechanover, A. (2015). The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol. 16, 322–324. doi: 10.1038/nrm3982
Cildir, G., Low, K. C., and Tergaonkar, V. (2016). Noncanonical NF-κB signaling in health and disease. Trends Mol. Med. 22, 414–429. doi: 10.1016/j.molmed.2016.03.002
Collaborators, G. B. D. R. F. (2020). Global burden of 87 risk factors in 204 countries and territories, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet 396, 1223–1249. doi: 10.1016/S0140-6736(20)30752-2
Correia, S. C., and Moreira, P. I. (2010). Hypoxia-inducible factor 1: a new hope to counteract neurodegeneration? J. Neurochem. 112, 1–12. doi: 10.1111/j.1471-4159.2009.06443.x
Delhase, M., Hayakawa, M., Chen, Y., and Karin, M. J. S. (1999). Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science 284, 309–313. doi: 10.1126/science.284.5412.309
Dengler, V. L., Galbraith, M., and Espinosa, J. M. (2014). Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 49, 1–15. doi: 10.3109/10409238.2013.838205
Delpeso, L., Castellanos, M. C., Temes, E., Martin-Puig, S., Cuevas, Y., Olmos, G., et al. (2003). The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. J. Biol. Chem. 278, 48690–48695. doi: 10.1074/jbc.M308862200
DiDonato, J. A., Mercurio, F., and Karin, M. (2012). NF−κB and the link between inflammation and cancer. Immunol. Rev. 246, 379–400. doi: 10.1111/j.1600-065X.2012.01099.x
Diseases, G. B. D., and Injuries, C. (2020). Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet 396, 1204–1222. doi: 10.1016/S0140-6736(20)30925-9
Doeppner, T. R., Mlynarczuk-Bialy, I., Kuckelkorn, U., Kaltwasser, B., Herz, J., Hasan, M. R., et al. (2012). The novel proteasome inhibitor BSc2118 protects against cerebral ischaemia through HIF1A accumulation and enhanced angioneurogenesis. Brain 135, 3282–3297. doi: 10.1093/brain/aws269
Durand, J., and Baldwin, A. S. (2017). Targeting IKK and NF-κB for therapy. Adv. Protein Chem. Struct. Biol. 107, 77–115. doi: 10.1016/bs.apcsb.2016.11.006
Eiyama, A., and Okamoto, K. (2015). PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 33, 95–101. doi: 10.1016/j.ceb.2015.01.002
Fao, L., Mota, S. I., and Rego, A. C. (2019). Shaping the Nrf2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing Res. Rev. 54:100942. doi: 10.1016/j.arr.2019.100942
Feehan, K. T., and Gilroy, D. W. (2019). Is resolution the end of inflammation? Trends Mol. Med. 25, 198–214. doi: 10.1016/j.molmed.2019.01.006
Fei, C., Li, Z., Li, C., Chen, Y., Chen, Z., He, X., et al. (2013). Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/beta-catenin signaling. Mol. Cell Biol. 33, 4095–4105. doi: 10.1128/MCB.00418-13
Fricker, L. D. (2020). Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 60, 457–476. doi: 10.1146/Annuurev-pharmtox-010919-023603
Gao, F., Shang, Y., Liu, W., and Li, W. (2016). The linkage specificity determination of Ube2g2-gp78 mediated polyubiquitination. Biochem. Biophys. Res. Commun. 473, 1139–1143. doi: 10.1016/j.bbrc.2016.04.029
Gatti, M., Pinato, S., Maiolica, A., Rocchio, F., Prato, M. G., Aebersold, R., et al. (2015). RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep. 10, 226–238. doi: 10.1016/j.celrep.2014.12.021
Ge, P., Luo, Y., Liu, C. L., and Hu, B. (2007). Protein aggregation and proteasome dysfunction after brain ischemia. Stroke 38, 3230–3236. doi: 10.1161/STROKEAHA.107.487108
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell. Biol. 12, 119–131. doi: 10.1038/ncb2012
