
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Aging Neurosci., 08 December 2022
Sec. Neurocognitive Aging and Behavior
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.1085406
This article is part of the Research TopicThe UN International Day of Families: Neurodegeneration as a Result of Genetic InheritanceView all 10 articles
 Giacomina Rossi1*
Giacomina Rossi1* Erika Salvi2
Erika Salvi2 Elkadia Mehmeti2Martina Ricci1
Elkadia Mehmeti2Martina Ricci1 Cristina Villa1
Cristina Villa1 Sara Prioni3
Sara Prioni3 Fabio Moda1
Fabio Moda1 Giuseppe Di Fede1Pietro Tiraboschi1Veronica Redaelli1
Giuseppe Di Fede1Pietro Tiraboschi1Veronica Redaelli1 Cinzia Coppola4
Cinzia Coppola4 Giacomo Koch5
Giacomo Koch5 Elisa Canu6
Elisa Canu6 Massimo Filippi6,7,8,9,10
Massimo Filippi6,7,8,9,10 Federica Agosta6,7,8
Federica Agosta6,7,8 Giorgio Giaccone1
Giorgio Giaccone1 Paola Caroppo1
Paola Caroppo1Semantic and right temporal variant of frontotemporal dementia (svFTD and rtvFTD) are rare clinical phenotypes in which, in most cases, the underlying pathology is TDP-43 proteinopathy. They are usually sporadic disorders, but recent evidences suggest a higher frequency of genetic mutations for the right temporal versus the semantic variant. However, the genetic basis of these forms is not clear. In this study we performed a genetic screening of a single-center cohort of svFTD and rtvFTD patients, aiming at identifying the associated genetic variants. A panel of 73 dementia candidate genes has been analyzed by NGS target sequencing including both causal and risk/modifier genes in 23 patients (15 svFTD and 8 rtvFTD) and 73 healthy age-matched controls. We first performed a single variant analysis considering rare variants and then a gene-based aggregation analysis to evaluate the cumulative effects of multiple rare variants in a single gene. We found 12 variants in nearly 40% of patients (9/23), described as pathogenic or classified as VUS/likely pathogenic. The overall rate was higher in svFTD than in rtvFTD. Three mutations were located in MAPT gene and single mutations in the following genes: SQSTM1, VCP, PSEN1, TBK1, OPTN, CHCHD10, PRKN, DCTN1. Our study revealed the presence of variants in genes involved in pathways relevant for the pathology, especially autophagy and inflammation. We suggest that molecular analysis should be performed in all svFTD and rtvFTD patients, to better understand the genotype–phenotype correlation and the pathogenetic mechanisms that could drive the clinical phenotypes in FTD.
The term semantic variant of frontotemporal dementia (svFTD) usually refers to the form of FTD characterized by an early impairment of verbal semantic knowledge, that manifests as anomia and impaired single word comprehension, linked to the predominant atrophy of the left anterior temporal lobe and the diagnosis is based on current clinical criteria (Gorno-tempini et al., 2011). The right temporal variant of FTD (rtvFTD), a rarer clinical phenotype, originally considered a right variant of svFTD, has distinct clinical features and predominantly right hemisphere involvement. Patients with rtvFTD exhibit early behavioral symptoms, particularly empathy deficits and psychiatric disorders (Ulugut Erkoyun et al., 2020), but the diagnosis is challenging, and patients are often misdiagnosed as behavioral variant of FTD (bvFTD; Younes et al., 2022). A motor neuron involvement is rare in svFTD, but it has been found in up to 28% of rtvFTD cases in a recent neuropathological cohort (Ulugut et al., 2021).
While pathology can be highly heterogeneous in bvFTD, most of patients affected by svFTD and rtvFTD has TDP-43 neuronal accumulation, particularly the Type C pathology (Rohrer et al., 2010). In a minority of patients with rtvFTD a recent study reported also tauopathy as pathological substrate of the disease (Ulugut et al., 2021).
FTD has a prominent genetic basis, with up to 40% of cases presenting a family history (Mann and Snowden, 2017). To date three major genes have been described as linked to FTD: microtubule-associated protein tau (MAPT), progranulin (GRN) and Chromosome 9 open reading frame 72 (C9ORF72; Mann and Snowden, 2017). Furthermore, other causative genes have been disclosed, underlining rarer familial cases: Sequestosome 1 (SQSTM1), Valosin-Containing Protein (VCP), Charged Multivesicular Body Protein 2B (CHMP2B), TAR DNA Binding Protein, 43-KD (TARDBP), Fused in Sarcoma (FUS), Triggering Receptor Expressed on Myeloid cells 2 (TREM2), Tank-Binding Kinase 1 (TBK1) (see Fenoglio et al., 2018 for review). Left and right variants of FTD are usually sporadic, but recent evidence suggests a higher frequency of genetic mutations for the right versus the left variant (Ulugut Erkoyun et al., 2021). However, the genetic basis of these forms is not clear and genotype–phenotype correlations need to be elucidated, to allow accurate genetic counseling.
In this study, we used a next generation sequencing (NGS) approach to perform a genetic screening of a single-center cohort of patients with svFTD and rtvFTD, aiming at evaluating the genetic contribution in those disorders, by confirming candidate or discovering new genetic variants that could be associated with these clinical phenotypes.
Twenty-three patients with clinical diagnosis of svFTD and rtvFTD (15 and 8 respectively) were retrospectively selected from FTD patients followed-up at Fondazione IRCCS Istituto Neurologico Carlo Besta from 2015 to 2020. One of them, carrying a MAPT mutation has been recently published (Villa et al., 2022). Clinical diagnosis of svFTD was made on the basis of current criteria (Gorno-Tempini et al., 2011). For the diagnosis of rtvFTD we followed the clinical and neuroimaging proposed framework (Ulugut Erkoyun et al., 2020). All patients underwent a complete clinical, neuropsychological and neuroimaging assessment. Analysis of markers of neurodegeneration (amyloid-β 1–42 – Aβ42, total tau and T181-phosphorylated-tau) was performed on cerebrospinal fluid (CSF) of 16 patients using LUMIPULSE G (Fujirebio, Malvern, PA, United States). Seventy-three age-matched healthy subjects from an in-house cohort were used as control group (healthy controls, HC). Written informed consent was obtained from all patient and healthy subjects.
