 Rodrigo Diaz-Espinoza
Rodrigo Diaz-Espinoza- Departamento de Biología, Facultad de Química y Biología, Universidad de Santiago de Chile, Santiago, Chile
Amyloids are highly ordered aggregates composed of proteins or peptides. They are involved in several pathologies, including hallmark neurodegenerative disorders such as Alzheimer’s (AD) and Parkinson’s (PD). Individuals affected by these diseases accumulate in their brains amyloids inclusions composed of misfolded forms of a peptide (Aβ) and a protein (Tau) in AD and α-synuclein protein (α-Sn) in PD. Tau and α-Sn aggregates are also present in other neurodegenerative diseases. The insoluble nature and heterogeneity of amyloids have hampered their study at the molecular level. However, the use of solid state NMR and Cryogenic-electron microscopy along with fine-tuned modulation of the aggregation in vitro and improved isolation methods of brain-derived amyloids has allowed the elucidation of these elusive conformations at high resolution. In this work, we review the latest progress on the recent amyloid structures reported for Aβ, Tau, and α-Sn. The two-fold symmetry emerges as a convergent feature in the tridimensional arrangement of the protofilaments in the fibrillary structure of these pathological amyloids, with many of them exhibiting a Greek-key topology as part of their overall architecture. These specific features can serve as novel guides to seek potential molecular targets in drug design efforts.
Introduction
The amyloid state of proteins is nowadays recognized as a convergent conformation that is accessible to most proteins and peptides under the appropriate conditions (Hartl and Hayer-Hartl, 2009). However, the proteinaceous nature of amyloids was recognized decades after their original descriptions (Kyle, 2001). Since then, amyloid deposits have been identified in different human pathologies and also in many organisms as functional assemblies (Chiti and Dobson, 2017). These aggregates are relevant pathological agents in neurodegenerative disorders such as AD and PD where they accumulate in the brain as inclusions with disease-dependent morphological features and location (Terry, 1963; Spillantini et al., 1997; Selkoe and Hardy, 2016). In AD, the affected individuals have two types of amyloid deposits that are composed of aggregated Aβ or misfolded Tau, whereas in PD misfolded α-Sn forms intracellular aggregates. Their specific pathological roles constitute active areas of research. However, their peculiar structural features have imposed important challenges to their study at the molecular level. Since these are unique pathological agents, elucidating their structures is crucial to design novel therapeutic approaches. We will review here the three iconic amyloids involved in neurodegenerative diseases along with their recently solved high-resolution structures.
Overview of Amyloid Structure
Amyloids are highly ordered aggregates stabilized by a beta-sheet core (Greenwald and Riek, 2010). Misfolded proteins or peptides are arranged in the beta-sheet in an intermolecular fashion through a hydrogen-bonded network in which the strands are transversally disposed with respect to the growing axis of the amyloid fibril, creating an overall “cross-beta” pattern (Riek, 2017). Beta-sheets can be arranged in a parallel or anti-parallel manner. Interestingly, regardless of the specific sequence all amyloids exhibit a similar overall architecture. This is indicative of a convergent misfolding pathway (Hartl and Hayer-Hartl, 2009). Moreover, amyloids are formed by the self-assembly of not only proteins but also small peptides and even single aromatic amino acids, indicating that the amyloid fold may be structurally and universally encrypted in the hydrocarbon chain of polypeptides (Balbach et al., 2000; Dobson, 2004; Adler-Abramovich et al., 2012). Still, the type and ordering of the amino acids within a given sequence is recognized to have a significant role in the aggregation propensity, with hydrophobic residues being the main contributors to the stability of the amyloid fold (Gazit, 2002; Hall et al., 2005; Greenwald and Riek, 2010). Despite their structural convergence, amyloids can still exhibit structural diversity at the molecular level (Gallardo et al., 2020). Such variations can be dependent on the specific polypeptide sequence as well as on the size.
Although amyloids are in general very stable, the non-covalent nature of the intermolecular interactions makes amyloid fibrils relatively fragile, which can fragment into smaller fibrils when subjected to mechanical or chemical stresses (Xue et al., 2010). Therefore, amyloids suffer from an inherent heterogeneity with respect to size distribution (Morales et al., 2016; Eisenberg and Sawaya, 2017). Such heterogeneity and their often-low solubility has made their structural characterization extremely challenging using classical structural biology techniques, let alone high-resolution approaches (Diaz-Espinoza and Soto, 2012; Fändrich et al., 2018). The first high-resolution amyloid structures were achieved using small peptides that formed amyloid-like structures when subjected to controlled dehydration (Nelson et al., 2005; Sawaya et al., 2007). The fine-tuning of experimental approaches aimed at increasing the homogeneity of the samples as well as implementing and combining novel and refined techniques such as sold-state NMR (ssNMR) and Cryogenic-Electron Microscopy (Cryo-EM) have allowed to obtain high-resolution structures of several amyloids, including those formed by Aβ, Tau, and α-Sn.
