 Afzal Misrani
Afzal Misrani Sidra Tabassum
Sidra Tabassum Li Yang
Li Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 18 February 2021
Sec. Alzheimer's Disease and Related Dementias
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.617588
This article is part of the Research Topic Mitochondrial Dysfunction: The Link Between Aging and Alzheimer’s View all 4 articles
Mitochondria play a pivotal role in bioenergetics and respiratory functions, which are essential for the numerous biochemical processes underpinning cell viability. Mitochondrial morphology changes rapidly in response to external insults and changes in metabolic status via fission and fusion processes (so-called mitochondrial dynamics) that maintain mitochondrial quality and homeostasis. Damaged mitochondria are removed by a process known as mitophagy, which involves their degradation by a specific autophagosomal pathway. Over the last few years, remarkable efforts have been made to investigate the impact on the pathogenesis of Alzheimer’s disease (AD) of various forms of mitochondrial dysfunction, such as excessive reactive oxygen species (ROS) production, mitochondrial Ca2+ dyshomeostasis, loss of ATP, and defects in mitochondrial dynamics and transport, and mitophagy. Recent research suggests that restoration of mitochondrial function by physical exercise, an antioxidant diet, or therapeutic approaches can delay the onset and slow the progression of AD. In this review, we focus on recent progress that highlights the crucial role of alterations in mitochondrial function and oxidative stress in the pathogenesis of AD, emphasizing a framework of existing and potential therapeutic approaches.
Alzheimer’s disease (AD) is a neurodegenerative disease that affects millions of people worldwide (Alzheimer’s Association, 2020), for which only symptomatic treatments are currently available. Current estimates indicate that, in the United States, around 5.8 million patients of age 65 years and older are living with AD in 2020 (Alzheimer’s Association, 2020). AD is characterized by the deposition of extracellular senile plaques, neurofibrillary tangles (NFTs), and neurodegeneration, leading to memory impairment and dementia. The exact mechanisms underlying AD remain unclear despite comprehensive attempts to understand its pathophysiology.
The most prominent theory postulates that, in AD, tau and Aβ negatively affect neuronal cells by compromising energy supply and the antioxidant response, causing mitochondrial and synaptic dysfunction. Neuronal activity is highly energy-dependent, and neurons are particularly sensitive to disruption in mitochondrial function (Kann and Kovacs, 2007; Cunnane et al., 2020). In addition, mitochondria produce cellular energy (adenosine triphosphate; ATP) and are also involved in many processes that are important for the life and death of the cell, including the control of second messenger levels, such as calcium ions (Ca2+) and reactive oxygen species (ROS) (Roger et al., 2017; Giorgi et al., 2018). Importantly, mitochondrial dysfunction contributes to reduced ATP production, Ca2+ dyshomeostasis, and ROS generation. Alterations in mitochondrial dynamics and mitophagy occur in early-stage AD, but the underlying mechanisms are poorly understood. Thus, studies elucidating the mechanisms of mitochondrial abnormalities in AD will facilitate a greater understanding of the pathogenesis of this neurodegenerative disease and potentially contribute to the advancement of therapeutic strategies to protect synaptic activity and subsequent cognitive function. Here, we review studies that suggest a role of mitochondrial dysfunction and the consequent ROS production in AD pathology and provide a context to explain current and future therapeutic approaches. We suggest that improving mitochondrial function should be considered an important therapeutic intervention against AD.
Oxidative stress is caused by an imbalance between the production and accumulation of ROS, which are inevitable by-products of metabolism that represent a double-edged sword in biological systems (Ferrer et al., 2010; Sies and Jones, 2020); under carefully regulated conditions, they can serve essential roles as, for example, signaling agents, but can also damage cells when produced in excessive amounts since they are capable of oxidizing all major biomolecules, including nucleic acids (DNA and RNA), proteins and lipids (Butterfield et al., 2010; Arimon et al., 2015). ROS are defined as chemically reactive oxygen free radicals as well as no radical derivatives of oxygen. Either enhanced ROS production or an impaired antioxidant system can tip the redox balance of the cell toward an oxidative state.
The brain is especially susceptible to oxidative damage due to its high rate of oxygen consumption, elevated levels of polyunsaturated fatty acids (which are easily targeted by free radicals), and relatively high levels of redox transition metal ions; besides, the brain has very low antioxidants levels (Butterfield et al., 2001; Llanos-Gonzalez et al., 2019; Cassidy et al., 2020). ROS has been proven to account for cellular injury in aging and neurodegenerative disorders (Valko et al., 2007). Indeed, the accumulation of Aβ protein induced by ROS in AD causes lysosome membrane degradation and eventually contributes to neuronal death (Zhang et al., 2009). A deficiency of cytochrome c oxidase is the most common defect in the mitochondrial electron transport chain (ETC) in AD, leading to an increase in ROS production, a decrease in energy stores, and a disruption of energy metabolism (Rak et al., 2016). Furthermore, ROS causes inhibition of phosphatase 2A (PP2A) (Elgenaidi and Spiers, 2019), which facilitates glycogen synthase kinase (GSK) 3β activation (one of the kinases involved in tau phosphorylation). Hence increased GSK3β activation might cause hyperphosphorylation of tau and neurofibrillary lesions in AD (Toral-Rios et al., 2020).
