 Biyao Wang
Biyao Wang Minghao Huang
Minghao Huang Dehao Shang
Dehao Shang Xu Yan
Xu Yan Baohong Zhao
Baohong Zhao Xinwen Zhang
Xinwen Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 08 March 2021
Sec. Alzheimer's Disease and Related Dementias
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.650038
This article is part of the Research Topic Central Nervous System: From Aging to Repair and Regeneration View all 10 articles
Mitochondria are organelles responsible for bioenergetic metabolism, calcium homeostasis, and signal transmission essential for neurons due to their high energy consumption. Accumulating evidence has demonstrated that mitochondria play a key role in axon degeneration and regeneration under physiological and pathological conditions. Mitochondrial dysfunction occurs at an early stage of axon degeneration and involves oxidative stress, energy deficiency, imbalance of mitochondrial dynamics, defects in mitochondrial transport, and mitophagy dysregulation. The restoration of these defective mitochondria by enhancing mitochondrial transport, clearance of reactive oxidative species (ROS), and improving bioenergetic can greatly contribute to axon regeneration. In this paper, we focus on the biological behavior of axonal mitochondria in aging, injury (e.g., traumatic brain and spinal cord injury), and neurodegenerative diseases (Alzheimer's disease, AD; Parkinson's disease, PD; Amyotrophic lateral sclerosis, ALS) and consider the role of mitochondria in axon regeneration. We also compare the behavior of mitochondria in different diseases and outline novel therapeutic strategies for addressing abnormal mitochondrial biological behavior to promote axonal regeneration in neurological diseases and injuries.
Mitochondria are dynamic organelles playing a pivotal role in energy generation, signaling, and calcium homeostasis (Han et al., 2016). Axonal mitochondria exhibit a linearly interspersed distribution. Approximately 87% of mitochondria are stationary, and the number of motile mitochondria decreases from the proximal to distal axon (Cheng and Sheng, 2020). Mitochondria are transported to specific sites in axons where high energy is in demand, such as in growth cones and axonal branches (Morris and Hollenbeck, 1993; Tao et al., 2014). These distribution events are stabilized when presynaptic structures and mature neurons are formed. Moreover, mitochondria will rapidly redistribute in response to physiological or pathological stress in order to maintain energy homeostasis. The vigorous response of axonal mitochondria redistribution is predominantly conducted by the cooperation of microtubule-based transport and anchoring substrates, including anchors, adaptors, and motors (Sheng, 2014, 2017; Misgeld and Schwarz, 2017; Guedes-Dias and Holzbaur, 2019). Impairment of these substrates and process will result in the inhibition of axonal mitochondrial transport and ultimately induce local energy depletion and axon degeneration (Sheng and Cai, 2012).
Axon degeneration has been observed during normal neuronal aging, injury (e.g., traumatic brain injury, TBI; spinal cord injury, SCI; Maxwell, 2015; Wang et al., 2019b) and neurodegenerative diseases (e.g., Alzheimer's disease, AD; Parkinson's disease, PD; Amyotrophic lateral sclerosis, ALS; Guo et al., 2020). Axon degeneration usually occurs before the neuronal soma's death at the early stage of the diseases. In most conditions above, there are two different processes of axon degeneration. The distal axons won't connect to soma undergoing Wallerian degeneration while the proximal axons will die back toward soma (Adalbert and Coleman, 2013; Gerdts et al., 2016). Axon degeneration has an intimate association with mitochondrial dysfunction, including energy deficits, oxidative stress, disequilibrium of mitochondrial fission and fusion, impaired axonal mitochondrial transport, and aberrant mitophagy (Court and Coleman, 2012; Sheng and Cai, 2012; Geden and Deshmukh, 2016). Therefore, analysis of the mitochondrial behavior in axons may be considered a novel target for therapeutic strategies.
The intrinsic capacity and process of axon regeneration are different between the peripheral and central nervous systems (PNS and CNS, respectively), and the capacity of the former is superior to that of the latter (Fawcett and Verhaagen, 2018; Mahar and Cavalli, 2018). Axon regeneration in PNS initiates a vigorous regenerative response via the permissive Schwann cell environment and form a new growth cone. This process is supported by a regenerative program through expression of the Regeneration-associated gene (Attwell et al., 2018; Mahar and Cavalli, 2018). Nevertheless, axotomy at the central branch has a minimal regenerative response and the Regeneration-associated gene response also seems to make no difference on axon regeneration (Chandran et al., 2016; Cartoni et al., 2017). While in the CNS, the ability of axon regeneration is dependent on the maturity of the neurons. Moreover, the environment of the injured neuron is full of molecules and structures which may suppress axon regeneration in CNS (Fawcett and Verhaagen, 2018). CNS axons cut from embryos exhibits a potential to grow for long distance after transplanted into an adult CNS via expressing a series of molecules inducing axon growth (Reier et al., 1986; Lu et al., 2012). Nevertheless, axons from the adult CNS respond to axotomy with a minimal regenerative response and a poor change of gene expression despite being in a permissive environment. These diverse response to injury in PNS and CNS (mature and immature) form the concept of intrinsic regenerative capacity of axon regeneration (Fawcett and Verhaagen, 2018). Because this process consumes a large amount of energy and molecules or substrates during restoration, alteration of mitochondria behavior may represent one of the prominent intrinsic regenerative capacities in axon regeneration; however, the mechanisms underlying this process have just begun to be elucidated.
In this review, we summarize the normal mechanism of mitochondrial quality control in axons. We also describe the behavior of mitochondria in axon degeneration under different conditions (i.e., aging, nerve trauma, and neurodegenerative diseases) and their role in axon regeneration and the treatment methods derived from these effects.
Mitochondrial transport is indispensable for mitochondrial quality control. Anterograde transport contributes to the energy supply and neuronal survival, and retrograde transport helps with damaged mitochondrial clearance (Chen et al., 2016). Only a small proportion of axonal mitochondria (30–40%) is motile in vivo and in vitro; most mitochondria are stalled and do not move for long periods (Zheng et al., 2019). Mitochondrial transport depends on interactions between microtubules, microtubule motor proteins, adaptor complexes that bind the motor proteins to mitochondria, and their anchors.
Mitochondria are transported along the microtubule filament by the motor proteins kinesin and dynein to the plus and minus ends, respectively (Lin and Sheng, 2015). Kinesin-1 (KIF5) is the major motor protein involved in anterograde transport, the other member of kinesin family such as KIF1Ba and KLP are also participants (Zheng et al., 2019). Mitochondria interact with motor proteins via the adaptor complex formed by Miro, a GTPase located in the outer mitochondrial membrane (OMM), and Milton/TRAK (TRAK 1 and 2 in mammals). TRAK1 binds both the heavy chain of KIF5 and dynein; however, TRAK2 preferentially binds dynein (Smith and Gallo, 2018). Compared to the preference for dynein in dendrites, axonal mitochondria bind either kinesin or dynein via the Miro-TRAK1 complex.
The axon-specific mitochondrial outer membrane protein syntaphilin (SNPH) is a representative cytoskeletal tether (i.e., anchor), which opposes motility by increasing the force between the mitochondria and microtubules and inhibiting ATPase activity (Chen and Sheng, 2013). Microtubule-associated protein, such as myosin V or myosin VI (Pathak et al., 2010), and actin filament-based mechanisms may also regulate stalling and transport in axons (Henderson et al., 2017). Cytosolic calcium is another key regulator of mitochondrial transport. There are two main mechanisms for calcium resulting in the stalling of mitochondria (Zheng et al., 2019). For one thing, SNPH can bind kinesin I and sequester motor protein in the context of calcium signaling. For another, Miro has two EF calcium binding domains and Miro will directly bind KIF5 when binding of calcium (Liu and Hajnóczky, 2009), which will prevent motor activity. Therefore, increased intracellular calcium levels are sufficient to inhibit this process (Woolums et al., 2020).
A dynamic process in mitochondria (i.e., mitochondrial fission and fusion) continuously occurs to maintain a steady mitochondrial morphology and population (Yan et al., 2020b). Mitochondrial fission helps a damaged mitochondrion segregate from the healthy one while mitochondrial fusion facilitates the interaction and fusion of two mitochondria, which repairs each other, demonstrating that the equilibrium of fission and fusion acts a pivotal part in axonal mitochondria (Yu et al., 2016; Pickles et al., 2018). Mitochondrial fission is mediated by the GTPase dynamin-related protein 1 (Drp1), which is recruited to the OMM and forms a contractile ring around the mitochondrion, dividing one mitochondrion into two separate mitochondria (Otera et al., 2013). GTPase dynamin-related protein 1 (Drp1)-independent mitochondrial fission can also occur (Rival et al., 2011; Stavru et al., 2013; Roy et al., 2016). Mitochondrial fusion involves two separate processes, the fusion of the OMM, which is regulated by mitofusins (Mfns), and the fusion of the inner mitochondrial membrane that is mediated by optic atrophy 1. The fusion of the OMM is regulated by mitofusins 1 and 2 (Mfn1 and Mfn2). Both mitochondrial fission and fusion take place along the axon (Yan et al., 2020b).
Mitophagy plays a key role in relieving oxidative stress and preventing axonal and cytosolic defects and subsequent cell death (Wang et al., 2018). To maintain mitochondrial quality control in axons and cells, mitophagy degrade and recycle damaged mitochondria undergoing physiological or pathological processes, such as mitochondria depolarization and oxidative stress (Martinez-Vicente, 2017). The PINK1/Parkin pathway is a canonical mechanism of mitophagy. In addition to the PINK1/PARKIN pathway, other OMM proteins directly interact with LC3 and promote mitophagy in mammals, including FUNDC1, NIX/BNIP3L, and Bcl2-L-13 (Di Rita et al., 2018).
Aging is a normal physiological phenomenon, and glaucoma and many neurodegenerative diseases (e.g., Alzheimer's disease, Parkinson's disease) are related to aging. Mitochondrial behavior plays a key role in axon degeneration. First, aging has been shown to be associated with mitochondrial transport. Glaucoma is a chronic and progressive neurodegenerative disease. Degeneration of ganglion cells and their axons has been shown to be related to aging (Liu et al., 2018). In ganglion cell neurons, with age the cross-section axon area enlarges characterized by axoplasm disorganization and accumulation of hyperphosphorylated neurofilaments which is indicative of axonopathy (Cooper et al., 2016). Further, there are more mitochondria-free regions and decreased lengths of mitochondrial transport in degenerative axons (Mao et al., 2016). The changes in these mitochondria with age are related to changes in the expression of Mfn2. Among the healthy mice in the control group, the level of Mfn2 in retina was slightly elevated in aged mice (Nivison et al., 2017). Considering the function of Mfn2 in maintaining mitochondrial morphology and participating in mitochondrial transport (Misko et al., 2010), this increase indicates the demand of mitochondrial fusion and transport. The increase was more pronounced in retina of DBA mice (a murine glaucoma model), which explained the mitochondria in glaucoma neurons were elongated or fused. However, Mfn2 is either damaged or cannot be transported to optic nerve, resulting in a decrease in the level of this protein in optic nerve, indicating that Mfn2 protein is not transported down axons to function in mitochondrial transport and repair, which leads to disease progression. Moreover, given that phosphorylated Mfn2 is a receptor for Parkin on depolarized mitochondria, the amount of phosphorylated Mfn2 in young mice are also higher than the aged control indicating that the mitochondria transport is altered (Nivison et al., 2017).
Pathological morphology of mitochondria has been reported in degenerative axons with age. The average diameter of axonal mitochondria increases, with few or no cristae observed in the aging axonal mitochondria (Cao et al., 2017). This phenomenon is attributed to reduced optic atrophy 1 and Mfn2 expression (Rebelo et al., 2018) and even the absence of expression at the aging axon terminal. This morphological change is also relevant to neuronal apoptosis, P53-dependent neuronal cell death is associated with increased mitochondrial length mediated by reduced Drp1 and Parkin expression during aging (Wang D. B. et al., 2013).
Different phenomena are observed for mitophagy in axons in vivo and in vitro. In vitro, Sung et al. (2016) demonstrated that autophagosomes co-localized with mitochondria in axons when neurons were treated with drugs to induce mitochondrial damage. However, evidence from in vivo imaging suggests that either PINK1/Parkin or Parkin-dependent or mitophagy is indispensable for mitochondria turnover and axon integrity (Cao et al., 2017). Further research is needed to determine whether the mitochondria in axons need to undergo mitophagy or are transported back to the soma.
Finally, all of the described changes in mitochondrial behavior may affect bioenergetics by influencing oxidative phosphorylation and producing excessive reactive oxidative species (ROS). The molecular mechanisms regulating this process in aging include increases in the levels of proteins related to mitochondria and oxidative phosphorylation, more specifically, Complex I (Kline et al., 2019).
The relationship between aging and axon regeneration is complex and affected by multiple factors. Both endogenous (Geoffroy et al., 2017) and exogenous factors (Sutherland and Geoffroy, 2020) of neurons play an important role in the ability of axons to regenerate after damage and this process is declining with aging (Geoffroy et al., 2016). Although young neurons have a strong ability for axon growth, mature neurons can usually not regenerate after injury because mature CNS axons lose their ability to regenerate due to most mitochondria being stationary, which is associated with high SNPH expression (Lewis et al., 2016). Therefore, without enough local mitochondria, sufficient ATP cannot be provided, and the process of axon regeneration, including re-sealing cell membranes, rearranging cytoskeletal structures, and reforming active growth cones, will be hindered (Bradke et al., 2012). However, mitochondrial transport can be enhanced by stimulating the cAMP/Protein Kinase A (PKA) pathway to upregulate kinesin-1 expression to counteract its decrease during aging (Vagnoni and Bullock, 2018). Interestingly, the effects of cAMP or PKA on increasing mitochondrial transport were more pronounced in the aging group than in the younger group.
