 Thomas A. Bayer
Thomas A. Bayer- Division of Molecular Psychiatry, Department of Psychiatry and Psychotherapy, University Medical Center Göttingen (UMG), Georg-August-University, Göttingen, Germany
The discussion of whether amyloid plaque Aβ is a valid drug target to fight Alzheimer’s disease (AD) has been a matter of scientific dispute for decades. This question can only be settled by successful clinical trials and the approval of disease-modifying drugs. However, many clinical trials with antibodies against different regions of the amyloid Aβ peptide have been discontinued, as they did not meet the clinical endpoints required. Recently, passive immunization of AD patients with Donanemab, an antibody directed against the N-terminus of pyroglutamate Aβ, showed beneficial effects in a phase II trial, supporting the concept that N-truncated Aβ is a relevant target for AD therapy. There is long-standing evidence that N-truncated Aβ variants are the main variants found in amyloid plaques besides full-length Aβ1–42, t, therefore their role in triggering AD pathology and as targets for drug development are of interest. While the contribution of pyroglutamate Aβ3–42 to AD pathology has been well studied in the past, the potential role of Aβ4–42 has been largely neglected. The present review will therefore focus on Aβ4–42 as a possible drug target based on human and mouse pathology, in vitro and in vivo toxicity, and anti-Aβ4-X therapeutic effects in preclinical models.
Introduction
Even though the field of Alzheimer’s disease (AD) research has rapidly developed over the last decade, there is still a lack of disease-modifying therapies. Passive immunization with Donanemab a pyroglutamate Aβ (AβpE3) specific antibody showed disease-modification on cognition and for the ability to perform the activities of daily living (Mintun et al., 2021). No biomarkers are yet available based on N-truncated Aβ although autoantibodies were identified in plasma (Marcello et al., 2011). Trieb et al. (1996) investigated whether amyloid-β peptides may be relevant targets for the immune system using peripheral blood lymphocytes from healthy blood donors and patients with AD. While healthy donors elicited normal proliferative responsiveness after stimulation, a significant reduction was observed using lymphocytes from AD patients. Meanwhile, lower levels of naturally occurring anti-Aβ auto-antibodies have also been reported in the CSF (Du et al., 2001) and sera (Weksler et al., 2002) of AD patients, and elevated serum levels were also reported (Nath et al., 2003). It is also of note that in plasma of patients with mild cognitive impairment (MCI) and AD reduced pools of autoantibodies of the IgM class directed against pyroglutamate Aβ3-X (AβpE3-X) have been reported (Marcello et al., 2009). In MCI patients, the level of the autoantibodies correlated with cognitive performance as evaluated by mini-mental state examination. N- and C-terminally truncated Aβ variants, their potential function and toxicity as well as their potential as drug targets were discussed recently (Bayer and Wirths, 2014; Dunys et al., 2018; Wirths and Zampar, 2019). The current mini-review, discusses the potential role of N-truncated Aβ in AD, focussing on N-truncated Aβ starting with position four Aβ4–42.
Discovery of N-Truncated Aβ4–42 and Prevalence in The Human Brain
As revealed by a study by Portelius et al. (2010), the relative prevalence of full-length and N-truncated Aβ is of significant interest within the Alzheimer field. The authors used Aβ antibodies binding to Aβ4–9 and Aβ8–22 for immunoprecipitation. This was followed by mass spectrometry for identification of all Aβ variants in post-mortem tissue from patients with sporadic AD, familial AD with mutations in the presenilin-1 (PS-1; PSEN-1), or amyloid precursor protein (APP) genes. The authors demonstrated that the dominating Aβ isoforms are Aβ1–42, AβpE3–42, Aβ4–42, and Aβ1–40. The most prevalent variants in the hippocampus and the cortex were Aβ1–42 and Aβ4–42. The importance of Aβ4–42 did not receive appropriate attention in the past, although N-truncated Aβ4-X has been discovered with the first sequencing endeavors of Aβ peptides isolated from plaque cores. This surprising finding puzzled Masters et al. (1985) as the most abundant variant of Aβ in the formic acid soluble fraction of plaque cores and subsequent peptide sequencing started with phenylalanine at position four (Aβ4-X) and not with the full-length Aβ1-X they had hoped.
Glenner and Wong (Glenner and Wong, 1984) published ground-breaking work showing the full-length sequence of Ab peptides derived from the vasculature of AD patients. A number of reports have discussed the dominant presence of N-truncated Aβ variants within amyloid plaques in AD and Down syndrome patients (Harigaya et al., 2000; Tekirian, 2001; Miravalle et al., 2005; Piccini et al., 2005; Jawhar et al., 2011; Bayer and Wirths, 2014), while others suggest that full-length Aβ1–42 is pathologically relevant (Haass et al., 1992; Näslund et al., 1994; Selkoe, 2001; Walsh et al., 2002). Haass et al. (1993) discovered that Aβ is produced as a normal physiological process in APP transfected cell lines and cultured cells. The cells were analyzed by epitope mapping and radiosequencing of secreting Aβ variants. They observed mainly full-length Aβ1–42 (aspartate-1), but also two other Aβ peptides starting with the amino acid phenylalanine at position four and glutamate at position 11.
Although different methodologies for extracting and solubilizing aggregated Aβ have the potential for over- or under-estimating the relative prevalence of the various Aβ pools within amyloid plaques, there is general agreement that the C-terminus mostly ends with AβX-42 (alanine-42) and less abundantly with AβX-40 (arginin-40). For example, Ancolio et al. (1999) have demonstrated a large elevation of N-truncated Aβx–42 in familial AD, postulating that all Aβx–42 variants are the main factors driving AD pathology.
The idea that N-truncated Aβ may represent a potential drug target to fight AD has been largely neglected but was brought into attention recently (Jawhar et al., 2011; Bayer and Wirths, 2014; Cabrera et al., 2018). After this original discovery (Masters et al., 1985), other research groups have verified the presence of Aβ4–42 by other methodologies. Miller et al. (1993) employed matrix-assisted, laser-desorption-time-of-flight mass spectrometry of Aβ peptides isolated from plaque cores or the cerebrovasculature obtained from patients with AD. The authors demonstrated that the C-terminus of Aβ peptides within plaques ended with Aβ42, whereas cerebrovascular Aβ ended at Aβ40. They also proved that N-truncated Aβ4-X represents a dominant fraction within plaque cores. Lewis et al. (2006) employed surface-enhanced laser desorption/ionization mass spectrometry for identifying the composition of Aβ peptides in AD and vascular dementia patients. In the brain of AD patients, Aβ started mostly with Phe-4, but other N-termini starting with Asp-1, Ala-2, pE-3, and Arg-5 were also detected in extractions from plaque cores. Using specific Aβ antibodies, Lemere et al. (1996) showed that patients with Down syndrome harbor Aβ starting at Asp-1 or pE-3 in a subset of plaques. Zampar et al. (2020) used capillary isoelectric focusing for showing Aβ antibody specificity of the N-terminus. Furthermore, the authors used immunohistochemistry with the verified N-terminal specific Aβ antibodies to stain post-mortem brain tissue from sporadic AD patients. They concluded that the staining signal for Aβ1-X was much weaker in plaques as compared to cerebrovascular amyloid. In contrast, the signal for Aβ4-X was much more evident in amyloid plaques.