Oxidized biomolecule products produced by ROS are far more stable and widely used as ROS markers. Besides, ROS may also be indirectly tested by measuring antioxidant levels or the activity of antioxidant enzymes. In fact, oxidative imbalance and a substantial increase in its by-products have been repeatedly reported in AD. A large body of research has demonstrated that lipid peroxidation, the process in which ROS attacks lipids to produce lipid peroxidation products via a free radical chain reaction mechanism, is greatly enhanced in AD (Pratico et al., 2001; Galbusera et al., 2004). The most extensive lipid peroxidation products studied in AD are reactive aldehydes, including 4-hydroxynonal, malondialdehyde (MDA), and 2-propenal (acrolein), and chemically and metabolically stable isoprostanoids including F2-isoprostanes and F4-neuroprostanes. A substantial increase in MDA was reported in the hippocampus, pyriform cortex (Lovell et al., 1995), and erythrocytes of AD patients (Bermejo et al., 1997; Dei et al., 2002). Measuring MDA levels, which is both easy and cheap to perform, might be of great importance in monitoring AD progression and treatments. Conversely, the markers of oxidative stress that are commonly used in biological samples include protein carbonyls and 3-nitrotyrosine (3-NT) for protein oxidation; thiobarbituric acid-reactive substances (TBARS), free fatty acid release, iso- and neuroprostane formation, 2-propen-1-al (acrolein), and 4-hydroxy-2-trans-nonenal (HNE) for lipid peroxidation; advanced glycation end products for carbohydrates; 8-OH-2’-deoxyguanosine, 8-OH-guanosine and other oxidized bases, and altered DNA repair mechanisms for DNA and RNA oxidation. Increased levels of toxic carbonyls, 3-NT, and HNE are among the earliest alterations seen after an oxidative insult in AD (Butterfield et al., 2007b, 2011; Gamba et al., 2019). Several lines of evidence indicate that oxidative and nitrosative stress can lead to changes in vital cellular elements such as nucleic acids, lipids, and proteins. ROS comprise of both radical and non-radical oxygen species produced by a partial reduction of the oxygen, such as superoxide radical anion (O2), hydrogen peroxide (H2O2), hydroxyl radical (HO), nitric oxide (NO), and peroxynitrite (ONOO-). A key source of free radicals is the mitochondrial-resident oxidative phosphorylation, in which electron leakage from the mitochondrial ETC triggers the production of O2 (Ray et al., 2012). In addition to these common markers of protein modifications, protein oxidation/nitrosylation can also result in S-nitrosylation and methionine oxidation (sulfoxidation). S-nitrosylation is the production of the reaction between cysteine moiety and N2O3 to form an S-nitrosothiol (SNO) (Broniowska and Hogg, 2012). The latter is important in redox-based intracellular signaling, and altered SNO-profile has been documented in AD (Zahid et al., 2014).
In aging and AD, progressive impairment of mitochondrial function has also been implicated as the primary cause of ROS generation; mitochondria are themselves also a major target of oxidative damage (Swerdlow, 2011). In view of the above phenomenon, numerous studies have documented mitochondrial dysfunction through the abnormal processing of ROS as an essential factor in AD pathogenesis (Ohta and Ohsawa, 2006; Selfridge et al., 2013; Wang et al., 2014; Tobore, 2019). Similarly, the insertion of Aβ as oligomers into the bilayer can lead to the development of ROS, thereby initiating lipid peroxidation of membranes, followed by intracellular protein and nucleic acid oxidation (Butterfield, 2002; Butterfield et al., 2007a, Cheignon et al., 2018). It is also important to note that oxidative stress is linked to mitochondrial function, not just because mitochondria generate ROS, but also because ROS can cause deterioration of mitochondrial function (Figure 1). For this reason, reducing ROS levels, using strategies such as diet, exercise, and antioxidant drugs, may protect neuronal mitochondria from oxidative damage and thus reduce the risk of AD.
