 Chih-Yuan Ko1,2,3,4†‡
Chih-Yuan Ko1,2,3,4†‡ Jian-Hua Xu5†Yu-Wei Chang6†
Jian-Hua Xu5†Yu-Wei Chang6† Yangming Martin Lo7James Swi-Bea Wu8‡
Yangming Martin Lo7James Swi-Bea Wu8‡ Wen-Chung Huang9
Wen-Chung Huang9 Szu-Chuan Shen6*‡
Szu-Chuan Shen6*‡
- 1Department of Clinical Nutrition, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, China
- 2Department of Respiratory and Critical Care Medicine, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, China
- 3School of Public Health, Fujian Medical University, Fuzhou, China
- 4Respiratory Medicine Center of Fujian Province, Quanzhou, China
- 5Department of Tumor Surgery, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, China
- 6Graduate Program of Nutrition Science, National Taiwan Normal University, Taipei, Taiwan
- 7Institute for Advanced Study, Shenzhen University, Shenzhen, China
- 8Graduate Institute of Food Science and Technology, National Taiwan University, Taipei, Taiwan
- 9Graduate Institute of Health Industry Technology, Chang Gung University of Science and Technology, Taoyuan, Taiwan
Background and objectives: This study aimed to investigate the enhancing effect of vitamin-like alpha-lipoic acid (ALA) on phagocytosis of oligomeric beta-amyloid (oAβ)1–42 in BV-2 mouse microglial cells.
Methods: An in vitro model was established to investigate phagocytosis of oAβ1–42 in BV-2 cells. Transmission electron microscopy images indicated that the morphology of prepared oAβ1–42 was spherical particles. BV-2 cells treated with ALA were incubated with 5(6)-carboxyfluorescein-labeled oAβ1–42 (FAM-oAβ1–42) for 24 h, followed by flow cytometer analysis, western blotting, real-time quantitative PCR, and immunocytochemistry (ICC) analysis to assess the in vitro phagocytosis ability of oAβ1–42.
Results: Alpha-lipoic acid significantly increased messenger RNA (mRNA) expression of the CD36 receptor in BV-2 cells. ICC analysis showed that ALA significantly elevated CD36 protein expression in BV-2 cells both with and without oAβ1–42 treatment. Results from the flow cytometry analysis indicated that the CD36 receptor inhibitor significantly attenuated ALA-promoted phagocytosis of FAM-oAβ1–42 in BV-2 cells. Moreover, ICC analysis revealed that ALA caused the translocation of peroxisome proliferator-activated receptor-γ (PPAR-γ), which is known to regulate the expression of CD36 mRNA in BV-2 cells. ALA also elevated both the mRNA and protein expression of cyclooxygenase-2 (COX-2), which is a key enzyme involved in the synthesis of 15-deoxy-Δ12,14-prostaglandin J2 in BV-2 cells.
Conclusion: We postulated that ALA enhances oAβ1–42 phagocytosis by upregulating the COX-2/15-deoxy-Δ12,14-prostaglandin J2/PPAR-γ/CD36 pathway in BV-2 cells. Finally, future studies should be conducted with an in vivo study to confirm the findings.
Introduction
Alzheimer’s disease (AD) is an irreversible neurodegenerative disease with cognitive impairment that accounts for more than half of patients with dementia (Knopman et al., 2021). To date, while the complexity of neurodegeneration continues to be explored and recognized, AD has been characterized in part by excessive extracellular beta-amyloid (Aβ) plaque accumulation, the presence of intracellular tau-containing neurofibrillary tangles, and neuroinflammation in the brain (Erika et al., 2018; Rai et al., 2020; Knopman et al., 2021; Singh et al., 2021). In the brains of patients with AD, synaptic loss can precede neurodegeneration, brain tissue gradually shrinks, which, in turn, leads to a decline in memory and learning abilities of patients, eventually causing dementia (Blennow et al., 2006; Srivastava et al., 2019; Tripathi et al., 2019). Synaptic alterations are actually a critical part of neurodegeneration that are strongly correlated with the morphological lesions of AD, neurotoxicities of tau and Aβ, and cognitive impairment, which induce various neurotoxic effects such as increased inflammation, increased oxidative pressure, and subsequently damages the nerve synapses of the brain (Jack et al., 2019; Srivastava et al., 2019; Knopman et al., 2021). These factors gradually alter the microenvironment system of the brain, which becomes unconducive to the survival of nerve cells. In addition, inflammation plays a hallmark role in progression of AD. Aβ accumulation facilitates activation of immune cells including microglia, astrocytes, and macrophages. Remarkably, when microglia are activated in AD, activated microglia contribute to degraded Aβ accumulation through autophagy or clearance. Additionally, chronically activated microglia stimulating to the release of cytokines, which initiate a proinflammatory cascade and subsequently induce synaptic alterations (Pomilio et al., 2020; Knopman et al., 2021). There is a strong correlation between oligomeric Aβ (oAβ) accumulation in the brain and cognitive ability (Benilova et al., 2012). The oAβ strongly inhibits long-term potentiation of the hippocampal gyrus and causes damage to the nerve synapse, which eventually results in memory deficits in the patient (Walsh et al., 2002; Haass and Selkoe, 2007; Querfurth and LaFerla, 2010; Benilova et al., 2012). Therefore, oAβ is a crucial cause of occurrence of AD; however, the mechanism is still unclear.
Microglial autophagy may be potentially affected for treating AD (Pomilio et al., 2020). Evidence has highlighted that autophagy and antioxidants play core roles in AD (Liu et al., 2015). According to the theory of oxidative stress, numerous antioxidants are associated with the prevention of progression of AD (Su et al., 2008). For example, 15-deoxy-Δ12,14-prostaglandin J2 (15-deoxy-Δ12,14-PGJ2), which is produced by cyclooxygenase (COX), has been widely accepted as an anti-inflammatory prostaglandin and its anti-inflammatory mechanism is related to peroxisome proliferator-activated receptor-γ (PPAR-γ) (Scher and Pillinger, 2005). PPAR-γ is the transcription factor of CD36 messenger RNA (mRNA). When the activity of PPAR-γ increases, the transcription of CD36 mRNA increases by cells (Shimizu et al., 2008; Yamanaka et al., 2012). Bujold et al. (2009) successfully linked the mechanisms of COX-2/15-deoxy-Δ12,14-PGJ2/PPAR-γ/CD36 to elucidate in atherosclerosis. This is a critical mechanism pathway for this study.
The vitamin-like compound, alpha-lipoic acid (ALA), is a thioctic acid- and sulfur-containing organic compound that is a natural biological antioxidant (Packer et al., 1995). It can combat various chronic diseases such as diabetes, its related complications, and hypertension (Thirunavukkarasu et al., 2004; Ko et al., 2021). ALA has also been proven to affect the central nervous system by penetrating the blood–brain barrier (BBB) (Packer et al., 1995). Thus, ALA has been used to study numerous neurodegenerative diseases. One study showed that administering 600 mg/day of ALA to patients with AD can delay the progression of AD (Hager et al., 2007). Study on ALA and AD has largely focused on antioxidation, anti-inflammation, and the promotion of brain glucose metabolism. However, there are limited investigations on the use of ALA to promote the clearance of Aβ by microglia, thereby reducing the neurotoxic effects of Aβ-induced brain oxidative stress, inflammation, and glucose uptake resistance (Holmquist et al., 2007; Maczurek et al., 2008; Farr et al., 2012; Dos Santos et al., 2019; Kim et al., 2019; Choi et al., 2020). Therefore, we evaluated the alleviating effect of ALA on progression of AD and investigated the possible mechanism of ALA in promoting phagocytosis of BV-2 cell oAβ1–42 and its association with the COX-2/15-deoxy-Δ12,14-PGJ2/PPAR-γ/CD36 pathway.
Materials and Methods
Cell Culture
The mouse microglial BV-2 cell line used in this study was kindly donated by Professor Fu’s laboratory of the College of Medicine, National Taiwan University. BV-2 cells were cultured in an F12 medium without phenol red, containing 100 U/ml penicillin, 100 g/ml streptomycin, and 10% fetal bovine serum (FBS) at 37°C and 5% carbon dioxide (CO2) (Lu et al., 2007).
Identification of oAβ1–42 Aggregation Pattern
The Aβ1–42 monomer was purchased from Biopeptide (San Diego, CA, United States). The preparation of oAβ1–42 was adopted by the previous study (Yang et al., 2011). The appearance of the prepared oAβ1–42 was observed using JEM-1200EX II transmission electron microscopy (TEM) (JEOL, Tokyo, Japan). OAβ1–42 was first stained with 1% phosphotungstic acid for 2 min and placed on a 200-mesh copper net for observation. Then, freshly dissolved and fibrillary Aβ1–42 was compared to observe whether oAβ1–42 had an ideal appearance. In addition, the molecular weight of oAβ1–42 was analyzed using XL-A analytical ultracentrifugation (AUC) (Beckman, Palo Alto, CA, United States); the sample was centrifuged at 60,000 rpm and the orientation of the Aβ1–42 peptide was detected at a wavelength of 280 nm (Yang et al., 2011). Results of AUC were fitted to the continuous c(s) distribution model using the SEDFIT software1 and the molecular weight and percentage of aggregation of oAβ1–42 were estimated.
Oligomeric Beta-Amyloid1–42 Uptake Analysis and Quantification
The fluorescent molecule 5(6)-carboxyfluorescein (FAM) was used as a calibrator to explore the uptake of oAβ1–42 in BV-2 cells. When cells uptake oAβ1–42, calibrated by FAM, they were excited by a light with a wavelength of 488 nm and emitted green fluorescence of approximately 517 nm. This fluorescence was detected and accurately quantified using the LSRFortessa Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, United States).
