 Peter Kramer
Peter Kramer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 23 December 2021
Sec. Aging Neuroscience Archive
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.776936
This article is part of the Research Topic The Gut-Brain Axis – Opportunities for a Novel Approach Against Neurodegeneration View all 9 articles
Alzheimer’s and Parkinson’s are the two best-known neurodegenerative diseases. Each is associated with the excessive aggregation in the brain and elsewhere of its own characteristic amyloid proteins. Yet the two afflictions have much in common and often the same amyloids play a role in both. These amyloids need not be toxic and can help regulate bile secretion, synaptic plasticity, and immune defense. Moreover, when they do form toxic aggregates, amyloids typically harm not just patients but their pathogens too. A major port of entry for pathogens is the gut. Keeping the gut’s microbe community (microbiota) healthy and under control requires that our cells’ main energy producers (mitochondria) support the gut-blood barrier and immune system. As we age, these mitochondria eventually succumb to the corrosive byproducts they themselves release, our defenses break down, pathogens or their toxins break through, and the side effects of inflammation and amyloid aggregation become problematic. Although it gets most of the attention, local amyloid aggregation in the brain merely points to a bigger problem: the systemic breakdown of the entire human superorganism, exemplified by an interaction turning bad between mitochondria and microbiota.
If we live long enough, we will degenerate; and because it demands a great deal of oxygen and energy, our brain’s decline is particularly conspicuous (Kramer and Bressan, 2018). The most notable neurodegenerative diseases are Alzheimer’s and Parkinson’s, which are so common that in 2015 they affected, respectively, 46 and 6 million people worldwide (Dorsey et al., 2007; GBD 2015 Disease and Injury Incidence and Prevalence Collaborators, 2016; Dorsey and Bloem, 2018; see also Alzheimer’s Association, 2019). On the face of it, the two diseases look quite different. The most notable symptoms of Alzheimer’s are progressive loss of memory and a worsening cognitive ability (Alzheimer’s Association, 2019). The most notable ones of Parkinson’s are tremors, stiffness, and instability when standing or walking (Kalia and Lang, 2015). Often, however, patients suffer from a range of symptoms that do not all neatly fit the diagnostic criteria of one disease or another, and often these patients are concurrently plagued by multiple health issues (Del Tredici and Braak, 2020).
Underneath, moreover, the two disorders involve broadly similar physiological processes, with a major role in both for amyloids – sticky proteins that can aggregate into nanoscale-tiny fibers (fibrils) and larger clumps. The aggregation is due to the folding of the amyloids in a way that causes similar nearby ones to fold in a similar way – a process akin to the propagation of prion amyloids in mad-cow disease and in its human counterpart, Creutzfeldt-Jakob disease (Goedert, 2015; Stopschinski and Diamond, 2017; but cf. Surmeier et al., 2017; Steiner et al., 2018; Kowalski and Mulak, 2019; Jankovska et al., 2021; Nguyen et al., 2021).
Soluble amyloid aggregates tend to be toxic; larger insoluble ones, although quite prominent, need not be (Bendor et al., 2013; D’Andrea, 2016; Li et al., 2018; Quntanilla and Tapia-Monsalves, 2020). In Parkinson’s, the soluble amyloid alpha-synuclein can aggregate, inside neurons, into insoluble Lewy bodies (Figure 1; Del Tredici and Braak, 2020). In Alzheimer’s, soluble beta-amyloid can do so into insoluble plaques outside neurons (Figure 1; Makin, 2018). Beta-amyloid is also present inside neurons, and while soluble, it can likely enter neurons from the outside (Bayer and Wirths, 2010; D’Andrea, 2016). Inside neurons, both alpha-synuclein (Guo et al., 2013) and beta-amyloid (Choi et al., 2014) can induce the soluble amyloid tau to aggregate into insoluble tangled fibrils of tau-compounds or truncated pieces of tau (Guo et al., 2017; Quntanilla and Tapia-Monsalves, 2020; Figure 1).
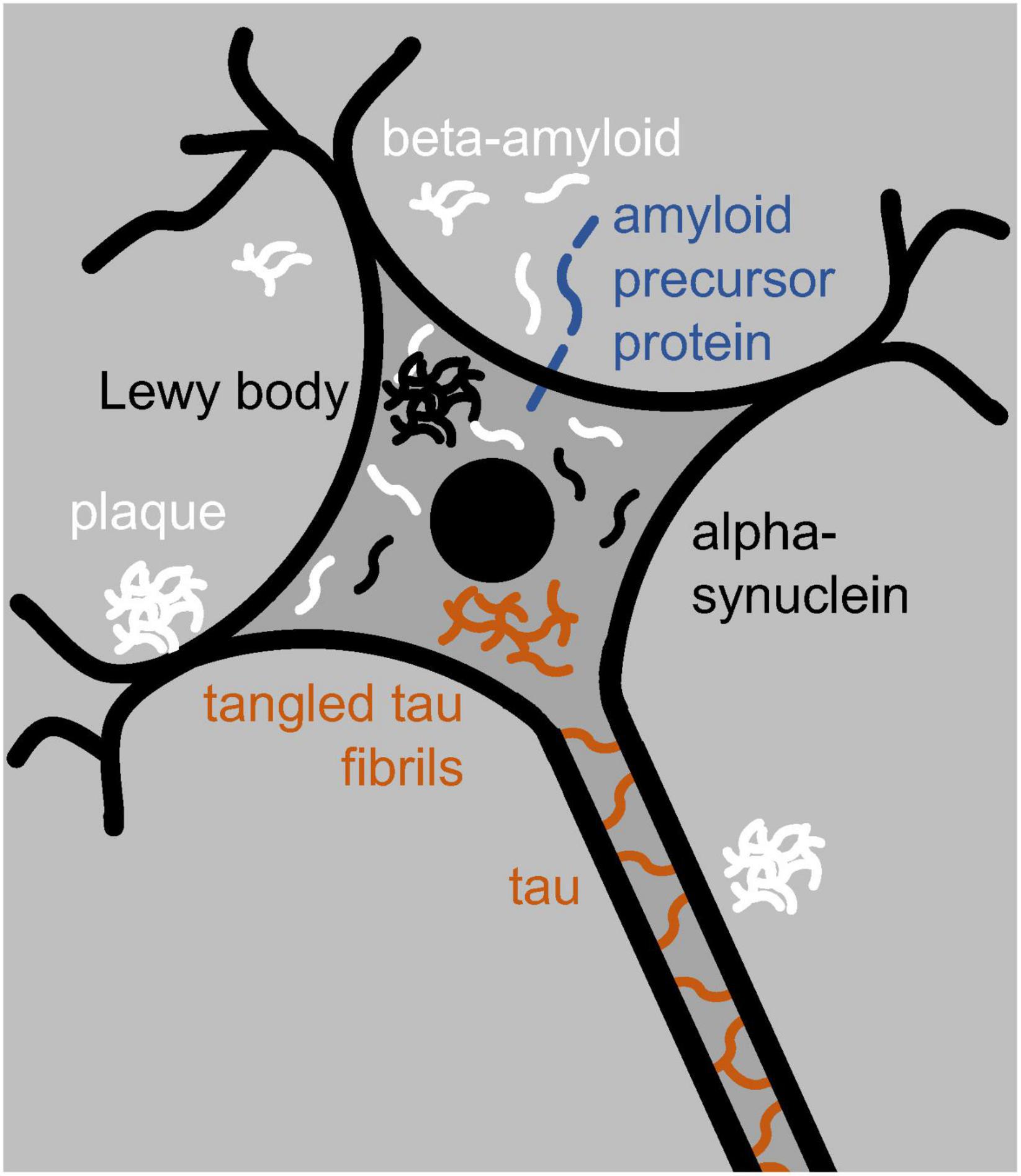
Figure 1. Neuron with a nucleus (black disk), dendrites (black branching), and an axon (running toward the bottom right). The neuron contains tau and tangled-tau fibrils (brown), alpha-synuclein and Lewy bodies (black), and beta-amyloid (white). Outside the neuron are beta-amyloid and amyloid plaques. Locked in the neuron’s membrane is amyloid precursor protein (blue) from which beta-amyloid is cut out.
Beta-amyloid, and especially tau, compounds have primarily been associated with Alzheimer’s disease; alpha-synuclein compounds and Lewy bodies instead with Parkinson’s disease (Del Tredici and Braak, 2020; Nguyen et al., 2021). Yet all three amyloids, and additional ones besides, can trigger one another’s aggregation, and mixed neurodegenerative disease is quite common (Nguyen et al., 2021). Alpha-synuclein, for example, contributes to the aggregation of beta-amyloid and tau compounds, tau compounds contribute to the aggregation of alpha-synuclein (Pan et al., 2021), and Lewy bodies have been observed not just in Parkinson’s but in other neurodegenerative disease as well (Bendor et al., 2013; Wong and Krainc, 2017). Moreover, besides alpha-synuclein, Lewy bodies can contain tau and be surrounded by tau fibrils (Pan et al., 2021). And conversely, besides beta-amyloid, plaques typically contain other amyloids (D’Andrea and Nagele, 2010), including alpha-synuclein (Bendor et al., 2013; Jankovska et al., 2021). In addition, the MAPT gene, which expresses tau, is implicated in not only Alzheimer’s (Zhou and Wang, 2017) but also Parkinson’s (Pan et al., 2021). It might thus be a mistake to treat the various degenerative diseases of the brain and body as categorically distinct rather than as interrelated (see also Nguyen et al., 2021).
Although both Alzheimer’s and Parkinson’s can progress toward dementia, accompanied by widespread damage (D’Andrea, 2016; Weil et al., 2017), they differ in which part of the brain they damage the most. Alzheimer’s is best known for degeneration of the hippocampus (Figure 2A) and resulting deficits in the formation of new memories (D’Andrea, 2016); Parkinson’s with that of the pars compacta portion of the substantia nigra (Figure 2B) and resulting deficits in one’s ability to move about (Dickson et al., 2009; Poewe et al., 2017). Yet these neurological differences actually highlight yet again how much the two diseases have in common. Most of the energy our body and brain run on is furnished by our mitochondria – entities inside our cells that evolved from bacteria (Lane, 2015). And it so happens that the hippocampus contains unusually low levels of a protein – implicated in Alzheimer’s disease (Acker et al., 2019) – that helps neurons and their mitochondria obtain oxygen (Burmester et al., 2000). This, then, leaves these hippocampal mitochondria particularly vulnerable to dysfunction (Acker et al., 2019). Likewise, the pars compacta hosts exceptionally large neurons, which require exceptionally large amounts of energy, and these neurons’ mitochondria may therefore also be particularly vulnerable to dysfunction (Pacelli et al., 2015; Rietdijk et al., 2017; Shlevkov and Schwarz, 2017; Gonzaìlez-Rodriìguez et al., 2021). This is especially the case because calcium levels in these neurons tend to be higher than elsewhere, and this stimulates alpha-synuclein buildup (Surmeier et al., 2017). In Parkinson’s, much smaller neurons in nearby brain areas tend to be spared (Pacelli et al., 2015; Rietdijk et al., 2017; Shlevkov and Schwarz, 2017).
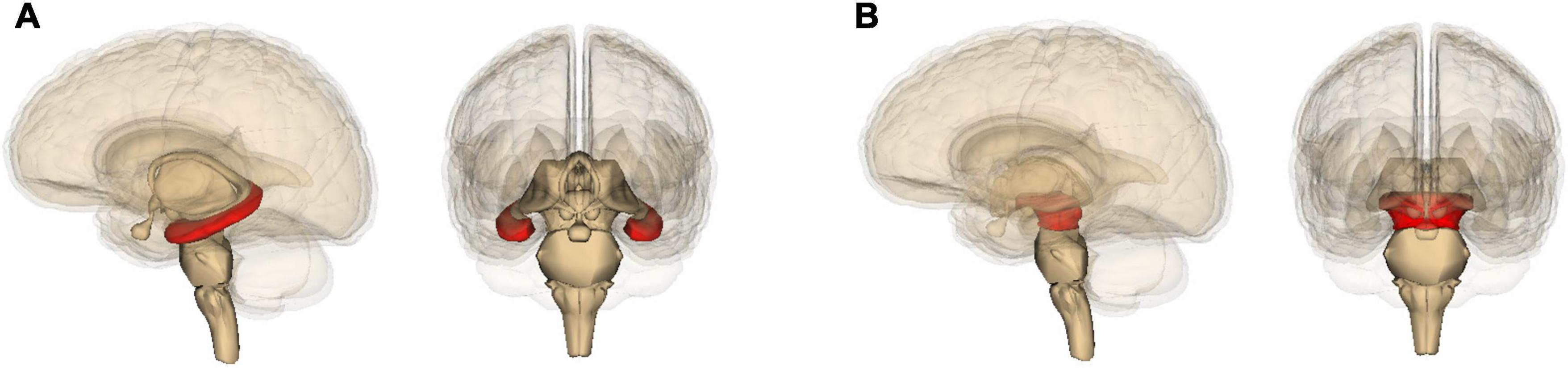
Figure 2. Location in the brain of (A) the hippocampus and (B) the midbrain (containing the substantia nigra), shown in red. The images are generated by Life Science Databases from the Anatomography website maintained by Life Science Databases (Creative Commons): https://commons.wikimedia.org/w/index.php?curid=7887124; https://commons.wikimedia.org/w/index.php?curid=7837965.
Although also heavily implicated in virtually every other mental affliction, from chronic psychological stress and fatigue to schizophrenia and autism (Kramer and Bressan, 2018), mitochondrial dysfunction typically both accompanies and precedes Alzheimer’s and Parkinson’s disease (see section “Poor Mitochondria Means a Poor Microbiota”). Such mitochondrial dysfunction can have various different causes, but one of them is an abnormal presence of amyloids (Wong and Krainc, 2017; Bernal-Conde et al., 2020; Huang et al., 2020; Quntanilla and Tapia-Monsalves, 2020; Szabo et al., 2020; Borsche et al., 2021; Feng et al., 2021; Nguyen et al., 2021; Zaretsky and Zaretskaia, 2021). The question arises, however, what causes this abnormal presence. An important part of the answer, it appears, is infection and inflammation (see section “The Main Problem Is Pathogens, Not Amyloids”).
Pathogens can invade us through various points of entry, but a major one is the gut. Primarily in the intestines, we house an estimated 38 trillion microbes (Sender et al., 2016), among which at least 2,172 known species (Hugon et al., 2015), comprising about as many (small) microbial cells as we have (large) human ones (Sender et al., 2016). In addition, these intestines leave space for an even more diverse and numerous collection of viruses (Cryan et al., 2019). Most of these are phages and prey on our gut microbes without affecting us directly (Cryan et al., 2019) but they nonetheless have been reported to affect their host’s gut-blood barrier (Fitzgerald et al., 2019). Even at the best of times, the community of microbes (microbiota) in the gut is not a harmless community of symbionts: we possess a protective gut-blood barrier to keep them out of our bloodstream, and because that is not good enough, we also have a blood-brain barrier to keep them out of our brain. To ease nutrients through, the gut-blood barrier is only one-cell-thick and semipermeable (Groschwitz and Hogan, 2009). In compensation, the intestine (especially the large one, the colon, in which most of the gut microbiota resides) is coated with a layer of sticky mucus, which helps protect the gut-blood barrier from pathogen invasion (von Martels et al., 2017; Kowalski and Mulak, 2019). Most importantly, the barriers of both the small and the large intestine are protected by a heavy presence of the immune system (Kowalski and Mulak, 2019).
This system, however, can only do its job if it is furnished with sufficient energy, a task entrusted mostly to mitochondria. Counterintuitively, some of these mitochondria have a sort of brake on their energy production and release these brakes only when needed (see section “Poor Mitochondria Means a Poor Microbiota”). If the mitochondria fail to deliver the energy the immune system needs, an unhealthy change in the gut microbiota results, known as dysbiosis (see section “Poor Mitochondria Means a Poor Microbiota”). The health of the microbiota thus depends on that of the mitochondria, but because mitochondria need nutrients and are harmed by pathogens, their health also depends on that of the microbiota. For how long the interaction between mitochondria and microbiota remains beneficial, and we can keep neurodegenerative disease at bay, depends in part on the quantity of food we eat (see section “Mitochondria’s Oxygen Consumption Benefits the Microbiota”). It also depends on how rich this food is in, for example, fibers (see section “The Microbiota’s Fatty Acids Benefit Mitochondria”), fatty acids (see section “The Microbiota’s Fatty Acids Benefit Mitochondria”), and protein (see section “The Microbiota’s Hydrogen Sulfide Can Either Benefit or Harm Mitochondria”). With age, both mitochondria and the microbiota deteriorate, and gut pathogens then have multiple options to either indirectly or directly wreak havoc on the brain (see section “Trouble in the Gut Is Trouble in the Brain”). Indeed, Alzheimer’s (Fulop et al., 2018; Moir et al., 2018; Ashraf et al., 2019; Cryan et al., 2019; Li et al., 2021) and Parkinson’s (Cardoso and Empadinhas, 2018; Nair et al., 2018; Cryan et al., 2019; Lubomski et al., 2019; Huang et al., 2021; Munoz-Pinto et al., 2021), along with many other mental afflictions (Vuong et al., 2017; Cryan et al., 2019), tend to be both accompanied and preceded by not only mitochondrial dysfunction but also dysbiosis and intestinal disease (see also Vogt et al., 2017; Villumsen et al., 2019; Shen et al., 2021).
