 ZhaoLei Ma1,2,3,4
ZhaoLei Ma1,2,3,4 Lan Chu
Lan Chu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci., 17 May 2022
Sec. Neuroinflammation and Neuropathy
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.806200
Acute ischemic stroke (AIS) is a common neurological disease that seriously endangers both the physical and mental health of human. After AIS, activated immune cells are recruited to the stroke site, where inflammatory mediators are released locally, and severe immune inflammatory reactions occur within a short time, which affects the progress and prognosis of IS. Circular RNA (circRNA) is a type of non-coding RNA (ncRNA) with a closed-loop structure and high stability. Studies have found that circRNA can affect the course of IS. However, there is no report on ceRNA’s pathogenesis in AIS that is mediated by circRNA. In this study, the CIBERSORT algorithm was used to analyze the distribution of immune cells in patients with AIS. mRNA dataset was downloaded from the GEO database, and the weighted gene co-expression network analysis (WGCNA) method was used to construct weighted gene co-expression to determine 668 target genes, using GO, KEGG enrichment analysis, construction of protein-protein interaction (PPI) network analysis, and molecular complex detection (MCODE) plug-in analysis. The results showed that the biological function of the target gene was in line with the activation and immune regulation of neutrophils; signal pathways were mostly enriched in immune inflammation-related pathways. A Venn diagram was used to obtain 52 intersection genes between target genes and disease genes. By analyzing the correlation between the intersection genes and immune cells, we found that the top 5 hub genes were TOM1, STAT3, RAB3D, MDM2, and FOS, which were all significantly positively correlated with neutrophils and significantly negatively correlated with eosinophils. A total of 52 intersection genes and the related circRNA and miRNA were used as input for Cytoscape software to construct a circRNA-mediated ceRNA competition endogenous network, where a total of 18 circRNAs were found. Further analysis of the correlation between circRNA and immune cells found that 4 circRNAs are positively correlated with neutrophils. Therefore, we speculate that there may be a regulatory relationship between circRNA-mediated ceRNA and the immune mechanism in AIS. This study has important guiding significance for the progress, outcome of AIS, and the development of new medicine.
Stroke is one of the major diseases that cause death and disability worldwide. Statistically, about 15 million people worldwide suffer from stroke every year (Li et al., 2018). With the rapid growth of the aging population, the incidence and mortality of stroke increase progressively each year. Notably, acute ischemic stroke (AIS) caused by cerebral artery thrombosis or embolism accounts for about 80–85% of stroke cases (Jian et al., 2019). So far, the most effective treatments for AIS are intravenous thrombolysis within the “time window” (Powers et al., 2018) and intravascular interventional therapy (Goyal et al., 2015). However, only a few patients have benefited from these treatments, and there is a high level of risks associated with these treatment methods. Therefore, the identification of the molecular mechanism of AIS pathogenesis, the screening of effective and reliable biological markers for early screening and AIS diagnosis, and timely and accurate intervention for patients with AIS are essential to reduce the incidence and the poor prognosis.
Recent studies have found that immune cell-mediated inflammatory response plays an important role in the pathogenesis of AIS. Following the stroke, the body’s innate immune system and adaptive immune system are activated. Inflammatory responses are also active during the entire pathophysiological process of AIS, which forms one of the important pathophysiological processes after AIS (Fu et al., 2015). The possible mechanism is through the damage-associated molecular patterns (DAMPs) released by damaged neurons after stroke, which triggers local immune responses that lead to glial cell activation and peripheral leukocytes recruitment to the damaged area, and the secretion of a variety of pro-inflammatory cytokines, chemokines, and matrix metalloproteinases (MMPs), which cause the damage to the blood-brain barrier, cerebral edema, hemorrhagic transformation, and neuronal necrosis (Rajkovic et al., 2018; Yang et al., 2019).
Non-coding RNAs (ncRNAs) include microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs), which can regulate protein coding through a variety of mechanisms. This has an important role in the pathophysiology and recovery of angiogenesis after AIS (Ding et al., 2020; Gan et al., 2021; Li J. et al., 2021). Competing endogenous RNA (ceRNA) has miRNA binding sites and can compete with mRNA to bind miRNA to construct a circRNA-miRNA-mRNA regulatory network, thereby inhibiting miRNA regulation of target genes. The mechanism of ceRNA is complicated. For example, circRNA can bind miRNA for ceRNA, thereby preventing miRNA from inhibiting target genes (Salmena et al., 2011). Studies have shown that circTLK1 as a ceRNA can inhibit the activity of miR-335-3p and cause neuronal damage. circTLK1 knockout can significantly reduce the cerebral infarct volume of MCAO mice, reduce neuronal damage, and improve subsequent long-term neurological deficits (Wu et al., 2019).
The role of lncRNA-mediated ceRNA in the immune pathogenesis of AIS has been reported previously (Li S. et al., 2021). However, there is no report on the immune mechanism of circRNA-mediated ceRNA in AIS. This study aims to explore the distribution characteristics of immune cells in patients with AIS, to explore the role of circRNA-mediated ceRNA regulatory network in the immune mechanism of AIS, and to provide a new path for the pathogenesis and treatment of IS research.
CIBERSORT is currently the most used immune cell infiltration estimation and analysis method (Newman et al., 2015). CIBERSORT1 algorithm was used to analyze RNA-seq data from different patient subgroups and to infer the composition ratio of 22 types of immune infiltrating cells. The default matrix was set to 1,000 permutations, and samples with p < 0.05 showed significant differences. The p-value of CIBERSORT can reflect the statistical significance of the deconvolution results of all cell subsets, which was used to filter out the deconvolution results with less significant fitting accuracy. To evaluate the difference of immune cell infiltration between the stroke group and the control group, “pheatmap” package was used to draw the heatmaps of immune cell infiltration to explore the distribution of immune cells between high- and low-risk groups. “corrplot” software package was used to analyze the interactions between immune cells and the impact of these interactions. “vioplot” software package was used to plot the relative content of immune cells.
In this study, the original circRNA, miRNA, and mRNA gene expression matrix datasets were downloaded from the gene expression database, i.e., GEO.2 circRNA dataset (GSE133768) (Yang et al., 2020) included 3 disease groups and 3 healthy control groups. The sequencing platform used was GPL21825[074301] Arraystar Human circRNA microarray V2 (Agilent Technologies, Rockville, MD, United States). The mRNA dataset (GSE58294) is composed of 92 mRNA sequencing data (Stamova et al., 2014), including 69 disease groups and 23 healthy control groups. The expression profile sequencing platform used was GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA, United States). R limma package (Smyth, 2005) was used to detect the differential expression of circRNAs and mRNA between the disease group and the healthy control group, respectively. ggplot2 package (Davis and Meltzer, 2007) and Complexheatmap package were used (Gu et al., 2016) for the visualization of heatmaps and volcano maps to show the significant expression of DEcircRNAs and DEmRNAs (|logFC| > 1 and p < 0.05 are considered as significant differences).
The DAVID database3 (Rameshwari and Prasad, 2012) that integrates biological data and analysis tool and performs functional enrichment analysis based on the DAVID database, including gene ontology (GO) annotations and gene encyclopedia (KEGG) enrichment analysis, was used in this study. GO analysis divides the gene function into three categories: biological process (BP), cell component (CC), and molecular function (MF). Based on the above three aspects, we can get gene annotation information (Harris et al., 2004). KEGG enrichment analysis can help us understand the signaling pathways in which the differentially expressed genes (DEGs) have been involved.
The study of mutual network between proteins helps to identify the core regulatory genes. Currently, the Search Tool for the Retrieval of Interaction Genes (STRING) database (von Mering et al., 2003)4 is the highest species coverage and the largest interaction. A protein-protein interaction (PPI) network was constructed based on STRING. In this study, the PPI network of DEGs was constructed based on the minimum interaction (>0.4). PPI network was uploaded to the Cytoscape software (version 3.7.2) for visualization (Shannon et al., 2003).
Molecular complex detection (MCODE) plug-in Cytoscape software was used to identify closely related modules in the PPI network: cutoff value was set at 2, and the node score cutoff value was 0.2. The advantage of the algorithm of this module is that it will not be affected by a high proportion of false positives caused by high-throughput technology. In addition, GO and KEGG analysis of genes in the PPI network module were performed through the “clusterprofiler” package in the R software. The value of p < 0.05 indicated a significant difference.
The final intersection genes through the Venn diagram have undergone GO and KEGG analysis using the Metascape database.
The NetworkAnalyzer in Cytoscape (version 3.7.1) was used to analyze the topological parameters in the network, select the best target according to the degree and the top 5 as hub genes, and analyze the correlation between the hub genes and the immune cells.
The pROC package in the R language was used to perform receiver operating characteristic (ROC) on the selected specific genes. The area under the curve (AUC) of the core gene was compared to verify the core gene’s ability to predict disease sensitivity and specificity; finally, ggplot2 package was used for ROC curve visualization.
CircInteractome data5 is a commonly used database for querying the correlation between circRNA and miRNA. Target miRNA prediction for AIS was based on the top 10 most significant positive and negative correlations circRNAs with a statistical difference. Prediction of the interactions between miRNA and mRNA was based on miRDB, miRTarBase, and TargetScan. Combining circRNA-miRNA interactions and visualization of mRNA-miRNA interaction through Cytoscape, a circRNA-miRNA-mRNA network was established. Analysis of the correlation between circRNA and immune cells.
The Shapiro–Wilk normality test was used to test the normality of the data for the correlation analysis. For data with normal distribution, Pearson analysis was used. For data without normal distribution, Spearman’s correlation analysis was used. Ggplot2 (3.3.3 version) was used for visualization. −1 ≤ r ≤ 1, where a positive r-value indicates a positive correlation and a negative r-value indicates a negative correlation.
All statistical analysis was performed in the R language (version 3.6). All statistical tests were two-sided, and p < 0.05 was considered statistically significant.
We used CIBERSORT to estimate the distribution ratio of 22 immune cell subsets in AIS, of which neutrophils accounted for the highest proportion (Figure 1A). The results of violin graph showed that the immune infiltration levels of naive B cells, CD8+ T cells, naive CD4+ T cells, and resting mast cells were downregulated in patients with AIS, while the immune infiltration levels of activated memory CD4+ T cells, macrophages M0, and neutrophils were upregulated. Resting dendritic cells, neutrophils, naive CD4+ T cells, and T follicular helper cells were significantly different in patients with AIS and normal controls (Figure 1B).
