 Ting Sun
Ting Sun Li Zeng
Li Zeng Zhongdi Cai
Zhongdi Cai Qingshan Liu
Qingshan Liu Zhuorong Li
Zhuorong Li Rui Liu
Rui Liu- 1Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2Key Laboratory of Ethnomedicine of Ministry of Education, School of Pharmarcy, Minzu University of China, Beijing, China
Introduction: Alzheimer’s disease (AD) causes a decline in cognitive function that poses a significant hazard to human health. However, the exact pathogenesis of AD and effective treatment have both proven elusive. Circular RNAs (circRNAs), which were initially deemed as meaningless non-coding RNAs, have been shown to participate in a variety of physiological and pathological processes. However, the variations and characteristics of circRNAs are not fairly well understood during the occurrence and development of AD.
Methods: In this study, we performed RNA sequencing analyses, identified circRNA expression profiles, and explored the circRNA-associated competing endogenous RNA (ceRNA) relationship in the hippocampus of five familial AD (5 × FAD) mice with cognitive dysfunction.
Results: The RNA sequencing results identified 34 dysregulated circRNAs in the hippocampus of 5 × FAD mice, including 17 upregulated and 17 downregulated circRNAs. The circRNA-miRNA interaction network for the dysregulated circRNAs was generated, and it was found to include 34 circRNAs and 711 miRNAs. Next, 2067 mRNAs potentially modulated by upregulated circRNA-interacting miRNAs and 2297 mRNAs potentially modulated by downregulated circRNA-interacting miRNAs were identified. Pathway enrichment analyses revealed that the circRNA-miRNA-mRNA network modulated AD development via multiple pathways, such as axon guidance, mitogen-activated protein kinase, and neurotrophin. The associated biological processes were mainly related to neuron projection development, cell morphogenesis, and head development. Their corresponding distributions were especially high in the axon, postsynapse, and neuronal body. We constructed a ceRNA network that included five circRNAs, four miRNAs, and 188 mRNAs. In this network, the differential expressions of three circRNAs (circRNA04655, circRNA00723, and circRNA01891), two miRNAs (miR-3470b and miR-6240), and 13 mRNAs (Vgll3, Nhsl2, Rab7, Tardbp, Vps33b, Fam107a, Tacr1, Ankrd40, Creb1, Snap23, Csnk1a1, Bmi1, and Bfar) in the hippocampus of 5 × FAD mice using qRT-PCR analyses were consistent with the RNA sequencing results. Another one circRNAs (circRNA00747) and two mRNAs (Zfp37 and Polr1e) had similar expression trends to the sequencing data, while circRNA03723 and Mapk10 had deviated expression trends to the sequencing data.
Conclusions: In conclusion, our study uncovered dysregulated circRNA expression profiles in the hippocampus of 5 × FAD mice, stretched comprehension of ceRNA biology, investigated the potential role of this ceRNA network in pathogenesis and progression, and identified potential biomarkers and therapeutic targets for AD.
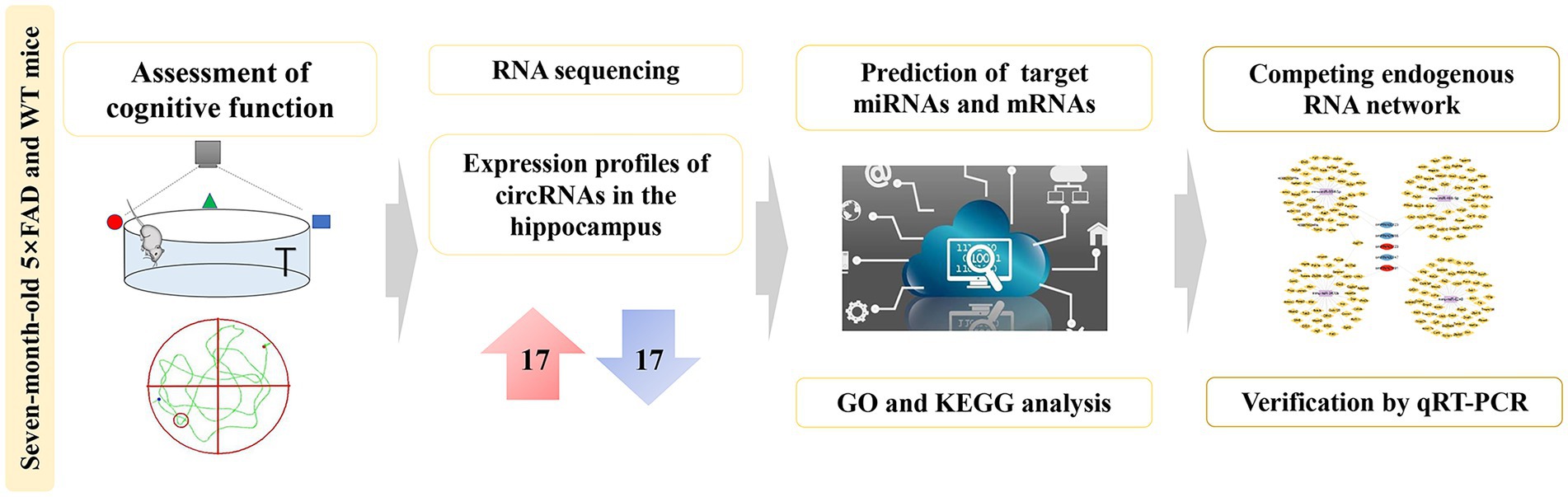
Graphical abstract. Comprehensive analysis of dysregulated circRNAs and construction of a ceRNA network involved in the pathology of Alzheimer’s disease in a 5 × FAD mouse model.
Introduction
Alzheimer’s disease (AD) is a critical degenerative disorder that causes progressive impairment of cognition and behavior (Gonzales et al., 2022). The main pathological features of AD are bound up with two protein abnormalities, plaques with excessive accumulation of amyloid and neurofibrillary tangles formed by tau protein hyperphosphorylation. In addition, neuronal loss, neuroinflammation, and synaptic dysfunction can be observed in AD brains (Scheltens et al., 2021). Although there are numerous strategies for slowing or preventing the rapid progression of AD, no feasible treatments are currently available (Mahaman et al., 2021). Therefore, the discovery of possible biomarkers and curative perspectives for AD is of great importance for clinical diagnosis and drug discovery.
Circular RNAs (circRNAs), linked with covalent bonds to form a closed ring, are a particular category of non-coding RNAs (Chen et al., 2021). Unlike linear RNAs, there are neither 5′ caps nor 3′ polyadenylated tails in circRNAs (Zhang L. et al., 2021). The structure types of circRNAs encompass single exons, multiple exons, introns, and combinations of introns and exons (Chen, 2020). Because their loop structure is fixed by covalent bonds, circRNAs are resistant to exonucleases and highly stable (Sanger et al., 1976). circRNAs were originally considered an artifact of alternative splicing caused by exon transcription errors and did not serve any physiological function (Hansen et al., 2013). However, evidence is emerging that circRNAs are implicated in the occurrence and development of multiple diseases, including nervous system disorders, cardiovascular diseases, cancer, and diabetes. Moreover, circRNAs are thought to play different functional roles depending on their location (Yang et al., 2017). Thus, circRNAs in the nucleus mainly transcribe their parental genes (Zhou et al., 2020), while those in the cytoplasm are mainly miRNA sponges, protein sponges, or templates for peptides translation (Piwecka et al., 2017).
circRNAs are abundantly distributed in the mammalian nervous system (compared with other organs), where they are involved in neuronal differentiation and synaptogenesis (Gokool et al., 2020). Research in Drosophila and mouse has revealed that circRNA expression is dramatically increased (relative to linear RNAs) in the central nervous system with aging (Westholm et al., 2014; Gruner et al., 2016). As a consequence, circRNAs may be indispensable signals in neurodegenerative diseases. In line with the broad spectrum and accuracy of RNA sequencing technologies, circRNAs functions and changes in their expression profile during AD progression have attracted increasing attention. In AD mice, circRNAs may function as miRNA sponges via the competing endogenous RNA (ceRNA) action. A study revealed that the serum of AD patients contains abnormally high levels of circLPAR1, which increases target gene levels by competitively binding to miR-212-3p (Wu et al., 2021). Likewise, ciRS-7 functions as a ceRNA to sponge miR-7 in the regulation of ubiquitin-protein ligase A, which is well-known to coordinate the scavenging of amyloid (Zhao et al., 2016). Currently, the role of circRNAs in the pathogenesis of AD and the complexity involved in the circRNA-centered ceRNA networks have not been widely studied.
In this study, we firstly reported the differentially expressed circRNA profiles in the hippocampus of five familial AD (5 × FAD) mice compared with wild-type (WT) mice using RNA sequencing analysis. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis presented the potential biological functions and signaling pathways of these dysregulated circRNAs. Furthermore, we established a circRNA-miRNA-mRNA network from the present sequencing data to obtain new potential biomarkers and drug targets for AD. Finally, we verified the expression of key circRNAs, miRNAs, and mRNAs within the network using qRT-PCR analysis (Figure 1). These findings might provide a better understanding of the characteristics and functions of circRNAs in the pathogenesis of AD.
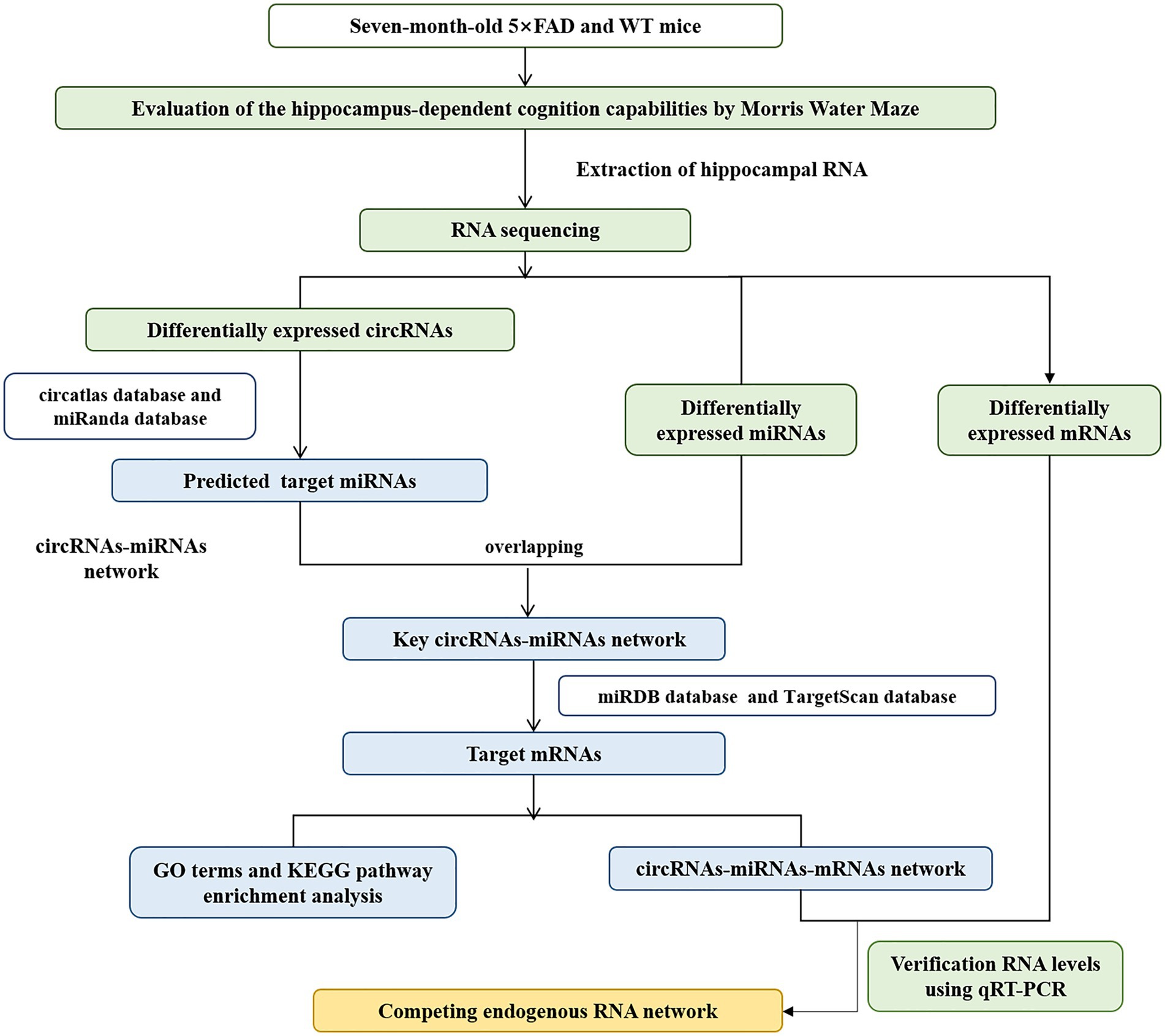
Figure 1. Workflow for circRNA analysis and ceRNA network construction.