In low concentrations, amyloids are not a threat but a blessing, and serve useful physiological functions. Beta-amyloid and alpha-synuclein, for example, are involved in fat metabolism, neurotransmitter release, synaptic plasticity, learning and memory (Golovko et al., 2009; Parihar and Brewer, 2010; Cheng et al., 2011; Bendor et al., 2013; D’Andrea, 2016; Fitzgerald et al., 2019; Panza et al., 2019). Tau has an important role in structural support in neurons and in the transport of mitochondria through axons to synapses in need of energy (Guo et al., 2017). Beta-amyloid has been preserved for at least 400 million years, which would be difficult to understand if it had not contributed to survival (Moir et al., 2018). Most Alzheimer’s or Parkinson’s researchers focus their attention on the production and aggregation of amyloids in and around neurons in the brain. Yet amyloids are also produced in other cell types and body parts (D’Andrea, 2016; Filippini et al., 2019), including various organs, the skin, muscles, blood vessels, and the gut (D’Andrea, 2016; Chalazonitis and Rao, 2018).
Both beta-amyloid (Moir et al., 2018) and alpha-synuclein (Barbut et al., 2019) share several characteristics with substances like LL-37 that have well-established antimicrobial properties. In fact, they have anti-pathogenic properties themselves and they, or their soluble aggregates, boost immunity further by triggering inflammation (D’Andrea, 2016; Park et al., 2016; Fulop et al., 2018; Li et al., 2018; beta-amyloid: Moir et al., 2018; Ashraf et al., 2019; alpha-synuclein: Barbut et al., 2019; Fitzgerald et al., 2019; Kowalski and Mulak, 2019; Li et al., 2021). In Alzheimer’s, beta-amyloid levels are typically rather high in the temporal lobes, which host the hippocampus (Soscia et al., 2010). And tellingly, after being injected into post-mortem samples of temporal lobes, Candida albicans (a fungus implicated in intestinal disease) grows less in samples taken from Alzheimer’s patients than in samples taken from people of a similar age without the disease (Soscia et al., 2010).
Likewise, after a brain injection of live Salmonella typhimurium (a bacterium implicated in food poisoning), mice engineered to overproduce human beta-amyloid displayed a lower pathogen load in the brain (Kumar et al., 2016). They also lost less weight and survived better than their control littermates (Kumar et al., 2016). Mice engineered to underproduce beta-amyloid survived marginally worse (Kumar et al., 2016). Injection with dead Salmonella had no such effects.
Like antimicrobial LL-37, beta-amyloid is sticky, latches on to pathogens like S. typhimurium, and entraps them in its fibrils (Figure 3; Kumar et al., 2016). Any antimicrobial property of beta-amyloid is only called for if such pathogens are indeed present. It thus makes sense that even mice that have been engineered to overproduce beta-amyloid have lower levels of beta-amyloid in their brain if they had been raised germ-free than if they had not (Harach et al., 2017; see also Minter et al., 2017). After injection into the bloodstream of live S. typhimurium, mice engineered to underproduce alpha-synuclein were also less likely to survive than control littermates (Tomlinson et al., 2017). Infection with a virus, rather than a bacterium, had a similar effect (Beatman et al., 2015; Tomlinson et al., 2017).

Figure 3. Pathogen entrapped by amyloid fibrils. Icon adapted from the Noun Project: Maxim Kulikov.
Small concentrations of antimicrobial substances like LL-37 provide a net benefit to the host. Large concentrations of its soluble aggregates can instead be toxic and are implicated in several aging-related diseases (Moir et al., 2018). The same is true for the amyloids typically implicated in Alzheimer’s and Parkinson’s (Moir et al., 2018; Barbut et al., 2019). Healthy mitochondria degrade damaged or misfolded proteins, including amyloids like alpha-synuclein and beta-amyloid, but they become overwhelmed when amyloid concentrations become too high (Lautenschäger and Schierle, 2019; Lautenschäger et al., 2020; Sivanesan et al., 2020). Moreover, mitochondria release free radicals – chemicals that are corrosive to their host but also to their own mitochondrial DNA. As a result, over time, more and more of these mitochondria become dysfunctional (Lane, 2015; Kramer and Bressan, 2019) and eventually unable to detoxify the amyloids (Sivanesan et al., 2020).
Inflammation and the formation and aggregation of amyloids and other antimicrobial substances are perhaps best seen as a necessary evil. When moderate and brief, these defenses offer a net benefit to the host and its mitochondria but when excessive and chronic, their side effects may become worse than the infections they were designed to quell (Wong and Krainc, 2017; Bernal-Conde et al., 2020; Huang et al., 2020; Quntanilla and Tapia-Monsalves, 2020; Szabo et al., 2020; Vezzani et al., 2020; Borsche et al., 2021; Feng et al., 2021; Zaretsky and Zaretskaia, 2021). Because amyloid production and aggregation serve a definite purpose, however, suppressing them may be risky. Indeed, drugs that lower beta-amyloid levels raise the risk of infection and have time and again either failed to improve the health of Alzheimer’s patients or made it worse (Green et al., 2009; Imbimbo, 2009; Makin, 2018).
By far the greatest risk factor of Alzheimer’s disease, but also implicated in some forms of Parkinson’s disease (Tsuang et al., 2013; Pang et al., 2018; Shi and Holtzman, 2018; Belloy et al., 2019), is the gene Apolipoprotein E (APOE), which is involved in the transport of cholesterol and other fatty acids or compounds (Urosevic and Martins, 2008; Shi and Holtzman, 2018; Belloy et al., 2019). APOE comes in four variants: APOE1 (extremely rare and not considered further), APOE2 (rare), APOE3 (most common), and APOE4 (common) (Belloy et al., 2019). APOE4 is evolutionarily the oldest (Belloy et al., 2019). Which APOE variant one carries affects tau and alpha-synuclein levels (Huynh et al., 2017; Hohman et al., 2018) and is likely involved in the production, and very clearly in the breakdown, of beta-amyloids (Belloy et al., 2019). Most studies suggest that beta-amyloid buildup is favored least by APOE2, a bit more by APOE3, and much more by APOE4 (Belloy et al., 2019). Mirroring this pattern, APOE2 is the least and APOE4 the most inflammatory (Moir et al., 2018; Sullivan, 2020).
In Western nations, with a diet rich in inflammatory fats and poor in anti-inflammatory ones and with high blood cholesterol levels (Sullivan, 2020), APOE4 is a major risk factor for early-onset Alzheimer’s (especially in women: Belloy et al., 2019) and so it has a bad reputation (Sullivan, 2020). Yet in places where, because of a better diet or for other reasons, high cholesterol is less common, APOE4 is either less of a risk factor for Alzheimer’s (like in East-Asians) or not at all (like in Africa’s Bushmen) (Notkola et al., 1998). The incidence of Alzheimer’s is also higher among Japanese Americans and African Americans than among the Japanese and Africans (here Nigerians) living in their native countries (Sullivan, 2020). And although APOE4 is a bigger risk factor for Alzheimer’s than is APOE3, and Alzheimer’s involves cognitive decline, among infected Tsimane Amazon-Indians, APOE4 is associated with better cognition than APOE3 (Trumble et al., 2017; see also van Exel et al., 2017; Zhao et al., 2020). APOE4’s more inflammatory effect is a burden in those in good health but apparently can be a boon during infection. More recently, beta-amyloid was found to impair, but APOE4 itself to slightly improve, short-term memory among some 400 British participants without dementia (Lu et al., 2021).
Beta-amyloid consists of a chain of amino acids that can differ in length, with the most common lengths being either 40 or 42 amino acids (by comparison, alpha-synuclein consists of some 140 amino acids and tau of 352 to 421; Nguyen et al., 2021). Beta-amyloid 42 tends to aggregate into soluble fibrils and insoluble plaques more easily than beta-amyloid 40 and is more toxic. Unsurprisingly therefore, like APOE4, beta-amyloid 42 also has a bad a reputation (D’Andrea, 2016; Jankovska et al., 2021). Yet beta-amyloid 42 is not only more toxic to us, it is also more toxic to pathogens (Soscia et al., 2010) and that can be a good thing. Moreover, as mentioned, beta-amyloid has other functions than just helping to fight off infection, and in small amounts both beta-amyloid 40 and beta-amyloid 42 promote neuronal survival (D’Andrea, 2016). What is bad or good about APOE variants and amyloids, then, depends on circumstances like diet and pathogen exposure.
The expression of various genes involved in neurodegenerative disease is regulated by epigenetic processes (Balmik and Chinnathambi, 2021) – processes that act upon (epi in Greek) genes. An important epigenetic process is methylation – the addition of a methyl group to a gene, or nearby protein, so that it can no longer be read. The methyl group is provided by S-adenosyl methionine, a chemical that in part is produced with the help of mitochondria (Wallace and Fan, 2010; Lopes, 2020). Mitochondria are also the prime source of the energy methylation requires. With age, as more and more mitochondria begin to malfunction, this methylation process can go awry. Indeed, diminished methylation can lead to abnormal formation and accumulation of tau compounds (Balmik and Chinnathambi, 2021) and likely of beta-amyloid too (Lin et al., 2009). Thus, even if amyloids help protect us from pathogens, as we age, mitochondrial dysfunction can eventually turn the genes that express these amyloids against us.
Before cognitive or motor symptoms emerge, metabolism invariably has already gone awry in both Alzheimer’s (Sun et al., 2020b) and Parkinson’s (Phillipson, 2017), as well as in risk factors of these two diseases: diabetes (Arnold et al., 2018; Cheong et al., 2020), obesity (Floud et al., 2020; Kim and Jun, 2020; Qu et al., 2020), impaired amyloid clearance (Phillipson, 2017; Sun et al., 2020b). Metabolism and amyloid clearance rely to a large extent on mitochondria, which during neurodegeneration are deteriorating (Phillipson, 2017; Sun et al., 2020b; Nguyen et al., 2021). One way to simulate the decline in mitochondrial health in lab animals is to expose mitochondria to a poison. When they are poisoned, mitochondria produce less energy and more free radicals, just like they do during aging. If its cells receive less energy and more free radicals, the gut-blood barrier becomes leaky and lets pathogens through into the bloodstream (Jackson and Theiss, 2020). Lowering free radical levels with antioxidants shores up the barrier, instead, and diminishes its leakiness (Jackson and Theiss, 2020), which helps prevent dysbiosis (Thevaranjan et al., 2017).
Mitochondria are not only important for the maintenance of protective barriers but also for the proper functioning of the immune system (Cardoso and Empadinhas, 2018; Munoz-Pinto et al., 2021). Each mitochondrion features an outer membrane that separates it from the rest of the cell, and an inner membrane that contains its machinery for energy production (Kramer and Bressan, 2019). Curiously, the inner membrane of the mitochondria that reside in a particular type of white blood cells produces much less energy than it is capable of Konjar et al. (2018). Upon recognizing viruses, bacteria, or harmful debris, immune cells release proinflammatory signaling molecules that, in defense of the host, trigger inflammation (Jackson and Theiss, 2020). When this happens, the properties of the inner membrane of the white-blood cells’ mitochondria abruptly change: the brakes come off and energy production shoots up (Konjar et al., 2018). This sudden boost of energy allows the white blood cells to proliferate fast and take immediate action against their foes. If mitochondria cannot produce the energy that white blood cells need to fight off infection, the gut-blood barrier suffers, and gut pathogens get a chance to thrive and invade their host’s bloodstream (Figure 4; Konjar et al., 2018). Mitochondrial dysfunction reduces the energy available to the immune system and impairs the immune system’s ability to keep the gut microbiota under control. This, then, leads to dysbiosis as well (Yardeni et al., 2019; Jackson and Theiss, 2020).
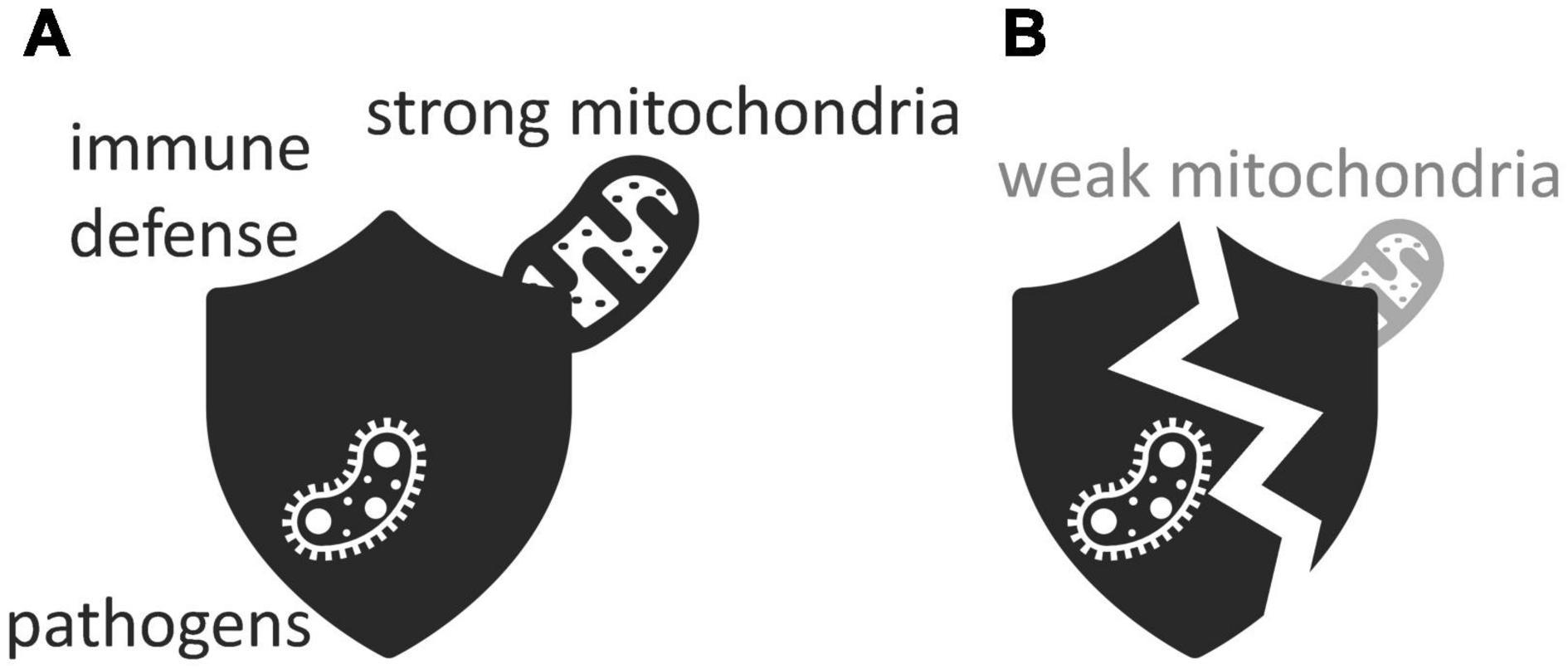
Figure 4. Immune defenses protect against pathogens but only if they are empowered by (A) strong mitochondria rather than (B) weak ones. Icons adapted from the Noun Project: Maxim Kulikov (bacterium), dDara (mitochondrion), and David Khai (shield).
Activation of the immune system comes with a surge of free radicals that is harmful not only to invading pathogens, but also to the tissues of the host itself and the mitochondria in the cells of these tissues (López-Armada et al., 2013; Angajala et al., 2018). Aside from the fact that they consume resources, inflammation and free-radical production should thus not be brought to bear more than strictly needed (Konjar et al., 2018). This, then, might be why mitochondria behave like gate keepers; they hold their guard dogs (immune cells) on a tight leash when all is fine and set them loose only when bad characters (pathogens) come charging in Kramer and Bressan (2019).
Although free radicals can harm mitochondria, the mitochondria release many of them themselves during energy production (Lane, 2015; Kramer and Bressan, 2019). Much of the DNA that mitochondria carried as independent bacteria was eventually incorporated into the nucleus of their host cells (Lane and Martin, 2010; Lane, 2015). Yet the mitochondria did hold on to 37 genes, of which 13 express proteins needed for the mitochondrial energy-production machinery (Johnston and Williams, 2016). To a large extent, mitochondrial genes determine how much energy mitochondria can produce (Johnston and Williams, 2016) and how many free radicals they release in the process (Moreno-Loshuertos et al., 2006, 2013). Lab animals have been bred or engineered that carry versions of mitochondrial DNA that increase free-radical production (Yardeni et al., 2019). The higher this production, the leakier the gut, offering opportunities to gut pathogens (Jackson and Theiss, 2020). In fact, mice made to produce more free radicals harbor a less diverse gut microbiota, with more of it consisting of Bacteroidales (a subset of Bacteroidetes; Figure 5) rather than Clostridia (a subset of Firmicutes; Figure 5; Yardeni et al., 2019). Consistently, after 10 weeks on a drug that lowers rather than raises free radical levels, normal mice ended up with 31% fewer Bacteroidales, and more than twice as many Clostridia, as before (Yardeni et al., 2019).