Aβ
Aβ are peptides of varying sizes released upon proteolytic cleavage of a membrane protein (amyloid precursor protein) (Kang et al., 1987). Some of these peptides can aggregate into fibrillary species, which then accumulates as plaques in the brain (Glenner and Wong, 1984; Selkoe and Hardy, 2016). These plaques typically contain Aβ fragments 1–40 (Aβ40) and 1–42 (Aβ42) among other components (Roher et al., 1993). Cellular toxicity associated to misfolded Aβ proceeds through diverse mechanisms including microglia-mediated localized inflammation around the plaques, impaired synapsis, membrane disruptions induced by Aβ oligomeric species, metals imbalance, etc (Cavallucci et al., 2012). Moreover, Aβ fibrils and oligomers (Aβ40 and Aβ42) prepared in vitro using chemically synthesized or recombinant Aβ peptide display dose-dependent cytotoxicity in cell cultures of diverse neuronal lines (Zagorski et al., 1999; Finder et al., 2010). The in vitro assembly of synthetic Aβ into amyloids has been characterized extensively in the literature and has enabled access to molecular details of Aβ aggregation, which may partially resemble the in vivo mechanisms during plaque formation (Caughey and Lansbury, 2003). Aβ42 is more abundant in the plaques than Aβ40 and hence it has been studied in greater detailed (Roher et al., 1993). The assembly into amyloids in vitro from monomeric Aβ42 is a sequential nucleation-polymerization (NP) process that involves the emergence of nucleating oligomers followed by the formation of small fibrillary species or proto-fibrils, which then associate to form mature fibrils (Hardy and Higgins, 1992; Chen et al., 2017). The plaques in AD contain mature-like Aβ42 fibrils and hence these fibrils can serve as an in vitro model for their study. Aβ42 fibrils subjected to mechanical disruption can give rise to small fragments that can act as seeds of Aβ42 polymerization, accelerating the formation of mature fibrils. This seeding mechanism along with the NP process is a general trait of amyloids (Jarrett and Lansbury, 1993; Ke et al., 2020).
Two recent works have produced the first structures of full length Aβ42 in the amyloid state and at atomic resolution using ssNMR combined with molecular dynamics (MD; Colvin et al., 2016; Wälti et al., 2016). Wälti et al. (2016) showed that serially seeding the in vitro aggregation of synthetic Aβ42 along with fine-tuning the solution conditions can produce highly homogenous Aβ42 fibrils, with the resulting fibrils being recognized by plaque-specific antibodies. Colvin et al. (2016) grew Aβ42 fibrils overnight at cooler temperatures without agitation to produce the homogenous fibrils. Interestingly, both approaches yielded very similar structures (Figure 1, pdb codes 2NAO and 5KK3). The amyloids are arranged by a two-fold symmetry, in which two protofilaments are stabilized mainly by lateral contacts. Each Aβ42 monomer is stacked in a parallel, in-register fashion along the fibril axis. In both models the Aβ42 N-terminal region (1–14) does not make specific contacts with the amyloid core probably due to its flexible and less hydrophobic nature. In a later work, Gremer et al. (2017) combined data from ssNMR with Cryo-EM to provide a novel structure for Aβ42 in a fibrillary state. Homogenous fibrils were obtained by growing Aβ42 at low pH in a solution containing a small fraction of acetonitrile and trifluoroacetic acid. The resulting fibrils were similarly toxic to PC12 rat brain cells as fibrils grown at neutral pH. Considering the fibrillary nature of this structure, the resulting model exhibited several significant