 Xiangyue Zhou
Xiangyue Zhou Hanmin Chen
Hanmin Chen Ling Wang2
Ling Wang2 Cameron Lenahan
Cameron Lenahan Lifei Lian
Lifei Lian Yibo Ou
Yibo Ou Yue He
Yue He
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 07 September 2021
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.721428
This article is part of the Research TopicMitochondrial Dysfunction in StrokeView all 12 articles
Stroke is one of the leading causes of death and disability worldwide. Brain injury after ischemic stroke involves multiple pathophysiological mechanisms, such as oxidative stress, mitochondrial dysfunction, excitotoxicity, calcium overload, neuroinflammation, neuronal apoptosis, and blood-brain barrier (BBB) disruption. All of these factors are associated with dysfunctional energy metabolism after stroke. Mitochondria are organelles that provide adenosine triphosphate (ATP) to the cell through oxidative phosphorylation. Mitochondrial dynamics means that the mitochondria are constantly changing and that they maintain the normal physiological functions of the cell through continuous division and fusion. Mitochondrial dynamics are closely associated with various pathophysiological mechanisms of post-stroke brain injury. In this review, we will discuss the role of the molecular mechanisms of mitochondrial dynamics in energy metabolism after ischemic stroke, as well as new strategies to restore energy homeostasis and neural function. Through this, we hope to uncover new therapeutic targets for the treatment of ischemic stroke.
Stroke is an acute cerebrovascular disease resulting in cerebral blood circulation disorders due to the sudden rupture or occlusion of blood vessels in the brain. Stroke is associated with high morbidity, mortality, and rates of disability (GBD 2016 Stroke Collaborators, 2019), and can be classified as either ischemic or hemorrhagic. Currently, stroke has become the second leading cause of death globally (Lindsay et al., 2019), and is the primary cause of death in China (Wang Y. et al., 2020). It has been a difficult endeavor to save more lives and improve neurological recovery after stroke. As such, this challenge emphasizes the growing need for therapeutic agents that can mitigate brain injury and promote neurological recovery after stroke.
Energy metabolism is an important basis for cellular function, as it is the process by which cells utilize nutrient substances, such as sugars and fats, and produce adenosine triphosphate (ATP). Additionally, ATP is broadly used in cellular activities, and is necessary for ensuring a normal cell lifespan. Mitochondria, which are commonly considered the powerhouse of the cell, are a major site of oxidative metabolism in eukaryotes, and are where sugars, fats, and amino acids are ultimately oxidized to release energy (Cardoso et al., 2010). The state of cellular energy metabolism is closely associated with mitochondrial dynamics, which refers to the dynamic process of mitochondrial fusion and division. Mitochondria maintain a steady state in the mitochondrial network through continuous fusion-division, thus maintaining the normal physiological function of cells (Dorn and Kitsis, 2015). Mitochondrial dynamics are involved in the formation and regulation of mitochondrial permeability transition pores (MPTPs), reactive oxygen species (ROS), and neuronal apoptosis (Roy et al., 2015). Mitochondrial dynamics can affect energy metabolism and post-stroke neuronal function by regulating the number, morphology, and function of mitochondria.
To identify potential interventional targets and novel diagnostic methods, it is crucial to understand the molecular mechanisms, especially those of mitochondrial dynamics after ischemic stroke. Herein, we will discuss the role of mitochondrial dynamics, as well as the energy metabolism involved in ischemic stroke. Moreover, an improved understanding of how mitochondrial dynamics affect energy metabolism will provide opportunities for the development of new therapeutic strategies targeting mitochondrial fusion and division after ischemic stroke.
Cell energy metabolism refers to the metabolic pathway of ATP synthesis associated with nicotinamide adenine dinucleotide (NADH) turnover (Rigoulet et al., 2020). This pathway mainly includes the decompositional metabolism of sugar (aerobic oxidation, glycolysis, and phosphate sugar pathway), the tricarboxylic acid (TCA) cycle, fatty acid oxidation and synthesis, amino acid metabolism, and vitamin metabolism. Mitochondria are “energy factories” in eukaryotic cells, and are key sites of oxidative phosphorylation. Mitochondria are organelles that are present in most cells and are coated by two layers of membrane. Mitochondria can be divided into four functional regions: the outer mitochondrial membrane (OMM), intermembrane space (IMS), inner mitochondrial membrane (IMM), and mitochondrial matrix (listed in order from outside to inside). The proton concentration gradient originating from the electron transport chain in the IMM drives ATP generation (Scheffler, 2001). Moreover, mitochondria are highly mobile. Mitochondrial dynamics include fusion, division, selective degradation, and transport processes. Dynamic changes in mitochondria are important for immunity, apoptosis, and the cell cycle. These dynamic transformations are mainly mediated by large GTPases that belong to the dynamin family (Tilokani et al., 2018). In addition to generating energy, mitochondria can also drive cell dysfunction or death either passively (through ROS toxicity) or actively (through programed necrosis and apoptosis). Mitochondrial division and fusion play central roles in these processes (Dorn and Kitsis, 2015).
Mitochondrial fusion refers to the merging of two mitochondria into a single mitochondrion (Figure 1). Because mitochondria have two layers of membrane, the process of mitochondrial fusion consists of outer membrane fusion and inner membrane fusion (Table 1). The tether is a physical connection between the two mitochondrial outer membranes, and is a prerequisite for actual membrane fusion (Koshiba et al., 2004). Mitofusin1 (Mfn1) and Mfn2 mediate fusion of the OMM, and optic atrophy protein 1 (Opa1) mediates fusion of the IMM (Chiurazzi et al., 2020). The overexpression of Mfn2 could increase mitochondrial fusion (Qin et al., 2020). However, the absence of Mfn1, Mfn2 (Hoppins, 2014), or Opa1 can lead to mitochondrial fragmentation (Song et al., 2007). Mitochondrial fusion can also facilitate the exchange of matrix contents among mitochondria through brief contact without resulting in a morphological merge. This is described as kiss-and-run fusion events (Chan, 2020). Mitochondrial fusion, and the material exchanged between mitochondria, optimize mitochondrial function and avoid damage accumulation due to mutations in mitochondrial DNA aging (Westermann, 2010; Chen et al., 2011; Cohen and Tareste, 2018).
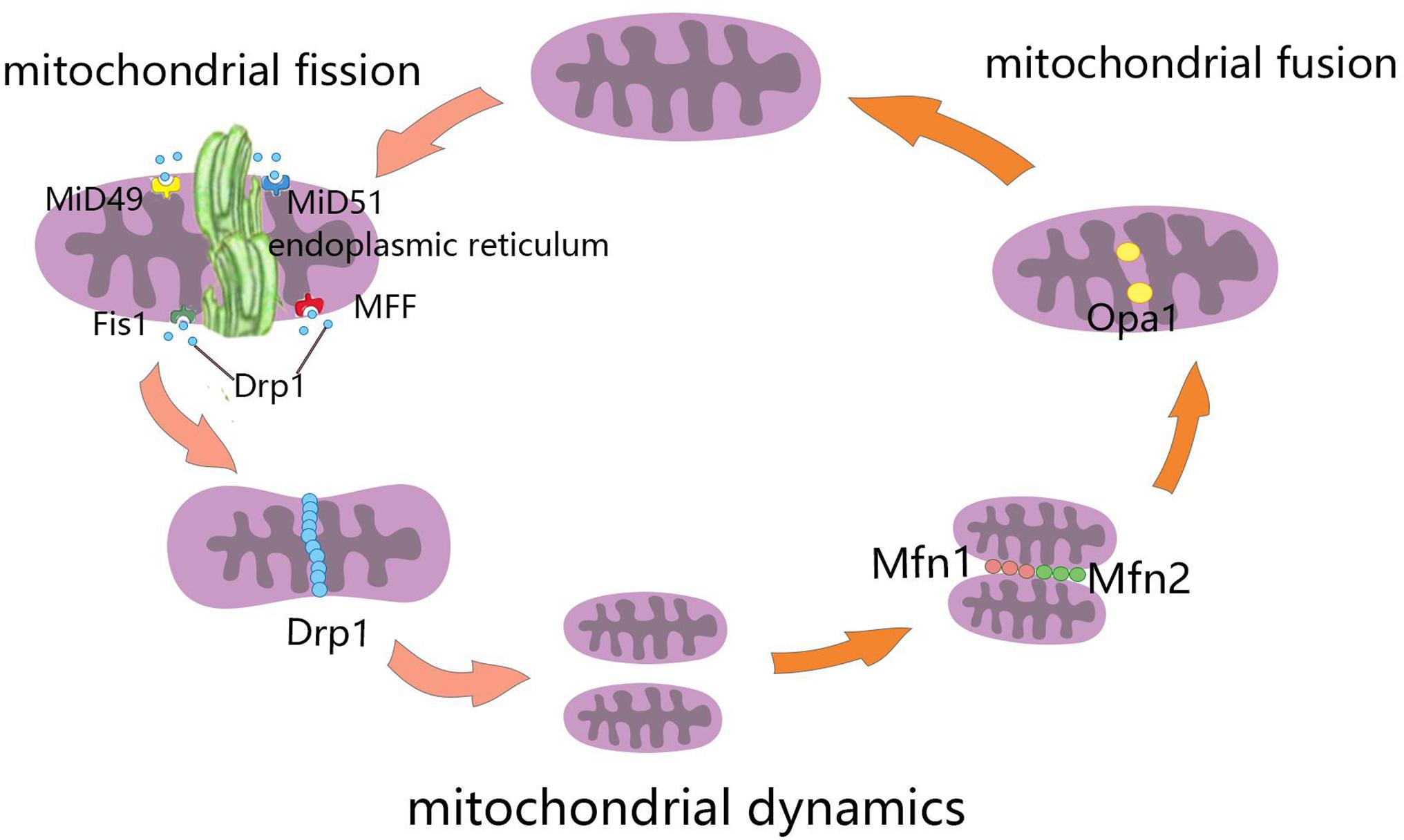
Figure 1. Schematic diagram of mitochondrial dynamics. Mitochondrial fission is the division of mitochondria into two smaller mitochondria, and the dynamin-related GTPase Drp1 plays a crucial role in this process. Drp1 was recruited through four single-way transmembrane Drp1 receptors anchored on the outer mitochondrial membrane (OMM): mitochondrial fission factors (Mff), mitochondrial kinetic proteins 49 and 51(MiD49 and MiD51), and Fis1. Mitochondrial fusion refers to the merging of two mitochondria into one. Mitofusin1 (Mfn1) and mitofusin2 (Mfn2) mediate mitochondrial outer membrane fusion, and optic atrophy protein 1 (Opa1) mediates the fusion of mitochondrial inner membrane.