Sergeant et al. (2003) verified that N-truncated Aβ represented the majority of all variants in both AD and pre-symptomatic AD with a substantial amount being Aβ4–42. The authors explored brain specimen from non-demented individuals with low amyloid load and tangle formation by Western blotting and mass spectrometry of the formic acid soluble fraction of amyloid plaques. Rosen et al. (2016) compared the composition of amyloid peptides isolated from AD neocortex and aged squirrel monkeys using immunochemical staining with an Aβ4-X specific antibody and by mass spectrometry. The authors confirmed the high prevalence of N-truncated Aβ peptides including Aβ4–42 in the AD brain, while the prevalence in the non-human primate brain was low.
Although it has been shown that Aβ4-X is generated and secreted in vitro, it was a matter of concern whether N-truncation and post-translational modifications of Aβ represent a post-mortem artifact due to long-term storage or tissue handling. Such an assumption can now be neglected with the therapeutic effect of an AβpE3-specific antibody in patients with AD after passive immunization (Mintun et al., 2021). Wildburger et al. (2017) employed high-resolution mass spectrometry to explore the question of whether N-truncation and other post-translational modifications of Aβ are found in AD brains due to post-mortem artifacts. The authors studied different Aβ pools depending on their solubility and concluded that the N-truncated variants did not correlate with post-mortem interval.
Aβ4-X Can Be Generated by Enzymatic Cleavage
The generation of N-terminal Aβ4-X by known enzymatic cleavages has been reviewed before in detail (Bayer and Wirths, 2014). It can be generated by a two-step process starting with β-site APP cleaving enzyme 1 (BACE-1) cutting APP between Met at postion-1 and Asp at position +1 liberating the N-terminus of full-length Aβ1-X (Vassar et al., 1999). ADAMTS4 (a disintegrin and metalloproteinase with thrombospondin motifs 4) and neprilysin (NEP) further cuts between Glu at position +3 and Phe at position +4 liberating the N-terminus of Aβ4-X (Bayer and Wirths, 2014; Walter et al., 2019; Figure 1A). NEP cleaves Aβ at multiple sites thereby detoxifying amyloid-β peptides (Bayer and Wirths, 2014). Walter et al. (2019) identified a recognition site for the secreted form of metalloprotease ADAMTS4 within the full-length Aβ sequence. The induction of ADAMTS4 expression in cell culture led to increased secretion of Aβ4–40 the levels of Aβ1-x were not altered. Furthermore, the authors identified adult oligodendrocytes as the only source of ADAM4TS triggered Aβ4-X generation in the murine brain. The main function of NEP on Aβ is degradation and catabolism of the peptide (Iwata et al., 2001; Leissring et al., 2003). The loss of NEP activity leads to enhanced levels of brain and plasma levels of full-length Aβ, the elevated half-life of soluble Aβ, and increased amyloid plaque pathology in the J9 mouse model of AD (Farris et al., 2007). Hama et al. (2004) showed that different intracellular compartments are involved in the degradation of amyloid peptides by NEP. Thus NEP is mainly responsible for the intra- and extracellular neuronal clearance of Aβ peptides and more importantly also at the presynaptic site (Iwata et al., 2004). Elevated NEP impaired hippocampal synaptic plasticity and cognitive function in the APP23 mouse model for AD (Huang et al., 2006). Besides general clearance of full-length Aβ peptides, NEP is also involved in the generation of N-truncated Aβ peptides Aβ4-X (Bayer and Wirths, 2014), and further clearing of Aβ4–42 in vivo and in vitro (Hornung et al., 2019). Aβ4–9, a main degradation product of NEP, is a major Cu2+ binding and has been suggested as a possible Cu2+ carrier in the brain (Bossak-Ahmad et al., 2019) and NEP modulation (Mital et al., 2018). Modulating Cu metabolism is discussed as a relevant therapeutic target (Lei et al., 2020). In APP23 mice, Cu supplementation lowered amyloid plaque load and stabilized Cu-dependent superoxide dismutase-1 activity (Bayer et al., 2003). In mild to moderate AD patients cognitive decline correlated with low plasma concentrations of Cu (Pajonk et al., 2005). However, treatment for 12 months with supplemental Cu had no effect on cognition in patients with mild AD in a phase 2 clinical trial (Kessler et al., 2008). Alternatively, N-truncated Aβ4-X may be generated directly by cutting APP between Glu at position 3 and Phe at position 4 by unknown enzymatic activity. Of note, N-truncated Aβ4-X is secreted together with Aβ1–42 in APP overexpressing cells in vitro, which may indicate unknown enzymatic activity in neurons (Haass et al., 1993). For example, N-truncated Aβ5-X is mainly produced from the caspase-cleaved form of APP and not from full-length Aβ (Murayama et al., 2007). Another potential alternative may be the generation the Aβ4-x peptide, a sequential cleavage of full-length Aβ by aminopeptidase A, meprin-β or dipeptidyl-peptidases (Sevalle et al., 2009; Antonyan et al., 2018; Schlenzig et al., 2018; Valverde et al., 2021). At least theoretically, Aβ4-x could be derived from Aβ2-x or Aβ3-x peptide (Figure 1B).
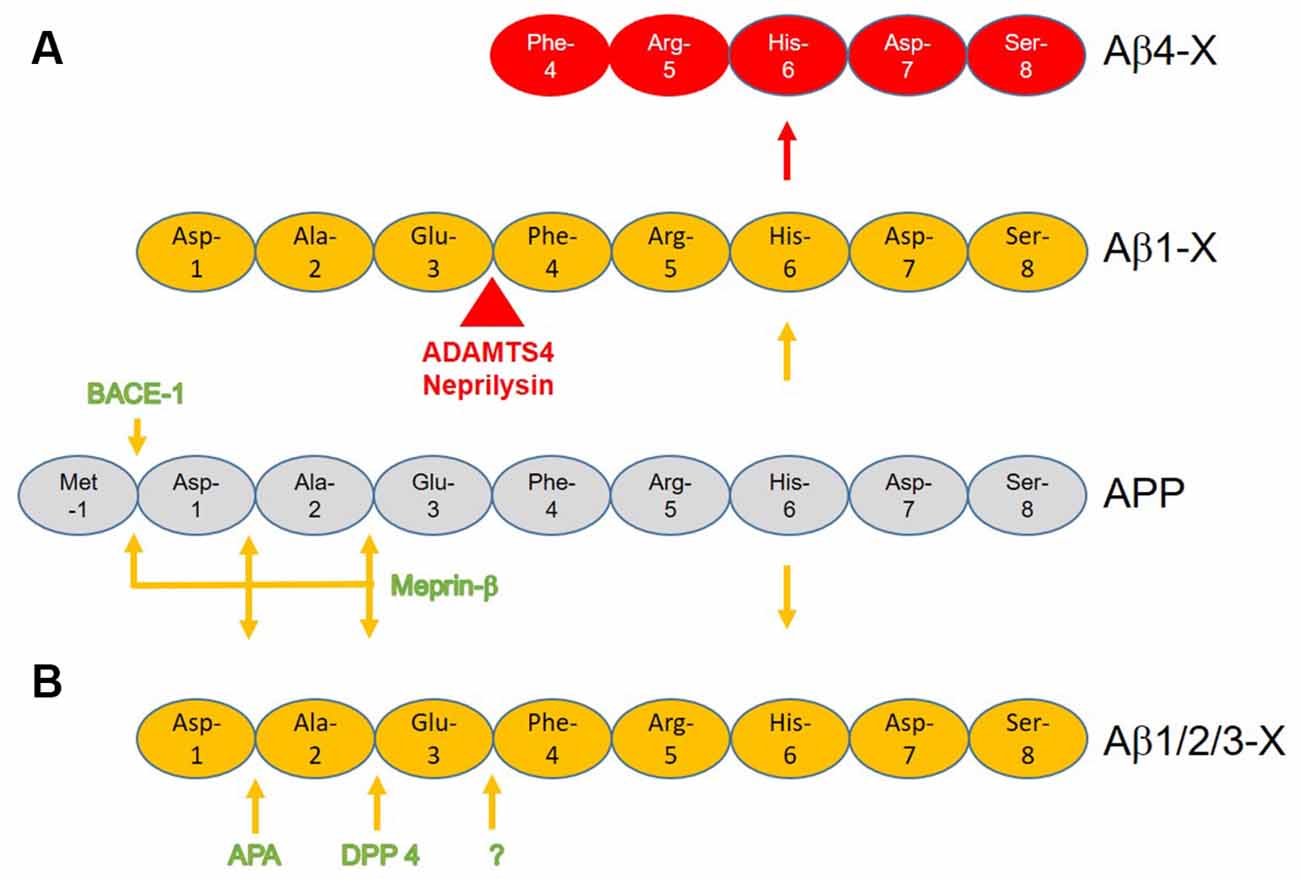
Figure 1. Schematic presentation of the potential generation of N-terminal Aβ4-X with known enzymatic cleavages of the amyloid precursor protein (APP) and Aβ1-X. (A) β-site APP cleaving enzyme 1 (BACE-1) or meprin-β cuts APP between amino acid position -1 and +1 liberating the N-terminus of full-length Aβ1-X. While ADAMTS4 further cuts between position +3 and +4 liberating Aβ4–40/42, the normal function of neprilysin is to detoxify Aβ as it has several N-terminal activities within Aβ with N-truncation of Aβ4-X being only one alternative. (B) Hypothetical pathway(s) by which N-truncated Aβ4-x is generated by sequential cleavage by meprin-β, aminopeptidase A (APA), and/or dipeptidyl peptidase 4 (DPP 4).