Gillette, T. G., Kumar, B., Thompson, D., Slaughter, C. A., and DeMartino, G. N. (2008). Differential roles of the COOH termini of AAA subunits of PA700 (19 S regulator) in asymmetric assembly and activation of the 26 S proteasome. J. Biol. Chem. 283, 31813–31822. doi: 10.1074/jbc.M805935200
Goetzke, C. C., Ebstein, F., and Kallinich, T. (2021). Role of proteasomes in inflammation. J. Clin. Med. 10:1783. doi: 10.3390/jcm10081783
Graham, S. H., and Liu, H. (2017). Life and death in the trash heap: the ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral Ischemia. Ageing Res. Rev. 34, 30–38. doi: 10.1016/j.arr.2016.09.011
Grice, G. L., Lobb, I. T., Weekes, M. P., Gygi, S. P., Antrobus, R., and Nathan, J. A. (2015). The proteasome distinguishes between heterotypic and homotypic lysine-11-linked polyubiquitin chains. Cell Rep. 12, 545–553. doi: 10.1016/j.celrep.2015.06.061
Hayes, J. D., and Dinkova-Kostova, A. T. (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218. doi: 10.1016/j.tibs.2014.02.002
Hicke, L. (2001). Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2, 195–201. doi: 10.1038/35056583
Hu, X., Tao, C., Gan, Q., Zheng, J., Li, H., and You, C. (2016). Oxidative Stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid. Med. Cell Longev. 2016:3215391. doi: 10.1155/2016/3215391
Hybertson, B. M., Gao, B., Bose, S. K., and McCord, J. M. (2011). Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med. 32, 234–246. doi: 10.1016/j.mam.2011.10.006
Hyder, F., Rothman, D. L., and Bennett, M. R. (2013). Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. U.S.A 110, 3549–3554. doi: 10.1073/pnas.1214912110
Imbriani, P., D’Angelo, V., Platania, P., Di Lazzaro, G., Scalise, S., Salimei, C., et al. (2020). Ischemic injury precipitates neuronal vulnerability in Parkinson’s disease: insights from PINK1 mouse model study and clinical retrospective data. Parkinsonism Relat. Disord. 74, 57–63. doi: 10.1016/j.parkreldis.2020.04.004
Jiang, S., Deng, C., Lv, J., Fan, C., Hu, W., Di, S., et al. (2017). Nrf2 Weaves an elaborate network of neuroprotection against stroke. Mol. Neurobiol. 54, 1440–1455. doi: 10.1007/s12035-016-9707-7
Kabaria, S., Choi, D. C., Chaudhuri, A. D., Jain, M. R., Li, H., and Junn, E. (2015). MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic. Biol. Med. 89, 548–556. doi: 10.1016/j.freeradbiomed.2015.09.010
Kane, E. I., Waters, K. L., and Spratt, D. E. (2021). Intersection of redox chemistry and ubiquitylation: post-translational modifications required for maintaining cellular homeostasis and neuroprotection. Cells 10:2121. doi: 10.3390/cells10082121
Kansanen, E., Kuosmanen, S. M., Leinonen, H., and Levonen, A. L. (2013). The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 1, 45–49. doi: 10.1016/j.redox.2012.10.001
Karin, M., and Ben-Neriah, Y. (2000). Phosphorylation meets ubiquitination: the control of NF-κB activity. Annuu. Rev. Immunol. 18, 621–663. doi: 10.1146/Annuurev.immunol.18.1.621
Karunatilleke, N. C., Fast, C. S., Ngo, V., Brickenden, A., Duennwald, M. L., KonermAnnu, L., et al. (2021). Nrf2, the major regulator of the cellular oxidative stress response, is partially disordered. Int. J. Mol. Sci. 22:7434. doi: 10.3390/ijms22147434
Kaspar, J. W., Niture, S. K., and Jaiswal, A. K. (2009). Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 47, 1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035