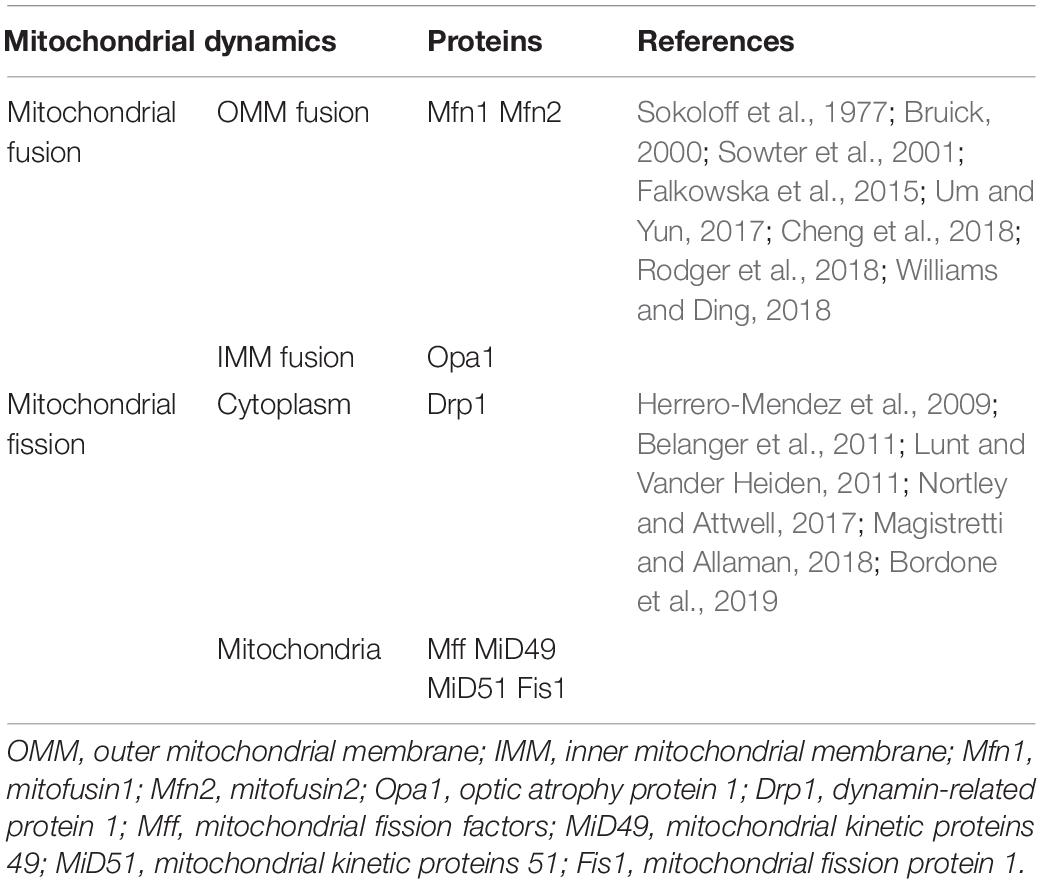
Table 1. Mitochondrial dynamics related proteins.
Mitochondrial fission is the division of a mitochondrion into two smaller mitochondria (Figure 1), and dynamin-related GTPase1 (Drp1) plays a crucial role in this process (Table 1). Drp1 is recruited through four single-way transmembrane Drp1 receptors anchored on the OMM: mitochondrial fission factors (Mffs), mitochondrial kinetic proteins 49 and 51(MiD49 and MiD51), and Fis1 (Pagliuso et al., 2018). Mitochondrial fission usually occurs at the endoplasmic reticulum (ER)-mitochondrial contact site (Lewis et al., 2016). Additionally, the contraction of mitochondria is Drp1-independent, and ER tubules are more important in defining the position of mitochondrial fission sites (Friedman et al., 2011). However, mitochondrial fusion and fission are colocalized in ER membrane contact sites (MCSs; Guo et al., 2018). Therefore, mitochondrial fission and fusion could be regulated by controlling certain enzyme-like nodes on the ER MCS (Abrisch et al., 2020). Mitochondrial fission can irreparably fragment mitochondria, remove organelles to maintain mitochondria quality, and protect the normal function of the mitochondrial network (Nunnari, 2007).
Mitochondrial dynamics can regulate mitochondrial morphology, promote mitochondrial substance exchange, maintain mitochondrial DNA and inheritance, and eliminate damaged mitochondria. Normal regulation of mitochondrial dynamics is important in maintaining regular cell activity. Dysregulation of Mitochondrial dynamics plays an important role in driving cell death. Mitochondrial dynamics also play a role in necroptosis. The mitochondrial phosphatase, PGAM5, dephosphorylates Drp1Ser-637, leading to increased ROS and mitochondrial division, as well as promotion of necroptosis (Wang et al., 2012; Yu et al., 2020). When the energy supply is decreased, the increased levels of ADP and AMP can promote mitochondrial division and induce autophagy by activating MiD51 and Mff, respectively (Ducommun et al., 2015; Toyama et al., 2016). In addition, the reduction in Opa1 leads to an increase in autophagy (White et al., 2009). Mitochondrial fission is a necessary step in apoptosis (Xie et al., 2018). Apoptosis is a unique and important mode of programed cell death (Elmore, 2007) that maintains the number of cells in tissues, and it functions as a defense mechanism in the immune response (Norbury and Hickson, 2001). Drp1 can promote Bax translocation to the mitochondria (Montessuit et al., 2010), change the permeability of the mitochondrial membrane, release cytochrome C (Cyt-c), cause a cascade reaction, and lead to apoptosis. Mitochondrial fusion can protect cells from apoptosis (Bueler, 2010). Opa1 maintains the stability of mitochondrial cristae (Varanita et al., 2015) and Mfn2 interferes with Bax translocation by promoting mitochondrial fusion (Neuspiel et al., 2005). However, both Drp1 and Opa1 can prevent apoptosis. A variety of cell death patterns, such as apoptosis, necrosis, phagoptosis, and autophagy, form a complex network with different molecular mechanisms after stroke (Fricker et al., 2018), which suggests that mitochondrial dynamics may play an important role in cell death after stroke. However, in this review we mainly focused on the molecular mechanisms of mitochondrial dynamics in apoptosis after ischemic stroke.
In addition, mitophagy is a defensive mechanism that selectively removes damaged or unnecessary mitochondria via autophagy, which plays an important role in maintaining mitochondrial quality control and homeostasis (Gustafsson and Dorn, 2019). Mitophagy is mediated by autophagy-related proteins. Autophagy-related proteins specifically recognize and bind to functionally defective mitochondria so that they become fused with lysosomes to complete the degradation of damaged organelles and proteins (Gustafsson and Dorn, 2019). Mitophagy is closely associated with many functions and physiological processes in cells, such as cell differentiation and development, cell programing, cell death, and the immune response (Um and Yun, 2017). Molecular mechanisms of mitophagy involve PTEN-induced putative kinase 1(PINK1)/Parkin, BCL2/adenovirus E1B 19kDa-interacting protein 3 (BNIP3)/BCL2/adenovirus E1B 19kDa-interacting protein 3-like (NIX), FUN14 domain containing 1 (FUNDC1), BCL2-like 13(BCL2L13), FK506-Binding Protein 8 (FKBP8), and Cardiolipin (Rodger et al., 2018; Williams and Ding, 2018). BNIP3/NIX is associated with hypoxia-induced mitophagy, and the levels of BNIP3 and NIX are upregulated by hypoxia-inducible factor (HIF-1α) transcription (Bruick, 2000; Sowter et al., 2001), suggesting that this pathway may be involved in the brain damage that occurs as a result of the hypoxic ischemia that manifests after stroke.
The weight of the brain accounts for only 2% of total body weight. However, the brain accounts for 25 and 20% of glucose and oxygen consumption in the body, respectively (Sokoloff et al., 1977). Under aerobic conditions, ATP in brain cells is primarily derived from glucose that is consumed in the TCA cycle occurring in the mitochondria for oxidative phosphorylation. Moreover, the uptake and utilization of glucose by cells in the brain are associated with specific features. This process requires several cell types, which comprise the neurovascular unit, to coordinate and complete it. The neurovascular unit is composed of brain capillary endothelial cells, pericytes, astrocytes, oligodendrocytes, microglia, and neurons (Cheng et al., 2018; Figure 2). Glucose enters cells through specific glucose transporters (GLUTs), such as GLUT1, GLUT2, and GLUT7 in astrocytes, GLUT1 in oligodendrocytes, and GLUT3 and GLUT4 in neurons. Additionally, glucose is phosphorylated by hexokinase to produce glucose-6-phosphate (Falkowska et al., 2015). Glucose 6-phosphate can be processed through various metabolic pathways (e.g., glycolysis, pentose phosphate pathway, and glycogenesis). Numerous metabolic intermediates formed by glucose in the brain, such as lactate, pyruvate, glutamate, or acetate, can also be utilized to produce energy (Belanger et al., 2011).
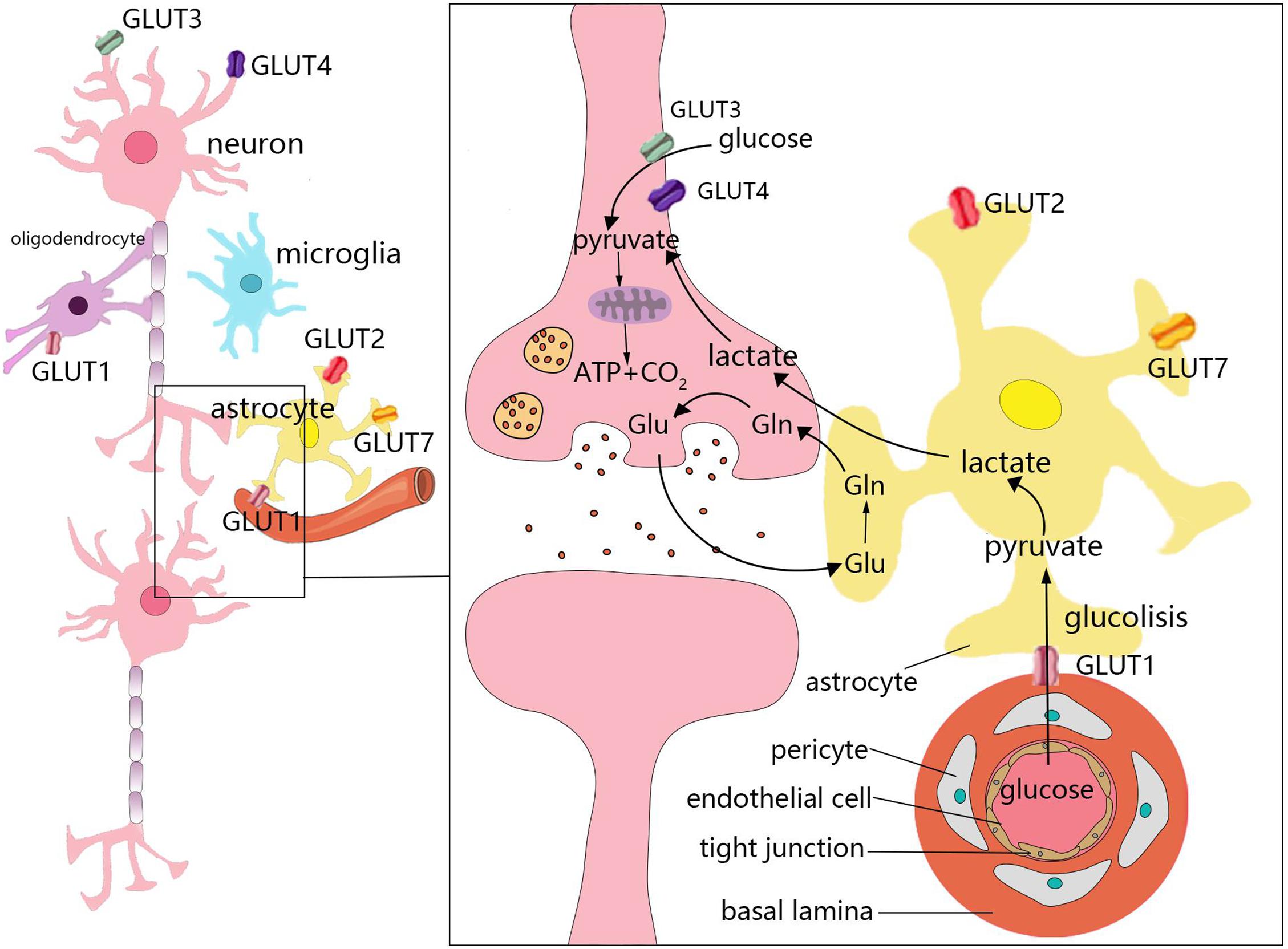
Figure 2. Energy metabolism in the brain. The uptake and utilization of glucose by cells in the brain requires the neurovascular unit, which is composed of brain capillary endothelial cells, pericytes, astrocytes, oligodendrocytes, microglia, and neurons. Glucose enters cells through specific glucose transporters (GLUTs), such as GLUT1, GLUT2, and GLUT7 in astrocytes, GLUT1 in oligodendrocytes, and GLUT3 and GLUT4 in neurons. Numerous metabolic intermediates formed by glucose in the brain can also be oxidized to produce energy, such as lactate, pyruvate, and glutamate. The dynamic regulatory mechanism of lactic acid metabolism between astrocytes and neurons is known as the astrocyte–neuron lactate shuttle. Neurons release glutamate to stimulate glucose uptake by astrocytes. Astrocytes produce lactic acid by aerobic glycolysis in the cytoplasm and then transport to neurons.
