
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 13 August 2021
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.695965
This article is part of the Research Topic The Neurovascular Unit as a Potential Biomarker and Therapeutic Target in Cerebrovascular Disease View all 13 articles
 Amanda C. Rosehart1
Amanda C. Rosehart1 Thomas A. Longden2
Thomas A. Longden2 Nick Weir2Jackson T. Fontaine1
Nick Weir2Jackson T. Fontaine1 Anne Joutel3,4
Anne Joutel3,4 Fabrice Dabertrand1,5*
Fabrice Dabertrand1,5*Prostaglandin E2 (PGE2) has been widely proposed to mediate neurovascular coupling by dilating brain parenchymal arterioles through activation of prostanoid EP4 receptors. However, our previous report that direct application of PGE2 induces an EP1-mediated constriction strongly argues against its direct action on arterioles during neurovascular coupling, the mechanisms sustaining functional hyperemia. Recent advances have highlighted the role of capillaries in sensing neuronal activity and propagating vasodilatory signals to the upstream penetrating parenchymal arteriole. Here, we examined the effect of capillary stimulation with PGE2 on upstream arteriolar diameter using an ex vivo capillary-parenchymal arteriole preparation and in vivo cerebral blood flow measurements with two-photon laser-scanning microscopy. We found that PGE2 caused upstream arteriolar dilation when applied onto capillaries with an EC50 of 70 nM. The response was inhibited by EP1 receptor antagonist and was greatly reduced, but not abolished, by blocking the strong inward-rectifier K+ channel. We further observed a blunted dilatory response to capillary stimulation with PGE2 in a genetic mouse model of cerebral small vessel disease with impaired functional hyperemia. This evidence casts previous findings in a different light, indicating that capillaries are the locus of PGE2 action to induce upstream arteriolar dilation in the control of brain blood flow, thereby providing a paradigm-shifting view that nonetheless remains coherent with the broad contours of a substantial body of existing literature.
As a leading cause of stroke and dementia, cerebral small vessel diseases (SVDs) pose a horrendous threat to the elderly population (Pantoni, 2010; Wardlaw et al., 2019). Despite their major contribution to age-related vascular cognitive impairment and disability (Iadecola et al., 2019), the disease processes and key biological mechanisms underlying these disorders remain largely unknown. Moreover, there are no specific treatments outside the management of vascular risk factors and use of anti-platelets after ischemic stroke. However, accumulating experimental evidence suggests that functional alterations in the cerebral microcirculation have early and deleterious consequences on functional hyperemia—the ability of the brain to increase local blood flow in response to local increases in neuronal activity (Huneau et al., 2018; Iadecola et al., 2019).
Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) is the most common genetic form of cerebral SVDs. Remarkably, CADASIL includes all clinical and MRI manifestations of sporadic SVDs, hence offering a lens through which to view more common forms of sporadic SVDs (Chabriat et al., 2009). Both CADASIL patients and the TgNotch3R169C mouse model, hereafter referred to as SVD mice, exhibit deficits in functional hyperemia at an early stage of the disease progression (Joutel et al., 2010; Huneau et al., 2018). We have recently demonstrated that activation of the epidermal growth factor receptor (EGFR) by its ligand heparin-binding EGF-like growth factor (HB-EGF) ameliorates the cerebral vascular deficits of the SVD mouse—including impaired functional hyperemia (Capone et al., 2016; Dabertrand et al., 2021). Part of this effect is mediated by reenabling capillary-to-arteriole signaling during the neurovascular coupling that underpins functional hyperemia (Dabertrand et al., 2021). In physiological conditions, action potentials increase extracellular K+ concentration which activates the strong inward-rectifier K+ (Kir2.1) channel in capillary endothelial cells (cECs). This creates a hyperpolarizing signal that rapidly propagates to upstream arterioles, driving vasodilation and local hyperemia (Longden et al., 2017; Harraz et al., 2018a; Moshkforoush et al., 2020). This mechanism is vulnerable to pathology, and we recently demonstrated that SVD lowers the synthesis of the phospholipid PIP2, which prevents Kir2.1 channels from acting as sensors of increases in external K+ (Dabertrand et al., 2021). Strikingly, we showed that both HB-EGF and systemic injection of exogenous PIP2 improved functional hyperemia deficits in SVD mice by restoring capillary-to-arteriole signaling.
Functional hyperemia is a vital process controlled by multiple mechanisms that provide layers of redundancy, and thus neurovascular coupling agents other than K+ ions have been postulated (Attwell et al., 2010; Kaplan et al., 2020). Among these, prostaglandin E2 (PGE2), produced by cyclooxygenase-2 (COX-2) from arachidonic acid, has been widely proposed to be released from excitatory neurons to relax arteriolar smooth muscle cells (SMCs) by activation of the GS protein-coupled prostanoid EP4 receptor and subsequent cyclic adenosine monophosphate-dependent pathway (Zonta et al., 2002; Takano et al., 2005; Gordon et al., 2008; Attwell et al., 2010; Lacroix et al., 2015). Yet, this interpretation appears incompatible with our previous demonstration that PGE2 causes constriction, rather than dilation, when applied directly to isolated cortical arterioles—an effect that occurs via activation of the Gq protein-coupled prostanoid EP1 receptor (Dabertrand et al., 2013). Consistent with these observations, the EP1 receptor is robustly expressed not only in the arteriolar SMCs, but also in cECs of the brain microcirculation (Vanlandewijck et al., 2018). We therefore hypothesized that PGE2 contributes to neurovascular coupling by initiating a vasodilatory signal from the capillaries to the upstream parenchymal arterioles, rather than by targeting SMCs directly. Using a combination of ex vivo and in vivo approaches, we report that capillary stimulation with PGE2 induces upstream arteriolar dilation and increases blood flow in vivo. Consistent with defective functional hyperemia, we further show that PGE2-induced capillary-to-arteriole signaling is blunted in the SVD mouse model and can be rescued by HB-EGF.
All experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado, Anschutz Medical Campus. Adult (2–3 months old) male C57/BL6J mice (Jackson Laboratories, United States), were housed on a 12-h light:dark cycle with environmental enrichment and free access to food and water. TgNotch3WT (WT) and TgNotch3R169C (SVD) lines have been previously described (Joutel et al., 2010) and were used at 6 months of age in order for the TgNotch3R169C mice to develop the SVD phenotype, and for consistency with our previous studies (Dabertrand et al., 2015, 2021; Capone et al., 2016; Fontaine et al., 2020). All mice were euthanized by i.p. injection of sodium pentobarbital (100 mg/kg) followed by rapid decapitation.
The CaPA preparation was obtained as previously described (Longden et al., 2017; Rosehart et al., 2019) by dissecting intracerebral arterioles arising from the M1 region of the middle cerebral artery, leaving the attached capillary bed intact. Arteriolar segments were cannulated on glass micropipettes with one end occluded by a tie and pressurized using a Living Systems Instrumentation (United States) pressure servo controller with a mini peristaltic pump. The ends of the capillaries were then sealed by the downward pressure of an overlying glass micropipette. CaPA preparations were superfused (4 mL/min) with prewarmed (36°C ± 1°C), gassed (5% CO2, 20% O2, and 75% N2) artificial cerebrospinal fluid (aCSF) for at least 30 min. The composition of aCSF was 125 mM NaCl, 3 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 1 mM MgCl2, 4 mM glucose, 2 mM CaCl2, pH 7.3 (after aeration with 5% CO2). Application of pressure (40 mmHg) to the cannulated parenchymal arteriole segment in this preparation pressurized the entire tree and induced myogenic tone in the arteriolar segment. Only viable CaPA preparations, defined as those that developed pressure-induced myogenic tone greater than 15%, were used in subsequent experiments. Arteriolar viability was validated by bath-applying NS309 (1 μM), which causes an endothelial-dependent vasodilation through activation of small- and intermediate-conductance, Ca2+-sensitive K+ (SK and IK, respectively) channels, or the thromboxane receptor agonist U46619 (1 μM), which causes robust vasoconstriction. Dilatory responses of the attached arteriole segment to K+ and PGE2 were obtained by applying aCSF containing 10 mM K+ or PGE2 onto capillary extremities by pressure ejection from a glass micropipette (tip diameter, ∼5 μm) attached to a Picospritzer III (Parker, United States) at ∼5 psi for 20 s. The dose-responses were performed using pipettes filled with the different concentrations of PGE2 and testing them sequentially starting with the lowest concentration. Contrary to arteriolar endothelial cells, cECs do not express functional IK and SK channels and spatial restriction of the drugs applied onto the capillary ends was validated by the lack of response to local stimulation with NS309 (1 μM), as previously described (Rosehart et al., 2019). In some control experiments, K+ and PGE2 were applied directly to the arteriole segment and the other drugs were applied via the bath perfusion. The luminal diameter of the parenchymal arteriole was acquired at 15 Hz using a CCD camera and IonWizard 6.2 edge-detection software (IonOptix, United States). Two regions were simultaneously recorded, zone 1 where the capillary tree sprouted from of the arteriole and zone 2 located 250 μm upstream of this, and diameter from both of these sites was averaged unless noted otherwise. Changes in arteriolar diameter were calculated from the average luminal diameter measured over the last 10 s of stimulation and were normalized to the maximum dilatory responses in 0 mM Ca2+ bath solution at the end of each experiment using the following equation: [(change in diameter)/(maximal diameter-initial diameter)] × 100.