Kaelin, W. G. Jr., and Ratcliffe, P. J. (2008). Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402. doi: 10.1016/j.molcel.2008.04.009
Ke, Q., and Costa, M. (2006). Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 70, 1469–1480. doi: 10.1124/mol.106.027029
Koh, Y. C., Yang, G., Lai, C. S., Weerawatanakorn, M., and Pan, M. H. (2018). Chemopreventive effects of phytochemicals and medicines on M1/M2 polarized macrophage role in inflammation-related diseases. Int. J Mol. Sci. 19:2208. doi: 10.3390/ijms19082208
Komander, D., and Rape, M. (2012). The ubiquitin code. Annuu. Rev. Biochem. 81, 203–229. doi: 10.1146/Annuurev-biochem-060310-170328
Kramer, L., Groh, C., and HerrmAnnu, J. M. (2021). The proteasome: friend and foe of mitochondrial biogenesis. FEBS Lett. 595, 1223–1238. doi: 10.1002/1873-3468.14010
Kwon, Y. T., and Ciechanover, A. (2017). The Ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 42, 873–886. doi: 10.1016/j.tibs.2017.09.002
Lando, D., Peet, D. J., Whelan, D. A., Gorman, J. J., and Whitelaw, M. L. (2002b). Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295, 858–861. doi: 10.1126/science.1068592
Lando, D., Peet, D. J., Gorman, J. J., Whelan, D. A., Whitelaw, M. L., and Bruick, R. K. (2002a). FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 16, 1466–1471. doi: 10.1101/gad.991402
Landowski, L. M., Niego, B., Sutherland, B. A., Hagemeyer, C. E., and Howells, D. W. (2020). Applications of nanotechnology in the diagnosis and therapy of stroke. Semin. Thromb. Hemost. 46, 592–605. doi: 10.1055/s-0039-3399568
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 22, 320–333. doi: 10.1016/j.devcel.2011.12.014
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Leu, T., Schutzhold, V., Fandrey, J., and Ferenz, K. B. (2019). When the brain yearns for oxygen. Neurosignals 27, 50–61. doi: 10.33594/000000199
Li, C., Sun, T., and Jiang, C. (2021). Recent advances in nanomedicines for the treatment of ischemic stroke. Acta Pharm. Sin. B 11, 1767–1788. doi: 10.1016/j.apsb.2020.11.019
Li, S., Wang, X., Li, Y., Kost, C. K. Jr., and Martin, D. S. (2013). Bortezomib, a proteasome inhibitor, attenuates angiotensin II-induced hypertension and aortic remodeling in rats. PLoS One 8:e78564. doi: 10.1371/journal.pone.0078564
Li, Y., Wu, J., Yu, S., Zhu, J., Zhou, Y., Wang, P., et al. (2020). Sestrin2 promotes angiogenesis to alleviate brain injury by activating Nrf2 through regulating the interaction between p62 and Keap1 following photothrombotic stroke in rats. Brain Res. 1745:146948. doi: 10.1016/j.brainres.2020.146948
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Ling, L., Cao, Z., and Goeddel, D. V. (1998). NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. U.S.A 95, 3792–3797. doi: 10.1073/pnas.95.7.3792
Linker, R. A., Lee, D. H., Ryan, S., van Dam, A. M., Conrad, R., Bista, P., et al. (2011). Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134, 678–692. doi: 10.1093/brain/awq386
Lipton, P. (1999). Ischemic cell death in brain neurons. Physiol. Rev. 79, 1431–1568. doi: 10.1152/physrev.1999.79.4.1431
Liu, F., Lu, J., Manaenko, A., Tang, J., and Hu, Q. (2018). Mitochondria in ischemic stroke: new insight and implications. Aging Dis. 9, 924–937. doi: 10.14336/AD.2017.1126
Liu, H., Wu, X., Luo, J., Wang, X., Guo, H., Feng, D., et al. (2019). Pterostilbene attenuates astrocytic inflammation and neuronal oxidative injury after ischemia-reperfusion by inhibiting NF-κB phosphorylation. Front. Immunol. 10:2408. doi: 10.3389/fimmu.2019.02408