differences with the previous models (Figure 1, pdb code 5OQV). First, the N-terminal region (1–15) appeared as part of the amyloid arrangement and engaged in specific contacts including intermolecular (1Asp-28Lys) and intramolecular salt bridges (5Arg-7Asp, 6His-13His-11Glu). Secondly, the C-terminal region (38–42) makes direct intermolecular, inter-subunit hydrophobic contacts that appears as key in stabilizing the two-fold symmetry, whereas in the previous models these residues extend outward the amyloid core. Interestingly, Aβ40 lacks the last two of these C-terminal residues and peptides containing the full C-terminal region (sequence GGVVIA) spontaneously self-assembly into amyloids in vitro, which may suggest that this fibril model may be capturing a key feature of plaque-like Aβ42 fibrils (Sawaya et al., 2007). Despite these differences, all the structures show a left-handed twist with a general two-fold symmetry formed by two interacting protofilaments.
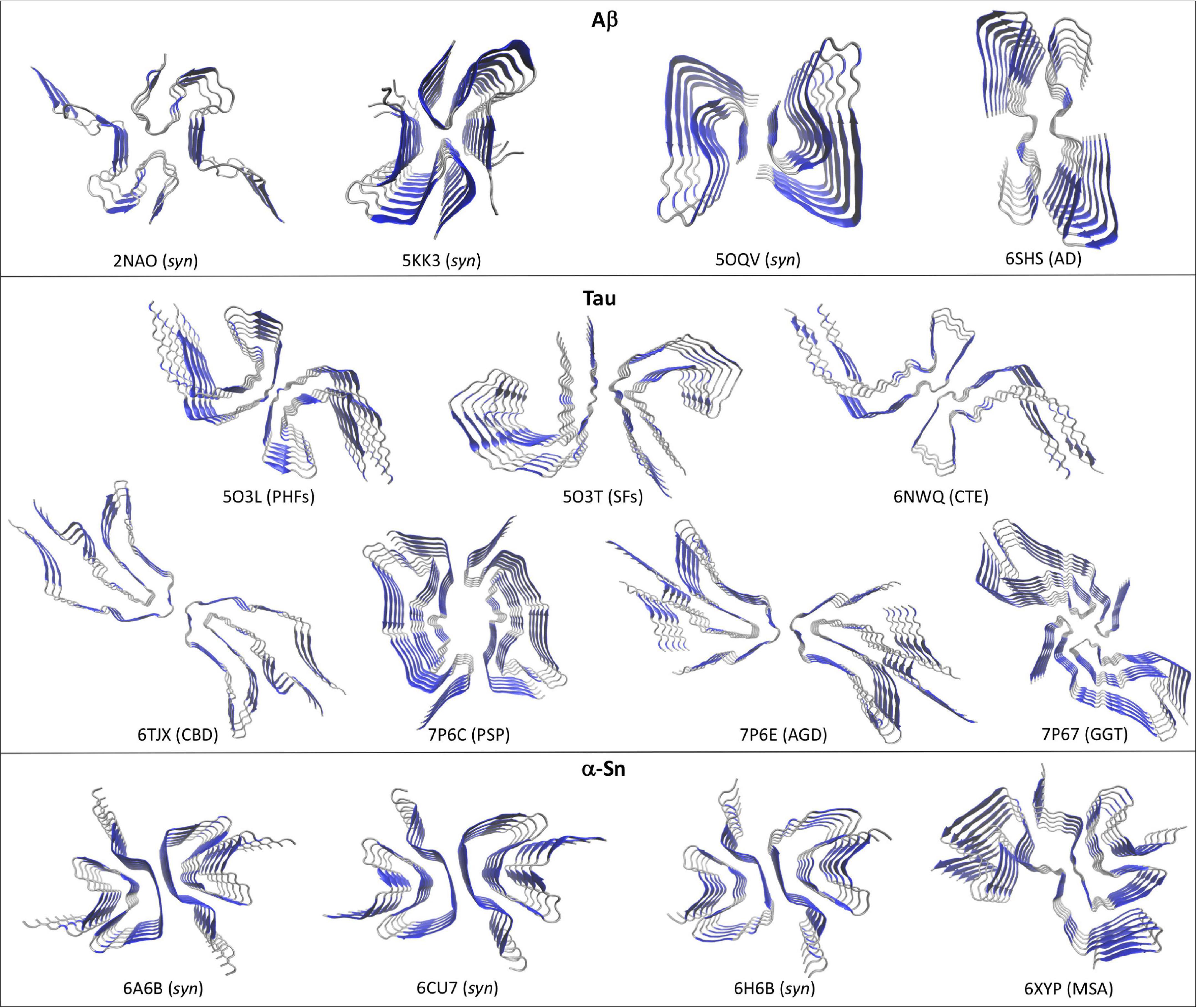
Figure 1. Different high-resolution structures of Aβ, Tau, and α-Sn in the amyloid state. Top views are shown to highlight the two-fold symmetry arrangement of the protofilaments. Aβ: 2NAO, 5KK3, and 5OQV are synthetic Aβ42 fibrils (syn); 6SHS are brain-derived Type II Aβ40 fibrils from AD. Tau: 5O3L and 5O3T are respectively, ex vivo PHFs and SFs from AD; 6NWQ, 6TJX, 7P6C, 7P6E, and 7P67 are ex vivo Type II filaments from CTE, CBD, PSP, AGD, and GGT, respectively. α-Sn: 6A6B, 6CU7, and 6H6B are synthetic fibrils (syn); 6XYP are brain-derived Type I fibrils from MSA. Beta-sheets are depicted in blue and coils and α-helices in silver.
Although high-resolution structures of full length Aβ42 fibrils isolated from diseased brains (ex vivo) are not yet available, fibrillary structures of brain-derived Aβ40 fibrils from an AD-affected patient were recently solved using Cryo-EM (Kollmer et al., 2019). These structures differed significantly from those obtained with synthetic Aβ42 fibrils in that they showed a right-handed twist, different stabilizing hydrophobic contacts and different arrangements of the protofilaments. A single protofilament fibrillary arrangement was dominant in the sample (Type I), in which two stacks of Aβ40 peptides are stabilized by hydrophobic contacts and arranged symmetrically. Additional fibrils were also observed in which two (Type II) or three (Type III) Type I protofilaments interact through lateral contacts mediated by two salt bridges (3Glu-5Arg), with Type II filaments showing a two-fold symmetry (Figure 1, pdb code 6H6S). It remains to be seen whether these structural features are exclusive of ex vivo Aβ40 fibrils or they rather represent a convergent trait of brain-derived Aβ fibrils.
Tau
In addition to Aβ42 plaques, AD-affected brains can also contain intracellular inclusions composed of misfolded Tau protein (Terry, 1963; Lee et al., 1991). These Tau filaments give rise to neurofibrillary lesions, which can accumulate in different areas of the affected brains, including neocortex and limbic system, among others. In the case of AD, two types of Tau inclusions are commonly: paired helical filaments (PHFs) and straight filaments (SFs; Kidd, 1963; Crowther, 1991). Tau-associated inclusions are not an exclusive feature of AD and similar depositions can be found in other neurodegenerative disorders such as Pick’s disease (PiD), chronic traumatic encephalopathy (CTE), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), argyrophilic grain disease (AGD) and globular glial tauopathy (GGT), among others (Crowther and Goedert, 2000; Ghetti et al., 2015).