DNA of patients was obtained, after informed consent, from peripheral blood lymphocytes. We designed a targeted enrichment panel to capture the coding and 25 bp flanking intron sequences of 73 dementia genes, including both causal genes and risk/modifier factors (Supplementary material S1). Nextera Flex for Enrichment system (Illumina, San Diego, CA, United States) coupled with gene-specific probes (Integrated DNA Technologies, Coralville, IA, United States) and the MiSeq instrument (Illumina, San Diego, CA, United States) were used for sequencing. A first quality control step was performed with FastQC software to identify base quality drops across cycles and adapter contamination, and to evaluate overall data quality. We performed both adapter trimming and quality trimming using Trimmomatic (version 0.36; Bolger et al., 2014). High-quality reads were mapped to the hg19 reference genome using bwa v. 0.7.17-r1188 (mem algorithm; Li and Durbin, 2010). Hence, we performed duplicated read marking, local realignment, and base quality score recalibration as suggested by GATK best practices (DePristo et al., 2011). We performed single-nucleotide variant (SNV) and insertion/deletion (INDEL) calling using the GATK module Haplotype Caller (version 4.1.9) over the target region. For the variant annotation we used the SnpEff software (Cingolani et al., 2012) that annotates and predicts the effects of genetic variants on genes and proteins. Variants that did not pass the variant filtering (total reads count≤20, alternative allele depth ≤ 10 and allele balance of 25%) were removed. Visual inspection of bam files was performed using IGV software (Thorvaldsdóttir et al., 2013).
In our cohort of 23 patients and 73 HC we first performed a single-variant analysis considering non-synonymous (missense, splicing, frameshift, STOP gain/loss) rare variants, that is variants with minor allele frequency (MAF) <0.01 in Genome Aggregation Database – Exomes – Non Finnish Europeans (GNOMEX_NFE). Franklin by Genoox1 and/or Varsome2 were used to report the classification according the criteria of the American College of Medical Genetics (ACMG).
Furthermore, we performed a gene-based aggregation analysis to evaluate the cumulative effects of multiple rare and low frequency (MAF <0.05) variants in a gene. All non-synonymous variants with MAF <0.05 in the studied cohort and reaching at least 80% of call rate were collapsed into single genes. Genes mapped by only one variant were not considered. To test whether there is an excess of variants in patients in comparison with HC, we applied both the combined burden and variance-component SKAT-O (Lee et al., 2012) and the simple burden method as implemented in EPACTS software (Efficient and Parallelizable Association Container Toolbox).3 Rho statistic indicates the direction of the effects, with rho = 1 referring to high percentage of causality in the same direction and rho = 0 to the simultaneous presence of causal and non-causal variants with opposing directions.
All the analyses were corrected for sex and age as covariates.
The GGGGCC hexanucleotide repeat in the C9ORF72 gene (DeJesus-Hernandez et al., 2011) was assessed by a repeat-primed PCR reaction using the AmplideX® PCR/CE C9orf72 Kit (Asuragen, Austin, TX, United States), according to the manufacturer’s instructions. PCR products were analyzed on an ABI3130xl Genetic Analyzer.
Demographic and clinical data of overall cohort are reported in Table 1. Patients and HC did not show significant difference in term of sex (p = 0.11) and age (p = 0.18). 39% (9/23) of patients had a family history of dementia (53% of svFTD).
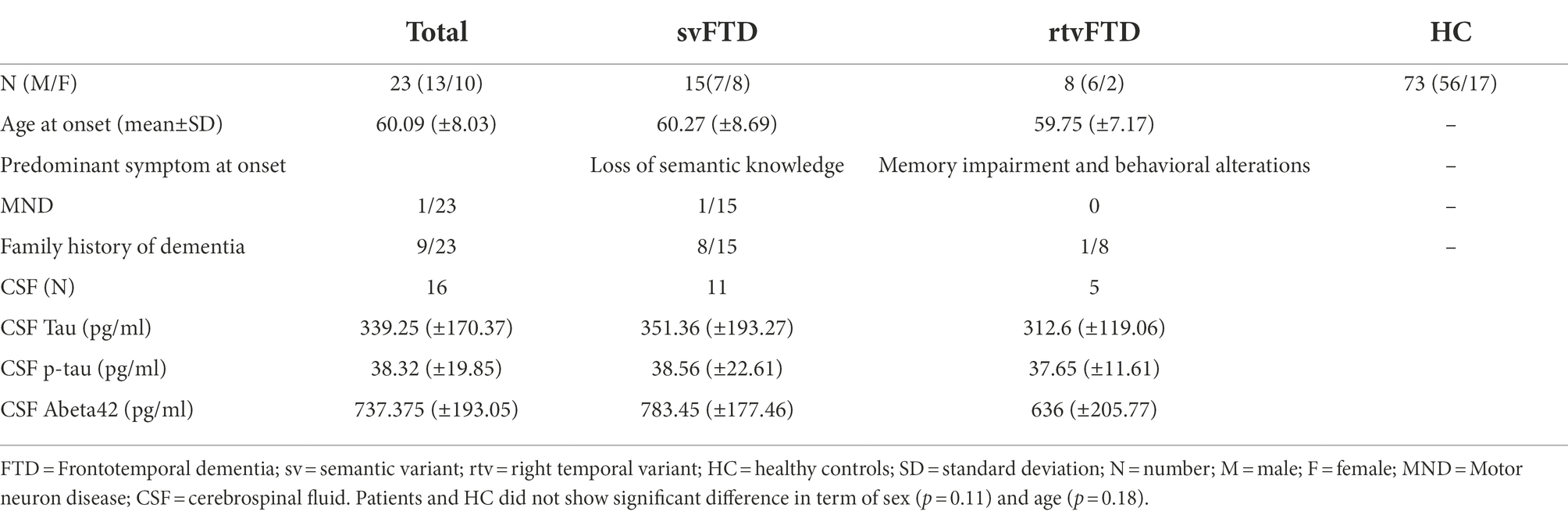
Table 1. Clinical and demographic features of study groups.
Semantic impairment with naming deficits was the first symptom of svFTD patients, while rtvFTD patients showed early changes in their personality and behaviour (among them, eating behavior changes, loss of empathy, compulsive behaviors), memory loss, prosopagnosia and topographical disorientation. According with clinical diagnosis, brain magnetic resonance imaging and fluorodeoxyglucose-positron emission tomography showed predominant atrophy and hypometabolism in left anterior temporal lobe in svFTD and a predominant right temporal involvement in all rtvFTD. CSF biomarkers were in the normal range in 13/16 patients, 3 patients had isolated reduction of Abeta42. Total tau was elevated in 2 patients and one of them had also elevated p-tau, but normal Abeta42.
By single variant analysis, we found 12 variants described as pathogenic in the scientific literature (reported in Human Gene Mutation Database, HGMD) or classified as variant of unknown significance (VUS)/likely pathogenic in 9 patients (39% of patients) and none in control subjects (Table 2).

Table 2. Variants present in patients.
We found three mutations in the microtubule-associated protein tau (MAPT) gene, which was the first causative gene to be linked to FTD: P301L, Q336H (Villa et al., 2022) and N286N. In addition, we discovered single mutations in the following genes: SQSTM1, VCP, presenilin 1 (PSEN1), TBK1, optineurin (OPTN), coiled-coil-helix-coiled-coil-helix domain containing protein 10 (CHCHD10), parkin (PRKN) and dynactin 1 (DCTN1). Three patients carried two mutations each: P01, N286N MAPT and M93V PSEN1; P06, Q314L OPTN and R795H DCTN1; P21, G376E VCP and I207T TBK1. All the variants are singletons (present only in 1 patient) and not present in control subjects. Clinical characteristics of the 9 patients carrying the variants are reported in Table 3.