As previously described (Longden et al., 2017), mice were anesthetized with isoflurane (5% induction and 2% maintenance) during the surgical procedure. The skull was exposed, cleaned, and a stainless-steel head plate was attached with a mixture of dental cement and superglue. Isoflurane was replaced with α-chloralose (50 mg/kg) and urethane (750 mg/kg) during recording. FITC-dextran (2000 kDa) was injected via the retro-orbital sinus to visualize the cerebral vasculature and for contrast imaging of RBCs. A penetrating arteriole, identified by the direction of the RBCs flowing into the brain, was followed and a downstream capillary was selected for study in cortical layers 2 or 3. A pipette was then maneuvered into the brain and positioned adjacent to the capillary under study, and aCSF containing, or not, 1 μM PGE2 was ejected (200–300 ms, 8 ± 1 psi, ∼4 picoliters). The ejected volume was monitored by including 0.2 mg/mL tetramethylrhodamine isothiocyanate (TRITC; 150 kDa)-labeled dextran in the pipette (Figure 1E). RBC flux data were collected by line scanning at 5 kHz. Images were acquired using a Zeiss LSM-7 multiphoton microscope (Zeiss, United States) equipped with a 20x Plan Apochromat 1.0 N.A. DIC VIS-IR water-immersion objective and coupled to a Coherent Chameleon Vision II Titanium-Sapphire pulsed infrared laser (Coherent, United States). FITC and TRITC were excited at 820 nm, and emitted fluorescence was separated through 500–550 and 570–610 nm bandpass filters, respectively.
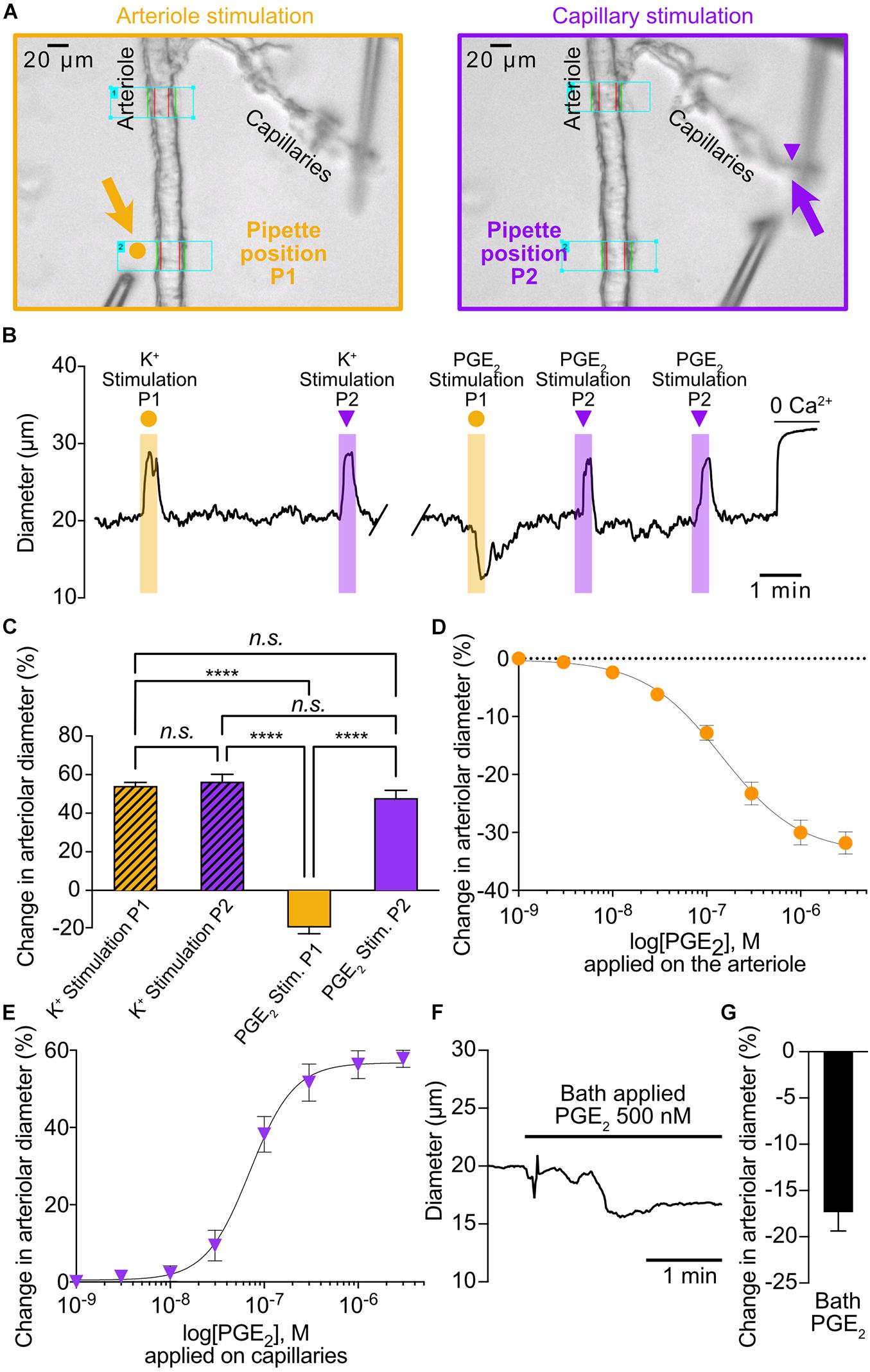
Figure 1. PGE2 causes upstream arteriolar dilation when applied onto capillaries. (A) Pipette positions for arteriole stimulation (left, 5 orange arrow) and capillary stimulation (right, purple arrow) in CaPA preparations. (B) Representative trace of arteriolar diameter showing effects of pressure ejection of 10 mM K+ or 1 μM PGE2 onto arteriole (P1, orange dot) and capillaries (P2, purple triangle). (C) Summary data from 6 mice (n.s., not significant; ****P < 0.0001, one-way ANOVA, Tukey’s test). (D) Concentration-response curve produced by locally applying PGE2 to the arteriolar segment over a concentration range of 1 nM to 3 μM (5 mice). An EC50 of 145 nM was calculated from the non-linear regression curve. (E) Concentration-response curve produced by locally applying PGE2 to capillary extremities over a concentration range of 1 nM to 3 μM (8 mice). An EC50 of 70 nM was calculated from the non-linear regression curve. (F) Representative trace of arteriolar diameter showing effects of bath application of 500 nM PGE2. (G) Summary data from 5 mice.
HC030031, HC067047, PGE2, NS309, and SC51322 were purchased from Tocris Bioscience (United States); All other chemicals and reagents were obtained from Sigma-Aldrich (United States). The vehicle for HB-EGF solutions was 0.2-μm–filtered PBS containing 0.1% BSA.
Data in figures and text are presented as means ± standard error of the mean (SEM). Statistical testing was performed using GraphPad Prism 8 software. All data passed the Kolmogorov–Smirnov test for normality. Statistical significance was determined using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test, unless otherwise stated.
To investigate the effect of capillary stimulation with PGE2 on upstream arteriolar diameter, we used our previously developed ex vivo capillary-parenchymal arteriole (CaPA) preparation that allows to apply vasoactive substances at specific points along the arteriole-capillary continuum by pressure ejection (Longden et al., 2017; Rosehart et al., 2019). Consistent with our previous report that PGE2 acts as a vasoconstrictor (Dabertrand et al., 2013), local application of 1 μM PGE2 directly on the arteriolar segment caused a decrease in diameter (Figures 1A–C). In contrast, and as expected (Longden et al., 2017; Dabertrand et al., 2021), 10 mM K+ also applied on the arteriole caused a robust dilation. Interestingly, either 10 mM K+ or 1 μM PGE2 caused upstream dilations when applied onto capillary extremities and the amplitudes of these were virtually identical (56.2% ± 3.9% and 47.8% ± 4.2%, respectively) (Figures 1A–C and Supplementary Movie 1). However, the kinetics of the responses appear significantly different with slower onset (5.27 ± 1.35 s) and time to peak (9.65 ± 1.5 s) for PGE2 compared to K+ responses, (1.7 ± 0.4 s and 3.87 ± 1.13 s, respectively).
We next tested the effect of increasing concentrations of PGE2 locally applied to the arteriolar segment and observed a concentration-dependent constriction with a calculated EC50 of 145 nM (Figure 1D). The arteriolar dilation in response to capillary stimulations with PGE2 was also concentration-dependent with a calculated EC50 of 70 nM and a maximal response at 1 μM, and thus we chose to use the latter concentration throughout the study for capillary stimulation (Figure 1E and Supplementary Movie 1). Finally, we tested the effect of a modest concentration of PGE2 (500 nM) applied via the bath perfusion, hence stimulating both the capillary ends and the arteriolar segment, and measured a small but clear constriction (Figures 1F,G).