The internal components of the mitochondria also affect axonal regeneration by modifying the biological behavior of mitochondria and oxidizing the respiratory chain. In addition to acting as a transcription factor for axon regeneration in the nucleus, STAT3 can also reach the inner mitochondrial membrane following the activation of mitogen-activated protein kinase (MAPK), which improves axon regeneration by enhancing cell bioenergetics in the spinal cord (Luo et al., 2016) in mature mice with a reduced regenerative capacity. The mitochondrial signature phospholipid cardiolipin present in the inner leaflet of the inner mitochondrial membrane is essential for mitochondrial dynamics, mitochondrial biogenesis, and energy metabolism in the mitochondria (Mårtensson et al., 2017).
Traumatic brain injury (TBI) leads to the dynamic deformation of the parenchyma, resulting in shear and stretch injuries to axons, commonly known as diffuse axonal injury. Diffuse axonal injury is demonstrated to result from the mechanical deformation of the axonal cell membranes via calpain-mediated proteolysis of sidearms or phosphorylation, triggering neurofilament compaction, calcium changes, microtubules destabilization, and metabolic dysfunction (Barkhoudarian et al., 2016). Subsequently, axoplasmic transport mechanisms are defective, which leads to the accumulation of transport products that cause axonal swelling, secondary disconnection, and Wallerian degeneration (Frati et al., 2017).
Transmembrane sodium channels are damaged in TBI, leading to an abnormal influx of calcium into the axonal plasma (Maxwell, 2015). Calcium is also released from mitochondria and the axoplasmic reticulum (Springer et al., 2018), leading to axonal mitochondria dysfunction. Under Transmission electron microscopy, the axoplasm contains damaged or lucent mitochondria and a disorganized cytoskeleton within injured nerve fibers (Maxwell, 2015). The axonal swellings of TBI are characterized by the aggregation of membranous organelles, especially mitochondria, resulted from the injury and loss of axonal microtubules, which interact with dynein and kinesin (Maxwell, 2015). It is reported that the loss of axonal microtubules in TBI results in the lack of neuronal mitochondria delivery, failing to replace those damaged by the destroyed calcium homeostasis and to provide vesicular profiles (Maxwell et al., 1997; Tuck and Cavalli, 2010). Within an injured mouse optic nerve fiber, which is far away from the secondary axotomy site, accumulating evidence demonstrated the dieback or withdrawal of the proximal and distal terminal swellings (Staal et al., 2010; Wang et al., 2011). In the latter study, the proximal swellings contained intact mitochondria while the distal swellings exhibited defected mitochondria short of cristae, which appeared consistent with an ongoing degenerative process (Wang et al., 2011). In addition, the distal swelling exhibits three types of mitochondria, including mitochondria aggregating pyroantimonate crystals, those unremarkable or intact, and those containing electron lucent spaces in the mitochondrial matrix (Wang et al., 2011).
Damaged mitochondria fail to generate adequate ATP and cannot support the essential physiological and biochemical processes and disturb the transmembrane transporter function (Maxwell, 2015). Furthermore, oxidative stress in TBI results from the uncontrolled influx of Ca2+, leading to calcium accumulation in the mitochondria. The subsequent activation of caspases and calpains correlates with the initiation of apoptosis (Frati et al., 2017). Calpain-mediated spectrin proteolysis reaction product, which is reported to be correlated with damaged mitochondria, spread widely in the axoplasm at foci of neurofilament compaction (Büki et al., 1999), suggesting the defection of neurofilaments (Dewar et al., 2003). Moreover, the release of cytochrome c in the damaged axons triggers retrograde signaling by “apoptosis-inducing factor,” resulting in the programmed cell death and apoptosis of injured neurons (Büki et al., 2000).
Mitochondrial transport and bioenergetics are enhanced to facilitate axon regeneration following TBI (Misgeld et al., 2007). Within axon regeneration in TBI, it is beneficial to elevate the density of mitochondria while decreasing the density is disadvantageous. Mitochondria are transported into the injured zone in order to elevate the average mitochondria density. Regenerating axons have a higher density of mitochondria than non-regenerating axons in Caenorhabditis elegans (Han et al., 2016). Accumulating evidence in the SNPH knockout mice demonstrates that mitochondrial transport is strengthened to clear impaired mitochondria, replenish healthy mitochondria in injured axons, and ultimately reverse the energy deficits in axon regeneration (Zhou et al., 2016; Cheng and Sheng, 2020). In contrast, the Armadillo Repeat Containing X-Linked 1 protein, responsible for connecting the Miro adaptor protein with the mitochondrial membrane, is overexpressed, leading to the acceleration of optic nerve regeneration following TBI (Cartoni et al., 2016). Moreover, the dual leucine zipper kinase 1 microtubule-associated protein kinase pathway in C. elegans can promote adequate mitochondrial transport into the regeneration zone independent of Miro to increase the mitochondria density in injured axons (Han et al., 2016). Overall, axon regeneration following TBI can be achieved by increasing the density of mitochondria by enhancing the transport of healthy mitochondria, which provide sufficient energy for axonal regrowth.
The damage caused by SCI is mainly divided into two stages. The initial trauma is mainly caused by a contusion or direct compression. The secondary injury is caused by disruption of blood vessels, microcirculation failure, ion homeostasis disorder, excessive production of free radicals, and inflammation (Wang S. et al., 2019).
After acute SCI and the accompanying depolarization of neurons, mitochondrial permeability transition pores are opened by calcium influx, which impairs ATP synthesis, destroys the mitochondrial outer membrane, and releases ROS and pro-apoptotic proteins into the cytoplasm (Pivovarova and Andrews, 2010). In an SCI rat model, swollen and disrupted mitochondria with disorganized cristae can be observed in both pre-apoptotic neurons and astrocytes (Xu et al., 2017).
Immunofluorescence staining of post-mortem tissue from Thy1YFP+ transgenic mice showed that a significant increase in TOMM20 signal reflected the density of mitochondria in the axonal spheroids and endbulbs (Rajaee et al., 2020). The accumulation of mitochondria and tubulin polyglutamylation indicates the disruption of axonal transport, an important characteristic of axonal degeneration. Moreover, the failure of mitochondria to migrate to the remote ends of axons indicates abnormal mitochondrial axonal localization that contributes to axon retraction.
In the early stage of SCI, mitochondria fuse to restore the respiratory chain and inhibit mitochondrial apoptosis. In contrast, mitochondrial fission resulting from mitochondrial dysfunction further inhibits microtubule stabilization and axonal regeneration and is associated with apoptosis (Csordás et al., 2018). The dysfunction of mitochondrial fission and fusion caused by SCI is associated with the upregulation of Drp1, which reduces mitochondrial membrane potential, releases cytochrome C and caspase3, and induces neuronal apoptosis (Jia et al., 2016).
Within a few days to a few weeks after a person suffers axon damage, the damaged spinal cord begins to recover. To solve the functional defect caused by the separation of the white matter tracts between the rostral and caudal spinal cords, axonal regeneration is required to reconnect the caudal/rostral neurons and form new neural circuits to restore signal conduction (O'Shea et al., 2017). However, observations in mice showed that the reduced growth capacity of mature neurons and scar tissue caused by reactive astrocytes form a physical barrier, and the upregulation of axon growth-inhibitory factors inhibit axon regeneration (Yiu and He, 2006). However, intravital imaging of murine spinal cord showed that the number of axons increases significantly from 7 to 14 days after injury compared to 3 days post-injury, indicating that a certain number of injured axons undergo endogenous repair (Rajaee et al., 2020), demonstrating that while the regeneration of axons is limited after SCI injury, it can still be improved.
Given the important role of axonal mitochondria in growth cone migration, mitochondrial transport is required for axonal regeneration after SCI. Spinal cord injury (SCI) itself does not directly affect mitochondrial transport in axons; however, enhancing the transport of mitochondria after SCI is helpful for this process. In vitro data showed that fibroblast growth factor-13 co-localizes with mitochondria, and its increased activity and density in axons may, in part, explain why fibroblast growth factor-13 promotes axon regeneration in SCI models (Li et al., 2018).
Changing mitochondrial dynamics after SCI, specifically by promoting mitochondrial fusion and inhibiting its fission, positively affects axon regeneration. In an SCI model, Loureirin B significantly increased both bcl-2 and Mfn1 and doubled the size of the mitochondria, which promoted fusion and facilitated axon regeneration (Wang et al., 2019a). Considering the adverse effects of Drp1 upregulation after SCI injury, mitochondrial division inhibitor-1, an inhibitor of Drp1, promotes animal recovery and reduces apoptosis (Jia et al., 2016).
Once the peripheral nerve is injured, the axon is divided into two segments; the proximal axon segment and the distal segment. The distal axon undergoes Wallerian degeneration, while the proximal axon possibly regenerates via a growth cone, triggering axon extension (Neukomm et al., 2017).
In the injured peripheral neuronal axon of C. elegans and ric-7 (resistant to inhibitors of cholinesterase) mutants, the density of healthy mitochondria is decreased while the density of defected mitochondria is elevated, leading to energetic failure and oxidative stress (Rawson et al., 2014). Furthermore, mitochondrial transport is similarly inhibited in the Thy1-mitoCFP transgenic mouse (Thy1-MitoCFP mice expressing CFP fused to the human cytochrome c oxidase mitochondrial targeting sequence were from Jackson Laboratories; Misgeld et al., 2007).
In response to injury, multiple proteins are inhibited or activated during distal axon degeneration. Together with increased calcium influx, decreased nicotinamide mononucleotide adenyltransferase (NMNAT) and NAD1 activates SARM1 (Sterile Alpha and TIR Motif Containing 1), which are key initiating factors in Wallerian degeneration (Osterloh et al., 2012). Sterile Alpha and TIR Motif Containing 1 (SARM1) activation results in mitochondrial dysfunction, calpain-dependent defects in the cytoskeleton, and subsequent axon degeneration (Adalbert et al., 2012; Ma et al., 2013; Yang et al., 2015). Wallerian degeneration slow fusion protein (WldS), comprising full NMNAT1, has been shown to alleviate degeneration in Zebrafish and Drosophila (Coleman et al., 1998; Conforti et al., 2000). NMNAT1 has a pivotal role in maintaining axon integrity (O'Donnell et al., 2013; Wang Y. et al., 2015), the activity of which is greatly inhibited by defects in healthy mitochondria, decreases in mitochondrial transport, and ROS accumulation during axon degeneration (Yahata et al., 2009; Avery et al., 2012; Fang et al., 2012; O'Donnell et al., 2013). The dual leucine zipper kinase (DLK) is a mitogen activated protein kinase (MAP3K), and contributes to axon degeneration by reducing energy production. Dual leucine zipper kinase (DLK) is an axonal integrity sensor and interacts with NMNAT and activated SARM1 (Cavalli et al., 2005; Xiong et al., 2012; Yang et al., 2015; Gerdts et al., 2016); subsequently, SARM1 interacts with Axundead to consume NAD1 (Essuman et al., 2017; Neukomm et al., 2017). Decreased NAD1 production reduces energy generation and ATP levels, causing defects in Na+/Ca2+ exchangers and Ca2+ channels, and the loss of mitochondrial membrane potential (Gerdts et al., 2015; Loreto et al., 2015). Thus collectively, these proteins damage axon integrity.
In contrast to the central nerve, after peripheral nerves are injured, axons have a strong regenerative ability to restore function. At the proximal segment of peripheral nerve injury, Golgi-derived vesicles transport anterogradely to aggregate near the axon end to repair the ruptured membrane, while increasing intracellular Ca2+ levels activate kinases and phosphatases assemble microtubules to support the formation of new growth cones, and drive axon elongation (Girouard et al., 2018). This process is relevant to Schwann cells (Jessen et al., 2015; Nocera and Jacob, 2020) and intrinsic signaling events induced by injury, such as growth-associated protein-43 (GAP-43) (Chung et al., 2020).
Mitochondria are essential for axon regeneration. Increased mitochondrial density in C. elegans effectively promotes axon regeneration (Han et al., 2016). Reduced oxidative stress and enhanced axon regeneration are observed in a sciatic nerve crush injury model when mitochondria are injected into injured nerves (Kuo et al., 2017). Thus, increasing mitochondrial density at the site of injured axons can be achieved, mainly by increasing mitochondrial transport in axons, and increasing mitochondrial fission. Miro overexpression enhances mitochondrial transport in proximal segments, whereas axon regeneration is significantly inhibited in individuals where Miro is suppressed (Han et al., 2016). In addition, by imaging axonal mitochondrial transport in vivo, and when the intercostal nerve is damaged, mitochondrial anterograde transport is increased (Misgeld et al., 2007), which may be associated with tyrosinated tubulin, and upregulated molecular motors (Yang et al., 2021). An in vivo sciatic nerve compression study indicates that enhanced mitochondrial transport, via SNPH knockout in mice, accelerates axon regeneration in the peripheral nervous system (Zhou et al., 2016). The early fission of mitochondria after axon damage is also associated with axon regeneration. For example, in mice, sciatic nerve transection generates significantly increased mitochondrial division in damaged axons (Kiryu-Seo et al., 2016). This is further confirmed by the inhibition of axon regeneration in individuals with mutations in the mitochondrial fusion gene, OPA1 (Knowlton et al., 2017).