Acute Effect of N-Truncated Aβ4–42
Exposure of soluble oligomers of Aβ4–42 preparations induced neuron degeneration after 7 days of cultured rat primary cortical neurons (Antonios et al., 2013, 2015). Injecting Aβ4–42 into the lateral ventricles of wildtype mice induced working memory deficits after 4 days (Antonios et al., 2013, 2015). In both assays, the neurodegenerative effect of Aβ4–42 was similar to the exposure of Aβ1–42 and AβpE3–42. In 1995, Pike et al. (1995) claimed that Aβ peptides with N-terminal truncations including Aβ4-X exhibited enhanced peptide aggregation relative to full-length Aβ species. Furthermore, they concluded from obtained CD (circular dichroism) spectra that β-sheets were the predominantly formed conformations by all Aβ variants. They exhibited fibrillary morphology viewed by transmission electron microscopy, and induced degeneration of cultured rat hippocampus neurons. Bouter et al. (2013) demonstrated that soluble oligomeric aggregates derived from Aβ4–42 and AβpE3–42 have specific structural features distinct from full-length Aβ1–42, although fibril formation as reported previously (Pike et al., 1995) was comparable. Spectral alterations using ultraviolet circular dichroism spectroscopy showed that Aβ4–42 forms a folded conformation upon heating (Bouter et al., 2013). Using the liquid state nuclear magnetic resonance technique Aβ4–42 and AβpE3–42 exhibited soluble and stable aggregates, which was less pronounced for Aβ1–42. However, the size of Aβ4–42 and AβpE3–42 aggregates were different from those formed by Aβ1–42 (Bouter et al., 2013). Even though Aβ1–42, AβpE3–42 and Aβ4–42 are unstructured in the monomeric state. After heating all Aβ peptides formed of folded structures. Interestingly, monomeric Aβ4–42 and AβpE3–42 rapidly converted into soluble oligomeric forms in contrast to full-length Aβ1–42, which stay in equilibrium for a longer time between monomers and oligomers (Bouter et al., 2013).
Parodi-Rullan et al. (2020) studied the effect of full-length and different N-terminal truncated Aβ variants on blood-brain barrier permeability, cerebral microvascular endothelial cell viability, and angiogenesis. The authors demonstrated that Aβ4–42 followed by Aβ4–40 was the most potent inhibitor of angiogenesis, and they were also much stronger than Aβ1–42 and Aβ1–40.
Chronic Effect of N-Truncated Aβ4–42
The development of the APP/PSEN-1 double-transgenic mouse model, APP/PS1KI, for AD revealed, besides massive neuron loss in the hippocampus, many N-truncated AβX-42 variants including Aβ4–42 elucidated by two-dimensional Western blotting, which were subsequently verified by mass spectrometry (Casas et al., 2004). The 5XFAD mouse model is more widely used in the scientific community expressing mutant APP and PSEN-1 transgenes (Oakley et al., 2006). Mass spectrometric analysis with N-terminal specific Aβ antibodies of 5XFAD mouse brain elucidated that the vast majority of Aβ peptides were full-length Aβ1–42 (Wittnam et al., 2012).
Using a highly specific antiserum against the N-terminus of Aβ4-X abundant plaque staining was observed in APP/PS1KI and 5XFAD transgenic mouse brain (Wirths et al., 2017). In the human AD brain, this antiserum demonstrated staining of plaque cores of senile plaques, but none in diffuse amyloid deposits. The peptide content of plaques from AD brain and an AD mouse model (presenilin-2/APP transgenic mice, PS2APP) was analyzed using laser dissection microscopy combined with mass spectrometry (Rufenacht et al., 2005). The authors described various N-terminal truncated Aβ peptides in PS2APP and AD amyloid plaques however with significantly elevated levels in AD brain.
Kawarabayashi et al. (2001) studied another AD mouse model with mutant APP (Tg2576) and elucidated that only a minor fraction of Aβ was N-terminally truncated in contrast to AD brain (Kawarabayashi et al., 2001). This was verified, by another study by Kalback et al. (2002). The authors employed size-exclusion and reverse-phase chromatography, amino acid sequencing, and mass spectrometry of amyloid plaques of Tg2576 mice. The authors found that the amyloid plaques differed in their physical and chemical properties from those isolated from the AD brain. In Tg2576 mice, most peptides were full-length Aβ1–42, whereas, in AD brains, most peptides were N-truncated. The brain tissue of the transgenic mouse models mentioned above were freshly prepared without any post-mortem delay, therefore changes in pH, long-storage artifacts, state of agony, and medication, etc. can be ruled out as an explanation of the appearance of N-truncated Aβ variants in transgenic mouse brain.