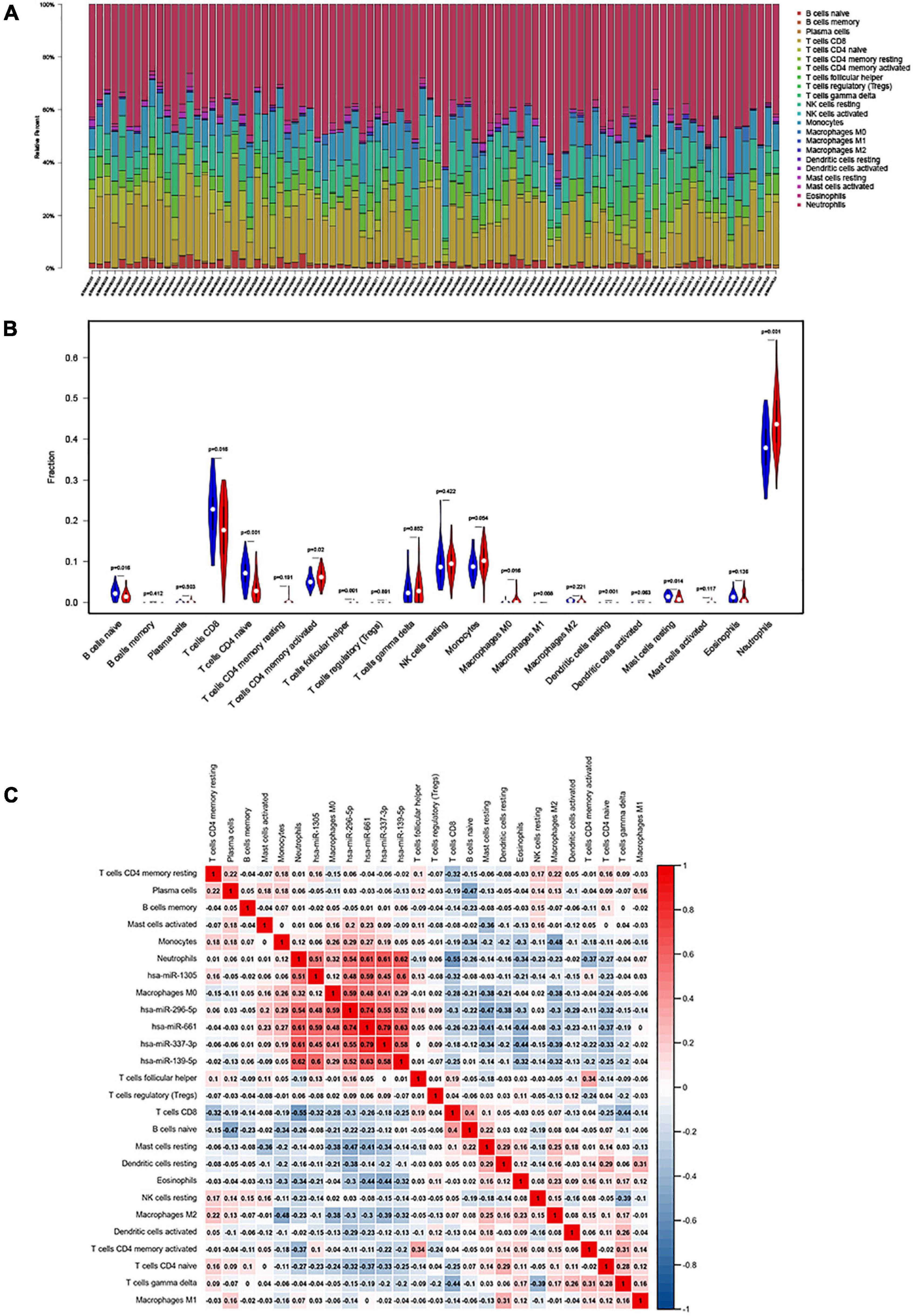
Figure 1. (A) The fraction of 22 subsets of immune cells in acute ischemic stroke (AIS), of which the highest proportion is neutrophils; x-axis: each GEO sample; y-axis: percentage of each kind of immune cells. (B) Violin plot showing differences in immune infiltration between normal and diseased groups. Blue showed the normal group and red showed the disease group. Resting dendritic cells, neutrophils, naive CD4+ naive T cells, and T follicular helper cell revealed the largest differences between the two groups (p < 0.001). (C) The co-expression heatmap illustrated significant co-expression patterns about key members in the ceRNA network and key members in the immune cells.
The Pearson’s correlation coefficient was used to analyze the correlation between immune cells (Figure 1C). Among them, CD8+ T cells were strongly positively correlated with naive B cells (0.43), and resting NK cells were moderately positively correlated with memory B cells (0.32). Memory CD4+ T cells are moderately positively correlated with gamma-delta T cells (0.25), naive B cells are moderately positively correlated with resting dendritic cells (0.26), CD8+ T cells are strongly negatively correlated with neutrophils (−0.6), CD8+ T cells and gamma-delta T cells were moderately negatively correlated (−0.43), and macrophages M2 and monocytes were moderately negatively correlated (−0.42).
The GSE133768 dataset was downloaded from the GEO database. A total of 559 differential circRNAs (213 upregulated and 346 downregulated) were screened out based on the set threshold (p < 0.05 and |Log2FC| > 1). The mRNA expression data come from 92 mRNA sequencing data of the GSE58294 dataset. Volcano maps of DEGcircRNAs and DEGmRNAs were shown in Figure 2.
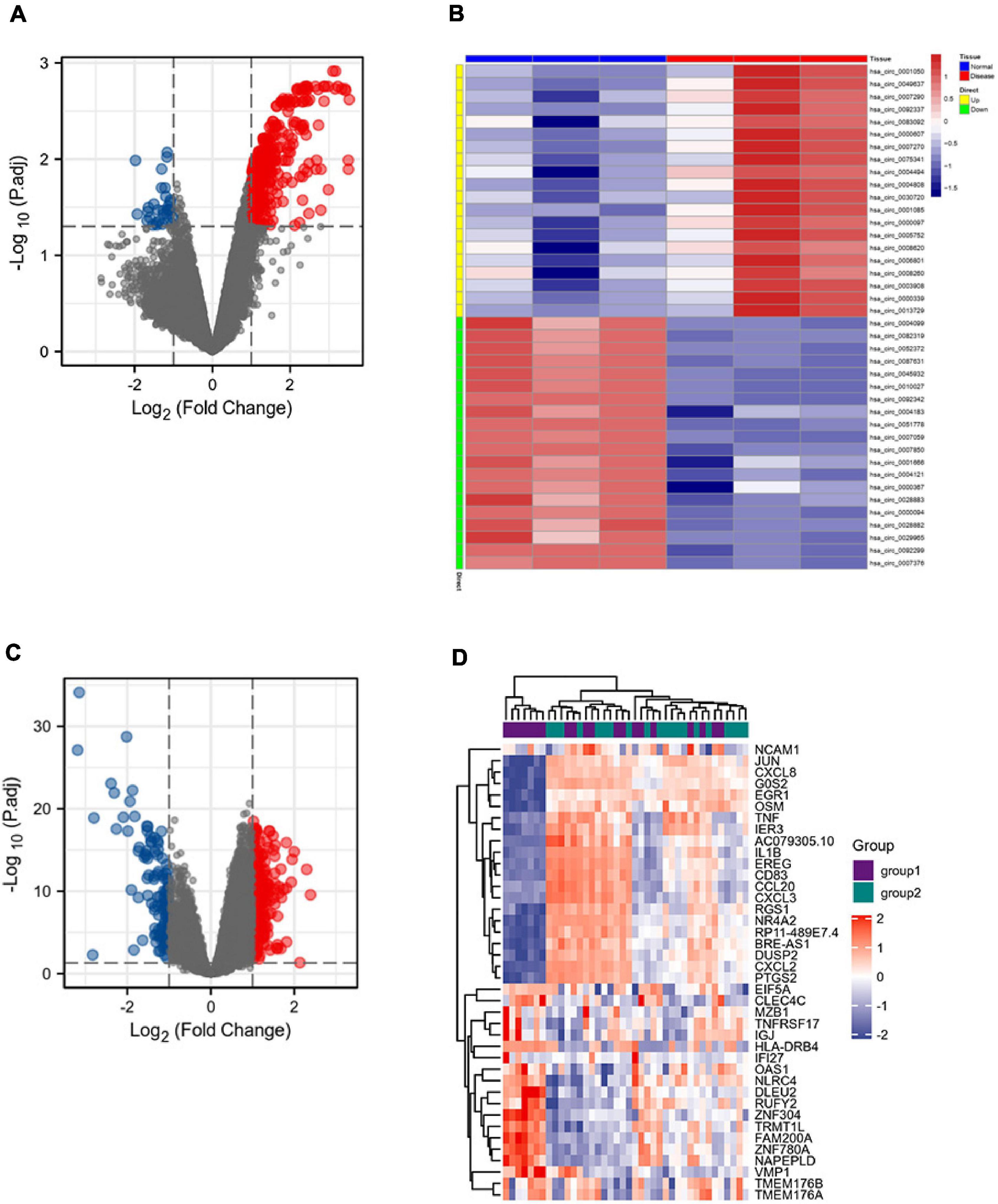
Figure 2. DEGcircRNAs gene expression shown in panel (A) volcano map and (B) heatmap, including 213 upregulated differential genes and 346 downregulated differential genes. DEmRNAs gene expression shown in panel (C) volcano map and (D) heatmap, including 198 upregulated differential genes and 121 downregulated differential genes.
The GSE58294 dataset, composed of a total of 92 mRNA sequencing data, was downloaded from the GEO database, and an mRNA expression matrix was obtained. The weighted gene co-expression network analysis (WGCNA) method was then used. In general, a weighted gene co-expression network (Figure 3A) was constructed; cooperatively expressed gene modules and the correlation among gene expression networks, phenotypes, and the core genes in the co-expression network were explored; and the WGCNA-R package was used to screen out the top 10,000 genes in variance for further analysis. The results showed that the blue gene modules were most relevant to the disease (Figures 3B–F).
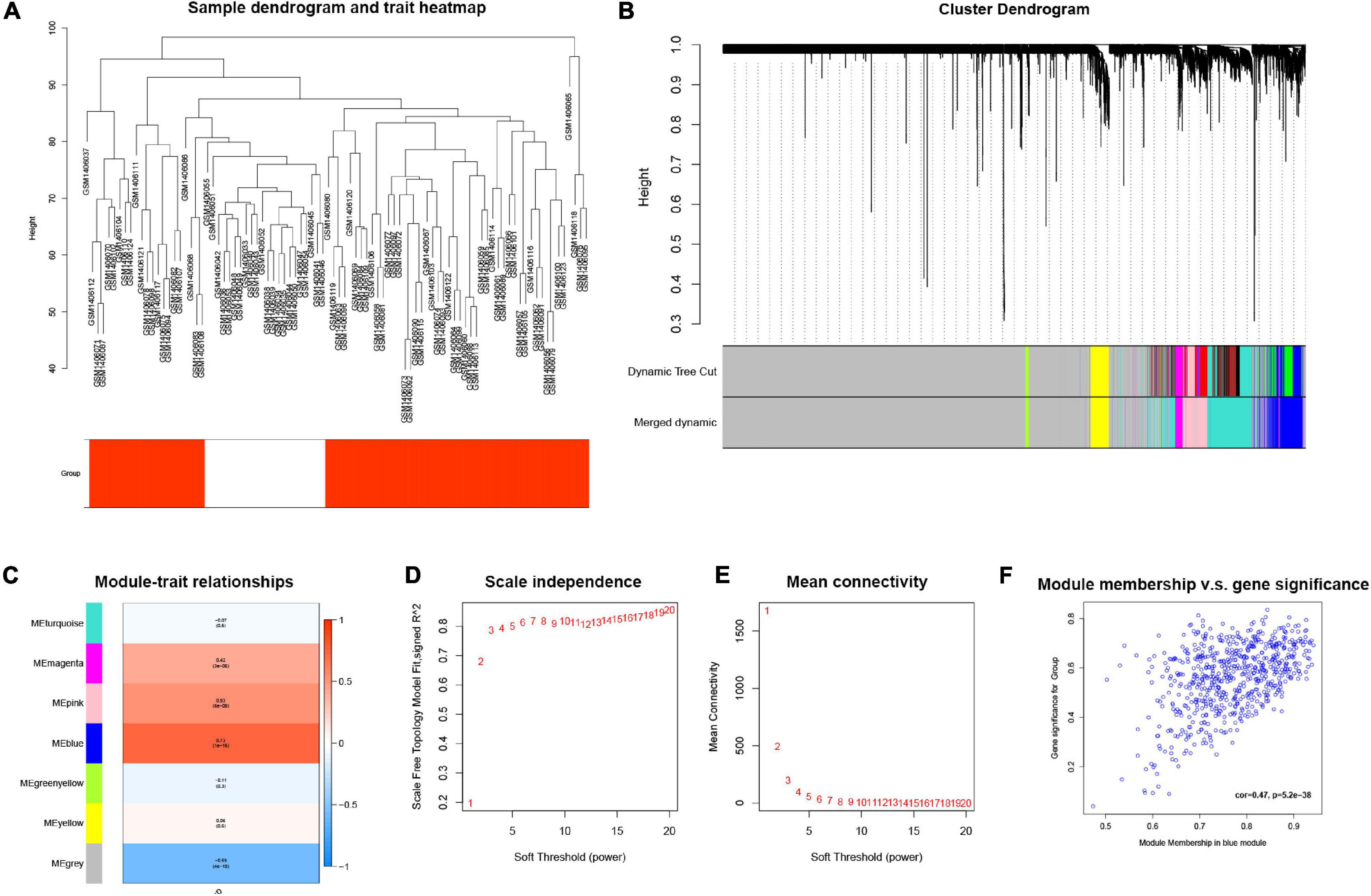
Figure 3. (A) Cluster tree drawn using the weighted gene co-expression network analysis method. Each branch represents a gene; y-axis represents the cluster distance. (B) Cluster dendrogram with soft threshold and 7 merged modules: 668 genes in blue module, 60 genes in green-yellow module, 7,130 genes in gray module, 153 genes in magenta module, 550 genes in pink module, 1,116 genes in turquoise module, and 323 genes in yellow module. (C) Module-trait relationships, where blue module has the highest correlation with diseases (cor = 0.73, p = 1e–16). (D) Scale independence; x-axis represents the value of power and y-axis represents R. (E) Mean connectivity; x-axis represents the value of power and y-axis represents the average connected number of all nodes. (F) Correlation between genes in blue module and corresponding traits (cor = 0.47, p = 5.2e–38).