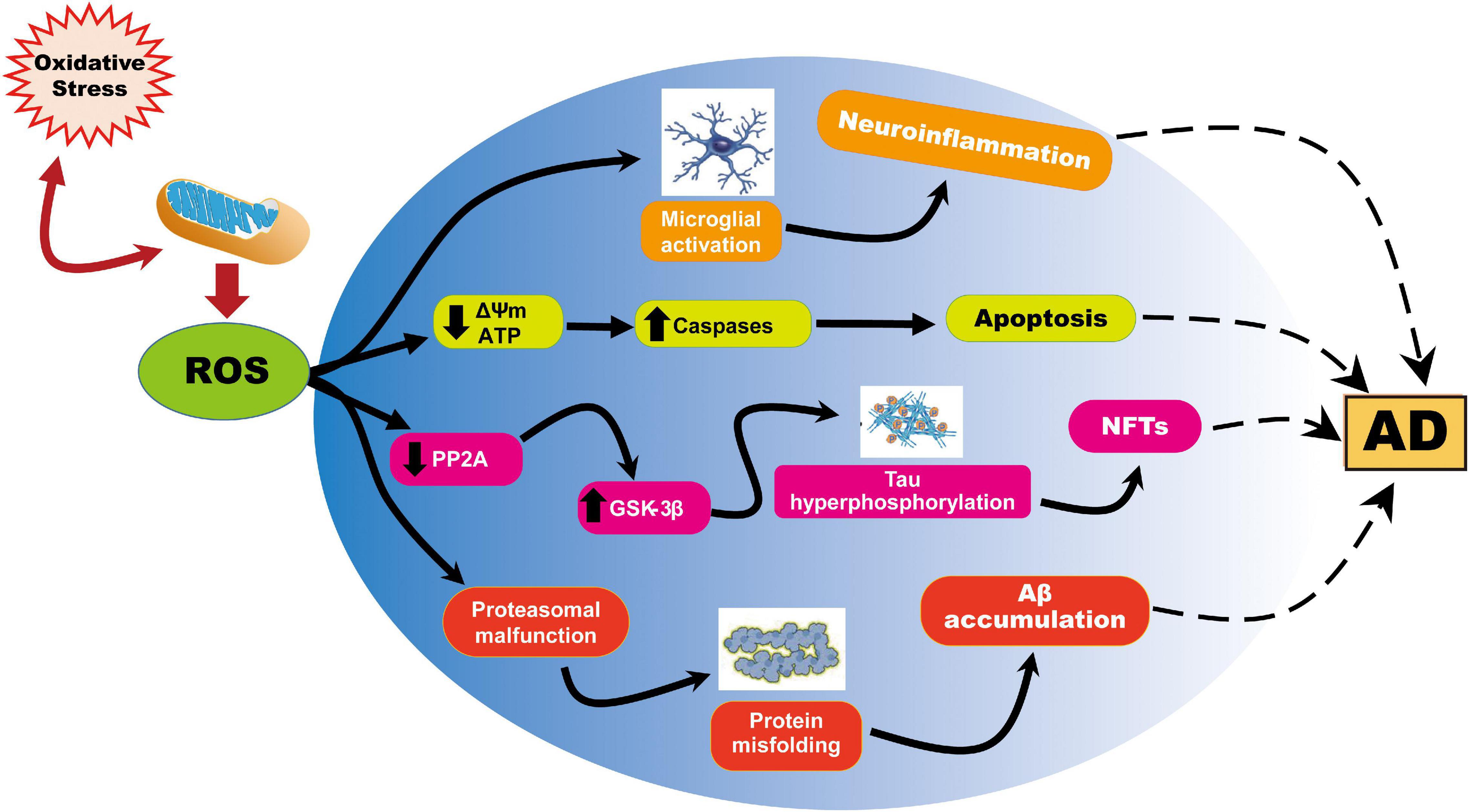
Figure 1. Representation of ROS-induced mitochondrial abnormalities in AD. ROS production or impaired antioxidant system results in the cellular redox balance to oxidative imbalance and cause ROS overproduction. ROS generated during cellular respiration has detrimental effects on mitochondria and neuronal function. Increased ROS causes reduction of mitochondrial ΔΨm and ATP generation through negatively affecting mitochondrial energy stores, disturbance in energy metabolism, and compromised dynamics and mitophagy. ROS further causes an increase in caspase activity and initiates apoptosis. On the other hand, ROS overproduction causes inhibition of phosphatase 2A (PP2A), which also activates glycogen synthase kinase (GSK) 3β causing tau hyperphosphorylation and neurofibrillary tangles accumulation.
Mitochondrial ATP production by oxidative phosphorylation (OXPHOS) is essential for cellular functions, such that mitochondria are known as the powerhouses of the cell (Verschueren et al., 2019). The mitochondrial ETC consists of five enzyme complexes in the inner membrane of the mitochondria. ETC generates a charge across the inner mitochondrial membrane, which drives ATP synthase (complex V) to synthesize ATP from ADP and inorganic phosphate.