Real-Time Quantitative PCR Analysis
According to the instructions of the manufacturer, the total RNA of cells was extracted using the Direct-zol RNA MiniPrep Kit (Zymo Research, Irvine, CA, United States). RNA concentrations were determined by the ND-1000 Spectrophotometer (NanoDrop®). Complementary DNA (cDNA) was synthesized by adding preisolated RNA and RNA to the cDNA EcoDry™ Premix (Oligo dT; Clontech Laboratories Incorporation, Mountain View, CA, United States). Real-time quantitative PCR was used to analyze genes such as scavenger receptor A1 (SR-A1), scavenger receptor B1 (SR-B1), CD36, receptor for advanced glycation endproducts (RAGE), COX-2, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Sequences for the primer sets used in quantitative PCR for each gene are given in Table 1. The amplification reactions were performed using the StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, United States). Data were described using the relative quantification method 2–ΔΔCt.

Table 1. Primer sequences for real-time PCR (RT-PCR).
Western Blot Analysis
The procedure for Western blot was carried out according to our previous study (Chen et al., 2021). Briefly, the protein concentration in the cell extract was determined using a Bio-Rad protein assay dye reagent (Richmond, Virginia, VA, United States). Aliquots of the supernatant containing 50 μg of protein were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred onto polyvinylidene difluoride membranes. The membranes were then incubated with anti-COX-2 (1:1,000; Cell Signaling Technology, Danvers, MA, United States) and anti-GAPDH (1:5,000; Signalway Antibody Corporation, College Park, MD, United States) antibodies at 4°C overnight. Next, the membranes were incubated with antirabbit immunoglobulin G and washed thrice for 5 min each time. The final images were detected and captured using the UVP Biospectrum Imaging System (Level, Cambridge, United Kingdom). Finally, all the relevant protein expressions were normalized with GAPDH.
Immunocytochemistry
A total of 4 × 104 cells were cultivated on a cover glass. After 24 h, the prepared oAβ1–42 was added and incubated for 24 h, fixed with 3% formaldehyde, punched with 0.1% Triton X-100, and placed in phosphate buffered saline with Tween (PBST) containing 5% bovine serum albumin (BSA) for blocking overnight. On the subsequent day, cells were stained with the primary antibody. After the invariable reaction overnight, cells were stained with a secondary antibody of fluorescent molecule Alexa 488 or 647. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) and observed under an upright fluorescence microscope (Carl Zeiss, Jena, Germany, United Kingdom). Cell fluorescence intensity was analyzed using MetaMorph (Molecular Devices, Sunnyvale, CA, United States).
Detection of Nitric Oxide
A total of 2 × 105 cells were cultured in six-well plates for 24 h, replaced with 1.5 ml Dulbecco’s Modified Eagle Medium (DMEM) without FBS and lipopolysaccharides treatment, and cocultured with cells for 24 h. The cell culture fluid was collected and mixed with Griess reagent in a 1:1 manner and reacted for 15 min and absorbance was detected at 540 nm using an ELISA reader.
Analysis of 15-Deoxy-Δ12,14-PGJ2
A total of 3 × 105 cells were cultured in six-well plates for 24 h, replaced with FBS-free DMEM, and treated with different concentrations of ALA. The cells were pretreated with ALA for 30 min and the vehicle or prepared oAβ1–42 was added and cocultivated for 24 h. An ELISA reader was used to analyze the culture medium. Standard of 15-deoxy-Δ12,14-PGJ2 was diluted with 400 μM ALA and the 4-parameters logical curve was used to analyze the 15-deoxy-Δ12,14-PGJ2 concentration of the sample.
Statistical Analyses
Values are presented as the mean ± SD using the SAS version 9.2 (SAS Institute Incorporation, Cary, NC, United States). The one-way ANOVA and Duncan’s multiple range tests were performed. p < 0.05 was accepted as statistically significant.
Results
Analysis of Transmission Electron Microscopy Appearance and Analytical Ultracentrifugation Physical Properties of Aβ1–42
The appearance of oAβ1–42 as a blank group (Figure 1A), freshly prepared Aβ1–42 (Figure 1B), oAβ1–42 solution (Figure 1C) after 24 h of aggregation at 4°C, and fibrillary Aβ1–42 with an apparent fibrous production group (Figure 1D) was examined using TEM. AUC was used to analyze the sedimentation coefficient of the prepared oAβ1–42 and the molecular weight and friction ratio of oAβ1–42 were further fitted using the SEDFIT software, as shown in Figure 1E. The molecular weight of the Aβ1–42 monomer was approximately 4.6 kDa. Approximately, 50% of Aβ1–42 aggregated into an oligomeric form and three primary oligomeric forms with molecular weights of 26.3, 115, and 28 kDa were observed.

Figure 1. Transmission electron microscopy (TEM), morphological and analytical ultracentrifugation (AUC), and physical characterization analysis of prepared beta-amyloid (Aβ1–42). TEM image of (A) blank [F12 medium + dimethyl sulfoxide (DMSO)], (B) freshly dissolved Aβ1–42, (C) oligomeric Aβ1–42, (D) fibrillar Aβ1–42, and (E) Sedimentation coefficient analysis of oligomeric Aβ1–42 using AUC.
Effect of Alpha-Lipoic Acid on Uptake of FAM-oAβ1–42 in BV-2 Cells
The fluorescence of BV-2 cells increased by 73.44 ± 8.99% under 100 μM ALA treatment (p < 0.05) and fluorescence intensity increased by 108.83 ± 51.07% under 400 μM ALA treatment (p < 0.05) compared with the ALA untreated group, which demonstrated a dose-dependent effect. The 400 μM ALA-treated group with untreated FAM-oAβ1–42 showed no significant difference in fluorescence from that of the blank group. Therefore, we confirmed that the increase in fluorescence of the BV-2 cells by ALA was not attributed to ALA itself—rather it was caused by an increased uptake of FAM-oAβ1–42 (Figure 2).
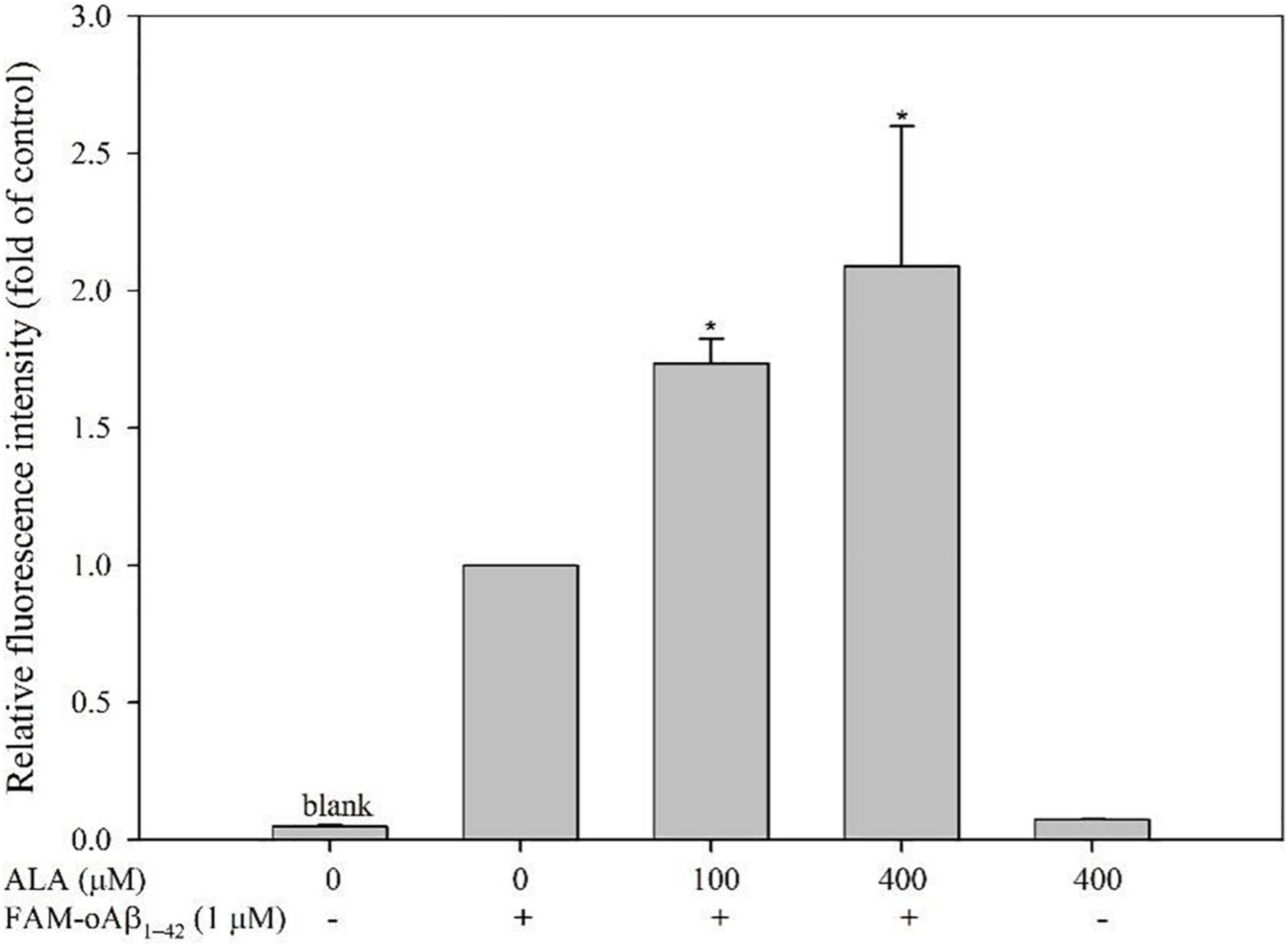
Figure 2. Effect of alpha-lipoic acid (ALA) on 5(6)-carboxyfluorescein (FAM)-oligomeric Aβ (oAβ1–42) uptake in BV-2 mouse microglial cells. BV-2 cells were pretreated with or without ALA (100 or 400 μM) for 30 min, followed by FAM-oAβ1–42 treatment for 24 h. Mean fluorescence intensity was detected using a flow cytometer. *p < 0.05 vs. the FAM-oAβ1–42 group.