A total of 668 genes in the blue module were analyzed for GO and KEGG enrichment analysis. Under the conditions of adjusted p-value < 0.1 and q-value < 0.2, there were 377 BP, 40 CC, 11 MF, and 31 KEGG. The specific functions are shown in Table 1 and Figure 4.
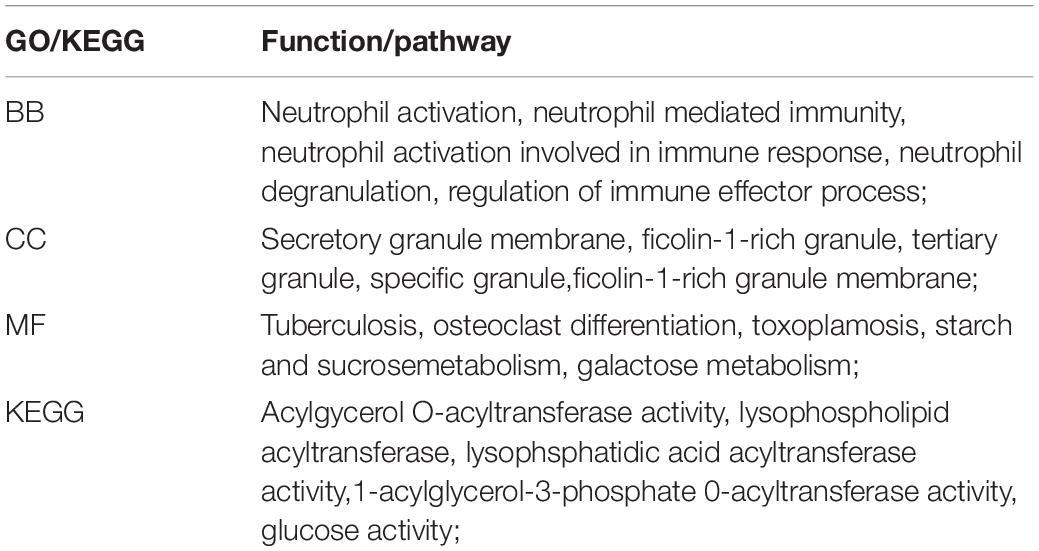
Table 1. Blue module gene biological function and pathway enrichment analysis.
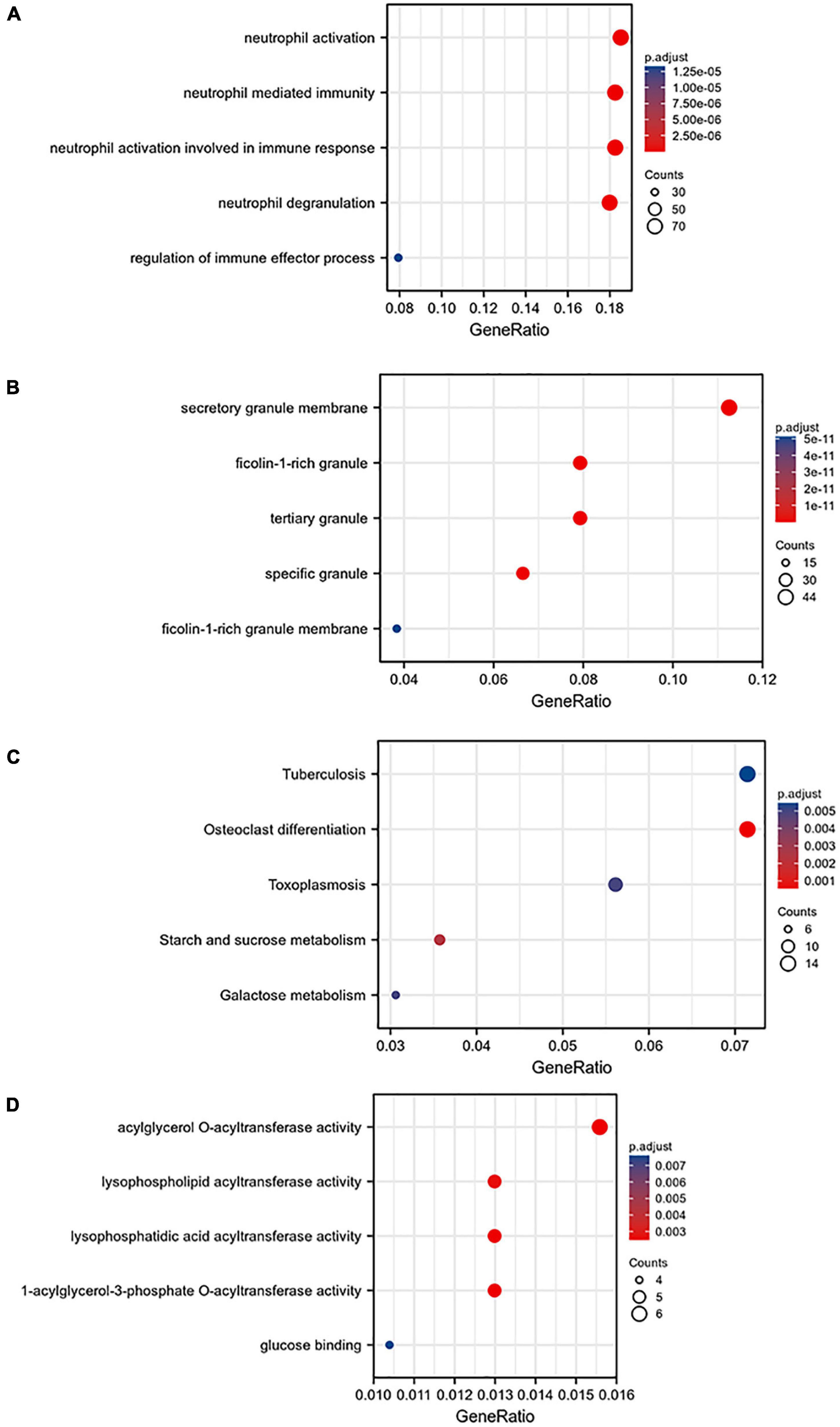
Figure 4. Gene ontology (GO) analysis of the blue module: (A) BP, (B) CC, (C) MF, and (D) KEGG pathway enrichment analysis of the blue module.
The 668 disease-related genes were used to construct PPI of 564 genes and 104 edge genes in the STRING database and were then visualized using Cytoscape software (Figure 5). In the PPI network, the average node degree was 0.36, and the average local clustering coefficient was 0.158. The analysis results showed that genes related to diseases were mainly expressed in neutrophil, phagocyte, granulocyte, mononuclear cell, and so on. The signaling pathways were mainly enriched in STAT5 activation, interleukin (IL)-10 signaling, neutrophil degranulation, IL-4 and IL-13 signaling, oxidative stress-induced senescence, and innate immune system.
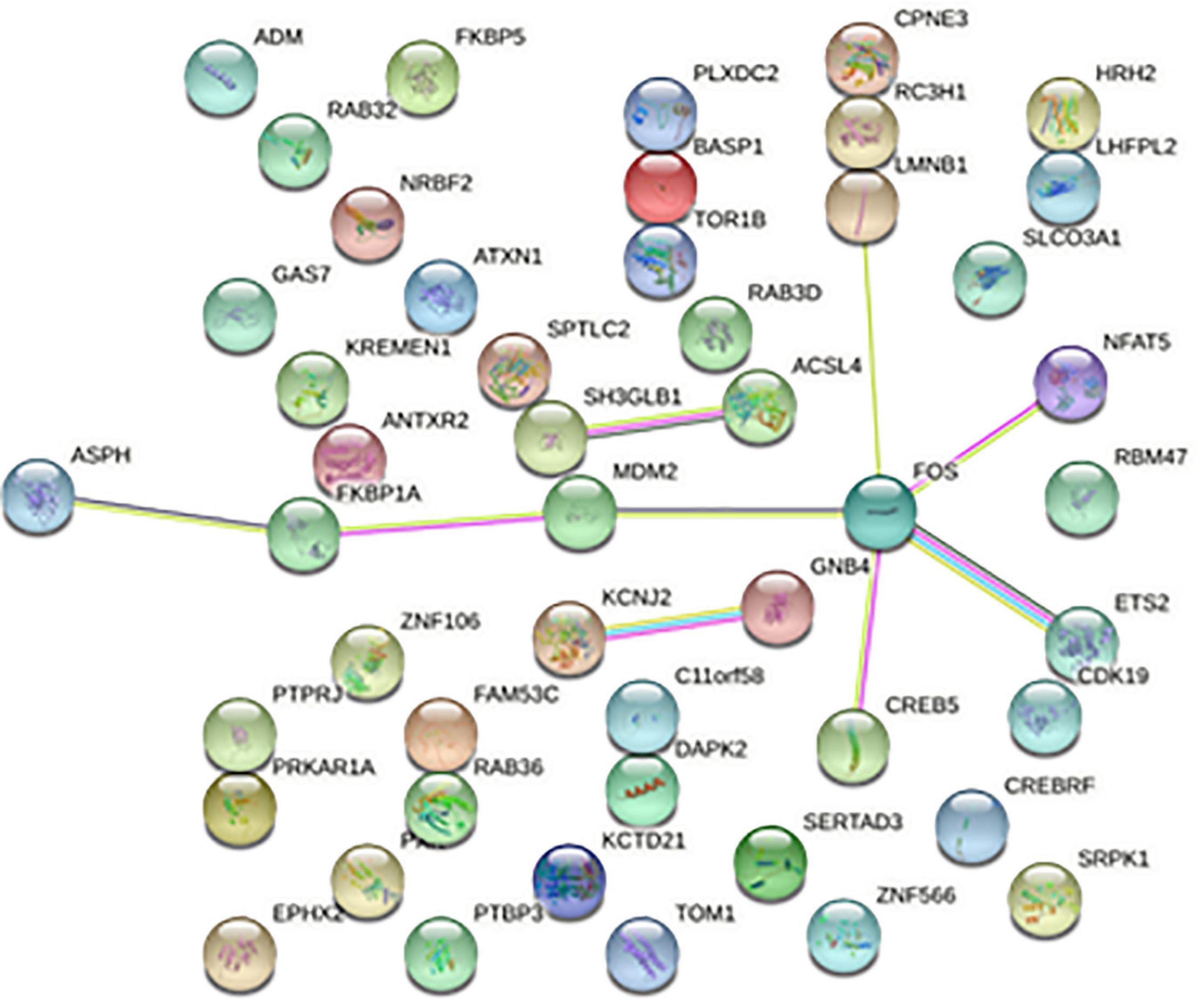
Figure 5. Protein-protein interaction network. Each node represents a protein, and the letter represented is the gene symbol of the corresponding gene. The lines between nodes represent the interaction between two proteins, and the multiple lines between two proteins indicate that there are multiple interaction relationships between the two proteins.