In the brain, different cell types have different metabolic characteristics. There is dynamic regulation between astrocytes and neurons known as the astrocyte–neuron lactate shuttle (Bordone et al., 2019; Figure 2). Neurons release glutamate to stimulate astrocytes to take up glucose (Magistretti and Allaman, 2018). Due to the low activity of pyruvate dehydrogenase in astrocytes, it is easier to produce lactic acid via aerobic glycolysis in the cytoplasm, which is also known as the Warburg effect in cancer cells (Lunt and Vander Heiden, 2011; Magistretti and Allaman, 2018). Astrocytes play a significant role in regulating the energy supply of the brain, but it remains unclear whether the lactic acid produced by astrocytes is fuel for neurons (Nortley and Attwell, 2017). Neurons lack fructose 6-phosphate-2-kinase/fructose-2,6- bisphosphatase-3(Pfkfb3), thus demonstrating a slower rate of glycolysis than other cells. However, neurons can efficiently utilize lactic acid (Herrero-Mendez et al., 2009; Bolanos, 2016). After glucose is transported through the blood-brain barrier (BBB) and the cell membrane, it is transformed into pyruvate via anaerobic glycolysis in the cytoplasm, which leads to the generation of 2 mol of ATP. Each mole of glucose pyruvate is subsequently transported to the mitochondria, and is then converted into acetyl coenzyme A, which then participates in the TCA cycle. During the subsequent steps of the TCA cycle, oxidative phosphorylation produces an additional 30 mol of ATP per mole of glucose. This process is closely associated with the TCA cycle, electron transfer in the respiratory chain, oxygen consumption, and the production of carbon dioxide and water. In addition to basic cell activities, the energy produced by various cells in the brain is mainly used to maintain and restore ion gradients that are dissipated by signaling processes (e.g., postsynaptic and action potentials), as well as participate in the uptake and recycling of neurotransmitters (Attwell and Laughlin, 2001).
Obviously, ischemic hypoxic brain injury after stroke is closely associated with energy metabolism disorder. However, there are various pathophysiological mechanisms involved in brain damage after stroke, such as oxidative stress, mitochondrial dysfunction, excitotoxicity, calcium overload, neuroinflammation, acidosis, neuronal apoptosis, and BBB disruption (Moskowitz et al., 2010; Sekerdag et al., 2018). The normal supply of glucose and oxygen is cut off during cerebral ischemia, although astrocytes can synthesize and store glycogen, as well as metabolize glycogen into lactic acid to provide energy for neurons (Mergenthaler et al., 2013; Falkowska et al., 2015). When the glucose supply is insufficient, ketone bodies and lactic acid can also become substrates for brain energy metabolism (Sokoloff, 1981). In early ischemia, when synaptic activity disappears, adenosine is released to block presynaptic Ca2+ influx and inhibit glutamate release (Hofmeijer and van Putten, 2012). This early inhibition of glutamate release prevents glutamate excitotoxicity. However, if the ischemic period is prolonged, neuronal cells cannot maintain the normal transmembrane concentration gradient, which causes neuronal signal impairment. Conversely, synaptic terminal depolarization releases the neurotransmitter glutamate. Furthermore, the reabsorption of glutamate clearance from the synaptic space leads to glutamic acid accumulation, excessive stimulation of N-methyl-D-aspartate (NMDA) receptors, high levels of calcium influx, mitochondrial depolarization, the release of Cyt-c, and neuronal apoptosis (Harris et al., 2012; Campbell et al., 2019).
After stroke, different regions comprising the stroke lesion will exhibit various metabolic characteristics. The location of the stroke lesion can be divided into three regions according to cerebral blood flow (CBF; Pushie et al., 2018). The ischemic core area is the central region of brain tissue infarct. The ischemic periphery region surrounding the core is divided into two components. One is close to the infarct area. Here, there are a large number of cells undergoing oxidative stress due to the reduced blood supply, but timely reperfusion therapy can save this section of tissue, known as the ischemic penumbra. The ischemic peripheral region is relatively far from the infarct area, and the blood circulation is relatively similar to that of normal tissue. The levels of glucose utilization, ATP, lactic acid, creatine phosphate, and pH varies according to CBF changes (Leigh et al., 2018). The ischemic penumbra cannot be rescued and it expands into the infarct area when CBF decreases beyond a certain threshold (Astrup et al., 1981). Currently, it is an important objective in stroke treatment to identify ischemic penumbral tissue based on PET/CT and related brain metabolism (Sarrafzadeh et al., 2010; Zenonos and Kim, 2010), and to treat it in a timely manner. Due to the different metabolic demands of grey matter (GM) and white matter (WM), the CBF and cerebral blood volume (CBV) thresholds of the damaged region are different (An et al., 2015; Leigh et al., 2018). Moreover, GM is more susceptible to ischemia than WM because GM has a higher CBF, CBV, and apparent diffusion coefficient (ADC), as well as a shorter mean transit time (MTT; Bristow et al., 2005). GM mainly includes neuronal cell bodies, dendrites, and axons, which are used for local information, while WM mainly includes axons, oligodendrocytes, and astrocytes. Neuronal apoptosis or necrosis results from excessive free radical production, calcium overload, and excitotoxicity following ischemia and hypoxia (Radak et al., 2017). The decreased blood supply after stroke leads to the destruction of axonal electrophysiological characteristics and nutritional dysfunction in WM. The effect of glutamate metabolism on astrocytes induces excitotoxicity in oligodendrocytes (Wang et al., 2016). After reperfusion, energy metabolism does not immediately return to baseline as expected. The activation of platelets and complement systems, the release of inflammatory mediators, and neuronal mitochondria in the ischemic penumbra overcompensate for ischemic injury by inducing metabolic pathways, which leads to the excessive release of ROS. All of these factors contribute to neuronal death and brain injury after ischemia-reperfusion (Al-Mufti et al., 2018).
The mechanism of mitochondrial division and fusion is complex, and not only affects energy metabolism in cells, but also induces apoptosis. After cerebral ischemia and hypoxia, changes in mitochondrial dynamics also greatly impact the survival of nerve cells. Seventy percent of neuronal energy is used to maintain the sodium and potassium pump on the cell membrane. The ATP supply is insufficient after stroke, which leads to depolarization of the neuronal plasma membrane, release of the excitatory neurotransmitter, glutamate, and causes glutamate excitotoxicity (Doyle et al., 2008). Ischemia induced an increase in glutamate release from neurons and astrocytes, which leads to overstimulation of NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors. Glutamate excitotoxicity and oxidative stress influence mitochondrial division and fusion, as well as the imbalance in mitochondrial division and fusion, leading to NMDA receptor upregulation and oxidative stress (Nguyen et al., 2011). Next, we will review the molecular mechanisms of mitochondrial dynamics in post-stroke injury (Figure 3).
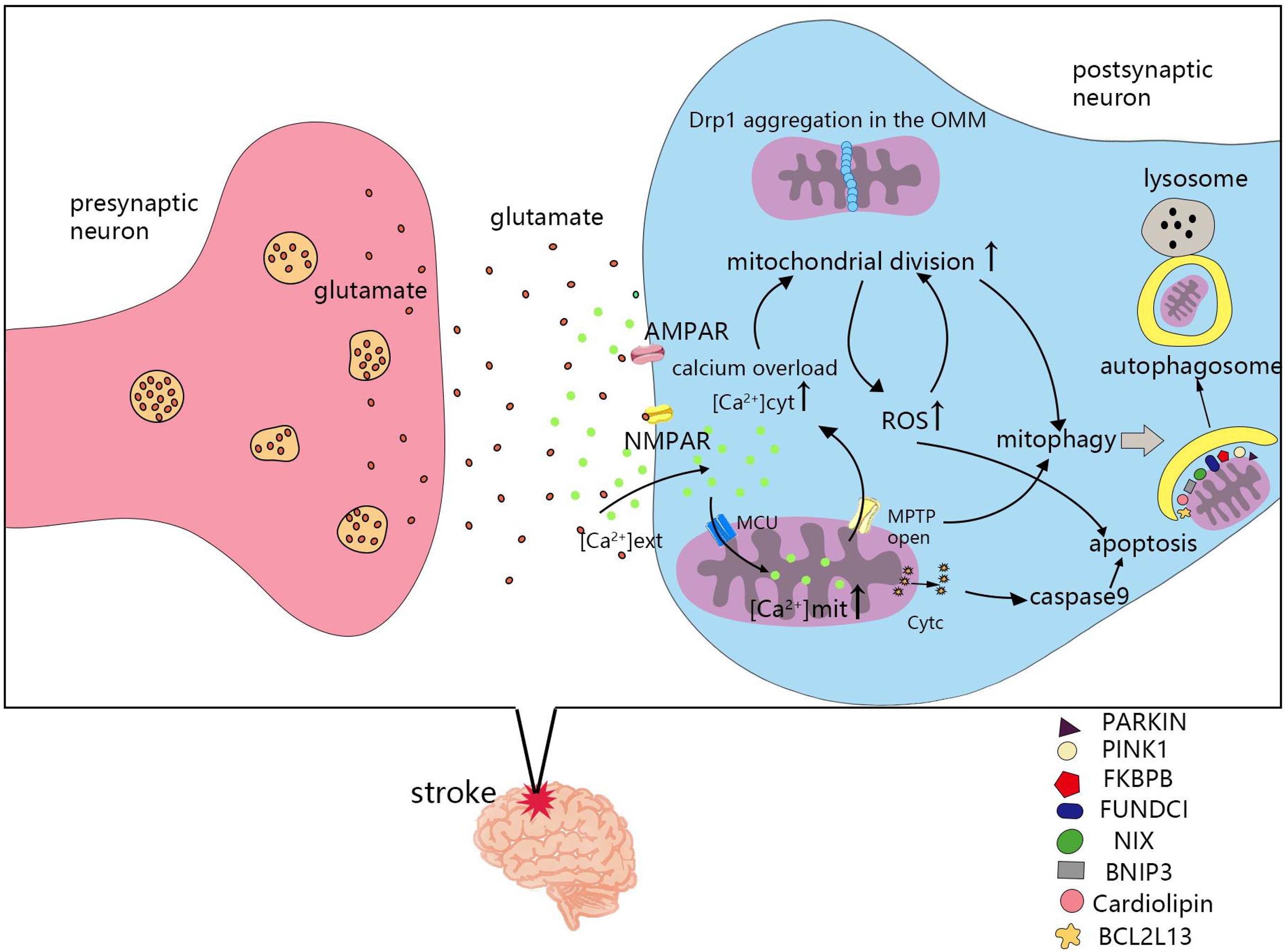
Figure 3. Molecular mechanisms of mitochondrial dynamics in post-ischemic stroke injury. Cerebral ischemia and hypoxia lead to the imbalance of mitochondrial division and fusion, which is related to calcium overload, reactive oxygen species (ROS), mitochondrial permeability transition pores(MPTP), apoptosis, and mitophagy. MCU, mitochondrial calcium uniporter; NMPAR, N-methyl-D-aspartate receptors; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors; [Ca2+] ext, calcium ion concentration in extracellular; [Ca2+] cyt, calcium ion concentration in cytoplasmic; [Ca2+] mit, calcium ion concentration in mitochondria.