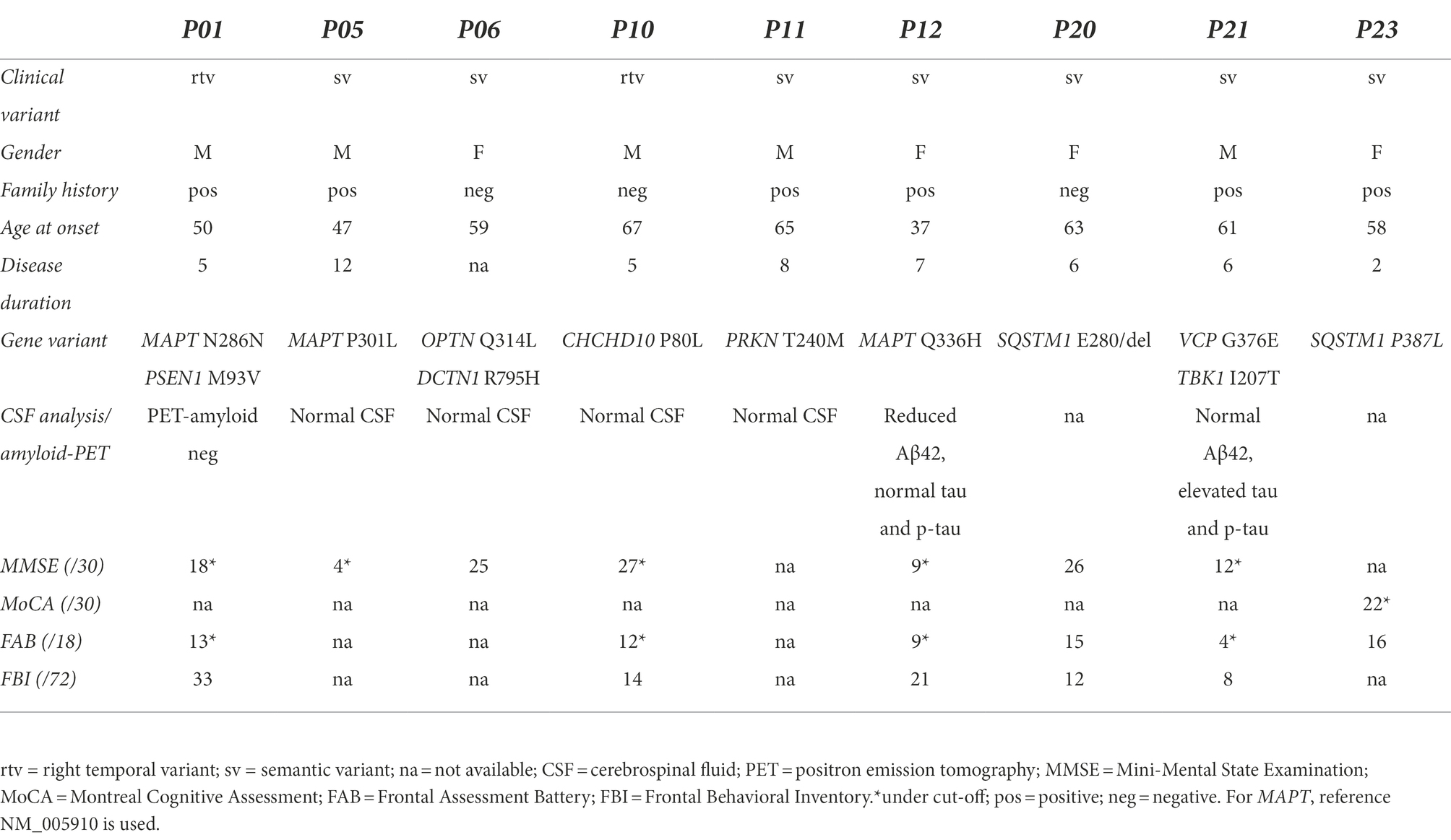
Table 3. Clinical and demographic characteristics of the 9 patients carrying the variants.
None of the genes reached the Bonferroni corrected threshold (for the total number of genes considered, p = 0.001). However, the SKAT-O test highlighted 4 genes with suggestive value of p <0.05, enriched for rare variants: CD33, PSEN1, OPTN and ABCA1 (Table 4).
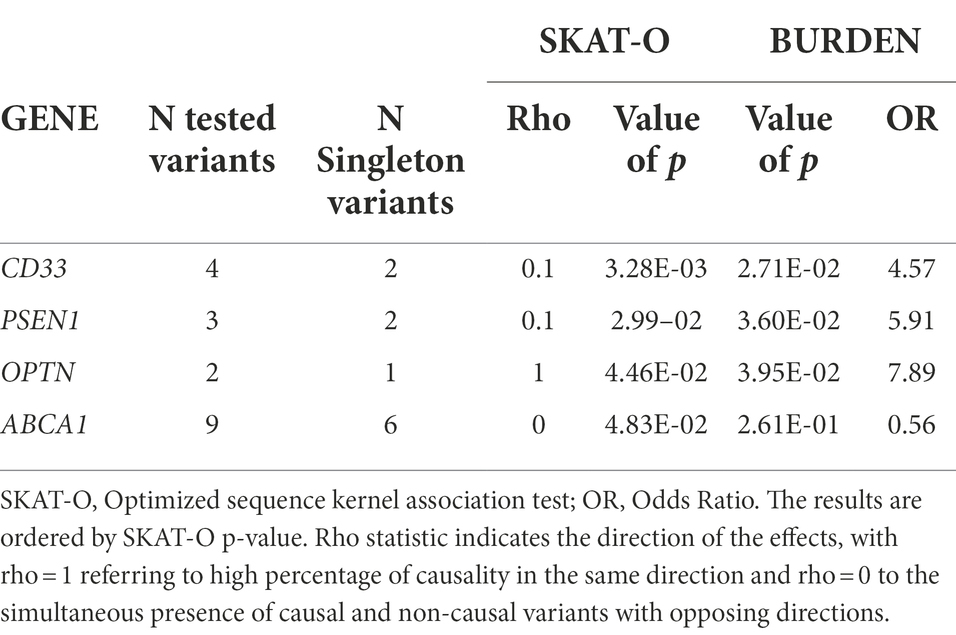
Table 4. Genes enriched by rare/low frequency variants after gene-based analysis with a SKAT-O p value <0.05.
OPTN with a rho value equal to 1 showed a unidirectional risk association, reflected by the fact that the two rare variants are exclusively present or more frequent in cases than in HC: M98K was present in 6.5% of patients and 2.1% of HC, whereas Q314L was carried by one patient (Table 5). The simple burden test showed OPTN as conferring a risk 7.89-fold higher in patients compared to HC (p = 0.0395, OR = 7.89).