The Tg4–42 mouse model for AD expresses only Aβ4–42, which represents a unique model system for studying the effect of chronic exposure of Aβ4–42 in the mouse brain (Bouter et al., 2013; Figure 2). The long-lasting exposure of Aβ4–42 induced an age-dependent neuron loss in the hippocampus, which correlated with hippocampus-dependent spatial reference memory deficits. Tg4–42 mice demonstrated synaptic hyper-excitability, changes in short-term synaptic plasticity but no effects on short- and long-term potentiation in the hippocampus (Dietrich et al., 2018). Busche et al. (2012) have demonstrated that full-length Aβ induced neuronal hyperactivity in brain slice cultures of wild-type mice. Synaptic hyperactivity of hippocampal pyramidal neurons is therefore an early event in AD pathology. 18F-Fluorodeoxyglucose (18F-FDG)-PET in combination with magnetic resonance imaging (MRI) in Tg4–42 mice was used for analyzing cerebral brain glucose metabolism in vivo (Hinteregger et al., 2021). Tg4–42 mice demonstrated lower glucose uptake correlating with neuron loss and memory deficits in an age-dependent manner (Bouter et al., 2018). The reduction of glucose metabolism was detected already in young Tg4–42 prior to neuron death and neurological deficits. In clinical settings, the quantification of brain glucose uptake using 18F-FDG-PET is an established diagnostic tool widely used for differential diagnosis of patients with dementia, including AD (Chetelat et al., 2020). The expert panel suggested a diagnostic algorithm with appropriate time-points using amyloid-PET and 18F-FDG-PET for better clinical management of patients with AD. 18F-FDG-PET signal is a powerful tool to monitor cerebral glucose consumption in vivo, a measure for synaptic activity (Sokoloff, 2008). It is also a valuable tool in preclinical research (Bouter and Bouter, 2019). A comparison between 5XFAD and Tg4–42 showed, that only young Tg4–42 developed robust neurological deficits, whereas in aged mice both models elicited similar memory deficits and fear conditioning tasks (Bouter et al., 2014). Reduced acoustic startle response, prepulse inhibition, and motor coordination were reported in Tg4–42 (Sichler et al., 2019; Wagner et al., 2019). Differentially expressed mRNAs in the brain of Tg4–42 and 5XFAD were identified by deep sequencing. A significant number of mRNAs were associated with memory deficits and neuron loss, which points to common disease pathways in both AD models (Bouter et al., 2014). Moreover, small RNA sequencing of the microRNAome in the hippocampus of Tg4–42 mice revealed microRNAs involved in learning, memory function, and synaptic signaling (Bouter et al., 2020). Metabolic changes of the glutamate/4-aminobutyrate-glutamine axis correlated with neurological deficits, neurodegeneration, and elevated CSF levels of neurofilament light chain in aged Tg4–42 mice (Hinteregger et al., 2021). Tg4–42 mice harbor more than 20 copies of the transgene in exon 2 of the retinoic acid receptor β (RARB) leading to decreased expression of RARB, which could have an (at least partial) effect on the phenotype (Hinteregger et al., 2020). Co-expression of Aβ4–42 with AβpE3–42 within the same neuron accelerated neurodegeneration in the hippocampus, enhanced loss of anxiety and motor deficits as well as sensori-motor deficits (Lopez-Noguerola et al., 2018).
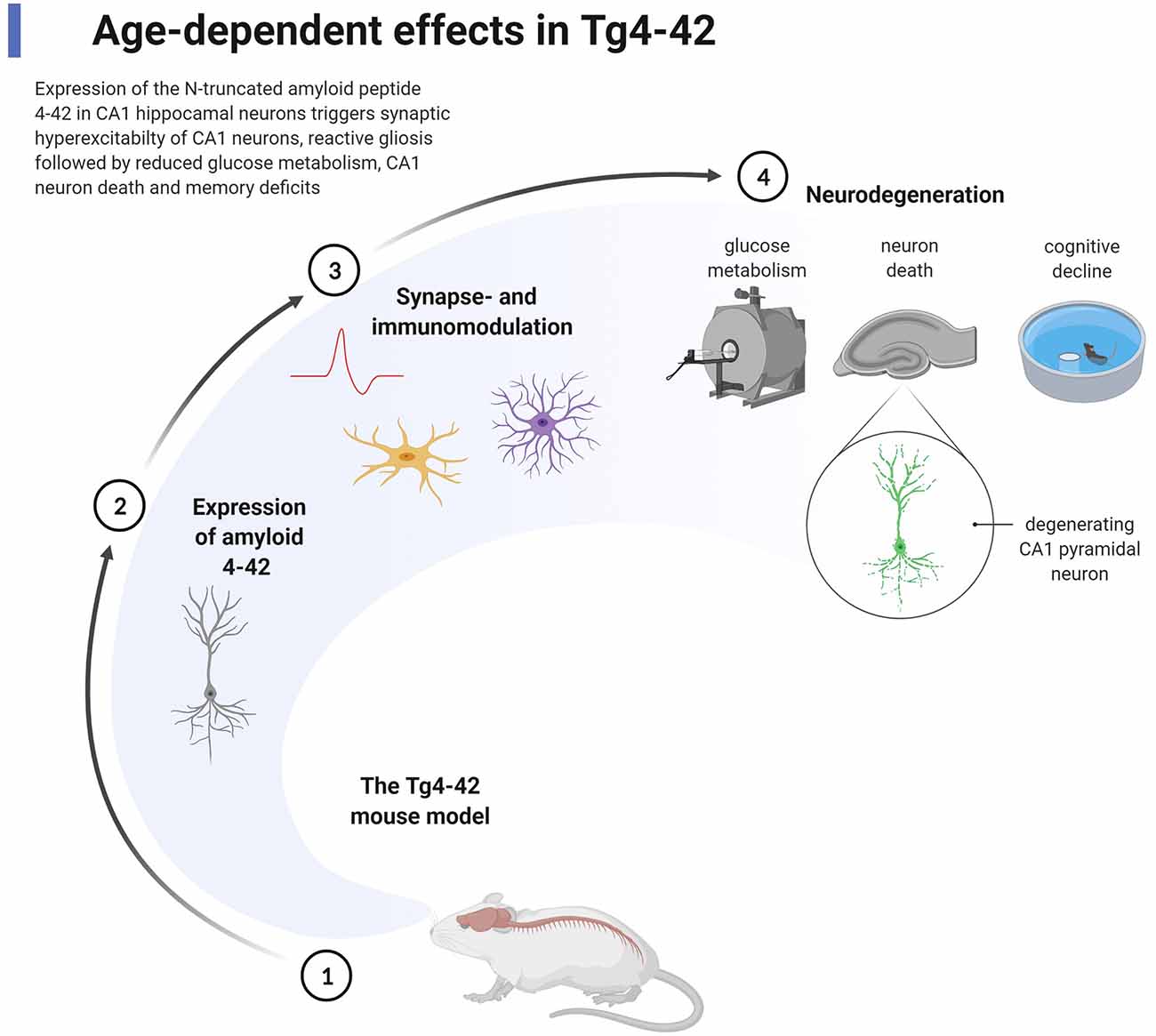
Figure 2. Chronic effect of the expression of Aβ4–42 in the Tg4–42 mouse model for Alzheimer’s disease (AD). The transgenic mouse model Tg4–42 (1) expresses exclusively normal wildtype human Aβ4–42 predominantly in pyramidal neurons in the CA1 area of the hippocampus (2). The expression of Aβ4–42 induces early (starting at 2–3 months of age) pathological effects highlighted by synaptic hyperexcitability of CA1 pyramidal neurons, reactive micro- and astroglia (3). As a consequence of chronic exposure of Aβ4–42 (4), massive reduction in glucose metabolism is detected by 18F-PET/magnetic resonance imaging (MRI), loss of degenerating CA1 pyramidal neurons, and loss of spatial reference memory analyzed by the Morris water maze test (starting at 4–6 months of age). The figure shows methodologies in addition to pathological events using appropriate symbols. Created with BioRender.com.