Excessive calcium enters cells, activates many calcium-dependent proteases, lipases, and deoxyribonucleases, and leads to cell death (Besancon et al., 2008). The mitochondrial calcium uniporter (MCU), a calcium transporter in mitochondria, plays a vital role in maintaining intracellular homeostasis by transporting Ca2+ from the cytoplasm into the mitochondrial matrix (Zhao et al., 2013; Kwong et al., 2015). Mitochondrial Ca2+ controls energy production and metabolism by regulating key enzymes and fatty acid oxidation in the TCA cycle. Calcium overload in the cytoplasm activates calcineurin, and dephosphorylates Drp1 at serine 637, leading to Drp1 accumulation from the cytoplasm to the OMM. Promoting mitochondrial division and ROS production, but inhibiting mitochondrial division by using mitochondrial division inhibitor 1(Mdivi-1), Drp1siRNA, or calcineurin inhibitors shows that mitochondrial morphology is retained, intracellular calcium ions are reduced, and cell death is prevented in cardiac ischemia-reperfusion injury (Sharp et al., 2014). Inhibiting Drp1 can participate in neuroprotection by combating glutamate toxicity in vitro and ischemic brain injury in vitro (Grohm et al., 2012). Calcium overload also causes mitochondria to release apoptotic factors and induce apoptosis (Pivovarova et al., 2004).
Reactive oxygen species and oxidative stress are important causes of tissue damage during cerebral ischemia (Crack and Taylor, 2005). Mitochondria are thought to be the main origin of intracellular ROS (Bayir and Kagan, 2008). Oxygen-free radicals are highly active and have the ability to destroy cellular components (Droge, 2002). Changes in ROS lead to changes in the expression or activity of proteins associated with mitochondrial dynamics that affect mitochondrial fusion and division (Cid-Castro et al., 2018). In general, elevated ROS levels trigger mitochondrial fission. It is worth mentioning that phosphorylation at different sites of Drp1 differentially affects mitochondrial dynamics. The increase in ROS promotes Drp1 activation through Ser616 phosphorylation, which promotes mitochondrial fission (Cho et al., 2012). However, Drp1 serine 637 phosphorylation inhibits mitochondrial fission. The phosphorylation of Drp1 at tyrosines 266, 368, and 449 leads to mitochondrial division and neuronal death (Zhou et al., 2017). The Drp1-mitochondrial fission ROS cycle may play a role in central nervous system (CNS) diseases (da Rosa et al., 2020). There is an interaction between mitochondrial dynamics and ROS production. On one hand, increased mitochondrial division will increase the production of ROS, but inhibition of mitochondrial division can restore ROS levels to normal (Zhang Y. K. et al., 2020). On the other hand, the increase of ROS will promote the activation of Drp1, leading to increased mitochondrial division (Youle and van der Bliek, 2012; Cid-Castro et al., 2018), thereby further increasing the production of ROS, whereas knocking out Drp1 reduces oxidative stress-induced mitochondrial fragmentation (Youle and van der Bliek, 2012). This cyclic reaction will aggravate mitochondrial dysfunction after stroke, increase the level of ROS, and increase brain damage after stroke. After inhibiting ROS levels in a mouse model of stroke, mitochondrial dysfunction, obstruction volume, and neurological deficits were reduced (Hwang et al., 2020). From this perspective, interrupting this cycle by reducing ROS levels or inhibiting mitochondrial division is a key part of stroke treatment. Studies have shown that inhibiting excessive mitochondrial division could protect against neurotoxicity (Rappold et al., 2014). Hyperbaric oxygen (HBO) can also reduce brain damage by regulating Drp1 phosphorylation (Ni et al., 2020). ROS can also activate OMA1 to promote the cleavage of L-Opa1 to S-Opa1, resulting in mitochondrial crest remodeling and Cyt-c release, further resulting in increased apoptosis (Zhang K. et al., 2014).
The MPTP is a channel that triggers a sudden increase in the permeability of the mitochondrial inner membrane when there is mitochondrial calcium overload, especially when combined with oxidative or nitrite stress and/or ATP depletion (Bernardi, 1999). When the MPTP is open, it can lead to a rapid increase in the osmotic pressure of the matrix solute, rupture of the OMM, collapse of the mitochondrial membrane potential (ΔΨm), and a reduction in intracellular ATP, which eventually leads to necrotic cell death (Kinnally et al., 2011). The MPTP can also be used as a physiological efflux pathway, and can open transiently (Altschuld et al., 1992). In cyclophilin D-knockout mice, transient activation of the MPTP could protect neurons from cytoplasmic Ca2+ overload (Barsukova et al., 2011). Mitochondrial matrix calcium overload is the main mechanism of MPTP opening, and plays an important role in regulating the MPTP (Hurst et al., 2017). Moreover, ROS can also regulate the activity of the MPTP (Halestrap et al., 1997). Hence, the opening of the MPTP is involved in the pathophysiological mechanism after stroke. The infarct size after transient occlusion of the artery can be reduced by the cyclosporine A analog, N-methyl-Val-4-cyclosporin A (Me ValCsA), which blocks the MPTP (Matsumoto et al., 1999). Melatonin and atorvastatin can also block the MPTP and the release of Cyt-c from mitochondria, thereby reducing neuronal apoptosis after ischemia-reperfusion (Andrabi et al., 2004; Song et al., 2014). Treatment of HL-1 cells with Drp-1 inhibitors reduces mitochondrial permeability transition pore sensitivity after cardiac ischaemia-reperfusion (Ong et al., 2010). In Mfn-2-deficient mice, MPTP opening is delayed. Therefore, Mfn-2 participates in the regulation of the MPTP and triggers death in cardiomyocytes (Papanicolaou et al., 2011).
After apoptotic stimulation, apoptotic members of the Bcl-2 protein family, such as Bax and Bak, insert into the mitochondrial outer membrane (Ader et al., 2019) and change the permeability of the OMM, thus mediating the release of apoptosis-related factors, such as Cyt-c, second mitochondria-derived activator of caspases (Smac), and apoptosis-inducing factor (AIF) from mitochondria into the cytoplasmic matrix (Jurgensmeier et al., 1998; Susin et al., 1999; Jeong and Seol, 2008). Apoptosis-related factors in the cytoplasm activate caspase-9, causing cascade reactions, consequently leading to apoptosis (Green and Reed, 1998). However, mitochondrial dynamics is involved in the regulation of apoptosis (Frank et al., 2001). The dominant-negative mutant of Drp1, as well as siRNA-mediated silencing of Drp1, could reduce apoptosis in Q111/0 cells in Huntington’s disease (Costa et al., 2010). By inhibiting Drp1 activation, mitochondrial debris and ROS can be reduced, and neuronal damage can be alleviated (Zhou et al., 2020). Using propofol to treat oxidation-injured neurons could reduce the expression of Fis1 and increase the expression of Mfn1, maintaining normal levels of these two proteins, thus reducing the expression of apoptotic proteins in neurons and alleviating neuronal damage induced by hypoxia (Zhang H. S. et al., 2020). Research has shown that inducing mitochondrial fission via activation of Drp1 and Mff results in apoptosis in hippocampal neuronal cells (Zhang C. et al., 2020).
The regulation of mitochondrial autophagy is associated with mitochondrial dynamics (Yoo and Jung, 2018), and is expected to be a new target for ischemic stroke treatment (Guan et al., 2018). In an acute model of mitochondrial damage induced by carbonyl cyanide-m-chlorophenylhydrazone (CCCP), the ubiquitination of mitofusin 1 (Mfn-1) and 2 (Mfn-2) is closely associated with the induction of mitophagy, and the ubiquitination of both proteins is dependent on parkin and PINK1, which play crucial roles in mitophagy (Gegg et al., 2010). In Parkin-null HeLa cells and in HEK293 cells with siRNA-mediated Parkin knockdown, glycoprotein 78 (Gp78) induction of mitophagy was Mfn1-dependent (Fu et al., 2013). In adult cardiac progenitor cells (CPCs), knockdown of either Fundc1 or Bnip3l had no effect on Mfn1/2 protein levels, but led to the activation of DNM1L and the promotion of mitochondrial fission during differentiation (Lampert et al., 2019). In proximal tubule-specific Fundc1-knockout mice, Drp1 is overactivated and leads to mitochondrial division (Wang J. et al., 2020). In traumatic brain injury (TBI) models, the Drp1 inhibitor, Mdivi-1, could inhibit the activation of PTEN-induced putative kinase 1 (PINK1)-Parkin mediated mitophagy (Wu et al., 2018).
Mitochondrial dynamics play various roles in different cells after stroke. Because of the length of axons and special energy requirements in neurons, the distribution, movement, and timely fusion and repair of mitochondria are particularly important in maintaining normal neuronal function (Mandal and Drerup, 2019). Oxygen-glucose deprivation (OGD) can induce mitochondrial division, leading to autophagy or apoptosis (Wu et al., 2017). Astrocytic endfeet are enriched in mitochondria-ER contact sites. These sites, as well as mitochondrial fusion, can promote vascular remodeling after brain injury (Gobel et al., 2020). After several hours of OGD, the length of the astrocytic endfeet were shortened, mitochondrial fission was delayed, and the mitochondria were lost (O’Donnell et al., 2016). Increased mitochondrial division in astrocytes during hypoxia may be a possible method of increasing mitochondrial energy production (Quintana et al., 2019). In microglia, the inflammatory response induced by OGD can be improved by inhibiting mitochondrial fission (Zhou et al., 2019b). Mitochondrial division can also promote the production of mitochondrial ROS in microglia by activating NF-κB and mitogen-activated protein kinase (MAPK), and by inducing the expression of proinflammatory mediators (Park et al., 2013). The neurovascular unit is composed of neurons, glial cells, such as astrocytes and microglia, vascular cells, such as endothelial cells and perivascular cells, and basement membrane (Iadecola, 2017). After inducing excessive mitochondrial division, endothelial cell death also increased (Rao et al., 2020). After stroke, changes in mitochondrial dynamics affect the cells of the neurovascular unit, thus destroying the BBB. Using Mdivi-1 to inhibit mitochondrial division can reduce damage to the BBB after TBI (Wu et al., 2018). Oligodendrocytes primarily wrap axons and form insulated myelin structures, which are essential in maximizing the conduction and velocity of the action potential. Unlike neurons, mitochondria are sparsely distributed in oligodendrocytes with poor fluidity, and glutamate activation can promote mitochondrial motility in oligodendrocytes (Rinholm et al., 2016). However, unlike in astrocytes, the use of mitochondrial division inhibitor 1 makes oligodendrocytes sensitive to excitotoxicity and ER stress due to their non-targeted effects, resulting in oxidative stress and apoptosis (Ruiz et al., 2020).