Contrary to Aβ, Tau is a constitutively expressed protein involved in cytoskeleton homeostasis through direct stabilizing interactions with microtubules (Desai and Mitchison, 1997). At the structural level, Tau is an intrinsically disorder protein that can be found in six isoforms with sequence lengths varying from 352 to 441 amino acids (Goedert et al., 1989; Schweers et al., 1994). The details of Tau aggregation in vivo are intricate and different mechanisms have been proposed to contribute, including post-translational modifications such as hyperphosphorylation, glycosylation and truncation, ligand interactions, dimerization events, etc (Goedert et al., 1992; Alquezar et al., 2020). Such complexity has been very difficult to recreate in vitro and hence synthetic Tau fibrils that fully mimic PHFs and SFs is an ongoing challenge. However, alternative approaches that rely on isolating Tau amyloid inclusions at high homogeneity from diseased brains has proven a valuable tool for structural studies. In fact, high resolution structures of Tau PHFs and SFs isolated from an AD-affected brain have been recently solved using Cryo-EM (Fitzpatrick et al., 2017). Fitzpatrick et al. (2017) isolated and purified sarkosyl-insoluble fractions that were enriched in PHFs and SFs, which were then used to obtain high resolution structures of both filaments (Figure 1). Both PHFs and SFs showed a two-fold symmetry similar to that observed with in vitro-prepared Aβ42 fibrils, in which Tau monomers are stacked through parallel beta-sheets with each molecule containing eight beta strands alternated with loop regions, forming two protofilaments running along the fibril axis. Interestingly, each Tau protofilament is arranged in a clear Greek-like key pattern (Figure 2). Compared to Aβ, only one of all the solved Aβ fibril structures showed this topology and with a much less organized fashion. Although Tau is not particularly hydrophobic as Aβ42, the main intramolecular hydrophobic contacts reside precisely at the end of the end hinge region of the Greek-key fold, which may help stabilize this particular shape. Intermolecular contacts between the two protofilaments in PHFs involve H-bonds among residues 332PGGGQ336 in an anti-parallel fashion. SFs intermolecular interactions are more asymmetrical, with contacts between region 321KCGS324 and 313VDLSK317, though the specific types of stabilizing interactions are not yet clear. Using a similar approach the same group have elucidated high resolution structures of brain-derived Tau amyloids from patients affected with PiD, CTE, CBD, PSP, GPT, or GGT (Falcon et al., 2018, 2019; Zhang et al., 2020; Shi et al., 2021). As observed with brain-derived Aβ40 amyloids, heterogeneous arrangements of the protofilaments were evident in several of the structures, particularly in those from CBD, CTE, PSP, and GGT. Still, the two-fold symmetry emerged again as a common feature in Type I and II filaments in CTE, and Type II filaments from CBD, PSP, AGD, and GGT, whereas single protofilament arrangements included Tau from PiD, PSP and Type I filaments from CBD, PSP, AGD, and GGT (Figure 1). Regardless of the disease, most Tau filaments have the Greek-key topology as a general feature, including Type I and II filaments from CTE, CBD, PSP, and AGD and in a less organized fashion the single protofilaments from PSP and Type I and II filaments from GGT (Figure 2).
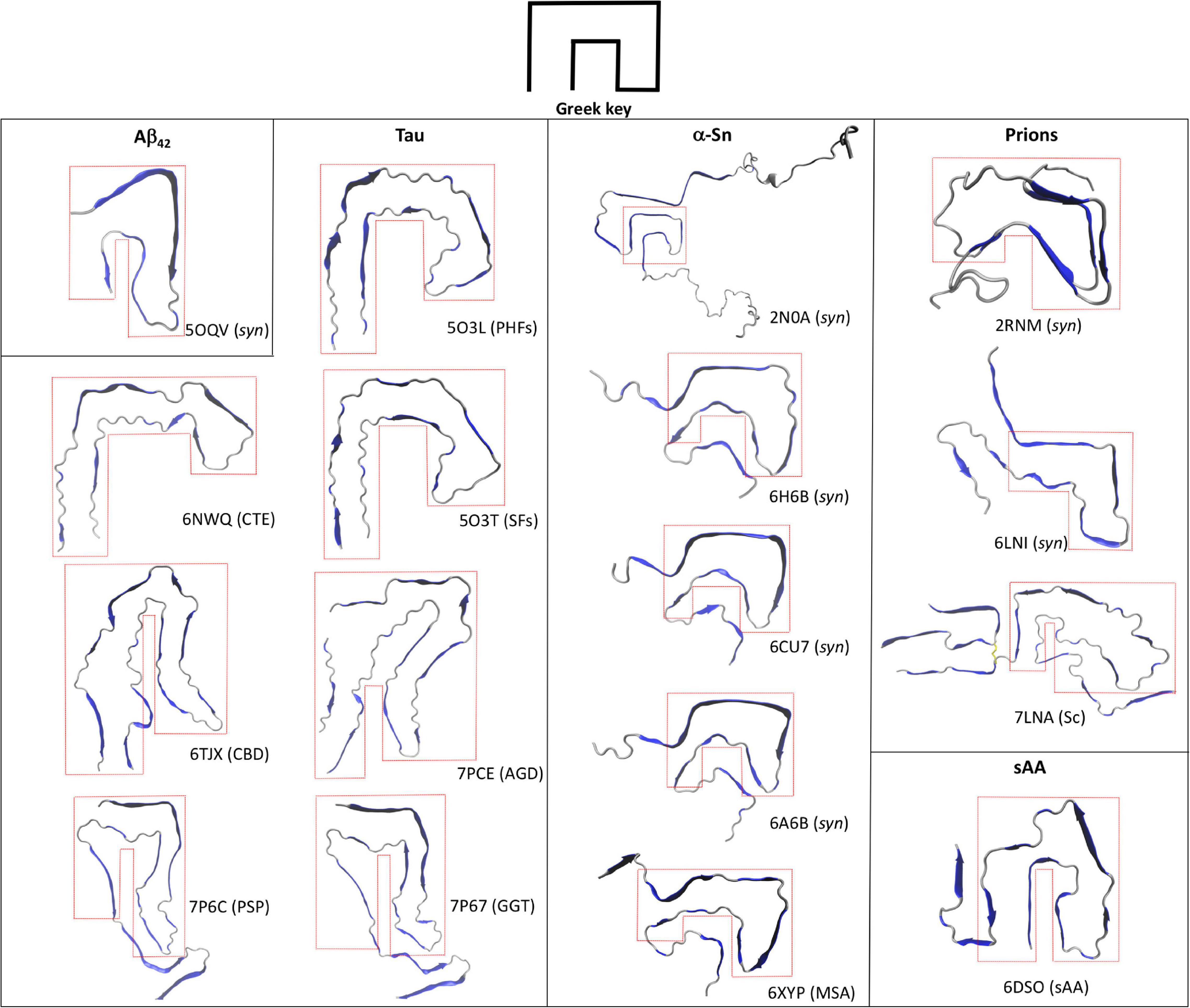
Figure 2. Greek-key topology in the amyloid fold. Top views of the single chains of several amyloid structures of Aβ42, Tau, and α-Sn are shown along with their associated disease or synthetic nature (syn). Synthetic HET prion (2RNM), human recombinant prion protein in the amyloid state (6LNI), infectious hamster brain-derived prion protein (7LNA), and mouse spleen-derived acute phase protein serum amyloid A1 (6DSO) are depicted for comparison. The enclosing red dotted lines highlight the Greek-key topology in each structure.