In our initial report of capillary-to-arteriole electrical signaling (Longden et al., 2017), we identified the cEC strong inward-rectifier K+ channel, Kir2.1, as the molecular cornerstone of the capillary-to-arteriole electrical mechanism elicited by capillary stimulation with 10 mM K+. Increasing extracellular K+ concentration from 3 to 10 mM activates Kir2.1 channels in cECs, which induces a regenerative hyperpolarization that travels retrogradely to dilate the upstream arteriole. Consistent with this model, 30 μM Ba2+—which, among the inward rectifier K+ channels expressed in the microcirculation, preferentially blocks Kir2 channels (Longden and Nelson, 2015)—completely abolished the arteriolar dilation in response to capillary-applied 10 mM K+. Therefore, we tested the effect of Ba2+ on arteriolar dilation induced by capillary stimulation with PGE2. Arteriolar diameter was recorded at two distinct zones: zone 1 was placed at the branching of the transitional segment from the arteriole and zone 2 was located 250 μm upstream of this (Figure 2A). PGE2 locally applied onto capillaries led to similar dilatory responses in zone 1 (49.1% ± 5.5%) and zone 2 (48.2% ± 2.9%) (Figures 2B,C). Remarkably, bath application of 30 μM Ba2+ decreased upstream arteriolar dilation at zone 1 in response to capillary stimulation with PGE2 by 63% and virtually eliminated dilation at zone 2. The onset of the remnant dilation observed at zone 1 was unchanged by Ba2+ (5.27 ± 1.35 s 6.49 ± 2.81 s). Taken together, these data suggest that the Kir2.1 channel is not required to initiate PGE2-induced upstream arteriolar dilation, but rather participates in the amplification and propagation of the vasodilatory signal, as evidenced by the difference between zone 1 and zone 2.
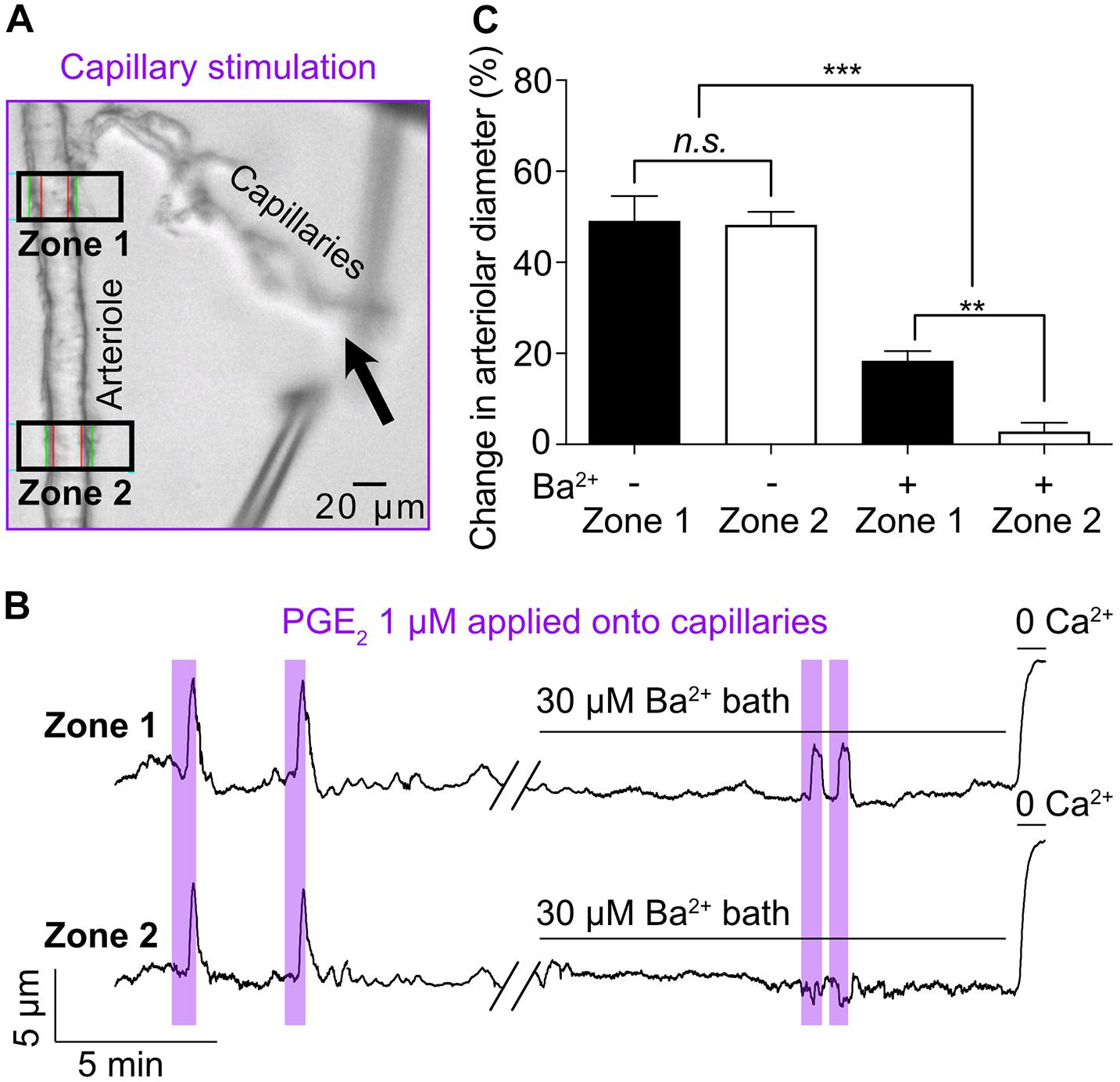
Figure 2. Kir channel blocker Ba2+ (30 μM) inhibits the propagation, not the initiation, of the PGE2-induced upstream arteriolar dilation. (A) Micrograph showing Zone 1 and Zone 2 positions for arteriolar diameter recording during capillaries stimulation (black arrow) with 1 μM PGE2. (B) Representative traces of arteriolar diameter at zone 1 and zone 2 showing the effect of 30 μM bath-applied Ba2+ on PGE2-induced upstream dilation. (C) Summary data from 5 mice (n.s., not significant; **P < 0.01, ***P < 0.001, one-way ANOVA, Tukey’s test).
To investigate the mechanistic underpinnings of the capillary-to-arteriole signaling induced by PGE2, we first superfused the ex vivo CaPA preparation with 1 μM SC51322, a prostanoid receptor antagonist specific for the EP1 receptor. SC51322 abolished the response to PGE2, while the dilation induced by 10 mM K+ remained intact (Figures 3A,B). This observation suggests that PGE2 acts through activation of G protein-coupled receptors of the Gq/11 subtype (GqPCR) and subsequent Ca2+ signaling. Depletion of plasma membrane phosphatidylinositol 4,5-bisphosphate (PIP2) following GqPCR activation is known to stimulate transient receptor potential (TRP) channels, a major pathway for extracellular Ca2+ influx (Kim et al., 2008; Harraz et al., 2018b). Moreover, recent work by Thakore et al. (2021) has revealed that activation of TRPA1 channel in cECs can initiate a biphasic, propagating retrograde signal that dilates upstream parenchymal arterioles during functional hyperemia. However, the TRPA1 antagonist HC030031 at 3 μM had no effect on the dilation induced by capillary stimulation with PGE2 (Figures 3A,B). Inhibition of TRPV4, another Ca2+-permeable TRP channel expressed by cECs (Harraz et al., 2018b), with 1 μM HC067047 also did not impact the effect of PGE2 (Figures 3A,B). These results suggest that PGE2 induces upstream arteriole dilation via activation of the EP1 receptor but independently of TRPV4 and TRPA1 channels.