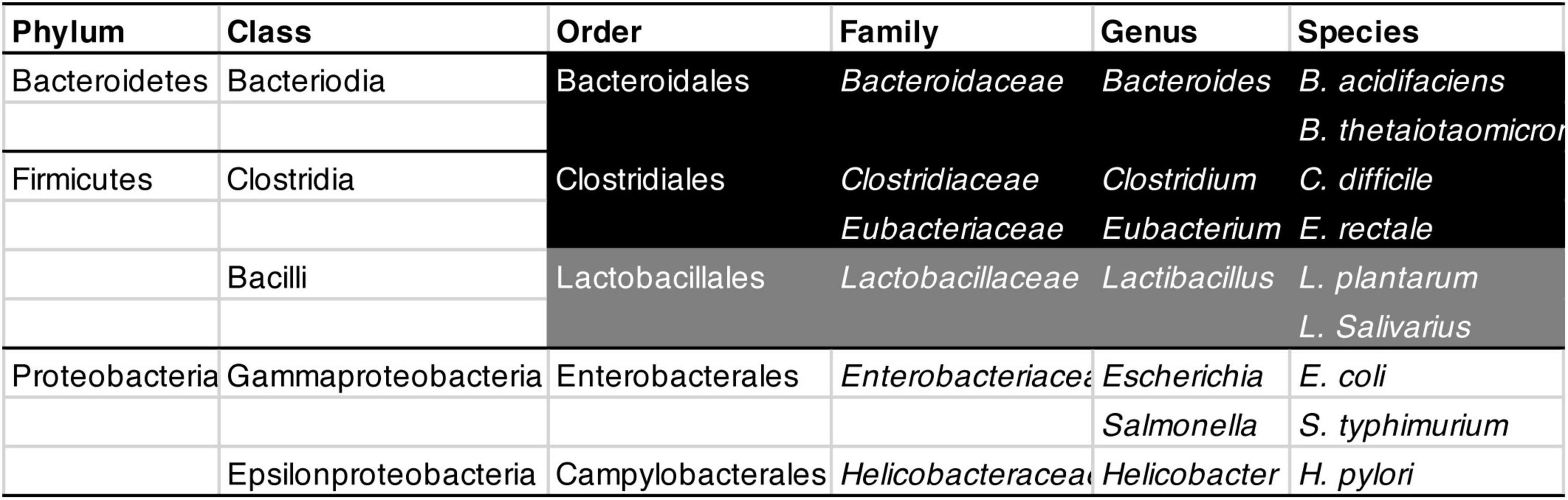
Figure 5. Classification of bacteria mentioned in this review, from Phylum (the widest category) to Species (the narrowest). From Order to Species, strictly anaerobic bacteria are shown in white on black, largely anaerobic ones in white on gray, and aerobic ones in black on white.
Bacteroidales need not necessarily be harmful. Bacteroides acidifaciens, for example, helps prevent obesity and improve insulin sensitivity (shown in mice: Yang et al., 2017). Conversely, Clostridia need not necessarily be beneficial; once ingested, Clostridium difficile can cause food poisoning and kills thousands each year in the US alone1 (Czepiel et al., 2019). Most studies do not investigate individual species or strains of bacteria but entire families or other broad categories. Substantial methodological differences between studies complicate matters further, with some studies controlling for medication whereas others do not (Romano et al., 2021). Unsurprisingly, they regularly come to different conclusions about which ones are beneficial or harmful (Cryan et al., 2019). Still, aging – the largest risk factor for both Alzheimer’s and Parkinson’s (Hou et al., 2019) – is generally accompanied by a less diverse microbiota (Cardoso and Empadinhas, 2018; Haran and McCormick, 2021; Romano et al., 2021), and in mice also by a proliferation of Bacteroidales at the expense of Clostridiales (Figure 5; Yardeni et al., 2019).
Alas, improving the gut microbiota requires more than just enriching or replacing it. Mice deficient in tumor necrosis factor (a trigger of inflammation) are protected not only from age-related inflammation and a leaky gut but also from dysbiosis (Thevaranjan et al., 2017). Reducing the levels of this signaling molecule with a drug reproduces some of these positive effects (Thevaranjan et al., 2017). This shows once again that the gut microbiota is affected by the immune system and its signaling molecules. Because the levels of tumor necrosis factor rise with age (Thevaranjan et al., 2017), dysbiosis should be expected to worsen with age, and indeed it does (Thevaranjan et al., 2017). Moreover, inflammation is typically accompanied by a rise in free radical levels, and in mice whose mitochondrial DNA has been engineered to engender excessive free-radical production, revealing effects were obtained with cross-fostering. In this procedure, the offspring of mothers engineered to produce relatively many free radicals are given the microbiota of a foster mother engineered to produce relatively few. Within 2 months, the adopted microbiota changed into one consistent with the biological mother’s and not the foster mother’s (see also Hirose et al., 2017 and for related human evidence: Ma et al., 2014; Yardeni et al., 2019). Keeping a healthy microbiota thus requires keeping healthy mitochondria too, and this means being lucky enough to carry good mitochondrial genes. As we get older, mitochondria accrue more and more mutations, release more and more inflammation provoking debris (Munoz-Pinto et al., 2021), countermeasures to mitigate the harm begin to fail (Lane, 2015), and keeping the microbiota healthy becomes less and less feasible. Of course, if someone’s dysbiosis is due not to genes but to circumstances, such as a poor diet or a treatable infection, hope remains for a more lasting cure.
To function well, mitochondria need to be fed. They can only metabolize food that has been digested. After having been chewed to bits, meals are first broken down with the help of acid in the stomach, and bile and enzymes in the small intestine; next, the process is taken over by the microbiota, mainly in the large intestine. The gut microbiota expands through cloning and with each of our meals; it diminishes with defecation. Among the bacteria frequently found in the gut, the majority by far use little or no oxygen (here collectively referred to as “anaerobes”), a minority can or must use larger quantities of this gas (here collectively referred to as “aerobes”) (von Martels et al., 2017). Perhaps because our well-oxygenated tissues are not ideal places for them, anaerobes are less inclined than aerobes to invade, or interfere with, these tissues (Shelton et al., 2020). Although anaerobes are by no means all harmless symbionts (the Bacteroidales and Clostridia mentioned before are all anaerobes), aerobes are more often associated with disease (Shoaie et al., 2021). A healthy gut is thus one in which anaerobes tend to do well and aerobes do not (Rigottier-Gois, 2013; Byndloss et al., 2017; Litvak et al., 2017).
Gut oxygen levels depend on the kind of metabolism the host engages in Byndloss et al. (2017) and Wang et al. (2019). In each of our cells except red blood cells, the conversion of digested food into energy (catabolism) typically involves mitochondria (Wang et al., 2019). In this process, food is effectively treated as fuel and burned down with the help of lots of oxygen (Wang et al., 2019). Instead, the conversion of food into either building blocks for growth, or fat or glycogen for energy storage (anabolism), occurs mostly without engaging mitochondria, and requires much less oxygen or none at all (Byndloss et al., 2017; Wang et al., 2019). So, cells extract far more oxygen from their surroundings when they catabolize food than when they anabolize it. The consequences are particularly interesting if one looks at the cells that compose the gut wall: when they burn food for energy (rather than fermenting it for building blocks or energy storage), the level of oxygen near the gut wall drops (von Martels et al., 2017), and the microbiota as a whole becomes more anaerobic and healthier (Byndloss et al., 2017; Litvak et al., 2017).
Whether food is catabolized or anabolized is regulated in part by hormones sent out by the hypothalamus and an adjacent organ directly under its control: the pituitary gland (Figure 6). In part, it is also regulated inside cells by various chemicals that act like sensors of nutrients, of pathogen invasion, and of other intracellular circumstances (Wang et al., 2019). If the hypothalamus and individual cells ascertain that food is abundant and circumstances suitable, they push for growth and reproduction, which rely heavily on anabolism. If the opposite is the case, they encourage maintenance operations, which rely heavily on catabolism (Wang et al., 2019). For the survival of genes, growth and reproduction are obviously paramount; for the survival of their host, however, maintenance is more important than either growth or reproduction. Likely to allow for growth and reproduction (and for the storage of energy for leaner times), we have evolved to enjoy eating abundantly. Eating no more than necessary, however, has repeatedly been found to improve the health of both mitochondria (Mattson and Arumugam, 2018; Mattson et al., 2018) and the gut microbiota (Rinninella et al., 2020). In addition, fasting lowers free radical levels and inflammation (Mattson and Arumugam, 2018; Mattson et al., 2018), improves leaky gut problems and gut disorders (Rinninella et al., 2020), diminishes overweight and diabetes (Anton et al., 2018), stimulates the release of brain-derived neurotrophic factor (which stimulates the growth of nerves) (Mattson et al., 2018), and helps stave off neurodegenerative diseases like Alzheimer’s and Parkinson’s (Mattson and Arumugam, 2018).
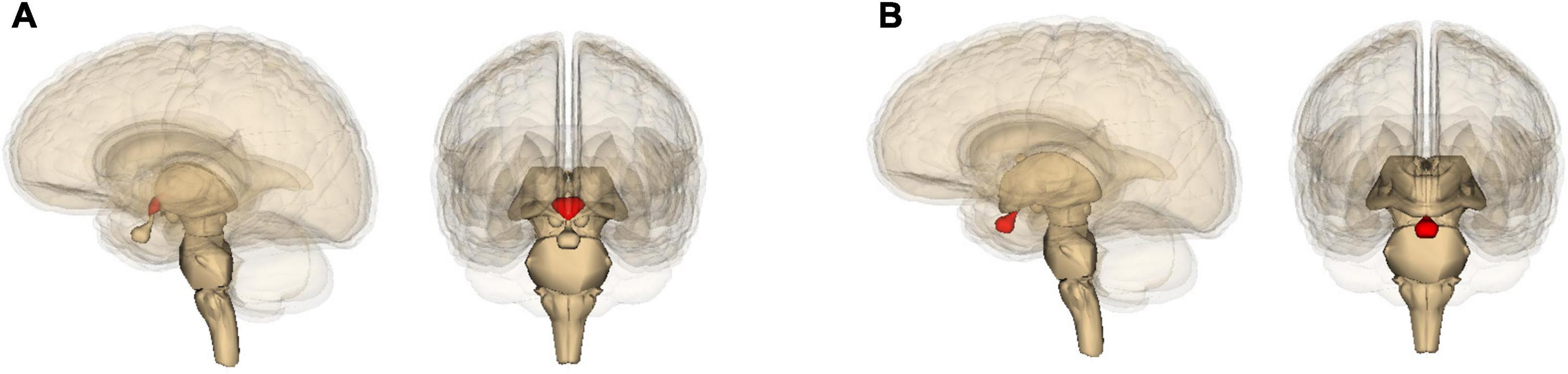
Figure 6. Location in the brain of (A) the hypothalamus and (B) the pituitary gland, shown in red. Images are generated by Life Science Databases from the Anatomography website maintained by Life Science Databases (Creative Commons): https://commons.wikimedia.org/w/index.php?curid=7848243; https://commons.wikimedia.org/w/index.php?curid=7833196.
Still, when facing pathogens, the immune system needs not only catabolism to energize immune cells but also anabolism to allow these cells to grow and multiply (Wang et al., 2019; Jackson and Theiss, 2020). Dieting should thus not be so extreme that there are no reserves left to fight infections or for essential growth of, for example, new neuronal connections. Indeed, in elderly individuals and others who suffer disproportionately from infections, inflammation, or weak mitochondria, dieting could be too stressful and hasten death rather than extending life (Forster et al., 2003; Mulvey et al., 2014; Lazic et al., 2020). For everyone else, however, adopting a diet that promotes mitochondrial catabolism and reduces extra-mitochondrial anabolism helps improve the health of the gut microbiota and stave off disease.
Among the beneficial bacteria that in the large intestine thrive on little or no oxygen are many that ferment soluble and insoluble fibers or starches that in the small intestine resist digestion (Sajilata et al., 2006; Canfora et al., 2015; Cryan et al., 2019; Silva et al., 2020; Gill et al., 2021). These fibers and starches can be found in abundance in, for example, fruits and vegetables and cooled-off boiled potatoes. During the fermentation of these fibers, fatty acids are produced by bacteria such as Bacteroides (Figure 5), and these fatty acids can then be consumed by the host or by other bacteria that, in turn, produce different fatty acids. Bacteroides thetaiotaomicron, for example, ferments dietary fibers and releases the fatty acid acetate; the firmicute Eubacterium rectale consumes this acetate and releases the fatty acid butyrate (Shoaie et al., 2013).
Butyrate is a prime fuel for the mitochondria of colon cells, which typically burn it down with lots of oxygen (Donohoe et al., 2011; Byndloss et al., 2017). The low oxygen levels in the gut that result from butyrate catabolism is, as mentioned, particularly harmful to gut pathogens. At the same time, mitochondria that are well-nourished with butyrate should be able to offer better support to the cells of the gut wall and gut-blood barrier (Jackson and Theiss, 2020), which in turn can then better prevent gut pathogens from invading the bloodstream. Whether mitochondria mediate the effect or not, increased butyrate production and exposure has indeed been associated with a better functioning gut-blood barrier (Lewis et al., 2010; Knudsen et al., 2018; but cf. Tabat et al., 2020).
Butyrate affects pathogens in a more direct way as well. The fatty acid consists of a very short chain of carbon atoms, and like other fatty acids of this kind, dissolves both the protective outer layer of viruses and the type of cell-wall that pathogenic microbes tend to have more often than do benign ones (Alcock et al., 2012). Likely because they reduce the need for the immune system to act, very-short-chain fatty acids, and butyrate in particular, have an anti-inflammatory effect (Fernández et al., 2016; Couto et al., 2020). Inflammation relies not only on catabolism but also on anabolism (Wang et al., 2019). So, the toning down of inflammation should, help reduce gut oxygen levels further and create a virtuous cycle.
One of the pathogens directly harmed by butyrate is Helicobacter pylori (Yonezawa et al., 2012), which is typically found in the mouth and digestive tract – especially the stomach – of about half the world’s population (Doulberis et al., 2018). H. pylori releases a toxin that induces mitochondria of white blood cells to kill their host cells so that they can no longer harm the pathogen (see also Foo et al., 2010; Akazawa et al., 2015). If H. pylori is abundant enough, it can cause the side effects of ulcers and stomach cancer (Foo et al., 2010; Akazawa et al., 2015). The bacterium is typically found far from the brain and it is unclear whether it can trigger amyloid deposition (Wang et al., 2015) or not (Albaret et al., 2020). Its eradication fails to improve already existing symptoms of neurodegeneration (Huang et al., 2018; Tan et al., 2020). Still, H. pylori does appear to inflame neurons (Albaret et al., 2020) and has been found to raise the risk of Parkinson’s disease (Çamcı and Oğuz, 2016; Shen X. et al., 2017; Huang et al., 2018, 2021). There are indications that, especially in combination with other pathogens (Beydoun et al., 2020), H. pylori is also a risk factor for Alzheimer’s and associated conditions (see also Shen L. et al., 2017; Doulberis et al., 2018; but cf. Fani et al., 2018). Injecting H. pylori in between the membranes lining the abdominal cavity has been found to lead to learning and memory deficits in rats (Wang et al., 2015).
Short-chain fatty acids not only benefit mitochondria and harm gut pathogens like H. pylori, but also have another remarkable effect: they strongly inhibit the aggregation of amyloids, or at least that of beta-amyloid (shown in vitro; Ho et al., 2018). The levels of butyrate in feces correlate with those in the brain (shown in mice: Zhang et al., 2017), and tellingly, the feces of patients with Parkinson’s contain abnormally few butyrate-producing bacteria (Unger et al., 2016; Cirstea et al., 2020). Moreover, concentrations of butyrate in feces have been associated not only with Parkinson’s in humans (Unger et al., 2016), but also with aging in humans in general (Duncan and Flint, 2013) and with an Alzheimer-like condition in mice (Zhang et al., 2017). A nutrient that is particularly good for our mitochondria (butyrate) thus happens to be particularly harmful to gut pathogens (like H. pylori) and helps prevent or delay neurodegenerative disease.
The production of fatty acids through bacterial fermentation of fibers in the colon can be improved by consuming probiotics (foods rich in beneficial bacteria such as fermented products like yoghurt or kimchi) and prebiotics (foods rich in soluble or insoluble fibers probiotic bacteria ferment) (LeBlanc et al., 2017). Fatty acids are an important nutrient for the mitochondria of colon cells, but also for those elsewhere. In addition, they can be converted into cholesterol and other fatty substances with a wide range of uses (LeBlanc et al., 2017). One can induce a Parkinson’s-like condition in rats by injecting a poison in their right medial forebrain bundle (Nurrahma et al., 2021). Feeding such rats the probiotic Lactobacillus salivarius improves their mitochondrial function, raises antioxidant levels, and prevents deterioration in movement, loss of muscle mass, and loss of dopamine neurons (Nurrahma et al., 2021). A prebiotic has a comparable, but more limited, effect (Nurrahma et al., 2021). Consistently, two somewhat similar studies that induced an Alzheimer’s-like, rather than a Parkinson’s-like, condition showed a protective effect of two probiotic strains of Peng et al., 2014; Divyashri et al., 2015; Lactobacillus plantarum Mallikarjuna et al., 2016; see also Cryan et al., 2019 for a review, and in addition Abraham et al., 2019).
A meta-analysis across several studies involving humans failed to find any benefit of probiotics on cognition (Marx et al., 2020). Yet this analysis did not consider the health status of participants and a probiotic might not benefit people who already have a healthy gut microbiota, as the authors themselves point out, or who have already incurred irreparable damage. Probiotics do ameliorate conditions like obesity and diabetes (Hampe and Roth, 2017), which typically involve disturbed metabolism and mitochondrial dysfunction and are risk factors of both Parkinson’s and Alzheimer’s (Arnold et al., 2018; Biosa et al., 2018; Vieira et al., 2018; Chornenkyy et al., 2019; Luca et al., 2019; Rigotto and Basso, 2019; Cheong et al., 2020; Carranza-Naval et al., 2021; Nguyen et al., 2021). Diabetes is, in fact, associated with a reduction in butyrate-producing bacteria (Maniar et al., 2017). And Metformin, a prime drug against diabetes, happens to benefit butyrate-producing bacteria (Hampe and Roth, 2017; Maniar et al., 2017), seems to improve Alzheimer’s disease, and might even delay aging more generally (Maniar et al., 2017). So, healthy mitochondria promote an oxygen-avoiding, butyrate-producing gut microbiota (see section “Mitochondria’s Oxygen Consumption Benefits the Microbiota”), and conversely, such a microbiota also promotes the health of mitochondria.