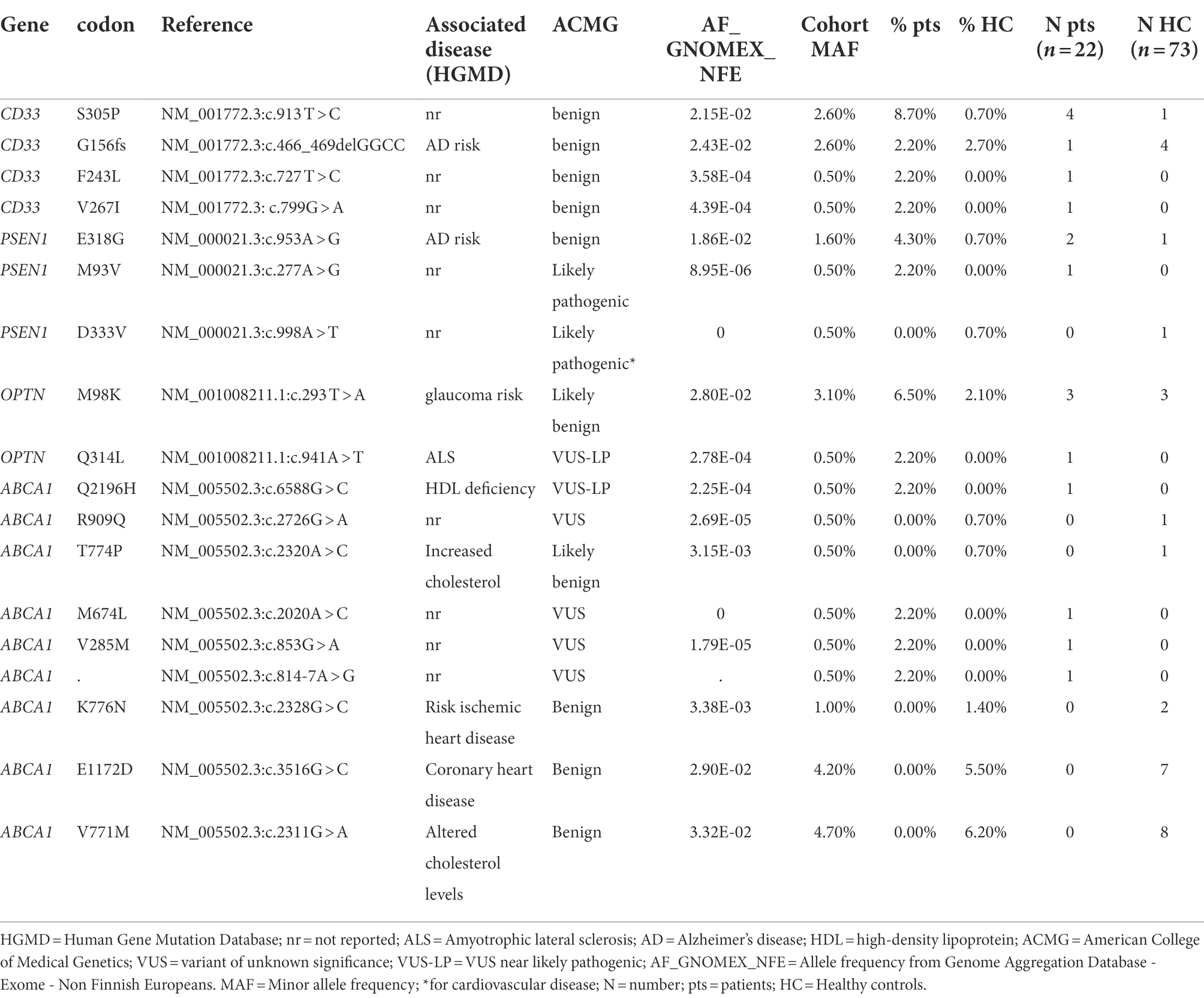
Table 5. Variants present in the enriched genes (gene-based aggregation analysis).
A rho value equal to 0.1, for CD33 and PSEN1, indicated a simultaneous presence of causal and non-causal variants with opposing directions. CD33 is enriched of 4 rare/low frequency variants in 7 patients and 5 HC: the frameshift G156fs was shared by patients and HC with a similar frequency, S305P was present in 4 of 23 patients (8.7%) and one HC (0.7%), F243L and V267I were exclusively carried by one patient (Table 5). In PSEN1, E318G was found in 4.3% of patients and 0.7% of controls, the M93V was exclusively carried by one patient whereas D333V was present in one control.
As for ABCA1 (Table 5), a rho value of 0 indicates the simultaneous presence of causal and non-causal variants with opposing directions. In fact, out of 9 variants, 4 were present in patients and 5 in a high number of HC, showing opposite directions in terms of risk association.
None of the patients carried an expanded allele (>30 repeats).
In this study we found several pathogenic or VUS/likely pathogenic variants, in nearly 40% of patients.
P301L, carried by P05, is one of the most known and studied MAPT mutations, whose pathogenicity has been demonstrated in several genetic, functional and neuropathological studies, as well as in animal models (Hutton et al., 1998; Barghorn et al., 2000; Lewis et al., 2000; Rossi et al., 2008). Different FTD clinical phenotypes have been associated with the mutation, including svFTD (Ishizuka et al., 2011). Q336H MAPT mutation, carried by patient P12, was recently published as associated with svFTD (Villa et al., 2022). This mutation had been previously functionally and neuropathologically characterized as pathogenic (Tacik et al., 2015). Patient P01 carried the N286N MAPT and M93V PSEN1 variants. As for the N286N MAPT variant, only once cited without additional details (Rohrer et al., 2009), the classification as likely benign does not take into account the pathogenetic mechanism typical of some silent MAPT mutations localized in exon 10, that is the alteration of an exon 10 splicing enhancer or silencer (D’Souza and Schellenberg, 2002; Qian and Liu, 2014). In fact, an in silico functional analysis demonstrated the effect on exon 10 splicing of this variant (Tubeuf et al., 2020). The M93V variant in PSEN1, so far never described, is very interesting as localized in a mutational hot spot in a critical region for the protein function, the transmembrane domain 1: in fact, the adjacent mutations C92S (Zhang et al., 2000; Tedde et al., 2003) and V94M (Arango et al., 2001; Somavarapu and Kepp, 2016) are associated with Alzheimer’s disease (AD). In addition, several other pathogenic AD mutations have been described in the same protein region. It is well known that PSEN1 mutations give rise to Alzheimer’s disease by affecting the beta amyloid levels; however, a few mutations have been reported as associated with a FTD phenotype, even pathologically confirmed as a tauopathy (Dermaut et al., 2004), although how loss of functional presenilin 1 could predispose to tauopathy has not been clarified. Our patient had a clinical phenotype of rtvFTD with early memory deficits and behavioural disturbances including lack of empathy, hyperphagia and loss of inhibition, the clinical phenotype suggesting the prevalence of FTD neurodegeneration pathway, while the AD pathway may cooperate in the pathology. Moreover, amyloid-PET was negative in our patient, making AD diagnosis unlikely.