N-Truncated Aβ4-X as The Target for Immunotherapy
The discovery by Schenk et al. (1999) represents an important development in the AD field because it was the first time that disease-modulation was shown to be possible. The authors showed that pre-aggregated synthetic Aβ1–42 drastically reduced amyloid plaques together with associated astrogliosis in the PDAPP transgenic mouse model for AD. This observation was supported by a subsequent report by Morgan et al. (2000) using another transgenic mouse model for AD (a cross of the Tg2576 and the PS1M146L transgenic line (Holcomb et al., 1998), demonstrating that memory decline assessed by the radial-arm water-maze test was stabilized by active immunization with full-length Aβ.
Janus et al. (2000) observed similar treatment effects of vaccination with Aβ1–42 in the TgCRND8 model. This was followed by exploring the effect of active immunization with AN1792 (pre-aggregated full-length Aβ1–42) in patients with AD. In phase I and IIa clinical trials a subset of the patients developed aseptic meningoencephalopathy and was therefore discontinued despite some hints that it might stabilize cognitive decline in a small subset of patients (Hock et al., 2002; reviewed in Morgan, 2011). Boche et al. (2008) demonstrated that amyloid plaques are solubilized by anti-Aβ antibodies after active immunization with pre-aggregated full-length Aβ1–42. Consequently, the solubilized amyloid-β is drained via the perivascular pathway and was found to be increased in the brain.
Passive immunization with antibodies against Aβ had beneficial treatment effects in AD mouse models and is, therefore, another promising approach to modulating amyloid pathology in vivo. Demattos et al. (2001) used the monoclonal antibody m266 directed against the central domain of Aβ for passive immunization of PDAPP mice leading to reduced amyloid load. Stabilizing memory deficits using an object recognition task and a holeboard learning and memory task without a treatment effect on amyloid load in PDAPP mice was also reported (Dodart et al., 2002). However, Solanezumab the humanized version of m266 did not reach clinical endpoints on cognitive or functional abilities in phase III clinical trials with AD patients (Doody et al., 2014). In clinical phase III trials, bapineuzumab, the humanized monoclonal antibody derived from murine 3D6 was directed against Aβ1–5 (Johnson-Wood et al., 1997) and did not improve clinical outcomes in patients with AD (Salloway et al., 2014). A comprehensive review of other antibody candidates for the treatment of AD has entered clinical trials and novel drug targets have recently been discussed in detail by Cummings et al. (2020).
Mclaurin et al. (2002) reported that active immunization with the full-length Aβ1–42 peptide of TgCRND8 mice reduced amyloid plaques and rescued memory deficits. Interestingly, the therapeutically active antibodies in this experimental setup recognized residues 4–10 of Aβ1–42, an indication that N-truncated Aβ4-X may be a relevant drug target. The therapeutic active anti-Aβ4–10 antibodies also influenced aggregation propensity and toxicity in vitro. We have generated novel murine monoclonal Aβ antibodies against N-truncated Aβ4-X (and AβpE3-X) using preparations of freshly prepared Aβ4–40 and used the antibody NT4X for further studies (Antonios et al., 2013, 2015). NT4X rescued the acute toxic effects of Aβ4–42 using rat primary neurons and in vivo by cerebroventricular injection into wildtype mice (Antonios et al., 2013). NT4X also rescued the chronic effects of Aβ4–42 on pyramidal neuron loss in the hippocampus and spatial reference memory deficits after passive immunization (Antonios et al., 2015). Therefore, besides AβpE3–42, Aβ4–42 might be another relevant drug target in AD.
The question of whether N-truncations of Aβ within plaques represent a post-mortem artifact or might even precede the symptomatology of AD was addressed by Russo et al. (1997), demonstrating that both Aβ1-X and AβpE-X can form stable water-soluble aggregates not related to aggregates amyloid within plaques. Moreover, Rijal Upadhaya et al. (2014) studied post-mortem brain tissue from AD cases with symptomatic and preclinical AD by Western blot and demonstrated that AβpE3-X is an informative biomarker for biochemical amyloid-β staging. AβpE-X and a phosphorylated Aβ variant were not only detectable in plaques but also in soluble aggregates. The authors concluded that the different Aβ variants occur in a hierarchical sequence that allows the distinction of three stages, and may therefore be relevant for therapeutic intervention (Rijal Upadhaya et al., 2014). Finally, Mintun et al. (2021) conducted a phase 2 clinical trial of Donanemab in patients with early symptomatic AD, an antibody that specifically detects AβpE3-X in plaques. The patients were selected based on the amount of tau and amyloid deposition on positron-emission tomography. The outcome of the study showed that a group of patients had a significantly better cognitive score than the placebo group as well as lower amyloid and tau load.
Author Contributions
TB wrote the article and designed the figures.
Conflict of Interest
The University Medicine of Göttingen holds patents, has submitted patent applications on the research tools as well as therapeutic antibodies against N-truncated Aβ.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ancolio, K., Dumanchin, C., Barelli, H., Warter, J. M., Brice, A., Campion, D., et al. (1999). Unusual phenotypic alteration of β amyloid precursor protein (β APP) maturation by a new Val715Met β APP-770 mutation responsible for probable early-onset alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 96, 4119–4124. doi: 10.1073/pnas.96.7.4119
Antonios, G., Borgers, H., Richard, B. C., Brauss, A., Meissner, J., Weggen, S., et al. (2015). Alzheimer therapy with an antibody against N-terminal Aβ 4-X and pyroglutamate Aβ 3-X. Sci. Rep. 5:17338. doi: 10.1038/srep17338
Antonios, G., Saiepour, N., Bouter, Y., Richard, B. C., Paetau, A., Verkkoniemi-Ahola, A., et al. (2013). N-truncated Aβ starting with position four: early intraneuronal accumulation and rescue of toxicity using NT4X-167, a novel monoclonal antibody. Acta Neuropathol. Commun. 1:56. doi: 10.1186/2051-5960-1-56
Antonyan, A., Schlenzig, D., Schilling, S., Naumann, M., Sharoyan, S., Mardanyan, S., et al. (2018). Concerted action of dipeptidyl peptidase IV and glutaminyl cyclase results in formation of pyroglutamate-modified amyloid peptides in vitro. Neurochem. Int. 113, 112–119. doi: 10.1016/j.neuint.2017.12.001
Bayer, T. A., and Wirths, O. (2014). Focusing the amyloid cascade hypothesis on N-truncated Aβ peptides as drug targets against Alzheimer’s disease. Acta Neuropathol. 127, 787–801. doi: 10.1007/s00401-014-1287-x