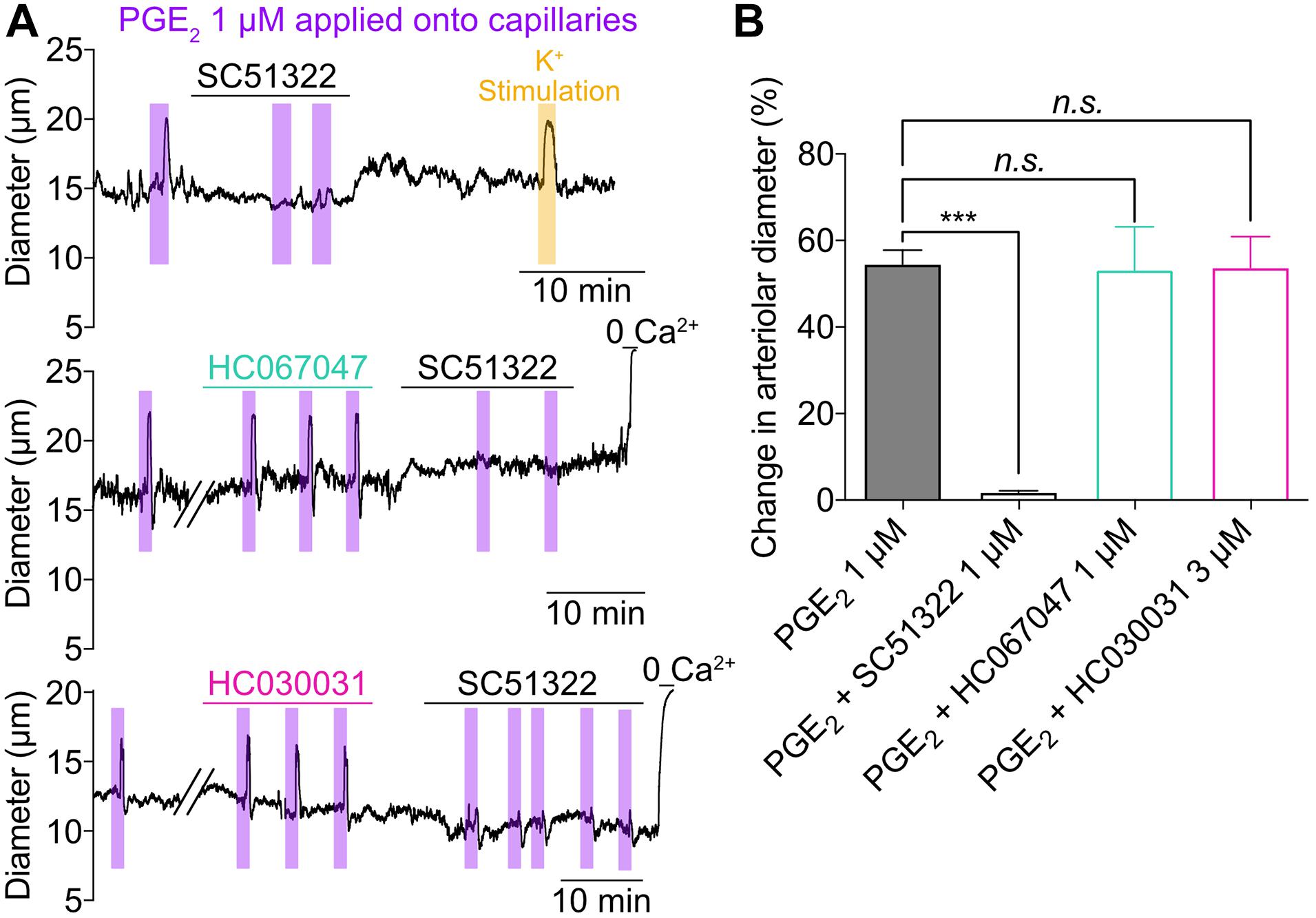
Figure 3. Activation of capillary EP1 receptor initiates PGE2-induced upstream dilation independently of TRPV4 and TRPA1 channels. (A) Representative traces of arteriolar diameter showing effects of pressure ejection of PGE2 1 μM onto capillaries in absence or presence of SC51322 (1 μM, EP1 antagonist), HC067047 (1 μM, TRPV4 antagonist), and HC030031 (3 μM, TRPA1 antagonist). (B) Summary data from 5 to 8 mice (n.s., not significant; ***P < 0.001, one-way ANOVA, Dunnett’s test).
We then tested the ability of PGE2 to dilate upstream arterioles in vivo by measuring capillary red blood cell (RBC) flux with two-photon laser-scanning microscopy. Fluorescein isothiocyanate (FITC)-labeled dextran was used to visualize the cortical microcirculation and a pipette containing 1 μM PGE2 was maneuvered into the brain through a cranial window and positioned next to a capillary of interest (Figure 4A). Pressure ejection (8 ± 1 psi) for up to 300 ms of PGE2 evoked a significant increase in capillary RBC flux (Δ = 12 ± 4 RBCs/s) consistent with the notion that the mechanisms we observed in CaPA preparations are also at play in the intact system (Figures 4B–D). In contrast, ejection of aCSF vehicle had no effect on capillary RBC flux (Figure 4E). To determine whether PGE2 also causes upstream dilation we next performed experiments in which we imaged at the parenchymal arteriole-transitional zone junction (Figure 4F). Here, we found that ejection of 1 μM PGE2 selectively onto capillaries routinely produced a small upstream dilation (Figures 4F,G and Supplementary Movie 2). In contrast, if we applied PGE2 onto both capillaries and arterioles in vivo by increasing the duration of ejection, causing spread of the ejected solution to the upstream arteriole along a paravascular route, we observed constriction (Figure 4H), consistent with our ex vivo data. As expected, ejection of aCSF vehicle alone had no effect on upstream arteriolar diameter (12.95 ± 0.57 μm baseline diameter vs. 12.96 ± 0.8 μm after aCSF ejection, n = 6 experiments, 3 mice, P = 0.97, paired Student’s t-test).
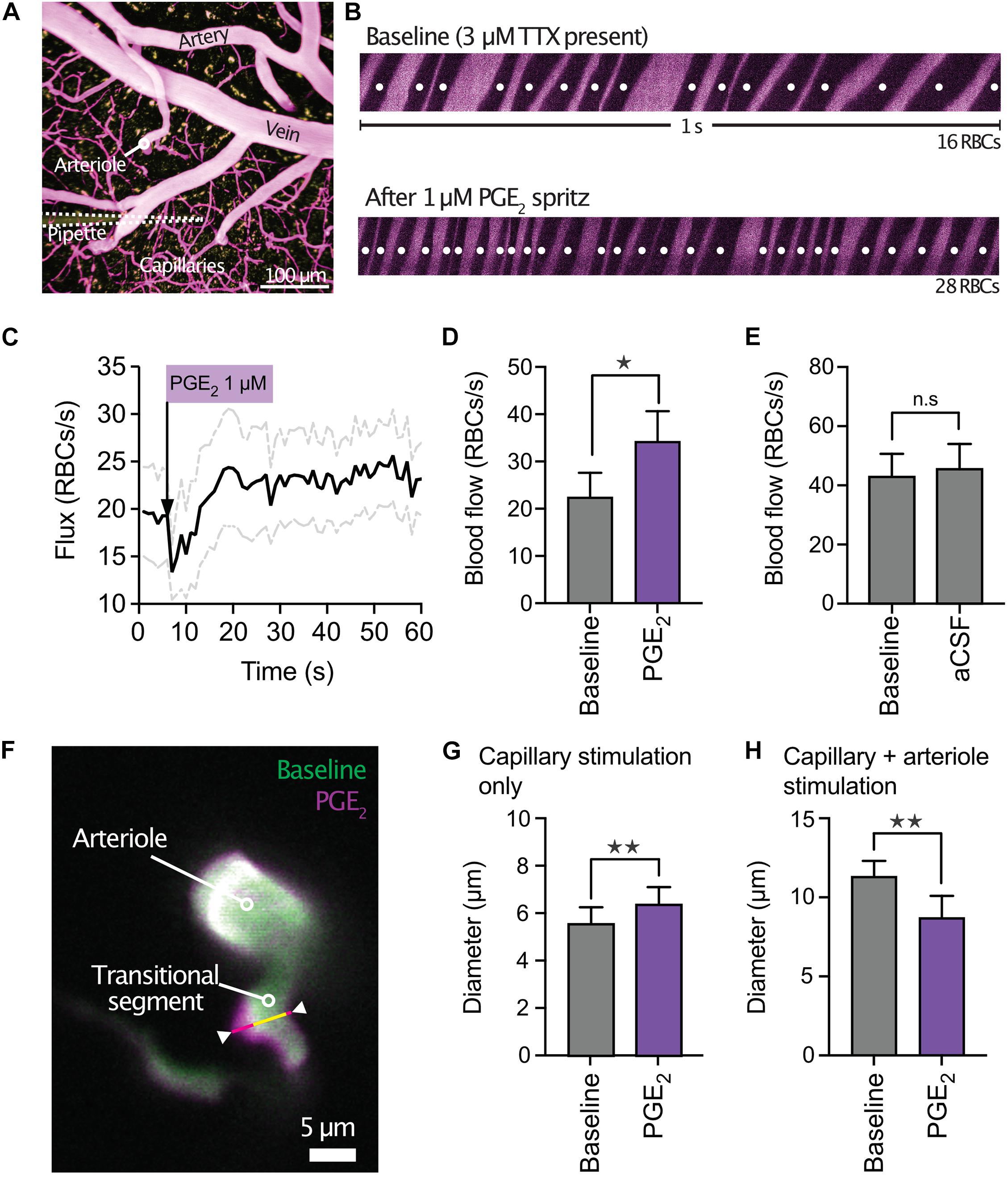
Figure 4. PGE2 causes capillary hyperemia in vivo. (A) Micrograph of mouse cortical vasculature showing a micropipette containing PGE2 and TRITC-dextran (yellow) in close apposition to a capillary (FITC-dextran, purple). (B) Red blood cell (RBC) flux was measured by high-frequency line scanning over a period of 1 min at baseline (top panel) and after application of 1 μM PGE2 (lower panel) onto a capillary. RBCs appear as black streaks in plasma (purple). (C) Average traces (black line) plus SEM (gray lines) showing the increase in RBC flux to PGE2. The dip immediately following the ejection of PGE2 is caused by momentary pressure on the capillary wall. (D) Summary data of RBC flux showing significant hyperemia following capillary stimulation with 1 μM PGE2 (n = 14 experiments, 6 mice, *P < 0.05, paired Student’s t-test). (E) In contrast, ejection of aCSF vehicle onto capillaries had no effect on RBC flux (n = 9 experiments, 5 mice, P > 0.05, paired Student’s t-test). (F) Overlay showing the diameter of a penetrating arteriole and the transitional segment to the capillary bed at baseline (green) and after PGE2 ejection onto a downstream capillary (magenta). The upstream dilation was most prominent in the region highlighted by the white arrowheads. (G) Summary data showing upstream dilation to capillary ejection of PGE2 (n = 5 experiments, 4 mice, **P < 0.01, paired Student’s t-test). (H) In contrast, simultaneous stimulation of both capillaries and arterioles with PGE2 in vivo led to constriction (n = 7 experiments, 3 mice, **P < 0.01, paired Student’s t-test).