Materials and methods
Animals
Seven-month-old 5 × FAD mice and age-matched WT mice, with one male and two females in each group, were used in this study. The 5 × FAD mice are typical transgenic mice for AD research that co-express a total of five familial AD mutations [APP K670N/M671L (Swedish) + I716V (Florida) + V717I (London) and PS1 M146L + L286V]. The Experimental Animal Care and Use Committee of Institute of Medicinal Biotechnology approved the experimental animal protocols (No. IMB-20210602D101). All mice were housed in an environment with a standard diet and clean water.
Cognitive behavior test
The Morris water maze (MWM) test was used to evaluate the impairment of spatial learning and memory of 5 × FAD mice (Liu et al., 2018; Jiang et al., 2022; Sun et al., 2022). A different cohort from the Cai et al. (2022) study was used for MWM test. Briefly, The testing included place navigation training and space memory trials. During the first 5 days, place navigation training of mice was conducted. Each mouse performed four tests from four quadrants as starting position, and each investigation was limited to 60 s. The time that mice found the platform was recorded as escape latency. Those mice that did not find the platform within 60 s were directed to the platform and stayed for 60 s. On the 6th and 7th days, spatial memory trials were performed in the pool without the platform. The number of crossings (as mice sought the original platform) and the duration within the target quadrant (where the original platform was located) were recorded.
RNA extraction
After the MWM, the mice were euthanized via cervical dislocation, and the obtained samples were preserved in liquid nitrogen. The total RNA of the hippocampus was isolated with TRIzol reagent (Invitrogen, CA, United States) according to the previous protocol (Zhao et al., 2022). The RNA sample used in this study was the same cohort as our reported investigation (Cai et al., 2022). Subsequently, the RNA integrity was determined using gel electrophoresis (Supplementary Figure 1). The RNA concentration was determined on Spark 20 M microplate reader (Tecan Group Ltd., Switzerland; Supplementary Table 1).
RNA sequencing
Ribosomal and linear RNAs were removed from total RNA using the Ribo-off rRNA Depletion kit and RNase R (Vazyme Biotech Co., Ltd., China). The rRNA free residue was cleaned up by ethanol precipitation. cDNA libraries were constructed from rRNA-depleted RNA using the VAHTS™ Stranded mRNA-seq Library Prep Kit for Illumina (Vazyme Biotech Co., Ltd., China). The quality of the libraries was then estimated using Agilent 2100 Bioanalyzer system and FastQC (Version 0.11.2). Mass shear was performed by Trimmomatic (Version 0.36) to obtain relatively accurate and effective data. An Illumina HiSeq 2500 platform was used to analyze the library sequences. CircRNAs were identified using CIRI2 (Version 2.06) after alignment with the reference genome using Burrows-Wheeler-Alignment Tool. Subsequently, variable shear analysis was performed using CIRI-AS (Version 1.2). BEDtools was used to determine the origin of circRNAs based on circRNA location information and gene annotations. The Reads Per Kilobase Million (RPKM) formula based on BSJ reads was used to calculate the expression of circRNA and DESeq2 (Version 1.12.4) was used for differential analysis of transcript expression. The RNA sequencing procedures were undertaken by Sangon Biotech (Shanghai, China). Data have been deposited in Gene Expression Omnibus (GEO) with the accession code GSE206562.
Prediction of miRNA targets of circRNAs and ceRNA network construction
The circatlas1 and miRanda2 were used to predict the miRNAs with the strongest binding to differently expressed circRNAs. The circatlas is a comprehensive resource database that integrated over one million circRNAs across multiple species and a variety of tissues, as well as the binding relationships between circRNAs and miRNAs. The miRanda database provided predictive information about miRNA targets in the genomes of multiple species and the expression profiles of miRNAs in different tissues. miRDB,3 and TargetScan4 databases were employed to predict the interactions of mRNA-miRNA. miRDB is a database for miRNA target prediction and functional annotations. TargetScan predicted biological targets of miRNAs by searching for the presence of conserved sites that match the seed region of each miRNA. A circRNA-miRNA-mRNA network was then formed and visualized using Cytoscape software (v3.7.2).
Enrichment analyses of GO enrichment and KEGG enrichment
KEGG enrichment was performed to analyze the enriched pathways associated with circRNA target genes (Chen et al., 2020; Shen et al., 2020). The GO knowledge base was used to describe information on the functions of genes, including biological processes (BP), molecular functions (MF), and cellular components (CC). GO and KEGG analyses were implemented using GENEONTOLOGY5 and KEGG6 databases.
Detection of RNA expression levels by qRT-PCR
The remaining RNA samples from the RNA sequencing were used to perform qRT-PCR. For circRNA, mRNA and GAPDH, cDNA was synthesized by HiscriptIII RT SuperMix 1st Strand cDNA synthesis kit and the qRT-PCR was completed using SYBR Master Mix kits (Vazyme, Nanjing, China). For miRNA and U6, miRNA Synthesis Kit and miRNA SYBR Master Mix were used (Vazyme, Nanjing, China). Sangon Biotech (Shanghai, China) produced all primers (Table 1). Relative gene expression levels were calculated using the 2−ΔΔCTmethod.

Table 1. Primer sequences of the RNAs in the present study.
Acquisition of circRNA datasets and data processing
To clarify the novelty of these key circRNAs in the hippocampus of 5 × FAD mice, we compared the circRNAs from different datasets in various AD models that were downloaded from the GEO, with the species limitation (mouse and rat) and the biological sample type (different brain regions). The circatlas7 and circBase8 were used to convert the circRNA ID and name. The GEO dataset containing the expression level of cicrRNA and miRNA associated with AD were selected, including GSE138382, GSE129053, GSE121769, GSE52023, GSE166393, GSE48028, GSE158995, GSE190880, GSE132177, GSE194137, GSE153180, GSE20447, GSE186929, and GSE129054. The significantly dysregulated RNAs were screened with|Fold Change| ≥ 1 and p-value <0.05.
Statistical analysis
Data analyses were conducted by SPSS software (Version 18.0, SPSS, Inc., Chicago, IL, United States). One-way Repeated Measures ANOVA was used to analyze escape latency and swimming speed. Student’s t-test was used to analyze the other data. Differentially expressed circRNAs, miRNAs, and mRNA following the RNA sequencing were screened with |Fold Change| ≥ 1 and p-value <0.05. Results are presented as mean ± standard deviation (S.D.). The statistical criterion used was p < 0.05.
Results
Spatial cognition dysfunction in 5 × FAD mice
We estimated the hippocampus-dependent cognition capabilities of 5 × FAD mice using the MWM test prior to RNA-sequencing detection. The results revealed that 5 × FAD mice exhibited impairments in recognition ability (compared with WT mice), as demonstrated by longer escape latencies (p = 0.0072; Figures 2A), less duration within the target quadrant (p = 0.0052 and p = 0.0001, respectively; Figures 2C), as well as fewer platform crossings (p = 0.02 and p = 0.03, respectively; Figures 2D). 5 × FAD mice did not exhibit motor dysfunction, reflected in the same swimming speeds as WT mice (Figure 2B). Representative traces of the spatial memory trials are shown in Figure 2E, and these indicate that 5 × FAD mice exhibited an aimless travel path. Collectively, the above results reveal that 7-month-old 5 × FAD mice present hippocampus-dependent spatial cognitive impairment.
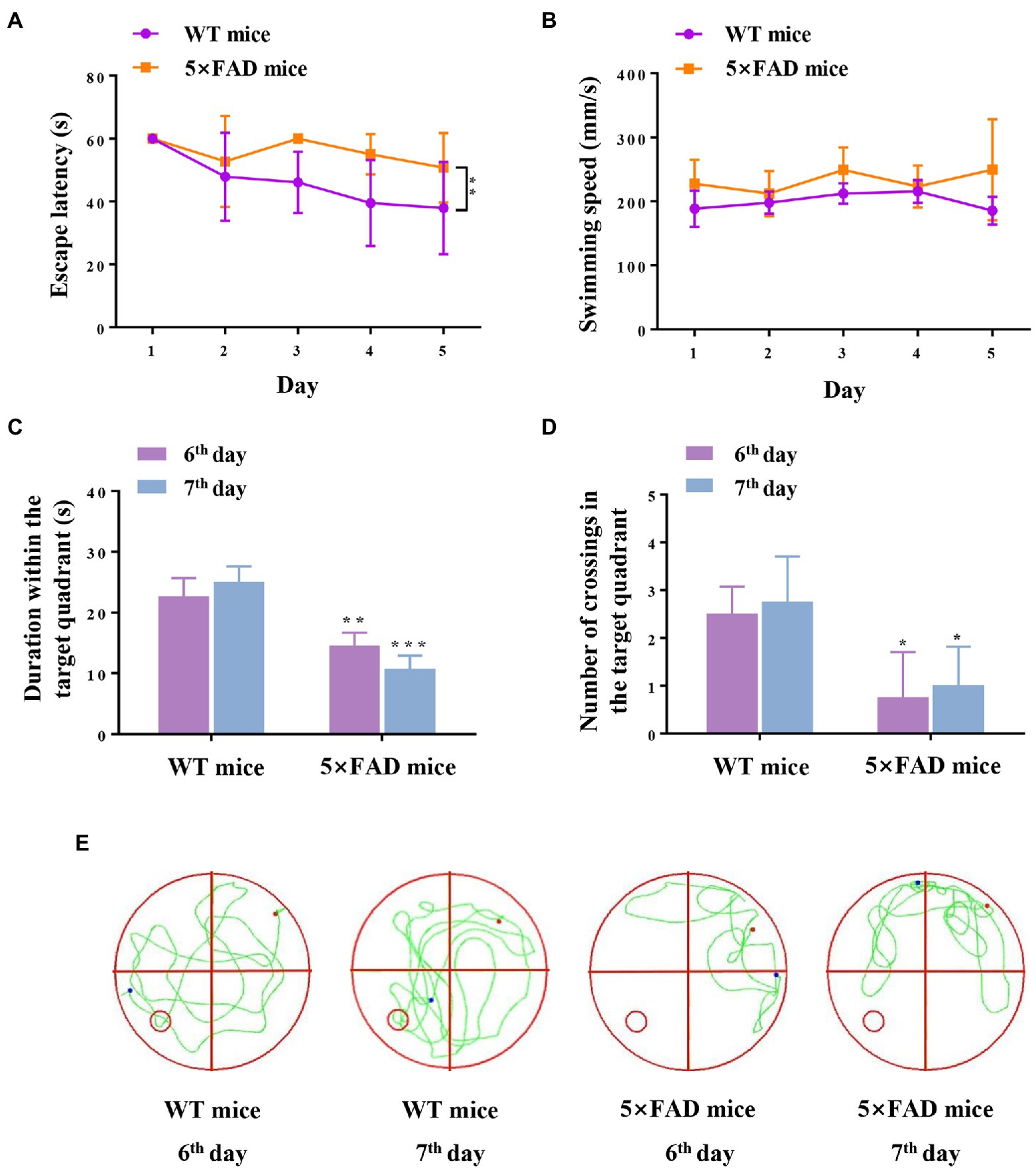
Figure 2. Cognition and memory capabilities in WT and 5 × FAD mice. (A) Escape latency of mice during five-day navigation training in MWM test. (B) Swimming speed during five-day navigation training in MWM test. (C) Duration within the target quadrant in space memory trials on the 6th and 7th days. (D) Number of crossings through the original platform position in space memory trials on the 6th and 7th days. (E) Representative traces of mice in the spatial probe test on the 6th and 7th days. The red dots indicate the position of the mouse at the beginning of the spatial exploration trial, the blue dots indicate the position of the mouse at the end of the 60-s trial, and the green line indicates the representative traces of mice. Data are displayed as mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT mice.