Bayer, T. A., Schafer, S., Simons, A., Kemmling, A., Kamer, T., Tepest, R., et al. (2003). Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Aβ production in APP23 transgenic mice. Proc. Natl. Acad. Sci. U S A 100, 14187–14192. doi: 10.1073/pnas.2332818100
Boche, D., Zotova, E., Weller, R. O., Love, S., Neal, J. W., Pickering, R. M., et al. (2008). Consequence of Aβ immunization on the vasculature of human Alzheimer’s disease brain. Brain 131, 3299–3310. doi: 10.1093/brain/awn261
Bossak-Ahmad, K., Mital, M., Plonka, D., Drew, S. C., and Bal, W. (2019). Oligopeptides generated by neprilysin degradation of β-amyloid have the highest Cu(II) affinity in the whole Aβ family. Inorg. Chem. 58, 932–943. doi: 10.1021/acs.inorgchem.8b03051
Bouter, C., and Bouter, Y. (2019). (18)F-FDG-PET in mouse models of Alzheimer’s disease. Front. Med. (Lausanne) 6:71. doi: 10.3389/fmed.2019.00071
Bouter, C., Henniges, P., Franke, T. N., Irwin, C., Sahlmann, C. O., Sichler, M. E., et al. (2018). (18)F-FDG-PET detects drastic changes in brain metabolism in the Tg4–42 model of Alzheimer’s disease. Front. Aging Neurosci. 10:425. doi: 10.3389/fnagi.2018.00425
Bouter, Y., Dietrich, K., Wittnam, J. L., Rezaei-Ghaleh, N., Pillot, T., Papot-Couturier, S., et al. (2013). N-truncated amyloid β (Aβ) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 126, 189–205. doi: 10.1007/s00401-013-1129-2
Bouter, Y., Kacprowski, T., Rossler, F., Jensen, L. R., Kuss, A. W., Bayer, T. A., et al. (2020). miRNA alterations elicit pathways involved in memory decline and synaptic function in the hippocampus of aged Tg4–42 mice. Front. Neurosci. 14:580524. doi: 10.3389/fnins.2020.580524
Bouter, Y., Kacprowski, T., Weissmann, R., Dietrich, K., Borgers, H., Brauss, A., et al. (2014). Deciphering the molecular profile of plaques, memory decline and neuron loss in two mouse models for Alzheimer’s disease by deep sequencing. Front. Aging Neurosci. 6:75. doi: 10.3389/fnagi.2014.00075
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 109, 8740–8745. doi: 10.1073/pnas.1206171109
Cabrera, E., Mathews, P., Mezhericher, E., Beach, T. G., Deng, J., Neubert, T. A., et al. (2018). Aβ truncated species: Implications for brain clearance mechanisms and amyloid plaque deposition. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 208–225. doi: 10.1016/j.bbadis.2017.07.005
Casas, C., Sergeant, N., Itier, J. M., Blanchard, V., Wirths, O., Van Der Kolk, N., et al. (2004). Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ 42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 165, 1289–1300. doi: 10.1016/s0002-9440(10)63388-3
Chetelat, G., Arbizu, J., Barthel, H., Garibotto, V., Law, I., Morbelli, S., et al. (2020). Amyloid-PET and (18)F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 19, 951–962. doi: 10.1016/S1474-4422(20)30314-8
Cummings, J., Lee, G., Ritter, A., Sabbagh, M., and Zhong, K. (2020). Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. (N Y) 6:e12050. doi: 10.1002/trc2.12050
Demattos, R. B., Bales, K. R., Cummins, D. J., Dodart, J. C., Paul, S. M., Holtzman, D. M., et al. (2001). Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 98, 8850–8855. doi: 10.1073/pnas.151261398
Dietrich, K., Bouter, Y., Muller, M., and Bayer, T. A. (2018). Synaptic alterations in mouse models for Alzheimer disease-A special focus on N-truncated Aβ 4–42. Molecules 23:718. doi: 10.3390/molecules23040718
Dodart, J. C., Bales, K. R., Gannon, K. S., Greene, S. J., Demattos, R. B., Mathis, C., et al. (2002). Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nat. Neurosci. 5, 452–457. doi: 10.1038/nn842
Doody, R. S., Thomas, R. G., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. doi: 10.1056/NEJMoa1312889
Du, Y., Dodel, R., Hampel, H., Buerger, K., Lin, S., Eastwood, B., et al. (2001). Reduced levels of amyloid β-peptide antibody in Alzheimer disease. Neurology 57, 801–805. doi: 10.1212/wnl.57.5.801
Dunys, J., Valverde, A., and Checler, F. (2018). Are N- and C-terminally truncated Aβ species key pathological triggers in Alzheimer’s disease? J. Biol. Chem. 293, 15419–15428. doi: 10.1074/jbc.R118.003999
Farris, W., Schutz, S. G., Cirrito, J. R., Shankar, G. M., Sun, X., George, A., et al. (2007). Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. 171, 241–251. doi: 10.2353/ajpath.2007.070105
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/s0006-291x(84)80190-4
Haass, C., Hung, A. Y., Schlossmacher, M. G., Oltersdorf, T., Teplow, D. B., Selkoe, D. J., et al. (1993). Normal cellular processing of the β-amyloid precursor protein results in the secretion of the amyloid β peptide and related molecules. Ann. N Y Acad. Sci. 695, 109–116. doi: 10.1111/j.1749-6632.1993.tb23037.x
Haass, C., Schlossmacher, M. G., Hung, A. Y., Vigo Pelfrey, C., Mellon, A., Ostaszewski, B. L., et al. (1992). Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359, 322–325. doi: 10.1038/359322a0
Hama, E., Shirotani, K., Iwata, N., and Saido, T. C. (2004). Effects of neprilysin chimeric proteins targeted to subcellular compartments on amyloid-β peptide clearance in primary neurons. J. Biol. Chem. 279, 30259–30264. doi: 10.1074/jbc.M401891200
Harigaya, Y., Saido, T. C., Eckman, C. B., Prada, C. M., Shoji, M., Younkin, S. G., et al. (2000). Amyloid β protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer’s disease brain. Biochem. Biophys. Res. Commun. 276, 422–427. doi: 10.1006/bbrc.2000.3490
Hinteregger, B., Loeffler, T., Flunkert, S., Neddens, J., Bayer, T. A., Madl, T., et al. (2021). Metabolic, phenotypic and neuropathological characterization of the Tg4–42 mouse model for Alzheimer’s disease. J. Alzheimers Dis. 80, 1151–1168. doi: 10.3233/JAD-201204
Hinteregger, B., Loeffler, T., Flunkert, S., Neddens, J., Birner-Gruenberger, R., Bayer, T. A., et al. (2020). Transgene integration causes RARB downregulation in homozygous Tg4–42 mice. Sci. Rep. 10:6377. doi: 10.1038/s41598-020-63512-8
Hock, C., Konietzko, U., Papassotiropoulos, A., Wollmer, A., Streffer, J., Von Rotz, R. C., et al. (2002). Generation of antibodies specific for β-amyloid by vaccination of patients with Alzheimer disease. Nat. Med. 8, 1270–1275. doi: 10.1038/nm783
Holcomb, L., Gordon, M. N., Mcgowan, E., Yu, X., Benkovic, S., Jantzen, P., et al. (1998). Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100. doi: 10.1038/nm0198-097
Hornung, K., Zampar, S., Engel, N., Klafki, H., Liepold, T., Bayer, T. A., et al. (2019). N-terminal truncated Aβ 4–42 Is a substrate for neprilysin degradation in vitro and in vivo. J. Alzheimers Dis. 67, 849–858. doi: 10.3233/JAD-181134