α-Sn
One of the hallmark and specific pathological features of PD is the formation of intracellular depositions called Lewy bodies (LB) and Lewy neurites (LN) in nerve cells (Yves, 1991). These inclusions are mainly composed of an abnormally folded α-Sn protein in a fibrillary state (Spillantini et al., 1998b) and hence the intracellular accumulation of fibrillary α-Sn is recognized as a pathological condition. Fibrils prepared in vitro using recombinant α-Sn can seed the formation of LB- and LN-like inclusions in cell culture models, reinforcing the role of α-Sn in PD (Luk et al., 2009; Volpicelli-Daley et al., 2011). Furthermore, intracerebral inoculation of these synthetic fibrils in wild-type mice can not only seed the aggregation of native brain α-Sn but also initiate a PD-like pathology in the mice (Luk et al., 2012). Therefore, these synthetic fibrils have served as important models for molecular studies.
Several high-resolution structures of synthetic α-Sn fibrils have been reported in the last years. Tuttle et al. (2016) prepared in vitro recombinant α-Sn fibrils (full length) that seeded in a dose-dependent manner the aggregation of α-Sn in primary hippocampal neuronal cells and also produced toxicity to the same cells at high protein concentrations. A high-resolution structure of a single α-Sn protofilament was then obtained using ssNMR, electron and X-ray fiber diffraction (Figure 1, pdb code 2NAO). Based on this structure, the amyloid Greek-key pattern was proposed for the first time (Figure 2). Interestingly, for α-Sn this topology exhibits specific intramolecular interactions that appear as poised for its overall stabilization, including a combination of hydrophobic contacts (88Ile-90Ala-94Phe), a salt bridge (46Glu-80Lys), and a H-bond (79Gln-90QAla). In a later work, Guerrero-Ferreira et al. (2018) reported the high-resolution structure of α-Sn in a fibrillary state by using Cryo-EM (Figure 1, pdb code 6H6B). They showed that a truncated recombinant α-Sn (residues 1–121) was the only preparation to yield homogenous fibrils upon self-assembly in vitro, as confirmed by EM. This truncated α-Sn is known to be highly prone to form amyloids in vitro and has been observed to naturally emerge in cells (Crowther et al., 1998; Li et al., 2005). The protofilaments are arranged in a two-fold symmetry (Figure 1). As observed with Tau filaments, α-Sn monomers are stacked onto the amyloid fold with eight beta strands alternated with loops in each of the protofilaments. The interface between both protofilaments is dominated by intermolecular hydrophobic interactions (53Ala-53Ala and 55Val-51Gly) and salt bridges (50His-57Glu). From the six familial mutations described for α-Sn, four occur directly in this interface, which suggest that the two-fold symmetry may have a key role in pathogenesis. The Greek-key pattern is again evident in the overall structure (Figure 2). Although the final Cryo-EM model was built using the structure from Tuttle et al. (2016) major refinements were needed for the protofilaments structure, which is indicative of structural differences. The corresponding interacting strands at the interface are positioned differently in the parent model since only a single protofilament was solved. Moreover, several key residues of the interface such as 53Ala are positioned inward. Two additional Cryo-EM structures for α-Sn fibrils were also reported in the same year (Figure 1, pdb codes 6A6B and 6CU7). These structures were obtained using full length recombinant α-Sn that upon in vitro aggregation was able to seed the misfolding of endogenous α-Sn in rat primary neurons (Li Y. et al., 2018) or HEK 293T cells (Li B. et al., 2018). Both preparations were toxic to different cell lines in a dose-dependent manner. The solved structures were very similar to that reported by Guerrero-Ferreira et al. (2018) showing again a Greek-key pattern (Figure 2). The 53Ala-53Ala interaction appears again as key for the stability of the protofilaments interface. Interestingly, an intramolecular hydrophobic contact (70Val-66Val) within the hinge of the Greek key is present in all of the reported structures, suggesting this interaction may be pivotal for stabilizing the topology. In a later work, Ni et al. (2019) reported several Cryo-EM structures using recombinant α-Sn with different truncations. The Greek-key topology was again evident in all of the structures, showing that this pattern is still maintained upon α-Sn truncation. The fact that all these synthetic fibrils exhibited similar overall structures is interesting, with a very similar overall two-fold symmetry, Greek-key topology and interface interactions.