Mitochondrial regeneration after peripheral nerve injury highlights the importance of axon transport, to not only provide structural components of cells such as lipid vesicles and microtubules, but to meet the high energy requirements of regenerating axons (Prior et al., 2017; Mahar and Cavalli, 2018). In an in vitro sciatic nerve injury study, Hwang et al. found that after sciatic nerve injury, axon CDK5 levels increased, they were translocated into the mitochondria and phosphorylated mitochondrial STAT3, thereby regulating mitochondrial activity (Hwang and Namgung, 2021). STAT3 phosphorylation affects microtubule assembly and energy production in axons (Selvaraj et al., 2012). When translocated to the mitochondria, STAT3 interacts with electron transport chain proteins, regulating ROS production and the release of cytochrome C (Szczepanek et al., 2011), and contributing to ATP energy supply to promote axon regeneration.
Mitochondria in the injured axon have pivotal roles in axon degeneration and regeneration. Therefore, mitochondria may be specific therapeutic targets for inhibiting axon degeneration and enhancing axon regeneration. Oxidative stress caused by dysfunctional mitochondria results in a variety of adverse effects. Thus, a potential therapeutic strategy would be to decrease this stress. In a TBI animal model, cyclosporin, an anti-oxidative drug, can decrease cytochrome c release from mitochondria and inhibit Ca2+ influx into mitochondria, ultimately alleviating mitochondrial dysfunction and axon damage (Kelsen et al., 2019). Additionally, ROS-mediated axonal degeneration in TBI caused by the aberrant axonal calcium homeostasis is reported to be alleviated via the application of calcium channel blocker (McAllister, 2011; Namjoshi et al., 2014). Moreover, Metformin can stabilize the microstructure in injured axons of SCI by activating the PI3K/Akt signaling pathway. Furthermore, activation of Akt/Nrf2 signaling could be a potential approach for axon regeneration treatment by inhibiting excessive oxidative stress and restoring mitochondrial function (Wang et al., 2020).
Enhancing mitochondrial transport and recruitment aids axon regeneration in injured axons and facilitates the removal of damaged mitochondria and replenishment of healthy ones to provide adequate ATP (Kaasik, 2016; Sheng, 2017; Smith and Gallo, 2018). Inhibiting the microtubule-severing protein Fidgetin (Matamoros et al., 2019) or non-muscle myosin II (Hur et al., 2011) promotes axon regeneration after SCI by facilitating the reorganization of microtubule and actin cytoskeletal proteins. Moreover, Loureirin B promotes mitochondrial fusion and suppresses ER stress by activating the Akt/GSK-3β pathway, facilitating axon regeneration (Wang et al., 2019a). In summary, combined approaches targeting mitochondria may provide a novel therapeutic strategy for alleviating axon degeneration and enhancing axon regeneration in TBI and SCI.
Alzheimer's disease is an age-related neurodegenerative disease characterized by progressive cognitive impairment, mobility disorder, and memory loss. Hallmarks of AD include aberrant amyloid-β (Aβ) metabolism and neurofibrillary tangles of hyperphosphorylated Tau protein, which are particularly significant in axon degeneration (Wang et al., 2017). β-site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) is the major neuronal Aβ-secretase for Aβ generation. Axon degeneration in AD involves a complicated mechanism, including glucose metabolism defects, mitochondrial dysfunction, aberrant calcium homeostasis, oxidative stress, and imbalances in energy homeostasis (Cieri et al., 2018; Mata, 2018; Swerdlow, 2018; Albensi, 2019).
Accumulating evidence has demonstrated that aggregation of pathological Tau and Aβ impairs mitochondria transport (anterograde and retrograde) in axons (Wang Z. X. et al., 2015; Sadleir et al., 2016; Cai and Tammineni, 2017; Zhang et al., 2018). One of the first researches in this area was carried out in primary neurons from Tg2576 mice with mutant human APP protein (Calkins et al., 2011). Compared to wild-type neurons, primary neurons from Tg2576 mice exhibited inhibited anterograde mitochondrial transport as well as promoted mitochondrial fission and inhibited mitochondrial fusion (Calkins et al., 2011). Mitochondria transport in axons requires a variety of substrates and molecular support, whose dysfunction can lead to negative impact on mitochondria transport. The defected transport results in the reduction of mitochondria density at the distal axons and the depletion of axonal NMAT protein 2 (NMAT2), which is continually supplemented in axons via fast axonal transport (Ljungberg et al., 2012; Ali et al., 2016). Following axotomy, the transport of NMNAT2 toward axons is inhibited, meanwhile, it has a rapid decline in axons before Wallerian degeneration occurs (Gilley and Coleman, 2010; Gerdts et al., 2016). Since the damaged mitochondria inhibited anterograde and promoted retrograde transport, the quantity of SNPH cargo vesicles is significant elevated in AD axons, similar to ALS. The SNPH-mediated response in axonal mitochondrial transport of AD-related cortical neurons from mutant human APP-expressing transgenic mice is summarized in the ALS section (Lin et al., 2017a,b; Cheng and Sheng, 2020). Ultimately, the impairment of mitochondrial axonal transport in AD inhibits OXPHOS complex activity, causing significant energy deficiency (Spires-Jones and Hyman, 2014).
These two AD hallmarks contribute to mitochondrial transport pathology in axons. Defects in axon transport can be initiated by soluble low molecular weight Aβ species in the plasma membrane in mice (Zhang et al., 2018). Moreover, inhibited axon transport does not promote mitochondrial dysfunction or ATP depletion at once (Zhang et al., 2018), suggesting that reconstruction of mitochondrial axon transport may be advantageous at early AD stages. The restoration of KIF5A, an isoform of kinesin-1, abrogates the impairment of mitochondria axonal transport by Aβ, particularly anterograde transport in 5 × FAD mice (Wang et al., 2019b). Additionally, it is reported that axon degeneration has an intimate correlation with mitochondrial dysfunction and mPTP (Barrientos et al., 2011). The deficiency of cyclophilin D (CypD), a mPTP regulator, inhibited Aβ-mediated permeability transition and mitochondrial swelling, and alleviated oxidative stress both in mice model and brain samples of AD. Notably, knockout of CypD gene appears to suppress the mPTP, which may improve cognitive and synaptic function in mouse model (Du et al., 2008, 2011). Moreover, the defection of mitochondrial axon transport triggered by Aβ is dependent on the activation of mPTP, which is mediated by CypD. Knockout of CypD inhibit the induction of the p38/MAPK signaling pathway mediated by Aβ, restore the dysfunction of axonal mitochondria and synapses (Guo et al., 2013). Furthermore, the defective dynein-snapin coupling mediated by Aβ seriously inhibits retrograde transport, resulting in the aggregation of amphisomes at axonal terminals, which can greatly contribute to mitophagy (Tammineni et al., 2017). Therefore, it seems that there is an intimate relationship between the impaired axonal transport and mitophagy, together consist of the complicated mechanism of mitochondrial dysfunction in axons of AD.
Overexpression or hyperphosphorylation of Tau not only causes defects in mitochondrial transport but also disrupts mitochondrial distribution and localization in mouse and cellular AD models (Cheng and Bai, 2018). It is reported that in tauopathies and AD, the integrity of microtubules is damaged via hyperphosphorylation and binding with protein Tau (Misko et al., 2010). Moreover, mitochondrial transport motor proteins and Tau (Ser199, Ser202, Thr205) are phosphorylated, which can lead to the dysfunction of mitochondrial transport in AD, mediated by a serine/threonine protein kinase, Glycogen synthase kinase 3 (Shahpasand et al., 2012). The aberrant aggregation of Tau is also demonstrated to induce microtubule instability, aggravating the impaired transport.
Amyloid-β (Aβ) oligomers and neurofibrillary tangles also elicit mitochondria dysfunction and oxidative stress (Butterfield and Boyd-Kimball, 2018; Mata, 2018), which may, in turn, impair axonal transport. It is demonstrated that oxidative stress is one of the typical event correlated with axon degeneration at the early stage of AD (Alavi Naini and Soussi-Yanicostas, 2015). Oxidative stress triggered by breaking the equilibrium between antioxidants and pro-oxidants may induce the excessive hyperphosphorylation of Tau. The aggregation of Tau is reported to inhibit microtubule transport, resulting in the decrease of peroxisome, which increase the incidence of oxidative stress (Stamer et al., 2002). Oxidative stress may in turn impair axon transport, which will aggravate tau phosphorylation in animal models and neuronal cultures of AD (Melov et al., 2007; Su et al., 2010). Interestingly, antioxidative treatment conducted in 3xTg-AD mice is found to alleviate oxidative stress, Tau hyperphosphorylation (Clausen et al., 2012). Therefore, the interconnection between oxidative stress and tau hyperphosphorylation seems to act a key role in axon degeneration of AD. Accumulating damaged mechanisms summarized above lead to the damage of axon homeostasis and finally, axonal degeneration in AD, which seems to provide some novel mitochondria-targeted therapeutic strategies in future.
Parkinson's disease is a common late-onset neurodegenerative disease characterized by the degenerative death of dopamine (DA) neurons in the substantia nigra region and the formation of Lewy bodies, cytoplasmic inclusion bodies that contain α-synuclein (α-Syn) (Gao et al., 2018). Mitochondrial dysfunction is an early event in PD. Indeed, some familial PD cases are caused by mutations in genes encoding mitochondria proteins (e.g., Pink, Parkin, and DJ-1) (Cheng et al., 2010).
The degeneration of DA neurons in PD originates from the distal axons of fragile neurons which is long and highly branched (Surmeier et al., 2017). The increased size of axonal arborization which resulted in elevated mitochondrial bioenergetics is considered of the reason for the vulnerability for DA neurons in the substantia nigra (Pacelli et al., 2015; Giguere et al., 2019). Highly branched axons needs increased energy to deal with protein delivery, oxidative stress and mitochondrial dysfunction (Bhaskar et al., 2020).
Dysfunction of mitochondrial biogenesis plays a key role in the pathogenesis of PD. For instance, the inactivation of Parkin, which plays an important role in the pathological changes of familial PD, suppresses PCG-1α, a key regulator of mitochondrial biogenesis, ultimately causing the loss of DA neurons (Lee et al., 2017). In contrast, neurodegeneration caused by α-Syn overexpression in zebrafish can be partially relieved by PCG-1α upregulation (O'Donnell et al., 2014). Given that the dopaminergic axons loss precedes the cell death in both PD patient and PD model constructed by exposure to rotenone, a complex I inhibitor linked to PD (Tagliaferro and Burke, 2016), mitochondrial homeostasis in distal axon may participate in initial changes resulting in later neuron death. The limited early increase in mitochondrial density induced by rotenone in distal axons is due to upregulated distal axonal mitochondrial biogenesis (Van Laar et al., 2018), which is likely a compensatory process to mitochondrial disruption in PD.
Analysis of the post-mortem brains of PD patients showed that the distance between the synaptic terminal and mitochondria in DA neurons increased with a decreased volume and number of mitochondria, suggesting that mitochondrial synaptic availability and biomass contribute to degenerative neurons (Mallach et al., 2019). Mutations in Parkin impair Complex I activity, mitochondrial fusion, and the plasticity of synapses (Goldberg et al., 2003). Another example of how dysfunction mitochondria act in PD by disrupting synaptic plasticity comes from DJ-1, which is located in mitochondria and plays a role in synaptic transmission (Wang et al., 2008; Yan et al., 2020b).
Aberrant axonal transport contributes to neurodegeneration in PD (Lamberts et al., 2015). PD can be induced in mice by treatment with 6-hydroxydopamine (6-OHDA), which leads to axonal degeneration in dopaminergic neurons by inducing dysfunction of mitochondrial transport and microtubule disruption (Fuku, 2016). Significant alterations in mitochondria motility were observed in neurons induced from pluripotent stem cells (iPSCs) derived from an LRRK2 mutant PD patient (Cooper et al., 2012). Prots et al. (2018) discovered that this alteration was associated with α-Syn oligomerization resulting from an increased α-Syn dosage, which resulted in reduced Miro and kinesin light chain-1 levels in axons and increased levels of Tau in neuronal soma.
PINK1 and Parkin are two proteins that can selectively remove damaged mitochondria. Mutations in these proteins can lead to PD and link mitophagy with this disease. Knockout of Atg5 or Atg7 causes loss of autophagy, resulting in swelling at the axon terminals and eventually cell death (Maday, 2016). Thus, autophagy plays an indispensable role in axon homeostasis. The accumulation of α-Syn and LRKK2 results from the loss of autophagy (Friedman et al., 2012), providing further proof that defects in autophagy cause the accumulation of abnormal organelles in axons (e.g., mitochondria), leading to oxidative damage and apoptotic cascades. In addition to PINK1/Parkin, LRRK2 is involved in initiating mitophagy by forming a complex with Miro. In pathologic LRRK2G2019S neurons, this function is disrupted but can be rescued by reducing Miro levels (Hsieh et al., 2016). While mitophagy has a protective effect on neurons, determining whether increased autophagy levels are the cause or effect of PD requires further study.