Messenger RNA Expression and oAβ1-42 Phagocytosis-Related Receptors in BV-2 Cells
Cells were treated with 50, 100, 200, and 400 μM ALA compared with the untreated ALA and oAβ group (control group); mRNA expression levels of CD36 were 1.81 ± 0.06, 2.36 ± 0.33, 3.63 ± 1.00, and 7.29 ± 0.39 times for the respective ALA treatment groups (50, 100, 200, and 400 μM; p < 0.05) (Figure 3A). However, mRNA expression levels of SR-A1, SR-B1, and RAGE showed no noticeable dose-dependent effect (Figures 3B–D). In addition, ALA increased the expression of CD36 mRNA in BV-2 cells that were not treated with oAβ1–42 (p < 0.05); this result was similar to that of the group treated with 1 μM oAβ1–42 (p < 0.05) (Figure 4).
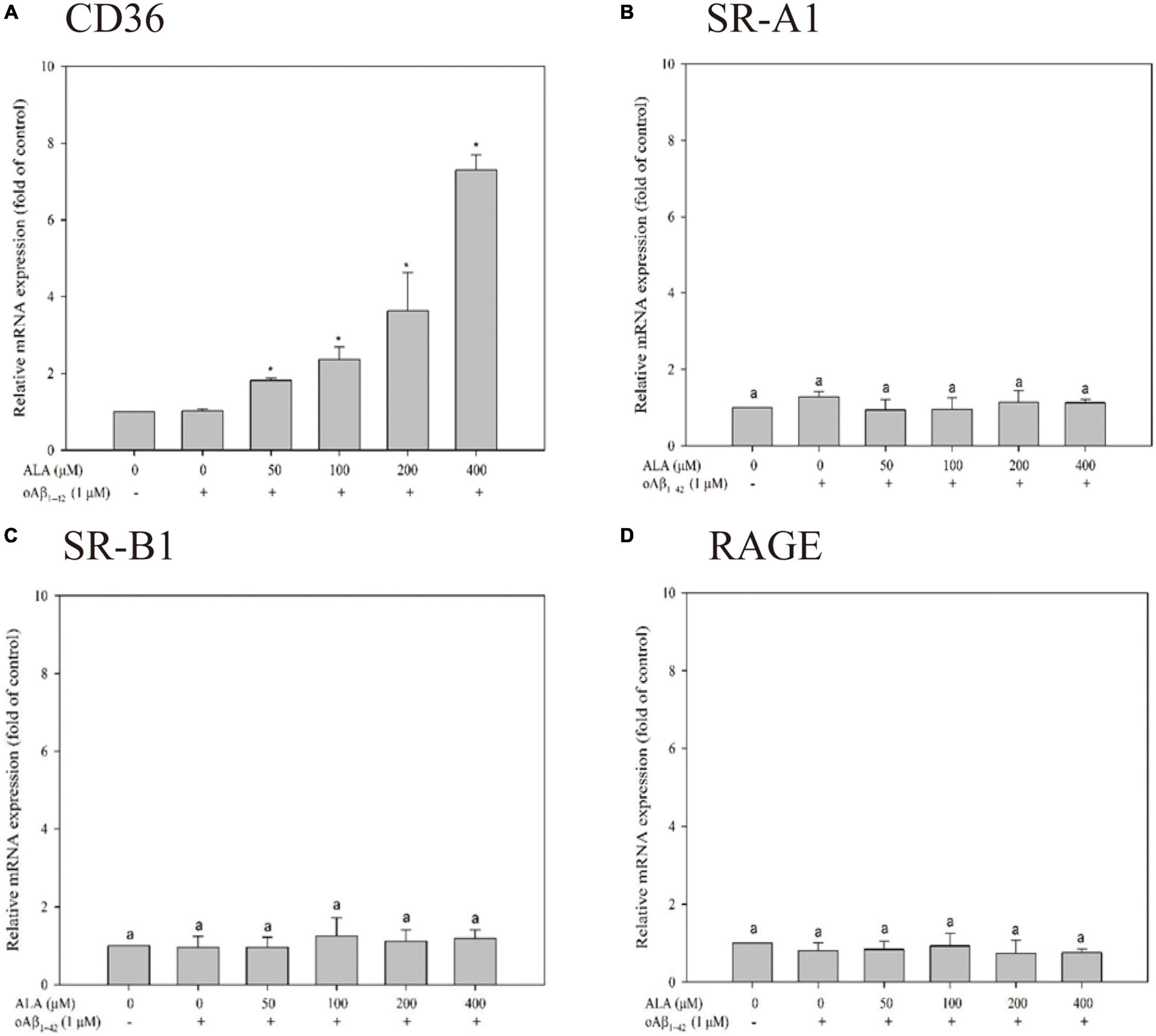
Figure 3. Messenger RNA (mRNA) expression of oAβ1–42 phagocytosis-related receptor including CD36 (A), scavenger receptor A1 (SR-A1) (B), scavenger receptor B1 (SR-B1) (C), and receptor for advanced glycation endproducts (RAGE) (D) in BV-2 mouse microglial cells. BV-2 cells were pretreated with or without different concentrations of ALA for 30 min, followed by oAβ1–42 treatment for 24 h. The relative mRNA expression of CD36, SR-A1, SR-B1, and RAGE was detected using real-time PCR. *p < 0.05 vs. the control group.
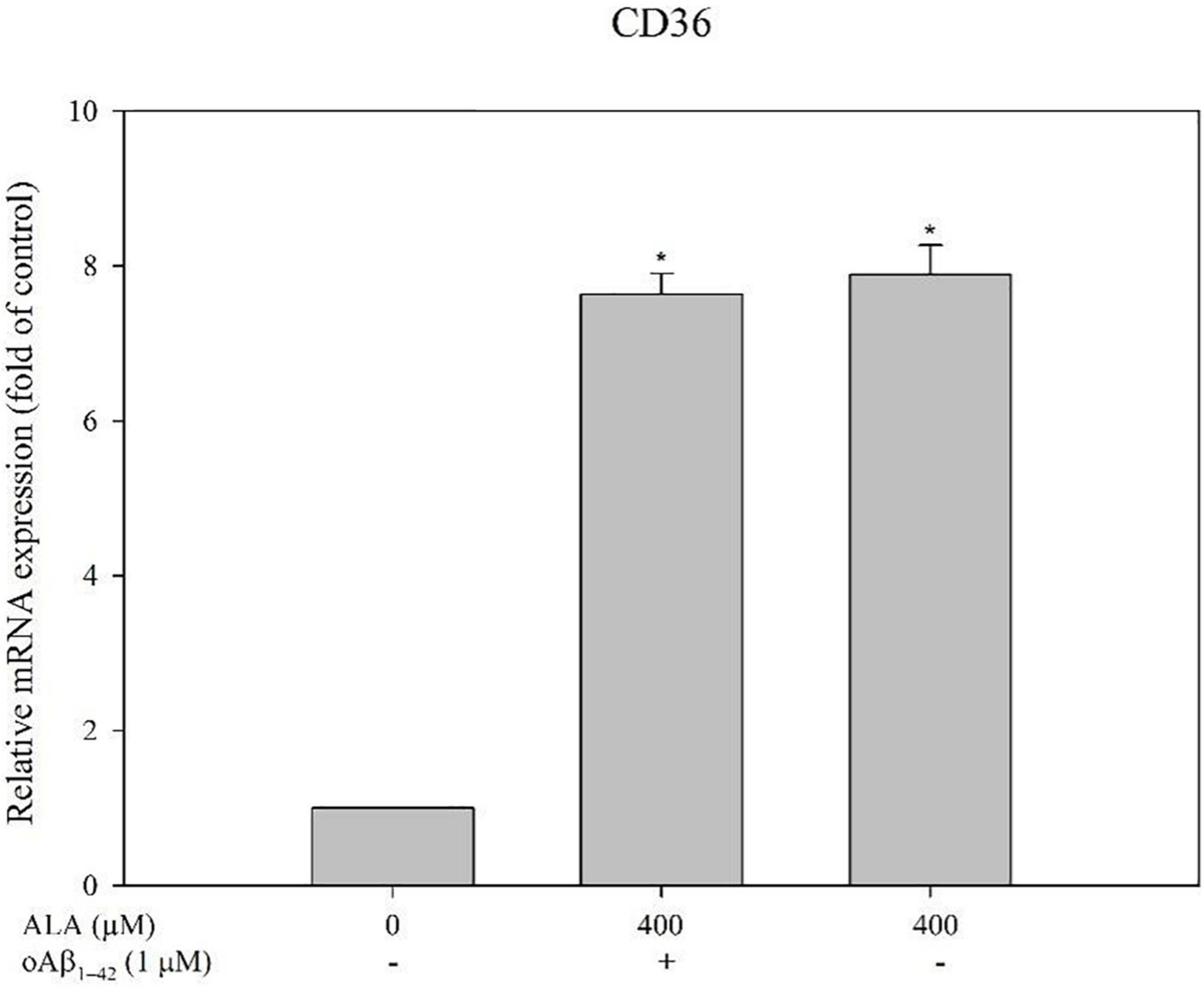
Figure 4. Alpha-lipoic acid elevated CD36 mRNA expression in BV-2 mouse microglial cells. BV-2 cells were pretreated with or without ALA (400 μM) for 30 min, followed by vehicle or oAβ1–42 treatment for 24 h. The relative mRNA expression of CD36 was detected using real-time PCR. *p < 0.05 vs. the control group.
The expression of ICC-measured CD36 protein is shown in Figure 5A. When Alexa 647 was used to stain CD36 on the cell membrane, it appeared red in the image, whereas the nuclei of the BV-2 cells appeared blue when stained with DAPI. The appearance of cells was analyzed to confirm the size and location of the cells using differential interference contrast light paths. Cells treated with ALA promoted the protein expression of CD36. Regardless of whether oAβ1–42 was stimulated, CD36 protein expression in BV-2 cells increased with ALA treatment. The fluorescence intensity of the 400 μM ALA treatment group was approximately 1.3 times that of the untreated group (p < 0.05) (Figure 5B).