As mentioned previously, the interaction between calcium overload, ROS production, and the MPTP leads to an increase in mitochondrial fission in ischemic stroke. Although increased mitochondrial fission during hypoxia may increase mitochondrial energy production, which is beneficial in the maintenance of neural function after stroke (Quintana et al., 2019), inducing mitochondrial fission is detrimental to neurons (Zhang C. et al., 2020). However, inhibiting Drp1 to restore the balance of mitochondrial fission and fusion could reduce Bax oligomerization and the release of apoptotic factors after ischemic stroke, thereby reducing the volume of cerebral infarction (Zhao et al., 2014). Exercise preconditioning could promote mitochondrial fusion after cerebral ischemia by up-regulating OPA1, thus reducing cerebral edema and improving neurologic function in ischemic stroke (Zhang L. et al., 2014). Mitochondrial fission can clear damaged mitochondria through autophagy, but excessive mitochondrial division affects the normal function of mitochondria so that the production of ATP is reduced (Wai and Langer, 2016; Sprenger and Langer, 2019). Dysfunction of mitochondrial dynamics can lead to obstacles in mitochondrial distribution and transport in neurons, which can lead to insufficient local energy in neurons (Sheng, 2014). Excessive mitochondrial fission also affects intracellular calcium homeostasis and exacerbates excitotoxicity (Wang et al., 2015). However, mitochondrial fusion can repair damaged mitochondria (Cohen and Tareste, 2018) and produce additional energy by upregulating the activity of ATP synthase through the remodeling of mitochondrial cristae (Gomes et al., 2011). Therefore, inhibiting excessive mitochondrial fission, properly promoting mitochondrial fusion, and restoring the balance of mitochondrial dynamics are beneficial in recovery after ischemic stroke.
The clinical treatment of ischemic stroke primarily includes acute stage treatment and subacute stage treatment. The acute stage occurs within 4.5 h. If patients meet admission criteria and have no thrombolytic contraindications, thrombolytic therapy is recommended (Powers et al., 2018). Currently, great progress has been made in endovascular treatment, such as mechanical thrombectomy, which has played a great role in the treatment of stroke (Prabhakaran et al., 2015). However, these treatments still have limitations, such as a strict time window for thrombolytic therapy, and there is a certain risk of complications, such as hemorrhagic transformation (Yaghi et al., 2017). Therefore, it is necessary to find new therapeutic targets and to develop corresponding drugs. The mitochondrial dynamics are closely associated with energy metabolism after stroke and pathophysiological mechanisms, such as ROS, apoptosis, and autophagy. Therefore, the molecular mechanisms associated with mitochondrial dynamics may represent new directions in ischemic stroke treatment. However, the excessive mitochondrial division plays an essential role in ischemic stroke. Therefore, an emphasis should be placed on inhibiting excessive mitochondrial fission and restoring the balance of mitochondrial dynamics when using mitochondrial dynamics as a focal point in the treatment of ischemic stroke (Table 2).
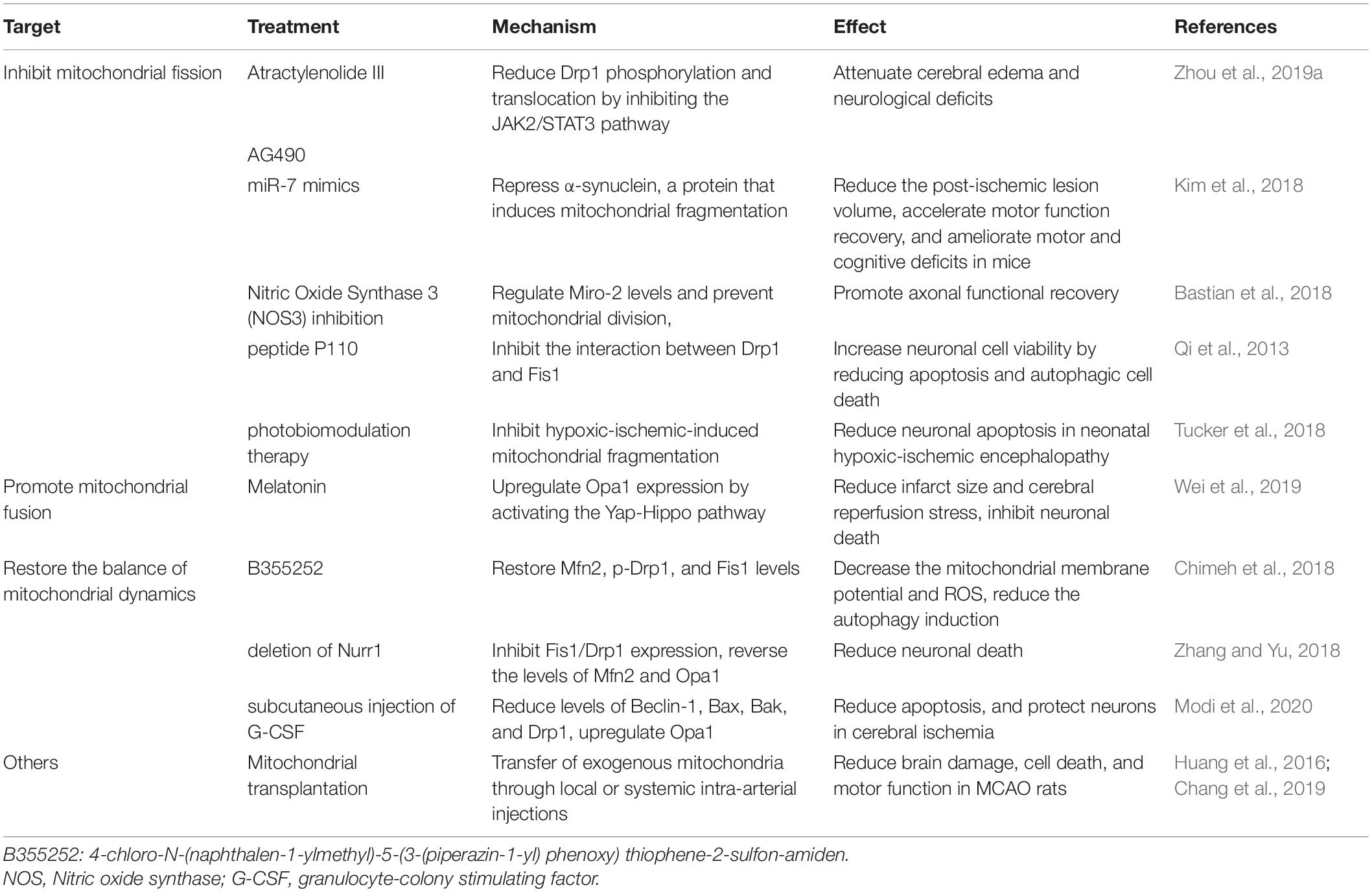
Table 2. Drugs targeting mitochondrial dynamics after ischemic stroke.
Atractylenolide III and AG490 (an inhibitor of jak2) therapy in middle cerebral artery occlusion (MCAO) mice could reduce Drp1 phosphorylation and translocation, as well as and mitochondrial division though the JAK2/STAT3 pathway, thereby attenuating cerebral edema and neurological deficits (Zhou et al., 2019a). Melatonin upregulates Opa1 expression by activating the Yap–Hippo pathway, thereby promoting mitochondrial fusion, reducing infarct size, inhibiting neuronal death, and reducing cerebral reperfusion stress (Wei et al., 2019). Opa1, Mfn2, p-Drp1, and FIS1 were decreased to varying degrees in the mouse model of CoCl2-induced cerebral hypoxia. However, 4-chloro-N-(naphthalen-1-ylmethyl)-5-(3-(piperazin-1-yl) phenoxy) thiophene-2-sulfon-amide (B355252) treatment could restore Mfn2, p-Drp1, and Fis1 levels, maintain mitochondrial stability, restore mitochondrial membrane potential, and reduce ROS (Chimeh et al., 2018). Injection of miR-7 mimic oligonucleotide after cerebral ischemia could repress α-synuclein, a protein that induces mitochondrial fragmentation, oxidative stress, autophagy, and the promotion of neuronal cell death, thus reducing brain injury after stroke (Kim et al., 2018). Nitric oxide synthase 3 (NOS3) inhibition regulates Miro-2 levels, prevents mitochondrial division, and promotes axonal functional recovery by protecting mitochondrial structure and movement (Bastian et al., 2018). Fis1 and Drp1 are increased, and both Mfn2 and Opa1 are downregulated after cerebral ischemia-reperfusion, indicating that cerebral ischemia-reperfusion induces excessive mitochondrial division and prevents mitochondrial fusion. However, deletion of Nurr1 could inhibit Fis1/Drp1 expression, reverse the levels of Mfn2 and Opa1, correct the imbalance in mitochondrial division and fusion, and reduce neuronal death (Zhang and Yu, 2018). In a mouse model of bilateral common carotid artery occlusion, subcutaneous injection of granulocyte-colony stimulating factor (G-CSF) was used as treatment. G-CSF reportedly reduced the levels of the autophagy marker, Beclin-1, and the proapoptotic proteins, Bax, Bak, and Drp1, but upregulated the mitochondrial fusion protein, Opa1. Therefore, G-CSF can maintain cellular homeostasis by maintaining the stability of mitochondrial dynamics, reducing apoptosis, and protecting neurons in cerebral ischemia (Modi et al., 2020).
However, mitochondrial fission is necessary for maintaining normal cellular function, and it is unrealistic to inhibit mitochondrial fission completely. Some studies have focused on the role of the interaction between Drp1 and its recruitment molecules, such as Mff (Kornfeld et al., 2018) and Fis1 (Qi et al., 2013), in physiological and pathological mitochondrial fission. One study showed that peptide P259-mediated inhibition of the Drp1-Mff interaction could influence normal mitochondrial morphology and basic function under physiological conditions (Kornfeld et al., 2018). The peptide P110 could inhibit the interaction between Drp1 and Fis1, thus reducing excessive mitochondrial fission under pathological conditions without affecting physiological fission (Qi et al., 2013). Moreover, some biomarkers, such as flavin adenine dinucleotide fluorescence (Berndt et al., 2020), can be used as early markers of mitochondrial damage during brain hypoxia, which helps in studying the optimal time to administer drugs targeting mitochondria. There are also some new therapies, such as photobiomodulation therapy, which can inhibit hypoxic-ischemia-induced mitochondrial fragmentation, alleviate mitochondrial dysfunction and oxidative stress, and ultimately reduce neuronal apoptosis in neonatal hypoxic-ischemic encephalopathy (Tucker et al., 2018). Mitochondrial transplantation is also on the rise, and the transfer of exogenous mitochondria through local or systemic intra-arterial injections reduces brain damage, cell death, and motor function in MCAO rats (Huang et al., 2016; Chang et al., 2019).
Energy metabolism after ischemic stroke is closely associated with mitochondrial dynamics. Targeting mitochondrial dynamics-related molecular mechanisms and associated processes, such as calcium overload, ROS, MPTP, apoptosis, and mitophagy, can alleviate brain injury after stroke by improving mitochondrial function and its effect on energy metabolism. This review shows that mitochondrial dynamics play essential roles in pathophysiological changes after stroke. However, the related studies of mitochondrial dynamics mainly focus on cardiac ischemia-reperfusion and neurodegenerative diseases. Mitochondrial dynamics should also be studied intensively in ischemic stroke to understand the specific regulatory mechanisms of mitochondrial dynamics on the clinical manifestations and prognosis of stroke. The clinical application and long-term prognosis of stroke patients are also worthy of further study. In addition, most drug-related effects have only been examined in laboratory studies. There have been few studies on clinical applications. Whether mitochondrial dynamics-targeted drugs could improve a stroke patient’s condition and prognosis in the clinic is a challenge to be explored in subsequent drug studies.