The MCODE plug-in was used to identify the 8 core modules (Figure 6), including 21 core genes and 36 edge genes, the average node degree was 3.43, and the average local clustering coefficient was 0.692. In addition, the functional analysis of DEGs in the module was performed (p < 0.05). In addition, the DEGs in the module were analyzed, and it was found that the core genes and immune functions of the 8 modules have a significant correlation (p < 0.05) (Table 2 and Figure 7).
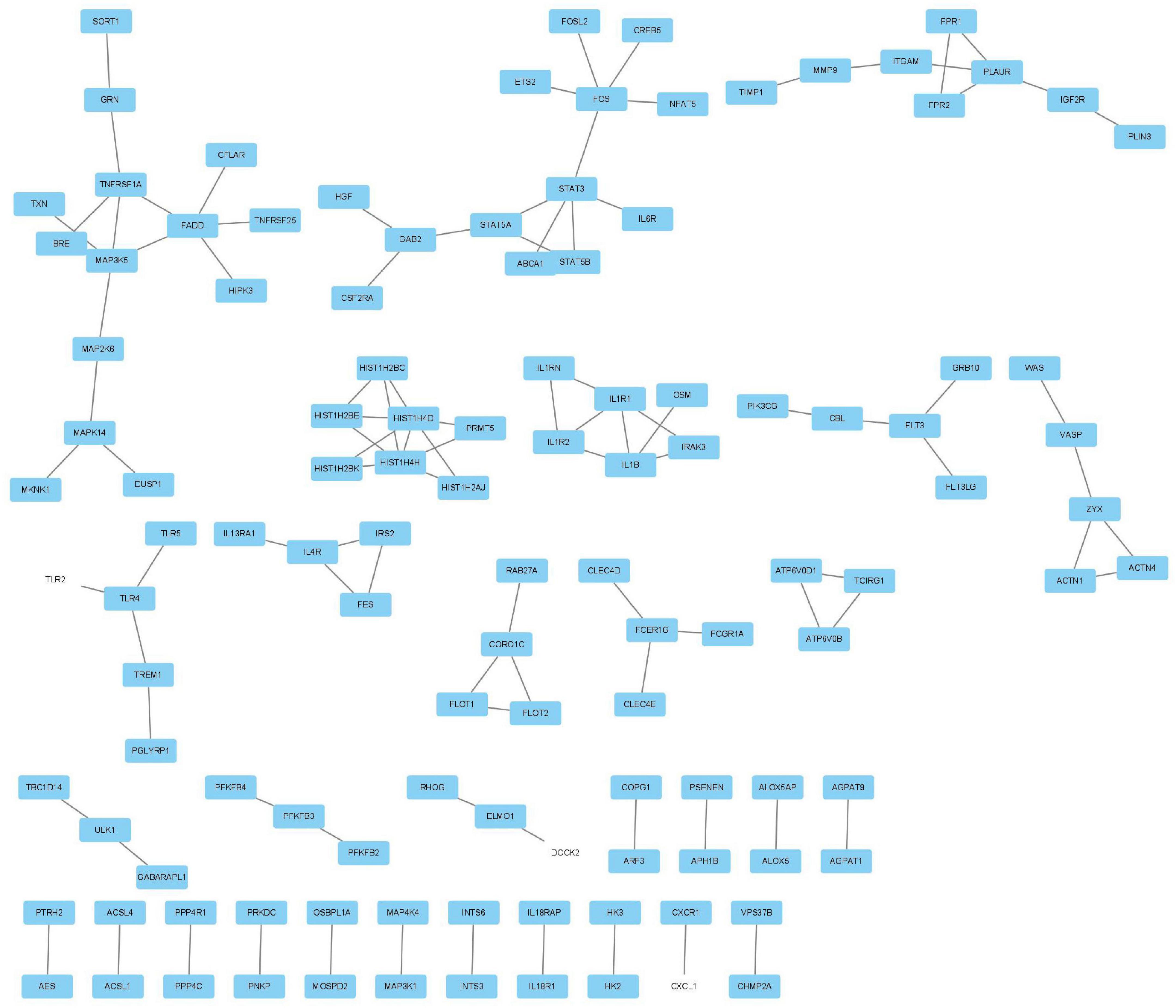
Figure 6. Eight modules, including 21 core genes and 36 marginal genes, were identified by using the molecular complex detection plug-in.

Table 2. Gene biology function and signal pathway enrichment analysis of eight modules.
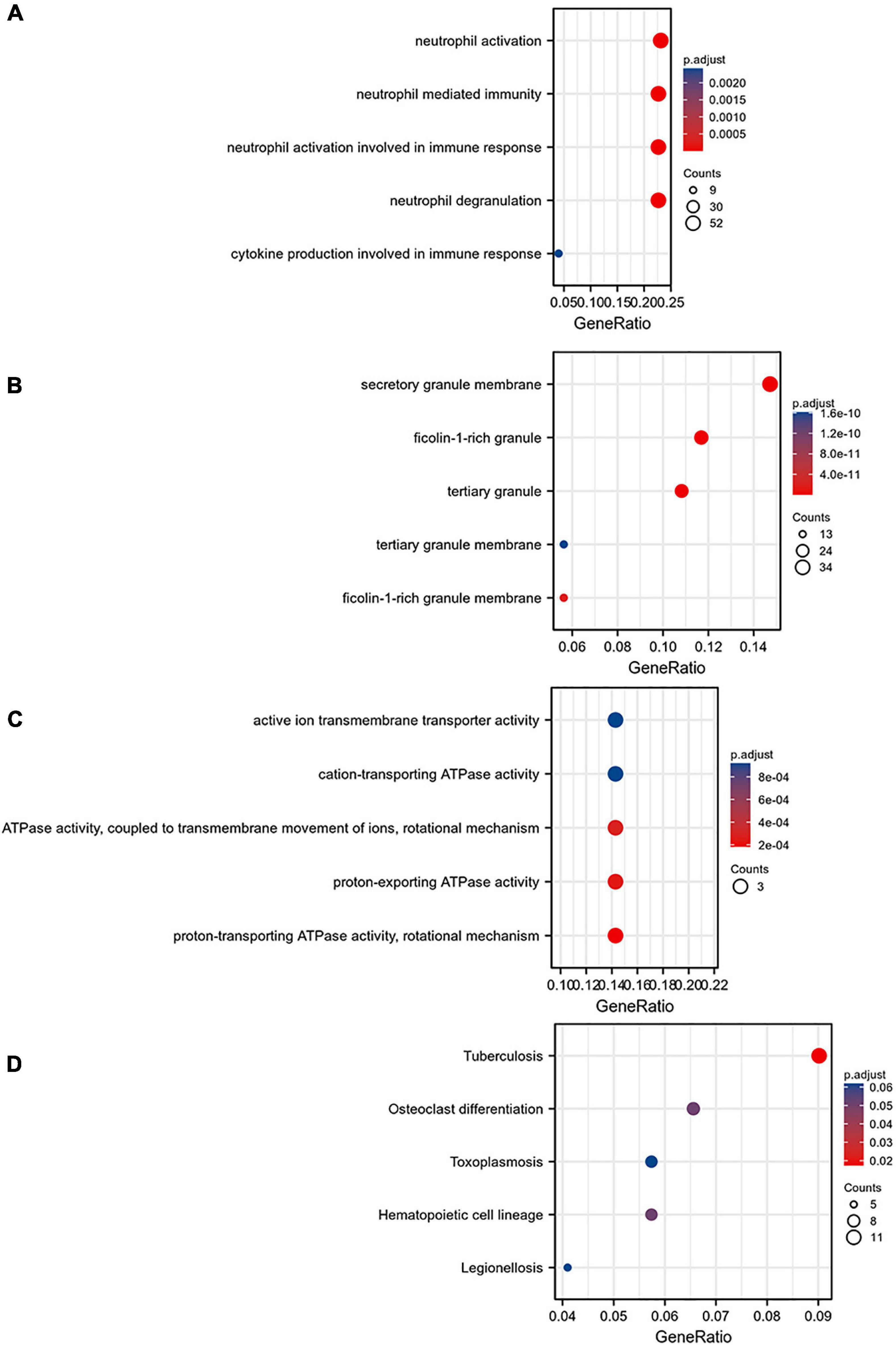
Figure 7. Gene ontology analysis of the eight core modules: (A) BP, (B) CC, (C) MF, and (D) KEGG pathway enrichment analysis of the eight core modules.
The Venn diagram analysis was used to extract 52 intersecting genes related to disease genes and target mRNAs (Figure 8A). Metascape database6 was used for annotation and visualization. Notably, 52 genes were analyzed for GO and KEGG pathway analysis. Min overlap >3 and p < 0.01 were considered statistically significant. The GO enrichment results showed that the differential genes were mainly enriched in chaperone-mediated protein folding, regulation of blood circulation, and leukocyte activation involved in immune response; the KEGG enrichment results showed that the differential genes were mainly enriched in the cholinergic synapse, human cytomegalovirus infection, and autophagy-animal pathways. (Figures 8B,C).
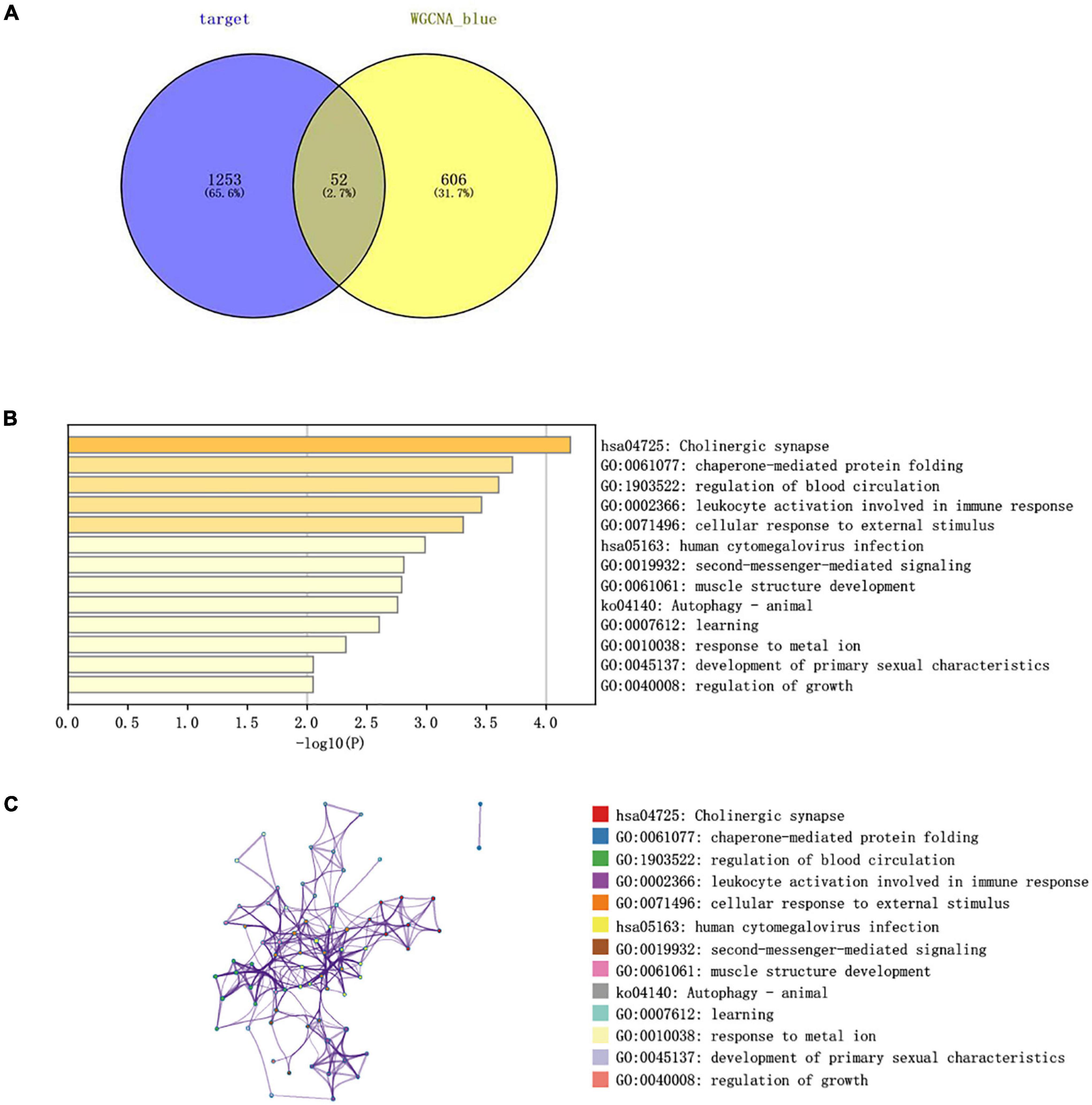
Figure 8. (A) Venn diagram constructed based on the blue module gene and the target gene. A total of 52 overlapping genes were identified. (B) Heatmap of the enrichment pathways. (C) Interaction relationship between the enrichment pathways.