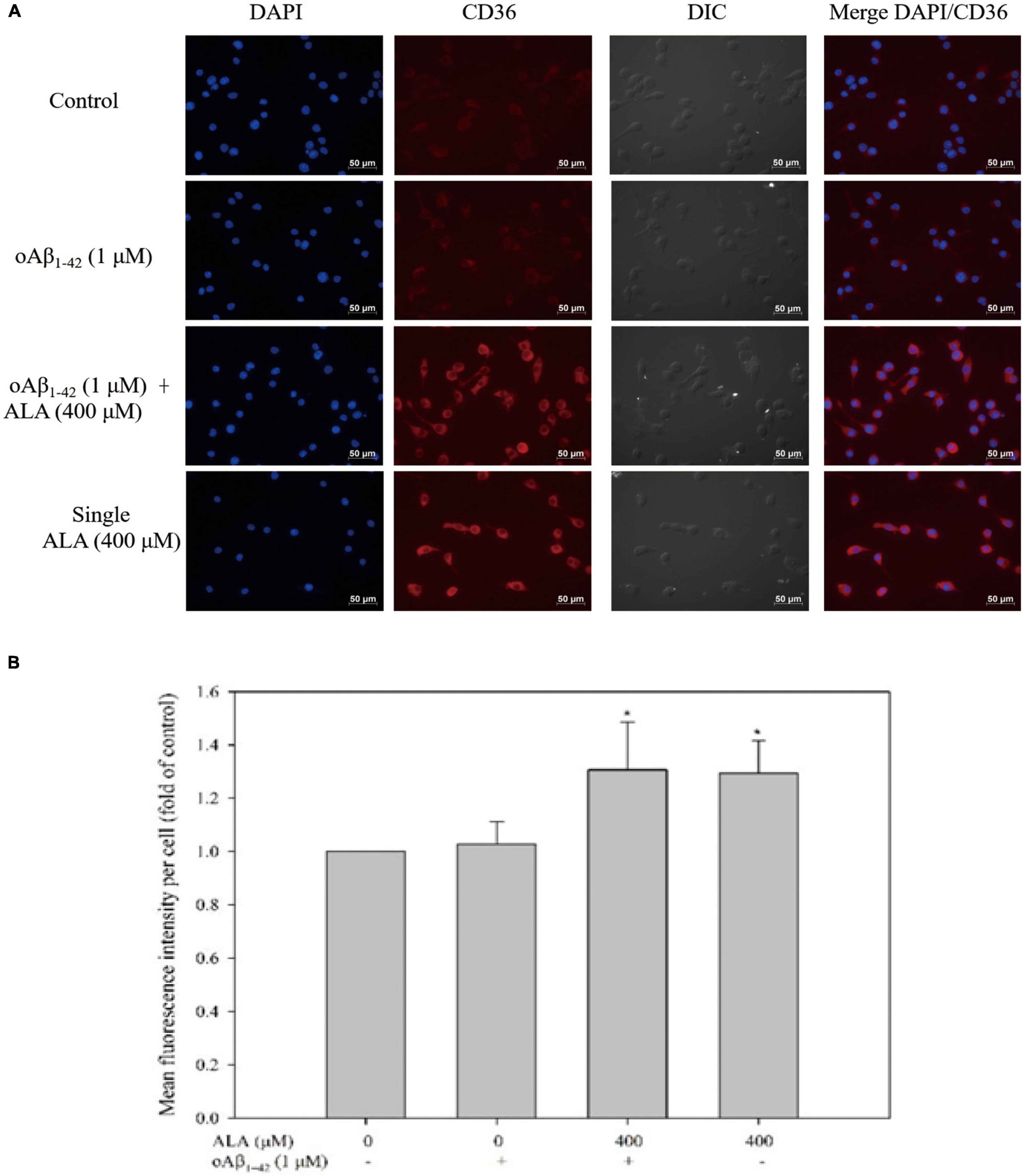
Figure 5. Alpha-lipoic acid elevated CD36 protein expression in BV-2 mouse microglial cells. BV-2 cells were pretreated with or without ALA (400 μM) for 30 min, followed by vehicle or oAβ1–42 treatment for 24 h. After treatment, BV-2 cells were immunostained with CD36 antibody (red) and the nuclei were stained with DAPI (blue). (A) Fluorescence image from immunocytochemistry. (B) Quantification of mean fluorescence intensity per cell. *p < 0.05 vs. the control group.
The addition of the CD36 antibody significantly reduced the uptake capacity of FAM-oAβ1–42 enhanced by ALA. The immunoglobulin A isotype control antibody did not affect the uptake of FAM-oAβ1–42 by BV-2 cells. In addition, the CD36 antibody did not affect the ability of BV-2 cells without ALA treatment to take up FAM-oAβ1–42. Results showed that ALA increased the uptake of oAβ1–42 by increasing the expression of CD36 (p < 0.05) (Figure 6).
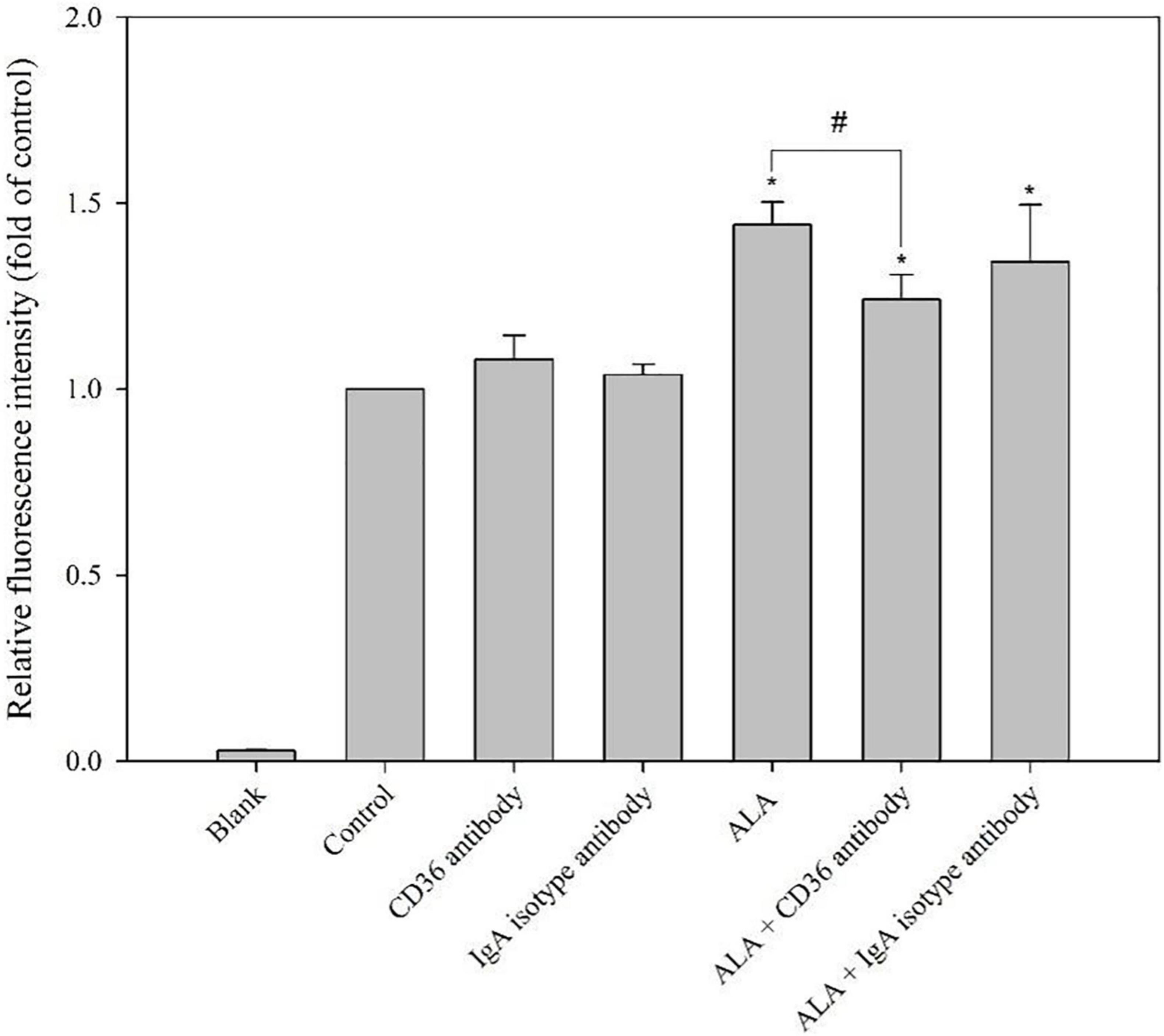
Figure 6. Blocking effect of CD36 antibody on ALA-enhanced phagocytosis ability of FAM-oAβ1–42 in BV-2 mouse microglial cells. BV-2 cells were pretreated with or without ALA (400 μM) in the presence of a CD36 antibody or isotype control for 30 min, followed by FAM-oAβ1–42 treatment for 24 h. Mean fluorescence intensity was detected using a flow cytometer. *p < 0.05 vs. the control group and #p < 0.05 vs. the ALA group.
Alpha-Lipoic Acid Promotes the Translocation of PPAR-γ in BV-2 Cells
When Alexa 488 was used to stain PPAR-γ, it appeared green in the image, whereas the nuclei of the BV-2 cells appeared blue when stained with DAPI, as shown in Figure 7. In cells without ALA treatment, most PPAR-γ were located outside the nuclei. Furthermore, cavities were evident in the nucleus, as shown by the white arrow in Figure 7. However, in cells treated with ALA, the transcription factor PPAR-γ was translocated and transferred from the cytoplasm to the nucleus and the void area in the nucleus was significantly reduced compared to that of the untreated group. This was observed regardless of whether oAβ1–42 was stimulated.