Amyotrophic lateral sclerosis (ALS) is the most common adult motor neuron disease with characteristic progressive motor neuron degeneration, leading to muscle atrophy, paralysis, and ultimately death (Taylor et al., 2016). Approximately 90% of cases are sporadic (sALS), while the remaining 10% are familial (fALS) (Turner et al., 2017). fALS is largely associated with gene mutations, particularly in TAR DNA Binding Protein 43 (TDP-43) and Cu/Zn superoxide dismutase (SOD1) (Chia et al., 2018). Accumulating evidence suggests that axon degeneration in fALS is significantly correlated with mitochondrial damage, impairment of mitochondria transport, and autophagy-lysosomal dysfunction. Within an early stage in SOD1G93A mice, distal axonal transport is reported to be impaired, meanwhile, the expression of kinesin and dynein is also inhibited (Warita et al., 1999). In both SOD1 patients (Boillée et al., 2006; De Vos et al., 2007) and SOD1G93A mice (Fischer et al., 2004; Damiano et al., 2006; Tallon et al., 2016), damaged mitochondria accumulate at distal sites, causing ATP deficiencies and aberrant calcium homeostasis at neuromuscular junctions, ultimately resulting in distal axon degeneration.
Enhancing mitochondria motility can remove the dysfunctional mitochondria from distal synapses meanwhile deliver the healthy ones to distal axons. Nevertheless, in motor neurons from hSOD1G93A mice, axonal mitochondria transport is damaged, and the defective mitochondria aberrantly accumulate in distal axons (De Vos et al., 2007; Magrané and Manfredi, 2009; Bilsland et al., 2010; Cozzolino et al., 2013). In order to address this defection, a research designed to cross hSOD1G93A and SNPH−/− mice, aiming to examine whether enhancing mitochondria transport could make effect on the hSOD1G93A mice (Zhu and Sheng, 2011). Although the total mitochondrial transport is increased in the crossed mice, it seems to have no differences in the disease course. Moreover, this study reveals that deficits in mitochondrial transport appears to be insufficient to cause axon degeneration in the fALS-linked motor neurons (Zhu and Sheng, 2011), supported by an in vitro study in mutant hSOD1 models (Marinkovic et al., 2012). In the motor neuron of fALS-linked SOD1G93A mice and the axons of AD-related cortical neurons from mutant human APP-expressing transgenic mice, impaired mitochondria are removed via the bulk release of SNPH cargo vesicles, which are mitochondria-derived and Parkin-independent, promoting mitochondria transport (Lin et al., 2017a). Recently, a study demonstrated that as the disease progresses in fALS-linked SOD1G93A and AD-linked mice, the quantity of SNPH cargo vesicles is also altered (Cheng and Sheng, 2020). It is interesting that at asymptomatic stages of fALS and AD, the number of SNPH cargo vesicles is greatly elevated in the axons of motor neurons (Lin et al., 2017a). Nevertheless, at the onset and late stages of the disease, SNPH is significantly depleted via mitochondrial impairment in these axons (Lin et al., 2017a). Furthermore, the study revealed that in response to these chronic pathological stresses, the quantity of SNPH cargo vesicles changes in distal axons. The defective mitochondria anchored by SNPH are transported to the soma for recovery or degeneration (Cheng and Sheng, 2020). The SNPH-mediated pathway acts as a protective mechanism during the early asymptomatic stages of fALS and AD and is independent of and acts before PINK1/Parkin mediated mitophagy (Lin et al., 2017b). Thus, it is regarded as a significant hallmark of diagnosis and an early target for treatment. However, simply increasing mitochondrial transport by turning off SNPH-mediated anchoring (Lin et al., 2017a) or eliminating SNPH (Zhu and Sheng, 2011) fails to replenish ATP deficiency in hSOD1G93A mutant mice. The autophagy-lysosomal function is also defective in fALS-linked mice at an early stage of disease both ex vivo and in vivo, preventing the elimination of damaged mitochondria from distal axons (Xie et al., 2015). Thus, the combination of enhanced mitochondria transport and increased defective mitochondria elimination may be an effective therapeutic strategy for fALS.
A study has reported that anterograde and retrograde transport of mitochondria in primary motor neurons axons are greatly defected via overexpression of wild-type TDP-43. Interestingly, the study demonstrated that the loss of TDP-43 also inhibits axonal transport of mitochondria, revealing that damaged mitochondria axonal transport may involve various pathways mediated by TDP-43 (Wang W. et al., 2013). Moreover, a recent study showed that specific protein synthesis is inhibited (i.e., transcripts involved in mitochondrial energy metabolism, cytoskeletal components, and translational machinery) within the axons of motor neurons in TDP-43-knockout mice (Briese et al., 2020). Thus, axonal transport and mitochondria function are significantly defected in TDP-43-knockout mice, leading to the impairment of axon growth as well as revealing that depletion of TDP-43 may act a predominant role in axon degeneration of ALS (Briese et al., 2020).
In neurodegenerative diseases, the degeneration of axons often occurs at early disease stages. This degeneration is often accompanied by the accumulation of damaged or fragmented mitochondria. Therefore, maintaining mitochondrial dynamics in axons is a common treatment strategy.
Amyloid-β (Aβ) toxicity and Tau dysfunction in AD, often accompanied by impaired axonal mitochondrial transport, jointly lead to disease progression. Wang et al. (2019b) demonstrated that mitochondrial transport defects can be corrected in Aβ-treated neurons by protecting KIF5A, suggesting that maintaining normal mitochondrial axon transport is a valuable treatment strategy in AD. There are many targeted treatments for reducing the mitochondrial content caused by defective mitochondrial trafficking, as represented by increased retrograde transport rates in axons (Schwab et al., 2017). In addition, glycolytic defects in oligodendrocytes induce axon degeneration in both AD mice and patients via the Drp1-hexokinase 1-NLRP3 (NLR family pyrin domain containing 3) signaling axis, which is considered to be a therapeutic target (Yan et al., 2020a; Zhang et al., 2020). Mdivi-1, Dynasore, and P110 are promising agents that maintain axonal mitochondrial turnover by inhibiting dynamin to prevent the production of fragmented mitochondria, maintain normal mitochondria morphology, and restore ATP levels in a PD model (Elfawy and Das, 2019). Additionally, deep brain stimulation (DBS) is a very effective intervention to treat patients with advanced PD and its mechanism is related to the restoration of the number and volume of mitochondria (Mallach et al., 2019). The selective peptide inhibitor, P110 inhibits the Drp1-Fis1 interaction and improves the structure and function of mitochondria in G93A SOD1 mutant mice, indicating an attractive target for ALS patients (Joshi et al., 2018). Nicotinamide mononucleotide adenylyl transferase1 (Nmnat1), an enzyme involved in nicotinamide adenine dinucleotide (NAD+) synthesis, has a protective effect on mitochondrial dynamics and morphology following vincristine-induced axon degeneration (Berbusse et al., 2016).
Reactive oxidative species (ROS) production caused by mitochondrial dysfunction leads to oxidative stress, an important factor in axonal degeneration. Therefore, many treatment strategies focus on mitigating local ROS production using novel antioxidants that directly target mitochondria. Some mitochondria-targeted compounds (e.g., latrepirdine, methylene blue, triterpenoids, and the mitochondrial-targeted coenzyme Q10 derivative MitoQ) have been extensively evaluated in in vivo and in vitro AD and PD models (Elfawy and Das, 2019). In addition, a significant reduction in ROS levels results from treatment with the selective peptide inhibitor P110 and improves motor capacity and survival rates in G93A SOD1 mutant mice (Joshi et al., 2018).
Axon regeneration is an attractive target in neurodegenerative diseases. Because growth cone formation requires a large ATP supply, targeted mitochondrial therapy is key to axonal regeneration. While mature CNS neurons almost lose their intrinsic capacity for axon regeneration, upregulation of axon cytoskeleton proteins was observed in the 6-OHDA-hemiparkinsonian rat model, indicating a high plasticity and regeneration potential in adult animals (Lessner et al., 2010).
The G2019S mutation in the conserved serine kinase MAPK kinase domain contained in LRRK2 is the cause of familial PD (Cookson, 2010). Therefore, reduced neurite outgrowth and increased growth cone size were observed in neurons of LRRK2 G2019S mutant mice and the level of F-actin in growth cone also increases (Parisiadou et al., 2009), suggesting that growth cone formation, an essential step for axon regeneration is seriously impacted in PD. However, in the 6-OHDA-induced PD animal model, there are many compensatory responses in early-stage PD, including the upregulation of some proteins related to axon regeneration. Dihydropyrimidinase-related protein 2 (DPYSL2), which is important for axon outgrowth and regeneration (Sun and Cavalli, 2010), first decreased and then gradually recovered in this model. Moreover, axon growth and mitochondrial transport-related proteins (e.g., dynein, dynamin, and myosin) were also overexpressed (Kuter et al., 2016). Thus, a potential therapy to promote axon regeneration would be to enhance the expression of mitochondrial transport-related proteins. A new promising therapeutic method involving exogenous mitochondrial transplant effectively attenuates the progression of neurologic disorders, including experimental Parkinson's disease (Shi et al., 2017). Supportive mitochondrial-targeting therapy is a promising approach for future therapeutic intervention in the CNS to promote axon regeneration. However, it should be noted that there are currently very limited ways to treat neurodegenerative diseases by promoting axon regeneration because of the diminishing intrinsic axonal regeneration in mature CNS neurons compared to PNS neurons, and there are no optimal treatments for these neurodegenerative diseases. Thus, researchers have focused on stopping the degeneration of these nerves. With the deepening understanding of the treatment of mitochondria to prevent neural degeneration, we believe that axonal mitochondria regeneration will become a better therapeutic target for treating neurodegenerative diseases.
Axon degeneration and regeneration are essential parts of CNS neuronal death and restoration of CNS neurons, respectively, in aging, injury, and neurodegenerative diseases. However, the decline in the intrinsic regrowth capacity of mature CNS axons leads to a failure to regenerate after neuronal impairment (Fawcett and Verhaagen, 2018). Accumulating evidence demonstrates that mitochondrial dysfunction is intimately correlated with the initiation of axon degeneration and inhibition of axon regeneration (Qian and Zhou, 2020). Despite the mechanism underlying is still elusive, it suggests that mitochondrial quality control is an important intrinsic capacity of axon regeneration. For axon degeneration, mitochondrial dysfunction not only causes energy deficits and oxidative stress but also comprises mitochondrial dynamics, axonal transport, and mitophagy (Court and Coleman, 2012; Vasic et al., 2019). In contrast, restoring and enhancing these processes during axon regeneration increases the capacity of intrinsic regrowth (Figure 1). It comes to a consensus that axon regeneration requires adequate energy production and mitochondrial transport both in aging and in diseases (Zhou et al., 2016; Sheng, 2017; Zheng et al., 2019). Defected mitochondria can be replaced via mitochondria injection, which is demonstrated to decrease oxidative stress and trigger axon regeneration both in vitro and in vivo (Kuo et al., 2017). Thus, further study of axonal mitochondrial behavior under these conditions is important, not only for the alleviation and treatment of axon degeneration but also for identifying potential targets to enhance axon regeneration.
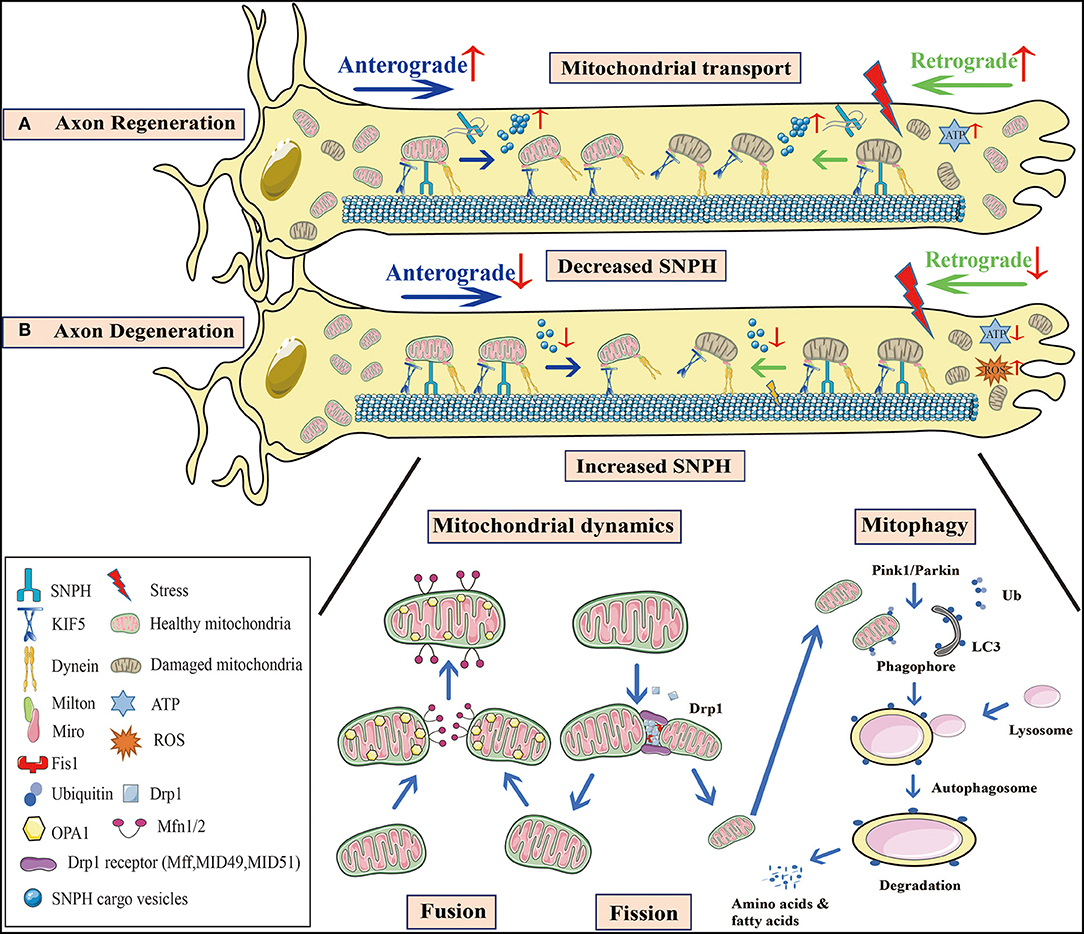
Figure 1. Mitochondrial behavior in axon degeneration and regeneration. (A) Axon regeneration: Since this process requires considerable energy, mitochondrial density at distal axons is elevated to provide sufficient ATP. Not only is anterograde transport of healthy mitochondria promoted, but so too is retrograde transport of damaged mitochondria, to increase mitochondrial density at regenerative zones. SNPH knockout in TBI models, or the bulk release of SNPH cargo vesicles at AD and fALS early stages, both enhance axonal mitochondrial transport. (B) Axon degeneration: Mitochondrial dysfunction and oxidative stress occur in injured axons. When axonal mitochondrial transport is seriously impaired, damaged mitochondria aggregate at distal zones and the healthy ones fail to transport from the proximal zones to replenish ATP insufficiency. A decrease in SNPH cargo vesicle release at later stages of AD and fALS, as well as increased SNPH expression in SCI models inhibit mitochondrial transport. Moreover, axonal microtubule loss contributes to impaired axonal transport in TBI. Mitochondrial dynamics are also defective during axon degeneration. Due to increased Drp1-mediated fission in SCI, mitochondrial fragmentation is aggravated, causing mitochondrial damage. These organelle fuse with healthy mitochondria, however, most undergo mitophagy. In PD, Pink1-Parkin dependent mitophagy is defective, facilitating damaged mitochondrial accumulation and apoptotic cascades. These processes are interconnected in response to stress, and collectively lead to axon degeneration. SNPH, syntaphilin; TBI, traumatic brain injury; AD, Alzheimer's disease; fALS, familial amyotrophic lateral sclerosis; SCI, spinal cord injury; PD, Parkinson's disease.