NetworkAnalyzer in the Cytoscape (version 3.7.1) was used to analyze the topological parameters in the network. The best target was chosen based on the degree. The top 5 genes were selected as hub genes, namely, TOM1, STAT3, RAB3D, MDM2, and FOS. The correlation between the top 5 genes and the level of immune infiltration was studied. The results showed that hub genes are related to most of the immune cells. For example, the 5 hub genes are significantly positively correlated with neutrophils and significantly negatively correlated with eosinophils (Figure 9A).
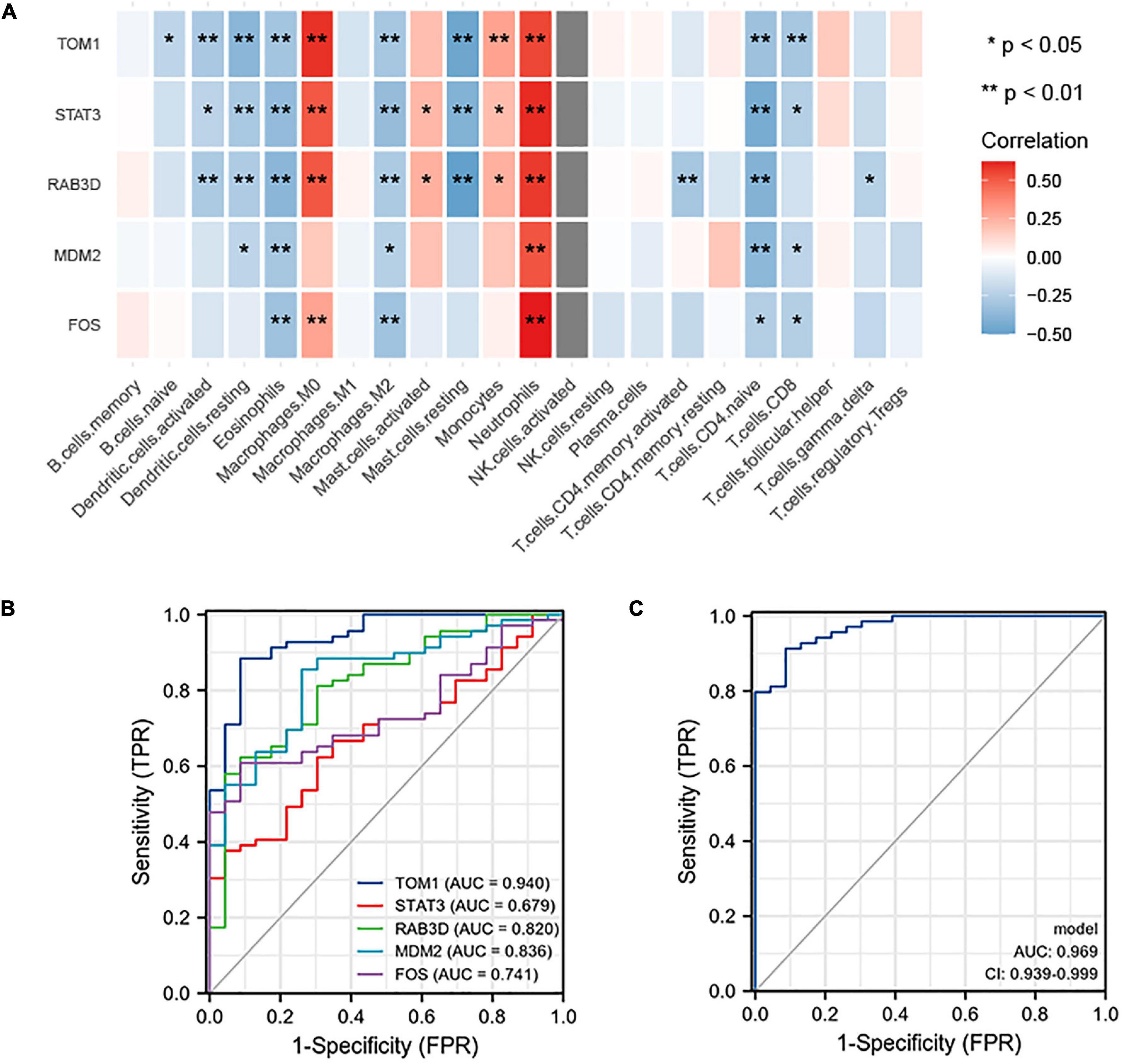
Figure 9. (A) Correlation between hub genes and immune infiltration. The top five genes (i.e., TOM1, STAT3, RAB3D, MDM2, and FOS) are all positively related to neutrophils and negatively related to eosinophils. (B) ROC prediction sensitivity of 5 core genes (TOM1 showed the highest sensitivity, followed by STAT3, RAB3D, MDM2, and FOS). (C) Sensitivity of joint prediction of 5 core genes (area under the curve = 0.969).
To understand the diagnostic value of the five core genes, ROC curve analysis was used to calculate the AUC of the gene expression levels in normal control groups and disease groups. In predicting the outcome of the normal control group and stroke, TOM1 has a higher accuracy (AUC = 0.940, CI = 0.887–0.992) in prediction, STAT3 has a lower accuracy (AUC = 0.679, CI = 0.565–0.793) in prediction, RAB3D has a certain accuracy (AUC = 0.820, CI = 0.723–0.916) in prediction, MDM2 has a certain accuracy (AUC = 0.836, CI = 0.747–0.924) in prediction, and FOS has a certain accuracy (AUC = 0.741, CI = 0.641–0.841) in prediction (Figure 9B). The abovementioned 5 hub genes were combined to obtain the ROC curve. In predicting the outcome of control and stroke, the predictive ability of the combined model has a higher accuracy (AUC = 0.969, CI = 0.939–0.999) (Figure 9C).
The CircInteractome online database was used to predict the miRNA targets of statistically different circRNAs. The results showed that there were 213 circRNA-related targeted miRNAs and a total of 405 circRNA-miRNA relationship pairs. By using TargetScan, miRDB, and miRTarBase databases to predict miRNA-related targeted mRNAs, 1,688 miRNA-mRNA relationship pairs were screened.
Combining the 52 genes in the Venn diagram, and taking the related circRNA and miRNA at the same time, a total of 132 circRNA-miRNA-mRNA relationship pairs are included. And through Cytoscape software analysis of core genes in circRNA and miRNA competition endogenous network, and successfully constructed circRNA-mediated ceRNA competition endogenous network diagram (Figure 10A).
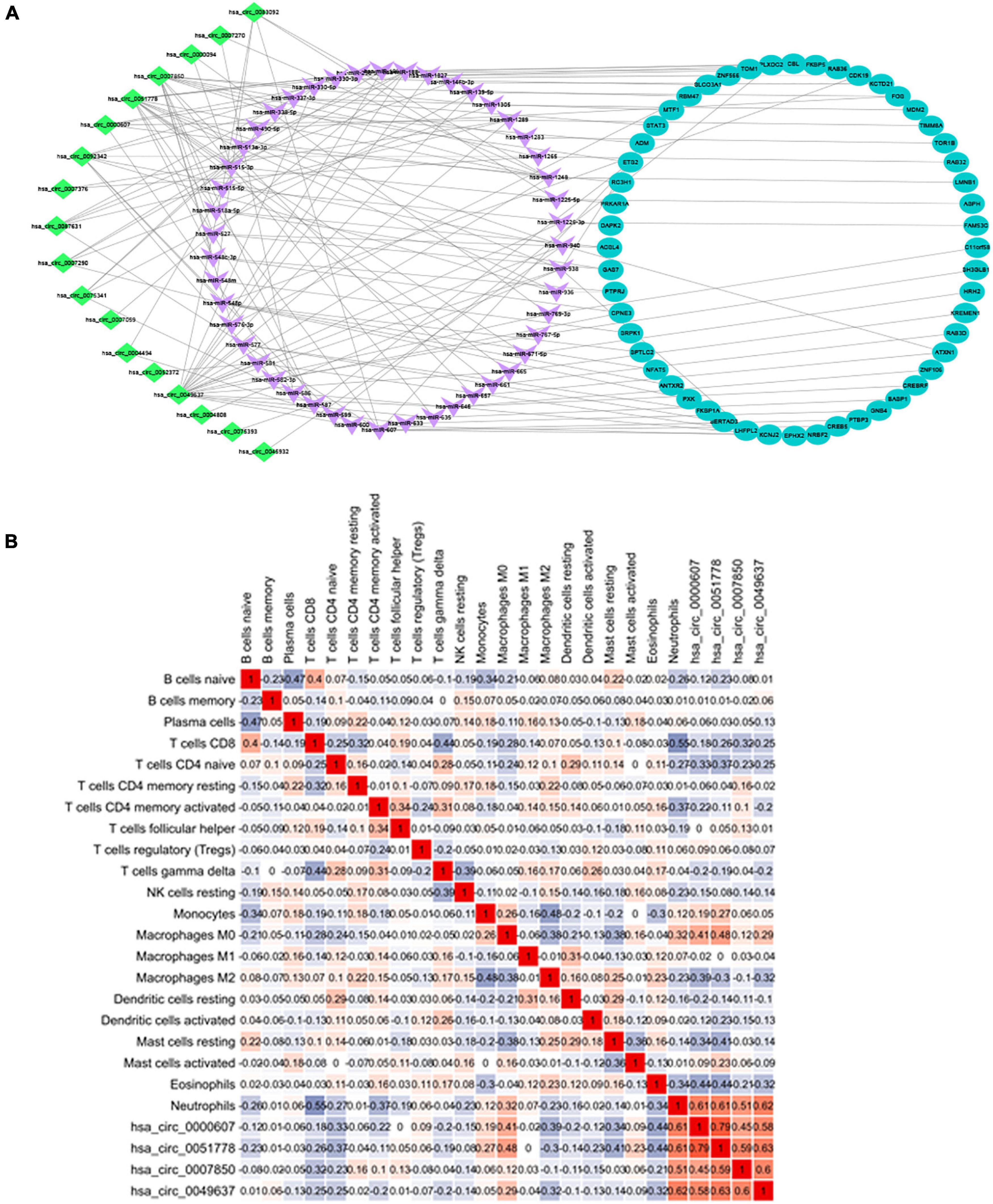
Figure 10. (A) ceRNA construction, including 18 circRNA, 47 hsa-miRNA, and 52 mRNA. ceRNA core gene was analyzed using the Metascape database for GO and KEGG enrichment analysis. (B) Correlation between immune cells in patients with IS. There is a strong positive correlation between CD8+ T cells and initial B cells (0.43), and a strong negative correlation between CD8+ T cells and neutrophils (–0.6).