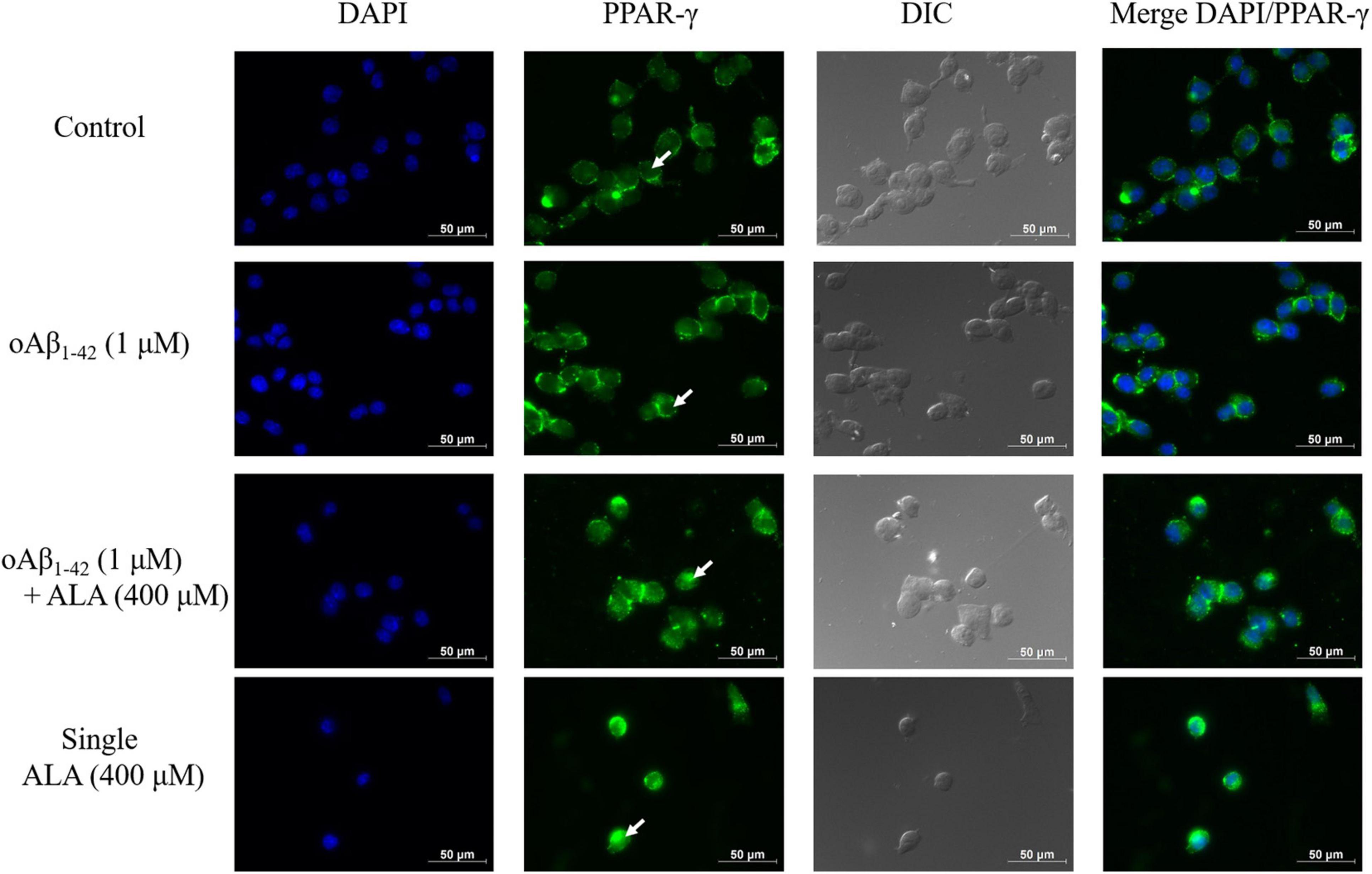
Figure 7. Alpha-lipoic acid caused PPAR-γ translocation from the cytoplasm to the nucleus. BV-2 cells were pretreated with or without ALA (400 μM) for 30 min, followed by vehicle or oAβ1–42 treatment for 24 h. After treatment, BV-2 cells were immunostained with PPAR-γ antibody (green) and the nuclei were stained with DAPI (blue).
Alpha-Lipoic Acid Causes PPAR-γ Translocation Through COX-2/15-Deoxy-Δ12,14-PGJ2 in BV-2 Cells
Alpha-lipoic acid promoted BV-2 cells and increased the production of 15-deoxy-Δ12,14-PGJ2 for the respective ALA treatment groups (50, 100, 200, and 400 μM; p < 0.05) (Figure 8). For example, in BV-2 cells that were treated with 400 μM ALA for 24 h, the amount of 15-deoxy-Δ12,14-PGJ2 produced was 6.92 ± 2.06 times that of the untreated group and there was no significant difference from the ALA group cocultured with oAβ1–42 (Figure 8).
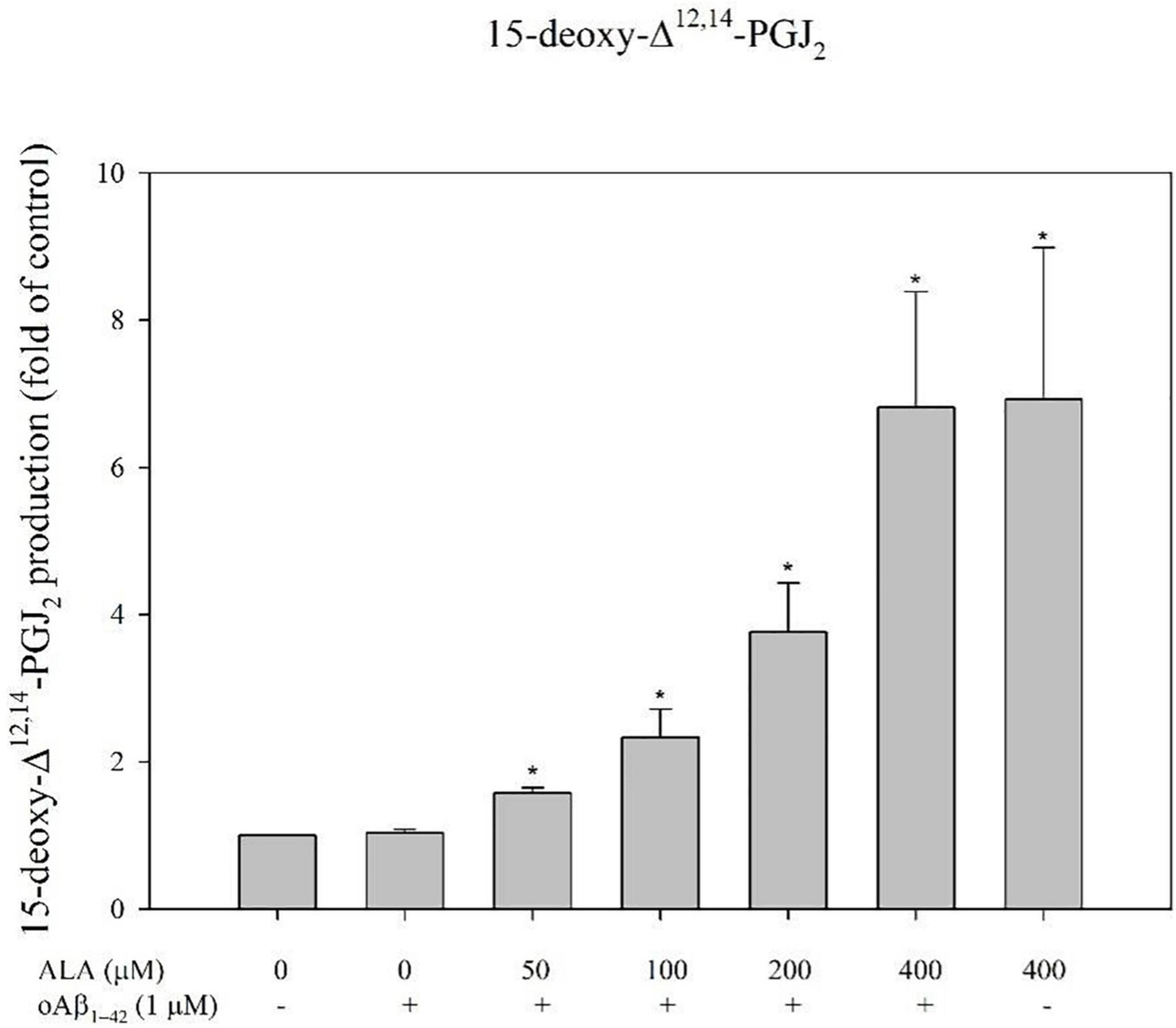
Figure 8. Alpha-lipoic acid promoted 15-deoxy-Δ12,14-PGJ2 production in BV-2 mouse microglial cells. BV-2 cells were pretreated with or without different concentrations of ALA for 30 min, followed by vehicle or oAβ1–42 treatment for 24 h. Relative 15-deoxy-Δ12,14-PGJ2 production was detected using ELISA. Data are presented as means ± SDs. *p < 0.05 vs. the vehicle control group.
After cocultured with oAβ1–42 in BV-2 cells for 24 h, the expression of the COX-2 protein did not differ significantly from that of the untreated oAβ1–42 group. However, the expression of the COX-2 protein in BV-2 cells increased with the increase in ALA treatment concentration (p < 0.05) (Figure 9A). In addition, the expression of COX-2 mRNA in BV-2 cells was similar to the expression of the protein (Figure 9B).