Given that supplementing diets with pro- or pre-biotics typically increases gut microbial fatty acid production, and that fats are fatty-acid compounds, it is interesting to note that an unsupplemented diet poor in carbohydrates, no more than adequate in protein, but high in fats, has repeatedly been found to produce similar benefits. The diet improves mitochondrial function after brain trauma (Greco et al., 2016) and can prevent or ameliorate cognitive deficits and Alzheimer’s and Parkinson’s disease (but cf. Bostock et al., 2017; Włodarek, 2019; Cunnane et al., 2020; Lilamand et al., 2020; Rawat et al., 2021). The diet is intended to mimic the positive health effects of those that restrict the consumption of food more broadly. Still, for elderly people who are degenerating and have already begun to lose weight, such dieting may no longer be advisable (Włodarek, 2019) (see also section “Mitochondria’s Oxygen Consumption Benefits the Microbiota”).
The highlight of a satisfying meal is typically some source of protein: meat, fish, cheese. Its component amino acids stimulate growth and reproduction at the expense of maintenance operations (Kim and Guan, 2019; Kitada et al., 2021). Essential amino acids like methionine, which are critical to normal growth, are obtained from food with the help of enzymes, the microbiota in the small intestine, and – to a small extent – also the microbiota in the large intestine (Ma and Ma, 2019). Methionine can be recycled from dysfunctional mitochondria or other cellular debris as well (Kitada et al., 2021). A chemical reaction of methionine with adenosine triphosphate (ATP), which is biology’s most important portable energy store, produces S-adenosyl-methionine (SAM), which is biology’s most important molecule for the silencing or expression of genes (Loenen, 2006; Janke et al., 2015; Kramer and Bressan, 2021). In fact, it is not methionine itself but SAM that promotes growth and reproduction and diminishes maintenance and recycling (Kitada et al., 2021). Mitochondria are the most important source of ATP and the production of SAM depends on mitochondria too, and in part on mitochondrial DNA. This means that normal growth depends on the health and genetic makeup of mitochondria (Kramer and Bressan, 2021).
Provided its intake remains sufficient, the less methionine is consumed, the more its metabolization is directed toward the production of glutathione and the gas hydrogen sulfide. Glutathione is an antioxidant and in small quantities so is hydrogen sulfide, but hydrogen sulfide is particularly important as a kind of neurotransmitter (Paul and Snyder, 2018; Kitada et al., 2021; Paul, 2021; Sokolov et al., 2021). The less methionine or SAM and the more hydrogen sulfide is available to a cell, the more of the cell’s dysfunctional mitochondria and other debris are recycled, the better its remaining mitochondria and cellular components perform, and the fewer free radicals its mitochondria release (Paul and Snyder, 2018; Wu et al., 2020; Kitada et al., 2021; Sokolov et al., 2021). This, in turn, allows fatty-acid producing gut bacteria to thrive at the expense of gut pathogens (Wu et al., 2020) and promotes a normal permeability of the gut-blood barrier (Ramalingam et al., 2010). In the process, homocysteine derived from methionine is also metabolized (Paul and Snyder, 2018; Kitada et al., 2021), and the resulting low levels of homocysteine have been found to promote the normal permeability of the blood-brain barrier (Kamath et al., 2006). Restricting the consumption of protein, and in particular of methionine, improves not only the health of mitochondria, the gut microbiota, and protective barriers, but also the health and lifespan of the host (Soultoukis and Partridge, 2016; Kitada et al., 2021; Sokolov et al., 2021).
Small amounts of hydrogen sulfide are anti-inflammatory (Blachier et al., 2007) and as a result promote the health of both our mitochondria and microbiota. In addition, small amounts of this gas inhibit the formation of abnormal amyloid compounds of alpha-synuclein (Hou et al., 2017), tau (Giovinazzo et al., 2021), and beta-amyloid (Rosario-Alomar et al., 2015) and helps keep at bay neurodegenerative disease (Hou et al., 2017; Paul and Snyder, 2018; Munoz-Pinto et al., 2021; Paul, 2021). Conversely, mutations in genes involved in methionine metabolism, dysregulated methionine metabolism, and abnormal hydrogen-sulfide and glutathione levels have all been implicated in neurodegenerative disease and in aging more generally (Paul, 2021; Sokolov et al., 2021).
Since primordial times, hydrogen sulfide has sustained life (Fu et al., 2012; Lane, 2015; Paul and Snyder, 2018), and in small amounts it still has a net beneficial effect on us today (Blachier et al., 2007; Paul and Snyder, 2018; Kitada et al., 2021; Sokolov et al., 2021). Many people, however, are exposed to larger quantities of this smelly gas, quantities that are more toxic than beneficial (Blachier et al., 2007; Sokolov et al., 2021). Some gut microbes in the colon metabolize either sulfate or – more importantly – sulfur-containing amino acids, which are both obtained from partially digested protein (Blachier et al., 2007). Among the amino acid metabolizers are familiar pathogens like particular strains of Escherichia coli and Salmonella (Jackson and Theiss, 2020). These microbes are more flexible than many others. They thrive on oxygen but can do without it too. At least one particular strain of pathogenic E. coli switches its metabolism during gut inflammation toward the fermentation of amino acids when these are available, whereas more beneficial strains do not. This flexibility gives the pathogen, and others like it, an edge over its less flexible competition (Kitamoto et al., 2020).
During the metabolism by gut bacteria of sulfate or sulfur-containing amino acids, hydrogen sulfide is released, and mitochondria – especially those of colon cells (Goubern et al., 2007) – can use this gas as a fuel (Blachier et al., 2007; Fu et al., 2012). Yet if too much protein has been consumed, which benefits gut pathogens, the concentration of hydrogen sulfide can become harmful to mitochondria (Blachier et al., 2007; Mottawea et al., 2016). As these mitochondria begin to malfunction, butyrate catabolism and energy production decrease; free-radical production, inflammation, gut-blood permeability increase; and intestinal disorders worsen (Blachier et al., 2007; Singh and Lin, 2015; Mottawea et al., 2016; Bajpai et al., 2018; Pal et al., 2018; Aguilar-López et al., 2020; Jackson and Theiss, 2020; Sokolov et al., 2021). Moreover, whereas low levels of hydrogen sulfide have a positive effect on the vascular system (Paul and Snyder, 2018) – and directly or indirectly diminish neuroinflammation, cognitive deficits, and the risk of Alzheimer’s and Parkinson’s – high levels exert the opposite effect (Paul and Snyder, 2018; Paul, 2021).
As an aside, note that like hydrogen sulfide, nitric oxide is also a gas that functions like a neurotransmitter. It is produced by both mitochondria (Litvinova et al., 2015) and the gut microbiota (Tse, 2017), its effects interact with those of hydrogen sulfide (Blachier et al., 2007), and it also has net beneficial consequences in small amounts and harmful ones in large amounts. And in large amounts, it too has been implicated in dysbiosis, mitochondrial dysfunction, and Alzheimer’s and Parkinson’s disease (Litvinova et al., 2015; Tse, 2017).
Let us return to protein, methionine, and hydrogen sulfide. Note that the health benefits that can be obtained through restricting food intake, including delayed neurodegeneration, can also be obtained by simply restricting protein, or even just methionine, intake (see also Wei et al., 2017; Thelen and Brown-Borg, 2020; Wu et al., 2020; Kitada et al., 2021; McCarty and Lerner, 2021). Like protein, vegetables like broccoli and cauliflower contain sulfur and during their digestion hydrogen sulfide is released too (Citi et al., 2014). This release, however, is slow (Citi et al., 2014) and as a result has a positive effect on our health (Whiteman et al., 2010). Broccoli and cauliflower, and members of the Brassicaceae family more generally, also contain sulforaphane. This sulfurous substance is toxic not only to a number of the vegetables’ own microbial enemies (Schillheim et al., 2018) but also to many of our gut pathogens, including H. pylori (Kellingray et al., 2017; Le et al., 2020). So, whereas healthy mitochondria promote an oxygen-avoiding, butyrate-producing gut microbiota, and conversely such a microbiota also promotes the health of mitochondria, protein- or methionine-fermenting gut microbiota harms them instead. And this harmful effect has downstream ones on both Alzheimer’s and Parkinson’s.
Besides low levels of oxygen and hydrogen sulfide, and high levels of fatty acids, a healthy gut requires the proper regulation of immunity, hormones, gut-blood permeability, bowel movements, and stomach-acid and bile secretion. The orchestration of the full system is demanding enough that the gut has dedicated to this task its own bespoke nervous system. Hinting at its importance, this “enteric” nervous system evolved before the central one did (Snoek et al., 2010; Furness, 2012; Cryan et al., 2019; Niesler et al., 2021) and comprises between 400 and 600 million neurons, covering the entire 32 m2 inside surface of the intestines (Helander and Fändriks, 2014).
Dysfunctional gut receptors of the enteric nervous system, as well as genetic aberrations of the enteric nervous system, the central nervous system, and the connections between them, have all been associated with gut disorders (Snoek et al., 2010; Cryan et al., 2019; Niesler et al., 2021). Alzheimer’s (Cui et al., 2018; Waziry et al., 2020) and Parkinson’s (Liu et al., 2020) raise the risk of stroke. Remarkably, this kind of trauma is associated not just with the death of neurons in the brain but also with subsequent degeneration of neurons in the gut (Cheng et al., 2016; Stanley et al., 2016; Blanke et al., 2021) and with dysbiosis (Cryan et al., 2019, 2020). Moreover, zebrafish genetically engineered to grow to maturity without an enteric nervous system become more susceptible to dysbiosis and gut inflammation (Rolig et al., 2017). A transplant of a healthy enteric nervous system corrects this situation, as does a simple probiotic (Rolig et al., 2017). That a probiotic can be effective suggests that the enteric nervous system is less important when the gut microbiota is healthy than when it is not.
The enteric nervous system is part of the autonomous nervous system along with the sympathetic nervous system, which is involved in urgent actions and psychological stress, and the parasympathetic nervous system, which is involved in maintaining homeostasis and calming down stress. The sympathetic and parasympathetic nervous systems connect the enteric nervous system to the central one and enable two-way signaling between them (Figure 7). A pair of parasympathetic nerves, together referred to as the vagus nerve, is particularly important (Bonaz et al., 2017). Gut microbes can affect the firing of this nerve through their production and release of nutrients like fatty acids and vitamins, neurotransmitters like serotonin and dopamine, and various toxins (LeBlanc et al., 2017; Cryan et al., 2019; Fülling et al., 2019). Changes in the composition of the gut microbiota can also, via the vagus nerve, be relayed to the brain by the host’s own enteric neurotransmitters, hormones, and immune signaling molecules (Bonaz et al., 2017; Cryan et al., 2019). Gut viruses and gut microbes that manage to slip or break through protective barriers, or the toxins or other chemicals of gut microbes that do so, can also physically travel up to the brain via either the bloodstream, the lymph system, or the sympathetic and parasympathetic nervous systems – including the vagus nerve (Zhan et al., 2018; Cryan et al., 2019; Fülling et al., 2019; Kowalski and Mulak, 2019; Huang et al., 2021; Figure 7). So, any battle between mitochondria and microbiota in the gut can easily have consequences as far away as in the brain.
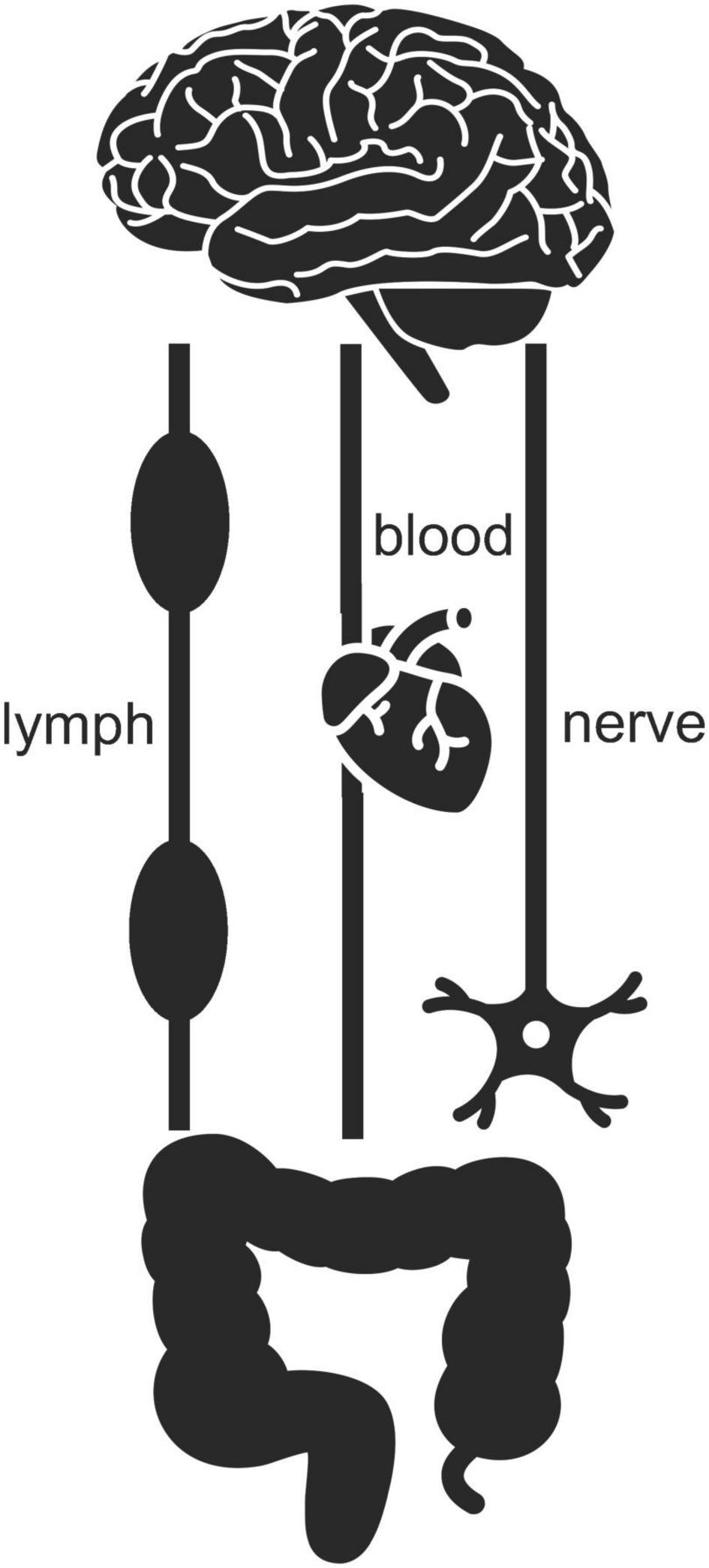
Figure 7. Two-way signaling between gut and brain can proceed via the lymph, blood, and nervous systems. Icons adapted from the Noun Project: Clockwise (brain), Tezar Tantular (heart), public domain (nerve), Bom Symbols (intestine).
Beside their hosts, gut bacteria can produce amyloids too (Friedland and Chapman, 2017; Tursi and Tükel, 2018; Van Gerven et al., 2018; Vidakovic et al., 2018). Curli, for example, is an amyloid produced by for example E. coli that binds these bacteria together in a biofilm that helps protect them from viruses and the host’s immune system (Friedland and Chapman, 2017; Fulop et al., 2018; Tursi and Tükel, 2018; Van Gerven et al., 2018; D’Argenio and Sarnataro, 2019; Kowalski and Mulak, 2019; Miraglia and Colla, 2019; Huang et al., 2021; Miller et al., 2021). The bacterial amyloids resemble those of the host and can trigger the folding and clumping of the host’s amyloids. One study compared aged rats of a kind that is particularly susceptible to alpha-synuclein buildup and that were force-fed either Curli-producing E. coli or nearly identical control E. coli that were incapable of producing Curli. As predicted, those that had received the Curli-producing E. coli via the mouth deposited more alpha-synuclein in the brain, and showed more signs of inflammation there, than those who had received the control E. coli (see also Lundmark et al., 2005; Chen et al., 2016). A polyphenol found in especially green tea blocks the formation of Curli and biofilm by E. coli and reduces alpha-synuclein aggregation and deposition in the striatum and midbrain of E. coli-infected mice that had been genetically engineered to overproduce human alpha-synuclein (Sampson et al., 2020). Consumption of the polyphenol reduced both gastrointestinal and motoric deficits.
After injecting bacterial amyloids into the wall of the stomach of mice, aggregates of fluorescent mouse beta-amyloid were found within hours in the enteric nervous system, within a month elsewhere in the gut as well, and within a year in the vagus nerve and brain, including the hippocampus (Sun et al., 2020a). The spread of beta-amyloid from the stomach to the brain subsequently compromised both spatial short-term, and spatial long-term, memory (see also Choi et al., 2018; Sun et al., 2020a).
Broadly similar results were obtained when aggregates of alpha-synuclein rather than beta-amyloid were orally administered or injected into various locations in and around the gut, with the alpha-synuclein affecting also specifically the substantia nigra (see also Braak et al., 2003; Holmqvist et al., 2014; Rietdijk et al., 2017; Barbut et al., 2019; Kim et al., 2019; Lohmann et al., 2019; Van Den Berge et al., 2019). One study investigated mice that had been engineered to overexpress human alpha-synuclein and that, in addition, had been raised germ free (Sampson et al., 2016; see also Sampson, 2020). Of these mice, those given a (microbe rich) fecal transplant from Parkinson’s patients, rather than healthy people, ended up with more dysbiosis, intestinal problems, and Parkinsonian motor deficits. Mice that did not overexpress human alpha-synuclein were unaffected by a such a fecal transplant. And mice that were not raised germ free benefited from antibiotic treatment. In line with these findings, human Parkinson’s patients with bacterial overgrowth in the small intestine show reduced Parkinsonian symptoms after antibiotic treatment (Fasano et al., 2013). Skepticism remains (Munoz-Pinto et al., 2021). Indeed, live imaging of mitochondrial function and calcium levels in neurons contained in small-intestine biopsies did not reveal any difference between Parkinson’s patients and their unaffected partners (Desmet et al., 2017). No differences with regard to alpha-synuclein were found either. With only 15 couples as participants, the study might have lacked sufficient power. Several other studies, however, also failed to find a difference between patients and controls in alpha-synuclein accumulation in the gut (Munoz-Pinto et al., 2021).