It is generally accepted that diminished intrinsic regeneration is a major barrier for axon regeneration in mature CNS neurons. Analysis of intrinsic axon degeneration and regeneration pathways and signaling networks can bring new insights for potential mitochondria-targeted therapeutic strategies. For example, a novel therapeutic strategy for enhancing the intrinsic capacity of axon regeneration may include genetic reprogramming through epigenome modifications and the transcriptome (Qian and Zhou, 2020). Mitochondrial damage-associated molecular patterns (mtDAMPs) released by injured axonal mitochondria can activate Schwann cell processes mediated by formylpeptide receptor 2 (FPR2) and toll-like receptor 9 (TLR9), which have a pivotal role in axon regeneration and cell migration (Korimová et al., 2018). Therefore, regulation of these mitochondria-targeted genetic reprogramming can be considered to be a promising molecular strategy for activating axon regeneration. At the cellular level, enhancing mitochondrial transport could greatly contribute to axon regeneration by removing defective mitochondria and replenishing healthy mitochondria at injured axon sites (Zheng et al., 2019). In addition, glial cells (astrocytes, oligodendrocytes, and microglia) contribute to mitochondrial dysfunction during axon growth inhibition and regeneration failure in the adult CNS (Brosius Lutz and Barres, 2014; Cregg et al., 2014; Silver and Silver, 2014). Astrocytes inhibit axonal mitochondrial energy metabolism by increasing nitric oxide production, glutamate levels, and intracellular calcium influx during Multiple Sclerosis (MS) (Correale and Farez, 2015). Similarly, astrocytes can release mitochondria containing particles, and transport them to defective axons following stroke (Babenko et al., 2015; Hayakawa et al., 2016). In vitro astrocyte-to-neuron mitochondrial delivery and in vivo astrocyte-derived mitochondrial transport improves neuronal survival, plasticity, and behavior outcomes (Hayakawa et al., 2016). Reactive oxygen and nitrogen species generated by microglia directly damage neurons by inhibiting cytochrome C oxidase and mitochondrial respiratory chain complex IV, causing axonal mitochondrial dysfunction in MS (Nikić et al., 2011). Furthermore, oligodendrocytes and astrocytes provide axonal mitochondria with pyruvate or lactate, which are imported into the mitochondria for energy metabolism during MS and AD (Fünfschilling et al., 2012; Correale et al., 2019; Zhang et al., 2020). Clearance of damaged mitochondria not only relies on mitophagy at the injury sites but also depends on mitochondrial retrograde transport, which involves SNPH and dual leucine zipper kinase 1 (Han et al., 2016; Cheng and Sheng, 2020). However, many molecules and substrates engaged in mitochondrial transport or dynamics need to be clarified. Moreover, to strengthen the intrinsic capacity for regrowth, improving the extrinsic inhibiting environment could make a critical difference in axon regeneration. Drugs targeted at oxidative stress via the clearance of ROS (Geden and Deshmukh, 2016) or directed at the restoration of calcium homeostasis by inhibiting calcium channels and calcium-activated enzymes (Mu et al., 2015) will aid axon regeneration. Furthermore, remodeling the neuronal cytoskeleton may be a novel mechanism to alleviate the extrinsic inhibitory cues (Qian and Zhou, 2020). In conclusion, combined approaches that target mitochondria, which increase the intrinsic capacity and decrease the extrinsic inhibiting environment, may provide an effective therapeutic strategy to enhance axon regeneration in aging, injury, and neurodegenerative diseases.
BW, MH, BZ, and XZ made substantial contributions to the conception and design of the study. BW, MH, DS, XY, BZ, and XZ participated in drafting the article. BW and MH created the figure. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (no. 81700977 to XY, no. 82071151 to BZ, and no. 81500858 to XZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adalbert, R., and Coleman, M. P. (2013). Review: axon pathology in age-related neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 39, 90–108. doi: 10.1111/j.1365-2990.2012.01308.x
Adalbert, R., Morreale, G., Paizs, M., Conforti, L., Walker, S. A., Roderick, H. L., et al. (2012). Intra-axonal calcium changes after axotomy in wild-type and slow Wallerian degeneration axons. Neuroscience 225, 44–54. doi: 10.1016/j.neuroscience.2012.08.056
Alavi Naini, S. M., and Soussi-Yanicostas, N. (2015). Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell. Longev. 2015:151979. doi: 10.1155/2015/151979
Albensi, B. C. (2019). Dysfunction of mitochondria: implications for Alzheimer's disease. Int. Rev. Neurobiol. 145, 13–27. doi: 10.1016/bs.irn.2019.03.001
Ali, Y. O., Allen, H. M., Yu, L., Li-Kroeger, D., Bakhshizadehmahmoudi, D., Hatcher, A., et al. (2016). NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 14:e1002472. doi: 10.1371/journal.pbio.1002472
Attwell, C. L., van Zwieten, M., Verhaagen, J., and Mason, M. R. J. (2018). The dorsal column lesion model of spinal cord injury and its use in deciphering the neuron-intrinsic injury response. Dev. Neurobiol. 78, 926–951. doi: 10.1002/dneu.22601
Avery, M. A., Rooney, T. M., Pandya, J. D., Wishart, T. M., Gillingwater, T. H., Geddes, J. W., et al. (2012). WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr. Biol. 22, 596–600. doi: 10.1016/j.cub.2012.02.043
Babenko, V. A., Silachev, D. N., Zorova, L. D., Pevzner, I. B., Khutornenko, A. A., Plotnikov, E. Y., et al. (2015). Improving the post-stroke therapeutic potency of mesenchymal multipotent stromal cells by cocultivation with cortical neurons: the role of crosstalk between cells. Stem Cells Transl. Med. 4, 1011–1020. doi: 10.5966/sctm.2015-0010
Barkhoudarian, G., Hovda, D. A., and Giza, C. C. (2016). The molecular pathophysiology of concussive brain injury - an update. Phys. Med. Rehabil. Clin. N. Am. 27, 373–393. doi: 10.1016/j.pmr.2016.01.003
Barrientos, S. A., Martinez, N. W., Yoo, S., Jara, J. S., Zamorano, S., Hetz, C., et al. (2011). Axonal degeneration is mediated by the mitochondrial permeability transition pore. J. Neurosci. 31, 966–978. doi: 10.1523/jneurosci.4065-10.2011
Berbusse, G. W., Woods, L. C., Vohra, B. P. S., and Naylor, K. (2016). Mitochondrial dynamics decrease prior to axon degeneration induced by vincristine and are partially rescued by overexpressed cytNmnat1. Front. Cell. Neurosci. 10:179. doi: 10.3389/fncel.2016.00179
Bhaskar, S., Gowda, J., Prasanna, J., and Kumar, A. (2020). Does altering proteasomal activity and trafficking reduce the arborization mediated specific vulnerability of SNpc dopaminergic neurons of Parkinson's disease? Med. Hypotheses 143:110062. doi: 10.1016/j.mehy.2020.110062
Bilsland, L. G., Sahai, E., Kelly, G., Golding, M., Greensmith, L., and Schiavo, G. (2010). Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 20523–20528. doi: 10.1073/pnas.1006869107
Boillée, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Bradke, F., Fawcett, J. W., and Spira, M. E. (2012). Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat. Rev. Neurosci. 13, 183–193. doi: 10.1038/nrn3176
Briese, M., Saal-Bauernschubert, L., Lüningschrör, P., Moradi, M., Dombert, B., Surrey, V., et al. (2020). Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 8:116. doi: 10.1186/s40478-020-00987-6
Brosius Lutz, A., and Barres, B. A. (2014). Contrasting the glial response to axon injury in the central and peripheral nervous systems. Dev. Cell 28, 7–17. doi: 10.1016/j.devcel.2013.12.002
Büki, A., Okonkwo, D. O., Wang, K. K., and Povlishock, J. T. (2000). Cytochrome c release and caspase activation in traumatic axonal injury. J. Neurosci. 20, 2825–2834. doi: 10.1523/jneurosci.20-08-02825.2000
Büki, A., Siman, R., Trojanowski, J. Q., and Povlishock, J. T. (1999). The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J. Neuropathol. Exp. Neurol. 58, 365–375. doi: 10.1097/00005072-199904000-00007
Butterfield, D. A., and Boyd-Kimball, D. (2018). Oxidative stress, amyloid-β peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer's disease. J. Alzheimers. Dis. 62, 1345–1367. doi: 10.3233/jad-170543
Cai, Q., and Tammineni, P. (2017). Mitochondrial aspects of synaptic dysfunction in Alzheimer's disease. J. Alzheimers. Dis. 57, 1087–1103. doi: 10.3233/jad-160726
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., and Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 20, 4515–4529. doi: 10.1093/hmg/ddr381
Cao, X., Wang, H., Wang, Z., Wang, Q., Zhang, S., Deng, Y., et al. (2017). In vivo imaging reveals mitophagy independence in the maintenance of axonal mitochondria during normal aging. Aging Cell 16, 1180–1190. doi: 10.1111/acel.12654
Cartoni, R., Norsworthy, M. W., Bei, F., Wang, C., Li, S., Zhang, Y., et al. (2016). The mammalian-specific protein Armcx1 regulates mitochondrial transport during axon regeneration. Neuron 92, 1294–1307. doi: 10.1016/j.neuron.2016.10.060
Cartoni, R., Norsworthy, M. W., Bei, F., Wang, C., Li, S., Zhang, Y., et al. (2017). The mammalian-specific protein Armcx1 regulates mitochondrial transport during axon regeneration. Neuron 94:689. doi: 10.1016/j.neuron.2017.04.028
Cavalli, V., Kujala, P., Klumperman, J., and Goldstein, L. S. (2005). Sunday driver links axonal transport to damage signaling. J. Cell Biol. 168, 775–787. doi: 10.1083/jcb.200410136
Chandran, V., Coppola, G., Nawabi, H., Omura, T., Versano, R., Huebner, E. A., et al. (2016). A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 89, 956–970. doi: 10.1016/j.neuron.2016.01.034
Chen, M., Li, Y., Yang, M., Chen, X., Chen, Y., Yang, F., et al. (2016). A new method for quantifying mitochondrial axonal transport. Protein Cell 7, 804–819. doi: 10.1007/s13238-016-0268-3
Chen, Y., and Sheng, Z.-H. (2013). Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 202, 351–364. doi: 10.1083/jcb.201302040
Cheng, A., Hou, Y., and Mattson, M. P. (2010). Mitochondria and neuroplasticity. ASN Neuro. 2:e00045. doi: 10.1042/an20100019
Cheng, X. T., and Sheng, Z. H. (2020). Developmental regulation of microtubule-based trafficking and anchoring of axonal mitochondria in health and diseases. Dev. Neurobiol. doi: 10.1002/dneu.22748. [Epub ahead of print].