Misfolded α-Sn forms are also present in other neurodegenerative diseases such as multiple system atrophy (MSA; Spillantini et al., 1998a). Schweighauser et al. (2020) recently reported the first high-resolution structure of brain-derived α-Sn fibrils using Cryo-EM, from a patient affected by MSA. As observed with ex vivo fibrillary structures of Aβ40 and Tau, the α-Sn protofilaments are arranged in different fashions that in this case was also patient-dependent. The described filament types (I and II) showed a very similar two-protofilament overall arrangement but organized without full symmetry (Figure 1, pdb code 6XYP). Despite the low two-fold symmetry, the Greek-key topology of each protofilament is in general remarkably similar to those observed with synthetic α-Sn fibrils (Figure 2).
Conclusion
Advances in state-of-the-art experimental techniques such as ssNMR and Cryo-EM have enabled access to the molecular details of different amyloid assemblies. By using these techniques in combination with reducing size and conformational heterogeneity of the aggregates as well as improving isolation methods of brain-derived amyloids, solving high-resolution structures of amyloids formed by synthetic protein or ex vivo material of Aβ42, Tau, and α-Sn has became feasible. Analysis of the structures shows that at the molecular level, amyloid structures can be heterogeneous, with a single primary sequence yielding different conformations and/or protofilaments arrangements in a disease-, sample- and in some cases patient-dependent manner. Still, the fibrillary states of these proteins show that two interacting protofilaments appear as a common amyloid arrangement emerging beyond the classical cross-beta pattern. In addition, the Greek-key topology can be observed in several of the structures though this is not as a convergent feature as the two-fold symmetry. In most of the structures reviewed, the hinge of this topology appears as the minimal and ubiquitous feature, which is stabilized mainly by hydrophobic interactions. Greek-key motifs are common in proteins that have beta-barrels and beta-sandwiches, which suggests that beta-sheets are a requirement for this fold (Zhang and Kim, 2000). Interestingly, high-resolution structures of amyloids prepared with synthetic prion proteins also exhibit architectures that resemble a Greek key (Figure 2; Wasmer et al., 2008; Wang et al., 2020). Furthermore, the same topology is described in a recently reported Cryo-EM structure of a mammalian prion isolated directly from scrapie-infected (Sc) brains and can be also observed in the amyloid fibril structure of acute phase protein serum amyloid A1 involved in systemic amyloidosis (sAA; Liberta et al., 2019; Kraus et al., 2021). Still, the fact that this topology is not observed in all of the samples serves as evidence of the different amyloid conformations that even a single primary sequence can reach. Contrary to the less complex and more homogeneous amyloid structures obtained with small peptides, all these unique structural arrangements may be only accessible for proteins or large peptides, which by combining beta strands with flexible loop-like regions can then reach more intricate amyloid conformations. Rational design of drugs aimed at disrupting the protofilament interface and/or the Greek-key fold may hold promise for novel therapeutic approaches. For a given protein, however, interactions stabilizing these arrangements show great variability depending not only on the specific disease but also on the type of sample utilized (ex vivo fibrils, synthetic fibrils, full-length, or different fragments, etc.). Therefore, each of these models can serve as potential targets in drug design efforts for exploring and validating their biological relevance in each pathology.
Author Contributions
RD-E conceived, wrote, revised, and approved the manuscript.
Funding
This work was funded by research grant ANID FONDECYT 1211821 and Dicyt-Usach program.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adler-Abramovich, L., Vaks, L., Carny, O., Trudler, D., Magno, A., Caflisch, A., et al. (2012). Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 8, 701–706. doi: 10.1038/nchembio.1002
Alquezar, C., Arya, S., and Kao, A. W. (2020). Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front. Neurol. 11:595532. doi: 10.3389/fneur.2020.595532
Balbach, J. J., Ishii, Y., Antzutkin, O. N., Leapman, R. D., Rizzo, N. W., Dyda, F., et al. (2000). Amyloid fibril formation by Aβ 16–22, a seven-residue fragment of the Alzheimer’s β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry 39, 13748–13759. doi: 10.1021/bi0011330
Caughey, B., and Lansbury, P. T. (2003). Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298. doi: 10.1146/annurev.neuro.26.010302.081142
Cavallucci, V., D’Amelio, M., and Cecconi, F. (2012). Aβ toxicity in Alzheimer’s disease. Mol. Neurobiol. 45, 366–378. doi: 10.1007/s12035-012-8251-3
Chen, G. F., Xu, T. H., Yan, Y., Zhou, Y. R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Chiti, F., and Dobson, C. M. (2017). Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. doi: 10.1146/annurev-biochem-061516-045115
Colvin, M. T., Silvers, R., Ni, Q. Z., Can, T. V., Sergeyev, I., Rosay, M., et al. (2016). Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 138, 9663–9674. doi: 10.1021/jacs.6b05129
Crowther, R. A. (1991). Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc. Natl. Acad. Sci. U.S.A. 88, 2288–2292. doi: 10.1073/pnas.88.6.2288
Crowther, R. A., and Goedert, M. (2000). Abnormal tau-containing filaments in neurodegenerative diseases. J. Struct. Biol. 30, 271–279. doi: 10.1006/jsbi.2000.4270
Crowther, R. A., Jakes, R., Spillantini, M. G., and Goedert, M. (1998). Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 436, 309–312. doi: 10.1016/S0014-5793(98)01146-6
Desai, A., and Mitchison, T. J. (1997). Microtubule polymerization dynamics. Annu. Rev. Cell. Dev. Biol. 13, 83–117. doi: 10.1146/annurev.cellbio.13.1.83
Diaz-Espinoza, R., and Soto, C. (2012). High-resolution structure of infectious prion protein: the final frontier. Nat. Struct. Mol. Biol. 19, 370–377. doi: 10.1038/nsmb.2266
Dobson, C. M. (2004). Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol. 15, 3–16. doi: 10.1016/j.semcdb.2003.12.008
Eisenberg, D. S., and Sawaya, M. R. (2017). Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 86, 69–95. doi: 10.1146/annurev-biochem-061516-045104
Falcon, B., Zhang, W., Murzin, A. G., Murshudov, G., Garringer, H. J., Vidal, R., et al. (2018). Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140. doi: 10.1038/s41586-018-0454-y
Falcon, B., Zivanov, J., Zhang, W., Murzin, A. G., Garringer, H. J., Vidal, R., et al. (2019). Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568, 420–423. doi: 10.1038/s41586-019-1026-5
Fändrich, M., Nyström, S., Nilsson, K. P. R., Böckmann, A., LeVine, H., and Hammarström, P. (2018). Amyloid fibril polymorphism: a challenge for molecular imaging and therapy. J. Intern. Med. 283, 218–237.