Identification of circRNAs in 5 × FAD mice
The next-generation sequencing was performed to profile circRNA expression in the hippocampus of 5 × FAD and WT mice. In total, 11,112 circRNAs were detected in the hippocampus through sequencing. The expression distribution of all circRNAs was presented in the volcano diagram, in which the expression of 34 circRNAs was significantly different between 5 × FAD and WT mice, including 17 upregulated and 17 downregulated circRNAs (Figure 3A). The details of these circRNAs are listed in Table 2. A clear distinction between the circRNA expression patterns in different species of mice was shown in the hierarchical clustering diagram (Figure 3B). In addition, we classified the aberrantly expressed circRNAs in the light of their chromosomal location (Figure 3C). The results reveal that 20.58 and 14.7% of circRNAs were derived from chromosome 9 and 2, respectively. Finally, we found that circRNAs with lengths between 400 and 600 bp accounted for the major proportion (Figure 3D).
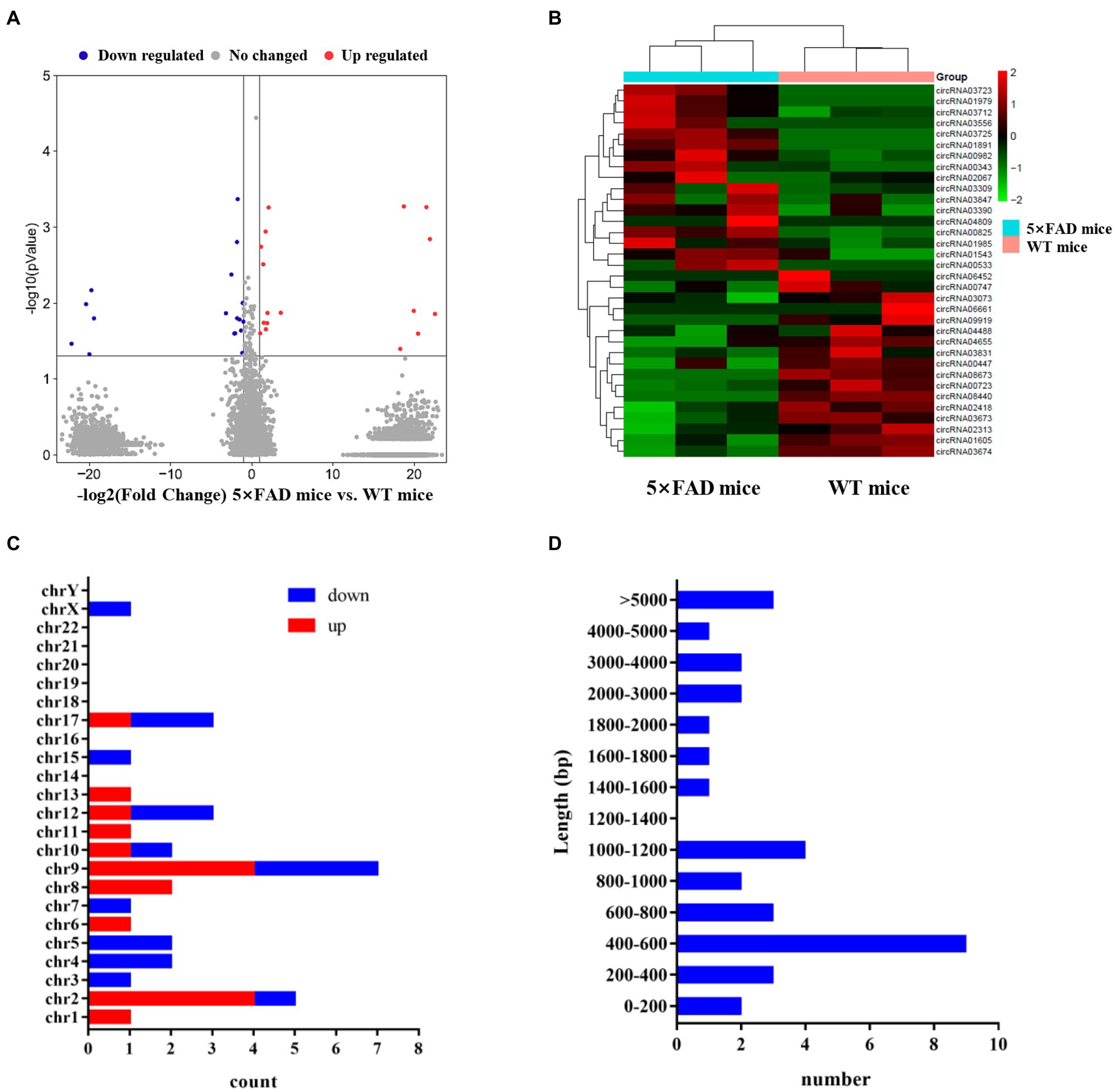
Figure 3. Expression profiles of circRNAs in 5 × FAD mice and WT mice. (A) Volcano plot of the circRNAs in the hippocampus of 5 × FAD mice and WT mice. The red dots indicate significantly upregulated circRNAs, the blue dots indicate the significantly downregulated circRNAs, and the gray dots indicate the circRNAs that have no change. (B) Heatmap of the aberrant circRNAs indicating the variation of significantly dysregulated circRNAs in the hippocampus of 5 × FAD mice. (C) Chromosomal locations of the aberrant circRNAs. The X-axis shows the number of circRNAs, and the Y-axis represents the chromosomal number. (D) The sequence length of the aberrant circRNAs. The X-axis indicates the number of circRNAs, and the Y-axis represents the length ranges.
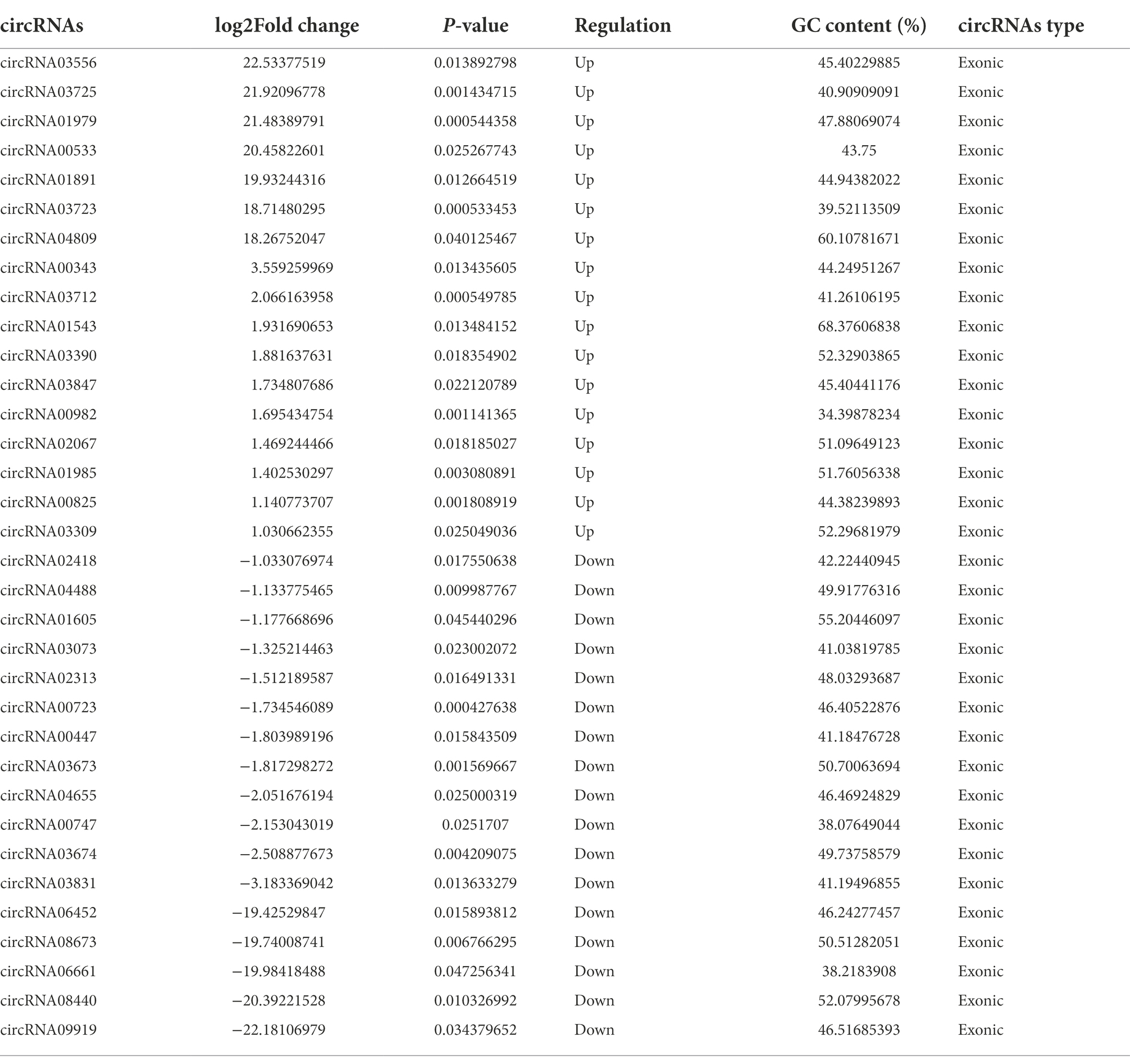
Table 2. Information of differentially expressed circRNAs.
Prediction of circRNA-miRNA interactions
circRNAs regulate gene expression via functioning as sponges that sequester specific target miRNAs (Kristensen et al., 2019). We utilized the circatlas database and the miRanda database to identify miRNAs that potentially correspond to the circRNAs of interest. There were 1,020 circRNA-miRNA interactions involving 34 circRNAs and 711 miRNAs (Figure 4A; Supplementary Table 2). For a more accurate prediction of potential miRNA targets, differentially expressed miRNAs identified in the RNA sequencing experiment were used for further analysis (Supplementary Table 3; Supplementary Figure 2). By intersecting the predicted target miRNAs with the miRNAs identified through sequencing, we identified 439 overlapping miRNAs in the hippocampus of 5 × FAD mice (Figure 4B; Supplementary Table 4). In the resulting network constructed using Cytoscape, we set the algorithm of the Degree to greater than three times the median to obtain 41 key miRNAs. Several types of interactions could be identified within this group of key miRNAs, including four upregulated circRNAs (circRNA03725, circRNA02067, circRNA03847, and circRNA03390) that interacted with downregulated miRNAs, one upregulated circRNA (circRNA03556) that interacted with upregulated miRNAs, three downregulated circRNAs (circRNA03073, circRNA02313, and circRNA04655) that interacted with upregulated miRNAs, five downregulated circRNAs (circRNA00447, circRNA00747, circRNA06661, circRNA00723, and circRNA01605) that interacted with downregulated miRNAs, and circRNAs that exhibited double interactions with upregulated or downregulated miRNAs (Figure 4C; Supplementary Table 5). Moreover, based on higher betweenness centrality and closeness centrality in the network, it could be deduced that a single circRNA might potentially interrelate with one or more miRNAs, and that one miRNA might be simultaneously affected by multiple circRNAs. For instance, circRNA00723 potentially interacted with miR-466i-5p, miR-5623-5p, miR-7058-5p, and miR-669f-5p. Conversely, miR-3971 might be co-connected with circRNA03673, circRNA06452, and circRNA08440.