Luo, T., Park, Y., Sun, X., Liu, C., and Hu, B. (2013). Protein misfolding, aggregation, and autophagy after brain ischemia. Transl. Stroke Res. 4, 581–588. doi: 10.1007/s12975-013-0299-5
Majmundar, A. J., Wong, W. J., and Simon, M. C. (2010). Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309. doi: 10.1016/j.molcel.2010.09.022
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Matyskiela, M. E., Lander, G. C., and Martin, A. (2013). Conformational switching of the 26S proteasome enables substrate degradation. Nat. Struct. Mol. Biol. 20, 781–788. doi: 10.1038/nsmb.2616
Mazzocchetti, P., Mancini, A., Sciaccaluga, M., Megaro, A., Bellingacci, L., Di Filippo, M., et al. (2020). Low doses of Perampanel protect striatal and hippocampal neurons against in vitro ischemia by reversing the ischemia-induced alteration of AMPA receptor subunit composition. Neurobiol. Dis. 140, 104848. doi: 10.1016/j.nbd.2020.104848
Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature 454, 428–435. doi: 10.1038/nature07201
Meller, R. (2009). The role of the ubiquitin proteasome system in ischemia and ischemic tolerance. Neuroscientist 15, 243–260. doi: 10.1177/1073858408327809
Miller, S., and Muqit, M. M. K. (2019). Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 705, 7–13. doi: 10.1016/j.neulet.2019.04.029
Mitchell, J. P., and Carmody, R. (2018). NF-κB and the transcriptional control of inflammation. Int. Rev. Cell Mol. Biol. 335, 41–84. doi: 10.1016/bs.ircmb.2017.07.007
Mitroshina, E. V., Savyuk, M. O., Ponimaskin, E., and Vedunova, M. V. (2021). Hypoxia-Inducible Factor (HIF) in Ischemic Stroke and Neurodegenerative Disease. Front. Cell Dev. Biol. 9:703084. doi: 10.3389/fcell.2021.703084
Mohyeldin, A., Garzon-Muvdi, T., and Quinones-Hinojosa, A. (2010). Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7, 150–161. doi: 10.1016/j.stem.2010.07.007
Mukhida, K., Kobayashi, N. R., and Mendez, I. (2005). A novel role for parkin in trauma-induced central nervous system secondary injury. Med. Hypotheses 64, 1120–1123. doi: 10.1016/j.mehy.2004.12.020
Narendra, D., Tanaka, A., Suen, D.-F., and Youle, R. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Ohh, M., Park, C. W., Ivan, M., Hoffman, M. A., Kim, T. Y., Huang, L. E., et al. (2000). Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2, 423–427. doi: 10.1038/35017054
Ohtake, F., and Tsuchiya, H. (2017). The emerging complexity of ubiquitin architecture. J. Biochem. 161, 125–133. doi: 10.1093/jb/mvw088
Oshima, Y., Verhoeven, N., Cartier, E., and Karbowski, M. (2021). The OMM-severed and IMM-ubiquitinated mitochondria are intermediates of mitochondrial proteotoxicity-induced autophagy in PRKN/parkin-deficient cells. Autophagy 17, 3884–3886. doi: 10.1080/15548627.2021.1964887
Park, J., Cho, J., and Song, E. (2020). Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 43, 1144–1161. doi: 10.1007/s12272-020-01281-8
Phillips, J. B., Williams, A. J., Adams, J., Elliott, P. J., and Tortella, F. C. J. S. (2000). Proteasome inhibitor PS519 reduces infarction and attenuates leukocyte infiltration in a rat model of focal cerebral ischemia. Stroke 31, 1686–1693. doi: 10.1161/01.str.31.7.1686
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Rahighi, S., Ikeda, F., Kawasaki, M., Akutsu, M., Suzuki, N., Kato, R., et al. (2009). Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 136, 1098–1109. doi: 10.1016/j.cell.2009.03.007