Even in young, healthy people, potentially harmful aggregates of alpha-synuclein are often found in the appendix (Killinger et al., 2018) – a protrusion of the colon that first and foremost serves as a safe house for beneficial bacteria (Bollinger et al., 2007). When illness harms the gut microbiota, the bacteria from the appendix can repopulate the gut and, if these bacteria are beneficial, they can help restore the health of the gut microbiota (Bollinger et al., 2007). Still, because the appendix can also harbor pathogens, its removal can sometimes have a positive effect too. An epidemiological study involving more than 1.6 million participants – and thus definitely not underpowered – showed, in fact, that removing the appendix diminishes the risk of Parkinson’s decades later, especially in people from rural areas who may have been more exposed than others to dysbiosis-causing pesticides (Killinger et al., 2018). The procedure also delayed Parkinson’s by up to several years among those who eventually did develop the disease (Killinger et al., 2018).
Severing the vagus nerve, rather than removing the appendix, can also hinder the propagation of alpha-synuclein from the gut to the brain in mice (Kim et al., 2019) and humans (Svensson et al., 2015) and improve a Parkinson’s-like affliction and Parkinson’s itself in, respectively, animals and humans (Borghammer, 2018; Liddle, 2018; Lionnet et al., 2018; Fitzgerald et al., 2019; Kim et al., 2019; Munoz-Pinto et al., 2021). This does not always happen, however, and overall the evidence so far is still mixed (Borghammer, 2018; Lionnet et al., 2018). Removal of the vagus nerve, of course, still leaves the bloodstream and lymph system open to amyloids, pathogens, or their toxins, and this could have confounded results. Indeed, in baboons, injected alpha-synuclein has been found to be transported from the gut to the brain and vice versa via the bloodstream (Arotcarena et al., 2020).
The bloodstream is affected early on in both Alzheimer’s (D’Andrea, 2016) and Parkinson’s (Berganzo et al., 2013; Pierzchlińska et al., 2021), so much so in fact that it has been argued that Alzheimer’s may be just as much a vascular disease as a neurological one (D’Andrea, 2016; Fulop et al., 2018). At least in Parkinson’s, however, the vascular problems are in part related to a disturbance of circadian rhythms, with high blood pressure being a problem particularly at night rather than during the day (Berganzo et al., 2013). Circadian rhythms are, in fact, disturbed in both Parkinson’s (Liu et al., 2021) and Alzheimer’s (Uddin et al., 2020; Sharma et al., 2021), are intertwined with cyclic changes in mitochondrial metabolism (Kramer and Bressan, 2019; Aguilar-López et al., 2020), and affect – and are affected by – the composition of the microbiota (Aguilar-López et al., 2020; Bishehsari et al., 2020; Sen et al., 2021). In sum, gut microbes need not necessarily leave the gut to trigger amyloid deposition in the brain. Whether this happens depends in part on mitochondrial metabolism and circadian rhythms. And to close the circle, these circadian rhythms are under the control of a central biological clock that is located in the brain (Kramer and Bressan, 2019; Aguilar-López et al., 2020).
At first sight, Alzheimer’s and Parkinson’s seem quite distinct diseases; they strike different areas of the brain in different ways and produce different behavioral deficits. It has been shown, for example, that experimentally inducing a specific dysfunction within the mitochondria (involving inner-membrane Complex I) of a specific kind of neuron (the type that uses dopamine as a neurotransmitter) in a specific part of the brain (the substantia nigra, and likely just the pars compacta part of it) is sufficient to induce progressively worsening symptoms of Parkinson’s disease that are not shared with Alzheimer’s disease (Gonzaìlez-Rodriìguez et al., 2021). Yet in natural settings, outside the lab, many patients suffer from a mix of the two diseases, with similar mitochondrial dysfunction affecting multiple cell types; with amyloid aggregates associated with either disease often found in the other as well; with similar accompanying infections, inflammation, and dysbiosis; and with similar effects of diet, lifestyle, trauma, and exposure to toxicants. Naturally developing Alzheimer’s and Parkinson’s may more accurately be described as different manifestations of one and the same systemic problem that involves not just certain areas of the brain, not just the brain as whole, not just the body as a whole, but the entire human superorganism, including even the non-human entities that reside inside us (see also Kramer and Bressan, 2015). Early players in neurodegeneration are deteriorating aerobic ex-bacteria (mitochondria), located throughout our body and brain, and thriving aerobic bacteria (unhealthy microbiota), located mostly as far away from the brain as the gut. Despite the diversity in neurodegenerative diseases, there is a common theme among them: a synergy between mitochondria and microbiota early in life is sooner or later disturbed and, late in life, turned into its very opposite.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
I am greatly indebted to Paola Bressan for major edits of an early version of this article, for insightful comments on a late version, for making me aware of various relevant articles, and for technical assistance.
Abraham, D., Feher, J., Scuderi, G. L., Szabo, D., Dobolyi, A., Cservenak, M., et al. (2019). Exercise and probiotics attenuate the development of Alzheimer’s disease in transgenic mice: Role of microbiome. Exp. Gerontol. 115, 122–131. doi: 10.1016/j.exger.2018.12.005
Acker, Z. P. V., Luyckx, E., and Dewilde, S. (2019). Neuroglobin expression in the brain: A story of tissue homeostasis preservation. Mol. Neurobiol. 56, 2101–2122. doi: 10.1007/s12035-018-1212-8
Aguilar-López, B. A., Moreno-Altamirano, M. M. B., Dockrell, H. M., Duchen, M. R., and Sánchez-García, F. J. (2020). Mitochondria: an integrative hub coordinating circadian rhythms, metabolism, the microbiome, and Immunity. Front. Cell Dev. Biol. 8:51. doi: 10.3389/fcell.2020.00051
Akazawa, Y., Matsuda, K., Isomoto, H., Matsushima, K., Kido, Y., Urabe, S., et al. (2015). BH3-only protein Bim is associated with the degree of Helicobacter pylori-induced gastritis and is localized to the mitochondria of inflammatory cells in the gastric mucosa. Int. J. Med. Microbiol. 305, 553–562. doi: 10.1016/j.ijmm.2015.07.002
Albaret, G., Sifré, E., Floch, P., Laye, S., Aubert, A., Dubus, P., et al. (2020). Alzheimer’s disease and Helicobacter pylori infection: Inflammation from stomach to brain. J. Alzheimers Dis. 73, 801–809. doi: 10.3233/JAD-190496
Alcock, J., Franklin, M. L., and Kuzawa, C. W. (2012). Nutrient signaling: Evolutionary origins of the immune-modulating effects of dietary fat. Q Rev. Biol. 87, 187–223. doi: 10.1086/666828
Alzheimer’s Association. (2019). 2019 Alzheimer’s disease facts and figures. Alzheimers Dem. 15, 321–387. doi: 10.1016/j.jalz.2019.01.010
Angajala, A., Lim, S., Phillips, J. B., Kim, J.-H., Yates, C., You, Z., et al. (2018). Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front. Immunol. 9:1605. doi: 10.3389/fimmu.2018.01605
Anton, S. D., Moehl, K., Donahoo, W. T., Marosi, K., Lee, S. A., Mainous, I. I. I. A. G., et al. (2018). Flipping the metabolic switch: understanding and applying the health benefits of fasting. Obesity 26, 254–268. doi: 10.1002/oby.22065
Arnold, S. E., Arvanitakis, Z., Macauley-Rambach, S. L., Koenig, A. M., Wang, H.-Y., Ahima, R. S., et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 14, 168–181. doi: 10.1038/nrneurol.2017.185
Arotcarena, M.-L., Dovero, S., Prigent, A., Bourdenx, M., Camus, S., Porras, G., et al. (2020). Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain 143, 1462–1475. doi: 10.1093/brain/awaa096
Ashraf, G. M., Tarasov, V. V., Makhmutovà, P., Chubarev, V. N., AvilaRodriguez, M., Bachurin, S. O., et al. (2019). The possibility of an infectious etiology of Alzheimer disease. Mol. Neurobiol. 56, 4479–4491. doi: 10.1007/s12035-018-1388-y
Bajpai, P., Darra, A., and Agrawal, A. (2018). Microbe-mitochondrion crosstalk and health: An emerging paradigm. Mitochondrion 39, 20–25. doi: 10.1016/j.mito.2017.08.008
Balmik, A. A., and Chinnathambi, S. (2021). Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun. Signal 19:51. doi: 10.1186/s12964-021-00732-z
Barbut, D., Stolzenberg, E., and Zasloff, M. (2019). Gastrointestinal immunity and alpha-synuclein. J. Parkinson Dis. 9, S313–S322. doi: 10.3233/JPD-191702
Bayer, T., and Wirths, O. (2010). Intracellular accumulation of amyloid-beta - a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci. 2:8. doi: 10.3389/fnagi.2010.00008
Beatman, E. L., Massey, A., Shives, K. D., Burrack, K. S., Chamanian, M., Morrison, T. E., et al. (2015). Alpha-synuclein expression restricts RNA viral infections in the brain. J. Virol. 90, 2767–2782. doi: 10.1128/JVI.02949-15
Belloy, M. E., Napolioni, V., and Greicius, M. D. (2019). A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 101, 820–838. doi: 10.1016/j.neuron.2019.01.056
Bendor, J. T., Logan, T. P., and Edwards, R. H. (2013). The function of α-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
Berganzo, K., Díez-Arrola, B., Tijero, B., Somme, J., Lezcano, E., Llorens, V., et al. (2013). Nocturnal hypertension and dysautonomia in patients with Parkinson’s disease: Are they related. J. Neurol. 260, 1752–1756. doi: 10.1007/s00415-013-6859-5
Bernal-Conde, L. D., Ramos-Acevedo, R., Reyes-Hernández, M. A., Balbuena-Olvera, A. J., Morales-Moreno, I. D., Argüero-Sánchez, R., et al. (2020). Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front. Neurosci. 13:1399. doi: 10.3389/fnins.2019.01399
Beydoun, M. A., Beydoun, H. A., Weiss, J., Hossain, S., El-Hajj, Z. W., and Zonderman, A. B. (2020). Helicobacter pylori, periodontal pathogens, and their interactive association with incident all-cause and Alzheimer’s disease dementia in a large national survey. Mol. Psychiatr. 2020, 1–16. doi: 10.1038/s41380-020-0736-2
Biosa, A., Outeiro, T. F., Bubacco, L., and Bisaglia, M. (2018). Diabetes mellitus as a risk factor for Parkinson’s disease: a molecular point of view. Mol. Neurobiol. 55, 8754–8763. doi: 10.1007/s12035-018-1025-9
Bishehsari, F., Voigt, R. M., and Keshavarzian, A. (2020). Circadian rhythms and the gut microbiota: from the metabolic syndrome to cancer. Nat. Rev. Endocrinol. 16, 731–739. doi: 10.1038/s41574-020-00427-4
Blachier, F., Mariotti, F., Huneau, J.-F., and Tomé, D. (2007). Effects of amino acid-derived luminal metabolites on the colonic epithelium and physiopathological consequences. Amino Acids 33, 547–562. doi: 10.1007/s00726-006-0477-9
Blanke, E. N., Holmes, G. M., and Besecker, E. M. (2021). Altered physiology of gastrointestinal vagal afferents following neurotrauma. Neural. Regen. Res. 16:254. doi: 10.4103/1673-5374.290883
Bollinger, R. R., Barbas, A. S., Bush, E. L., Lin, S. S., and Parker, W. (2007). Biofilms in the large bowel suggest an apparent function of the human vermiform appendix. J. Theor. Biol. 249, 826–831. doi: 10.1016/j.jtbi.2007.08.032
Bonaz, B., Sinniger, V., and Pellissier, S. (2017). The vagus nerve in the neuro-immune axis: implications in the pathology of the gastrointestinal tract. Front. Immunol. 8:1452. doi: 10.3389/fimmu.2017.01452
Borghammer, P. (2018). How does Parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov. Disord. 33, 48–57. doi: 10.1002/mds.27138
Borsche, M., Pereira, S. L., Klein, C., and Grünewald, A. (2021). Mitochondria and Parkinson’s disease: clinical, molecular, and translational aspects. J. Parkinson Dis. 11, 45–60. doi: 10.3233/JPD-201981
Bostock, E. C. S., Kirkby, K. C., and Taylor, B. V. M. (2017). The current status of the ketogenic diet in psychiatry. Front. Psychiatry 8:43. doi: 10.3389/fpsyt.2017.00043
Braak, H., Tredici, K. D., Rüb, U., de Vos, R. A. I., Jansen Steur, E. N. H., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Burmester, T., Weich, B., Reinhardt, S., and Hankeln, T. (2000). A vertebrate globin expressed in the brain. Nature 407, 520–523. doi: 10.1038/35035093
Byndloss, M. X., Olsan, E. E., Rivera-Chávez, F., Tiffany, C. R., Cevallos, S. A., Lokken, K. L., et al. (2017). Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575. doi: 10.1126/science.aam9949
Çamcı, G., and Oğuz, S. (2016). Association between Parkinson’s disease and Helicobacter pylori. J. Clin. Neurol. 12, 147–150. doi: 10.3988/jcn.2016.12.2.147
Canfora, E. E., Jocken, J. W., and Blaak, E. E. (2015). Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 11, 577–591. doi: 10.1038/nrendo.2015.128
Cardoso, S. M., and Empadinhas, N. (2018). The microbiome-mitochondria dance in prodromal Parkinson’s disease. Front. Physiol. 9:471. doi: 10.3389/fphys.2018.00471
Carranza-Naval, M. J., Vargas-Soria, M., Hierro-Bujalance, C., Baena-Nieto, G., Garcia-Alloza, M., Infante-Garcia, C., et al. (2021). Alzheimer’s disease and diabetes: Role of diet, microbiota and inflammation in preclinical models. Biomolecules 11:262. doi: 10.3390/biom11020262
Chalazonitis, A., and Rao, M. (2018). Enteric nervous system manifestations of neurodegenerative disease. Brain Res. 1693, 207–213. doi: 10.1016/j.brainres.2018.01.011
Chen, S. G., Stribinskis, V., Rane, M. J., Demuth, D. R., Gozal, E., Roberts, A. M., et al. (2016). Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci. Rep. 6, 1–10. doi: 10.1038/srep34477
Cheng, F., Vivacqua, G., and Yu, S. (2011). The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 42, 242–248. doi: 10.1016/j.jchemneu.2010.12.001
Cheng, X., Boza-Serrano, A., Turesson, M. F., Deierborg, T., Ekblad, E., and Voss, U. (2016). Galectin-3 causes enteric neuronal loss in mice after left sided permanent middle cerebral artery occlusion, a model of stroke. Sci. Rep. 6:32893. doi: 10.1038/srep32893
Cheong, J. L. Y., de Pablo-Fernandez, E., Foltynie, T., and Noyce, A. J. (2020). The association between type 2 diabetes mellitus and Parkinson’s disease. J. Parkinson Dis. 2020, 1–15. doi: 10.3233/JPD-191900
Choi, J. G., Kim, N., Ju, I. G., Eo, H., Lim, S.-M., Jang, S.-E., et al. (2018). Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Sci. Rep. 8:1275. doi: 10.1038/s41598-018-19646-x
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D’Avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278. doi: 10.1038/nature13800
Chornenkyy, Y., Wang, W., Wei, A., and Nelson, P. T. (2019). Alzheimer’s disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 29, 3–17. doi: 10.1111/bpa.12655
Cirstea, M. S., Yu, A. C., Golz, E., Sundvick, K., Kliger, D., Radisavljevic, N., et al. (2020). Microbiota composition and metabolism are associated with gut function in parkinson’s disease. Mov. Disord. 35, 1208–1217. doi: 10.1002/mds.28052
Citi, V., Martelli, A., Testai, L., Marino, A., Breschi, M. C., and Calderone, V. (2014). Hydrogen sulfide releasing capacity of natural isothiocyanates: is it a reliable explanation for the multiple biological effects of Brassicaceae. Planta Med. 80, 610–613. doi: 10.1055/s-0034-1368591
Couto, M. R., Gonçalves, P., Magro, F., and Martel, F. (2020). Microbiota-derived butyrate regulates intestinal inflammation: focus on inflammatory bowel disease. Pharmacol. Res. 2020:104947. doi: 10.1016/j.phrs.2020.104947
Cryan, J. F., O’Riordan, K. J., Cowan, C. S. M., Sandhu, K. V., Bastiaanssen, T. F. S., Boehme, M., et al. (2019). The microbiota-gut-brain axis. Physiol. Rev. 99, 1877–2013. doi: 10.1152/physrev.00018.2018
Cryan, J. F., O’Riordan, K. J., Sandhu, K., Peterson, V., and Dinan, T. G. (2020). The gut microbiome in neurological disorders. Lancet Neurol. 19, 179–194. doi: 10.1016/S1474-4422(19)30356-4
Cui, P., Ma, X., Li, H., Lang, W., and Hao, J. (2018). Shared biological pathways between Alzheimer’s disease and ischemic stroke. Front. Neurosci. 12:605. doi: 10.3389/fnins.2018.00605
Cunnane, S. C., Trushina, E., Morland, C., Prigione, A., Casadesus, G., Andrews, Z. B., et al. (2020). Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 19, 609–633. doi: 10.1038/s41573-020-0072-x
Czepiel, J., Dróżdż, M., Pituch, H., Kuijper, E. J., Perucki, W., Mielimonka, A., et al. (2019). Clostridium difficile infection: review. Eur. J. Clin. Microbiol. 38, 1211–1221. doi: 10.1007/s10096-019-03539-6
D’Andrea, M. R. (2016). Intracellular Consequences of Amyloid in Alzheimer’s Disease. Amsterdam: Elsevier.