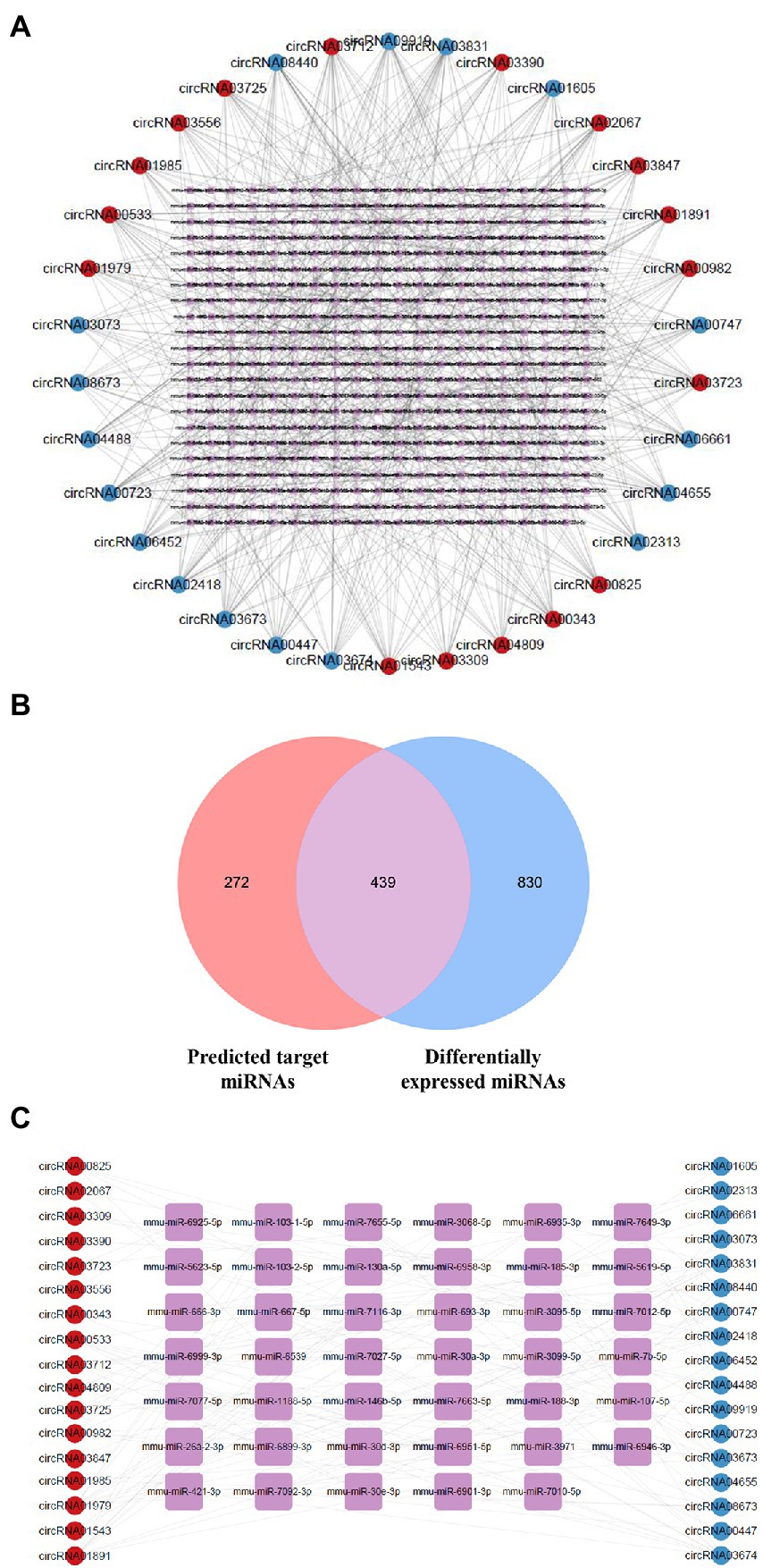
Figure 4. Network of dysregulated circRNAs and target miRNAs. (A) Network of dysregulated circRNAs and their predicted miRNAs using the database. Upregulated circRNAs are shown using red circles, downregulated circRNAs using blue circles, and potential miRNAs using purple squares. (B) Venn diagram of predicted target miRNAs and abnormally expressed miRNAs in 5 × FAD mice. The number of predicted miRNAs is shown in red, and the number of differentially expressed miRNAs is shown in blue. (C) Network of dysregulated circRNAs and key miRNAs selected by the Degree algorithm. Upregulated circRNAs are shown using red circles, downregulated circRNAs are shown using blue circles, and key miRNAs are shown using purple squares.
Functional enrichment analyses
To analyze the functional annotation of these circRNAs, the differentially upregulated and downregulated circRNAs were separately implemented to GO and KEGG analysis. There were 417 target miRNAs of upregulated circRNAs, and 2067 target mRNAs were obtained by merging predicted mRNAs from the miRDB and TargetScan databases. In addition, 2,297 overlapping mRNAs from the above two databases were identified for the 413 target miRNAs of downregulated circRNAs.
The top 10 GO terms from the BP sub-ontologies showed that the aberrantly upregulated circRNAs were mostly involved in neuron projection development (GO:0031175), cell morphogenesis (GO:0000902), and head development (GO:0060322; Figure 5A). The MF sub-ontologies were mainly related to transcription factor binding (GO:0008134), DNA-binding transcription activator activity (GO:0001216), and DNA-binding transcription factor binding for upregulated circRNAs (GO:0140297; Figure 5B). Moreover, CC sub-ontologies were mostly enriched in the axon (GO:0030424), the postsynapse (GO:0098794), and the glutamatergic synapse for aberrantly upregulated circRNAs (GO:0098978; Figure 5C). The downregulated circRNAs were mostly enriched in cell morphogenesis (GO:0000902), plasma membrane-bounded cell projection morphogenesis (GO:0120039), and cellular component morphogenesis (GO:0032989) in the BP sub-ontologies (Figure 5D), DNA-binding transcription activator activity (GO:0001216), transcription factor binding (GO:0008134), and protein kinase binding (GO:0004672) in the MF sub-ontologies (Figure 5E), and the axon (GO:0030424), post-synapse (GO:0098794), and neuronal cell body (GO:0043025) in the CC sub-ontologies (Figure 5F).
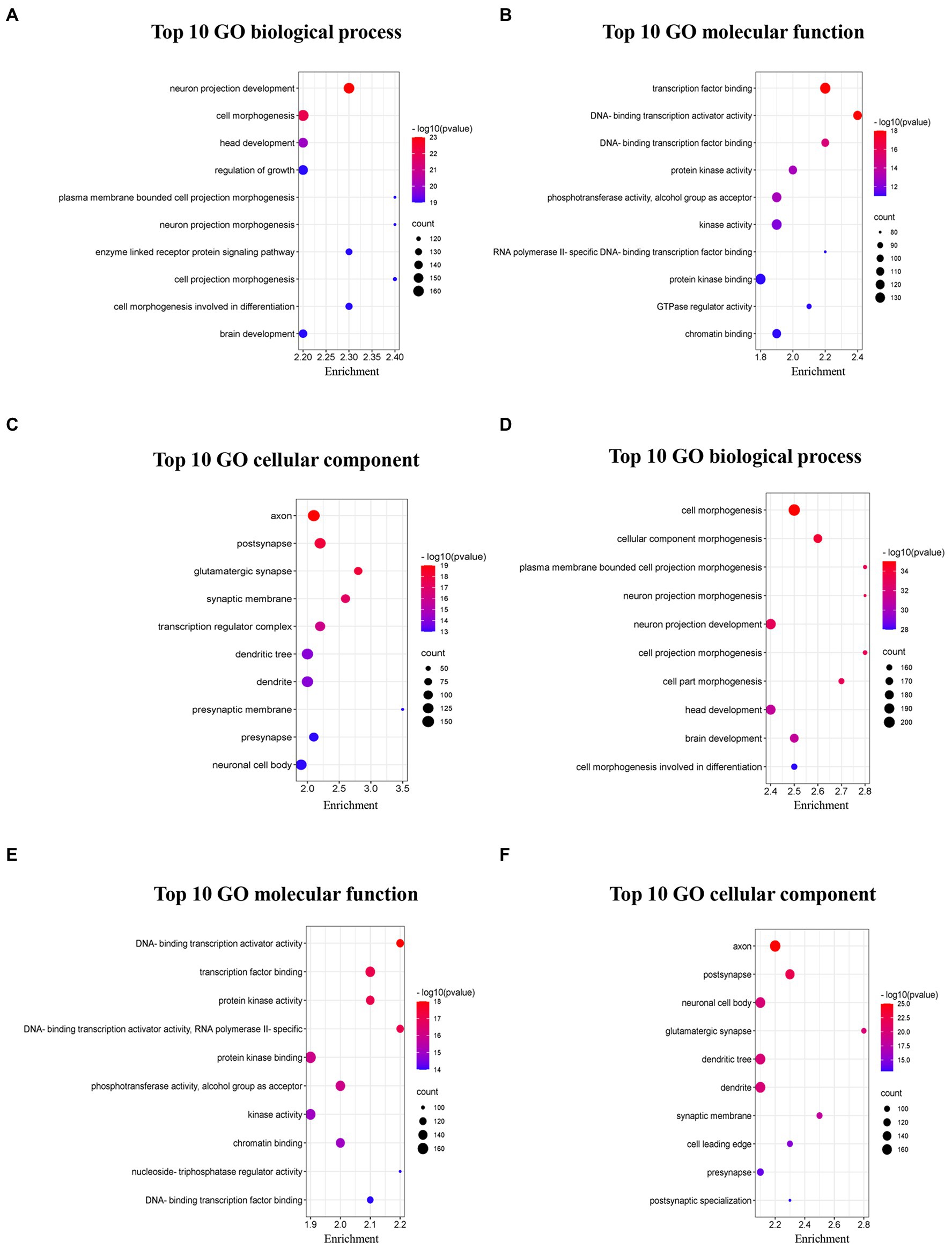
Figure 5. Top 10 GO pathways of dysregulated circRNAs in 5 × FAD mice. (A) Biological processes of upregulated circRNAs. (B) Molecular function of upregulated circRNAs. (C) Cellular component of upregulated circRNAs. (D) Biological processes of downregulated circRNAs. (E) Molecular function of downregulated circRNAs. (F) Cellular component of downregulated circRNAs. The bubble size represents the number of enriched genes, while the bubble color represents the significance of enrichment based on the corrected value of p.
KEGG pathway analysis revealed that axon guidance (mmu04360), MAPK signaling pathway (mmu04010), and Neurotrophin signaling pathway (mmu04722) were closely linked to the target gene of abnormally upregulated circRNAs (Figure 6A; Supplementary Table 6). Additionally, axon guidance (mmu04360), autophagy (mmu04140), and the mTOR signaling pathway (mmu04150) were closely connected with the target gene of abnormally downregulated circRNAs (Figure 6B; Supplementary Table 6).