MAPT mutations have been previously identified in 4 rtvFTD cases (Ulugut Erkoyun et al., 2021). However, in our cohort, both svFTD (P05 and P12) and rtvFTD (P01) patients carried MAPT mutations, not confirming a prevalent association of MAPT mutations with rtvFTD.
Patient P06 had Q314L OPTN and R795H DCTN1 variants. OPTN, earlier reported as causative of primary open-angle glaucoma, was afterwards linked to ALS (Maruyama et al., 2010), or to FTD (Dominguez et al., 2021), with dominant or recessive transmission. Heterozygous Q314L in OPTN was described in some sporadic ALS cases (Del Bo et al., 2011; Pensato et al., 2020). DCTN1, firstly linked to ALS and Perry syndrome, was then associated with progressive supranuclear palsy and FTD phenotypes (Caroppo et al., 2014). R795H variant was only described in a case of Charcot–Marie-Tooth. These variants are classified as VUS-near likely pathogenic and may contribute to the FTD pathology in our patient, although with very different mechanisms, as OPTN is involved in autophagy whereas DCTN1, binding to microtubules and to dynein, has a role in retrograde axonal transport (LaMonte et al., 2002). Patient P21 carried the G376E VCP and I207T TBK1 variants. Mutations in VCP were identified as the cause of inclusion body myopathy with Paget disease of bone and frontotemporal dementia (IBMPF); (Watts et al., 2004). Afterwards, VCP mutations were also found in cases of pure FTD (Saracino et al., 2018). The G376E variant has never been described and is not present in population databases, constituting a possible new mutation. In fact, it is adjacent to the ATPase domain 1, where other mutations have been described (Mol et al., 2021), and it is classified as VUS-near likely pathogenic, having at least 19 pathogenicity scores. I207T in TBK1 was described in a patient affected by sporadic ALS (Tohnai et al., 2018). The mutation is within the serine/threonine kinase domain and has 10 pathogenicity scores. TBK1 is now recognized also as a FTD gene (van Mossevelde et al., 2018). Since VCP has several functions, including the maintenance of lysosomal homeostasis (Arhzaouy et al., 2019), it may cooperate with TBK1, whose principal role is in the autophagy. The coexistence of variants in genes involved in the same pathway reinforces their pathogenic role and may explain the pathology. Both P06 and P21 patients were svFTD, without any sign of ALS.
Patients P20 and P23, both svFTD, carried a SQSTM1 mutation. SQSTM1 mutations were firstly identified as causative of Paget Disease of Bone (PDB; Laurin et al., 2002), and afterwards this gene was disclosed to be also causative of ALS (Fecto et al., 2011) and FTD (Le Ber et al., 2013). The P387L mutation, carried by patient P23, was previously described in patients affected by PDB (Johnson-Pais et al., 2003), and also in familiar cases of FTD, where a segregation with the disease was demonstrated (Le Ber et al., 2013). The amino acid deletion E280, carried by patient P20, was described as a susceptibility factor for FTD (Van Der Zee et al., 2014).
Patient P10, rtvFTD, had the P80L CHCHD10 mutation, previously reported in patients affected by familial or sporadic ALS (Ronchi et al., 2015; Zhang et al., 2015). CHCHD10 codes for a mitochondrial protein associated not only with ALS but also with FTD (Chaussenot et al., 2014).
While homozygous or heterozygous compound mutations in PRKN lead to early-onset Parkinson’s disease (PD; Lücking et al., 2000), the role of heterozygous mutations is still controversial, ranging from benign condition to susceptibility factor (Marder et al., 2010; Moura et al., 2013). PRKN T240M mutation, carried by the svFTD patient P11, is predicted to eliminate a phosphorylation site for casein kinase II and occurs in the same codon as other mutations (T240R and T240K), indicating that this is an important functional residue. Furthermore, functional studies in neuronal cell cultures demonstrated that this mutation impairs glutamatergic signaling (Zhu et al., 2018). Biallelic mutations in PKRN can lead to FTD (Zimmermann et al., 2018), whereas we suppose that heterozygous mutations may only constitute a susceptibility factor.
In summary, as for single variant findings, we found 12 mutations in 9 patients out of 23, evidencing a strong genetic component in FTD phenotypes usually regarded as sporadic. We also showed that the mutations appeared to be more associated with svFTD (7/15, 47%) than with rtvFTD (2/8, 25%) at variance with what suggested by others (Ulugut Erkoyun et al., 2021). Our data show that a gene strongly involved in our cohort of patients is MAPT, one of the three major genes causative of the FTD spectrum. Along with DCTN1, the involvement of this gene clearly demonstrates the relevance of the correct microtubule dynamics and transport in the nervous system. The mutations found in our patients outline cell pathways whose alterations are common to different neurodegenerative diseases such as FTD, ALS and Parkinson’s disease (PD). Lysosomal and autophagic functions are sustained by VCP, which plays a role in lysosome homeostasis, and by SQSTM1, TBK1 and OPTN, which interact in the autophagy (Abramzon et al., 2020; Fleming et al., 2022), all mutated in ALS and FTD. In addition, mitophagy, a specialized form of autophagy concerning mitocondria, is carried out by the interaction of TBK1, PRKN and OPTN (Harding et al., 2021). Mitochondrial homeostasis is the function of CHCHD10, linked to FTD, ALS and PD (Jiang et al., 2022).
We also performed a gene-level association analysis (burden analysis) in order to disclose genes which may contain a significantly higher number of variants in patients than in controls, suggesting the gene to be relevant to the pathology. We are aware of the small size of our cohorts, partly justified by the rarity of the disease, and in fact we did not achieved the Bonferroni significant threshold, but only suggestive p values highlighting 4 genes: CD33, PSEN1 and ABCA1, showing a simultaneous presence of causal and non-causal variants with opposing directions, and OPTN, showing unidirectional risk association with rare variants exclusively present or more frequent in patients than in HC.
CD33 is a transmembrane protein and a sialic acid-binding immunoglobulin-like lectin that regulates innate immunity. In the CNS, it is expressed by the microglia. CD33 has an immunoreceptor tyrosine-based inhibitory motif (ITIM) and one ITIM-like domain that facilitates an inhibitory signal. When CD33 is activated by sialic-acid-containing glycoproteins and glycolipids, it results in inhibition of microglia phagocytosis. Genome-wide association studies (GWAS) identified several genetic loci associated with increased susceptibility to late onset Alzheimer’s disease, including CD33 (Naj et al., 2011). Some polymorphisms increase CD33 expression, giving rise to reduced microglial phagocytosis and amyloid clearance. Although most polymorphisms were related to AD dementia, others were described as associated with a FTD phenotype (Rendina et al., 2020). In our cohorts we found 4 CD33 rare or low frequency variants. In particular, S305P was found in 9.1% of patients and 0.7% of control subjects, while F243L and V267I were only found in one patient each and no controls. G156fs was found approximately with the same frequency in patients and controls. All these variants are classified as benign. Besides the involvement in amyloid clearance, a pathological feature of AD, CD33 may be regarded as a gene more generally involved in immune and inflammatory pathways, which are also altered in FTD (Ferrari et al., 2014, 2015).