Rakovic, A., Ziegler, J., Mårtensson, C. U., Prasuhn, J., Shurkewitsch, K., König, P., et al. (2019). PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 26, 1428–1441. doi: 10.1038/s41418-018-0219-z
Rami, A., Agarwal, R., Botez, G., and Winckler, J. (2000). mu-Calpain activation, DNA fragmentation, and synergistic effects of caspase and calpain inhibitors in protecting hippocampal neurons from ischemic damage. Brain Res. 866, 299–312. doi: 10.1016/s0006-8993(00)02301-5
Ruas, J. L., Poellinger, L., and Pereira, T. (2002). Functional analysis of hypoxia-inducible factor-1 alpha-mediated transactivation. Identification of amino acid residues critical for transcriptional activation and/or interaction with CREB-binding protein. J. Biol. Chem. 277, 38723–38730. doi: 10.1074/jbc.M205051200
Sajadimajd, S., and Khazaei, M. (2018). Oxidative stress and cancer: the Role of Nrf2. Curr. Cancer Drug Targets 18, 538–557. doi: 10.2174/1568009617666171002144228
Semenza, G. L. (2004). Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 19, 176–182. doi: 10.1152/physiol.00001.2004
Semenza, G. L. (2009). Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 24, 97–106. doi: 10.1152/physiol.00045.2008
Shang, F., and Taylor, A. (2011). Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 51, 5–16. doi: 10.1016/j.freeradbiomed.2011.03.031
Shang, Y., Dai, S., Chen, X., Wen, W., and Liu, X. J. C. C. (2019). MicroRNA-93 regulates the neurological function, cerebral edema and neuronal apoptosis of rats with intracerebral hemorrhage through TLR4/NF-κB signaling pathway. Cell Cycle 18, 3160–3176. doi: 10.1080/15384101.2019.1670509
Sharpe, L. J., Coates, H. W., and Brown, A. J. (2020). Post-translational control of the long and winding road to cholesterol. J. Biol. Chem. 295, 17549–17559. doi: 10.1074/jbc.REV120.010723
Shen, J. L., Fortier, T. M., Wang, R., and Baehrecke, E. H. (2021). Vps13D functions in a Pink1-dependent and Parkin-independent mitophagy pathway. J. Cell Biol. 220, e202104073. doi: 10.1083/jcb.202104073
Shi, H. (2009). Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr. Med. Chem. 16, 4593–4600. doi: 10.2174/092986709789760779
Shu, S., Wang, Y., Zheng, M., Liu, Z., Cai, J., Tang, C., et al. (2019). Hypoxia and hypoxia-inducible factors in kidney injury and repair. Cells 8:207. doi: 10.3390/cells8030207
Sims, N. R., and Muyderman, H. (2010). Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 1802, 80–91. doi: 10.1016/j.bbadis.2009.09.003
Song, L., and Rape, M. (2010). Regulated degradation of spindle assembly factors by the anaphase-promoting complex. Mol. Cell 38, 369–382. doi: 10.1016/j.molcel.2010.02.038
Sun, X., Ou, Z., Chen, R., Niu, X., Chen, D., Kang, R., et al. (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. doi: 10.1002/hep.28251
Sun-Wang, J. L., Yarritu-Gallego, A., Ivanova, S., and Zorzano, A. (2021). The ubiquitin-proteasome system and autophagy: self-digestion for metabolic health. Trends Endocrinol. Metab. 32, 594–608. doi: 10.1016/j.tem.2021.04.015
Sun, S.-C. (2011). Non-canonical NF-κB signaling pathway. Cell Res. 21, 71–85. doi: 10.1038/cr.2010.177
Suzuki, T., and Yamamoto, M. (2015). Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 88, 93–100. doi: 10.1016/j.freeradbiomed.2015.06.006
Tanaka, K. (2020). The PINK1-Parkin axis: an overview. Neurosci. Res. 159, 9–15. doi: 10.1016/j.neures.2020.01.006