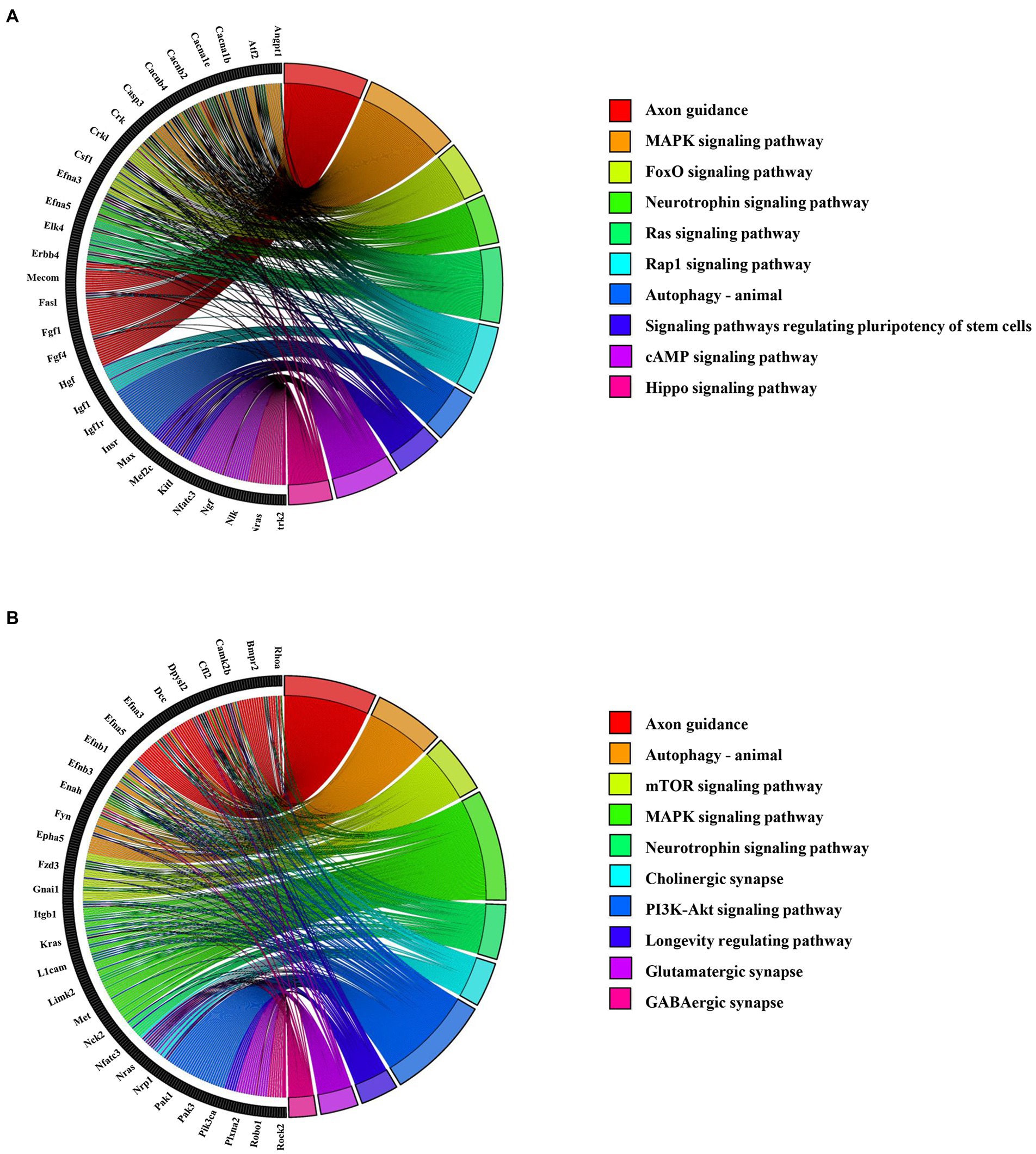
Figure 6. Top 10 KEGG enrichment pathways for the abnormally expressed circRNAs. (A) KEGG pathways for upregulated circRNA-related mRNAs. The left section shows the circRNA-related mRNA name, and the right section shows the top 10 pathways. (B) KEGG pathways for downregulated circRNA-related mRNAs. The left section shows the circRNA-related mRNAs, and the right section shows the top 10 KEGG pathways.
Construction of ceRNA network
ceRNA networks accurately and intuitively reflect the interactions among circRNAs, miRNAs, and mRNA. We established a ceRNA network using the top five circRNAs with the highest intermediate centrality and intimacy centrality values that have not been reported to have an association with AD. To establish a ceRNA network centered on circRNA00723, circRNA04655, circRNA01891, circRNA03723, and circRNA00747, we screened for aberrantly expressed miRNAs in RNA sequencing (Table 3). We identified six circRNA-miRNA interactions involving four miRNAs and five circRNAs (Table 4). Next, we identified 188 target mRNAs by merging the predicted mRNAs for these four miRNAs using the miRDB and TargetScan databases. In addition, we integrated the five circRNAs, four miRNAs, and 188 target mRNAs into a circRNA–miRNA–mRNA network (Figure 7).
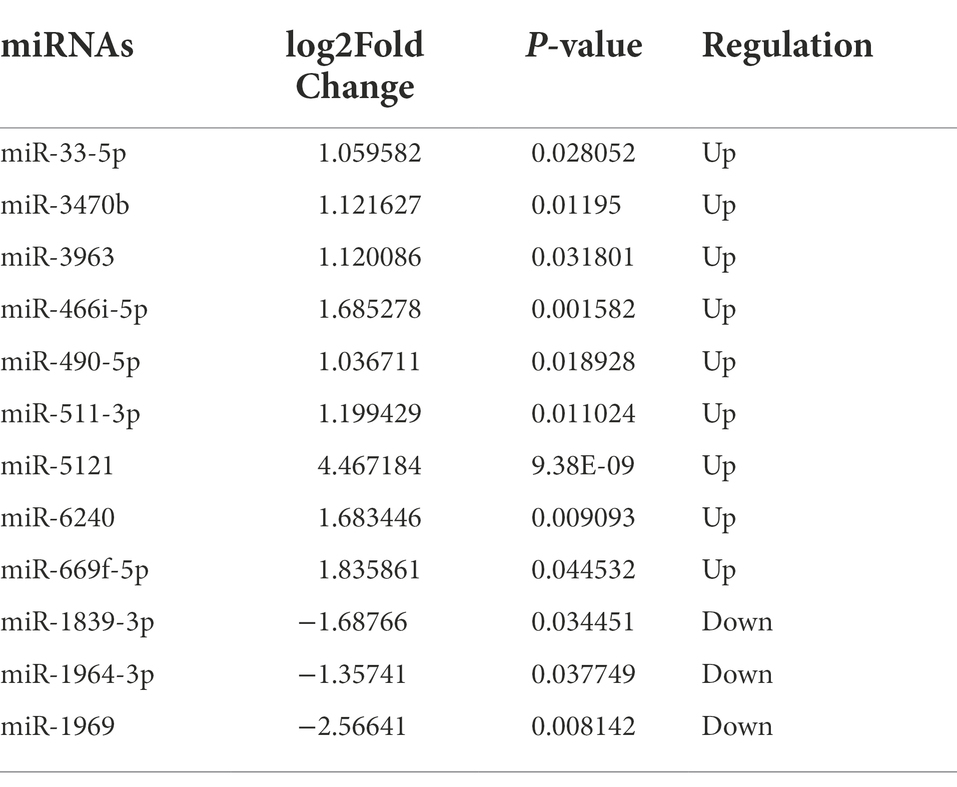
Table 3. Information of differentially expressed miRNAs.
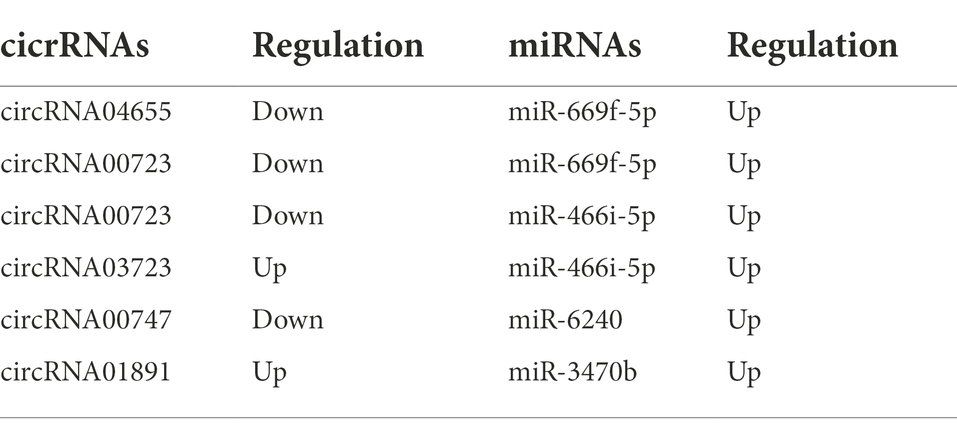
Table 4. Interactions of aberrantly expressed circRNAs and miRNAs.
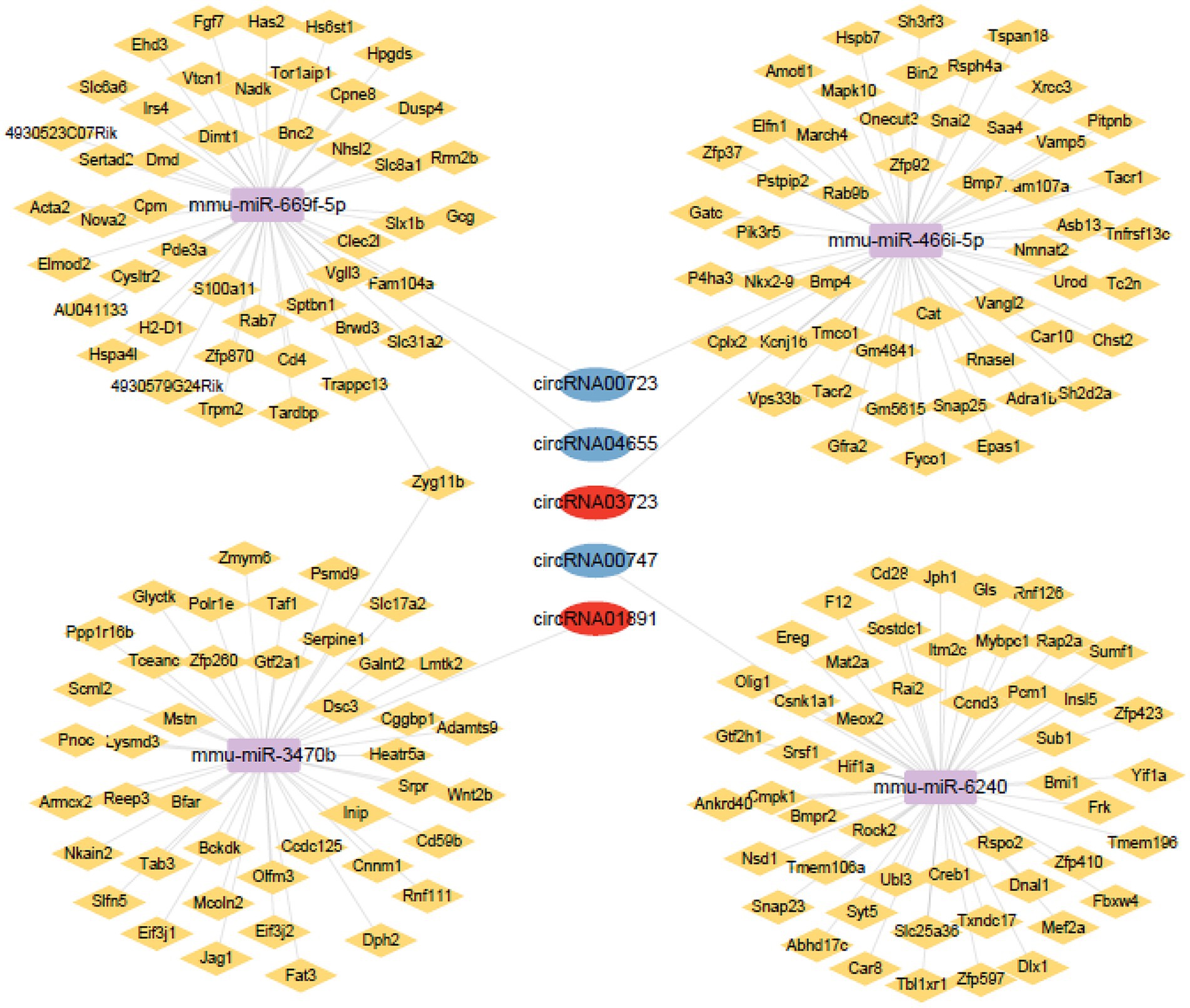
Figure 7. Network of circRNAs-miRNAs-mRNAs. Upregulated circRNAs are shown using red circles, downregulated circRNAs are shown using blue circles, potential miRNAs are shown using purple squares, and potential mRNAs are shown using yellow rhombus.