D’Andrea, M. R., and Nagele, R. G. (2010). Morphologically distinct types of amyloid plaques point the way to a better understanding of Alzheimer’s disease pathogenesis. Biotech. Histochem. 85, 133–147. doi: 10.3109/10520290903389445
D’Argenio, V., and Sarnataro, D. (2019). Microbiome influence in the pathogenesis of prion and Alzheimer’s diseases. Int. J. Mol. Sci. 20:4704. doi: 10.3390/ijms20194704
Del Tredici, K., and Braak, H. (2020). To stage, or not to stage. Curr. Opin. Neurobiol. 61, 10–22. doi: 10.1016/j.conb.2019.11.008
Desmet, A.-S., Cirillo, C., Tack, J., Vandenberghe, W., and Vanden Berghe, P. (2017). Live calcium and mitochondrial imaging in the enteric nervous system of Parkinson patients and controls. eLife 6:e26850. doi: 10.7554/eLife.26850
Dickson, D. W., Braak, H., Duda, J. E., Duyckaerts, C., Gasser, T., Halliday, G. M., et al. (2009). Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 8, 1150–1157. doi: 10.1016/S1474-4422(09)70238-8
Divyashri, G., Krishna, G., and Muralidhara Prapulla, S. G. (2015). Probiotic attributes, antioxidant, anti-inflammatory and neuromodulatory effects of Enterococcus faecium CFR 3003: in vitro and in vivo evidence. J. Med. Microbiol. 64, 1527–1540. doi: 10.1099/jmm.0.000184
Donohoe, D. R., Garge, N., Zhang, X., Sun, W., O’Connell, T. M., Bunger, M. K., et al. (2011). The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 13, 517–526. doi: 10.1016/j.cmet.2011.02.018
Dorsey, E. R., and Bloem, B. R. (2018). The parkinson pandemic – a call to action. JAMA Neurol. 75, 9–10. doi: 10.1001/jamaneurol.2017.3299
Dorsey, E. R., Constantinescu, R., Thompson, J. P., Biglan, K. M., Holloway, R. G., Kieburtz, K., et al. (2007). Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68, 384–386. doi: 10.1212/01.wnl.0000247740.47667.03
Doulberis, M., Kotronis, G., Thomann, R., Polyzos, S. A., Boziki, M., Gialamprinou, D., et al. (2018). Impact of Helicobacter pylori on Alzheimer’s disease: what do we know so far. Helicobacter 23, e12454. doi: 10.1111/hel.12454
Duncan, S. H., and Flint, H. J. (2013). Probiotics and prebiotics and health in ageing populations. Maturitas 75, 44–50. doi: 10.1016/j.maturitas.2013.02.004
Fani, L., Wolters, F. J., Ikram, M. K., Bruno, M. J., Hofman, A., Koudstaal, P. J., et al. (2018). Helicobacter pylori and the risk of dementia: a population-based study. Alzheimers Dement 14, 1377–1382. doi: 10.1016/j.jalz.2018.05.005
Fasano, A., Bove, F., Gabrielli, M., Petracca, M., Zocco, M. A., Ragazzoni, E., et al. (2013). The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov. Disord. 28, 1241–1249. doi: 10.1002/mds.25522
Feng, S.-T., Wang, Z.-Z., Yuan, Y.-H., Sun, H.-M., Chen, N.-H., and Zhang, Y. (2021). Update on the association between alpha-synuclein and tau with mitochondrial dysfunction: Implications for Parkinson’s disease. Eur. J. Neurosci. 53, 2946–2959. doi: 10.1111/ejn.14699
Fernández, J., Redondo-Blanco, S., Gutiérrez-del-Río, I., Miguélez, E. M., Villar, C. J., and Lombo, F. (2016). Colon microbiota fermentation of dietary prebiotics towards short-chain fatty acids and their roles as anti-inflammatory and antitumour agents: a review. J. Funct. Foods 25, 511–522. doi: 10.1016/j.jff.2016.06.032
Filippini, A., Gennarelli, M., and Russo, I. (2019). α-Synuclein and glia in parkinson’s disease: a beneficial or a detrimental duet for the endo-lysosomal system. Cell Mol. Neurobiol. 39, 161–168. doi: 10.1007/s10571-019-00649-9
Fitzgerald, E., Murphy, S., and Martinson, H. A. (2019). Alpha-synuclein pathology and the role of the microbiota in Parkinson’s disease. Front. Neurosci. 13:369. doi: 10.3389/fnins.2019.00369
Floud, S., Simpson, R. F., Balkwill, A., Brown, A., Goodill, A., Gallacher, J., et al. (2020). Body mass index, diet, physical inactivity, and the incidence of dementia in 1 million UK women. Neurology 94, e123–e132. doi: 10.1212/WNL.0000000000008779
Foo, J. H., Culvenor, J. G., Ferrero, R. L., Kwok, T., Lithgow, T., and Gabriel, K. (2010). Both the p33 and p55 subunits of the Helicobacter pylori VacA toxin are targeted to mammalian mitochondria. J. Mol. Biol. 401, 792–798. doi: 10.1016/j.jmb.2010.06.065
Forster, M. J., Morris, P., and Sohal, R. S. (2003). Genotype and age influence the effect of caloric intake on mortality in mice. FASEB J. 17, 690–692. doi: 10.1096/fj.02-0533fje
Friedland, R. P., and Chapman, M. R. (2017). The role of microbial amyloid in neurodegeneration. PLoS Pathog. 13:e1006654. doi: 10.1371/journal.ppat.1006654
Fu, M., Zhang, W., Wu, L., Yang, G., Li, H., and Wang, R. (2012). Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. P Natl. Acad. Sci. USA 109:2943. doi: 10.1073/pnas.1115634109
Fülling, C., Dinan, T. G., and Cryan, J. F. (2019). Gut Microbe to Brain Signaling: What Happens in Vagus. Neuron 101, 998–1002. doi: 10.1016/j.neuron.2019.02.008
Fulop, T., Witkowski, J. M., Bourgade, K., Khalil, A., Zerif, E., Larbi, A., et al. (2018). Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer’s Disease. Front. Aging Neurosci. 10:224. doi: 10.3389/fnagi.2018.00224
Furness, J. B. (2012). The enteric nervous system and neurogastroenterology. Nat. Rev. Gastro. Hepat. 9, 286–294. doi: 10.1038/nrgastro.2012.32
GBD 2015 Disease and Injury Incidence and Prevalence Collaborators (2016). Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388, 1545–1602. doi: 10.1016/s0140-6736(16)31678-6
Gill, S. K., Rossi, M., Bajka, B., and Whelan, K. (2021). Dietary fibre in gastrointestinal health and disease. Nat. Rev. Gastro. Hepat. 18, 101–116. doi: 10.1038/s41575-020-00375-4
Giovinazzo, D., Bursac, B., Sbodio, J. I., Nalluru, S., Vignane, T., Snowman, A. M., et al. (2021). Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation. P. Natl. Acad. Sci. USA 118:e2017225118. doi: 10.1073/pnas.2017225118
Goedert, M. (2015). Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349:1255555. doi: 10.1126/science.1255555
Golovko, M. Y., Barceló-Coblijn, G., Castagnet, P. I., Austin, S., Combs, C. K., and Murphy, E. J. (2009). The role of α-synuclein in brain lipid metabolism: a downstream impact on brain inflammatory response. Mol. Cell Biochem. 326, 55–66. doi: 10.1007/s11010-008-0008-y
Gonzaìlez-Rodriìguez, P., Zampese, E., Stout, K. A., Guzman, J. N., Ilijic, E., Yang, B., et al. (2021). Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 599, 650–656. doi: 10.1038/s41586-021-04059-0
Goubern, M., Andriamihaja, M., Nübel, T., Blachier, F., and Bouillaud, F. (2007). Sulfide, the first inorganic substrate for human cells. FASEB J. 21, 1699–1706. doi: 10.1096/fj.06-7407com
Greco, T., Glenn, T. C., Hovda, D. A., and Prins, M. L. (2016). Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cerebr. Blood F Met. 36, 1603–1613. doi: 10.1177/0271678X15610584
Green, R. C., Schneider, L. S., Amato, D. A., Beelen, A. P., Wilcock, G., Swabb, E. A., et al. (2009). Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild alzheimer disease: a randomized controlled trial. J. Amer. Med. Assoc. 302, 2557–2564. doi: 10.1001/jama.2009.1866
Groschwitz, K. R., and Hogan, S. P. (2009). Intestinal barrier function: molecular regulation and disease pathogenesis. J. Allergy Clin. Immun. 124, 3–22. doi: 10.1016/j.jaci.2009.05.038
Guo, J. L., Covell, D. J., Daniels, J. P., Iba, M., Stieber, A., Zhang, B., et al. (2013). Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117. doi: 10.1016/j.cell.2013.05.057
Guo, T., Noble, W., and Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704. doi: 10.1007/s00401-017-1707-9
Hampe, C. S., and Roth, C. L. (2017). Probiotic strains and mechanistic insights for the treatment of type 2 diabetes. Endocrine 58, 207–227. doi: 10.1007/s12020-017-1433-z
Harach, T., Marungruang, N., Duthilleul, N., Cheatham, V., Mc Coy, K. D., Frisoni, G., et al. (2017). Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 7:41802. doi: 10.1038/srep41802
Haran, J. P., and McCormick, B. A. (2021). Aging, frailty, and the microbiome—how dysbiosis influences human aging and disease. Gastroenterology 160, 507–523. doi: 10.1053/j.gastro.2020.09.060
Helander, H. F., and Fändriks, L. (2014). Surface area of the digestive tract–revisited. Scand. J. Gastroentero. 49, 681–689. doi: 10.3109/00365521.2014.898326
Hirose, M., Künstner, A., Schilf, P., Sünderhauf, A., Rupp, J., Jöhren, O., et al. (2017). Mitochondrial gene polymorphism is associated with gut microbial communities in mice. Sci. Rep. 7:15293. doi: 10.1038/s41598-017-15377-7
Ho, L., Ono, K., Tsuji, M., Mazzola, P., Singh, R., and Pasinetti, G. M. (2018). Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 18, 83–90. doi: 10.1080/14737175.2018.1400909
Hohman, T. J., Dumitrescu, L., Barnes, L. L., Thambisetty, M., Beecham, G., Kunkle, B., et al. (2018). Sex-specific association of apolipoprotein E with cerebrospinal fluid levels of tau. JAMA Neurol. 75, 989–998. doi: 10.1001/jamaneurol.2018.0821
Holmqvist, S., Chutna, O., Bousset, L., Aldrin-Kirk, P., Li, W., Björklund, T., et al. (2014). Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 128, 805–820. doi: 10.1007/s00401-014-1343-6
Hou, X., Yuan, Y., Sheng, Y., Yuan, B., Wang, Y., Zheng, J., et al. (2017). GYY4137, an H2S slow-releasing donor, prevents nitrative stress and α-synuclein nitration in an MPTP mouse model of Parkinson’s disease. Front. Pharmacol. 8:741. doi: 10.3389/fphar.2017.00741
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Huang, H.-K., Wang, J.-H., Lei, W.-Y., Chen, C.-L., Chang, C.-Y., and Liou, L.-S. (2018). Helicobacter pylori infection is associated with an increased risk of Parkinson’s disease: a population-based retrospective cohort study. Parkinsonism Rel. D 47, 26–31. doi: 10.1016/j.parkreldis.2017.11.331
Huang, Y., Liao, J., Liu, X., Zhong, Y., Cai, X., and Long, L. (2021). Review: the role of intestinal dysbiosis in parkinson’s disease. Front. Cell Infect. Microbiol. 11:615075. doi: 10.3389/fcimb.2021.615075
Huang, Z., Yan, Q., Wang, Y., Zou, Q., Li, J., Liu, Z., et al. (2020). Role of mitochondrial dysfunction in the pathology of amyloid-β. J. Alzheimers Dis. 2020, 1–10. doi: 10.1038/s41598-020-68882-7
Hugon, P., Dufour, J.-C., Colson, P., Fournier, P.-E., Sallah, K., and Raoult, D. (2015). A comprehensive repertoire of prokaryotic species identified in human beings. Lancet Infect. Dis. 15, 1211–1219. doi: 10.1016/S1473-3099(15)00293-5
Huynh, T.-P. V., Davis, A. A., Ulrich, J. D., and Holtzman, D. M. (2017). Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid. Res. 58, 824–836. doi: 10.1194/jlr.R075481
Imbimbo, B. P. (2009). Why did tarenflurbil fail in alzheimer’s disease. J. Alzheimers Dis. 17, 757–760. doi: 10.3233/JAD-2009-1092
Jackson, D. N., and Theiss, A. L. (2020). Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut. Microbes. 11, 285–304. doi: 10.1080/19490976.2019.1592421
Janke, R., Dodson, A. E., and Rine, J. (2015). Metabolism and epigenetics. Annu. Rev. Cell Dev. Biol. 31, 473–496. doi: 10.1146/annurev-cellbio-100814-125544
Jankovska, N., Olejar, T., and Matej, R. (2021). Extracellular amyloid deposits in alzheimer’s and creutzfeldt–jakob disease: similar behavior of different proteins. Int. J. Mol. Sci. 22:7. doi: 10.3390/ijms22010007
Johnston, I. G., and Williams, B. P. (2016). Evolutionary inference across eukaryotes identifies specific pressures favoring mitochondrial gene retention. Cell Syst. 2, 101–111. doi: 10.1016/j.cels.2016.01.013
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kamath, A. F., Chauhan, A. K., Kisucka, J., Dole, V. S., Loscalzo, J., Handy, D. E., et al. (2006). Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 107, 591–593. doi: 10.1182/blood-2005-06-2506
Kellingray, L., Tapp, H. S., Saha, S., Doleman, J. F., Narbad, A., and Mithen, R. F. (2017). Consumption of a diet rich in Brassica vegetables is associated with a reduced abundance of sulphate-reducing bacteria: a randomised crossover study. Mol. Nutr. Food Res. 61:1600992. doi: 10.1002/mnfr.201600992
Killinger, B. A., Madaj, Z., Sikora, J. W., Rey, N., Haas, A. J., Vepa, Y., et al. (2018). The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci. Transl. Med. 10:eaar5280. doi: 10.1126/scitranslmed.aar5280
Kim, J., and Guan, K.-L. (2019). mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 21, 63–71. doi: 10.1038/s41556-018-0205-1
Kim, R., and Jun, J. S. (2020). Impact of overweight and obesity on functional and clinical outcomes of early parkinson’s disease. J. Am. Med. Dir. Assoc. 21, 697–700. doi: 10.1016/j.jamda.2019.11.019
Kim, S., Kwon, S.-H., Kam, T.-I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103, 627–641. doi: 10.1016/j.neuron.2019.05.035
Kitada, M., Ogura, Y., Monno, I., Xu, J., and Koya, D. (2021). Effect of methionine restriction on aging: its relationship to oxidative stress. Biomedicines 9:130. doi: 10.3390/biomedicines9020130
Kitamoto, S., Alteri, C. J., Rodrigues, M., Nagao-Kitamoto, H., Sugihara, K., Himpsl, S. D., et al. (2020). Dietary L-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat. Microbiol. 5, 116–125. doi: 10.1038/s41564-019-0591-6
Knudsen, K. E. B., Lærke, H. N., Hedemann, M. S., Nielsen, T. S., Ingerslev, A. K., Gundelund Nielsen, D. S., et al. (2018). Impact of diet-modulated butyrate production on intestinal barrier function and inflammation. Nutrients 10:1499. doi: 10.3390/nu10101499
Konjar, S., Frising, U. C., Ferreira, C., Hinterleitner, R., Mayassi, T., Zhang, Q., et al. (2018). Mitochondria maintain controlled activation state of epithelial-resident T lymphocytes. Sci. Immunol. 3:eaan2543. doi: 10.1126/sciimmunol.aan2543
Kowalski, K., and Mulak, A. (2019). Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. 25, 48–60. doi: 10.5056/jnm18087
Kramer, P., and Bressan, P. (2015). Humans as superorganisms: how microbes, viruses, imprinted genes, and other selfish entities shape our behavior. Perspect. Psychol. Sci. 10, 464–481. doi: 10.1177/1745691615583131
Kramer, P., and Bressan, P. (2018). Our (mother’s) mitochondria and our mind. Perspect. Psychol. Sci. 13, 88–100. doi: 10.1177/1745691617718356
Kramer, P., and Bressan, P. (2019). Mitochondria inspire a lifestyle. Adv. Anat. Embryol. Cell Biol. 231, 105–126. doi: 10.1007/102_2018_5
Kramer, P., and Bressan, P. (2021). Infection threat shapes our social instincts. Behav. Ecol. Sociobiol. 75, 1–18. doi: 10.1007/s00265-021-02975-9
Kumar, D. K. V., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., Ghofrani, J., et al. (2016). Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 8:340ra72. doi: 10.1126/scitranslmed.aaf1059
Lane, N. (2015). The Vital Question: Energy, Evolution, and the Origins of Complex Life. London: WW Norton & Company.