Tarhonskaya, H., Hardy, A. P., Howe, E. A., Loik, N. D., Kramer, H. B., McCullagh, J. S., et al. (2015). Kinetic investigations of the role of factor inhibiting hypoxia-inducible factor (FIH) as an oxygen sensor. J. Biol. Chem. 290, 19726–19742. doi: 10.1074/jbc.M115.653014
Thacker, G., Mishra, M., Sharma, A., Singh, A. K., Sanyal, S., and Trivedi, A. K. (2020). E3 ligase SCF(SKP2) ubiquitinates and degrades tumor suppressor C/EBPalpha in acute myeloid leukemia. Life Sci. 257:118041. doi: 10.1016/j.lfs.2020.118041
Tonelli, C., Chio, I. I. C., and Tuveson, D. A. (2018). Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 29, 1727–1745. doi: 10.1089/ars.2017.7342
Tong, K. I., Katoh, Y., Kusunoki, H., Itoh, K., Tanaka, T., Yamamoto, M. J. M., et al. (2006). Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell Biol. 26, 2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006
Tsakiri, E. N., and Trougakos, I. P. (2015). The amazing ubiquitin-proteasome system: structural components and implication in aging. Int. Rev. Cell Mol. Biol. 314, 171–237. doi: 10.1016/bs.ircmb.2014.09.002
Uckelmann, M., and Sixma, T. K. (2017). Histone ubiquitination in the DNA damage response. DNA Repair (Amst) 56, 92–101. doi: 10.1016/j.dnarep.2017.06.011
Unno, M., Mizushima, T., Morimoto, Y., Tomisugi, Y., Tanaka, K., Yasuoka, N., et al. (2002). The structure of the mammalian 20S proteasome at 2.75 a resolution. Structure 10, 609–618. doi: 10.1016/s0969-2126(02)00748-7
Villeneuve, N. F., Lau, A., and Zhang, D. D. (2010). Regulation of the Nrf2–Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 13, 1699–1712. doi: 10.1089/ars.2010.3211
Virani, S. S., Alonso, A., Aparicio, H. J., Benjamin, E. J., Bittencourt, M. S., Callaway, C. W., et al. (2021). heart disease and stroke statistics-2021 update: a report from the american heart association. Circulation 143, e254–e743. doi: 10.1161/CIR.0000000000000950
Wang, G. L., Jiang, B. H., Rue, E. A., and Semenza, G. L. (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U.S.A 92, 5510–5514. doi: 10.1073/pnas.92.12.5510
Wang, L., Cho, Y.-L., Tang, Y., Wang, J., Park, J.-E., Wu, Y., et al. (2018a). PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1–Parkin-mediated mitophagy. Cell Res. 28, 787–802. doi: 10.1038/s41422-018-0056-0
Wang, M., and Pickart, C. M. (2005). Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 24, 4324–4333. doi: 10.1038/sj.emboj.7600895
Wang, Y., Huang, Y., Xu, Y., Ruan, W., Wang, H., Zhang, Y., et al. (2018b). A dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid. Redox Signal. 28, 141–163. doi: 10.1089/ars.2017.7003
Wang, Y., Wang, L., Wen, X., Hao, D., Zhang, N., He, G., et al. (2019). NF-κB signaling in skin aging. Mech. Ageing Dev. 184:111160. doi: 10.1016/j.mad.2019.111160
Wilck, N., and Ludwig, A. (2014). Targeting the ubiquitin-proteasome system in atherosclerosis: status quo, challenges, and perspectives. Antioxid. Redox Signal. 21, 2344–2363. doi: 10.1089/ars.2013.5805
Wojcik, C., and Di Napoli, M. (2004). Ubiquitin-proteasome system and proteasome inhibition: new strategies in stroke therapy. Stroke 35, 1506–1518. doi: 10.1161/01.STR.0000126891.93919.4e
Wu, G., Zhu, L., Yuan, X., Chen, H., Xiong, R., Zhang, S., et al. (2017). Britanin ameliorates cerebral ischemia-reperfusion injury by inducing the Nrf2 protective pathway. Antioxid. Redox Signal. 27, 754–768.