Finally, we investigated the effect of SVD on PGE2-initiated capillary-to-arteriole signaling. Ex vivo stimulation of capillaries from SVD mice with PGE2 induced an upstream arteriolar dilation that was 58% smaller at Zone 1 and 64.8% smaller at Zone 2 compared with that in TgNotch3WT control (WT) mice, revealing attenuated PGE2-mediated signaling (Figure 5). The difference between zone 1 and 2 dilations were not significant. We previously reported that activation of the epidermal growth factor receptor (EGFR) by its ligand heparin-binding EGF-like growth factor (HB-EGF) ameliorates the cerebral vascular deficits of the SVD mouse—including neurovascular coupling and functional hyperemia deficits (Capone et al., 2016; Dabertrand et al., 2021). We then directly tested the effect of HB-EGF on PGE2-induced upstream vasodilation in ex vivo preparations from SVD mice. Bath-applied 30 ng/mL HB-EGF dramatically enhanced the upstream arteriolar dilation induced by capillary stimulation with PGE2, increasing the average dilation from 19.8% ± 1% to 48.7% ± 4.3% at Zone 1, and from 16.8% ± 1.4% to 45.3% ± 5.1% at Zone 2 (Figure 5 and Supplementary Movie 3). HB-EGF completely restored PGE2-induced upstream dilation, abolishing the differences measured between WT and SVD animals.
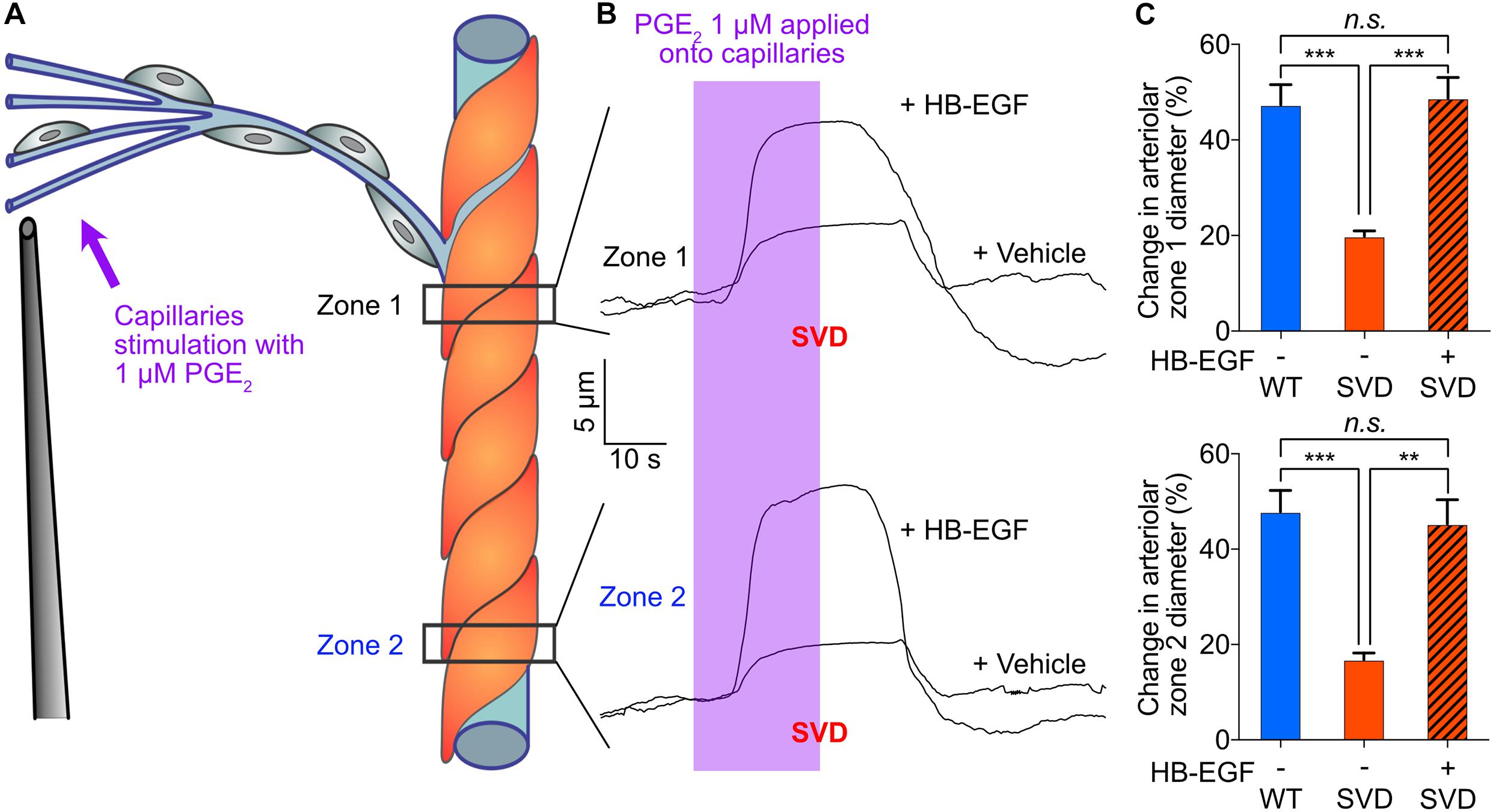
Figure 5. In the SVD mouse model, blunted upstream arteriolar dilation in response to capillary stimulation with PGE2 is completely restored by bath application of 30 ng/ml HB-EGF. (A) Cartoon showing Zone 1 and Zone 2 positions for arteriolar diameter recording during capillaries stimulation with 1 μM PGE2. (B) Representative traces of arteriolar diameter showing the effect of pressure ejection of PGE2 1 μM onto capillaries in absence or presence of 30 ng/ml HB-EGF in a preparation from SVD mouse model. (C) Summary data from 5 to 6 mice (n.s., not significant; **P < 0.01, ***P < 0.001, one-way ANOVA Dunnett’s test).
Our progress in understanding functional hyperemia in health and disease has been hampered by large gaps in our comprehension of the mechanism underlying this basic physiological response—and persistent controversies surrounding it (Kaplan et al., 2020). Our recent work identified Kir2.1 channels in cECs as the molecular cornerstone initiating and propagating a retrograde hyperpolarizing vasodilatory signal from capillaries to arterioles (Longden et al., 2017; Harraz et al., 2018a; Moshkforoush et al., 2020; Dabertrand et al., 2021). The present study extends this capillary-based paradigm, providing support for a new signaling modality that posits a central role for the GqPCR EP1 receptor in mediating PGE2-induced vasodilatory signal that propagates upstream to cause dilation of feeding parenchymal arterioles (Figure 6). Importantly, these findings hold the promise of resolving controversies surrounding how PGE2, a widely proposed mediator of neurovascular coupling (Zonta et al., 2002; Takano et al., 2005; Gordon et al., 2008; Attwell et al., 2010; Watkins et al., 2014), can promote functional hyperemia despite evidence that it directly constricts arterioles (Dabertrand et al., 2013).