Lane, N., and Martin, W. (2010). The energetics of genome complexity. Nature 467, 929–934. doi: 10.1038/nature09486
Lautenschäger, J., and Schierle, G. S. K. (2019). Mitochondrial degradation of amyloidogenic proteins—A new perspective for neurodegenerative diseases. Prog. Neurobiol. 181:101660. doi: 10.1016/j.pneurobio.2019.101660
Lautenschäger, J., Wagner-Valladolid, S., Stephens, A. D., Fernández-Villegas, A., Hockings, C., Mishra, A., et al. (2020). Intramitochondrial proteostasis is directly coupled to α-synuclein and amyloid β1-42 pathologies. J. Biol. Chem. 295, 10138–10152. doi: 10.1074/jbc.RA119.011650
Lazic, D., Tesic, V., Jovanovic, M., Brkic, M., Milanovic, D., Zlokovic, B. V., et al. (2020). Every-other-day feeding exacerbates inflammation and neuronal deficits in 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Dis. 136:104745. doi: 10.1016/j.nbd.2020.104745
Le, T. N., Chiu, C.-H., and Hsieh, P.-C. (2020). Bioactive compounds and bioactivities of brassica oleracea l. var. italica sprouts and microgreens: An updated overview from a nutraceutical perspective. Plants 9:946. doi: 10.3390/plants9080946
LeBlanc, J. G., Chain, F., Martín, R., Bermúdez-Humarán, L. G., Courau, S., and Langella, P. (2017). Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb. Cell Fact. 16:79. doi: 10.1186/s12934-017-0691-z
Lewis, K., Lutgendorff, F., Phan, V., Söderholm, J. D., Sherman, P. M., and McKay, D. M. (2010). Enhanced translocation of bacteria across metabolically stressed epithelia is reduced by butyrate. Inflamm. Bowel. Dis. 16, 1138–1148. doi: 10.1002/ibd.21177
Li, F., Hearn, M., and Bennett, L. E. (2021). The role of microbial infection in the pathogenesis of Alzheimer’s disease and the opportunity for protection by anti-microbial peptides. Crit. Rev. Microbiol. 47, 240–253. doi: 10.1080/1040841X.2021.1876630
Li, H. M., Liu, C. C., Zheng, H., and Huang, T. Y. (2018). Amyloid, tau, pathogen infection and antimicrobial protection in Alzheimer’s disease–conformist, nonconformist, and realistic prospects for AD pathogenesis. Transl. Neurodegen. 7:34. doi: 10.1186/s40035-018-0139-3
Liddle, R. A. (2018). Parkinson’s disease from the gut. Brain Res. 1693, 201–206. doi: 10.1016/j.brainres.2018.01.010
Lilamand, M., Porte, B., Cognat, E., Hugon, J., Mouton-Liger, F., and Paquet, C. (2020). Are ketogenic diets promising for Alzheimer’s disease? a translational review. Alzheimers Res. Ther. 12, 1–10. doi: 10.1186/s13195-020-00615-4
Lin, H.-C., Hsieh, H.-M., Chen, Y.-H., and Hu, M.-L. (2009). S-Adenosylhomocysteine increases β-amyloid formation in BV-2 microglial cells by increased expressions of β-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. NeuroToxicology 30, 622–627. doi: 10.1016/j.neuro.2009.03.011
Lionnet, A., Leclair-Visonneau, L., Neunlist, M., Murayama, S., Takao, M., Adler, C. H., et al. (2018). Does Parkinson’s disease start in the gut. Acta Neuropathol. 135, 1–12. doi: 10.1007/s00401-017-1777-8
Litvak, Y., Byndloss, M. X., Tsolis, R. M., and Bäumler, A. J. (2017). Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 39, 1–6. doi: 10.1016/j.mib.2017.07.003
Litvinova, L., Atochin, D. N., Fattakhov, N., Vasilenko, M., Zatolokin, P., and Kirienkova, E. (2015). Nitric oxide and mitochondria in metabolic syndrome. Front. Physiol. 6:20. doi: 10.3389/fphys.2015.00020
Liu, Y., Niu, L., Liu, X., Cheng, C., and Le, W. (2021). Recent progress in non-motor features of parkinson’s disease with a focus on circadian rhythm dysregulation. Neurosci. Bull. 2021, 1–15. doi: 10.1007/s12264-021-00711-x
Liu, Y., Xue, L., Zhang, Y., and Xie, A. (2020). Association between stroke and parkinson’s disease: a meta-analysis. J. Mol. Neurosci. 70, 1169–1176. doi: 10.1007/s12031-020-01524-9
Loenen, W. A. (2006). S-Adenosylmethionine: jack of all trades and master of everything? Biochem. Soc. T 34, 330–333. doi: 10.1042/BST20060330
Lohmann, S., Bernis, M. E., Tachu, B. J., Ziemski, A., Grigoletto, J., and Tamgüney, G. (2019). Oral and intravenous transmission of α-synuclein fibrils to mice. Acta Neuropathol. 138, 515–533. doi: 10.1007/s00401-019-02037-5
Lopes, A. F. C. (2020). Mitochondrial metabolism and DNA methylation: a review of the interaction between two genomes. Clin. Epigen. 12:182. doi: 10.1186/s13148-020-00976-5
López-Armada, M. J., Riveiro-Naveira, R. R., Vaamonde-García, C., and Valcárcel-Ares, M. N. (2013). Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13, 106–118. doi: 10.1016/j.mito.2013.01.003
Lu, K., Nicholas, J. M., Pertzov, Y., Grogan, J., Husain, M., Pavisic, I. M., et al. (2021). Dissociable effects of APOE ε4 and β-amyloid pathology on visual working memory. Nat. Aging 1, 1002–1009. doi: 10.1038/s43587-021-00117-4
Lubomski, M., Tan, A. H., Lim, S.-Y., Holmes, A. J., Davis, R. L., and Sue, C. M. (2019). Parkinson’s disease and the gastrointestinal microbiome. J. Neurol. 2019, 1–17. doi: 10.1007/s00415-019-09320-1
Luca, M., Di Mauro, M., Di Mauro, M., and Luca, A. (2019). Gut microbiota in alzheimer’s disease, depression, and type 2 diabetes mellitus: the role of oxidative stress. Oxid. Med. Cell Longev. 2019:4730539. doi: 10.1155/2019/4730539
Lundmark, K., Westermark, G. T., Olsén, A., and Westermark, P. (2005). Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. P. Natl. Acad. Sci. USA 102, 6098–6102. doi: 10.1073/pnas.0501814102
Ma, J., Coarfa, C., Qin, X., Bonnen, P. E., Milosavljevic, A., Versalovic, J., et al. (2014). mtDNA haplogroup and single nucleotide polymorphisms structure human microbiome communities. BMC Genomics 15:257. doi: 10.1186/1471-2164-15-257
Ma, N., and Ma, X. (2019). Dietary amino acids and the gut-microbiome-immune axis: physiological metabolism and therapeutic prospects. Compr. Rev. Food Sci. F 18, 221–242. doi: 10.1111/1541-4337.12401
Makin, S. (2018). The amyloid hypothesis on trial. Nature 559, S4–S4. doi: 10.1038/d41586-018-05719-4
Mallikarjuna, N., Praveen, K., and Yellamma, K. (2016). Role of lactobacillus plantarum MTCC1325 in membrane-bound transport ATPases system in Alzheimer’s disease-induced rat brain. Bioimpacts 6, 203–209. doi: 10.15171/bi.2016.27
Maniar, K., Moideen, A., Mittal, A., Patil, A., Chakrabarti, A., and Banerjee, D. (2017). A story of metformin-butyrate synergism to control various pathological conditions as a consequence of gut microbiome modification: genesis of a wonder drug. Pharmacol. Res. 117, 103–128. doi: 10.1016/j.phrs.2016.12.003
Marx, W., Scholey, A., Firth, J., D’Cunha, N. M., Lane, M., Hockey, M., et al. (2020). Prebiotics, probiotics, fermented foods and cognitive outcomes: A meta-analysis of randomized controlled trials. Neurosci. Biobehav. R 118, 472–484. doi: 10.1016/j.neubiorev.2020.07.036
Mattson, M. P., and Arumugam, T. V. (2018). Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell metab. 27, 1176–1199. doi: 10.1016/j.cmet.2018.05.011
Mattson, M. P., Moehl, K., Ghena, N., Schmaedick, M., and Cheng, A. (2018). Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 19:63. doi: 10.1038/nrn.2017.156
McCarty, M. F., and Lerner, A. (2021). Perspective: low risk of parkinson’s disease in quasi-vegan cultures may reflect GCN2-mediated upregulation of parkin. Adv. Nutr. 12, 355–362. doi: 10.1093/advances/nmaa112
Miller, A. L., Bessho, S., Grando, K., and Tukel, C. (2021). Microbiome or infections: amyloid-containing biofilms as a trigger for complex human diseases. Front. Immunol. 12:514. doi: 10.3389/fimmu.2021.638867
Minter, M. R., Hinterleitner, R., Meisel, M., Zhang, C., Leone, V., Zhang, X., et al. (2017). Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1ΔE9 murine model of Alzheimer’s disease. Sci. Rep. 7:10411. doi: 10.1038/s41598-017-11047-w
Miraglia, F., and Colla, E. (2019). Microbiome, parkinson’s disease and molecular mimicry. Cells 8:222. doi: 10.3390/cells8030222
Moir, R. D., Lathe, R., and Tanzi, R. E. (2018). The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 14, 1602–1614. doi: 10.1016/j.jalz.2018.06.3040
Moreno-Loshuertos, R., Acin-Perez, R., Fernandez-Silva, P., Movilla, N., Perez-Martos, A., de Cordoba, S. R., et al. (2006). Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 38, 1261–1268. doi: 10.1038/ng1897
Moreno-Loshuertos, R., Perez Martos, A., Fernandez Silva, P., and Enriquez, J. A. (2013). Length variation in the mouse mitochondrial tRNAA rg DHU loop size promotes oxidative phosphorylation functional differences. FEBS J. 280, 4983–4998. doi: 10.1111/febs.12466
Mottawea, W., Chiang, C.-K., Muhlbauer, M., Starr, A. E., Butcher, J., Abujamel, T., et al. (2016). Altered intestinal microbiota–host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 7:13419. doi: 10.1038/ncomms13419
Mulvey, L., Sinclair, A., and Selman, C. (2014). Lifespan modulation in mice and the confounding effects of genetic background. J. Genet. Genomics 41, 497–503. doi: 10.1016/j.jgg.2014.06.002
Munoz-Pinto, M. F., Empadinhas, N., and Cardoso, S. M. (2021). The neuromicrobiology of Parkinson’s disease: a unifying theory. Ageing Res. Rev. 70:101396. doi: 10.1016/j.arr.2021.101396
Nair, A. T., Ramachandran, V., Joghee, N. M., Antony, S., and Ramalingam, G. (2018). Gut microbiota dysfunction as reliable non-invasive early diagnostic biomarkers in the pathophysiology of Parkinson’s disease: a critical review. J. Neurogastroenterol. 24:30. doi: 10.5056/jnm17105
Nguyen, P. H., Ramamoorthy, A., Sahoo, B. R., Zheng, J., Faller, P., Straub, J. E., et al. (2021). Amyloid oligomers: a joint experimental/computational perspective on Alzheimer’s disease, Parkinson’s disease, Type II diabetes, and amyotrophic lateral sclerosis. Chem. Rev. 121, 2545–2647. doi: 10.1021/acs.chemrev.0c01122
Niesler, B., Kuerten, S., Demir, I. E., and Schäfer, K.-H. (2021). Disorders of the enteric nervous system—a holistic view. Nat. Rev. Gastro. Hepat. 18, 393–410. doi: 10.1038/s41575-020-00385-2
Notkola, I. L., Sulkava, R., Pekkanen, J., Erkinjuntti, T., Ehnholm, C., Kivinen, P., et al. (1998). Serum total cholesterol, apolipoprotein E (Notkola et al., 1998) e4 allele, and Alzheimer’s disease. Neuroepidemiology 17, 14–20. doi: 10.1159/000026149
Nurrahma, B. A., Tsao, S.-P., Wu, C.-H., Yeh, T.-H., Hsieh, P.-S., Panunggal, B., et al. (2021). Probiotic supplementation facilitates recovery of 6-OHDA-induced motor deficit via improving mitochondrial function and energy metabolism. Front. Aging Neurosci. 13:230. doi: 10.3389/fnagi.2021.668775
Pacelli, C., Giguere, N., Bourque, M. J., Levesque, M., Slack, R. S., and Trudeau, L.-E. (2015). Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 25, 2349–2360. doi: 10.1016/j.cub.2015.07.050
Pal, V. K., Bandyopadhyay, P., and Singh, A. (2018). Hydrogen sulfide in physiology and pathogenesis of bacteria and viruses. IUBMB Life 70, 393–410. doi: 10.1002/iub.1740
Pan, L., Meng, L., He, M., and Zhang, Z. (2021). Tau in the pathophysiology of parkinson’s disease. J. Mol. Neurosci. 2021, 1–13. doi: 10.1007/s12031-020-01776-5
Pang, S., Li, J., Zhang, Y., and Chen, J. (2018). Meta-analysis of the relationship between the apoe gene and the onset of parkinson’s disease dementia. Parkinsons Dis. 2018:9497147. doi: 10.1155/2018/9497147
Panza, F., Lozupone, M., Logroscino, G., and Imbimbo, B. P. (2019). A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 15, 73–88. doi: 10.1038/s41582-018-0116-6
Parihar, M. S., and Brewer, G. J. (2010). Amyloid-β as a modulator of synaptic plasticity. J. Alzheimers Dis. 22, 741–763. doi: 10.3233/JAD-2010-101020
Park, S.-C., Moon, J. C., Shin, S. Y., Son, H., Jung, Y. J., Kim, N.-H., et al. (2016). Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem. Bioph. Res. Co. 478, 924–928. doi: 10.1016/j.bbrc.2016.08.052
Paul, B. D. (2021). Neuroprotective roles of the reverse transsulfuration pathway in alzheimer’s disease. Front. Aging Neurosci. 13:659402. doi: 10.3389/fnagi.2021.659402
Paul, B. D., and Snyder, S. H. (2018). Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 149, 101–109. doi: 10.1016/j.bcp.2017.11.019
Peng, X., Meng, J., Chi, T., Liu, P., Man, C., Liu, S., et al. (2014). Lactobacillus plantarum NDC 75017 alleviates the learning and memory ability in aging rats by reducing mitochondrial dysfunction. Exp. Ther. Med. 8, 1841–1846. doi: 10.3892/etm.2014.2000
Phillipson, O. T. (2017). Alpha-synuclein, epigenetics, mitochondria, metabolism, calcium traffic, & circadian dysfunction in Parkinson’s disease. An integrated strategy for management. Ageing Res. Rev. 40, 149–167. doi: 10.1016/j.arr.2017.09.006
Pierzchlińska, A., Kwasniak-Butowska, M., Sławek, J., Drozdzik, M., and Białecka, M. (2021). Arterial blood pressure variability and other vascular factors contribution to the cognitive decline in parkinson’s disease. Molecules 26:1523. doi: 10.3390/molecules26061523
Poewe, W., Seppil, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Qu, Y., Hu, H.-Y., Ou, Y.-N., Shen, X.-N., Xu, W., Wang, Z.-T., et al. (2020). Association of body mass index with risk of cognitive impairment and dementia: A systematic review and meta-analysis of prospective studies. Neurosci. Biobehav. R 115, 189–198. doi: 10.1016/j.neubiorev.2020.05.012
Quntanilla, R. A., and Tapia-Monsalves, C. (2020). The role of mitochondrial impairment in alzheimer’s disease neurodegeneration: the tau connection. Curr. Neuropharmacol. 18, 1076–1091. doi: 10.2174/1570159X18666200525020259
Ramalingam, A., Wang, X., Gabello, M., Valenzano, M. C., Soler, A. P., Ko, A., et al. (2010). Dietary methionine restriction improves colon tight junction barrier function and alters claudin expression pattern. Am. J. Physiol. Cell Ph. 299, C1028–C1035. doi: 10.1152/ajpcell.00482.2009
Rawat, K., Singh, N., Kumari, P., and Saha, L. (2021). A review on preventive role of ketogenic diet (KD) in CNS disorders from the gut microbiota perspective. Rev. Neurosci. 32, 143–157. doi: 10.1515/revneuro-2020-0078
Rietdijk, C. D., Perez-Pardo, P., Garssen, J., Wezel, R. J. A. V., and Kraneveld, A. D. (2017). Exploring Braak’s hypothesis of Parkinson’s disease. Front. Neurol. 8:37. doi: 10.3389/fneur.2017.00037
Rigottier-Gois, L. (2013). Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 7, 1256–1261. doi: 10.1038/ismej.2013.80
Rigotto, G., and Basso, E. (2019). Mitochondrial dysfunctions: a thread sewing together alzheimer’s disease, diabetes, and obesity. Oxid. Med. Cell Longev. 2019:7210892. doi: 10.1155/2019/7210892
Rinninella, E., Cintoni, M., Raoul, P., Ianiro, G., Laterza, L., Lopetuso, L. R., et al. (2020). Gut microbiota during dietary restrictions: new insights in non-communicable diseases. Microorganisms 8:1140. doi: 10.3390/microorganisms8081140
Rolig, A. S., Mittge, E. K., Ganz, J., Troll, J. V., Melancon, E., Wiles, T. J., et al. (2017). The enteric nervous system promotes intestinal health by constraining microbiota composition. PLoS Biol. 15:e2000689. doi: 10.1371/journal.pbio.2000689
Romano, S., Savva, G. M., Bedarf, J. R., Charles, I. G., Hildebrand, F., and Narbad, A. (2021). Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinson’s Dis. 7:27. doi: 10.1038/s41531-021-00156-z
Rosario-Alomar, M. F., Quiñones-Ruiz, T., Kurouski, D., Sereda, V., Ferreira, E. B., Jesús-Kim, L. D., et al. (2015). Hydrogen sulfide inhibits amyloid formation. J. Phys. Chem. B 119, 1265–1274. doi: 10.1021/jp508471v
Sajilata, M. G., Singhal, R. S., and Kulkarni, P. R. (2006). Resistant starch–a review. Compr. Rev. Food Sci. F 5, 1–17. doi: 10.1111/j.1541-4337.2006.tb00076.x
Sampson, T. (2020). The impact of indigenous microbes on Parkinson’s disease. Neurobiol. Dis. 135:104426. doi: 10.1016/j.nbd.2019.03.014
Sampson, T., Challis, C., Jain, N., Moisevenko, A., Landinsky, M. S., Shastri, G. G., et al. (2020). A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. eLife 9:e53111. doi: 10.7554/eLife.53111
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of parkinson’s disease. Cell 167, 1469–1480. doi: 10.1016/j.cell.2016.11.018
Schillheim, B., Jansen, I., Baum, S., Beesley, A., Bolm, C., and Conrath, U. (2018). Sulforaphane modifies histone H3, unpacks chromatin, and primes defense. Plant Physiol. 176, 2395–2405. doi: 10.1104/pp.17.00124
Sen, P., Molinero-Perez, A., O’Riordan, K. J., McCafferty, C. P., O’Halloran, K. D., and Cryan, J. F. (2021). Microbiota and sleep: awakening the gut feeling. Trends Mol. Med. 2020:4. doi: 10.1016/j.molmed.2021.07.004
Sender, R., Fuchs, S., and Milo, R. (2016). Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340. doi: 10.1016/j.cell.2016.01.013
Sharma, A., Sethi, G., Tambuwala, M. M., Aljabali, A. A. A., Chellappan, D. K., Dua, K., et al. (2021). Circadian rhythm disruption and Alzheimer’s disease: the dynamics of a vicious cycle. Curr. Neuropharmacol. 19, 248–264. doi: 10.2174/1570159X18666200429013041
Shelton, C. D., Byndloss, M. X., and Richardson, A. R. (2020). Gut epithelial metabolism as a key driver of intestinal dysbiosis associated with noncommunicable diseases. Infect. Immun. 88, e939–e919. doi: 10.1128/IAI.00939-19
Shen, L., Liu, L., and Ji, H.-F. (2017). Alzheimer’s disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. J. Alzheimers Dis. 56, 385–390. doi: 10.3233/JAD-160884
Shen, T., Yue, Y., He, T., Huang, C., Qu, B., Lv, W., et al. (2021). The association between the gut microbiota and Parkinson’s disease, a meta-analysis. Front. Aging Neurosci. 13:40. doi: 10.3389/fnagi.2021.636545
Shen, X., Yang, H., Wu, Y., Zhang, D., and Jiang, H. (2017). Meta-analysis: association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 22:e12398. doi: 10.1111/hel.12398
Shi, Y., and Holtzman, D. M. (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 18, 759–772. doi: 10.1038/s41577-018-0051-1
Shlevkov, E., and Schwarz, T. L. (2017). Axonal Mitochondrial Transport Parkinson’s Disease. San Diego: Academic Press, 113–137.