XZ and YH conceived the main outline. XZ wrote the manuscript. LW and HC made the figures. CL, LL, and YO took charge for the manuscript revision in English. YH participated in the correction and finally reviewed of this manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to acknowledge the help of all the staff involved in contributing to the manuscript writing, editing, and literature review.
Abrisch, R. G., Gumbin, S. C., Wisniewski, B. T., Lackner, L. L., and Voeltz, G. K. (2020). Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 219:e201911122. doi: 10.1083/jcb.201911122
Ader, N. R., Hoffmann, P. C., Ganeva, I., Borgeaud, A. C., Wang, C., Youle, R. J., et al. (2019). Molecular and topological reorganizations in mitochondrial architecture interplay during Bax-mediated steps of apoptosis. eLife 8:e40712. doi: 10.7554/eLife.40712
Al-Mufti, F., Amuluru, K., Roth, W., Nuoman, R., El-Ghanem, M., and Meyers, P. M. (2018). Cerebral ischemic reperfusion injury following recanalization of large vessel occlusions. Neurosurgery 82, 781–789. doi: 10.1093/neuros/nyx341
Altschuld, R. A., Hohl, C. M., Castillo, L. C., Garleb, A. A., Starling, R. C., and Brierley, G. P. (1992). Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. 262, H1699–H1704. doi: 10.1152/ajpheart.1992.262.6.H1699
An, H., Ford, A. L., Chen, Y., Zhu, H., Ponisio, R., Kumar, G., et al. (2015). Defining the ischemic penumbra using magnetic resonance oxygen metabolic index. Stroke 46, 982–988. doi: 10.1161/Str.0000000000000063
Andrabi, S. A., Sayeed, I., Siemen, D., Wolf, G., and Horn, T. F. (2004). Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 18, 869–871. doi: 10.1096/fj.03-1031fje
Astrup, J., Siesjo, B. K., and Symon, L. (1981). Thresholds in cerebral ischemia – the ischemic penumbra. Stroke 12, 723–725. doi: 10.1161/01.str.12.6.723
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Barsukova, A., Komarov, A., Hajnoczky, G., Bernardi, P., Bourdette, D., and Forte, M. (2011). Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur. J. Neurosci. 33, 831–842. doi: 10.1111/j.1460-9568.2010.07576.x
Bastian, C., Zaleski, J., Stahon, K., Parr, B., McCray, A., Day, J., et al. (2018). NOS3 inhibition confers post-ischemic protection to young and aging white matter integrity by conserving mitochondrial dynamics and Miro-2 levels. J. Neurosci. 38, 6247–6266. doi: 10.1523/JNEUROSCI.3017-17.2018
Bayir, H., and Kagan, V. E. (2008). Bench-to-bedside review: mitochondrial injury, oxidative stress and apoptosis–there is nothing more practical than a good theory. Crit. Care 12:206. doi: 10.1186/cc6779
Belanger, M., Allaman, I., and Magistretti, P. J. (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. doi: 10.1016/j.cmet.2011.08.016
Bernardi, P. (1999). Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 79, 1127–1155. doi: 10.1152/physrev.1999.79.4.1127
Berndt, N., Kovacs, R., Rosner, J., Wallach, I., Dreier, J. P., and Liotta, A. (2020). Flavin adenine dinucleotide fluorescence as an early marker of mitochondrial impairment during brain hypoxia. Int. J. Mol. Sci. 21:3977. doi: 10.3390/ijms21113977
Besancon, E., Guo, S., Lok, J., Tymianski, M., and Lo, E. H. (2008). Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol. Sci. 29, 268–275. doi: 10.1016/j.tips.2008.02.003
Bolanos, J. P. (2016). Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 139, (Suppl. 2) 115–125. doi: 10.1111/jnc.13486
Bordone, M. P., Salman, M. M., Titus, H. E., Amini, E., Andersen, J. V., Chakraborti, B., et al. (2019). The energetic brain – a review from students to students. J. Neurochem. 151, 139–165. doi: 10.1111/jnc.14829
Bristow, M. S., Simon, J. E., Brown, R. A., Eliasziw, M., Hill, M. D., Coutts, S. B., et al. (2005). MR perfusion and diffusion in acute ischemic stroke: human gray and white matter have different thresholds for infarction. J. Cereb. Blood Flow Metab. 25, 1280–1287. doi: 10.1038/sj.jcbfm.9600135
Bruick, R. K. (2000). Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. U.S.A. 97, 9082–9087. doi: 10.1073/pnas.97.16.9082
Bueler, H. (2010). Mitochondrial dynamics, cell death and the pathogenesis of Parkinson’s disease. Apoptosis 15, 1336–1353. doi: 10.1007/s10495-010-0465-0
Campbell, B. C. V., De Silva, D. A., Macleod, M. R., Coutts, S. B., Schwamm, L. H., Davis, S. M., et al. (2019). Ischaemic stroke. Nat. Rev. Dis. Prim. 5:70. doi: 10.1038/s41572-019-0118-8
Cardoso, A. R., Queliconi, B. B., and Kowaltowski, A. J. (2010). Mitochondrial ion transport pathways: role in metabolic diseases. Biochim. Biophys. Acta 1797, 832–838. doi: 10.1016/j.bbabio.2009.12.017
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 15, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chang, C. Y., Liang, M. Z., and Chen, L. (2019). Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl. Neurodegener. 8:17. doi: 10.1186/s40035-019-0158-8
Chen, Y., Liu, Y., and Dorn, G. W. II (2011). Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331. doi: 10.1161/CIRCRESAHA.111.258723
Cheng, J., Korte, N., Nortley, R., Sethi, H., Tang, Y., and Attwell, D. (2018). Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 136, 507–523. doi: 10.1007/s00401-018-1893-0
Chimeh, U., Zimmerman, M. A., Gilyazova, N., and Li, P. A. (2018). B355252, a novel small molecule, confers neuroprotection against cobalt chloride toxicity in mouse hippocampal cells through altering mitochondrial dynamics and limiting autophagy induction. Int. J. Med. Sci. 15, 1384–1396. doi: 10.7150/ijms.24702
Chiurazzi, M., Di Maro, M., Cozzolino, M., and Colantuoni, A. (2020). Mitochondrial dynamics and microglia as new targets in metabolism regulation. Int. J. Mol. Sci. 21:3450. doi: 10.3390/ijms21103450
Cho, M. H., Kim, D. H., Choi, J. E., Chang, E. J., and Seung, Y. (2012). Increased phosphorylation of dynamin-related protein 1 and mitochondrial fission in okadaic acid-treated neurons. Brain Res. 1454, 100–110. doi: 10.1016/j.brainres.2012.03.010
Cid-Castro, C., Hernandez-Espinosa, D. R., and Moran, J. (2018). ROS as regulators of mitochondrial dynamics in neurons. Cell. Mol. Neurobiol. 38, 995–1007. doi: 10.1007/s10571-018-0584-7
Cohen, M. M., and Tareste, D. (2018). Recent insights into the structure and function of Mitofusins in mitochondrial fusion. F1000Research 7:1983. doi: 10.12688/f1000research.16629.1
Costa, V., Giacomello, M., Hudec, R., Lopreiato, R., Ermak, G., Lim, D., et al. (2010). Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2, 490–503. doi: 10.1002/emmm.201000102
Crack, P. J., and Taylor, J. M. (2005). Reactive oxygen species and the modulation of stroke. Free Radic. Biol. Med. 38, 1433–1444. doi: 10.1016/j.freeradbiomed.2005.01.019
da Rosa, M. S., da Rosa-Junior, N. T., Parmeggiani, B., Glanzel, N. M., de MouraAlvorcem, L., Ribeiro, R. T., et al. (2020). 3-Hydroxy-3-methylglutaric acid impairs redox and energy homeostasis, mitochondrial dynamics, and endoplasmic reticulum-mitochondria crosstalk in rat brain. Neurotox. Res. 37, 314–325. doi: 10.1007/s12640-019-00122-x
Dorn, G. W. II, and Kitsis, R. N. (2015). The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ. Res. 116, 167–182. doi: 10.1161/circresaha.116.303554
Doyle, K. P., Simon, R. P., and Stenzel-Poore, M. P. (2008). Mechanisms of ischemic brain damage. Neuropharmacology 55, 310–318. doi: 10.1016/j.neuropharm.2008.01.005
Droge, W. (2002). Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95. doi: 10.1152/physrev.00018.2001
Ducommun, S., Deak, M., Sumpton, D., Ford, R. J., Galindo, A. N., Kussmann, M., et al. (2015). Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal 27, 978–988. doi: 10.1016/j.cellsig.2015.02.008
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi: 10.1080/01926230701320337
Falkowska, A., Gutowska, I., Goschorska, M., Nowacki, P., Chlubek, D., and Baranowska-Bosiacka, I. (2015). Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int. J. Mol. Sci. 16, 25959–25981. doi: 10.3390/ijms161125939
Frank, S., Gaume, B., Bergmann-Leitner, E. S., Leitner, W. W., Robert, E. G., Catez, F., et al. (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525. doi: 10.1016/s1534-5807(01)00055-7
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Fu, M., St-Pierre, P., Shankar, J., Wang, P. T., Joshi, B., and Nabi, I. R. (2013). Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol. Biol. Cell 24, 1153–1162. doi: 10.1091/mbc.E12-08-0607
GBD 2016 Stroke Collaborators (2019). Global, regional, and national burden of stroke, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 439–458. doi: 10.1016/S1474-4422(19)30034-1
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Gobel, J., Engelhardt, E., Pelzer, P., Sakthivelu, V., Jahn, H. M., Jevtic, M., et al. (2020). Mitochondria-endoplasmic reticulum contacts in reactive astrocytes promote vascular remodeling. Cell Metab. 31, 791–808.e8. doi: 10.1016/j.cmet.2020.03.005
Gomes, L. C., Di Benedetto, G., and Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598. doi: 10.1038/ncb2220
Green, D. R., and Reed, J. C. (1998). Mitochondria and apoptosis. Science 281, 1309–1312. doi: 10.1126/science.281.5381.1309
Grohm, J., Kim, S. W., Mamrak, U., Tobaben, S., Cassidy-Stone, A., Nunnari, J., et al. (2012). Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 19, 1446–1458. doi: 10.1038/cdd.2012.18
Guan, R. Q., Zou, W., Dai, X. H., Yu, X. P., Liu, H., Chen, Q. X., et al. (2018). Mitophagy, a potential therapeutic target for stroke. J. Biomed. Sci. 25:87. doi: 10.1186/s12929-018-0487-4
Guo, Y., Li, D., Zhang, S., Yang, Y., Liu, J. J., Wang, X., et al. (2018). Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 175, 1430–1442.e17. doi: 10.1016/j.cell.2018.09.057
Gustafsson, A. B., and Dorn, G. W. (2019). Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol. Rev. 99, 853–892. doi: 10.1152/physrev.00005.2018
Halestrap, A. P., Woodfield, K. Y., and Connern, C. P. (1997). Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem. 272, 3346–3354. doi: 10.1074/jbc.272.6.3346
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Herrero-Mendez, A., Almeida, A., Fernandez, E., Maestre, C., Moncada, S., and Bolanos, J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747–752. doi: 10.1038/ncb1881
Hofmeijer, J., and van Putten, M. J. (2012). Ischemic cerebral damage: an appraisal of synaptic failure. Stroke 43, 607–615. doi: 10.1161/STROKEAHA.111.632943
Hoppins, S. (2014). The regulation of mitochondrial dynamics. Curr. Opin. Cell Biol. 29, 46–52. doi: 10.1016/j.ceb.2014.03.005
Huang, P. J., Kuo, C. C., Lee, H. C., Shen, C. I., Cheng, F. C., Wu, S. F., et al. (2016). Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transplant. 25, 913–927. doi: 10.3727/096368915X689785
Hurst, S., Hoek, J., and Sheu, S. S. (2017). Mitochondrial Ca(2+) and regulation of the permeability transition pore. J. Bioenerget. Biomembr. 49, 27–47. doi: 10.1007/s10863-016-9672-x
Hwang, J. A., Shin, N., Shin, H. J., Yin, Y., Kwon, H. H., Park, H., et al. (2020). Protective effects of ShcA protein silencing for photothrombotic cerebral infarction. Transl. Stroke Res. doi: 10.1007/s12975-020-00874-1