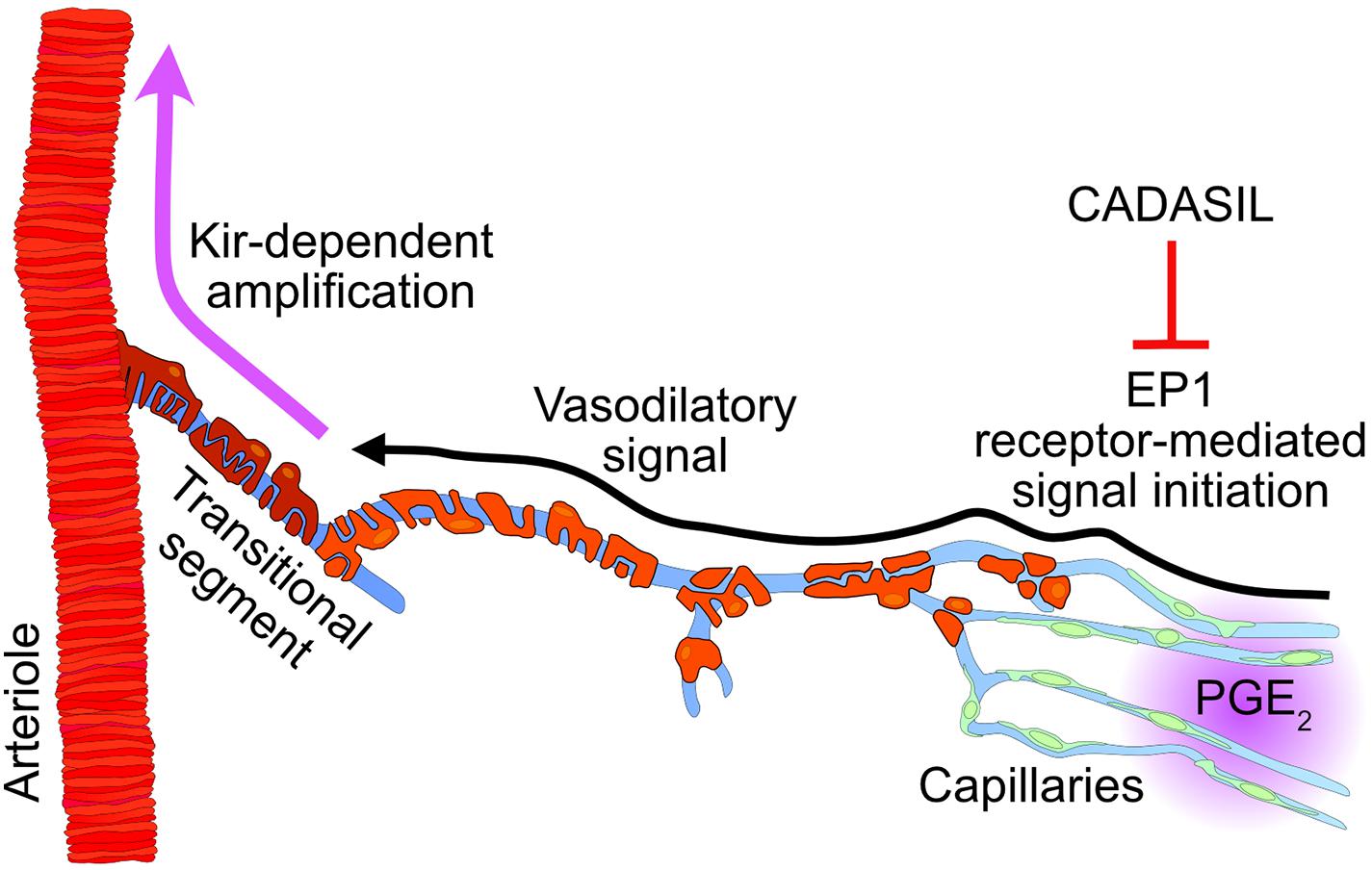
Figure 6. Proposed mechanism involving PGE2 during functional hyperemia and its inhibition in cerebral small vessel disease (CADASIL). Intracellular increases in Ca2+ caused by phospholipase C activation during EP1 signaling triggers a propagating vasodilatory signal that is amplified by Kir channel in the transitional segment region. CADASIL, which lowers the pool of phospholipase C substrate PIP2, would limit the synthesis of the second messenger IP3, resulting in a reduced PGE2-evoked upstream dilation during functional hyperemia.
Our observations raise the immediate question of which effect of PGE2, capillary-mediated vasodilation or direct arteriolar constriction, predominates during neurovascular coupling. We calculated an EC50 of 145 nM for the PGE2-induced constriction of the parenchymal arteriole, while the vasodilation induced by capillary stimulation displayed an EC50 of 70 nM. In our experimental conditions, a simultaneous stimulation of capillaries and the arteriole with 500 nM PGE2 led to a constriction, suggesting that the arteriolar effect prevails when both vascular segments are exposed to PGE2. A possible explanation for these observations is that the microvascular response to PGE2 has a kinetic component in which the fast constriction predominates over the slower capillary-mediated response. However, this stimulation via the bath perfusion likely does not reflect in vivo conditions. Accordingly, we tested the effect of increasing our ejection duration in vivo such that PGE2 not only stimulated the targeted capillaries but also spread upstream to the arteriole. Here too this maneuver produced arteriolar constriction, again suggesting that this response will predominate when the arteriole is exposed directly to PGE2. Since the dense capillary network within the brain lies in close proximity to all neurons (Nishimura et al., 2007; Blinder et al., 2013), it is expected that cECs are the primary sensors of neuronal activity and any neurally derived PGE2. Therefore, the vasodilatory effect would be expected to predominate during neurovascular coupling in physiological conditions and our data suggest that this occurs through local exposure of the capillaries to PGE2. However, a variety of brain conditions, including ischemia and neurodegeneration (Minghetti, 2004), are known to up-regulate COX-2 expression in excitatory neurons which results in an EP1-dependent neurotoxicity of PGE2 (Kawano et al., 2006). In this pathological situation, higher PGE2 concentrations could lead to arteriolar constriction and then contribute to PGE2 neurotoxicity by limiting local blood supply.
Neuronal activation leads to rapid increases in blood flow within, and on the surface, of the brain. Hillman and colleagues elegantly provided evidence for involvement of the endothelium in stimulus-evoked, conducted vasodilation from the brain parenchyma to arterioles and pial arteries in vivo (Chen et al., 2014). Our previous study on neurovascular coupling demonstrated how capillary endothelium is capable of transmitting an electrical signal to cause upstream vasodilation in support of functional hyperemia (Longden et al., 2017). We showed that activation of the Kir2.1 channel in cECs by extracellular K+ ions propagates a regenerative hyperpolarization from cell-to-cell up to the feeding arteriole to cause vasodilation (Longden et al., 2017; Harraz et al., 2018a; MacMillan and Evans, 2018). Interestingly, recent work from Thakore et al. (2021) showed that 4-hydroxynonenal (4-HNE), an endogenous product of lipid peroxidation, activates transient receptor potential ankyrin 1 (TRPA1) channel in cECs to cause upstream arteriolar dilation during functional hyperemia. These recent findings introduce the concept that a slowly propagating short-range Ca2+ signal is initiated in the capillary endothelium and converted into the fast-propagating hyperpolarization that causes dilation of upstream arterioles. The conversion is proposed to occur in the transitional region between the capillaries and the arteriole (Ratelade et al., 2020) by activation of the small- and intermediate-conductance Ca2+-activated K+ channels (SK and IK, respectively) and amplification of the hyperpolarization by Kir channel (Thakore et al., 2021). The transitional region refers to the first segment sprouting out of the arteriole, visible on Figure 1 micrographs, while local stimulations are applied onto the 3rd and 4th order capillary branches after the transitional region. A vast body of literature reports (de Wit et al., 1999; Domeier and Segal, 2007; Bagher and Segal, 2011) supports the concept that a propagating Ca2+ signal is capable of acting through IK/SK channels, which are not present in cECs (Longden et al., 2017) but are expressed by transitional and arteriolar ECs (Hannah et al., 2011; Thakore et al., 2021). The generated hyperpolarizing signal is further amplified via Kir channel activation (Sonkusare et al., 2016) and conveyed through myoendothelial junctions to adjacent SMCs. In the dilation induced by capillary stimulation with PGE2, inhibition of the Kir2.1 channel with Ba2+ had a profound effect, particularly measurable on the propagation of the dilation, which is consistent with the biphasic propagative model proposed by Thakore et al. (2021). Interestingly, inhibition of TRPA1 channels did not prevent PGE2 from causing upstream dilation, suggesting a different initiation mechanism, likely involving Gq protein activation and inositol trisphosphate (IP3)-mediated Ca2+ release, as opposed to direct Ca2+ entry across the plasma membrane. Our previous work also highlighted the role of GqPCR activation in breaking down phosphatidylinositol 4,5-bisphosphate (PIP2), resulting in decreased Kir2.1 channel activity and increased open probability of the Ca2+/Na+-permeable TRPV4 channel (Harraz et al., 2018a,b, 2020). However, blocking TRPV4 channels had no effect on the dilation induced by PGE2 either. Given these data, we propose that EP1-initiated IP3-dependent Ca2+ signals arriving in the transitional segment activate endothelial IK/SK channels, and the ensuing membrane potential hyperpolarization activates Kir channels, converting the incoming Ca2+ signal into a Kir-dependent hyperpolarizing signal. The characterization of such a Ca2+ signal will require more extensive investigation, but the vasodilation induced by capillary stimulation with PGE2 is clearly central to the postulated role of this molecule as a neurovascular coupling agent.
Cerebral SVDs have emerged as a central link between two major co-morbidities. They account for more than 30% of strokes worldwide and at least 40% of dementia cases (Pantoni, 2010; Iadecola, 2013). CADASIL is caused by dominant mutations in the NOTCH3 receptor, expressed by SMCs and pericytes, that stereotypically lead to the extracellular deposition of the NOTCH3 ectodomain (NOTCH3ECD), which recruits and aggregates other proteins on vessels, ultimately forming deposits termed granular osmiophilic material (GOM) (Joutel et al., 2000, 2016; Chabriat et al., 2009). One of these proteins is the tissue inhibitor of metalloproteinases 3 (TIMP3), which directly complexes with NOTCHECD and abnormally accumulates in the extra cellular matrix of brain vessels in patients and mice with CADASIL (Monet-Leprêtre et al., 2013). A deficit in CBF hemodynamics, including functional hyperemia, is an early disease manifestation in patients (Chabriat et al., 2000; Pfefferkorn et al., 2001; Liem et al., 2009; Huneau et al., 2018) and a prominent feature of the well-established TgNotch3R169C CADASIL mouse model used in the present study (Joutel et al., 2010; Capone et al., 2016; Dabertrand et al., 2021). Our recent work indicates that TIMP3 effects on cerebrovascular reactivity are attributable to inhibition of ADAM17 and subsequent suppression of EGFR signaling by inhibition of ectodomain shedding of its ligand HB-EGF (Dabertrand et al., 2015, 2021; Capone et al., 2016).