Heatmap was constructed to show the correlation between ceRNA and immune infiltration (Figure 10B). The Pearson’s correlation coefficient was used to analyze the correlation between the key factors in ceRNA and immune cells (non-normal distribution was analyzed by the Spearman’s method), CD8+ T cells and hsa-miR-1305 (r = −0.310, p < 0.005), resting mast cells and hsa-miR-296-5p (r = −0.47, p < 0.005), macrophages M0 and hsa-miR-296-5p (r = 0.59, p < 0.005), and neutrophils and hsa-miR-139-5p (r = 0.62, p < 0.005).
The GSE22255 dataset was downloaded from the GEO database for verification purposes. The dataset was composed of 20 cases of AIS and 20 healthy controls. The age and gender of the two groups were matched. The 5 genes (i.e., TOM1, STAT3, RAB3D, MDM2, and FOS) were retrieved in the GSE22255 dataset. It was found that there were only 4 genes, i.e., TOM1, MDM2, RAB3D, and FOS. Then, the data were downloaded and analyzed by t-test. The result (Figure 11) suggested that FOS genes were upregulated in the AIS group, and there were differences in expression between the AIS group and the control group.
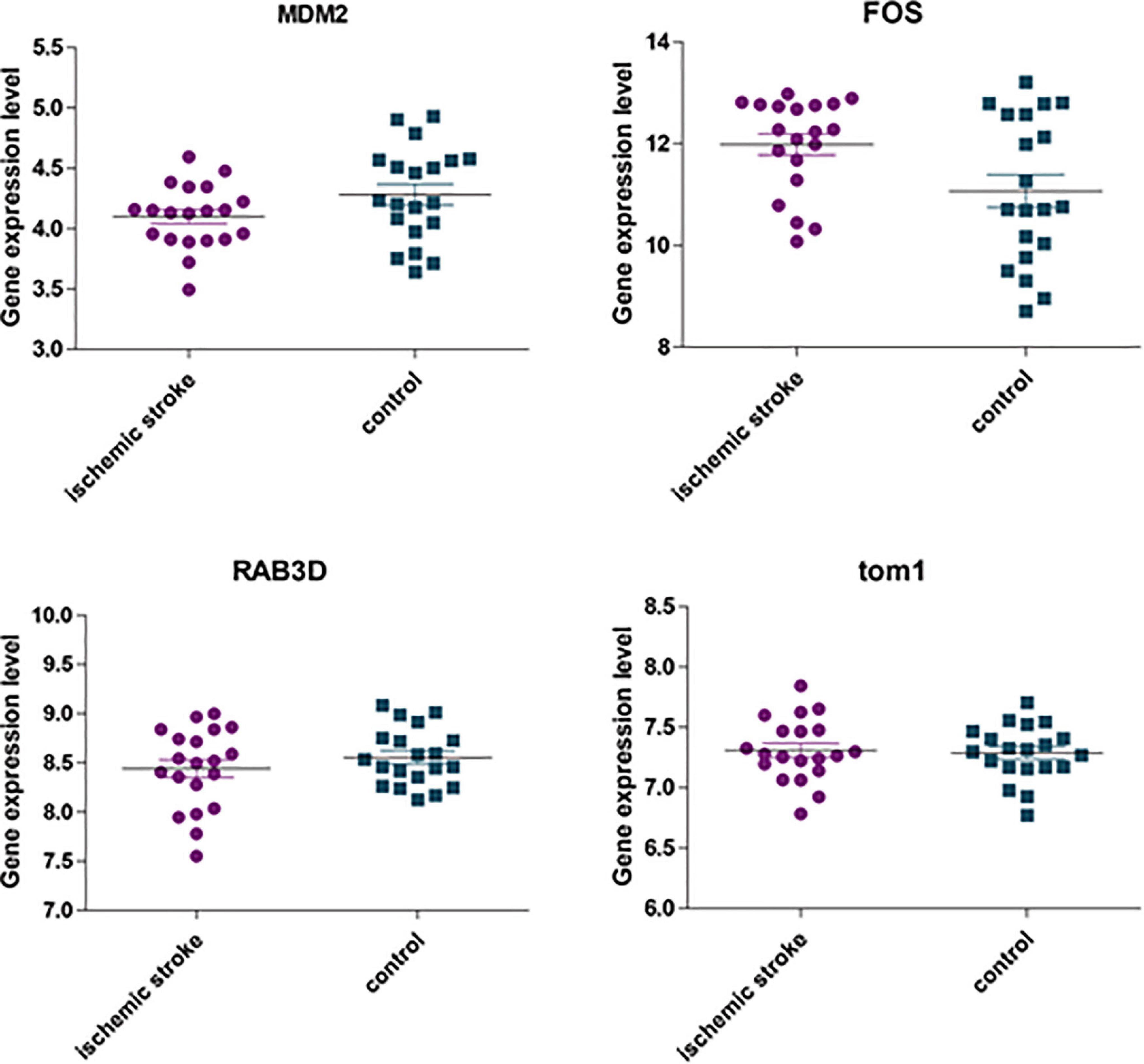
Figure 11. The expression of four hub genes was different between the AIS group and the healthy control group. Among them, p = 0.092 in the MDM2 group; p = 0.022 in the FOS group; p = 0.764 in the TOM1 group; p = 0.32 in the RAB3D group.
After AIS, neurons in the damaged area release DAMPs, which can trigger the immune response of the infarcted part and recruit/activate glial cells and peripheral white blood cells to the damaged brain area. This can further secrete a variety of pro-inflammatory cytokines, chemokines, and MMPs, resulting in the damage of the blood-brain barrier, and can cause brain edema, hemorrhage transformation, and neuronal necrosis (Rajkovic et al., 2018; Yang et al., 2019). During this process, different immune cells play various regulatory functions at different time points, which affect the prognosis of stroke.
Immune cell infiltration is related to the treatment and clinical outcome of various types of cancer. Evaluating the immune infiltration and determining the differences in immune infiltration components are crucial for the development of new immunotherapeutic drugs that target these cells. This has inspired us to think whether there is a correlation between immune cell infiltration and AIS pathogenesis and treatment. This study found that the immune infiltration levels of naive B cells, naive CD8+ T cells, naive CD4+ T cells, and resting mast cells were downregulated in patients with AIS, while the levels of CD4 memory-activated T cells, macrophages M0, and neutrophils immune infiltration were upregulated. Notably, the changes of CD8+ T-cell level were strongly positively correlated with naive B cells and were in a synergistic relationship. Resting NK cells were moderately positively correlated with memory B cells. Memory CD4+ T cells were moderately positively correlated with gamma-delta T cells. Naive B cells were correlated with resting dendritic cells and showed a moderately positive correlation change, where their possible mechanism of relationships is rarely reported. We infer that the changes of CD8+ T cells and the naïve B cells enhance each other’s effects during stroke, where further study on the correction of these immune cells is needed.
In the early stage of AIS, the activated M1 macrophages secrete a large amount of pro-inflammatory cytokines to promote the adhesion and exudation of neutrophils, lymphocytes, and monocytes. In the late stage of AIS, the replacement of activated M2 macrophages plays a major role, which secretes protective remodeling factors and promotes axon growth and angiogenesis (Ritzel et al., 2018); 24 h after AIS, the specific immune response in the body is activated (Haeusler et al., 2012), when the course of the disease enters the advanced stage. Due to the increased permeability of the blood-brain barrier in the early stage of stroke, lymphocytes enter the central nervous system after stroke and induce autoimmune reactions by recognizing antigens in the central nervous system. Studies have shown that the levels of CD4+, CD8+ T cells, and CD4+/CD8+ in the peripheral blood of patients with stroke are closely related to the severity and prognosis of AIS (Dolati et al., 2018). CD4+ T cells secrete cytokines, such as interferon (IFN)-γ and IL-17, in a non-antigen-dependent manner and promote the expression of chemokines by glial cells in the brain, enhancing neutrophils infiltration and aggravating the early nerve damage of AIS. In addition, T lymphocytes can mediate neurocytotoxicity. Regulatory T cells can be detected in large quantities in infarct tissues on 7 and 14 days after cerebral ischemia. The mechanism is that regulatory T cells release immunosuppression factors that regulate brain damage and maintain the body’s immune homeostasis, which forms the foundation for the reconstruction of the structure and function of the later stage of brain injury parts (Zhang et al., 2020; Shi et al., 2021). Animal studies found that regulatory T cells infiltrated the brain of mice 1–5 weeks after stroke. The selective depletion of regulatory T cells reduces oligodendrocyte production, white matter repair, and functional recovery after stroke. Increasing the levels of IL-2 and IL-2 antibody complexes after stroke can increase the number of regulatory T cells, which can improve white matter integrity in the long term and protect nerve functions (Li et al., 2020).
In addition, B lymphocytes are also involved in the immune response after stroke, but the mechanism is not clear. It is proposed that B lymphocytes secrete IL-10, which reduces the entry of activated inflammatory T cells into the ischemic brain tissue and increases CD8+CD122+Treg cells, and play a role in nerve protection (Stamova et al., 2014). After the stroke, the number of immune cells in the ischemic brain tissue and peripheral organs changes dynamically, and the expression levels of cell surface markers indicate that the brain is at the post-stroke inflammatory response state.
CircRNA-regulated ceRNA is a circRNA-miRNA-mRNA regulatory network constructed based on the regulatory and regulated relationship between different RNAs (Qu et al., 2015), and its possible mechanism is that there are a variety of miRNA response elements (MREs). miRNA binds to targeted mRNA through MRE and then plays a regulatory role in mRNA by inhibiting the translation of mRNA or reducing mRNA. Studies have shown that MRE also exists on circRNA. When circRNA and mRNA have the same MRE, the two will compete for the same type of miRNA after the disease occurs (Bosson et al., 2014). Another important biological function of circRNA is to act as the sponge of miRNA and indirectly regulate gene expression (Salmena et al., 2011). As a new form of post-transcriptional regulation, ceRNA has a great potential in disease research. Several studies have shown that the ceRNA regulatory network may be associated with cancer, atherosclerosis, heart disease, diabetes, and other diseases (Chan and Tay, 2018; Yan et al., 2020; Dai and Zhong, 2021; Liu et al., 2021), especially in AIS disease, where it plays an important regulatory role.
In our study, 559 differential circRNAs were identified in the AIS group and the normal group, including 213 upregulated circRNA and 346 downregulated circRNA. A total of 668 core genes were obtained from the GSE58294 dataset, as described previously. Three different methods were then used to find and verify the core genes and study their biological functions (1) using direct GO and KEGG analysis, (2) constructing a PPI network, and (3) using the MCODE plug-in to identify the 8 core modules. Its main biological functions are enriched in the activation of neutrophils, immune regulation of neutrophils, and other inflammatory factors. Most of the biological pathways involved are rich in immune and inflammatory response-related pathways, including primary immune-related pathways and immune diseases, and cytokine activation-related pathways. After multiple verifications of the target gene in this study, it is found that its biological function and signal pathway enrichment are mostly related to immune-inflammatory factors. It is speculated that the target gene plays an important function in the immune response mechanism of AIS.
The Venn diagram analysis was used to identify the disease gene and target mRNA-related intersection genes (a total of 52), and GO and KEGG pathway analysis were used to find that they are related to leukocyte activation involved in immune response and autophagy-animal pathway.