Expression verification of the circRNAs, miRNAs, and mRNAs in the hippocampus of 5 × FAD mice
To investigate the potential relationship within the predicted circRNA–miRNA–mRNA network, qRT-PCR analysis was used to verify circRNA, miRNA, and mRNA expression in the hippocampus of 5 × FAD mice. Consistent with RNA sequencing results, the expression of circRNA04655 and circRNA00723 in the hippocampus of 5 × FAD mice was significantly decreased (p = 0.045, p = 0.023 vs. WT mice; Figure 8A), and the expression of circRNA01891 was increased (p = 0.048 vs. WT mice; Figure 8A). The expression of circRNA00747 was reduced in the hippocampus of 5 × FAD mice, which had a similar trend to the sequencing data, but the statistical difference compared to WT mice was insignificant (Figure 8A). The expression of circRNA03723 was insignificantly reduced in the hippocampus of 5 × FAD mice, and the expression trend was inconsonant with its RNA sequencing result (Figure 8A). miR-3470b and miR-6240 were significantly upregulated in the hippocampus of 5 × FAD mice (p = 0.0049, p = 0.0062 vs. WT mice; Figure 8B), and the expression alteration was in line with the sequencing results. However, miR-669f-5p and miR-466i-5p were significantly downregulated in the hippocampus of 5 × FAD mice (p = 0.0018, p = 0.0239 vs. WT mice; Figure 8B), deviating from the sequencing results. By intersecting the mRNAs in the circRNA–miRNA–mRNA network (Figure 7) with the differential expressed mRNAs identified through sequencing, we identified 16 overlapping mRNAs, which presented the opposite trend with miRNAs. Consistent with sequencing results, the expression level of 13 mRNAs was significantly decreased in the hippocampus of 5 × FAD mice, including Vgll3, Nhsl2, Rab7, Tardbp, Vps33b, Fam107a, Tacr1, Ankrd40, Creb1, Snap23, Csnk1a1, Bmi1, and Bfar (p = 0.041, p = 0.035, p = 0.003, p = 0.041, p = 0.025, p = 0.019, p = 0.019, p = 0.035, p = 0.048, p = 0.024, p = 0.022, p = 0.008, p = 0.034, vs. WT mice; Figure 8C). Among the remaining three mRNAs, Zfp37 and Polr1e were downregulated and Mapk10 was upregulated in the hippocampus of 5 × FAD mice, but the differences were insignificant (Figure 8C). According to the qRT-PCR analysis, the Zfp37 and Polr1e had similar expression trends to the sequencing data, while Mapk10 had an inconsonant expression trend to the sequencing data. Overall, the qRT-PCR results of three circRNAs, two miRNAs, and 13 mRNAs were consistent with the RNA sequencing data.
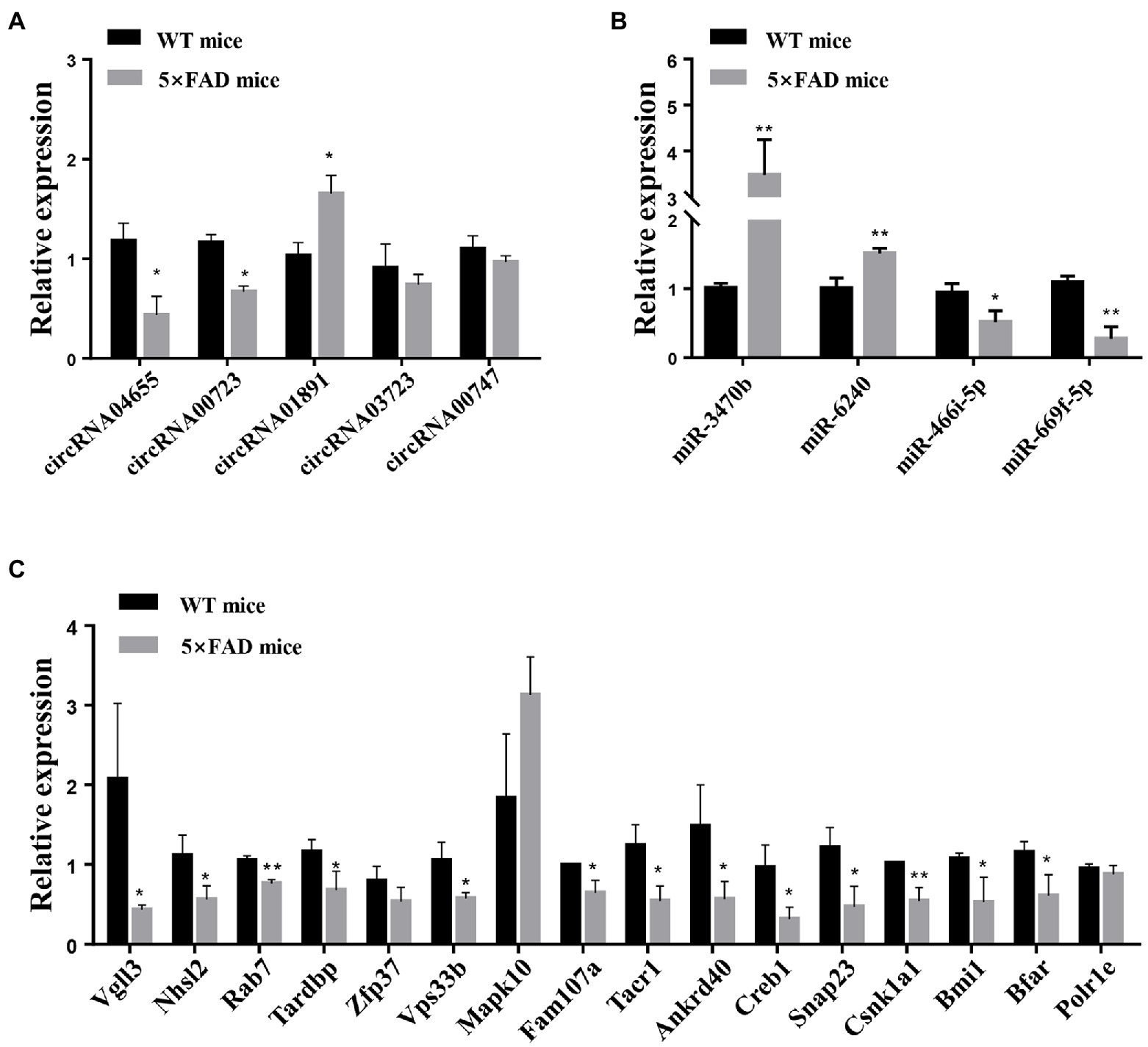
Figure 8. qRT-PCR analysis of circRNAs, miRNAs, and mRNAs. (A) Expression levels of circRNAs, including circRNA04655, circRNA00723, circRNA01891, circRNA03723, and circRNA00747, in the hippocampus of 5 × FAD mice and WT mice. (B) Expression of miRNAs, including miR-3470b, miR-6240, miR-466i-5p, and miR-669f-5p, in the hippocampus of 5 × FAD mice and WT mice. (C) Expression of mRNAs, including Vgll3, Nhsl2, Rab7, Tardbp, Zfp37, Vps33b, Mapk10, Fam107a, Tacr1, Ankrd40, Creb1, Snap23, Csnk1a1, Bmi1, Bfar, and Polr1e, in the hippocampus of 5 × FAD mice and WT mice. Results are shown as mean ± SD (n = 3). *p < 0.05, **p < 0.01 vs. WT mice.
Discussion
AD has enormous and detrimental impacts on the elderly, not only limited to cognitive impairment but also the shadow of developing dementia (Xia et al., 2018). While the etiopathogenesis of AD is complex with many contributing factors, including (but not limited to) genetic and environmental factors, many important details of AD pathogenesis remain unknown. Recently, the properties of non-coding RNAs in AD pathogenesis have attracted considerable attention, especially miRNAs and long non-coding RNAs. However, the properties of circRNAs during AD progression are still poorly understood. We reasoned that RNA sequencing in conjunction with a systematic analysis of the profiles of circRNAs dysregulated in AD could support novel foresight into comprehending the pathophysiology of AD. This study presents for the first time a profile of circRNA in the hippocampus of 5 × FAD mice, an investigation of the functional roles of these dysregulated circRNAs, and the construction of a circRNA-related ceRNA network.
Although dysregulated circRNAs in the senescence-accelerated mouse-prone 8 (SAMP8) mice and the precursor protein (APP) and presenilin 1 (PS1) double transgenic mice have been revealed in several investigations using RNA sequencing or microarrays (Zhang et al., 2017; Ma et al., 2019; Zhang Y. et al., 2021), circRNA expression profiles have not been determined in the brain of 5 × FAD mice, the transgenic mice that co-express a total of five familial AD mutations (Oakley et al., 2006). Because of these mutations, 5 × FAD mice present very similar clinicopathological characteristics to clinical patients. Furthermore, 5 × FAD mice demonstrate clinicopathological characteristics of AD at a younger age (in comparison with other transgenic mice). In 5 × FAD mice, aggregation of amyloid-beta peptide42 (Aβ42) in neurons occurs at 6 weeks of age, amyloid deposition and gliosis appear in the subiculum and deep cortical layers at 2 months, loss of synaptic proteins and damage to the hippocampus begin at 4 months, and impaired cognition is observed thereafter (Oakley et al., 2006). In line with these observations, 5 × FAD mice exhibit restricted learning capabilities and a decline in hippocampus-dependent memory (compared to age-matched WT mice) in our finding. Hence, 5 × FAD mice are an ideal model for RNA sequencing analyses of AD pathogenesis.
So far as we know, there has never been a report on RNA sequencing of circRNA profiles in 5 × FAD mice. Our results reveal that 34 circRNAs were significantly dysregulated in the hippocampus of 5 × FAD mice, including 17 upregulated and 17 downregulated circRNAs. Since circRNAs are transformed from host linear RNAs, the biological function of the host gene may provide clues concerning the function of the circRNA. Some host genes of circRNAs directly or indirectly participated in the pathological process of AD, including circRNA03556 (Hepacam), circRNA00723 (Myt1), circRNA02418 (Cntln), and circRNA04655 (Elavl2). Research on the AppNL-F mouse model of AD provided evidence that the hepacam protein has a marked increase in early-stage AD mice, as well as that there was a significant increase in its colocalization with Aβ (Aladeokin et al., 2019). Some study has proposed that Myt1 affects cholesterol 24S-hydroxylase (CYP46A1) gene transcription and is correlated with AD (Li et al., 2010). Hohman’s group revealed that the gene levels of CNTLN in the heart are associated with cognitive decline dependent on amyloid (Hohman et al., 2017). Lastly, neuronal ELAVL proteins modulate the regulation of Aβ precursor protein steady-state levels by inducing neuron-specific alternative splicing, alterations that have been linked to AD (Fragkouli et al., 2017).
Several differentially expressed circRNAs and miRNAs were found to be altered in the brain of different AD model mice (Supplementary Table 7). circRNA00825 and circRNA01985 were aberrantly upregulated in the brain of APP/PS1 mice (Ma et al., 2019; Wei et al., 2022). In aged C57BL/6J mice, circRNA03073 and circRNA08440 were found to have aberrant downregulation in the hippocampus of mice (Ran et al., 2022). However, the abnormal circRNAs found in the brains of SAMP8 mice and Tg2576 transgenic AD mice are different from those in the hippocampus of 5 × FAD mice in our study(Lee et al., 2019; Zhang Y. et al., 2021). This phenomenon may be related to the species differences of the animals. Although there are no identical circRNAs found in our study and other circRNA analyses, the KEGG pathways are found to be highly similar to the present study, including axon guidance, cAMP signaling pathway, and Hippo signaling pathway (Zhang Y. et al., 2021). Additionally, the aberrantly expressed miRNAs in the present study were found to be dysregulated in the hippocampus of APP/PS1 mice, Tg2576 mice, and Tg6799 mice, including miR-669f-5p, miR-6240, miR-1969, miR-3963, and miR-490-5p (Noh et al., 2014; Ma et al., 2019).