Consistent with this model, we previously found that EGFR activation with exogenous soluble HB-EGF restores cerebral arterial tone and functional hyperemia (Dabertrand et al., 2015, 2021; Capone et al., 2016). Here, we found that PGE2-induced dilation was impaired in the CADASIL mouse model and fully restored by HB-EGF.
We previously identified two downstream consequences of the suppressed TIMP3-ADAM17-EGFR signaling module: (i) the upregulation of voltage gated K+ (KV1.5) channels in the arteriolar SMCs (Dabertrand et al., 2015; Capone et al., 2016); and (ii) the partial inhibition of Kir2.1 channels in cECs, but not in arteriolar ECs and SMCs (Dabertrand et al., 2021). Here, we report a third consequence: the disruption of PGE2-induced capillary-to-arteriole signaling, reinforcing the concept that extracellular matrix alterations have profound impacts on cerebrovascular dynamics in SVDs. Using computational modeling, we previously investigated the impact of KV channel upregulation on membrane potential dynamics in the context of concurrent activation of myocyte Kir channels (Koide et al., 2018). Interestingly, while these analyses showed that a higher KV channel current density would reduce the membrane potential range over which Kir channels can be activated to cause and propagate dilation, the more hyperpolarized resting membrane potential (9 mV) actually facilitates Kir channel activation. Thus, arterioles from the CADASIL mouse model would still hyperpolarize and dilate in response to Kir channel opening, as observed experimentally (Dabertrand et al., 2015; Koide et al., 2018), but at the cost of a smaller vasodilatory reserve. At the capillary level, we previously described, and modeled, how a 50% reduction in cECs Kir2.1 current is sufficient to completely abolish the capillary-to-arteriole electrical signaling in response to 10 mM K+ (Harraz et al., 2018a; Moshkforoush et al., 2020), and then strongly reduces functional hyperemia in the CADASIL mouse model (Dabertrand et al., 2021). We attributed this endothelial dysfunction to a reduced cEC metabolism caused by inhibition of the EGFR pathway (Dabertrand et al., 2021). The resulting lower ATP/ADP ratio in CADASIL compared to WT cECs decreases the synthesis of PIP2 and its availability to act as an essential cofactor for Kir2.1 channel—hence reducing the channel activity (Huang et al., 1998; Hansen et al., 2011; Harraz et al., 2018a, 2020). Of particular note, PIP2 is also a substrate for phospholipase C during EP1 signaling, and a reduced pool of PIP2 would certainly limit the synthesis of the second messenger IP3, which mobilizes Ca2+ from endoplasmic reticulum stores through its action on its cognate receptor. The observation that HB-EGF restores both Kir2.1- and PGE2-initiated signaling is consistent with this concept. The impact of CADASIL on neurovascular coupling is thus multifaceted, involving disruption of two different propagating signals sharing connections to the EGFR pathway in cECs, and Kir channel activation in the transitional and arteriolar segments.
We have made major progress in establishing potential contributing mechanisms to cerebral hemodynamics impairment observed at an early stage of CADASIL, a Mendelian paradigm of SVDs (Chabriat et al., 2009) and the most common hereditary cause of stroke (Pantoni, 2010) and dementia (Schmidt et al., 2012). Here we demonstrate that functional hyperemia deficits in this SVD involve a PGE2-initiated capillary-to-arteriole signal that is normally regulated by the TIMP3-ADAM17-EGFR signaling module. Furthermore, the evidence that PGE2 induces arteriolar dilation via capillary stimulation has the potential to reconcile disparate findings in neurovascular studies. Our mechanistic studies thus lay the groundwork for novel targeted strategies for treating CADASIL and other cerebrovascular diseases.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado, Anschutz Medical Campus.
AR, JF, and FD performed ex vivo experiments, data collection, and analysis. TL and NW performed in vivo experiments, data collection, and analysis. AJ contributed to the study design. FD designed and directed the research and wrote the manuscript. All authors edited the manuscript and approved its submission.
This study was supported by the National Institute on Aging R01AG066645 to TL, and National Institute of Neurological Disorders and Stroke DP2OD02944801 to TL, a grant from the National Research Agency, France (ANR-16-RHUS-0004) to AJ, awards from the CADASIL Together We Have Hope Non-profit Organization, a research grant from the Center for Women’s Health Research located at the University of Colorado Anschutz Medical Campus, a research grant from the University of Pennsylvania Orphan Disease Center in partnership with the cureCADASIL, and National Heart, Lung, and Blood Institute R01HL136636 to FD.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GW declared a shared affiliation, with no collaboration, with one of the authors AJ to the handling editor at the time of the review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2021.695965/full#supplementary-material
Supplementary Movie 1 | Capillary stimulation with PGE2 1 μM evokes upstream arterial dilation in ex vivo CaPA preparation.
Supplementary Movie 2 | Capillary stimulation with PGE2 causes upstream dilation of the arteriole and transitional segment in vivo.
Supplementary Movie 3 | Exogenous application of 30 ng/ml HB-EGF fully restores upstream arterial dilation in response to capillary stimulation with 1 μM PGE2 in CaPA preparation from a SVD mouse.
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., MacVicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. doi: 10.1038/nature09613
Bagher, P., and Segal, S. S. (2011). Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol. 202, 271–284. doi: 10.1111/j.1748-1716.2010.02244.x
Blinder, P., Tsai, P. S., Kaufhold, J. P., Knutsen, P. M., Suhl, H., and Kleinfeld, D. (2013). The cortical angiome: an interconnected vascular network with noncolumnar patterns of blood flow. Nat. Neurosci. 16, 889–897. doi: 10.1038/nn.3426
Capone, C., Dabertrand, F., Baron-Menguy, C., Chalaris, A., Ghezali, L., Domenga-Denier, V., et al. (2016). Mechanistic insights into a TIMP3-sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics. Elife 5:e17536. doi: 10.7554/eLife.17536
Chabriat, H., Joutel, A., Dichgans, M., Tournier-Lasserve, E., and Bousser, M.-G. (2009). CADASIL. Lancet Neurol. 8, 643–653. doi: 10.1016/S1474-4422(09)70127-9
Chabriat, H., Pappata, S., Ostergaard, L., Clark, C. A., Pachot-Clouard, M., Vahedi, K., et al. (2000). Cerebral hemodynamics in CADASIL before and after acetazolamide challenge assessed with MRI bolus tracking. Stroke 31, 1904–1912. doi: 10.1161/01.str.31.8.1904
Chen, B. R., Kozberg, M. G., Bouchard, M. B., Shaik, M. A., and Hillman, E. M. C. (2014). A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J. Am. Heart Assoc. 3:e000787. doi: 10.1161/JAHA.114.000787
Dabertrand, F., Hannah, R. M., Pearson, J. M., Hill-Eubanks, D. C., Brayden, J. E., and Nelson, M. T. (2013). Prostaglandin E2, a postulated astrocyte-derived neurovascular coupling agent, constricts rather than dilates parenchymal arterioles. J. Cereb. Blood Flow Metab. 33, 479–482. doi: 10.1038/jcbfm.2013.9
Dabertrand, F., Harraz, O. F., Koide, M., Longden, T. A., Rosehart, A. C., Hill-Eubanks, D. C., et al. (2021). PIP2 corrects cerebral blood flow deficits in small vessel disease by rescuing capillary Kir2.1 activity. Proc. Natl. Acad. Sci. U. S. A. 118:e2025998118. doi: 10.1073/pnas.2025998118/pnas.1420765112
Dabertrand, F., Krøigaard, C., Bonev, A. D., Cognat, E., Dalsgaard, T., Domenga-Denier, V., et al. (2015). Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease. Proc. Natl. Acad. Sci. U. S. A. 112, E796–E805. doi: 10.1073/pnas.1420765112
de Wit, C., Esser, N., Lehr, H. A., Bolz, S. S., and Pohl, U. (1999). Pentobarbital-sensitive EDHF comediates ACh-induced arteriolar dilation in the hamster microcirculation. Am. J. Physiol. 276, H1527–H1534. doi: 10.1152/ajpheart.1999.276.5.H1527
Domeier, T. L., and Segal, S. S. (2007). Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J. Physiol. 579, 175–186. doi: 10.1113/jphysiol.2006.124529
Fontaine, J. T., Rosehart, A. C., Joutel, A., and Dabertrand, F. (2020). HB-EGF depolarizes hippocampal arterioles to restore myogenic tone in a genetic model of small vessel disease. Mech. Ageing Dev. 192:111389. doi: 10.1016/j.mad.2020.111389