We have already discussed the contribution of a PSEN1 likely pathogenic variant, M93V, to FTD phenotype (see above); in addition, the gene-based analysis revealed the presence of the E318G variant, previously described as a risk factor for AD but afterwards producing conflicting results (Dermaut et al., 1999). The D333V variant, present in a control subject, was never reported before and it is classified as likely pathogenic for cardiovascular disease (Li et al., 2006). As for OPTN, we have already described the Q341L variant (see above), while the M98K is reported in databases as likely benign.
ABCA1 is an ATP-binding cassette transporter that controls whole brain cholesterol homeostasis. Polymorphisms in ABCA1 have been demonstrated to influence susceptibility to dementia, in particular to AD (Lupton et al., 2014; Chen et al., 2016). In our cohorts we found 9 ABCA1 rare or low frequency variants. Four of them were found in patients and the others in controls, showing opposite directions in terms of risk association, that is not giving a true causal significance to this gene as regards to FTD phenotypes. Literature data about ABCA1 concern primarily cardiovascular disease and variants classified as benign due to their relatively high frequency in the population are often associated with a certain level of risk for these diseases. As for dementias, in experimental models, ABCA1 activities appear to influence neuroinflammation and neurodegeneration (Karasinska et al., 2013), thus possibly affecting different forms of dementia, including FTD.
In conclusion, our study revealed the presence of variants in genes involved in pathways relevant for the pathology, especially autophagy and inflammation, common to different neurodegenerative diseases such as FTD, ALS, AD and PD. The findings support the role of these pathways in semantic phenotypes of FTD, as recently suggested (Pascual et al., 2021). Molecular analysis of dementia-related genes should be performed in all patients with svFTD and rtvFTD, to better understand the genotype–phenotype correlations and to study the pathogenetic mechanisms that could drive the clinical phenotypes in FTD.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human subjects were reviewed and approved by Ethics Committee of Fondazione IRCCS Istituto Neurologico Carlo Besta (Prot. n.39–05/04/2017). All the subjects provided their written informed consent to participate in this study.
GR and PC: conceptualization. ES, EM, and MR: formal analysis. CV, SP, FM, GF, PT, VR, CC, GK, EC, MF, and FA: investigation. FA and GG: funding acquisition. GR, ES, and EM: methodology. GR, PC, and ES: writing – original draft. GR, PC, and FA: writing – review and editing. All authors contributed to the article and approved the submitted version.
This research was funded by the European Research Council (StG-2016_714388_NeuroTRACK) and the Italian Ministry of Health, Ricerca Corrente RRC.
EC has received research supports from the Italian Ministry of Health. MF is Editor-in-Chief of the Journal of Neurology; received compensation for consulting services and/or speaking activities from Bayer, Biogen Idec, Merck-Serono, Novartis, Roche, Sanofi Genzyme, Takeda, and Teva Pharmaceutical Industries; and receives research support from Biogen Idec, Merck-Serono, Novartis, Roche, Teva Pharmaceutical Industries, Italian Ministry of Health, Fondazione Italiana Sclerosi Multipla, and ARiSLA (Fondazione Italiana di Ricerca per la SLA). FA is Section Editor of NeuroImage: Clinical; has received speaker honoraria from Biogen Idec, Roche and Zambon; and receives or has received research supports from the Italian Ministry of Health, AriSLA (Fondazione Italiana di Ricerca per la SLA), the European Research Council and the Foundation Research on Alzheimer Disease.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.1085406/full#supplementary-material
Abramzon, Y. A., Fratta, P., Traynor, B. J., and Chia, R. (2020). The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci. 14:42. doi: 10.3389/fnins.2020.00042
Arango, D., Cruts, M., Torres, O., Backhovens, H., Serrano, M. L., Villareal, E., et al. (2001). Systematic genetic study of Alzheimer disease in Latin America: mutation frequencies of the amyloid beta precursor protein and presenilin genes in Colombia. Am. J. Med. Genet. 103, 138–143. doi: 10.1002/1096-8628(20011001)103:2<138::aid-ajmg1529>3.0.co;2-8
Arhzaouy, K., Papadopoulos, C., Schulze, N., Pittman, S. K., Meyer, H., and Weihl, C. C. (2019). VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy 15, 1082–1099. doi: 10.1080/15548627.2019.1569933
Barghorn, S., Zheng-Fischhöfer, Q., Ackmann, M., Biernat, J., von Bergen, M., Mandelkow, E. M., et al. (2000). Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 39, 11714–11721. doi: 10.1021/bi000850r
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Caroppo, P., Le Ber, I., Clot, F., Rivaud-Péchoux, S., Camuzat, A., De Septenville, A., et al. (2014). Dctn1 mutation analysis in families with progressive supranuclear palsy–like phenotypes. JAMA Neurol. 71, 208–215. doi: 10.1001/jamaneurol.2013.5100
Chaussenot, A., Le Ber, I., Ait-El-Mkadem, S., Camuzat, A., de Septenville, A., and Bannwarth, S. (2014). Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging 35, 2884.e1–2884.e4. doi: 10.1016/j.neurobiolaging.2014.07.022
Chen, Q., Liang, B., Wang, Z., Cheng, X., Huang, Y., Liu, Y., et al. (2016). Influence of four polymorphisms in ABCA1 and PTGS2 genes on risk of Alzheimer's disease: a meta-analysis. Neurol. Sci. 37, 1209–1220. doi: 10.1007/s10072-016-2579-9
Cingolani, P., Platts, A., Wang, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
D’Souza, I., and Schellenberg, G. D. (2002). Tau exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5′ and 3′ splice sites. J. Biol. Chem. 277, 26587–26599. doi: 10.1074/jbc.M203794200
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Del Bo, R., Tiloca, C., Pensato, V., Corrado, L., Ratti, A., Ticozzi, N., et al. (2011). Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 1239–1243. doi: 10.1136/jnnp.2011.242313
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Dermaut, B., Cruts, M., Slooter, A. J., Van Gestel, S., De Jonghe, C., Vanderstichele, H., et al. (1999). The Glu318Gly substitution in presenilin 1 is not causally related to Alzheimer disease. Am. J. Hum. Genet. 64, 290–292. doi: 10.1086/302200