Shoaie, S., Karlsson, F., Mardinoglu, A., Nookaew, I., Bordel, S., and Nielsen, J. (2013). Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 3, 1–10. doi: 10.1038/srep02532
Shoaie, S., Lee, S., Almeida, M., Bidkhori, G., Pons, N., Onate, F., et al. (2021). Global and temporal state of the human gut microbiome in health and disease. Under Rev 2021:339282. doi: 10.21203/rs.3.rs-339282/v1
Silva, Y. P., Bernardi, A., and Frozza, R. L. (2020). The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 11:25. doi: 10.3389/fendo.2020.00025
Singh, S. B., and Lin, H. C. (2015). Hydrogen sulfide in physiology and diseases of the digestive tract. Microorganisms 3:866. doi: 10.3390/microorganisms3040866
Sivanesan, S., Chang, E., Howell, M. D., and Rajadas, J. (2020). Amyloid protein aggregates: new clients for mitochondrial energy production in the brain. FEBS J. 287, 3386–3395. doi: 10.1111/febs.15225
Snoek, S. A., Verstege, M. I., Boeckxstaens, G. E., van den Wijngaard, R. M., and de Jonge, W. J. (2010). The enteric nervous system as a regulator of intestinal epithelial barrier function in health and disease. Expert Rev. Gastroent. 4, 637–651. doi: 10.1586/EGH.10.51
Sokolov, A. S., Nekrasov, P. V., Shaposhnikov, M. V., and Moskalev, A. A. (2021). Hydrogen sulfide in longevity and pathologies: inconsistency is malodorous. Ageing Res. Rev. 67:101262. doi: 10.1016/j.arr.2021.101262
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505. doi: 10.1371/journal.pone.0009505
Soultoukis, G. A., and Partridge, L. (2016). Dietary protein. metabolism, and aging Annu. Rev. Biochem. 85, 5–34. doi: 10.1146/annurev-biochem-060815-014422
Stanley, D., Mason, L. J., Mackin, K. E., Srikhanta, Y. N., Lyras, D., Prakash, M. D., et al. (2016). Translocation and dissemination of commensal bacteria in post-stroke infection. Nat. Med. 22, 1277–1284. doi: 10.1038/nm.4194
Steiner, J. A., Quansah, E., and Brundin, P. (2018). The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res. 373, 161–173. doi: 10.1007/s00441-018-2814-1
Stopschinski, B. E., and Diamond, M. I. (2017). The prion model for progression and diversity of neurodegenerative diseases. Lancet Neurol. 16, 323–332. doi: 10.1016/S1474-4422(17)30037-6
Sullivan, P. M. (2020). Influence of Western diet and APOE genotype on Alzheimer’s disease risk. Neurobiol. Dis. 138:104790. doi: 10.1016/j.nbd.2020.104790
Sun, Y., Ma, C., Sun, H., Wang, H., Peng, W., Zhou, Z., et al. (2020b). Metabolism: a novel shared link between diabetes mellitus and Alzheimer’s disease. J. Diab. Res. 2020:4981814. doi: 10.1155/2020/4981814
Sun, Y., Sommerville, N. R., Liu, J. Y. H., Ngan, M. P., Poon, D., Ponomarev, E. D., et al. (2020a). Intra-gastrointestinal amyloid-β1–42 oligomers perturb enteric function and induce Alzheimer’s disease pathology. J. Physiol. 598, 4209–4223. doi: 10.1113/JP279919
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Parkinson’s disease is not simply a prion disorder. J. Neurosci. 37, 9799–9807. doi: 10.1523/JNEUROSCI.1787-16.2017
Svensson, E., Horváth-Puhó, E., Thomsen, R. W., Djurhuus, J. C., Pedersen, L., Borghammer, P., et al. (2015). Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 78, 522–529. doi: 10.1002/ana.24448
Szabo, L., Eckert, A., and Grimm, A. (2020). Insights into disease-associated tau impact on mitochondria. Int. J. Mol. Sci. 21:21176344. doi: 10.3390/ijms21176344
Tabat, M. W., Marques, T. M., Markgren, M., Löfvendahl, L., Brummer, R. J., and Wall, R. (2020). Acute effects of butyrate on induced hyperpermeability and tight junction protein expression in human colonic tissues. Biomolecules 10:766. doi: 10.3390/biom10050766
Tan, A. H., Lim, S.-Y., Mahadeva, S., Loke, M. F., Tan, J. Y., Ang, B. H., et al. (2020). Helicobacter pylori eradication in parkinson’s disease: a randomized placebo-controlled trial. Mov. Disord. 35, 2250–2260. doi: 10.1002/mds.28248
Thelen, M., and Brown-Borg, H. M. (2020). Does diet have a role in the treatment of alzheimer’s disease. Front. Aging Neurosci. 12:473. doi: 10.3389/fnagi.2020.617071
Thevaranjan, N., Puchta, A., Schulz, C., Naidoo, A., Szamosi, J. C., Verschoor, C. P., et al. (2017). Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. 21, 455–466. doi: 10.1016/j.chom.2017.03.002
Tomlinson, J. J., Shutinoski, B., Dong, L., Meng, F., Elleithy, D., Lengacher, N. A., et al. (2017). Holocranohistochemistry enables the visualization of α-synuclein expression in the murine olfactory system and discovery of its systemic anti-microbial effects. J. Neural. Transm. 124, 721–738. doi: 10.1007/s00702-017-1726-7
Trumble, B. C., Stieglitz, J., Blackwell, A. D., Allayee, H., Beheim, B., Finch, C. E., et al. (2017). Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden. FASEB J. 31, 1508–1515. doi: 10.1096/fj.201601084R
Tse, J. K. Y. (2017). Gut microbiota, nitric oxide, and microglia as prerequisites for neurodegenerative disorders. ACS Chem. Neurosci. 8, 1438–1447. doi: 10.1021/acschemneuro.7b00176
Tsuang, D., Leverenz, J. B., Lopez, O. L., Hamilton, R. L., Bennett, D. A., Schneider, J. A., et al. (2013). APOE ϵ4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 70, 223–228. doi: 10.1001/jamaneurol.2013.600
Tursi, S. A., and Tükel, C. (2018). Curli-containing enteric biofilms inside and out: matrix composition, immune recognition, and disease implications. Microbiol. Mol. Biol. Rev. 82:e00028. 00028-18 doi: 10.1128/MMBR
Uddin, M. S., Tewari, D., Al Mamun, A., Kabir, M. T., Niaz, K., Wahed, M. I. I., et al. (2020). Circadian and sleep dysfunction in Alzheimer’s disease. Ageing Res. Rev. 60:101046. doi: 10.1016/j.arr.2020.101046
Unger, M. M., Spiegel, J., Dillmann, K.-U., Grundmann, D., Philippeit, H., Bürmann, J., et al. (2016). Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Dis. 32, 66–72. doi: 10.1016/j.parkreldis.2016.08.019
Urosevic, N., and Martins, R. N. (2008). Infection and Alzheimer’s disease: the apoE ε4 connection and lipid metabolism. J. Alzheimers Dis. 13, 421–435. doi: 10.3233/jad-2008-13407
Van Den Berge, N., Ferreira, N., Gram, H., Mikkelsen, T. W., Alstrup, A. K. O., Casadei, N., et al. (2019). Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 138, 535–550. doi: 10.1007/s00401-019-02040-w
van Exel, E., Koopman, J. J. E., Bodegom, D. V., Meij, J. J., Knijff, P. D., Ziem, J. B., et al. (2017). Effect of APOE ε4 allele on survival and fertility in an adverse environment. PLoS One 12:e0179497. doi: 10.1371/journal.pone.0179497
Van Gerven, N., Van der Verren, S. E., Reiter, D. M., and Remaut, H. (2018). The role of functional amyloids in bacterial virulence. J. Mol. Biol. 430, 3657–3684. doi: 10.1016/j.jmb.2018.07.010
Vezzani, B., Carinci, M., Patergnani, S., Pasquin, M. P., Guarino, A., Aziz, N., et al. (2020). The dichotomous role of inflammation in the CNS: a mitochondrial point of view. Biomolecules 10:1437. doi: 10.3390/biom10101437
Vidakovic, L., Singh, P. K., Hartmann, R., Nadell, C. D., and Drescher, K. (2018). Dynamic biofilm architecture confers individual and collective mechanisms of viral protection. Nat. Microbiol. 3, 26–31. doi: 10.1038/s41564-017-0050-1
Vieira, M. N. N., Lima-Filho, R. A. S., and De Felice, F. G. (2018). Connecting Alzheimer’s disease to diabetes: underlying mechanisms and potential therapeutic targets. Neuropharmacology 136, 160–171. doi: 10.1016/j.neuropharm.2017.11.014
Villumsen, M., Aznar, S., Pakkenberg, B., Jess, T., and Brudek, T. (2019). Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut 68, 18–24. doi: 10.1136/gutjnl-2017-315666
Vogt, N. M., Kerby, R. L., Dill-McFarland, K. A., Harding, S. J., Merluzzi, A. P., Johnson, S. C., et al. (2017). Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 7:13537. doi: 10.1038/s41598-017-13601-y
von Martels, J. Z. H., Sadabad, M. S., Bourgonje, A. R., Blokzijl, T., Dijkstra, G., Faber, K. N., et al. (2017). The role of gut microbiota in health and disease: in vitro modeling of host-microbe interactions at the aerobe-anaerobe interphase of the human gut. Anaerobe 44, 3–12. doi: 10.1016/j.anaerobe.2017.01.001
Vuong, H. E., Yano, J. M., Fung, T. C., and Hsiao, E. Y. (2017). The microbiome and host behavior. Annu. Rev. Neurosci. 40, 21–49. 031347 doi: 10.1146/annurev-neuro-072116-
Wallace, D. C., and Fan, W. (2010). Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10, 12–31. doi: 10.1016/j.mito.2009.09.006
Wang, A., Luan, H. H., and Medzhitov, R. (2019). An evolutionary perspective on immunometabolism. Science 363:eaar3932. doi: 10.1126/science.aar3932
Wang, X. L., Zeng, J., Yang, Y., Xiong, Y., Zhang, Z.-H., Qiu, M., et al. (2015). Helicobacter pylori filtrate induces Alzheimer-like tau hyperphosphorylation by activating glycogen synthase kinase-3β. J. Alzheimers Dis. 43, 153–165. doi: 10.3233/JAD-140198
Waziry, R., Chibnik, L. B., Bos, D., Ikram, M. K., and Hofman, A. (2020). Risk of hemorrhagic and ischemic stroke in patients with Alzheimer disease: a synthesis of the literature. Neurology 94, 265–272. doi: 10.1212/WNL.0000000000008924
Wei, M., Brandhorst, S., Shelehchi, M., Mirzaei, H., Cheng, C. W., Budniak, J., et al. (2017). Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 9:eaai8700. doi: 10.1126/scitranslmed.aai8700
Weil, R. S., Lashley, T. L., Bras, J., Schrag, A. E., and Schott, J. M. (2017). Current concepts and controversies in the pathogenesis of Parkinson’s disease dementia and Dementia with Lewy Bodies. F1000 Res. 6:1604. doi: 10.12688/f1000research.11725.1
Whiteman, M., Li, L., Rose, P., Tan, C.-H., Parkinson, D. B., and Moore, P. K. (2010). The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox. Sign. 12, 1147–1154. doi: 10.1089/ars.2009.2899
Włodarek, D. (2019). Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). Nutrients 11:169. doi: 10.3390/nu11010169
Wong, Y. C., and Krainc, D. (2017). α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1–13. doi: 10.1038/nm.4269
Wu, G., Shi, Y., Han, L., Feng, C., Ge, Y., Yu, Y., et al. (2020). Dietary methionine restriction ameliorated fat accumulation, systemic inflammation, and increased energy metabolism by altering gut microbiota in middle-aged mice administered different fat diets. J. Agr. Food Chem. 68, 7745–7756. doi: 10.1021/acs.jafc.0c02965
Yang, J. Y., Lee, Y. S., Kim, Y., Lee, S. H., Ryu, S., Fukuda, S., et al. (2017). Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal. Immunol. 10, 104–116. doi: 10.1038/mi.2016.42
Yardeni, T., Tanes, C. E., Bittinger, K., and Mattei, L. M. (2019). Host mitochondria influence gut microbiome diversity: a role for ROS. Sci. Signal 12:eaaw3159. doi: 10.1126/scisignal.aaw3159
Yonezawa, H., Osaki, T., Hanawa, T., Kurata, S., Zaman, C., Woo, T. D. H., et al. (2012). Destructive effects of butyrate on the cell envelope of Helicobacter pylori. J. Med. Microbiol. 61, 582–589. doi: 10.1099/jmm.0.039040-0
Zaretsky, D. V., and Zaretskaia, M. V. (2021). Mini-review: Amyloid degradation toxicity hypothesis of Alzheimer’s disease. Neurosci. Lett. 2021:135959. doi: 10.1016/j.neulet.2021.135959
Zhan, X., Stamova, B., and Sharp, F. R. (2018). Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: a review. Front. Aging Neurosci. 10:42. doi: 10.3389/fnagi.2018.00042
Zhang, L., Wang, Y., Xiayu, X., Shi, C., Chen, W., Song, N., et al. (2017). Altered gut microbiota in a mouse model of alzheimer’s disease. J. Alzheimers Dis. 60, 1241–1257. doi: 10.3233/JAD-170020
Zhao, C., Strobino, K., Moon, Y. P., Cheung, Y. K., Sacco, R. L., Stern, Y., et al. (2020). APOE ϵ4 modifies the relationship between infectious burden and poor cognition. Neurol. Genet. 6:e462. doi: 10.1212/NXG.0000000000000462
Keywords: microbiota, mitochondria, Alzheimer’s disease, Parkinson’s disease, gut-blood barrier, butyrate, hydrogen sulfide, methionine
Citation: Kramer P (2021) Mitochondria-Microbiota Interaction in Neurodegeneration. Front. Aging Neurosci. 13:776936. doi: 10.3389/fnagi.2021.776936
Received: 14 September 2021; Accepted: 30 November 2021;
Published: 23 December 2021.
Edited by:
Małgorzata Kujawska, Poznań University of Medical Sciences, PolandReviewed by:
Sandra Morais Cardoso, University of Coimbra, PortugalCopyright © 2021 Kramer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Kramer, cGV0ZXIua3JhbWVyQHVuaXBkLml0, orcid.org/0000-0003-4807-7077
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.