Gordon, G. R. J., Choi, H. B., Rungta, R. L., Ellis-Davies, G. C. R., and MacVicar, B. A. (2008). Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456, 745–749. doi: 10.1038/nature07525
Hannah, R. M., Dunn, K. M., Bonev, A. D., and Nelson, M. T. (2011). Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J. Cereb. Blood Flow Metab. 31, 1175–1186. doi: 10.1038/jcbfm.2010.214
Hansen, S. B., Tao, X., and MacKinnon, R. (2011). Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498. doi: 10.1038/nature10370
Harraz, O. F., Hill-Eubanks, D., and Nelson, M. T. (2020). PIP2: a critical regulator of vascular ion channels hiding in plain sight. Proc. Natl. Acad. Sci. U. S. A. 117, 20378–20389. doi: 10.1073/pnas.2006737117
Harraz, O. F., Longden, T. A., Dabertrand, F., Hill-Eubanks, D., and Nelson, M. T. (2018a). Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc. Natl. Acad. Sci. U. S. A. 115, E3569–E3577. doi: 10.1073/pnas.1800201115
Harraz, O. F., Longden, T. A., Hill-Eubanks, D., and Nelson, M. T. (2018b). PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. Elife 7:351. doi: 10.7554/eLife.38689
Huang, C. L., Feng, S., and Hilgemann, D. W. (1998). Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391, 803–806. doi: 10.1038/35882
Huneau, C., Houot, M., Joutel, A., Béranger, B., Giroux, C., Benali, H., et al. (2018). Altered dynamics of neurovascular coupling in CADASIL. Ann. Clin. Transl. Neurol. 5, 788–802. doi: 10.1002/acn3.574
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Iadecola, C., Duering, M., Hachinski, V., Joutel, A., Pendlebury, S. T., Schneider, J. A., et al. (2019). Vascular cognitive impairment and dementia: JACC scientific expert panel. J. Am. Coll. Cardiol. 73, 3326–3344. doi: 10.1016/j.jacc.2019.04.034
Joutel, A., Andreux, F., Gaulis, S., Domenga, V., Cecillon, M., Battail, N., et al. (2000). The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Invest. 105, 597–605. doi: 10.1172/JCI8047
Joutel, A., Haddad, I., Ratelade, J., and Nelson, M. T. (2016). Perturbations of the cerebrovascular matrisome: a convergent mechanism in small vessel disease of the brain? J. Cereb. Blood Flow Metab. 36, 143–157. doi: 10.1038/jcbfm.2015.62
Joutel, A., Monet-Leprêtre, M., Gosele, C., Baron-Menguy, C., Hammes, A., Schmidt, S., et al. (2010). Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J. Clin. Invest. 120, 433–445. doi: 10.1172/JCI39733DS1
Kaplan, L., Chow, B. W., and Gu, C. (2020). Neuronal regulation of the blood– brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 21, 1–17. doi: 10.1038/s41583-020-0322-2
Kawano, T., Anrather, J., Zhou, P., Park, L., Wang, G., Frys, K. A., et al. (2006). Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat. Med. 12, 225–229. doi: 10.1038/nm1362
Kim, D., Cavanaugh, E. J., and Simkin, D. (2008). Inhibition of transient receptor potential A1 channel by phosphatidylinositol-4,5-bisphosphate. Am. J. Physiol. Cell Physiol. 295, C92–C99. doi: 10.1152/ajpcell.00023.2008
Koide, M., Moshkforoush, A., Tsoukias, N. M., Hill-Eubanks, D. C., Wellman, G. C., Nelson, M. T., et al. (2018). The yin and yang of KV channels in cerebral small vessel pathologies. Microcirculation 25, 1–10. doi: 10.1111/micc.12436
Lacroix, A., Toussay, X., Anenberg, E., Lecrux, C., Ferreiros, N., Karagiannis, A., et al. (2015). COX-2-derived prostaglandin E2 produced by pyramidal neurons contributes to neurovascular coupling in the rodent cerebral cortex. J. Neurosci. 35, 11791–11810. doi: 10.1523/JNEUROSCI.0651-15.2015
Liem, M. K., Lesnik Oberstein, S. A. J., Haan, J., Boom, R. V. D., Ferrari, M. D., Buchem, M. A. V., et al. (2009). cerebrovascular reactivity is a main determinant of white matter hyperintensity progression in CADASIL. Am. J. Neuroradiol. 30, 1244–1247. doi: 10.3174/ajnr.A1533
Longden, T. A., Dabertrand, F., Koide, M., Gonzales, A. L., Tykocki, N. R., Brayden, J. E., et al. (2017). Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 20, 717–726. doi: 10.1038/nn.4533
Longden, T. A., and Nelson, M. T. (2015). Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 22, 183–196. doi: 10.1111/micc.12190
MacMillan, S., and Evans, A. M. (2018). AMPK-α1 or AMPK-α2 Deletion in smooth muscles does not affect the hypoxic ventilatory response or systemic arterial blood pressure regulation during hypoxia. Front. Physiol. 9:655. doi: 10.3389/fphys.2018.00655
Minghetti, L. (2004). Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 63, 901–910. doi: 10.1093/jnen/63.9.901
Monet-Leprêtre, M., Haddad, I., Baron-Menguy, C., Fouillot-Panchal, M., Riani, M., Domenga-Denier, V., et al. (2013). Abnormal recruitment of extracellular matrix proteins by excess Notch3ECD: a new pathomechanism in CADASIL. Brain 136, 1830–1845. doi: 10.1093/brain/awt092
Moshkforoush, A., Ashenagar, B., Harraz, O. F., Dabertrand, F., Longden, T. A., Nelson, M. T., et al. (2020). The capillary Kir channel as sensor and amplifier of neuronal signals: modeling insights on K+-mediated neurovascular communication. Proc. Natl. Acad. Sci. U. S. A. 117, 16626–16637. doi: 10.1073/pnas.2000151117
Nishimura, N., Schaffer, C. B., Friedman, B., Lyden, P. D., and Kleinfeld, D. (2007). Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc. Natl. Acad. Sci. U. S. A. 104, 365–370. doi: 10.1073/pnas.0609551104
Pantoni, L. (2010). Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 9, 689–701. doi: 10.1016/S1474-4422(10)70104-6
Pfefferkorn, T., von Stuckrad-Barre, S., Herzog, J., Gasser, T., Hamann, G. F., and Dichgans, M. (2001). Reduced cerebrovascular CO2 reactivity in CADASIL: a transcranial Doppler sonography study. Stroke 32, 17–21. doi: 10.1161/01.str.32.1.17
Ratelade, J., Klug, N. R., Lombardi, D., Angelim, M. K. S. C., Dabertrand, F., Domenga-Denier, V., et al. (2020). Reducing hypermuscularization of the transitional segment between arterioles and capillaries protects against spontaneous intracerebral hemorrhage. Circulation 141, 2078–2094. doi: 10.1161/CIRCULATIONAHA.119.040963
Rosehart, A. C., Johnson, A. C., and Dabertrand, F. (2019). Ex vivo pressurized hippocampal capillary-parenchymal arteriole preparation for functional study. J. Vis. Exp. 1–7. doi: 10.3791/60676
Schmidt, H., Freudenberger, P., Seiler, S., and Schmidt, R. (2012). Genetics of subcortical vascular dementia. Exp. Gerontol. 47, 873–877. doi: 10.1016/j.exger.2012.06.003
Sonkusare, S. K., Dalsgaard, T., Bonev, A. D., and Nelson, M. T. (2016). Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J. Physiol. 594, 3271–3285. doi: 10.1113/JP271652
Takano, T., Tian, G.-F., Peng, W., Lou, N., Libionka, W., Han, X., et al. (2005). Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 9, 260–267. doi: 10.1038/nn1623
Thakore, P., Alvarado, M. G., Ali, S., Mughal, A., Pires, P. W., Yamasaki, E., et al. (2021). Brain endothelial cell TRPA1 channels initiate neurovascular coupling. Elife 10:e63040. doi: 10.7554/eLife.63040
Vanlandewijck, M., He, L., Mäe, M. A., Andrae, J., Ando, K., Del Gaudio, F., et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. doi: 10.1038/nature25739
Wardlaw, J. M., Smith, C., and Dichgans, M. (2019). Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 18, 684–696. doi: 10.1016/S1474-4422(19)30079-1
Watkins, S., Robel, S., Kimbrough, I. F., Robert, S. M., Ellis-Davies, G., and Sontheimer, H. (2014). Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 5:4196. doi: 10.1038/ncomms5196
Keywords: functional hyperemia, cerebral small vessel diseases, CADASIL, microcirculation, neurovascular coupling, potassium channel, prostaglandin E2, epidermal growth factor receptor
Citation: Rosehart AC, Longden TA, Weir N, Fontaine JT, Joutel A and Dabertrand F (2021) Prostaglandin E2 Dilates Intracerebral Arterioles When Applied to Capillaries: Implications for Small Vessel Diseases. Front. Aging Neurosci. 13:695965. doi: 10.3389/fnagi.2021.695965
Received: 15 April 2021; Accepted: 15 June 2021;
Published: 13 August 2021.
Edited by:
Anusha Mishra, Oregon Health and Science University, United StatesReviewed by:
Cam Ha Tran, University of Nevada, Reno, United StatesCopyright © 2021 Rosehart, Longden, Weir, Fontaine, Joutel and Dabertrand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrice Dabertrand, RmFicmljZS5EYWJlcnRyYW5kQENVQW5zY2h1dHouZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.