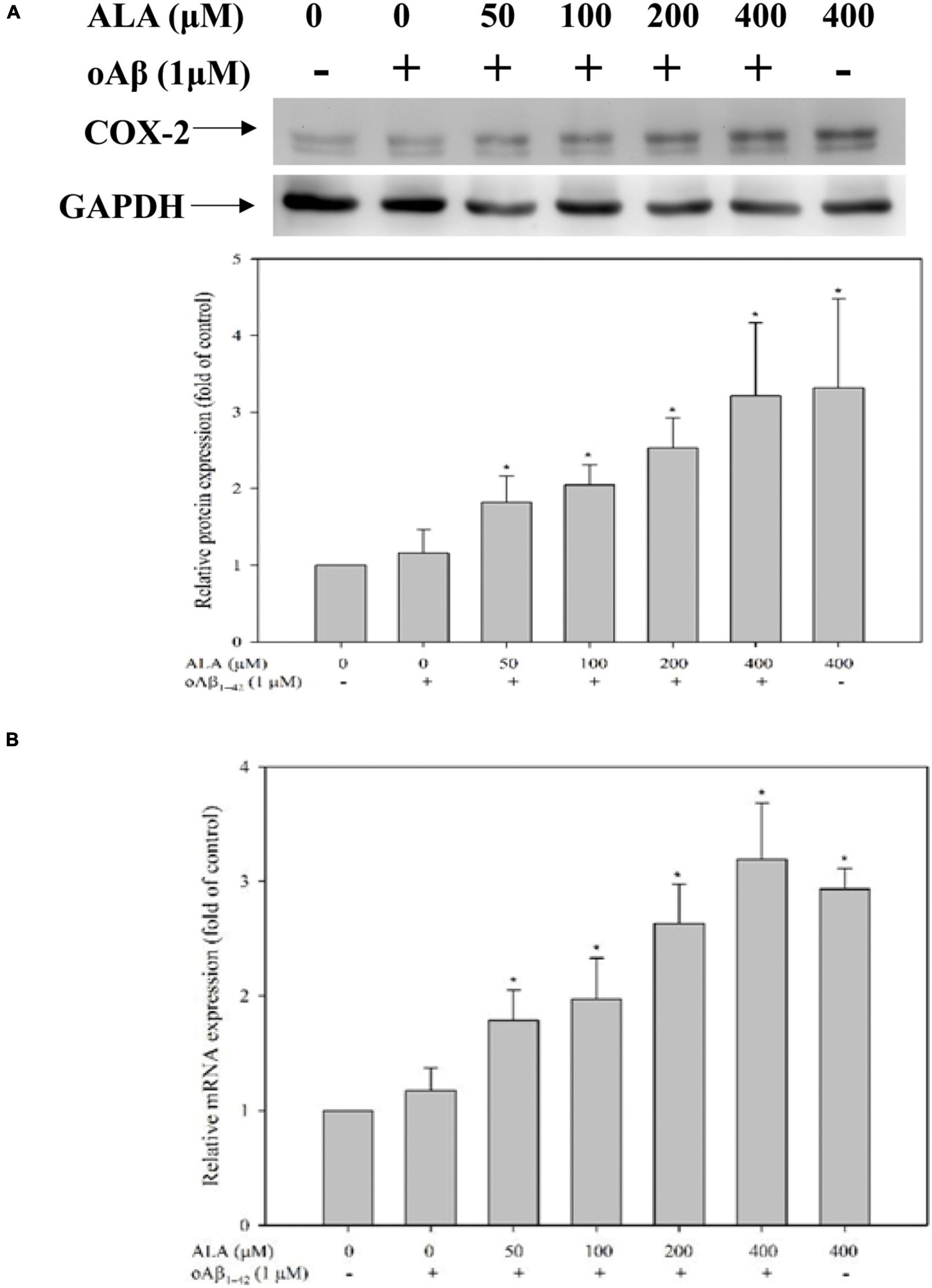
Figure 9. Alpha-lipoic acid elevated cyclooxygenase-2 (COX-2) expression in BV-2 mouse microglial cells. BV-2 cells were pretreated with or without different concentrations of ALA for 30 min, followed by vehicle or oAβ1–42 treatment for 24 h. (A) The relative COX-2 protein expression was detected using western blotting. (B) The relative mRNA expression of COX-2 was determined using real-time PCR. *p < 0.05 vs. the vehicle control group.
Discussion
This study demonstrated the possible mechanism of ALA on promoting oAβ1–42 uptake in mouse microglial BV-2 cells. ALA treatment increased both the expression of COX-2 mRNA and the COX-2 protein in BV-2 cells. The increase in COX-2 protein expression indirectly accelerated the production of 15-deoxy-Δ12,14-PGJ2 and acted as a PPAR-γ transcription factor ligand, which resulted in the translocation and combination of PPAR-γ with peroxisome proliferator hormone response elements. In addition, ALA increased the transcription of CD36 mRNA and increased the translation of CD36 mRNA to the CD36 receptor, which recognized oAβ1–42 and triggered the phagocytosis of oAβ1–42 in BV-2 cells (Figure 10). Thus, our findings suggested that ALA possesses significant potential for regulating the uptake of FAM-oAβ1–42 in BV-2 cells. A previous study demonstrated that supplementing 600 mg ALA per day ameliorated the progression of AD (Hager et al., 2007; Fava et al., 2013). However, the mechanism is not well understood. Numerous studies on the alleviating effect of ALA on AD have focused on its antioxidative and anti-inflammatory properties (Maczurek et al., 2008; Farr et al., 2012; Dos Santos et al., 2019; Kim et al., 2019; Choi et al., 2020). We revealed that in addition to antioxidant and anti-inflammatory properties, ALA may also enhance the ability of microglia to eliminate oAβ1–42 and subsequently reduce the neurotoxic effect induced by oAβ1–42, which may offer a possible mechanism underlying the prevention of dementia.
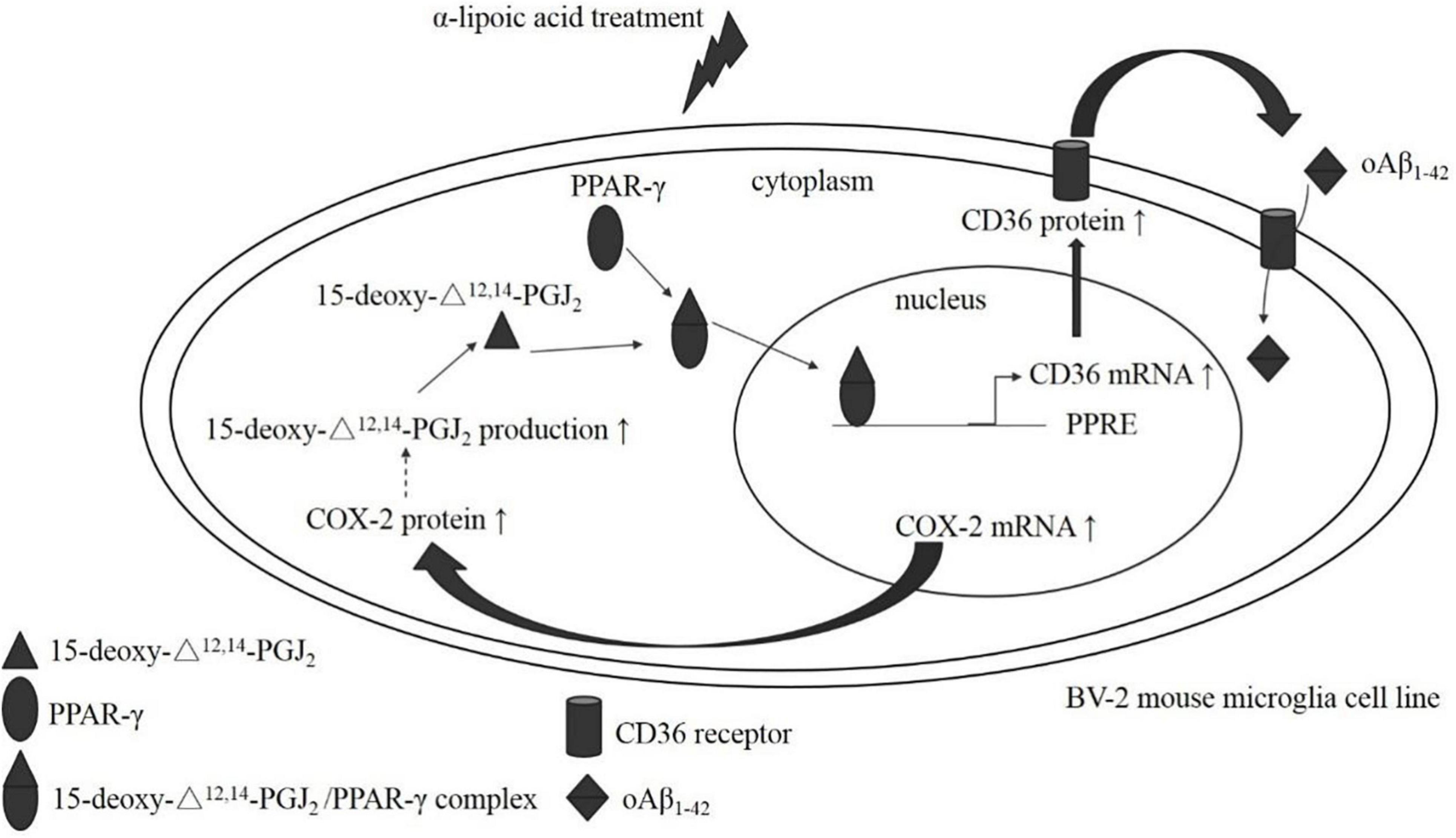
Figure 10. The postulated mechanism of ALA in enhancing oAβ1–42 phagocytosis in BV-2 mouse microglial cells. BV-2 mouse microglial cells made a difference after ALA stimulation, which caused the elevation of COX-2 mRNA and the increased translation of the COX-2 protein. Furthermore, the COX-2 protein induced 15-deoxy-Δ12,14-PGJ2 production and the 15-deoxy-Δ12,14-PGJ2 acted on the PPAR-γ ligand. Simultaneously, the 15-deoxy-Δ12,14-PGJ2/PPAR-γ complex translocated from the cytoplasm to the nucleus. PPAR-γ that located the nucleus stimulated the transcription of the CD36 gene, which promoted the synthesis of CD36 mRNA. The increase in CD36 mRNA promoted the translation of the CD36 protein. Finally, the elevated level of the CD36 protein enhanced the phagocytosis of oAβ1–42 in BV-2 mouse microglial cells.