After AIS, the white blood cell count increases significantly (Emsley et al., 2003), and it is closely related to the patient’s poor prognosis (Furlan et al., 2014). As the main subtype of white blood cells, neutrophils accumulate in the cerebral blood vessels within a few hours under the action of attracting chemokines, such as CCL5, cxcl1, cxcl2, and cxcl3 (Cai et al., 2020; Kang et al., 2020), which may cause the expansion of the infarct and block the capillaries (McColl et al., 2008). In contrast, neutrophils regulate neuroinflammation after stroke through various inflammatory mediators, such as neutrophil protease (NE), myeloperoxidase (MPO), MMP, tissue inhibitors of metalloproteinase (TIMP), IFN, and IL (Cai et al., 2020), especially the expression of MMP can directly destroy the blood-brain barrier, leading to secondary brain injury or hemorrhagic transformation (McColl et al., 2008). Another feature of neutrophils is the formation of neutrophil extracellular traps (NETs). Studies have confirmed that the presence of NETs is a predictor of poor outcomes in myocardial infarction (Mangold et al., 2021), and inhibiting the formation of NETs can effectively prevent myocardial ischemia (Vajen et al., 2018). When a stroke occurs, neutrophils accumulate in the ischemic area and release toxic signals, such as NETs, thereby inhibiting revascularization and vascular repair after stroke (Kang et al., 2020). Studies have found that when patients with acute cerebral infarction are admitted to the hospital, their severity is related to a higher total number of white blood cells and neutrophil counts (Tarkanyi et al., 2020).
This study further analyzed the correlation between the intersection genes and the level of immune infiltration. The top 5 hub genes were TOM1, STAT3, RAB3D, MDM2, and FOS, and they were all positively correlated with neutrophils and negatively correlated with eosinophils. FOS, Rab3D (Nishio et al., 1999), and MDM-2 (Wang H. et al., 2021) genes have been reported to be correlated with neutrophils. There are few studies on the relationship between eosinophils and stroke, but there is evidence that the eosinophil count is valuable for predicting the recurrence and prognosis of cerebral infarction, and its decrease is associated with the AIS severity and clinical outcome at 3 months (Wang et al., 2017; Zhao et al., 2019), although the specific mechanism is unclear. The author speculates that eosinophils may be related to stroke through these 5 hub genes, but further research is needed. To verify the sensitivity of the 5 hub genes to disease diagnosis, ROC curve diagnostic test shows that the 5 hub genes have good performance in disease diagnosis, and the joint prediction of 5 hub genes has a higher sensitivity and a positive prediction rate. It also provides theoretical support for clinical diagnosis.
We further used the GSE22255 dataset to verify the 5 hub genes. Among them, STAT3 was not found. Among the other genes, only FOS was statistically different between the AIS group and the control group. It may be related to the dataset we selected. The GSE58294 dataset has many cases and is representative, but AIS is a cardiogenic stroke. Other datasets do not specify which type of stroke it is, which will lead to certain limitations in the conclusion. However, from the abovementioned analysis results, we speculate that whether it is cardiogenic AIS or other types of stroke, FOS is a gene worthy of attention and may play a regulatory role in the immune infiltration mechanism in IS.
By constructing a circRNA-mediated ceRNA regulatory network, 18 circRNAs related to AIS immune mechanisms were identified, among which hsa-circ-0000607, hsa-circ-0051778, hsa-circ-0007850, and hsa-circ-0049637 showed the strongest correlation with neutrophils. At present, there are few related studies on these four circRNAs, and the relationship with immunity has not been reported. However, through the ceRNA regulatory network, we found that hsa-circ-0049637 and FOS gene are correlated, and hsa-circ-0000607, hsa-circ-0051778, hsa-circ-0007850, and LMNB1 are correlated.
The FOS protein is a cell transcriptional regulatory factor. After the cell is stimulated, the c-fos gene in the nucleus is activated, is transcribed into mRNA, enters the cytoplasm, and is translated and synthesized into nucleophosphate protein. As an immediate early gene, c-fos is activated instantaneously and rapidly in response to a variety of cell stimuli. In different rat models, it can reach its peak level in 2–4 h after cerebral ischemia. The C-fos level can then gradually return to normal levels within 24 h (Karch et al., 2013; Nikoletopoulou et al., 2013). Studies have shown that its expression is parallel to the death or apoptosis of neurons after cerebral ischemia, that is, the degree of expression reflects the degree of neuronal death or apoptosis after stroke. The higher the c-fos expression level, the more serious the corresponding brain tissue damage would be (Whitfield et al., 2001). The main function of FOS is that it forms heterodimer FOS-jun complex with jun nuclear protein, which becomes the main form of transcriptional activator protein-1 (AP-1) when the cell is stimulated. The latter is in the AP-1-specific sites of the promoter and enhancer regions. It can regulate gene expression by binding to DNA and transforming extracellular signals into the nucleus (Bejjani et al., 2019). While neutrophils can be recruited from blood vessels to the site of inflammation, chemokines play an important role. In acute lung injury (ALI), the p38MAPK signaling pathway can regulate various inflammatory response processes. AP-1 is a downstream transcription factor of p38MAPK. The activated p38MAPK further activates AP-1 in the nucleus to regulate the release of downstream inflammatory mediators (e.g., chemokines, IL-1β, tumor necrosis factor-α, and adhesion molecules), which then activate many neutrophils, causing them to infiltrate at the inflammation site, and damage alveolar epithelial cells and lung capillary endothelial cells (Kumari et al., 2015).
LMNB1 is a nuclear lamin, which is closely related to the nuclear membrane, chromatin, nuclear pore complex, and nuclear matrix in the structure. It not only provides a structural scaffolding for nuclear membrane and chromatin but also plays a role in DNA replication, mRNA sorting, and cell mitosis. During the process, the nuclear membrane’s disintegration and reformation play an important role (Gruenbaum et al., 2005; Lees-Miller, 2006). Current studies have shown that Lmnb1, as a substrate for autophagy in primary human cells, can interact with SIRT1 (Wang L. et al., 2021) and autophagy protein LC3/Atg8 (Dou et al., 2015), which plays a regulatory function in human aging. When using the Metascape database for GO and KEGG enrichment analysis of 52 target genes, it was found that one of the pathways was enriched in the autophagy-animal signaling pathway. Studies have shown that in pMCAO rats, the use of glycogen synthase inhibitor-3β (GSK-3β) can reduce the inflammatory response caused by ischemic injury and detect a higher level of autophagy. In cells cultured in vitro, the use of autophagy inhibitor Beclin-1 siRNA can increase the inflammatory response of microglia. Studies have shown that GSK-3β can inhibit neuroinflammation induced by ischemic brain injury in rats by activating autophagy (Zhou et al., 2011). In the MCAO/R model, GSK-3β can increase the activity of autophagy by inhibiting the inflammatory protein NLRP3, thereby reducing ischemia-reperfusion injury (Wang et al., 2019). Many other studies (Cho et al., 2014; Yang et al., 2015; Hong et al., 2018) have proved that in IS, autophagy is closely related to the inflammatory response (Green et al., 2011), and this correlation may provide a new direction for the treatment of IS. Although the four circRNAs have not been reported to be related to inflammation, the corresponding proteins have been previously reported to be related to inflammationOur method also has some limitations. Due to the inconsistent circRNA naming, some circRNA will be lost during the data integration process, which may reduce our results. Another limitation is that due to the small number of samples from AIS patients, some of the identified circRNAs may be false positives, and the choice of the dataset may cause the results to have certain limitations. In future studies, we will use more samples to confirm the role of circRNA in the immune mechanism of AIS.
In summary, after patients with AIS, the initial B cells, initial CD8+ T cells, initial CD4+ T cells, and resting mast cell immune infiltration levels were downregulated, while CD4 memory activated T cells, macrophages M0, and neutrophils immune infiltration levels were increased. The hub genes related to the level of immune cell infiltration are TOM1, STAT3, RAB3D, MDM2, and FOS, and they have a significant positive correlation with neutrophils and a negative correlation with eosinophils. By constructing a circRNA-mediated ceRNA regulatory network, 18 circRNAs related to the immune mechanism of AIS were identified, of which four circRNAs, namely, hsa-circ-0000607, hsa-circ-0051778, hsa-circ-0007850, and hsa-circ-0049637, have the strongest positive correlation with neutrophils. The distribution characteristics of immune infiltration in patients with AIS discovered in this study and the identified circRNA may provide new research ideas for IS treatment, prevention, and new drug development.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
ZM contributed to the data analysis, manuscript writing, and submission. C-FL, LZ, NX, and YZ contributed to the review and revision of the manuscript. LC contributed to the study design. All authors contributed to the article and approved the submitted version.
This work was supported by Guizhou Provincial Health Commission Science and Technology Fund, fund number: 2020XMSB00028276 for the purchase of related software; Guiyang Science and Technology Bureau planned project, fund number: Zhuke Contract [2019]-9-14-8 to cover the cost of literature access; Science and Technology Project of Baiyun District, Guiyang City, fund number: Baike Contract [2020] No. 7 for the purchase of related software; Science and Technology Fund of Guizhou Provincial Health Commission, fund number: 2020XMSB00026375; and Science and Technology Project of Baiyun District, Guiyang City, Guizhou Province, fund numbers: Baike Contract [2020] No. 11 and Baike Contract [2020] No. 14.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bejjani, F., Evanno, E., Zibara, K., Piechaczyk, M., and Jariel-Encontre, I. (2019). The AP-1 transcriptional complex: local switch or remote command? Biochim. Biophys. Acta Rev. Cancer 1872, 11–23. doi: 10.1016/j.bbcan.2019.04.003
Bosson, A. D., Zamudio, J. R., and Sharp, P. A. (2014). Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol. Cell 56, 347–359. doi: 10.1016/j.molcel.2014.09.018
Cai, W., Liu, S., Hu, M., Huang, F., Zhu, Q., Qiu, W., et al. (2020). Functional dynamics of neutrophils after ischemic stroke. Transl. Stroke Res. 11, 108–121. doi: 10.1007/s12975-019-00694-y
Chan, J. J., and Tay, Y. (2018). Noncoding RNA:RNA regulatory networks in cancer. Int. J. Mol. Sci. 19:1310. doi: 10.3390/ijms19051310
Cho, M. H., Cho, K., Kang, H. J., Jeon, E. Y., Kim, H. S., Kwon, H. J., et al. (2014). Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10, 1761–1775. doi: 10.4161/auto.29647
Dai, M., and Zhong, S. (2021). A pilot study to construct the ceRNA regulatory network of a specific lncRNA in coronary heart disease. Int. J. Cardiol. 330:177. doi: 10.1016/j.ijcard.2021.01.034
Davis, S., and Meltzer, P. S. (2007). GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847. doi: 10.1093/bioinformatics/btm254