Dermaut, B., Kumar-Singh, S., Engelborghs, S., Theuns, J., Rademakers, R., Saerens, J., et al. (2004). A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann. Neurol. 55, 617–626. doi: 10.1002/ana.20083
Dominguez, J., Yu, J. T., Tan, Y. J., Ng, A., De Guzman, M. F., Natividad, B., et al. (2021). Novel optineurin frameshift insertion in a family with frontotemporal dementia and parkinsonism without amyotrophic lateral sclerosis. Front. Neurol. 12:645913. doi: 10.3389/fneur.2021.645913
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Fenoglio, C., Scarpini, E., Serpente, M., and Galimberti, D. (2018). Role of genetics and epigenetics in the pathogenesis of alzheimer’s disease and frontotemporal dementia1. J. Alzheimers Dis. 62, 913–932. doi: 10.3233/JAD-170702
Ferrari, R., Grassi, M., Salvi, E., Borroni, B., Palluzzi, F., Pepe, D., et al. (2015). A genome-wide screening and SNPs-to-genes approach to identify novel genetic risk factors associated with frontotemporal dementia. Neurobiol. Aging 36, 2904.e13–2904.e26. doi: 10.1016/j.neurobiolaging.2015.06.005
Ferrari, R., Hernandez, D. G., Nalls, M. A., Rohrer, J. D., Ramasamy, A., Kwok, J. B. J., et al. (2014). Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 13, 686–699. doi: 10.1016/S1474-4422(14)70065-1
Fleming, A., Bourdenx, M., Fujimaki, M., Karabiyik, C., Krause, G. J., Lopez, A., et al. (2022). The different autophagy degradation pathways and neurodegeneration. Neuron 110, 935–966. doi: 10.1016/j.neuron.2022.01.017
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Harding, O., Evans, C. S., Ye, J., Cheung, J., Maniatis, T., and Holzbaur, E. L. F. (2021). ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. U.S.A 118:e2025053118. doi: 10.1073/pnas.2025053118
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Ishizuka, T., Nakamura, M., Ichiba, M., and Sano, A. (2011). Familial semantic dementia with P301L mutation in the tau gene. Dement. Geriatr. Cogn. Disord. 31, 334–340. doi: 10.1159/000328412
Jiang, T., Wang, Y., Wang, X., and Xu, J. (2022). CHCHD2 and CHCHD10: future therapeutic targets in cognitive disorder and motor neuron disorder. Front. Neurosci. 16:988265. doi: 10.3389/fnins.2022.988265
Johnson-Pais, T. L., Wisdom, J. H., Weldon, K. S., Cody, J. D., Hansen, M. F., Singer, F. R., et al. (2003). Three novel mutations in SQSTM1 identified in familial Paget's disease of bone. J. Bone Miner. Res. 18, 1748–1753. doi: 10.1359/jbmr.2003.18.10.1748
Karasinska, J. M., de Haan, W., Franciosi, S., Ruddle, P., Fan, J., Kruit,, et al. (2013). ABCA1 influences neuroinflammation and neuronal death. Neurobiol. Dis. 54, 445–455. doi: 10.1016/j.nbd.2013.01.018
LaMonte, B. H., Wallace, K. E., Holloway, B. A., Shelly, S. S., Ascaño, J., Tokito, M., et al. (2002). Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 34, 715–727. doi: 10.1016/s0896-6273(02)00696-7
Laurin, N., Brown, J. P., Morissette, J., and Raymond, V. (2002). Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588. doi: 10.1086/340731
Le Ber, I., Camuzat, A., Guerreiro, R., Bouya-Ahmed, K., Bras, J., Nicolas, G., et al. (2013). SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 70, 1403–1410. doi: 10.1001/jamaneurol.2013.3849
Lee, S., Emond, M. J., Bamshad, M. J., Barnes, K. C., Rieder, M. J., Nickerson, D. A., et al. (2012). Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237. doi: 10.1016/j.ajhg.2012.06.007
Lewis, J., McGowan, E., Rockwood, J., Melrose, H., Nacharaju, P., Van Slegtenhorst, M., et al. (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301l) tau protein. Nat. Genet. 25, 402–405. doi: 10.1038/78078
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, D., Parks, S. B., Kushner, J. D., Nauman, D., Burgess, D., Ludwigsen, S., et al. (2006). Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 79, 1030–1039. doi: 10.1086/509900
Lücking, C. B., Dürr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T., et al. (2000). Association between early-onset Parkinson's disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Lupton, M. K., Proitsi, P., Lin, K., Hamilton, G., Daniilidou, M., Tsolaki, M., et al. (2014). The role of ABCA1 gene sequence variants on risk of Alzheimer's disease. J. Alzheimers Dis. 38, 897–906. doi: 10.3233/JAD-131121
Mann, D. M. A., and Snowden, J. S. (2017). Frontotemporal lobar degeneration: pathogenesis, pathology and pathways to phenotype: frontotemporal lobar degeneration. Brain Pathol. 27, 723–736. doi: 10.1111/bpa.12486
Marder, K. S., Tang, M. X., Mejia-Santana, H., Rosado, L., Louis, E. D., Comella, C. L., et al. (2010). Predictors of parkin mutations in early-onset parkinson disease: the consortium on risk for early-onset parkinson disease study. Arch. Neurol. 67, 731–738. doi: 10.1001/archneurol.2010.95
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., and Watanabe, Y. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Mol, M. O., van Rooij, J. G. J., Wong, T. H., Melhem, S., Verkerk, A. J. M. H., Kievit, A. J. A., et al. (2021). Underlying genetic variation in familial frontotemporal dementia: sequencing of 198 patients. Neurobiol. Aging 97, 148.e9–148.e16. doi: 10.1016/j.neurobiolaging.2020.07.014
Moura, K. C. V., Campos Junior, M., de Rosso, A. L. Z., Nicaretta, D. H., Pereira, J. S., Silva, D. J., et al. (2013). Genetic analysis of park2 and pink1 genes in brazilian patients with early-onset parkinson’s disease. Dis. Markers 35, 181–185. doi: 10.1155/2013/597158
Naj, A. C., Jun, G., Beecham, G. W., Wang, L.-S., Vardarajan, B. N., Buros, J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441. doi: 10.1038/ng.801
Pascual, B., Funk, Q., Zanotti-Fregonara, P., Cykowski, M. D., Veronese, M., Rockers, E., et al. (2021). Neuroinflammation is highest in areas of disease progression in semantic dementia. Brain 144, 1565–1575. doi: 10.1093/brain/awab057
Pensato, V., Magri, S., Bella, E. D., Tannorella, P., Bersano, E., Sorarù, G., et al. (2020). Sorting rare als genetic variants by targeted re-sequencing panel in italian patients: Optn, vcp, and sqstm1 variants account for 3% of rare genetic forms. J. Clin. Med. 9:E412. doi: 10.3390/jcm9020412