Finder, V. H., Vodopivec, I., Nitsch, R. M., and Glockshuber, R. (2010). The recombinant amyloid-beta peptide Abeta1-42 aggregates faster and is more neurotoxic than synthetic Abeta1-42. J. Mol. Biol. 396, 9–18. doi: 10.1016/j.jmb.2009.12.016
Fitzpatrick, A. W. P., Falcon, B., He, S., Murzin, A. G., Murshudov, G., Garringer, H. J., et al. (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. doi: 10.1038/nature23002
Gallardo, R., Ranson, N. A., and Radford, S. E. (2020). Amyloid structures: much more than just a cross-β fold. Curr. Opin. Struct. Biol. 60, 7–16. doi: 10.1016/j.sbi.2019.09.001
Gazit, E. (2002). A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 16, 77–83. doi: 10.1096/fj.01-0442hyp
Ghetti, B., Oblak, A. L., Boeve, B. F., Johnson, K. A., Dickerson, B. C., and Goedert, M. (2015). Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46. doi: 10.1111/nan.12213
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135. doi: 10.1016/0006-291x(84)91209-9
Goedert, M., Spillantini, M. G., Cairns, N. J., and Crowther, R. A. (1992). Tauproteinsof Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8, 159–168. doi: 10.1016/0896-6273(92)90117-v
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526. doi: 10.1016/0896-6273(89)90210-9
Greenwald, J., and Riek, R. (2010). Biology of amyloid: structure, function, and regulation. Structure 18, 1244–1260. doi: 10.1016/j.str.2010.08.009
Gremer, L., Schölzel, D., Schenk, C., Reinartz, E., Labahn, J., Ravelli, R. B. G., et al. (2017). Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science 358, 116–119. doi: 10.1126/science.aao2825
Guerrero-Ferreira, R., Taylor, N. M., Mona, D., Ringler, P., Lauer, M. E., Riek, R., et al. (2018). Cryo-EM structure of alpha-synuclein fibrils. Elife 7:e36402. doi: 10.7554/eLife.36402
Hall, D., Hirota, N., and Dobson, C. M. (2005). A toy model for predicting the rate of amyloid formation from unfolded protein. J. Mol. Biol. 351, 195–205. doi: 10.1016/j.jmb.2005.05.013
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hartl, F. U., and Hayer-Hartl, M. (2009). Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 6, 574–581. doi: 10.1038/nsmb.1591
Jarrett, J. T., and Lansbury, P. T. Jr. (1993). Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73, 1055–1058. doi: 10.1016/0092-8674(93)90635-4
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., et al. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Ke, P. C., Zhou, R., Serpell, L. C., Riek, R., Knowles, T. P. J., Lashuel, H. A., et al. (2020). Half a century of amyloids: past, present and future. Chem. Soc. Rev. 49, 5473–5509. doi: 10.1039/c9cs00199a
Kidd, M. (1963). Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 197, 192–193. doi: 10.1038/197192b0
Kollmer, K., Close, W., Funk, L., Rasmussen, J., Bsoul, A., Schierhorn, A., et al. (2019). Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 10:4760. doi: 10.1038/s41467-019-12683-8
Kraus, A., Hoyt, F., Schwartz, C. L., Hansen, B., Artikis, E., Hughson, A. G., et al. (2021). High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 81, 4540–4551. doi: 10.1016/j.molcel.2021.08.011
Kyle, R. A. (2001). Amyloidosis: a convoluted story. Br. J. Haematol. 3, 529–538. doi: 10.1046/j.1365-2141.2001.02999.x
Lee, V. M., Balin, B. J., Otvos, L. Jr., and Trojanowski, J. Q. (1991). A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678. doi: 10.1126/science.1899488
Li, B., Ge, P., Murray, K. A., Sheth, P., Zhang, M., Nair, G., et al. (2018). Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 9:3609. doi: 10.1038/s41467-018-05971-2
Li, W., West, N., Colla, E., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2005). Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 2162–2167. doi: 10.1073/pnas.0406976102
Li, Y., Zhao, C., Luo, F., Liu, Z., Gui, X., Luo, Z., et al. (2018). Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell. Res. 28, 897–903. doi: 10.1038/s41422-018-0075-x
Liberta, F., Loerch, S., Rennegarbe, M., Schierhorn, A., Westermark, P., Westermark, G. T., et al. (2019). Cryo-EM fibril structures from systemic AA amyloidosis reveal the species complementarity of pathological amyloids. Nat. Commun. 10:1104. doi: 10.1038/s41467-019-09033-z
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luk, K. C., Song, C., O’Brien, P., Stieber, A., Branch, J. R., Brunden, K. R., et al. (2009). Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20051–20056. doi: 10.1073/pnas.0908005106
Morales, R., Hu, P., Duran-Aniotz, C., Moda, F., Diaz-Espinoza, R., Chen, B., et al. (2016). Strain-dependent profile of misfolded prion protein aggregates. Sci. Rep. 6:20526. doi: 10.1038/srep20526