Huang, S.-M., Mouri, A., Kokubo, H., Nakajima, R., Suemoto, T., Higuchi, M., et al. (2006). Neprilysin-sensitive synapse-associated Aβ oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. 281, 17941–17951. doi: 10.1074/jbc.M601372200
Iwata, N., Mizukami, H., Shirotani, K., Takaki, Y., Muramatsu, S.-I., Lu, B., et al. (2004). Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-β peptide in mouse brain. J. Neurosci. 24, 991–998. doi: 10.1523/JNEUROSCI.4792-03.2004
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., et al. (2001). Metabolic regulation of brain Aβ by neprilysin. Science 292, 1550–1552. doi: 10.1126/science.1059946
Janus, C., Pearson, J., Mclaurin, J., Mathews, P. M., Jiang, Y., Schmidt, S. D., et al. (2000). A β peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408, 979–982. doi: 10.1038/35050110
Jawhar, S., Wirths, O., and Bayer, T. A. (2011). Pyroglutamate Aβ - a hatchet man in Alzheimer disease. J. Biol. Chem. 286, 38825–38832. doi: 10.1074/jbc.R111.288308
Johnson-Wood, K., Lee, M., Motter, R., Hu, K., Gordon, G., Barbour, R., et al. (1997). Amyloid precursor protein processing and A β42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. U S A 94, 1550–1555. doi: 10.1073/pnas.94.4.1550
Kalback, W., Watson, M. D., Kokjohn, T. A., Kuo, Y. M., Weiss, N., Luehrs, D. C., et al. (2002). APP transgenic mice Tg2576 accumulate Aβ peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer’s disease senile plaques. Biochemistry 41, 922–928. doi: 10.1021/bi015685+
Kawarabayashi, T., Younkin, L., Saido, T., Shoji, M., Ashe, K., Younkin, S., et al. (2001). Age-dependent changes in brain, CSF and plasma amyloid (β) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 21, 372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001
Kessler, H., Bayer, T. A., Bach, D., Schneider-Axmann, T., Supprian, T., Herrmann, W., et al. (2008). Intake of copper has no effect on cognition in patients with mild Alzheimer’s disease: a pilot phase 2 clinical trial. J. Neural Transm. 115, 1181–1187. doi: 10.1007/s00702-008-0080-1
Lei, P., Ayton, S., and Bush, A. I. (2020). The essential elements of Alzheimer’s disease. J. Biol. Chem. 296:100105. doi: 10.1074/jbc.REV120.008207
Leissring, M. A., Farris, W., Chang, A. Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology and premature death. Neuron 40, 1087–1093. doi: 10.1016/s0896-6273(03)00787-6
Lemere, C. A., Blusztajn, J. K., Yamaguchi, H., Wisniewski, T., Saido, T. C., Selkoe, D. J., et al. (1996). Sequence of deposition of heterogeneous amyloid β-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis. 3, 16–32. doi: 10.1006/nbdi.1996.0003
Lewis, H., Beher, D., Cookson, N., Oakley, A., Piggott, M., Morris, C. M., et al. (2006). Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-β (42) peptide in vascular dementia. Neuropathol. Appl. Neurobiol. 32, 103–118. doi: 10.1111/j.1365-2990.2006.00696.x
Lopez-Noguerola, J. S., Giessen, N. M. E., Ueberuck, M., Meissner, J. N., Pelgrim, C. E., Adams, J., et al. (2018). Synergistic effect on neurodegeneration by N-truncated Aβ 4–42 and pyroglutamate Aβ 3–42 in a mouse model of Alzheimer’s disease. Front. Aging Neurosci. 10:64. doi: 10.3389/fnagi.2018.00064
Marcello, A., Wirths, O., Schneider-Axmann, T., Degerman-Gunnarsson, M., Lannfelt, L., Bayer, T. A., et al. (2009). Circulating immune complexes of Aβ and IgM in plasma of patients with Alzheimer’s disease. J. Neural Transm. 116, 913–920. doi: 10.1007/s00702-009-0224-y
Marcello, A., Wirths, O., Schneider-Axmann, T., Degerman-Gunnarsson, M., Lannfelt, L., Bayer, T. A., et al. (2011). Reduced levels of IgM autoantibodies against N-truncated pyroglutamate Aβ in plasma of patients with Alzheimer’s disease. Neurobiol. Aging 32, 1379–1387. doi: 10.1016/j.neurobiolaging.2009.08.011
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., Mcdonald, B. L., Beyreuther, K., et al. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
Mclaurin, J., Cecal, R., Kierstead, M. E., Tian, X., Phinney, A. L., Manea, M., et al. (2002). Therapeutically effective antibodies against amyloid-β peptide target amyloid-β residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat. Med. 8, 1263–1269. doi: 10.1038/nm790
Miller, D. L., Papayannopoulos, I. A., Styles, J., Bobin, S. A., Lin, Y. Y., Biemann, K., et al. (1993). Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch. Biochem. Biophys. 301, 41–52. doi: 10.1006/abbi.1993.1112
Mintun, M. A., Lo, A. C., Duggan Evans, C., Wessels, A. M., Ardayfio, P. A., Andersen, S. W., et al. (2021). Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 384, 1691–1704. doi: 10.1056/NEJMoa2100708
Miravalle, L., Calero, M., Takao, M., Roher, A. E., Ghetti, B., Vidal, R., et al. (2005). Amino-terminally truncated Aβ peptide species are the main component of cotton wool plaques. Biochemistry 44, 10810–10821. doi: 10.1021/bi0508237
Mital, M., Bal, W., Fraczyk, T., and Drew, S. C. (2018). Interplay between copper, neprilysin and N-truncation of β-Amyloid. Inorg. Chem. 57, 6193–6197. doi: 10.1021/acs.inorgchem.8b00391
Morgan, D. (2011). Immunotherapy for Alzheimer’s disease. J. Intern. Med. 269, 54–63. doi: 10.1111/j.1365-2796.2010.02315.x
Morgan, D., Diamond, D. M., Gottschall, P. E., Ugen, K. E., Dickey, C., Hardy, J., et al. (2000). Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408, 982–985. doi: 10.1038/35050116
Murayama, K. S., Kametani, F., Tabira, T., and Araki, W. (2007). A novel monoclonal antibody specific for the amino-truncated β-amyloid Aβ 5–40/42 produced from caspase-cleaved amyloid precursor protein. J. Neurosci. Methods 161, 244–249. doi: 10.1016/j.jneumeth.2006.11.010
Näslund, J., Schierhorn, A., Hellman, U., Lannfelt, L., Roses, A. D., Tjernberg, L. O., et al. (1994). Relative abundance of Alzheimer A β amyloid peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. 91, 8378–8382. doi: 10.1073/pnas.91.18.8378
Nath, A., Hall, E., Tuzova, M., Dobbs, M., Jons, M., Anderson, C., et al. (2003). Autoantibodies to amyloid β-peptide (Aβ) are increased in Alzheimer’s disease patients and Aβ antibodies can enhance Aβ neurotoxicity: implications for disease pathogenesis and vaccine development. Neuromol. Med. 3, 29–39. doi: 10.1385/nmm:3:1:29
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal β-amyloid aggregates, neurodegeneration and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Pajonk, F., Kessler, H., Supprian, T., Hamzei, P., Bach, D., Schweickhardt, J., et al. (2005). Cognitive decline correlates with low plasma concentrations of copper in patients with mild to moderate Alzheimer’s disease. J. Alzheimers Dis. 8, 23–27. doi: 10.3233/jad-2005-8103