It is well established that phagocytosis of oAβ by microglia is accomplished via a variety of receptors on the cell membrane such as CD36, SR-A1, SR-B1, and RAGE (Yan et al., 1996; El Khoury et al., 1998; Sokolowski and Mandell, 2011). The scavenger receptor CD36 has been identified as essential in the Aβ-recognition microglia activation (Doens et al., 2017; Dobri et al., 2021). In this study, the mRNA expression of CD36 increased with the increase in the concentration of ALA. We speculate that ALA increases phagocytosis of oAβ1–42 in BV-2 cells via the CD36 receptor. Additionally, the increase in CD36 mRNA expression in BV-2 cells treated with ALA did not appear to be related to oAβ1–42. In this study, ALA inhibited CD36 receptor expression and subsequently the uptake of FAM-oAβ1–42, but was unable to reverse this progression in BV-2 cells. We speculate that this may be executed solely through CD36, alongside the involvement of other receivers such as Toll-like receptor (TLR)-4 and TLR6 (Stewart et al., 2010). CD36 activation by different ligands is another reason; ligands of microglia in rodents are apoptotic bodies and oxidized low-density protein (Dobri et al., 2021). The CD36 antibody did not inhibit the uptake of FAM-oAβ1–42 in BV-2 cells, which is consistent with a previous study (Yang et al., 2011). Increasing the expression of CD36 may promote the ability of microglia to take up Aβ. Results of the Morris water maze test in transplant amyloid precursor protein 23 (APP23) mice showed that the Aβ deposition in the brains of mice in the interleukin-4/interleukin-13 (IL-4/IL-13) treatment group was lower than that in the control group and spatial learning and memory abilities improved significantly more in the treatment group compared with the untreated group, which may be attributed to the increased expression of CD36 by microglia (Kawahara et al., 2012). In vitro, it was found that increased expression of CD36 promoted the uptake of oAβ1–42 in rat primary type 2 microglia treated with IL-4 (Shimizu et al., 2008). Therefore, we speculate that ALA increases CD36 expression and, subsequently, the CD36 receptor identifies oAβ1–42 and initiates downstream phagocytosis.
Peroxisome proliferator-activated receptor-γ has been confirmed as a transcription factor of CD36 mRNA (Shimizu et al., 2008; Yamanaka et al., 2012). In this study, PPAR-γ moved from the cytoplasm to the nucleus under ALA treatment. Thus, the increase in CD36 mRNA facilitated by ALA is related to the increase in the translocation of transcription factor PPAR-γ. The treatment of ALA may increase the activity of PPAR-γ and prevents a variety of diseases (Golbidi et al., 2011; Wang et al., 2013). In a previous study using a PPAR-γ activator, when pioglitazone and DSP-8658 processed rat primary microglia, the expression of CD36 significantly increased and the increase in the CD36 receptor was shown to be the key receptor for pioglitazone and DSP-8658, which promoted the uptake of Aβ1–42 by rat primary microglia (Yamanaka et al., 2012). This illustrated the importance of PPAR-γ/CD36 in regulating the uptake of Aβ1–42. PPAR-γ must be combined with its ligand to be translocated. The anti-inflammatory prostaglandin 15-deoxy-Δ12,14-PGJ2 produced in COX-2 metabolizing polyunsaturated fatty acids is one of the key ligands that activate PPAR-γ (Scher and Pillinger, 2005; Ulivi et al., 2011; Ramer et al., 2013). To date, there has been no evidence demonstrating that ALA promotes the production of 15-deoxy-Δ12,14-PGJ2 in cells. Therefore, we speculate that anti-inflammatory response mechanism of ALA is linked to the production of 15-deoxy-Δ12,14-PGJ2 and is involved in PPAR-γ translocation in BV-2 cells. We used the anti-inflammatory properties of ALA to explore PPAR-γ activation and found that BV-2 cells increased the production of 15-deoxy-Δ12,14-PGJ2 under ALA treatment. This also increased the upstream expression of COX-2, which showed a trend increase with the increase in ALA treatment concentrations. Furthermore, we found that BV-2 cells increased the production of COX-2 mRNA under ALA stimulation, which suggested that ALA increases the translocation of PPAR-γ in BV-2 cells through COX-2/15-deoxy-Δ12,14-PGJ2.
Study with respect to the effect of ALA on COX-2 expression in microglia is limited. ALA is considered to be a substance with anti-inflammatory activity and COX-2 has a close relationship with inflammation (Wang et al., 2018). Diethylnitrosamine and choline-methionine-deficient diet-induced liver damage and ALA administration in Fischer rats showed that ALA increases hepatic COX-2 protein expression when the liver is damaged. However, this result was not found in the choline-supplemented diet group (Perra et al., 2008). Moreover, ALA did not increase the protein expression of COX-2 in RAW 264.7 cells (Hwang et al., 2015). We believe that this issue has been inadequately explored; although prostaglandin produced by COX-2-metabolizing polyunsaturated fatty acids is partially related to the inflammatory response, it is also partially related to the anti-inflammatory effect. Few studies have focused on the ability of ALA itself to induce COX-2. The best example is 15-deoxy-Δ12,14-PGJ2, which is a well-known anti-inflammatory prostaglandin and currently, studies on the increase of PPAR-γ translocation by ALA are limited. The results of this study clearly showed a correlation among ALA, 15-deoxy-Δ12,14-PGJ2, PPAR-γ, and anti-inflammatory effects. Taken together, we speculate that ALA increases the expression of CD36 via COX-2/15-deoxy-Δ12,14-PGJ2/PPAR-γ activation, which may be one of the major mechanisms of oAβ phagocytosis in BV-2 cells. A future study should be conducted with an in vivo study to confirm our in vitro findings.
Conclusion
This study demonstrated that ALA increases phagocytosis of oAβ1–42 possibly via modulating the COX-2/15-deoxy-Δ12,14-PGJ2/PPAR-γ/CD36 pathway in microglia. We postulated that ALA enhances mRNA and protein expression of COX-2, increases the production of 15-deoxy-Δ12,14-PGJ2, acts as a PPAR-γ transcription to elevate CD36 mRNA translation into CD36 protein, and subsequently enhances oAβ1–42 phagocytosis in BV-2 cells. These study findings suggest that ALA possesses antidementia potential and offers new insight into the mechanisms underlying the prevention of progression of AD.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author Contributions
C-YK and S-CS participated in the design of the study and writing the protocol. Y-WC carried out all the experiments. C-YK, J-HX, YL, JW, W-CH, and S-CS conducted kinds of literature searches and data analyses and wrote drafts of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Ministry of Science and Technology of the Republic of China (ROC), Taiwan, under contract no. MOST 107-2320-B-003-004-MY3, and academic funding of the Second Affiliated Hospital of Fujian Medical University (serial no. BS201902).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Special thanks are due to Fu in the College of Medicine, National Taiwan University for her kindly donation of BV-2 cells.
Footnotes
References
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer’s disease. Lancet 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Bujold, K., Rhainds, D., Jossart, C., Febbraio, M., Marleau, S., and Ong, H. (2009). CD36-mediated cholesterol efflux is associated with PPARgamma activation via a MAPK-dependent COX-2 pathway in macrophages. Cardiovasc. Res. 83, 457–464. doi: 10.1093/cvr/cvp118
Chen, H. J., Ko, C. Y., Xu, J. H., Huang, Y. C., Wu, J. S. B., and Shen, S. C. (2021). Alleviative effect of Ruellia tuberosa L. on insulin resistance and abnormal lipid accumulation in TNF-α-treated FL83B mouse hepatocytes. Evid. Based Complement. Alternat. Med. 2021:9967910. doi: 10.1155/2021/9967910
Choi, H. R., Ha, J. S., Kim, I. S., and Yang, S. J. (2020). Metformin or α-lipoic acid attenuate inflammatory response and NLRP3 inflammasome in BV-2 microglial cells. Korean J. Clin. Lab. Sci. 52, 253–260. doi: 10.15324/kjcls.2020.52.3.253
Dobri, A. M., Dudău, M., Enciu, A. M., and Hinescu, M. E. (2021). CD36 in Alzheimer’s disease: an overview of molecular mechanisms and therapeutic targeting. Neuroscience 453, 301–311. doi: 10.1016/j.neuroscience.2020.11.003
Doens, D., Valiente, P. A., Mfuh, A. M., Vo, A. X. T., Tristan, A., Carreño, L., et al. (2017). Identification of inhibitors of CD36-amyloid beta binding as potential agents for Alzheimer’s disease. ACS Chem. Neurosci. 8, 1232–1241. doi: 10.1021/acschemneuro.6b00386
Dos Santos, S. M., Romeiro, C. F. R., Rodrigues, C. A., Cerqueira, A. R. L., and Monteiro, M. C. (2019). Mitochondrial dysfunction and Alpha-lipoic acid: beneficial or harmful in Alzheimer’s disease? Oxid. Med. Cell. Longev. 2019:8409329. doi: 10.1155/2019/8409329
El Khoury, J., Hickman, S. E., Thomas, C. A., Loike, J. D., and Silverstein, S. C. (1998). Microglia, scavenger receptors, and the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 19, S81–S84. doi: 10.1016/s0197-4580(98)00036-0