Nelson, R., Sawaya, M. R., Balbirnie, M., Madsen, A. Ø, Riekel, C., Grothe, R., et al. (2005). Structure of the cross-beta spine of amyloid-like fibrils. Nature 435, 773–778. doi: 10.1038/nature03680
Ni, X., McGlinchey, R. P., Jiang, J., and Lee, J. C. (2019). Structural insights into α-synuclein fibril polymorphism: effects of Parkinson’s disease-related C-terminal truncations. J. Mol. Biol. 431, 3913–3919. doi: 10.1016/j.jmb.2019.07.001
Riek, R. (2017). The three-dimensional structures of amyloids” in Perspectives in Prion Biology. Cold Spring Harb. Perspect. Biol. 9:a023572. doi: 10.1101/cshperspect.a023572
Roher, A. E., Lowenson, J. D., Clarke, S., Woods, A. S., Cotter, R. J., Gowing, E., et al. (1993). beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 10836–10840. doi: 10.1073/pnas.90.22.10836
Sawaya, M. R., Sambashivan, S., Nelson, R., Ivanova, M. I, Sievers, S. A., and Apostol, M. I, et al. (2007). Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 447, 453–457. doi: 10.1038/nature05695
Schweers, O., Schönbrunn-Hanebeck, E., Marx, A., and Mandelkow, E. (1994). Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 269, 24290–24297.
Schweighauser, M., Shi, Y., Tarutani, A., Kametani, F., Murzin, A. G., Ghetti, B., et al. (2020). Structures of α-synuclein filaments from multiple system atrophy. Nature 585, 464–469. doi: 10.1038/s41586-020-2317-6
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 6, 595–608. doi: 10.15252/emmm.201606210
Shi, Y., Zhang, W., Yang, Y., Murzin, A. G., Falcon, B., Kotecha, A., et al. (2021). Structure-based classification of tauopathies. Nature 598, 359–363. doi: 10.1038/s41586-021-03911-7
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998b). Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spillantini, M. G., Crowther, R. A., Jakes, R., Cairns, N. J., Lantos, P. L., and Goedert, M. (1998a). Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 251, 205–208. doi: 10.1016/s0304-3940(98)00504-7
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Terry, R. D. (1963). The fine structure of neurofibrillary tangles in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 22, 629–642. doi: 10.1097/00005072-196310000-00005
Tuttle, M. D., Comellas, G., Nieuwkoop, A. J., Covell, D. J., Berthold, D. A., Kloepper, K. D., et al. (2016). Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 23, 409–415. doi: 10.1038/nsmb.3194
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Wälti, M. A., Ravotti, F., Arai, H., Glabe, C. G., Wall, J. S., Böckmann, A., et al. (2016). Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc. Natl. Acad. Sci. U.S.A. 113, E4976–E4984. doi: 10.1073/pnas.1600749113
Wang, L. Q., Zhao, K., Yuan, H. Y., Wang, Q., Guan, Z., Tao, T., et al. (2020). Cryo-EM structure of an amyloid fibril formed by full-length human prion protein. Nat. Struct. Mol. Biol. 27, 598–602. doi: 10.1038/s41594-020-0441-5
Wasmer, C., Lange, A., Melckebeke, H. V., Siemer, A. B., Riek, R., and Meier, B. H. (2008). Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science 319, 1523–1526. doi: 10.1126/science.1151839
Xue, W. F., Hellewell, A. L., Hewitt, E. W., and Radford, S. E. (2010). Fibril fragmentation in amyloid assembly and cytotoxicity. Prio 4, 20–25. doi: 10.4161/pri.4.1.11378
Yves, A. (1991). Parkinson’s disease: pathophysiology. Lancet 337, 1321–1324. doi: 10.1016/0140-6736(91)92989-f
Zagorski, M. G., Yang, J., Shao, H., Ma, K., Zeng, H., and Hong, A. (1999). Methodological and chemical factors affecting amyloid beta peptide amyloidogenicity. Methods Enzymol. 309, 189–204. doi: 10.1016/s0076-6879(99)09015-1
Zhang, C., and Kim, S. H. (2000). A comprehensive analysis of the Greek key motifs in protein beta-barrels and beta-sandwiches. Proteins 40, 409–419.
Keywords: neurodegenerative diseases, misfolding, amyloid, structure, Aβ, Tau, α-Sn
Citation: Diaz-Espinoza R (2021) Recent High-Resolution Structures of Amyloids Involved in Neurodegenerative Diseases. Front. Aging Neurosci. 13:782617. doi: 10.3389/fnagi.2021.782617
Received: 24 September 2021; Accepted: 28 October 2021;
Published: 19 November 2021.
Edited by:
Natalia Salvadores, Universidad Mayor, ChileReviewed by:
Victor Banerjee, University of Texas Health Science Center at Houston, United StatesRodrigo Gallardo, University of Leeds, United Kingdom
Copyright © 2021 Diaz-Espinoza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodrigo Diaz-Espinoza, cm9kcmlnby5kaWF6LmVAdXNhY2guY2w=