Qian, W., and Liu, F. (2014). Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 30, 367–377. doi: 10.1007/s12264-013-1411-2
Rendina, A., Drongitis, D., Donizetti, A., Fucci, L., Milan, G., Tripodi, F., et al. (2020). Cd33 and siglecl1 immunoglobulin superfamily involved in dementia. J. Neuropathol. Exp. Neurol. 79, 891–901. doi: 10.1093/jnen/nlaa055
Rohrer, J. D., Geser, F., Zhou, J., Gennatas, E. D., Sidhu, M., Trojanowski, J. Q., et al. (2010). TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology 75, 2204–2211. doi: 10.1212/WNL.0b013e318202038c
Rohrer, J. D., Guerreiro, R., Vandrovcova, J., Uphill, J., Reiman, D., Beck, J., et al. (2009). The heritability and genetics of frontotemporal lobar degeneration. Neurology 73, 1451–1456. doi: 10.1212/WNL.0b013e3181bf997a
Ronchi, D., Riboldi, G., Del Bo, R., Ticozzi, N., Scarlato, M., Galimberti, D., et al. (2015). CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain 138:e372. doi: 10.1093/brain/awu384
Rossi, G., Dalprà, L., Crosti, F., Lissoni, S., Sciacca, F. L., Catania, M., et al. (2008). A new function of microtubule-associated protein tau: involvement in chromosome stability. Cell Cycle 7, 1788–1794. doi: 10.4161/cc.7.12.6012
Saracino, D., Clot, F., Camuzat, A., Anquetil, V., Hannequin, D., Guyant-Maréchal, L., et al. (2018). Novel VCP mutations expand the mutational spectrum of frontotemporal dementia. Neurobiol. Aging 72, 187.e11–187.e14. doi: 10.1016/j.neurobiolaging.2018.06.037
Somavarapu, A. K., and Kepp, K. P. (2016). Loss of stability and hydrophobicity of presenilin 1 mutations causing Alzheimer’s disease. J. Neurochem. 137, 101–111. doi: 10.1111/jnc.13535
Tacik, P., DeTure, M., Hinkle, K. M., Lin, W. L., Sanchez-Contreras, M., Carlomagno, Y., et al. (2015). A novel tau mutation in exon 12, p.Q336H, causes hereditary pick disease. J. Neuropathol. Exp. Neurol. 74, 1042–1052. doi: 10.1097/NEN.0000000000000248
Tedde, A., Nacmias, B., Ciantelli, M., Forleo, P., Cellini, E., Bagnoli, S., et al. (2003). Identification of new presenilin gene mutations in early-onset familial Alzheimer disease. Arch. Neurol. 60, 1541–1544. doi: 10.1001/archneur.60.11.1541
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Tohnai, G., Nakamura, R., Sone, J., Nakatochi, M., Yokoi, D., and Katsuno, M. (2018). Frequency and characteristics of the TBK1 gene variants in Japanese patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 64, 158.e15–158.e19. doi: 10.1016/j.neurobiolaging.2017.12.005
Tubeuf, H., Charbonnier, C., Soukarieh, O., Blavier, A., Lefebvre, A., Dauchel, H., et al. (2020). Large-scale comparative evaluation of user-friendly tools for predicting variant-induced alterations of splicing regulatory elements. Hum. Mut. 41, 1811–1829. doi: 10.1002/humu.24091
Ulugut, H., Dijkstra, A. A., Scarioni, M., Netherlands Brain Bank, Barkhof, F., Scheltens, P., et al. (2021). Right temporal variant frontotemporal dementia is pathologically heterogeneous: a case-series and a systematic review. Acta Neuropathol. Commun. 9:131. doi: 10.1186/s40478-021-01229-z
Ulugut Erkoyun, H., Groot, C., Heilbron, R., Nelissen, A., van Rossum, J., Jutten, R., et al. (2020). A clinical-radiological framework of the right temporal variant of frontotemporal dementia. Brain 143, 2831–2843. doi: 10.1093/brain/awaa225
Ulugut Erkoyun, H., van der Lee, S. J., Nijmeijer, B., van Spaendonk, R., Nelissen, A., Scarioni,, et al. (2021). The right temporal variant of frontotemporal dementia is not genetically sporadic: a case series. J. Alzheimers Dis. 79, 1195–1201. doi: 10.3233/JAD-201191
van der Zee, J., Van Langenhove, T., Kovacs, G. G., Dillen, L., Deschamps, W., Engelborghs,, et al. (2014). Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 128, 397–410. doi: 10.1007/s00401-014-1298-7
Van Mossevelde, S., Engelborghs, S., van der Zee, J., and Van Broeckhoven, C. (2018). Genotype–phenotype links in frontotemporal lobar degeneration. Nat. Rev. Neurol. 14, 363–378. doi: 10.1038/s41582-018-0009-8
Villa, C., Rossi, G., Bizzozero, I., Prioni, S., Boiocchi, C., Agosta, F., et al. (2022). MAPT Q336H mutation: Intrafamilial phenotypic heterogeneity in a new Italian family. Eur. J. Neurol. 29, 1529–1533. doi: 10.1111/ene.15250
Watts, G. D. J., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi: 10.1038/ng1332
Younes, K., Borghesani, V., Montembeault, M., Spina, S., Mandelli, M. L., Welch, A. E., et al. (2022). Right temporal lobe and socioemotional semantics: semantic behavioural variant frontotemporal dementia. Brain 145, 4080–4096. doi: 10.1093/brain/awac217
Zhang, D. M., Levitan, D., Yu, G., Nishimura, M., Chen, F., Tandon, A., et al. (2000). Mutation of the conserved N-terminal cysteine (Cys92) of human presenilin 1 causes increased a beta42 secretion in mammalian cells but impaired notch/lin-12 signalling in C. elegans. Neuroreport 11, 3227–3230. doi: 10.1097/00001756-200009280-00035
Zhang, M., Xi, Z., Zinman, L., Bruni, A. C., Maletta, R. G., Curcio, S. A., et al. (2015). Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 138:e380. doi: 10.1093/brain/awv082
Zhu, M., Cortese, G. P., and Waites, C. L. (2018). Parkinson’s disease-linked Parkin mutations impair glutamatergic signaling in hippocampal neurons. BMC Biol. 16:100. doi: 10.1186/s12915-018-0567-7
Keywords: frontotemporal dementia, semantic variant, right temporal variant, next generation sequencing, genetic variant, pathogenic, mutation
Citation: Rossi G, Salvi E, Mehmeti E, Ricci M, Villa C, Prioni S, Moda F, Di Fede G, Tiraboschi P, Redaelli V, Coppola C, Koch G, Canu E, Filippi M, Agosta F, Giaccone G and Caroppo P (2022) Semantic and right temporal variant of FTD: Next generation sequencing genetic analysis on a single-center cohort. Front. Aging Neurosci. 14:1085406. doi: 10.3389/fnagi.2022.1085406
Edited by:
Prabhjyot Saini, Emory University, United StatesReviewed by:
Emanuele Buratti, International Centre for Genetic Engineering and Biotechnology, ItalyCopyright © 2022 Rossi, Salvi, Mehmeti, Ricci, Villa, Prioni, Moda, Di Fede, Tiraboschi, Redaelli, Coppola, Koch, Canu, Filippi, Agosta, Giaccone and Caroppo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giacomina Rossi, Z2lhY29taW5hLnJvc3NpQGlzdGl0dXRvLWJlc3RhLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.