Wu, X., Li, X., Liu, Y., Yuan, N., Li, C., Kang, Z., et al. (2018). Hydrogen exerts neuroprotective effects on OGD/R damaged neurons in rat hippocampal by protecting mitochondrial function via regulating mitophagy mediated by PINK1/Parkin signaling pathway. Brain Res. 1698, 89–98. doi: 10.1016/j.brainres.2018.06.028
Xiao, G., Harhaj, E. W., and Sun, S. C. (2001). NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 7, 401–409. doi: 10.1016/s1097-2765(01)00187-3
Xiao, L., Xu, X., Zhang, F., Wang, M., Xu, Y., Tang, D., et al. (2017). The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11, 297–311. doi: 10.1016/j.redox.2016.12.022
Xu, C., Ge, H., Wang, T., Qin, J., Liu, D., Liu, Y., et al. (2018). Increased expression of T cell immunoglobulin and mucin domain 3 on CD14+ monocytes is associated with systemic inflammatory reaction and brain injury in patients with spontaneous intracerebral hemorrhage. J. Stroke Cerebrovasc. Dis. 27, 1226–1236. doi: 10.1016/j.jstrokecerebrovasdis.2017.11.041
Yamano, K., and Youle, R. (2013). PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. doi: 10.4161/auto.24633
Yamazaki, S., Muta, T., and Takeshige, K. (2001). A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J. Biol. Chem. 276, 27657–27662. doi: 10.1074/jbc.M103426200
Yan, J., Zhou, B., Taheri, S., and Shi, H. (2011). Differential effects of HIF-1 inhibition by YC-1 on the overall outcome and blood-brain barrier damage in a rat model of ischemic stroke. PLoS One 6:e27798. doi: 10.1371/journal.pone.0027798
Yau, R., and Rape, M. (2016). The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586. doi: 10.1038/ncb3358
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640. doi: 10.1074/jbc.M110.209338
Youn, D. H., Kim, Y., Kim, B. J., Jeong, M. S., Lee, J., Rhim, J. K., et al. (2021). Mitochondrial dysfunction associated with autophagy and mitophagy in cerebrospinal fluid cells of patients with delayed cerebral ischemia following subarachnoid hemorrhage. Sci. Rep. 11:16512. doi: 10.1038/s41598-021-96092-2
Zhang, D. D., and Hannink, M. (2003). Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003
Zhang, Q., Lenardo, M. J., and Baltimore, D. J. C. (2017). 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57. doi: 10.1016/j.cell.2016.12.012
Zhang, T., Wu, P., Budbazar, E., Zhu, Q., Sun, C., Mo, J., et al. (2019). Mitophagy reduces oxidative stress via Keap1 (Kelch-Like epichlorohydrin-associated protein 1)/Nrf2 (Nuclear Factor-E2-Related Factor 2)/PHB2 (Prohibitin 2) pathway after subarachnoid hemorrhage in rats. Stroke 50, 978–988. doi: 10.1161/STROKEAHA.118.021590
Zhang, X., Liu, X., Hu, G., Zhang, G., Zhao, G., and Shi, M. (2020). Ginsenoside Rd attenuates blood-brain barrier damage by suppressing proteasome-mediated signaling after transient forebrain ischemia. Neuroreport 31, 466–472. doi: 10.1097/WNR.0000000000001426
Zhang, Z., Yan, J., Chang, Y., ShiDu Yan, S., and Shi, H. (2011). Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr. Med. Chem. 18, 4335–4343. doi: 10.2174/092986711797200426
Zheng, N., and Shabek, N. (2017). Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 86, 129–157. doi: 10.1146/Annuurev-biochem-060815-014922
Zhou, M., Wang, H., Zeng, X., Yin, P., Zhu, J., Chen, W., et al. (2019). Mortality, morbidity, and risk factors in China and its provinces, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet 394, 1145–1158. doi: 10.1016/S0140-6736(19)30427-1
Zhou, X., Chen, H., Wang, L., Lenahan, C., Lian, L., Ou, Y., et al. (2021). Mitochondrial dynamics: a potential therapeutic target for ischemic stroke. Front. Aging Neurosci. 13:721428. doi: 10.3389/fnagi.2021.721428
Keywords: UPS, stroke, ubiquitination, mitochondrial dysfunction, oxidative stress, inflammation
Citation: Li Y-C, Wang Y and Zou W (2022) Exploration on the Mechanism of Ubiquitin Proteasome System in Cerebral Stroke. Front. Aging Neurosci. 14:814463. doi: 10.3389/fnagi.2022.814463
Received: 13 November 2021; Accepted: 14 March 2022;
Published: 07 April 2022.
Edited by:
Benoit LAURENT, Université de Sherbrooke, CanadaReviewed by:
Yomna Badawi, University of Pittsburgh, United StatesNur M. Kocaturk, MRC Protein Phosphorylation and Ubiquitylation Unit (MRC), United Kingdom
Copyright © 2022 Li, Wang and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Zou, d2VpejIwMjFAMTI2LmNvbQ==