Iadecola, C. (2017). The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96, 17–42. doi: 10.1016/j.neuron.2017.07.030
Jeong, S. Y., and Seol, D. W. (2008). The role of mitochondria in apoptosis. BMB Rep. 41, 11–22. doi: 10.5483/bmbrep.2008.41.1.011
Jurgensmeier, J. M., Xie, Z., Deveraux, Q., Ellerby, L., Bredesen, D., and Reed, J. C. (1998). Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. U.S.A. 95, 4997–5002. doi: 10.1073/pnas.95.9.4997
Kim, T., Mehta, S. L., Morris-Blanco, K. C., Chokkalla, A. K., Chelluboina, B., Lopez, M., et al. (2018). The microRNA miR-7a-5p ameliorates ischemic brain damage by repressing alpha-synuclein. Sci. Signal. 11:eaat4285. doi: 10.1126/scisignal.aat4285
Kinnally, K. W., Peixoto, P. M., Ryu, S. Y., and Dejean, L. M. (2011). Is mPTP the gatekeeper for necrosis, apoptosis, or both? BBA Mol. Cell Res. 1813, 616–622. doi: 10.1016/j.bbamcr.2010.09.013
Kornfeld, O. S., Qvit, N., Haileselassie, B., Shamloo, M., Bernardi, P., and Mochly-Rosen, D. (2018). Interaction of mitochondrial fission factor with dynamin related protein 1 governs physiological mitochondrial function in vivo. Sci. Rep. 8:14034. doi: 10.1038/s41598-018-32228-1
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi: 10.1126/science.1099793
Kwong, J. Q., Lu, X., Correll, R. N., Schwanekamp, J. A., Vagnozzi, R. J., Sargent, M. A., et al. (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 12, 15–22. doi: 10.1016/j.celrep.2015.06.002
Lampert, M. A., Orogo, A. M., Najor, R. H., Hammerling, B. C., Leon, L. J., Wang, B. J., et al. (2019). BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15, 1182–1198. doi: 10.1080/15548627.2019.1580095
Leigh, R., Knutsson, L., Zhou, J., and van Zijl, P. C. (2018). Imaging the physiological evolution of the ischemic penumbra in acute ischemic stroke. J. Cereb. Blood Flow Metab. 38, 1500–1516. doi: 10.1177/0271678X17700913
Lewis, S. C., Uchiyama, L. F., and Nunnari, J. (2016). ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353:aaf5549. doi: 10.1126/science.aaf5549
Lindsay, M. P., Norrving, B., Sacco, R. L., Brainin, M., Hacke, W., Martins, S., et al. (2019). World stroke organization (WSO): global stroke fact sheet 2019. Int. J. Stroke 14, 806–817. doi: 10.1177/1747493019881353
Lunt, S. Y., and Vander Heiden, M. G. (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464. doi: 10.1146/annurev-cellbio-092910-154237
Magistretti, P. J., and Allaman, I. (2018). Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 19, 235–249. doi: 10.1038/nrn.2018.19
Mandal, A., and Drerup, C. M. (2019). Axonal transport and mitochondrial function in neurons. Front. Cell. Neurosci. 13:373. doi: 10.3389/fncel.2019.00373
Matsumoto, S., Friberg, H., Ferrand-Drake, M., and Wieloch, T. (1999). Blockade of the mitochondrial permeability transition pore diminishes infarct size in the rat after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 19, 736–741. doi: 10.1097/00004647-199907000-00002
Mergenthaler, P., Lindauer, U., Dienel, G. A., and Meisel, A. (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. doi: 10.1016/j.tins.2013.07.001
Modi, J., Menzie-Suderam, J., Xu, H., Trujillo, P., Medley, K., Marshall, M. L., et al. (2020). Mode of action of granulocyte-colony stimulating factor (G-CSF) as a novel therapy for stroke in a mouse model. J. Biomed. Sci. 27:19. doi: 10.1186/s12929-019-0597-7
Montessuit, S., Somasekharan, S. P., Terrones, O., Lucken-Ardjomande, S., Herzig, S., Schwarzenbacher, R., et al. (2010). Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901. doi: 10.1016/j.cell.2010.08.017
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.07.002
Neuspiel, M., Zunino, R., Gangaraju, S., Rippstein, P., and McBride, H. (2005). Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J. Biol. Chem. 280, 25060–25070. doi: 10.1074/jbc.M501599200
Nguyen, D., Alavi, M. V., Kim, K. Y., Kang, T., Scott, R. T., Noh, Y. H., et al. (2011). A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2:e240. doi: 10.1038/cddis.2011.117
Ni, X. X., Nie, J., Xie, Q. Y., Yu, R. H., Su, L., and Liu, Z. F. (2020). Protective effects of hyperbaric oxygen therapy on brain injury by regulating the phosphorylation of Drp1 through ROS/PKC pathway in heatstroke rats. Cell. Mol. Neurobiol. 40, 1253–1269. doi: 10.1007/s10571-020-00811-8
Norbury, C. J., and Hickson, I. D. (2001). Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 41, 367–401. doi: 10.1146/annurev.pharmtox.41.1.367
Nortley, R., and Attwell, D. (2017). Control of brain energy supply by astrocytes. Curr. Opin. Neurobiol. 47, 80–85. doi: 10.1016/j.conb.2017.09.012
Nunnari, J. (2007). The machines that divide and fuse mitochondria. FASEB J. 21:A96. doi: 10.1146/annurev.biochem.76.071905.090048
O’Donnell, J. C., Jackson, J. G., and Robinson, M. B. (2016). Transient oxygen/glucose deprivation causes a delayed loss of mitochondria and increases spontaneous calcium signaling in astrocytic processes. J. Neurosci. 36, 7109–7127. doi: 10.1523/Jneurosci.4518-15.2016
Ong, S. B., Subrayan, S., Lim, S. Y., Yellon, D. M., Davidson, S. M., and Hausenloy, D. J. (2010). Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121, 2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610
Pagliuso, A., Cossart, P., and Stavru, F. (2018). The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 75, 355–374. doi: 10.1007/s00018-017-2603-0
Papanicolaou, K. N., Khairallah, R. J., Ngoh, G. A., Chikando, A., Luptak, I., O’Shea, K. M., et al. (2011). Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 31, 1309–1328. doi: 10.1128/MCB.00911-10
Park, J., Choi, H., Min, J. S., Park, S. J., Kim, J. H., Park, H. J., et al. (2013). Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 127, 221–232. doi: 10.1111/jnc.12361
Pivovarova, N. B., Nguyen, H. V., Winters, C. A., Brantner, C. A., Smith, C. L., and Andrews, S. B. (2004). Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J. Neurosci. 24, 5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004
Powers, W. J., Rabinstein, A. A., Ackerson, T., Adeoye, O. M., Bambakidis, N. C., Becker, K., et al. (2018). 2018 Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American heart association/American stroke association. Stroke 49, e46–e110. doi: 10.1161/STR.0000000000000158
Prabhakaran, S., Ruff, I., and Bernstein, R. A. (2015). Acute stroke intervention: a systematic review. JAMA 313, 1451–1462. doi: 10.1001/jama.2015.3058
Pushie, M. J., Crawford, A. M., Sylvain, N. J., Hou, H., Hackett, M. J., George, G. N., et al. (2018). Revealing the penumbra through imaging elemental markers of cellular metabolism in an ischemic stroke model. ACS Chem. Neurosci. 9, 886–893. doi: 10.1021/acschemneuro.7b00382
Qi, X., Qvit, N., Su, Y. C., and Mochly-Rosen, D. (2013). A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 126, 789–802. doi: 10.1242/jcs.114439
Qin, Y., Li, A., Liu, B., Jiang, W., Gao, M., Tian, X., et al. (2020). Mitochondrial fusion mediated by fusion promotion and fission inhibition directs adult mouse heart function toward a different direction. FASEB J. 34, 663–675. doi: 10.1096/fj.201901671R
Quintana, D. D., Garcia, J. A., Sarkar, S. N., Jun, S. J., Engler-Chiurazzi, E. B., Russell, A. E., et al. (2019). Hypoxia-reoxygenation of primary astrocytes results in a redistribution of mitochondrial size and mitophagy. Mitochondrion 47, 244–255. doi: 10.1016/j.mito.2018.12.004
Radak, D., Katsiki, N., Resanovic, I., Jovanovic, A., Sudar-Milovanovic, E., Zafirovic, S., et al. (2017). Apoptosis and acute brain ischemia in ischemic stroke. Curr. Vasc. Pharmacol. 15, 115–122. doi: 10.2174/1570161115666161104095522
Rao, G., Murphy, B., Dey, A., Dwivedi, S. K. D., Zhang, Y., Roy, R. V., et al. (2020). Cystathionine beta synthase regulates mitochondrial dynamics and function in endothelial cells. FASEB J. 34, 9372–9392. doi: 10.1096/fj.202000173R
Rappold, P. M., Cui, M., Grima, J. C., Fan, R. Z., de Mesy-Bentley, K. L., Chen, L., et al. (2014). Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 5:5244. doi: 10.1038/ncomms6244
Rigoulet, M., Bouchez, C. L., Paumard, P., Ransac, S., Cuvellier, S., Duvezin-Caubet, S., et al. (2020). Cell energy metabolism: an update. Biochim. Biophys. Acta Bioenerget. 1861:148276. doi: 10.1016/j.bbabio.2020.148276
Rinholm, J. E., Vervaeke, K., Tadross, M. R., Tkachuk, A. N., Kopek, B. G., Brown, T. A., et al. (2016). Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia 64, 810–825. doi: 10.1002/glia.22965
Rodger, C. E., McWilliams, T. G., and Ganley, I. G. (2018). Mammalian mitophagy – from in vitro molecules to in vivo models. FEBS J. 285, 1185–1202. doi: 10.1111/febs.14336
Roy, M., Reddy, P. H., Iijima, M., and Sesaki, H. (2015). Mitochondrial division and fusion in metabolism. Curr. Opin. Cell Biol. 33, 111–118. doi: 10.1016/j.ceb.2015.02.001
Ruiz, A., Quintela-López, T., Sánchez-Gómez, M. V., Gaminde-Blasco, A., Alberdi, E., and Matute, C. (2020). Mitochondrial division inhibitor 1 disrupts oligodendrocyte Ca(2+)homeostasis and mitochondrial function. Glia 68, 1743–1756. doi: 10.1002/glia.23802
Sarrafzadeh, A. S., Nagel, A., Czabanka, M., Denecke, T., Vajkoczy, P., and Plotkin, M. (2010). Imaging of hypoxic-ischemic penumbra with F-18-fluoromisonidazole PET/CT and measurement of related cerebral metabolism in aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 30, 36–45. doi: 10.1038/jcbfm.2009.199
Scheffler, I. E. (2001). Mitochondria make a come back. Adv. Drug Deliv. Rev. 49, 3–26. doi: 10.1016/S0169-409x(01)00123-5
Sekerdag, E., Solaroglu, I., and Gursoy-Ozdemir, Y. (2018). Cell death mechanisms in stroke and novel molecular and cellular treatment options. Curr. Neuropharmacol. 16, 1396–1415. doi: 10.2174/1570159X16666180302115544
Sharp, W. W., Fang, Y. H., Han, M., Zhang, H. J., Hong, Z., Banathy, A., et al. (2014). Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 28, 316–326. doi: 10.1096/fj.12-226225
Sheng, Z. H. (2014). Mitochondrial trafficking and anchoring in neurons: new insight and implications. J. Cell Biol. 204, 1087–1098. doi: 10.1083/jcb.201312123
Sokoloff, L. (1981). Localization of functional activity in the central nervous system by measurement of glucose utilization with radioactive deoxyglucose. J. Cereb. Blood Flow Metab. 1, 7–36. doi: 10.1038/jcbfm.1981.4
Sokoloff, L., Reivich, M., Kennedy, C., Des Rosiers, M. H., Patlak, C. S., Pettigrew, K. D., et al. (1977). The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 28, 897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x
Song, T., Liu, J., Tao, X., and Deng, J. G. (2014). Protection effect of atorvastatin in cerebral ischemia-reperfusion injury rats by blocking the mitochondrial permeability transition pore. Genet. Mol. Res. 13, 10632–10642. doi: 10.4238/2014.December.18.5
Song, Z., Chen, H., Fiket, M., Alexander, C., and Chan, D. C. (2007). OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 178, 749–755. doi: 10.1083/jcb.200704110
Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H., and Harris, A. L. (2001). HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673.