Erika, N. C., Ma’ıra, A. B., Kirsten, L. V., and William, L. K. (2018). The Amyloid-β oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis. 64, S567–S610. doi: 10.3233/JAD-179941
Farr, S. A., Price, T. O., Banks, W. A., Ercal, N., and Morley, J. E. (2012). Effect of alpha-lipoic acid on memory, oxidation, and lifespan in SAMP8 mice. J. Alzheimers Dis. 32, 447–455. doi: 10.3233/JAD-2012-120130
Fava, A., Pirritano, D., Plastino, M., Cristiano, D., Puccio, G., Colica, C., et al. (2013). The effect of lipoic acid therapy on cognitive functioning in patients with Alzheimer’s disease. J. Neurodegener. Dis. 2013:454253. doi: 10.1155/2013/454253
Golbidi, S., Badran, M., and Laher, I. (2011). Diabetes and alpha lipoic Acid. Front. Pharmacol. 2:69. doi: 10.3389/fphar.2011.00069
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hager, K., Kenklies, M., McAfoose, J., Engel, J., and Munch, G. (2007). Alpha-lipoic acid as a new treatment option for Alzheimer’s disease–a 48 months follow-up analysis. J. Neural Transm. Suppl. 72, 189–193. doi: 10.1007/978-3-211-73574-9_24
Holmquist, L., Stuchbury, G., Berbaum, K., Muscat, S., Young, S., Hager, K., et al. (2007). Lipoic acid as a novel treatment for Alzheimer’s disease and related dementias. Pharmacol. Ther. 113, 154–164. doi: 10.1016/j.pharmthera.2006.07.001
Hwang, J. S., An, J. M., Cho, H., Lee, S. H., Park, J. H., and Han, I. O. (2015). A dopamine-alpha-lipoic acid hybridization compound and its acetylated form inhibit LPS-mediated inflammation. Eur. J. Pharmacol. 746, 41–49. doi: 10.1016/j.ejphar.2014.10.052
Jack, C. R. Jr., Wiste, H. J., Therneau, T. M., Weigand, S. D., Knopman, D. S., Mielke, M. M., et al. (2019). Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA 321, 2316–2325. doi: 10.1001/jama.2019.7437
Kawahara, K., Suenobu, M., Yoshida, A., Koga, K., Hyodo, A., Ohtsuka, H., et al. (2012). Intracerebral microinjection of interleukin-4/interleukin-13 reduces beta-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 207, 243–260. doi: 10.1016/j.neuroscience.2012.01.049
Kim, S. M., Ha, J. S., Han, A. R., Cho, S. W., and Yang, S. J. (2019). Effects of α-lipoic acid on LPS-induced neuroinflammation and NLRP3 inflammasome activation through the regulation of BV-2 microglial cells activation. BMB Rep. 52, 613–618. doi: 10.5483/BMBRep.2019.52.10.026
Knopman, D. S., Amieva, H., Petersen, R. C., Chételat, G., Holtzman, D. M., Hyman, B. T., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Primers 7:33. doi: 10.1038/s41572-021-00269-y
Ko, C. Y., Lo, Y. M., Xu, J. H., Chang, W. C., Huang, D. W., Wu, J. S., et al. (2021). Alpha-lipoic acid alleviates NAFLD and triglyceride accumulation in liver via modulating hepatic NLRP3 inflammasome activation pathway in type 2 diabetic rats. Food Sci. Nutr. 9, 2733–2742. doi: 10.1002/fsn3.2235
Liu, Z., Li, T., Li, P., Wei, N., Zhao, Z., Liang, H., et al. (2015). The ambiguous relationship of oxidative stress, Tau hyperphosphorylation, and autophagy dysfunction in Alzheimer’s disease. Oxid. Med. Cell. Longev. 2015:352723. doi: 10.1155/2015/352723
Lu, D. Y., Tang, C. H., Liou, H. C., Teng, C. M., Jeng, K. C., Kuo, S. C., et al. (2007). YC-1 attenuates LPS-induced proinflammatory responses and activation of nuclear factor-kappaB in microglia. Br. J. Pharmacol. 151, 396–405. doi: 10.1038/sj.bjp.0707187
Maczurek, A., Hager, K., Kenklies, M., Sharman, M., Martins, R., Engel, J., et al. (2008). Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer’s disease. Adv. Drug Deliv. Rev. 60, 1463–1470. doi: 10.1016/j.addr.2008.04.015
Packer, L., Witt, E. H., and Tritschler, H. J. (1995). alpha-Lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 19, 227–250. doi: 10.1016/0891-5849(95)00017-r
Perra, A., Pibiri, M., Sulas, P., Simbula, G., Ledda-Columbano, G. M., and Columbano, A. (2008). Alpha-lipoic acid promotes the growth of rat hepatic pre-neoplastic lesions in the choline-deficient model. Carcinogenesis 29, 161–168. doi: 10.1093/carcin/bgm205
Pomilio, C., Gorojod, R. M., Riudavets, M., Vinuesa, A., Presa, J., Gregosa, A., et al. (2020). Microglial autophagy is impaired by prolonged exposure to β-amyloid peptides: evidence from experimental models and Alzheimer’s disease patients. Geroscience 42, 613–632. doi: 10.1007/s11357-020-00161-9
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Rai, S. N., Singh, C., Singh, A., Singh, M. P., and Singh, B. K. (2020). Mitochondrial dysfunction: a potential therapeutic target to treat Alzheimer’s disease. Mol. Neurobiol. 57, 3075–3088. doi: 10.1007/s12035-020-01945-y
Ramer, R., Heinemann, K., Merkord, J., Rohde, H., Salamon, A., Linnebacher, M., et al. (2013). COX-2 and PPAR-gamma confer cannabidiol-induced apoptosis of human lung cancer cells. Mol. Cancer Ther. 12, 69–82. doi: 10.1158/1535-7163.MCT-12-0335
Scher, J. U., and Pillinger, M. H. (2005). 15d-PGJ2: the anti-inflammatory prostaglandin? Clin. Immunol. 114, 100–109. doi: 10.1016/j.clim.2004.09.008
Shimizu, E., Kawahara, K., Kajizono, M., Sawada, M., and Nakayama, H. (2008). IL-4-induced selective clearance of oligomeric beta-amyloid peptide (1-42) by rat primary type 2 microglia. J. Immunol. 181, 6503–6513. doi: 10.4049/jimmunol.181.9.6503
Singh, A. K., Rai, S. N., Maurya, A., Mishra, G., Awasthi, R., Shakya, A., et al. (2021). Therapeutic potential of phytoconstituents in management of Alzheimer’s disease. Evid. Based Complement. Alternat. Med. 2021:5578574. doi: 10.1155/2021/5578574
Sokolowski, J. D., and Mandell, J. W. (2011). Phagocytic clearance in neurodegeneration. Am. J. Pathol. 178, 1416–1428. doi: 10.1016/j.ajpath.2010.12.051
Srivastava, P., Tripathi, P. N., Sharma, P., Rai, S. N., Singh, S. P., Srivastava, R. K., et al. (2019). Design and development of some phenyl benzoxazole derivatives as a potent acetylcholinesterase inhibitor with antioxidant property to enhance learning and memory. Eur. J. Med. Chem. 163, 116–135. doi: 10.1016/j.ejmech.2018.11.049
Stewart, C. R., Stuart, L. M., Wilkinson, K., van Gils, J. M., Deng, J., Halle, A., et al. (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161. doi: 10.1038/ni.1836
Su, B., Wang, X., Nunomura, A., Moreira, P. I., Lee, H. G., Perry, G., et al. (2008). Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 5, 525–532. doi: 10.2174/156720508786898451
Thirunavukkarasu, V., Anitha Nandhini, A. T., and Anuradha, C. V. (2004). Lipoic acid attenuates hypertension and improves insulin sensitivity, kallikrein activity and nitrite levels in high fructose-fed rats. J. Comp. Physiol. B 174, 587–592. doi: 10.1007/s00360-004-0447-z
Tripathi, P. N., Srivastava, P., Sharma, P., Tripathi, M. K., Seth, A., Tripathi, A., et al. (2019). Biphenyl-3-oxo-1,2,4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory. Bioorg. Chem. 85, 82–96. doi: 10.1016/j.bioorg.2018.12.017
Ulivi, V., Lenti, M., Gentili, C., Marcolongo, G., Cancedda, R., and Descalzi Cancedda, F. (2011). Anti-inflammatory activity of monogalactosyldiacylglycerol in human articular cartilage in vitro: activation of an anti-inflammatory cyclooxygenase-2 (COX-2) pathway. Arthritis Res. Ther. 13:R92. doi: 10.1186/ar3367
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wang, K. C., Tsai, C. P., Lee, C. L., Chen, S. Y., Lin, G. J., Yen, M. H., et al. (2013). α-Lipoic acid enhances endogenous peroxisome-proliferator-activated receptor-gamma to ameliorate experimental autoimmune encephalomyelitis in mice. Clin. Sci. 125, 329–340. doi: 10.1042/CS20120560
Wang, Q., Lv, C., Sun, Y., Han, X., Wang, S., Mao, Z., et al. (2018). The role of alpha-lipoic acid in the pathomechanism of acute ischemic stroke. Cell. Physiol. Biochem. 48, 42–53. doi: 10.1159/000491661
Yamanaka, M., Ishikawa, T., Griep, A., Axt, D., Kummer, M. P., and Heneka, M. T. (2012). PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 32, 17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., et al. (1996). RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691. doi: 10.1038/382685a0
Keywords: α-lipoic acid, microglia, oligomeric beta-amyloid, phagocytosis, Alzheimer’s disease (AD)
Citation: Ko C-Y, Xu J-H, Chang Y-W, Lo YM, Wu JS-B, Huang W-C and Shen S-C (2022) Effects of α-Lipoic Acid on Phagocytosis of Oligomeric Beta-Amyloid1–42 in BV-2 Mouse Microglial Cells. Front. Aging Neurosci. 13:788723. doi: 10.3389/fnagi.2021.788723
Received: 03 October 2021; Accepted: 15 December 2021;
Published: 12 January 2022.
Edited by:
Woon-Man Kung, Chinese Culture University, TaiwanReviewed by:
Sachchida Nand Rai, University of Allahabad, IndiaDouglas F. Watt, Lesley University, United States
Nobuyuki Kimura, National Center for Geriatrics and Gerontology (NCGG), Japan
Copyright © 2022 Ko, Xu, Chang, Lo, Wu, Huang and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Szu-Chuan Shen, c2NzQG50bnUuZWR1LnR3
†These authors share first authorship
‡ORCID: Chih-Yuan Ko, orcid.org/0000-0002-5767-3041; James Swi-Bea Wu, orcid.org/0000-0002-2855-5716; Szu-Chuan Shen, orcid.org/0000-0001-9005-0455