According to KEGG enrichment analysis, circRNAs that were dysregulated in the hippocampus of 5 × FAD mice are involved in numerous signaling pathways, several of which have been shown to be necessary for AD pathogenesis, including neurotrophin signaling pathway, axon guidance, the MAPK signaling pathway, and the FoxO signaling pathway. Moreover, a broad spectrum of cellular functions (e.g., proliferation, differentiation, migration, apoptosis, and autophagy) are regulated by these signaling pathways. Axon guidance molecules are essential in neural circuit formation (Charron, 2018). A recent study found that axon guidance molecules have effects on Aβ expression and tau protein hyperphosphorylation during the occurrence and process of AD (Zhang L. et al., 2021). Our research showed that upregulated and downregulated circRNAs both modulated the neurotrophin signaling pathway, which has been known to be affected in AD. During the neurotrophin signaling pathway, brain-derived neurotrophic factors ameliorate cognitive impairment and neuronal damage in AD (Gao et al., 2022; Liu et al., 2022). Enriched GO terms uncovered that dysregulated circRNAs were mainly involved in neuron projection development, cell morphogenesis, head development, and cellular component morphogenesis. The factors that influence neuron projection development and cell morphogenesis may contribute to AD occurrence.
circRNAs and miRNAs are both potentially key regulators of multiple aspects of AD. circRNAs have strong effects on regulating transcription and translation by sponging miRNAs. Considering the importance of the ceRNA hypothesis for non-coding RNAs, we first identified circRNA−miRNA interactions in 5 × FAD mice. We revealed that many circRNAs could co-associate with more than one miRNA. For example, circRNA00533 might interact with both miR-146a and miR-34c-3p, and the signaling pathways associated with these miRNAs were predicted to have a significant dysregulation in AD (Müller et al., 2014; Nikolac Perkovic et al., 2021). Next, potential target genes were added to generate a circRNA-miRNA-mRNA ceRNA network that consisted of five circRNAs, four miRNAs, and 188 mRNAs for AD. qRT-PCR was utilized to measure the level of circRNAs (circRNA00723, circRNA04655, circRNA01891, circRNA03723, and circRNA00747), miRNAs (miR-3470b, miR-6240, miR-669f-5p, and miR-466i-5p) and mRNAs (Vgll3, Nhsl2, Rab7, Tardbp, Vps33b, Fam107a, Tacr1, Ankrd40, Creb1, Snap23, Csnk1a1, Zfp37, Polr1e, Mapk10, Bmi1, and Bfar). The differential expression of circRNA00723, circRNA04655, and circRNA01891 in the hippocampus of 5 × FAD mice using qRT-PCR analyses coincided with their RNA sequencing results. Although these differentially expressed circRNAs (circRNA00723, circRNA04655, circRNA03723, and circRNA00747) have not previously been reported, their potential regulatory roles in AD could be inferred from the signaling pathways of their interacting transcripts. These pathways included the cGMP-PKG signaling pathway, hippo signaling pathway, and the Wnt signaling pathway. The expression changes of two miRNAs (miR-3470b and miR-6240) and 13 mRNAs (Vgll3, Nhsl2, Rab7, Tardbp, Vps33b, Fam107a, Tacr1, Ankrd40, Creb1, Snap23, Csnk1a1, Bmi1, and Bfar) in the hippocampus of 5 × FAD mice using qRT-PCR analyses had consistency with the RNA sequencing results, while the expression changes of miR-669f-5p and miR-466i-5p were inconsonant. The statistical differences of another two circRNAs (circRNA03723 and circRNA00747) and three mRNAs (Zfp37, Polr1e, and Mapk10) expression levels were insignificant in the hippocampus of 5 × FAD mice compared to WT mice using qRT-PCR analyses. Among them, circRNA00747, Zfp37, and Polr1e had similar expression trends to the sequencing data. Most of the RNAs for expression verification using qRT-PCR were in line with the RNA sequencing results, and a few were biased. We speculate that this discordant expression of a few RNAs may be related to the differential experimental steps and analytical methods between the two methods and the small sample size. The data obtained from both methods underwent multiple experimental and analytical steps, such as library construction, sequencing, and data processing. The qRT-PCR results can be affected by amplification region and efficiency, and RNA sequencing results can be affected by GC preference, exon number, and gene length. As a small sample size was included in this study, the error caused by different methods were not eliminated. It is generally accepted that there is 80% concordance in RNA sequencing and qRT-PCR assay (Consortium, 2014; Robert and Watson, 2015; Everaert et al., 2017). The qRT-PCR analyses demonstrated that the majority of RNAs in the circRNA–miRNA–mRNA ceRNA network had good consistency with the sequencing results regarding the differential expressions and alteration trends.
miR-6240, a predicted target of circRNA00747 (contrary to the expression trend for circRNA00747), is aberrantly upregulated in 5 × FAD mice. A recent study revealed that miR-6240 showed functional conservation in cardiomyocyte proliferation and mitosis (Adamowicz et al., 2018). Many target genes of miR-6240 have been proposed to be related to AD etiopathogenesis, including serine and arginine-rich splicing factor 1 (SRSF1), hypoxia-inducible factor-1α (HIF1α), mesenchyme homeobox 2 (MEOX2), cAMP-responsive element-binding protein 1 (CREB1), pericentriolar material-1 (PCM1), coiled-coil containing protein kinases (ROCK2), synaptosomal-associated protein 23 (SNAP23), and myocyte enhancer factor 2A (MEF2A). These genes are involved in plaque deposition, tau protein biosynthesis, neuronal dysfunction, neuroinflammation, autophagy, and other pathological processes related to AD (Chakravarthy et al., 2013; Soto et al., 2016; Muhammad et al., 2019; Adeyemi et al., 2021; Li et al., 2021; Weber and Herskowitz, 2021). SRSF1 is a member of the serine/arginine-rich proteins family, which is involved in the process of exon selection and mRNA output and localization. Recent studies have revealed that SRSF1 promotes tau mRNA formation (Jakubauskienė et al., 2021). Microtubule-associated protein tau, expressed mainly in axons, regulates the aggregation and stability of microtubules (Gao et al., 2018). When tau protein is hyperphosphorylated, it dissociates from the axon microtubule, misfolds, and aggregates, eventually forming neurofibrillary tangles, considered one of the emblematic hallmarks of AD (Chong et al., 2018; Naseri et al., 2019). Nevertheless, regulation of these interactions by circRNAs-miRNAs-mRNAs networks in AD has not been investigated. Additional research on ceRNA networks will help expand our knowledge of their biological functions and help us to understand their regulatory roles in AD pathogenesis.
Our study has two limitations. Firstly, to obtain a more accurate expression profile of circRNAs, more samples should be analyzed, including samples from different animal models of AD, and blood and tissue samples from AD patients. Secondly, the interactions and underlying mechanisms of these identified circRNAs, miRNAs, and mRNAs need to be further investigated.
Conclusion
For the first time, this study revealed dysregulated expression profiles of circRNAs in the hippocampus of 5 × FAD mice using RNA sequencing. From this data, we identified the potential biological functions of these circRNAs and their signaling pathways in AD. In addition, we have constructed a ceRNA network centered on circRNAs and verified key circRNA, miRNA, and mRNA expressions using qRT-PCR. Our findings should broaden our knowledge of circRNA regulatory action in the pathogenesis of AD.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: Data have been deposited in Gene Expression Omnibus (GEO) with the accession code GSE206562.
Ethics statement
The animal study was reviewed and approved by Experimental Animal Care and Use Committee of the Institute of Medicinal Biotechnology.
Author contributions
RL and ZL: concept and design, manuscript editing, and manuscript review. RL: definition of intellectual content. TS: literature search and manuscript preparation. TS and LZ: experimental studies. TS and ZC: data acquisition and analysis. QL: statistical analysis. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. U1803281, 82173806, and 82204366), Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (2021-1-I2M-030 and 2022-I2M-2-002), and Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2022-JKCS-08 and 3332022054).
Acknowledgments
We are indebted to Zengqiang Yuan and Yajin Liao, Academy of Military Medical Sciences, China, for providing 5 × FAD mice.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.1020699/full#supplementary-material
Footnotes
1. ^http://circatlas.biols.ac.cn/
4. ^http://www.targetscan.org/
5. ^http://www.geneontology.org
References
Adamowicz, M., Morgan, C. C., Haubner, B. J., Noseda, M., Collins, M. J., Abreu Paiva, M., et al. (2018). Functionally conserved noncoding regulators of cardiomyocyte proliferation and regeneration in mouse and human. Circ. Genom. Precis. Med. 11:e001805. doi: 10.1161/circgen.117.001805
Adeyemi, O. S., Awakan, O. J., Afolabi, L. B., Rotimi, D. E., Oluwayemi, E., Otuechere, C. A., et al. (2021). Hypoxia and the kynurenine pathway: implications and therapeutic prospects in Alzheimer's disease. Oxid. Med. Cell. Longev. 2021:5522981. doi: 10.1155/2021/5522981
Aladeokin, A. C., Akiyama, T., Kimura, A., Kimura, Y., Takahashi-Jitsuki, A., Nakamura, H., et al. (2019). Network-guided analysis of hippocampal proteome identifies novel proteins that colocalize with Aβ in a mice model of early-stage Alzheimer’s disease. Neurobiol. Dis. 132:104603. doi: 10.1016/j.nbd.2019.104603
Cai, Z., Zhao, K., Zeng, L., Liu, M., Sun, T., Li, Z., et al. (2022). The relationship between the aberrant long non-coding RNA-mediated competitive endogenous RNA network and Alzheimer’s disease pathogenesis. Int. J. Mol. Sci. 23:8497. doi: 10.3390/ijms23158497
Chakravarthy, B., Ménard, M., Brown, L., Hewitt, M., Atkinson, T., and Whitfield, J. (2013). A synthetic peptide corresponding to a region of the human pericentriolar material 1 (PCM-1) protein binds β-amyloid (Aβ1-42) oligomers. J. Neurochem. 126, 415–424. doi: 10.1111/jnc.12208
Charron, F. (2018). Axon guidance: gained in translation. Neuron 99, 1–2. doi: 10.1016/j.neuron.2018.06.040
Chen, L. L. (2020). The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 21, 475–490. doi: 10.1038/s41580-020-0243-y
Chen, G., Lin, Q.-M., Zeng, L., and Zou, Y.-P. (2020). Mining lycodine-type alkaloid biosynthetic genes and genetic markers from transcriptome of Lycopodiastrum casuarinoides. Chin. Herb. Med. 12, 133–141. doi: 10.1016/j.chmed.2020.03.001