Sprenger, H. G., and Langer, T. (2019). The good and the bad of mitochondrial breakups. Trends Cell Biol. 29, 888–900. doi: 10.1016/j.tcb.2019.08.003
Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., et al. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446. doi: 10.1038/17135
Tilokani, L., Nagashima, S., Paupe, V., and Prudent, J. (2018). Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 62, 341–360. doi: 10.1042/EBC20170104
Toyama, E. Q., Herzig, S., Courchet, J., Lewis, T. L., Loson, O. C., Hellberg, K., et al. (2016). AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. doi: 10.1126/science.aab4138
Tucker, L. D., Lu, Y., Dong, Y., Yang, L., Li, Y., Zhao, N., et al. (2018). Photobiomodulation therapy attenuates hypoxic-ischemic injury in a neonatal rat model. J. Mol. Neurosci. 65, 514–526. doi: 10.1007/s12031-018-1121-3
Um, J. H., and Yun, J. (2017). Emerging role of mitophagy in human diseases and physiology. BMB Rep. 50, 299–307. doi: 10.5483/BMBRep.2017.50.6.056
Varanita, T., Soriano, M. E., Romanello, V., Zaglia, T., Quintana-Cabrera, R., Semenzato, M., et al. (2015). The Opa1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 21, 834–844. doi: 10.1016/j.cmet.2015.05.007
Wai, T., and Langer, T. (2016). Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 27, 105–117. doi: 10.1016/j.tem.2015.12.001
Wang, J., Zhu, P., Li, R., Ren, J., and Zhou, H. (2020). Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 30:101415. doi: 10.1016/j.redox.2019.101415
Wang, W. Z., Zhang, F., Li, L., Tang, F. Q., Siedlak, S. L., Fujioka, H., et al. (2015). MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J. Biol. Chem. 290, 168–182. doi: 10.1074/jbc.M114.617167
Wang, Y., Liu, G., Hong, D. D., Chen, F. H., Ji, X. M., and Cao, G. D. (2016). White matter injury in ischemic stroke. Prog. Neurobiol. 141, 45–60. doi: 10.1016/j.pneurobio.2016.04.00
Wang, Y., Zhou, L., Guo, J., Wang, Y., Yang, Y., Peng, Q., et al. (2020). Secular trends of stroke incidence and mortality in China, 1990 to 2016: the global burden of disease study 2016. J. Stroke Cerebrovasc. Dis. 29:104959. doi: 10.1016/j.jstrokecerebrovasdis.2020.104959
Wang, Z. G., Jiang, H., Chen, S., Du, F. H., and Wang, X. D. (2012). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243. doi: 10.1016/j.cell.2011.11.030
Wei, N., Pu, Y., Yang, Z., Pan, Y., and Liu, L. (2019). Therapeutic effects of melatonin on cerebral ischemia reperfusion injury: role of Yap-OPA1 signaling pathway and mitochondrial fusion. Biomed. Pharmacother. 110, 203–212. doi: 10.1016/j.biopha.2018.11.060
Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884. doi: 10.1038/nrm3013
White, K. E., Davies, V. J., Hogan, V. E., Piechota, M. J., Nichols, P. P., Turnbull, D. M., et al. (2009). OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Invest. Ophth. Vis. Sci. 50, 2567–2571. doi: 10.1167/iovs.08-2913
Williams, J. A., and Ding, W. X. (2018). Mechanisms, pathophysiological roles and methods for analyzing mitophagy – recent insights. Biol. Chem. 399, 147–178. doi: 10.1515/hsz-2017-0228
Wu, B., Luo, H., Zhou, X., Cheng, C. Y., Lin, L., Liu, B. L., et al. (2017). Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: therapeutical effects of kaempferol. BBA Mol. Basis Dis. 1863, 2307–2318. doi: 10.1016/j.bbadis.2017.06.011
Wu, Q., Gao, C., Wang, H., Zhang, X., Li, Q., Gu, Z., et al. (2018). Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int. J. Biochem. Cell Biol. 94, 44–55. doi: 10.1016/j.biocel.2017.11.007
Xie, L. L., Shi, F., Tan, Z., Li, Y., Bode, A. M., and Cao, Y. (2018). Mitochondrial network structure homeostasis and cell death. Cancer Sci. 109, 3686–3694. doi: 10.1111/cas.13830
Yaghi, S., Willey, J. Z., Cucchiara, B., Goldstein, J. N., Gonzales, N. R., Khatri, P., et al. (2017). Treatment and outcome of hemorrhagic transformation after intravenous alteplase in acute ischemic stroke: a scientific statement for healthcare professionals from the American heart association/American stroke association. Stroke 48, e343–e361. doi: 10.1161/STR.0000000000000152
Yoo, S. M., and Jung, Y. K. (2018). A molecular approach to mitophagy and mitochondrial dynamics. Mol. Cells 41, 18–26. doi: 10.14348/molcells.2018.2277
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Yu, B., Ma, J., Li, J., Wang, D. Z., Wang, Z. G., and Wang, S. S. (2020). Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat. Commun. 11:2549. doi: 10.1038/s41467-020-16312-7
Zenonos, G., and Kim, J. E. (2010). 18F-fluoromisonidazole PET/CT and related gauges of cerebral metabolism identify salvageable tissue in ischemic penumbra following aneurysmal subarachnoid hemorrhage. Neurosurgery 66, N13–N15. doi: 10.1227/01.neu.0000367839.75141.e6
Zhang, C., Wang, J., Zhu, J., Chen, Y., and Han, X. (2020). Microcystin-leucine-arginine induced neurotoxicity by initiating mitochondrial fission in hippocampal neurons. Sci. Total Environ. 703:134702. doi: 10.1016/j.scitotenv.2019.134702
Zhang, H. S., Liu, C. D., Zheng, M. C., Zhao, H. T., and Liu, X. J. (2020). Propofol alleviates hypoxic neuronal injury by inhibiting high levels of mitochondrial fusion and fission. Eur. Rev. Med. Pharmacol. Sci. 24, 9650–9657. doi: 10.26355/eurrev_202009_23054
Zhang, K., Li, H., and Song, Z. (2014). Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 15, 576–585. doi: 10.1002/embr.201338240
Zhang, L., He, Z., Zhang, Q., Wu, Y., Yang, X., Niu, W., et al. (2014). Exercise pretreatment promotes mitochondrial dynamic protein OPA1 expression after cerebral ischemia in rats. Int. J. Mol. Sci. 15, 4453–4463. doi: 10.3390/ijms15034453
Zhang, Y. K., Sun, R. J., Li, X. L., and Fang, W. H. (2020). Porcine circovirus 2 induction of ROS is responsible for mitophagy in PK-15 cells via activation of Drp1 phosphorylation. Viruses Basel 12:289. doi: 10.3390/v12030289
Zhang, Z., and Yu, J. (2018). Nurr1 exacerbates cerebral ischemia-reperfusion injury via modulating YAP-INF2-mitochondrial fission pathways. Int. J. Biochem. Cell Biol. 104, 149–160. doi: 10.1016/j.biocel.2018.09.014
Zhao, Q., Wang, S., Li, Y., Wang, P., Li, S., Guo, Y., et al. (2013). The role of the mitochondrial calcium uniporter in cerebral ischemia/reperfusion injury in rats involves regulation of mitochondrial energy metabolism. Mol. Med. Rep. 7, 1073–1080. doi: 10.3892/mmr.2013.1321
Zhao, Y. X., Cui, M., Chen, S. F., Dong, Q., and Liu, X. Y. (2014). Amelioration of ischemic mitochondrial injury and Bax-dependent outer membrane permeabilization by Mdivi-1. CNS Neurosci. Ther. 20, 528–538. doi: 10.1111/cns.12266
Zhou, K., Chen, J., Wu, J., Wu, Q., Jia, C., Xu, Y. X. Z., et al. (2019a). Atractylenolide III ameliorates cerebral ischemic injury and neuroinflammation associated with inhibiting JAK2/STAT3/Drp1-dependent mitochondrial fission in microglia. Phytomedicine 59:152922. doi: 10.1016/j.phymed.2019.152922
Zhou, K., Wu, J. Y., Chen, J., Zhou, Y., Chen, X. L., Wu, Q. Y., et al. (2019b). Schaftoside ameliorates oxygen glucose deprivation-induced inflammation associated with the TLR4/Myd88/Drp1-related mitochondrial fission in BV2 microglia cells. J. Pharmacol. Sci. 139, 15–22. doi: 10.1016/j.jphs.2018.10.012
Zhou, L., Zhang, Q., Zhang, P., Sun, L., Peng, C., Yuan, Z., et al. (2017). c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. 8:e3117. doi: 10.1038/cddis.2017.524
Keywords: energy metabolism, ischemic stroke, molecular mechanisms, mitochondrial dynamics, therapeutic target
Citation: Zhou X, Chen H, Wang L, Lenahan C, Lian L, Ou Y and He Y (2021) Mitochondrial Dynamics: A Potential Therapeutic Target for Ischemic Stroke. Front. Aging Neurosci. 13:721428. doi: 10.3389/fnagi.2021.721428
Received: 07 June 2021; Accepted: 16 August 2021;
Published: 07 September 2021.
Edited by:
Lin Wang, Zhejiang University, ChinaCopyright © 2021 Zhou, Chen, Wang, Lenahan, Lian, Ou and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue He, ZHJoeXdpbmRAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.