Cheng, Y., and Bai, F. (2018). The association of tau with mitochondrial dysfunction in Alzheimer's disease. Front. Neurosci. 12:163. doi: 10.3389/fnins.2018.00163
Chia, R., Chiò, A., and Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. doi: 10.1016/s1474-4422(17)30401-5
Chung, D., Shum, A., and Caraveo, G. (2020). GAP-43 and BASP1 in axon regeneration: implications for the treatment of neurodegenerative diseases. Front. Cell Dev. Biol. 8:567537. doi: 10.3389/fcell.2020.567537
Cieri, D., Vicario, M., Vallese, F., D'Orsi, B., Berto, P., Grinzato, A., et al. (2018). Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca(2+) handling. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3247–3256. doi: 10.1016/j.bbadis.2018.07.011
Clausen, A., Xu, X., Bi, X., and Baudry, M. (2012). Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer's disease: protection against and interruption of progression of amyloid and tau pathology and cognitive decline. J. Alzheimers. Dis. 30, 183–208. doi: 10.3233/jad-2012-111298
Coleman, M. P., Conforti, L., Buckmaster, E. A., Tarlton, A., Ewing, R. M., Brown, M. C., et al. (1998). An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc. Natl. Acad. Sci. U.S.A. 95, 9985–9990. doi: 10.1073/pnas.95.17.9985
Conforti, L., Tarlton, A., Mack, T. G., Mi, W., Buckmaster, E. A., Wagner, D., et al. (2000). A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc. Natl. Acad. Sci. U.S.A. 97, 11377–11382. doi: 10.1073/pnas.97.21.11377
Cookson, M. R. (2010). The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat. Rev. Neurosci. 11, 791–797. doi: 10.1038/nrn2935
Cooper, M. L., Crish, S. D., Inman, D. M., Horner, P. J., and Calkins, D. J. (2016). Early astrocyte redistribution in the optic nerve precedes axonopathy in the DBA/2J mouse model of glaucoma. Exp. Eye Res. 150, 22–33. doi: 10.1016/j.exer.2015.11.016
Cooper, O., Seo, H., Andrabi, S., Guardia-Laguarta, C., Graziotto, J., Sundberg, M., et al. (2012). Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson's disease. Sci. Transl. Med. 4:141ra190. doi: 10.1126/scitranslmed.3003985
Correale, J., and Farez, M. F. (2015). The role of astrocytes in multiple sclerosis progression. Front. Neurol. 6:180. doi: 10.3389/fneur.2015.00180
Correale, J., Marrodan, M., and Ysrraelit, M. C. (2019). Mechanisms of neurodegeneration and axonal dysfunction in progressive multiple sclerosis. Biomedicines 7:14. doi: 10.3390/biomedicines7010014
Court, F. A., and Coleman, M. P. (2012). Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 35, 364–372. doi: 10.1016/j.tins.2012.04.001
Cozzolino, M., Ferri, A., Valle, C., and Carrì, M. T. (2013). Mitochondria and ALS: implications from novel genes and pathways. Mol. Cell. Neurosci. 55, 44–49. doi: 10.1016/j.mcn.2012.06.001
Cregg, J. M., DePaul, M. A., Filous, A. R., Lang, B. T., Tran, A., and Silver, J. (2014). Functional regeneration beyond the glial scar. Exp. Neurol. 253, 197–207. doi: 10.1016/j.expneurol.2013.12.024
Csordás, G., Weaver, D., and Hajnóczky, G. (2018). Endoplasmic reticulum–mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 28, 523–540. doi: 10.1016/j.tcb.2018.02.009
Damiano, M., Starkov, A. A., Petri, S., Kipiani, K., Kiaei, M., Mattiazzi, M., et al. (2006). Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 96, 1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x
De Vos, K. J., Chapman, A. L., Tennant, M. E., Manser, C., Tudor, E. L., Lau, K. F., et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728. doi: 10.1093/hmg/ddm226
Dewar, D., Underhill, S. M., and Goldberg, M. P. (2003). Oligodendrocytes and ischemic brain injury. J. Cereb. Blood Flow Metab. 23, 263–274. doi: 10.1097/01.wcb.0000053472.41007.f9
Di Rita, A., Peschiaroli, A., D′Acunzo, P., Strobbe, D., Hu, Z., Gruber, J., et al. (2018). HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat. Commun. 9:3755. doi: 10.1038/s41467-018-05722-3
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Du, H., Guo, L., Zhang, W., Rydzewska, M., and Yan, S. (2011). Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 32, 398–406. doi: 10.1016/j.neurobiolaging.2009.03.003
Elfawy, H. A., and Das, B. (2019). Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: etiologies and therapeutic strategies. Life Sci. 218, 165–184. doi: 10.1016/j.lfs.2018.12.029
Essuman, K., Summers, D. W., Sasaki, Y., Mao, X., DiAntonio, A., and Milbrandt, J. (2017). The SARM1 Toll/Interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334.e1335–1343.e1335. doi: 10.1016/j.neuron.2017.02.022
Fang, Y., Soares, L., Teng, X., Geary, M., and Bonini, N. M. (2012). A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr. Biol. 22, 590–595. doi: 10.1016/j.cub.2012.01.065
Fawcett, J. W., and Verhaagen, J. (2018). Intrinsic determinants of axon regeneration. Dev. Neurobiol. 78, 890–897. doi: 10.1002/dneu.22637
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Frati, A., Cerretani, D., Fiaschi, A. I., Frati, P., Gatto, V., La Russa, R., et al. (2017). Diffuse axonal injury and oxidative stress: a comprehensive review. Int. J. Mol. Sci. 18:2600. doi: 10.3390/ijms18122600
Friedman, L. G., Lachenmayer, M. L., Wang, J., He, L., Poulose, S. M., Komatsu, M., et al. (2012). Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J. Neurosci. 32, 7585–7593. doi: 10.1523/JNEUROSCI.5809-11.2012
Fuku, K. (2016). Reactive oxygen species induce neurite degeneration before induction of cell death. J. Clin. Biochem. Nutr. 59, 155–159. doi: 10.3164/jcbn.16-34
Fünfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J., et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. doi: 10.1038/nature11007
Gao, Y., Wilson, G. R., Stephenson, S. E. M., Bozaoglu, K., Farrer, M. J., and Lockhart, P. J. (2018). The emerging role of Rab GTPases in the pathogenesis of Parkinson's disease. Mov. Disord. 33, 196–207. doi: 10.1002/mds.27270
Geden, M. J., and Deshmukh, M. (2016). Axon degeneration: context defines distinct pathways. Curr. Opin. Neurobiol. 39, 108–115. doi: 10.1016/j.conb.2016.05.002
Geoffroy, C. G., Hilton, B. J., Tetzlaff, W., and Zheng, B. (2016). Evidence for an age-dependent decline in axon regeneration in the adult mammalian central nervous system. Cell Rep. 15, 238–246. doi: 10.1016/j.celrep.2016.03.028
Geoffroy, C. G., Meves, J. M., and Zheng, B. (2017). The age factor in axonal repair after spinal cord injury: a focus on neuron-intrinsic mechanisms. Neurosci. Lett. 652, 41–49. doi: 10.1016/j.neulet.2016.11.003
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457. doi: 10.1126/science.1258366
Gerdts, J., Summers, D. W., Milbrandt, J., and DiAntonio, A. (2016). Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89, 449–460. doi: 10.1016/j.neuron.2015.12.023
Giguere, N., Delignat-Lavaud, B., Herborg, F., Voisin, A., Li, Y., Jacquemet, V., et al. (2019). Increased vulnerability of nigral dopamine neurons after expansion of their axonal arborization size through D2 dopamine receptor conditional knockout. PLoS Genet. 15:e1008352. doi: 10.1371/journal.pgen.1008352
Gilley, J., and Coleman, M. P. (2010). Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8:e1000300. doi: 10.1371/journal.pbio.1000300
Girouard, M. P., Bueno, M., Julian, V., Drake, S., Byrne, A. B., and Fournier, A. E. (2018). The molecular interplay between axon degeneration and regeneration. Dev. Neurobiol. 78, 978–990. doi: 10.1002/dneu.22627
Goldberg, M. S., Fleming, S. M., Palacino, J. J., Cepeda, C., Lam, H. A., Bhatnagar, A., et al. (2003). Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 278, 43628–43635. doi: 10.1074/jbc.M308947200
Guedes-Dias, P., and Holzbaur, E. L. F. (2019). Axonal transport: driving synaptic function. Science 366:eaaw9997. doi: 10.1126/science.aaw9997
Guo, L., Du, H., Yan, S., Wu, X., McKhann, G. M., Chen, J. X., et al. (2013). Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer's neurons. PLoS ONE 8:e54914. doi: 10.1371/journal.pone.0054914
Guo, W., Stoklund Dittlau, K., and Van Den Bosch, L. (2020). Axonal transport defects and neurodegeneration: molecular mechanisms and therapeutic implications. Semin. Cell Dev. Biol. 99, 133–150. doi: 10.1016/j.semcdb.2019.07.010
Han, S. M., Baig, H. S., and Hammarlund, M. (2016). Mitochondria localize to injured axons to support regeneration. Neuron 92, 1308–1323. doi: 10.1016/j.neuron.2016.11.025
Hayakawa, K., Esposito, E., Wang, X., Terasaki, Y., Liu, Y., Xing, C., et al. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. doi: 10.1038/nature18928
Henderson, C. A., Gomez, C. G., Novak, S. M., Mi-Mi, L., and Gregorio, C. C. (2017). Overview of the muscle cytoskeleton. Compr. Physiol. 7, 891–944. doi: 10.1002/cphy.c160033
Hsieh, C. H., Shaltouki, A., Gonzalez, A. E., Bettencourt da Cruz, A., Burbulla, L. F., St Lawrence, E., et al. (2016). Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson's disease. Cell Stem Cell 19, 709–724. doi: 10.1016/j.stem.2016.08.002
Hur, E. M., Yang, I. H., Kim, D. H., Byun, J., Saijilafu Xu, W. L., et al. (2011). Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc. Natl. Acad. Sci. U.S.A. 108, 5057–5062. doi: 10.1073/pnas.1011258108
Hwang, J., and Namgung, U. (2021). Phosphorylation of STAT3 by axonal Cdk5 promotes axonal regeneration by modulating mitochondrial activity. Exp. Neurol. 335:113511. doi: 10.1016/j.expneurol.2020.113511
Jessen, K. R., Mirsky, R., and Lloyd, A. C. (2015). Schwann cells: development and role in nerve repair. Cold Spring Harb. Perspect. Biol. 7:a020487. doi: 10.1101/cshperspect.a020487
Jia, Z.-Q., Li, G., Zhang, Z.-Y., Li, H.-T., Wang, J.-Q., Fan, Z.-K., et al. (2016). Time representation of mitochondrial morphology and function after acute spinal cord injury. Neural Regen. Res. 11, 137–143. doi: 10.4103/1673-5374.175061
Joshi, A. U., Saw, N. L., Vogel, H., Cunnigham, A. D., Shamloo, M., and Mochly-Rosen, D. (2018). Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 10:e8166. doi: 10.15252/emmm.201708166
Kaasik, A. (2016). Mitochondrial mobility and neuronal recovery. N. Engl. J. Med. 375, 1295–1296. doi: 10.1056/NEJMcibr1607955
Kelsen, J., Karlsson, M., Hansson, M. J., Yang, Z., Fischer, W., Hugerth, M., et al. (2019). Copenhagen head injury ciclosporin study: a phase IIa safety, pharmacokinetics, and biomarker study of ciclosporin in severe traumatic brain injury patients. J. Neurotrauma 36, 3253–3263. doi: 10.1089/neu.2018.6369
Kiryu-Seo, S., Tamada, H., Kato, Y., Yasuda, K., Ishihara, N., Nomura, M., et al. (2016). Mitochondrial fission is an acute and adaptive response in injured motor neurons. Sci. Rep. 6:28331. doi: 10.1038/srep28331
Kline, R. A., Dissanayake, K. N., Hurtado, M. L., Martínez, N. W., Ahl, A., Mole, A. J., et al. (2019). Altered mitochondrial bioenergetics are responsible for the delay in Wallerian degeneration observed in neonatal mice. Neurobiol. Dis. 130:104496. doi: 10.1016/j.nbd.2019.104496
Knowlton, W. M., Hubert, T., Wu, Z., Chisholm, A. D., and Jin, Y. (2017). A select subset of electron transport chain genes associated with optic atrophy link mitochondria to axon regeneration in Caenorhabditis elegans. Front. Neurosci. 11:263. doi: 10.3389/fnins.2017.00263
Korimová, A., Klusáková, I., Hradilová-SvíŽenská, I., Kohoutková, M., Joukal, M., and Dubový, P. (2018). Mitochondrial damage-associated molecular patterns of injured axons induce outgrowth of schwann cell processes. Front. Cell. Neurosci. 12:457. doi: 10.3389/fncel.2018.00457
Kuo, C. C., Su, H. L., Chang, T. L., Chiang, C. Y., Sheu, M. L., Cheng, F. C., et al. (2017). Prevention of axonal degeneration by perineurium injection of mitochondria in a sciatic nerve crush injury model. Neurosurgery 80, 475–488. doi: 10.1093/neuros/nyw090
Kuter, K., Kratochwil, M., Marx, S.-H., Hartwig, S., Lehr, S., Sugawa, M. D., et al. (2016). Native DIGE proteomic analysis of mitochondria from substantia nigra and striatum during neuronal degeneration and its compensation in an animal model of early Parkinson's disease. Arch. Physiol. Biochem. 122, 238–256. doi: 10.1080/13813455.2016.1197948