Chen, L., Wang, C., Sun, H., Wang, J., Liang, Y., Wang, Y., et al. (2021). The bioinformatics toolbox for circRNA discovery and analysis. Brief. Bioinform. 22, 1706–1728. doi: 10.1093/bib/bbaa001
Chong, F. P., Ng, K. Y., Koh, R. Y., and Chye, S. M. (2018). Tau proteins and Tauopathies in Alzheimer’s disease. Cell. Mol. Neurobiol. 38, 965–980. doi: 10.1007/s10571-017-0574-1
Consortium, S. M.-I. (2014). A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 32, 903–914. doi: 10.1038/nbt.2957
Everaert, C., Luypaert, M., Maag, J. L. V., Cheng, Q. X., Dinger, M. E., Hellemans, J., et al. (2017). Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci Rep. 7:1559. doi: 10.1038/s41598-017-01617-3
Fragkouli, A., Koukouraki, P., Vlachos, I. S., Paraskevopoulou, M. D., Hatzigeorgiou, A. G., and Doxakis, E. (2017). Neuronal ELAVL proteins utilize AUF-1 as a co-partner to induce neuron-specific alternative splicing of APP. Sci. Rep. 7:44507. doi: 10.1038/srep44507
Gao, Y., Tan, L., Yu, J. T., and Tan, L. (2018). Tau in Alzheimer’s disease: mechanisms and therapeutic strategies. Curr. Alzheimer Res. 15, 283–300. doi: 10.2174/1567205014666170417111859
Gao, L., Zhang, Y., Sterling, K., and Song, W. (2022). Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 11:4. doi: 10.1186/s40035-022-00279-0
Gokool, A., Loy, C. T., Halliday, G. M., and Voineagu, I. (2020). Circular RNAs: the brain transcriptome comes full circle. Trends Neurosci. 43, 752–766. doi: 10.1016/j.tins.2020.07.007
Gonzales, M. M., Garbarino, V. R., Pollet, E., Palavicini, J. P., Kellogg, D. L. Jr., Kraig, E., et al. (2022). Biological aging processes underlying cognitive decline and neurodegenerative disease. J. Clin. Invest. 132:e158453. doi: 10.1172/jci158453
Gruner, H., Cortés-López, M., Cooper, D. A., Bauer, M., and Miura, P. (2016). CircRNA accumulation in the aging mouse brain. Sci. Rep. 6:38907. doi: 10.1038/srep38907
Hansen, T. B., Jensen, T. I., Clausen, B. H., Bramsen, J. B., Finsen, B., Damgaard, C. K., et al. (2013). Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388. doi: 10.1038/nature11993
Hohman, T. J., Dumitrescu, L., Cox, N. J., and Jefferson, A. L. (2017). Genetic resilience to amyloid related cognitive decline. Brain Imaging Behav. 11, 401–409. doi: 10.1007/s11682-016-9615-5
Jakubauskienė, E., Vilys, L., Pečiulienė, I., and Kanopka, A. (2021). The role of hypoxia on Alzheimer's disease-related APP and tau mRNA formation. Gene 766:145146. doi: 10.1016/j.gene.2020.145146
Jiang, H., Liu, J., Guo, S., Zeng, L., Cai, Z., Zhang, J., et al. (2022). miR-23b-3p rescues cognition in Alzheimer’s disease by reducing tau phosphorylation and apoptosis via GSK-3β signaling pathways. Mol. Ther. Nucleic Acids 28, 539–557. doi: 10.1016/j.omtn.2022.04.008
Kristensen, L. S., Andersen, M. S., Stagsted, L. V. W., Ebbesen, K. K., Hansen, T. B., and Kjems, J. (2019). The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20, 675–691. doi: 10.1038/s41576-019-0158-7
Lee, W. J., Moon, J., Jeon, D., Shin, Y. W., Yoo, J. S., Park, D. K., et al. (2019). Possible epigenetic regulatory effect of dysregulated circular RNAs in Alzheimer’s disease model. Sci. Rep. 9:11956. doi: 10.1038/s41598-019-48471-z
Li, Y., Chu, L. W., Wang, B., Yik, P. Y., Huriletemuer,, Jin, D. Y., et al. (2010). CYP46A1 functional promoter haplotypes decipher genetic susceptibility to Alzheimer’s disease. J. Alzheimers Dis. 21, 1311–1323. doi: 10.3233/jad-2010-100765
Li, H., Wang, F., Guo, X., and Jiang, Y. (2021). Decreased MEF2A expression regulated by its enhancer methylation inhibits autophagy and may play an important role in the progression of Alzheimer’s disease. Front. Neurosci. 15:682247. doi: 10.3389/fnins.2021.682247
Liu, S., Fan, M., Xu, J. X., Yang, L. J., Qi, C. C., Xia, Q. R., et al. (2022). Exosomes derived from bone-marrow mesenchymal stem cells alleviate cognitive decline in AD-like mice by improving BDNF-related neuropathology. J. Neuroinflamm. 19:35. doi: 10.1186/s12974-022-02393-2
Liu, Q. S., Jiang, H. L., Wang, Y., Wang, L. L., Zhang, J. X., He, C. H., et al. (2018). Total flavonoid extract from Dracocephalum moldavica L. attenuates β-amyloid-induced toxicity through anti-amyloidogenesic and neurotrophic pathways. Life Sci. 193, 214–225. doi: 10.1016/j.lfs.2017.10.041
Ma, N., Pan, J., Ye, X., Yu, B., Zhang, W., and Wan, J. (2019). Whole-transcriptome analysis of APP/PS1 mouse brain and identification of circRNA-miRNA-mRNA networks to investigate AD pathogenesis. Mol. Ther. Nucleic Acids 18, 1049–1062. doi: 10.1016/j.omtn.2019.10.030
Mahaman, Y. A. R., Embaye, K. S., Huang, F., Li, L., Zhu, F., Wang, J. Z., et al. (2021). Biomarkers used in Alzheimer’s disease diagnosis, treatment, and prevention. Ageing Res. Rev. 74:101544. doi: 10.1016/j.arr.2021.101544
Muhammad, T., Ali, T., Ikram, M., Khan, A., Alam, S. I., and Kim, M. O. (2019). Melatonin rescue oxidative stress-mediated neuroinflammation/neurodegeneration and memory impairment in scopolamine-induced amnesia mice model. J. Neuroimmune Pharmacol. 14, 278–294. doi: 10.1007/s11481-018-9824-3
Müller, M., Kuiperij, H. B., Claassen, J. A., Küsters, B., and Verbeek, M. M. (2014). MicroRNAs in Alzheimer’s disease: differential expression in hippocampus and cell-free cerebrospinal fluid. Neurobiol. Aging 35, 152–158. doi: 10.1016/j.neurobiolaging.2013.07.005
Naseri, N. N., Wang, H., Guo, J., Sharma, M., and Luo, W. (2019). The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 705, 183–194. doi: 10.1016/j.neulet.2019.04.022
Nikolac Perkovic, M., Videtic Paska, A., Konjevod, M., Kouter, K., Svob Strac, D., Nedic Erjavec, G., et al. (2021). Epigenetics of Alzheimer’s disease. Biomol. Ther. 11:195. doi: 10.3390/biom11020195
Noh, H., Park, C., Park, S., Lee, Y. S., Cho, S. Y., and Seo, H. (2014). Prediction of miRNA-mRNA associations in Alzheimer’s disease mice using network topology. BMC Genomics 15:644. doi: 10.1186/1471-2164-15-644
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/jneurosci.1202-06.2006
Piwecka, M., Glažar, P., Hernandez-Miranda, L. R., Memczak, S., Wolf, S. A., Rybak-Wolf, A., et al. (2017). Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 357:eaam8526. doi: 10.1126/science.aam8526
Ran, W., Liang, N., Yuan, R., Wang, Z., and Gao, J. (2022). Identification of potential key circRNAs in aged mice with postoperative delirium. Front. Mol. Neurosci. 15:836534. doi: 10.3389/fnmol.2022.836534
Robert, C., and Watson, M. (2015). Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome Biol. 16:177. doi: 10.1186/s13059-015-0734-x
Sanger, H. L., Klotz, G., Riesner, D., Gross, H. J., and Kleinschmidt, A. K. (1976). Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. U. S. A. 73, 3852–3856. doi: 10.1073/pnas.73.11.3852
Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, H., Chételat, G., Teunissen, C. E., et al. (2021). Alzheimer’s disease. Lancet 397, 1577–1590. doi: 10.1016/s0140-6736(20)32205-4
Shen, Q., Zhang, D., Zhang, T.-Y., Xu, Y.-Y., and Zhao, D.-G. (2020). Comparative transcriptome and co-expression analysis reveal key genes involved in leaf margin serration in Perilla frutescens. Chin. Herb. Med. 12, 265–272. doi: 10.1016/j.chmed.2019.10.001
Soto, I., Grabowska, W. A., Onos, K. D., Graham, L. C., Jackson, H. M., Simeone, S. N., et al. (2016). Meox2 haploinsufficiency increases neuronal cell loss in a mouse model of Alzheimer’s disease. Neurobiol. Aging 42, 50–60. doi: 10.1016/j.neurobiolaging.2016.02.025
Sun, T., Zhao, K., Liu, M., Cai, Z., Zeng, L., Zhang, J., et al. (2022). miR-30a-5p induces Aβ production via inhibiting the nonamyloidogenic pathway in Alzheimer’s disease. Pharmacol. Res. 178:106153. doi: 10.1016/j.phrs.2022.106153
Weber, A. J., and Herskowitz, J. H. (2021). Perspectives on ROCK2 as a therapeutic target for Alzheimer’s disease. Front. Cell. Neurosci. 15:636017. doi: 10.3389/fncel.2021.636017
Wei, R., Hu, Q., Lu, Y., and Wang, X. (2022). ceRNA network analysis reveals AP-1 tanscription factor components as potential biomarkers for Alzheimer’s disease. Curr. Alzheimer Res. 19, 387–406. doi: 10.2174/1567205019666220613142303
Westholm, J. O., Miura, P., Olson, S., Shenker, S., Joseph, B., Sanfilippo, P., et al. (2014). Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 9, 1966–1980. doi: 10.1016/j.celrep.2014.10.062
Wu, L., Du, Q., and Wu, C. (2021). CircLPAR1/miR-212-3p/ZNF217 feedback loop promotes amyloid β-induced neuronal injury in Alzheimer’s disease. Brain Res. 1770:147622. doi: 10.1016/j.brainres.2021.147622
Xia, X., Jiang, Q., McDermott, J., and Han, J.-D. J. (2018). Aging and Alzheimer’s disease: comparison and associations from molecular to system level. Aging Cell 17:e12802. doi: 10.1111/acel.12802
Yang, Y., Fan, X., Mao, M., Song, X., Wu, P., Zhang, Y., et al. (2017). Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 27, 626–641. doi: 10.1038/cr.2017.31
Zhang, L., Qi, Z., Li, J., Li, M., Du, X., Wang, S., et al. (2021). Roles and mechanisms of axon-guidance molecules in Alzheimer’s disease. Mol. Neurobiol. 58, 3290–3307. doi: 10.1007/s12035-021-02311-2
Zhang, Y., Qian, L., Liu, Y., Liu, Y., Yu, W., and Zhao, Y. (2021). CircRNA-ceRNA network revealing the potential regulatory roles of CircRNA in Alzheimer’s disease involved the cGMP-PKG signal pathway. Front. Mol. Neurosci. 14:665788. doi: 10.3389/fnmol.2021.665788
Zhang, S., Zhu, D., Li, H., Li, H., Feng, C., and Zhang, W. (2017). Characterization of circRNA-associated-ceRNA networks in a senescence-accelerated mouse prone 8 brain. Mol. Ther. 25, 2053–2061. doi: 10.1016/j.ymthe.2017.06.009
Zhao, Y., Alexandrov, P. N., Jaber, V., and Lukiw, W. J. (2016). Deficiency in the ubiquitin conjugating enzyme UBE2A in Alzheimer’s disease (AD) is linked to deficits in a natural circular miRNA-7 sponge (circRNA; ciRS-7). Genes 7:116. doi: 10.3390/genes7120116
Zhao, K., Zeng, L., Cai, Z., Liu, M., Sun, T., Li, Z., et al. (2022). RNA sequencing-based identification of the regulatory mechanism of microRNAs, transcription factors, and corresponding target genes involved in vascular dementia. Front. Neurosci. 16:917489. doi: 10.3389/fnins.2022.917489
Keywords: Alzheimer’s disease, circular RNAs, competing endogenous RNA, 5 × FAD mice, microRNAs
Citation: Sun T, Zeng L, Cai Z, Liu Q, Li Z and Liu R (2022) Comprehensive analysis of dysregulated circular RNAs and construction of a ceRNA network involved in the pathology of Alzheimer’s disease in a 5 × FAD mouse model. Front. Aging Neurosci. 14:1020699. doi: 10.3389/fnagi.2022.1020699
Edited by:
Timothy Y. Huang, Sanford Burnham Prebys Medical Discovery Institute, United StatesReviewed by:
Siranjeevi Nagaraj, Université libre de Bruxelles, BelgiumXiaoming Zhang, Harvard Medical School, United States
Copyright © 2022 Sun, Zeng, Cai, Liu, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuorong Li, bGl6aHVvcm9uZ0BpbWIucHVtYy5lZHUuY24=; Rui Liu, bGl1cnVpQGltYi5wdW1jLmVkdS5jbg==