Parodi-Rullan, R., Ghiso, J., Cabrera, E., Rostagno, A., and Fossati, S. (2020). Alzheimer’s amyloid β heterogeneous species differentially affect brain endothelial cell viability, blood-brain barrier integrity and angiogenesis. Aging Cell 19:e13258. doi: 10.1111/acel.13258
Piccini, A., Russo, C., Gliozzi, A., Relini, A., Vitali, A., Borghi, R., et al. (2005). β-amyloid is different in normal aging and in Alzheimer disease. J. Biol. Chem. 280, 34186–34192. doi: 10.1074/jbc.M501694200
Pike, C. J., Overman, M. J., and Cotman, C. W. (1995). Amino-terminal deletions enhance aggregation of β-amyloid peptides in vitro. J. Biol. Chem. 270, 23895–23898. doi: 10.1074/jbc.270.41.23895
Portelius, E., Bogdanovic, N., Gustavsson, M. K., Volkmann, I., Brinkmalm, G., Zetterberg, H., et al. (2010). Mass spectrometric characterization of brain amyloid β isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 120, 185–193. doi: 10.1007/s00401-010-0690-1
Rijal Upadhaya, A., Kosterin, I., Kumar, S., Von Arnim, C. A., Yamaguchi, H., Fandrich, M., et al. (2014). Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 137, 887–903. doi: 10.1093/brain/awt362
Rosen, R. F., Tomidokoro, Y., Farberg, A. S., Dooyema, J., Ciliax, B., Preuss, T. M., et al. (2016). Comparative pathobiology of β-amyloid and the unique susceptibility of humans to Alzheimer’s disease. Neurobiol. Aging 44, 185–196. doi: 10.1016/j.neurobiolaging.2016.04.019
Rufenacht, P., Guntert, A., Bohrmann, B., Ducret, A., and Dobeli, H. (2005). Quantification of the Aβ peptide in Alzheimer’s plaques by laser dissection microscopy combined with mass spectrometry. J. Mass Spectrom. 40, 193–201. doi: 10.1002/jms.739
Russo, C., Saido, T. C., Debusk, L. M., Tabaton, M., Gambetti, P., Teller, J. K., et al. (1997). Heterogeneity of water-soluble amyloid β-peptide in Alzheimer’s disease and Down’s syndrome brains. FEBS Lett. 409, 411–416. doi: 10.1016/s0014-5793(97)00564-4
Salloway, S., Sperling, R., Fox, N. C., Blennow, K., Klunk, W., Raskind, M., et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333. doi: 10.1056/NEJMoa1304839
Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., et al. (1999). Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. doi: 10.1038/22124
Schlenzig, D., Cynis, H., Hartlage-Rubsamen, M., Zeitschel, U., Menge, K., Fothe, A., et al. (2018). Dipeptidyl-peptidase activity of meprin β links N-truncation of Aβ with glutaminyl cyclase-catalyzed pGlu-Aβ formation. J. Alzheimers Dis. 66, 359–375. doi: 10.3233/JAD-171183
Selkoe, D. J. (2001). Alzheimer’s disease: genes, proteins and therapy. Physiol. Rev. 81, 741–766. doi: 10.1152/physrev.2001.81.2.741
Sergeant, N., Bombois, S., Ghestem, A., Drobecq, H., Kostanjevecki, V., Missiaen, C., et al. (2003). Truncated β-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J. Neurochem. 85, 1581–1591. doi: 10.1046/j.1471-4159.2003.01818.x
Sevalle, J., Amoyel, A., Robert, P., Fournie-Zaluski, M. C., Roques, B., Checler, F., et al. (2009). Aminopeptidase A contributes to the N-terminal truncation of amyloid β-peptide. J. Neurochem. 109, 248–256. doi: 10.1111/j.1471-4159.2009.05950.x
Sichler, M. E., Low, M. J., Schleicher, E. M., Bayer, T. A., and Bouter, Y. (2019). Reduced acoustic startle response and prepulse inhibition in the Tg4–42 model of Alzheimer’s disease. J. Alzheimers Dis. Rep. 3, 269–278. doi: 10.3233/ADR-190132
Sokoloff, L. (2008). The physiological and biochemical bases of functional brain imaging. Cogn. Neurodyn 2, 1–5. doi: 10.1007/s11571-007-9033-x
Tekirian, T. L. (2001). Commentary: Aβ N- terminal isoforms: critical contributors in the course of AD pathophysiology. J. Alzheimers Dis. 3, 241–248. doi: 10.3233/jad-2001-3209
Trieb, K., Ransmayr, G., Sgonc, R., Lassmann, H., and Grubeck-Loebenstein, B. (1996). APP peptides stimulate lymphocyte proliferation in normals, but not in patients with Alzheimer’s disease. Neurobiol. Aging 17, 541–547.
Valverde, A., Dunys, J., Lorivel, T., Debayle, D., Gay, A. S., Lacas-Gervais, S., et al. (2021). Aminopeptidase A contributes to biochemical, anatomical and cognitive defects in Alzheimer’s disease (AD) mouse model and is increased at early stage in sporadic AD brain. Acta Neuropathol. 141, 823–839. doi: 10.1007/s00401-021-02308-0
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Wagner, J. M., Sichler, M. E., Schleicher, E. M., Franke, T. N., Irwin, C., Low, M. J., et al. (2019). Analysis of motor function in the Tg4–42 mouse model of Alzheimer’s disease. Front. Behav. Neurosci. 13:107. doi: 10.3389/fnbeh.2019.00107
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Walter, S., Jumpertz, T., Huttenrauch, M., Ogorek, I., Gerber, H., Storck, S. E., et al. (2019). The metalloprotease ADAMTS4 generates N-truncated Aβ4-x species and marks oligodendrocytes as a source of amyloidogenic peptides in Alzheimer’s disease. Acta Neuropathol. 137, 239–257. doi: 10.1007/s00401-018-1929-5
Weksler, M. E., Relkin, N., Turkenich, R., Larusse, S., Zhou, L., Szabo, P., et al. (2002). Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp. Gerontol. 37, 943–948. doi: 10.1016/s0531-5565(02)00029-3
Wildburger, N. C., Esparza, T. J., Leduc, R. D., Fellers, R. T., Thomas, P. M., Cairns, N. J., et al. (2017). Diversity of amyloid-β proteoforms in the Alzheimer’s disease brain. Sci. Rep. 7:9520. doi: 10.1038/s41598-017-10422-x
Wirths, O., and Zampar, S. (2019). Emerging roles of N- and C-terminally truncated Aβ species in Alzheimer’s disease. Expert Opin. Ther. Targets 23, 991–1004. doi: 10.1080/14728222.2019.1702972
Wirths, O., Walter, S., Kraus, I., Klafki, H. W., Stazi, M., Oberstein, T. J., et al. (2017). N-truncated Aβ4-x peptides in sporadic Alzheimer’s disease cases and transgenic Alzheimer mouse models. Alzheimers Res. Ther. 9:80. doi: 10.1186/s13195-017-0309-z
Wittnam, J. L., Portelius, E., Zetterberg, H., Gustavsson, M. K., Schilling, S., Koch, B., et al. (2012). Pyroglutamate amyloid β (Aβ) aggravates behavioral deficits in transgenic amyloid mouse model for Alzheimer disease. J. Biol. Chem. 287, 8154–8162. doi: 10.1074/jbc.M111.308601
Keywords: N-truncated Aβ, Tg4–42, transgenic mouse model, immunotherapy, neuron loss, PET, in vivo imaging, memory decline
Citation: Bayer TA (2021) N-Truncated Aβ Starting at Position Four—Biochemical Features, Preclinical Models, and Potential as Drug Target in Alzheimer’s Disease. Front. Aging Neurosci. 13:710579. doi: 10.3389/fnagi.2021.710579
Received: 16 May 2021; Accepted: 29 July 2021;
Published: 20 August 2021.
Edited by:
Nibaldo C. Inestrosa, Pontificia Universidad Católica de Chile, ChileReviewed by:
Mark Stephen Kindy, James A. Haley Veterans’ Hospital, United StatesFrédéric Checler, UMR7275 Institut de pharmacologie moléculaire et cellulaire (IPMC), France
Copyright © 2021 Bayer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas A. Bayer, dGJheWVyQGd3ZGcuZGU=