Lamberts, J. T., Hildebrandt, E. N., and Brundin, P. (2015). Spreading of α-synuclein in the face of axonal transport deficits in Parkinson's disease: a speculative synthesis. Neurobiol. Dis. 77, 276–283. doi: 10.1016/j.nbd.2014.07.002
Lee, Y., Stevens, D. A., Kang, S.-U., Jiang, H., Lee, Y.-I., Ko, H. S., et al. (2017). PINK1 primes Parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep. 18, 918–932. doi: 10.1016/j.celrep.2016.12.090
Lessner, G., Schmitt, O., Haas, S. J.-P., Mikkat, S., Kreutzer, M., Wree, A., et al. (2010). Differential proteome of the striatum from hemiparkinsonian rats displays vivid structural remodeling processes. J. Proteome Res. 2010, 9, 4671–4687. doi: 10.1021/pr100389u
Lewis, T. L., Turi, G. F., Kwon, S.-K., Losonczy, A., and Polleux, F. (2016). Progressive decrease of mitochondrial motility during maturation of cortical axons in vitro and in vivo. Curr. Biol. 26, 2602–2608. doi: 10.1016/j.cub.2016.07.064
Li, J., Wang, Q., Wang, H., Wu, Y., Yin, J., Chen, J., et al. (2018). Lentivirus mediating FGF13 enhances axon regeneration after spinal cord injury by stabilizing microtubule and improving mitochondrial function. J. Neurotrauma 35, 548–559. doi: 10.1089/neu.2017.5205
Lin, M.-Y., and Sheng, Z.-H. (2015). Regulation of mitochondrial transport in neurons. Exp. Cell Res. 334, 35–44. doi: 10.1016/j.yexcr.2015.01.004
Lin, M. Y., Cheng, X. T., Tammineni, P., Xie, Y., Zhou, B., Cai, Q., et al. (2017a). Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94, 595.e596–610.e596. doi: 10.1016/j.neuron.2017.04.004
Lin, M. Y., Cheng, X. T., Xie, Y., Cai, Q., and Sheng, Z. H. (2017b). Removing dysfunctional mitochondria from axons independent of mitophagy under pathophysiological conditions. Autophagy 13, 1792–1794. doi: 10.1080/15548627.2017.1356552
Liu, B., McNally, S., Kilpatrick, J. I., Jarvis, S. P., and O'Brien, C. J. (2018). Aging and ocular tissue stiffness in glaucoma. Surv. Ophthalmol. 63, 56–74. doi: 10.1016/j.survophthal.2017.06.007
Liu, X., and Hajnóczky, G. (2009). Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int. J. Biochem. Cell Biol. 41, 1972–1976. doi: 10.1016/j.biocel.2009.05.013
Ljungberg, M. C., Ali, Y. O., Zhu, J., Wu, C. S., Oka, K., Zhai, R. G., et al. (2012). CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum. Mol. Genet. 21, 251–267. doi: 10.1093/hmg/ddr492
Loreto, A., Di Stefano, M., Gering, M., and Conforti, L. (2015). Wallerian degeneration is executed by an NMN-SARM1-dependent late Ca(2+) influx but only modestly influenced by mitochondria. Cell Rep. 13, 2539–2552. doi: 10.1016/j.celrep.2015.11.032
Lu, P., Wang, Y., Graham, L., McHale, K., Gao, M., Wu, D., et al. (2012). Long-distance growth and connectivity of neural stem cells after severe spinal cord injury. Cell 150, 1264–1273. doi: 10.1016/j.cell.2012.08.020
Luo, X., Ribeiro, M., Bray, E. R., Lee, D. H., Yungher, B. J., Mehta, S. T., et al. (2016). Enhanced transcriptional activity and mitochondrial localization of STAT3 co-induce axon regrowth in the adult central nervous system. Cell Rep. 15, 398–410. doi: 10.1016/j.celrep.2016.03.029
Mårtensson, C. U., Doan, K. N., and Becker, T. (2017). Effects of lipids on mitochondrial functions. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1862, 102–113. doi: 10.1016/j.bbalip.2016.06.015
Ma, M., Ferguson, T. A., Schoch, K. M., Li, J., Qian, Y., Shofer, F. S., et al. (2013). Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 56, 34–46. doi: 10.1016/j.nbd.2013.03.009
Maday, S. (2016). Mechanisms of neuronal homeostasis: autophagy in the axon. Brain Res. 1649(Pt B), 143–150. doi: 10.1016/j.brainres.2016.03.047
Magrané, J., and Manfredi, G. (2009). Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid. Redox Signal. 11, 1615–1626. doi: 10.1089/ars.2009.2604
Mahar, M., and Cavalli, V. (2018). Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 19, 323–337. doi: 10.1038/s41583-018-0001-8
Mallach, A., Weinert, M., Arthur, J., Gveric, D., Tierney, T. S., and Alavian, K. N. (2019). Post mortem examination of Parkinson's disease brains suggests decline in mitochondrial biomass, reversed by deep brain stimulation of subthalamic nucleus. FASEB J. 33, 6957–6961. doi: 10.1096/fj.201802628R
Mao, X. R., Kaufman, D. M., and Crowder, C. M. (2016). Nicotinamide mononucleotide adenylyltransferase promotes hypoxic survival by activating the mitochondrial unfolded protein response. Cell Death Dis. 7:e2113. doi: 10.1038/cddis.2016.5
Marinkovic, P., Reuter, M. S., Brill, M. S., Godinho, L., Kerschensteiner, M., and Misgeld, T. (2012). Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 109, 4296–4301. doi: 10.1073/pnas.1200658109
Martinez-Vicente, M. (2017). Neuronal mitophagy in neurodegenerative diseases. Front. Mol. Neurosci. 10:64. doi: 10.3389/fnmol.2017.00064
Mata, A. M. (2018). Functional interplay between plasma membrane Ca(2+)-ATPase, amyloid β-peptide and tau. Neurosci. Lett. 663, 55–59. doi: 10.1016/j.neulet.2017.08.004
Matamoros, A. J., Tom, V. J., Wu, D., Rao, Y., Sharp, D. J., and Baas, P. W. (2019). Knockdown of fidgetin improves regeneration of injured axons by a microtubule-based mechanism. J. Neurosci. 39, 2011–2024. doi: 10.1523/JNEUROSCI.1888-18.2018
Maxwell, W. L. (2015). “Development of concepts in the pathology of traumatic axonal and traumatic brain injury,” in Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects, ed F. H. Kobeissy (Boca Raton, FL: CRC Press/Taylor and Francis).
Maxwell, W. L., Povlishock, J. T., and Graham, D. L. (1997). A mechanistic analysis of nondisruptive axonal injury: a review. J. Neurotrauma 14, 419–440. doi: 10.1089/neu.1997.14.419
McAllister, T. W. (2011). Neurobiological consequences of traumatic brain injury. Dialogues Clin. Neurosci. 13, 287–300. doi: 10.31887/DCNS.2011.13.2/tmcallister
Melov, S., Adlard, P. A., Morten, K., Johnson, F., Golden, T. R., Hinerfeld, D., et al. (2007). Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2:e536. doi: 10.1371/journal.pone.0000536
Misgeld, T., Kerschensteiner, M., Bareyre, F. M., Burgess, R. W., and Lichtman, J. W. (2007). Imaging axonal transport of mitochondria in vivo. Nat. Methods 4, 559–561. doi: 10.1038/nmeth1055
Misgeld, T., and Schwarz, T. L. (2017). Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666. doi: 10.1016/j.neuron.2017.09.055
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J., and Baloh, R. H. (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 30, 4232–4240. doi: 10.1523/jneurosci.6248-09.2010
Morris, R. L., and Hollenbeck, P. J. (1993). The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 104 (Pt 3), 917–927.
Mu, J., Song, Y., Zhang, J., Lin, W., and Dong, H. (2015). Calcium signaling is implicated in the diffuse axonal injury of brain stem. Int. J. Clin. Exp. Pathol. 8, 4388–4397.
Namjoshi, D. R., Cheng, W. H., McInnes, K. A., Martens, K. M., Carr, M., Wilkinson, A., et al. (2014). Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol. Neurodegener. 9:55. doi: 10.1186/1750-1326-9-55
Neukomm, L. J., Burdett, T. C., Seeds, A. M., Hampel, S., Coutinho-Budd, J. C., Farley, J. E., et al. (2017). Axon death pathways converge on axundead to promote functional and structural axon disassembly. Neuron 95, 78.e75–91.e75. doi: 10.1016/j.neuron.2017.06.031
Nikić, I., Merkler, D., Sorbara, C., Brinkoetter, M., Kreutzfeldt, M., Bareyre, F. M., et al. (2011). A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 17, 495–499. doi: 10.1038/nm.2324
Nivison, M. P., Ericson, N. G., Green, V. M., Bielas, J. H., Campbell, J. S., and Horner, P. J. (2017). Age-related accumulation of phosphorylated mitofusin 2 protein in retinal ganglion cells correlates with glaucoma progression. Exp. Neurol. 296, 49–61. doi: 10.1016/j.expneurol.2017.07.001
Nocera, G., and Jacob, C. (2020). Mechanisms of Schwann cell plasticity involved in peripheral nerve repair after injury. Cell. Mol. Life Sci. 77, 3977–3989. doi: 10.1007/s00018-020-03516-9
O'Donnell, K. C., Lulla, A., Stahl, M. C., Wheat, N. D., Bronstein, J. M., and Sagasti, A. (2014). Axon degeneration and PGC-1α-mediated protection in a zebrafish model of alpha-synuclein toxicity. Dis. Model. Mech. 7, 571–582. doi: 10.1242/dmm.013185
O'Donnell, K. C., Vargas, M. E., and Sagasti, A. (2013). WldS and PGC-1α regulate mitochondrial transport and oxidation state after axonal injury. J. Neurosci. 33, 14778–14790. doi: 10.1523/jneurosci.1331-13.2013
O'Shea, T. M., Burda, J. E., and Sofroniew, M. V. (2017). Cell biology of spinal cord injury and repair. J. Clin. Invest. 127, 3259–3270. doi: 10.1172/JCI90608
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Otera, H., Ishihara, N., and Mihara, K. (2013). New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 1833, 1256–1268. doi: 10.1016/j.bbamcr.2013.02.002
Pacelli, C., Giguère, N., Bourque, M.-J., Lévesque, M., Slack, R. S., and Trudeau, L.-É. (2015). Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 25, 2349–2360. doi: 10.1016/j.cub.2015.07.050
Parisiadou, L., Xie, C., Cho, H. J., Lin, X., Gu, X.-L., Long, C.-X., et al. (2009). Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 29, 13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009
Pathak, D., Sepp, K. J., and Hollenbeck, P. J. (2010). Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J. Neurosci. 30, 8984–8992. doi: 10.1523/JNEUROSCI.1621-10.2010
Pickles, S., Vigie, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x
Prior, R., Van Helleputte, L., Benoy, V., and Van Den Bosch, L. (2017). Defective axonal transport: a common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. 105, 300–320. doi: 10.1016/j.nbd.2017.02.009
Prots, I., Grosch, J., Brazdis, R. M., Simmnacher, K., Veber, V., Havlicek, S., et al. (2018). α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. U.S.A. 115, 7813–7818. doi: 10.1073/pnas.1713129115
Qian, C., and Zhou, F. Q. (2020). Updates and challenges of axon regeneration in the mammalian central nervous system. J. Mol. Cell Biol. 12, 798–806. doi: 10.1093/jmcb/mjaa026
Rajaee, A., Geisen, M. E., Sellers, A. K., and Stirling, D. P. (2020). Repeat intravital imaging of the murine spinal cord reveals degenerative and reparative responses of spinal axons in real-time following a contusive SCI. Exp. Neurol. 327:113258. doi: 10.1016/j.expneurol.2020.113258
Rawson, R. L., Yam, L., Weimer, R. M., Bend, E. G., Hartwieg, E., Horvitz, H. R., et al. (2014). Axons degenerate in the absence of mitochondria in C. elegans. Curr. Biol. 24, 760–765. doi: 10.1016/j.cub.2014.02.025
Rebelo, P., Giacomello, M., and Scorrano, L. (2018). Shipping calpastatin to the rescue: prevention of neuromuscular degeneration through Mitofusin 2. Cell Metab. 28, 536–538. doi: 10.1016/j.cmet.2018.09.017
Reier, P. J., Bregman, B. S., and Wujek, J. R. (1986). Intraspinal transplantation of embryonic spinal cord tissue in neonatal and adult rats. J. Comp. Neurol. 247, 275–296. doi: 10.1002/cne.902470302
Rival, T., Macchi, M., Arnauné-Pelloquin, L., Poidevin, M., Maillet, F., Richard, F., et al. (2011). Inner-membrane proteins PMI/TMEM11 regulate mitochondrial morphogenesis independently of the DRP1/MFN fission/fusion pathways. EMBO Rep. 12, 223–230. doi: 10.1038/embor.2010.214
Roy, M., Itoh, K., Iijima, M., and Sesaki, H. (2016). Parkin suppresses Drp1-independent mitochondrial division. Biochem. Biophys. Res. Commun. 475, 283–288. doi: 10.1016/j.bbrc.2016.05.038
Sadleir, K. R., Kandalepas, P. C., Buggia-Prévot, V., Nicholson, D. A., Thinakaran, G., and Vassar, R. (2016). Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer's disease. Acta Neuropathol. 132, 235–256. doi: 10.1007/s00401-016-1558-9
Schwab, A. J., Sison, S. L., Meade, M. R., Broniowska, K. A., Corbett, J. A., and Ebert, A. D. (2017). Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-derived dopaminergic neurons. Stem Cell Rep. 9, 1839–1852. doi: 10.1016/j.stemcr.2017.10.010