Ding, M. H., Lozoya, E. G., Rico, R. N., and Chew, S. A. (2020). The role of angiogenesis-inducing microRNAs in vascular tissue engineering. Tissue Eng. Part A. 26, 1283–1302. doi: 10.1089/ten.TEA.2020.0170
Dolati, S., Ahmadi, M., Khalili, M., Taheraghdam, A. A., Siahmansouri, H., Babaloo, Z., et al. (2018). Peripheral Th17/Treg imbalance in elderly patients with ischemic stroke. Neurol Sci. 2018 Apr;39(4):647-654. Erratum in Neurol. Sci. 39:655. doi: 10.1007/s10072-018-3250-4
Dou, Z., Xu, C., Donahue, G., Shimi, T., Pan, J. A., Zhu, J., et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109. doi: 10.1038/nature15548
Emsley, H. C., Smith, C. J., Gavin, C. M., Georgiou, R. F., Vail, A., Barberan, E. M., et al. (2003). An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J. Neuroimmunol. 139, 93–101. doi: 10.1016/s0165-5728(03)00134-6
Fu, Y., Liu, Q., Anrather, J., and Shi, F. D. (2015). Immune interventions in stroke. Nat. Rev. Neurol. 11, 524–535. doi: 10.1038/nrneurol.2015.144
Furlan, J. C., Vergouwen, M. D., Fang, J., and Silver, F. L. (2014). White blood cell count is an independent predictor of outcomes after acute ischaemic stroke. Eur. J. Neurol. 21, 215–222. doi: 10.1111/ene.12233
Gan, L., Liao, S., Xing, Y., and Deng, S. (2021). The regulatory functions of lncRNAs on angiogenesis following ischemic stroke. Front Mol Neurosci. 13:613976. doi: 10.3389/fnmol.2020.613976
Goyal, M., Demchuk, A. M., Menon, B. K., Eesa, M., Rempel, J. L., Thornton, J., et al. (2015). Randomized assessment of rapid endovascular treatment of ischemic stroke. N. Engl. J. Med. 372, 1019–1030. doi: 10.1056/NEJMoa1414905
Green, D. R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333, 1109–1112. doi: 10.1126/science.1201940
Gruenbaum, Y., Margalit, A., Goldman, R. D., Shumaker, D. K., and Wilson, K. L. (2005). The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 6, 21–31. doi: 10.1038/nrm1550
Gu, Z., Eils, R., and Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Haeusler, K. G., Schmidt, W. U., Foehring, F., Meisel, C., Guenther, C., Brunecker, P., et al. (2012). Immune responses after acute ischemic stroke or myocardial infarction. Int. J. Cardiol. 155, 372–377. doi: 10.1016/j.ijcard.2010.10.053
Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., et al. (2004). The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261. doi: 10.1093/nar/gkh036
Hong, Y., Liu, Y., Zhang, G., Wu, H., and Hou, Y. (2018). Progesterone suppresses Aβ42-induced neuroinflammation by enhancing autophagy in astrocytes. Int. Immunopharmacol. 54, 336–343. doi: 10.1016/j.intimp.2017.11.044
Jian, Z., Liu, R., Zhu, X., Smerin, D., Zhong, Y., Gu, L., et al. (2019). The involvement and therapy target of immune cells after ischemic stroke. Front. Immunol. 10:2167. doi: 10.3389/fimmu.2019.02167
Kang, L., Yu, H., Yang, X., Zhu, Y., Bai, X., Wang, R., et al. (2020). Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 11:2488. doi: 10.1038/s41467-020-16191-y
Karch, J., Kwong, J. Q., Burr, A. R., Sargent, M. A., Elrod, J. W., Peixoto, P. M., et al. (2013). Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2:e00772. doi: 10.7554/eLife.00772
Kumari, A., Tyagi, N., Dash, D., and Singh, R. (2015). Intranasal curcumin ameliorates lipopolysaccharide-induced acute lung injury in mice. Inflammation 38, 1103–1112. doi: 10.1007/s10753-014-0076-y
Lees-Miller, S. P. (2006). Dysfunction of lamin A triggers a DNA damage response and cellular senescence. DNA Repair (Amst). 2006 5(2):286-9. Erratum in DNA Repair. (Amst) 5:649. doi: 10.1016/j.dnarep.2005.10.007
Li, J., Wang, J., and Wang, Z. (2021). Circ_0006768 upregulation attenuates oxygen-glucose deprivation/reoxygenation-induced human brain microvascular endothelial cell injuries by upregulating VEZF1 via miR-222-3p inhibition. Metab. Brain Dis. 36, 2521–2534. doi: 10.1007/s11011-021-00775-8
Li, S., Cao, Y., Zhang, H., Lu, X., Wang, T., Xu, S., et al. (2021). Construction of lncRNA-mediated ceRNA network for investigating immune pathogenesis of ischemic stroke. Mol. Neurobiol. 58, 4758–4769. doi: 10.1007/s12035-021-02426-6
Li, Y., Wang, Y., Yao, Y., Griffiths, B. B., Feng, L., Tao, T., et al. (2020). Systematic study of the immune components after ischemic stroke using CyTOF techniques. J. Immunol. Res. 2020:9132410. doi: 10.1155/2020/9132410
Li, Z., Bai, Y., Li, W., Gao, F., Kuang, Y., Du, L., et al. (2018). Carotid vulnerable plaques are associated with circulating leukocytes in acute ischemic stroke patients: an clinical study based on contrast-enhanced ultrasound. Sci. Rep. 8:8849. doi: 10.1038/s41598-018-27260-0
Liu, Y., Liu, N., and Liu, Q. (2021). Constructing a ceRNA-immunoregulatory network associated with the development and prognosis of human atherosclerosis through weighted gene co-expression network analysis. Aging (Albany NY) 13, 3080–3100. doi: 10.18632/aging.202486
Mangold, A., Alias, S., Scherz, T., Hofbauer, M., Jakowitsch, J., Panzenböck, A., et al. (2021). Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res. 2015 116(7):1182-92. Erratum in Circ. Res. 128:e26. doi: 10.1161/CIRCRESAHA.116.304944
McColl, B. W., Rothwell, N. J., and Allan, S. M. (2008). Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J. Neurosci. 28, 9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008
Newman, A. M., Liu, C. L., Green, M. R., Gentles, A. J., Feng, W., Xu, Y., et al. (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. doi: 10.1038/nmeth.3337
Nikoletopoulou, V., Markaki, M., Palikaras, K., and Tavernarakis, N. (2013). Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 1833, 3448–3459. doi: 10.1016/j.bbamcr.2013.06.001
Nishio, H., Suda, T., Sawada, K., Miyamoto, T., Koike, T., and Yamaguchi, Y. (1999). Molecular cloning of cDNA encoding human Rab3D whose expression is upregulated with myeloid differentiation. Biochim. Biophys. Acta 1444, 283–290. doi: 10.1016/s0167-4781(98)00279-6
Powers, W. J., Rabinstein, A. A., Ackerson, T., Adeoye, O. M., Bambakidis, N. C., Becker, K., et al. (2018). 2018 guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the american heart association/american stroke association. Stroke. 2018 Mar;49(3):e46-e110. Erratum in Stroke 49:e138. doi: 10.1161/STR.0000000000000158
Qu, S., Yang, X., Li, X., Wang, J., Gao, Y., Shang, R., et al. (2015). Circular RNA: a new star of noncoding RNAs. Cancer Lett. 365, 141–148. doi: 10.1016/j.canlet.2015.06.003
Rajkovic, O., Potjewyd, G., and Pinteaux, E. (2018). Regenerative medicine therapies for targeting neuroinflammation after stroke. Front. Neurol. 9:734. doi: 10.3389/fneur.2018.00734
Rameshwari, R., and Prasad, T. V. (2012). Systematic and integrative analysis of proteomic data using bioinformatics tools. Int. J. Adv. Comp. Sci. Appl. 2, 29–35.
Ritzel, R. M., Lai, Y. J., Crapser, J. D., Patel, A. R., Schrecengost, A., Grenier, J. M., et al. (2018). Aging alters the immunological response to ischemic stroke. Acta Neuropathol. 136, 89–110. doi: 10.1007/s00401-018-1859-2
Salmena, L., Poliseno, L., Tay, Y., Kats, L., and Pandolfi, P. P. (2011). A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell 146, 353–358. doi: 10.1016/j.cell.2011.07.014
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shi, L., Sun, Z., Su, W., Xu, F., Xie, D., Zhang, Q., et al. (2021). Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity 54, 1527–1542.e8. doi: 10.1016/j.immuni.2021.04.022
Smyth, G. K. (2005). “Limma: linear models for microarray data,” in Bioinformatics and Computational Biology Solutions Using R and Bioconductor, eds R. Gentleman, V. J. Carey, W. Huber, R. A. Irizarry, and S. Dudoit (New York, NY: Springer), 397–420. doi: 10.1007/0-387-29362-0_23
Stamova, B., Jickling, G. C., Ander, B. P., Zhan, X., Liu, D., Turner, R., et al. (2014). Gene expression in peripheral immune cells following cardioembolic stroke is sexually dimorphic. PLoS One 9:e102550. doi: 10.1371/journal.pone.0102550
Tarkanyi, G., Karadi, Z. N., Szabo, Z., Szegedi, I., Csiba, L., and Szapary, L. (2020). Relationship between leukocyte counts and large vessel occlusion in acute ischemic stroke. BMC Neurol. 20:440. doi: 10.1186/s12883-020-02017-3
Vajen, T., Koenen, R. R., Werner, I., Staudt, M., Projahn, D., Curaj, A., et al. (2018). Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci. Rep. 8:10647. doi: 10.1038/s41598-018-29026-0
von Mering, C., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., and Snel, B. (2003). STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261. doi: 10.1093/nar/gkg034
Wang, H., Zhou, K., Li, W., Du, J., and Xiao, J. (2021). Ctnnb1 transcriptional upregulation compensates for Mdm2/p53-mediated β-catenin degradation in neutrophils following cardioembolic stroke. Gene 766:145022. doi: 10.1016/j.gene.2020.145022
Wang, J., Ma, L., Lin, T., Li, S. J., Chen, L. L., and Wang, D. Z. (2017). The significance of eosinophils in predicting the severity of acute ischemic stroke. Oncotarget 8, 104238–104246. doi: 10.18632/oncotarget.22199
Wang, L., Xu, C., Johansen, T., Berger, S. L., and Dou, Z. (2021). SIRT1 - a new mammalian substrate of nuclear autophagy. Autophagy 17, 593–595. doi: 10.1080/15548627.2020.1860541
Wang, Y., Meng, C., Zhang, J., Wu, J., and Zhao, J. (2019). Inhibition of GSK-3β alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy. Int. Immunopharmacol. 68, 234–241. doi: 10.1016/j.intimp.2018.12.042
Whitfield, J., Neame, S. J., Paquet, L., Bernard, O., and Ham, J. (2001). Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29, 629–643. doi: 10.1016/s0896-6273(01)00239-2
Wu, F., Han, B., Wu, S., Yang, L., Leng, S., Li, M., et al. (2019). Circular RNA TLK1 aggravates neuronal injury and neurological deficits after ischemic stroke via miR-335-3p/TIPARP. J. Neurosci. 39, 7369–7393. doi: 10.1523/JNEUROSCI.0299-19.2019
Yan, Q., He, X., Kuang, G., and Ou, C. (2020). CircRNA cPWWP2A: an emerging player in diabetes mellitus. J. Cell Commun. Signal. 14, 351–353. doi: 10.1007/s12079-020-00570-7
Yang, C., Hawkins, K. E., Doré, S., and Candelario-Jalil, E. (2019). Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 316, C135–C153. doi: 10.1152/ajpcell.00136.2018
Yang, L., Han, B., Zhang, Z., Wang, S., Bai, Y., Zhang, Y., et al. (2020). Extracellular vesicle-mediated delivery of circular RNA SCMH1 promotes functional recovery in rodent and nonhuman primate ischemic stroke models. Circulation 142, 556–574. doi: 10.1161/CIRCULATIONAHA.120.045765
Yang, Z., Zhong, L., Zhong, S., Xian, R., and Yuan, B. (2015). Hypoxia induces microglia autophagy and neural inflammation injury in focal cerebral ischemia model. Exp. Mol. Pathol. 98, 219–224. doi: 10.1016/j.yexmp.2015.02.003
Zhang, J., Liu, G., Chen, D., Wang, Z., Chen, D., Liu, Y., et al. (2020). The combination of CC chemokine receptor type 5(CCR5) and Treg cells predicts prognosis in patients with ischemic stroke. J. Neuroimmunol. 349:577404. doi: 10.1016/j.jneuroim.2020.577404
Zhao, H. M., Qin, W. Q., Wang, P. J., and Wen, Z. M. (2019). Eosinopenia is a predictive factor for the severity of acute ischemic stroke. Neural Regen. Res. 14, 1772–1779. doi: 10.4103/1673-5374.258411
Keywords: ischemic stroke, ceRNA, immune infiltration, inflammation, neutrophils
Citation: Ma ZL, Liu C-F, Zhang L, Xiang N, Zhang Y and Chu L (2022) The Construction and Analysis of Immune Infiltration and Competing Endogenous RNA Network in Acute Ischemic Stroke. Front. Aging Neurosci. 14:806200. doi: 10.3389/fnagi.2022.806200
Received: 31 October 2021; Accepted: 28 February 2022;
Published: 17 May 2022.
Edited by:
Guo-Yuan Yang, Shanghai Jiao Tong University, ChinaCopyright © 2022 Ma, Liu, Zhang, Xiang, Zhang and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Chu, Y2h1bGFuODk5OUB5ZWFoLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.