Selvaraj, B. T., Frank, N., Bender, F. L. P., Asan, E., and Sendtner, M. (2012). Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J. Cell Biol. 199, 437–451. doi: 10.1083/jcb.201203109
Shahpasand, K., Uemura, I., Saito, T., Asano, T., Hata, K., Shibata, K., et al. (2012). Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J. Neurosci. 32, 2430–2441. doi: 10.1523/jneurosci.5927-11.2012
Sheng, Z. H. (2014). Mitochondrial trafficking and anchoring in neurons: new insight and implications. J. Cell Biol. 204, 1087–1098. doi: 10.1083/jcb.201312123
Sheng, Z. H. (2017). the interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 27, 403–416. doi: 10.1016/j.tcb.2017.01.005
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Shi, X., Zhao, M., Fu, C., and Fu, A. (2017). Intravenous administration of mitochondria for treating experimental Parkinson's disease. Mitochondrion 34, 91–100. doi: 10.1016/j.mito.2017.02.005
Silver, D. J., and Silver, J. (2014). Contributions of chondroitin sulfate proteoglycans to neurodevelopment, injury, and cancer. Curr. Opin. Neurobiol. 27, 171–178. doi: 10.1016/j.conb.2014.03.016
Smith, G. M., and Gallo, G. (2018). The role of mitochondria in axon development and regeneration. Dev. Neurobiol. 78, 221–237. doi: 10.1002/dneu.22546
Spires-Jones, T. L., and Hyman, B. T. (2014). The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron 82, 756–771. doi: 10.1016/j.neuron.2014.05.004
Springer, J. E., Prajapati, P., and Sullivan, P. G. (2018). Targeting the mitochondrial permeability transition pore in traumatic central nervous system injury. Neural Regen. Res. 13, 1338–1341. doi: 10.4103/1673-5374.235218
Staal, J. A., Dickson, T. C., Gasperini, R., Liu, Y., Foa, L., and Vickers, J. C. (2010). Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J. Neurochem. 112, 1147–1155. doi: 10.1111/j.1471-4159.2009.06531.x
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063. doi: 10.1083/jcb.200108057
Stavru, F., Palmer, A. E., Wang, C., Youle, R. J., and Cossart, P. (2013). Atypical mitochondrial fission upon bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 110, 16003–16008. doi: 10.1073/pnas.1315784110
Su, B., Wang, X., Lee, H. G., Tabaton, M., Perry, G., Smith, M. A., et al. (2010). Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 468, 267–271. doi: 10.1016/j.neulet.2009.11.010
Sun, F., and Cavalli, V. (2010). Neuroproteomics approaches to decipher neuronal regeneration and degeneration. Mol. Cell. Proteomics 9, 963–975. doi: 10.1074/mcp.R900003-MCP200
Sung, H., Tandarich, L. C., Nguyen, K., and Hollenbeck, P. J. (2016). Compartmentalized regulation of Parkin-mediated mitochondrial quality control in the Drosophila nervous system in vivo. J. Neurosci. 36, 7375–7391. doi: 10.1523/JNEUROSCI.0633-16.2016
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1038/nrn.2016.178
Sutherland, T. C., and Geoffroy, C. G. (2020). The influence of neuron-extrinsic factors and aging on injury progression and axonal repair in the central nervous system. Front. Cell Dev. Biol. 8:190. doi: 10.3389/fcell.2020.00190
Swerdlow, R. H. (2018). Mitochondria and mitochondrial cascades in Alzheimer's disease. J. Alzheimers. Dis. 62, 1403–1416. doi: 10.3233/jad-170585
Szczepanek, K., Chen, Q., Derecka, M., Salloum, F. N., Zhang, Q., Szelag, M., et al. (2011). Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 286, 29610–29620. doi: 10.1074/jbc.M111.226209
Tagliaferro, P., and Burke, R. E. (2016). Retrograde axonal degeneration in Parkinson disease. J. Parkin. Dis. 6, 1–15. doi: 10.3233/JPD-150769
Tallon, C., Russell, K. A., Sakhalkar, S., Andrapallayal, N., and Farah, M. H. (2016). Length-dependent axo-terminal degeneration at the neuromuscular synapses of type II muscle in SOD1 mice. Neuroscience 312, 179–189. doi: 10.1016/j.neuroscience.2015.11.018
Tammineni, P., Ye, X., Feng, T., Aikal, D., and Cai, Q. (2017). Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer's disease neurons. Elife 6:e21776. doi: 10.7554/eLife.21776
Tao, K., Matsuki, N., and Koyama, R. (2014). AMP-activated protein kinase mediates activity-dependent axon branching by recruiting mitochondria to axon. Dev. Neurobiol. 74, 557–573. doi: 10.1002/dneu.22149
Taylor, J. P., Brown, R. H. Jr., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Tuck, E., and Cavalli, V. (2010). Roles of membrane trafficking in nerve repair and regeneration. Commun. Integr. Biol. 3, 209–214. doi: 10.4161/cib.3.3.11555
Turner, M. R., Al-Chalabi, A., Chio, A., Hardiman, O., Kiernan, M. C., Rohrer, J. D., et al. (2017). Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 88, 1042–1044. doi: 10.1136/jnnp-2017-315995
Vagnoni, A., and Bullock, S. L. (2018). A cAMP/PKA/kinesin-1 axis promotes the axonal transport of mitochondria in aging Drosophila neurons. Curr. Biol. 28, 1265-1272 e1264. doi: 10.1016/j.cub.2018.02.048
Van Laar, V. S., Arnold, B., Howlett, E. H., Calderon, M. J., St. Croix, C. M., Greenamyre, J. T., et al. (2018). Evidence for compartmentalized axonal mitochondrial biogenesis: mitochondrial DNA replication increases in distal axons as an early response to Parkinson's disease-relevant stress. J. Neurosci. 38, 7505–7515. doi: 10.1523/jneurosci.0541-18.2018
Vasic, V., Barth, K., and Schmidt, M. H. H. (2019). Neurodegeneration and neuro-regeneration-Alzheimer's disease and stem cell therapy. Int. J. Mol. Sci. 20:4272. doi: 10.3390/ijms20174272
Wang, D. B., Garden, G. A., Kinoshita, C., Wyles, C., Babazadeh, N., Sopher, B., et al. (2013). Declines in Drp1 and Parkin expression underlie DNA damage-induced changes in mitochondrial length and neuronal death. J. Neurosci. 33, 1357–1365. doi: 10.1523/JNEUROSCI.3365-12.2013
Wang, H., Zheng, Z., Han, W., Yuan, Y., Li, Y., Zhou, K., et al. (2020). Metformin promotes axon regeneration after spinal cord injury through inhibiting oxidative stress and stabilizing microtubule. Oxid. Med. Cell Longev. 2020:9741369. doi: 10.1155/2020/9741369
Wang, J., Gu, B. J., Masters, C. L., and Wang, Y. J. (2017). A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 13, 612–623. doi: 10.1038/nrneurol.2017.111
Wang, J., Hamm, R. J., and Povlishock, J. T. (2011). Traumatic axonal injury in the optic nerve: evidence for axonal swelling, disconnection, dieback, and reorganization. J. Neurotrauma 28, 1185–1198. doi: 10.1089/neu.2011.1756
Wang, Q., Cai, H., Hu, Z., Wu, Y., Guo, X., Li, J., et al. (2019a). Loureirin B promotes axon regeneration by inhibiting endoplasmic reticulum stress: induced mitochondrial dysfunction and regulating the Akt/GSK-3β pathway after spinal cord injury. J. Neurotrauma 36, 1949–1964. doi: 10.1089/neu.2018.5966
Wang, Q., Tian, J., Chen, H., Du, H., and Guo, L. (2019b). Amyloid beta-mediated KIF5A deficiency disrupts anterograde axonal mitochondrial movement. Neurobiol. Dis. 127, 410–418. doi: 10.1016/j.nbd.2019.03.021
Wang, S., Smith, G. M., Selzer, M. E., and Li, S. (2019). Emerging molecular therapeutic targets for spinal cord injury. Expert Opin Ther Targets 23, 787–803. doi: 10.1080/14728222.2019.1661381
Wang, W., Li, L., Lin, W. L., Dickson, D. W., Petrucelli, L., Zhang, T., et al. (2013). The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet. 22, 4706–4719. doi: 10.1093/hmg/ddt319
Wang, Y., Chandran, J. S., Cai, H., and Mattson, M. P. (2008). DJ-1 is essential for long-term depression at hippocampal CA1 synapses. Neuromol. Med. 10, 40–45. doi: 10.1007/s12017-008-8023-4
Wang, Y., Song, M., and Song, F. (2018). Neuronal autophagy and axon degeneration. Cell Mol Life Sci. 75, 2389–2406. doi: 10.1007/s00018-018-2812-1
Wang, Y., Zhao, D., Pan, B., Song, Z., Shah, S. Z. A., Yin, X., et al. (2015). Death receptor 6 and caspase-6 regulate prion peptide-induced axonal degeneration in rat spinal neurons. J. Mol. Neurosci. 56, 966–976. doi: 10.1007/s12031-015-0562-1
Wang, Z. X., Tan, L., and Yu, J. T. (2015). Axonal transport defects in Alzheimer's disease. Mol. Neurobiol. 51, 1309–1321. doi: 10.1007/s12035-014-8810-x
Warita, H., Itoyama, Y., and Abe, K. (1999). Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 819, 120–131. doi: 10.1016/s0006-8993(98)01351-1
Woolums, B. M., McCray, B. A., Sung, H., Tabuchi, M., Sullivan, J. M., Ruppell, K. T., et al. (2020). TRPV4 disrupts mitochondrial transport and causes axonal degeneration via a CaMKII-dependent elevation of intracellular Ca(2). Nat. Commun. 11:2679. doi: 10.1038/s41467-020-16411-5
Xie, Y., Zhou, B., Lin, M. Y., and Sheng, Z. H. (2015). Progressive endolysosomal deficits impair autophagic clearance beginning at early asymptomatic stages in fALS mice. Autophagy 11, 1934–1936. doi: 10.1080/15548627.2015.1084460
Xiong, T., Tang, J., Zhao, J., Chen, H., Zhao, F., Li, J., et al. (2012). Involvement of the Akt/GSK-3β/CRMP-2 pathway in axonal injury after hypoxic-ischemic brain damage in neonatal rat. Neuroscience 216, 123–132. doi: 10.1016/j.neuroscience.2012.04.052
Xu, J., Long, H., Chen, W., Cheng, X., Yu, H., Huang, Y., et al. (2017). Ultrastructural features of neurovascular units in a rat model of chronic compressive spinal cord injury. Front. Neuroanat. 11:136. doi: 10.3389/fnana.2017.00136
Yahata, N., Yuasa, S., and Araki, T. (2009). Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J. Neurosci. 29, 6276–6284. doi: 10.1523/jneurosci.4304-08.2009
Yan, X., Hu, Y., Wang, B., Wang, S., and Zhang, X. (2020a). Metabolic dysregulation contributes to the progression of Alzheimer's disease. Front. Neurosci. 14:530219. doi: 10.3389/fnins.2020.530219
Yan, X., Wang, B., Hu, Y., Wang, S., and Zhang, X. (2020b). Abnormal mitochondrial quality control in neurodegenerative diseases. Front. Cell. Neurosci. 14:138. doi: 10.3389/fncel.2020.00138
Yang, J., Wu, Z., Renier, N., Simon, D. J., Uryu, K., Park, D. S., et al. (2015). Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160, 161–176. doi: 10.1016/j.cell.2014.11.053
Yang, X., Liu, R., Xu, Y., Ma, X., and Zhou, B. (2021). The mechanisms of peripheral nerve preconditioning injury on promoting axonal regeneration. Neural Plast. 2021:6648004. doi: 10.1155/2021/6648004
Yiu, G., and He, Z. (2006). Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 7, 617–627. doi: 10.1038/nrn1956
Yu, Y., Lee, H. C., Chen, K. C., Suhan, J., Qiu, M., Ba, Q., et al. (2016). Inner membrane fusion mediates spatial distribution of axonal mitochondria. Sci Rep 6:18981. doi: 10.1038/srep18981
Zhang, L., Trushin, S., Christensen, T. A., Tripathi, U., Hong, C., Geroux, R. E., et al. (2018). Differential effect of amyloid beta peptides on mitochondrial axonal trafficking depends on their state of aggregation and binding to the plasma membrane. Neurobiol. Dis. 114, 1–16. doi: 10.1016/j.nbd.2018.02.003
Zhang, X., Wang, R., Hu, D., Sun, X., Fujioka, H., Lundberg, K., et al. (2020). Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer's disease. Sci. Adv. 6:eabb8680. doi: 10.1126/sciadv.abb8680
Zheng, Y. R., Zhang, X. N., and Chen, Z. (2019). Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 25, 876–886. doi: 10.1111/cns.13122
Zhou, B., Yu, P., Lin, M.-Y., Sun, T., Chen, Y., and Sheng, Z.-H. (2016). Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J. Cell Biol. 214, 103–119. doi: 10.1083/jcb.201605101
Keywords: mitochondria, axon regeneration, aging, traumatic brain injury, spinal cord injury, Alzheimer's disease, Parkinson's disease, Amyotrophic lateral sclerosis
Citation: Wang B, Huang M, Shang D, Yan X, Zhao B and Zhang X (2021) Mitochondrial Behavior in Axon Degeneration and Regeneration. Front. Aging Neurosci. 13:650038. doi: 10.3389/fnagi.2021.650038
Received: 06 January 2021; Accepted: 18 February 2021;
Published: 08 March 2021.
Edited by:
Homaira Nawabi, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Janelle Drouin-Ouellet, Université de Montréal, CanadaCopyright © 2021 Wang, Huang, Shang, Yan, Zhao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baohong Zhao, Ymh6aGFvQGNtdS5lZHUuY24=; Xinwen Zhang, emhhbmd4aW53ZW5AY211LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.