Several studies have shown impairments of all five complexes in multiple areas of the AD brain (Kim et al., 2000, 2001; Liang et al., 2008). Mitochondrial dysfunction in AD is apparent from a decrease in neuronal ATP levels, which is associated with the overproduction of ROS, and indicates that mitochondria may fail to maintain cellular energy. A substantial amount of ATP is consumed in the brain due to the high energy requirements of neurons and glia. Since an energy reserve (such as fat or glucose) is not available in the central nervous system (CNS), brain cells must continuously generate ATP to sustain neuronal function (Khatri and Man, 2013). Mitochondria are the primary source of cellular energy production, but aged or damaged mitochondria produce excess free radicals, which can reduce the supply of ATP and contribute to energy loss and mitochondrial dysfunction in AD. Importantly, oxidative damage of the promoter of the gene encoding subunit of the mitochondrial ATP synthase results in reduced levels of the corresponding protein, leading to decreased ATP production, nuclear DNA damage to susceptible genes, and loss of function (Lu et al., 2004; Reed et al., 2008).
In advanced stages of AD, substantial nitration of ATP synthase subunits can take place, leading to the irregular function of the respiratory chain (Castegna et al., 2003; Sultana et al., 2006; Reed et al., 2009). Likewise, ATP-synthase lipoxidation occurs in the hippocampus and parietal cortex of patients with mild cognitive impairment (Reed et al., 2008). Compromised OXPHOS contributes to a characteristic mitochondrial dysfunction in AD brains, leading to decreased ATP production, elevated oxidative stress, and ultimately cell death (Reddy, 2006; Reddy and Beal, 2008; Du et al., 2012). The specific mechanisms of OXPHOS deficiency in AD remain a long-standing scientific question, but the role of mitochondrial F1Fo ATP synthase dysfunction in AD-related mitochondrial OXPHOS failure is emphasized by emerging evidence (Beck et al., 2016; Gauba et al., 2019). Therefore, it is important to note that ATP-synthase deregulation caused by ROS is a hallmark of mitochondrial dysfunction in AD, and the strategies to block ROS should prove beneficial by restoring ATP synthase activity.
Mitochondria play a key role in cellular Ca2+ homeostasis, and Ca2+ is an important regulator of vital neuronal processes, such as secretion, motility, metabolic regulation, synaptic plasticity, proliferation, gene expression, and apoptosis. The hypothesis that dysregulation of Ca2+ homeostasis is a critical factor in accelerating AD pathology is well known. Mitochondrial Ca2+ can certainly become a death factor via induction of the permeability transition (PT). The PT is an increase in the permeability of the inner mitochondrial membrane (IMM) to ions and solutes mediated by the PT pore (mPTP), a high-conductance, a voltage-dependent channel that needs a permissive Ca2+ matrix load for opening. Persistent opening of mPTP is accompanied by depolarization of Ca2+ release, cessation of OXPHOS, matrix swelling with IMM remodeling, and eventually, rupture of the outer mitochondrial membrane (OMM) with the release of cytochrome c and other apoptogenic proteins (Zago et al., 2000; Bernardi et al., 2006; Vercesi et al., 2006; Assaly et al., 2012; Boyman et al., 2019; Carraro and Bernardi, 2020; Nesci, 2020). Understanding the process by which Ca2+ is converted from a physiological signal into a pathological effector is one of the outstanding concerns in AD pathology.
Mitochondria contribute to intracellular Ca2+ signaling as modulators, buffers, and sensors (Rizzuto et al., 2012). After a cytosolic rise in [Ca2+], mitochondria quickly take up Ca2+ to avoid an overload of Ca2+ in the cytosol. When excessive Ca2+ is absorbed into mitochondria, such that they become overloaded, there is a consequent increase in the production of ROS, inhibition of ATP synthesis, the opening of the mPTP, release of cytochrome c, and activation of caspases and apoptosis (Figure 2). A recent study indicates that Aβ accumulation induces in vivo mitochondrial Ca2+ overload via the mitochondrial Ca2+ uniporter (MCU) complex, leading to neuronal death, and suggests that MCU complex inhibition and blocking the activation of mPTP might represent novel therapeutic approaches toward AD (Calvo-Rodriguez et al., 2020). The increase in mitochondrial Ca2+ concentration could contribute to neurotoxicity, but monitoring mitochondrial Ca2+ in individual neurons is challenging. Furthermore, mitochondrial Ca2+ overload and the resulting dysfunction are a critical cause of apoptosis following ischemic and traumatic brain injury (Rao et al., 2015; Nichols et al., 2018; Novorolsky et al., 2020), as well as in multiple neurodegenerative diseases, including AD, Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) (Gibson et al., 2010; Pchitskaya et al., 2018; Cortes et al., 2020).
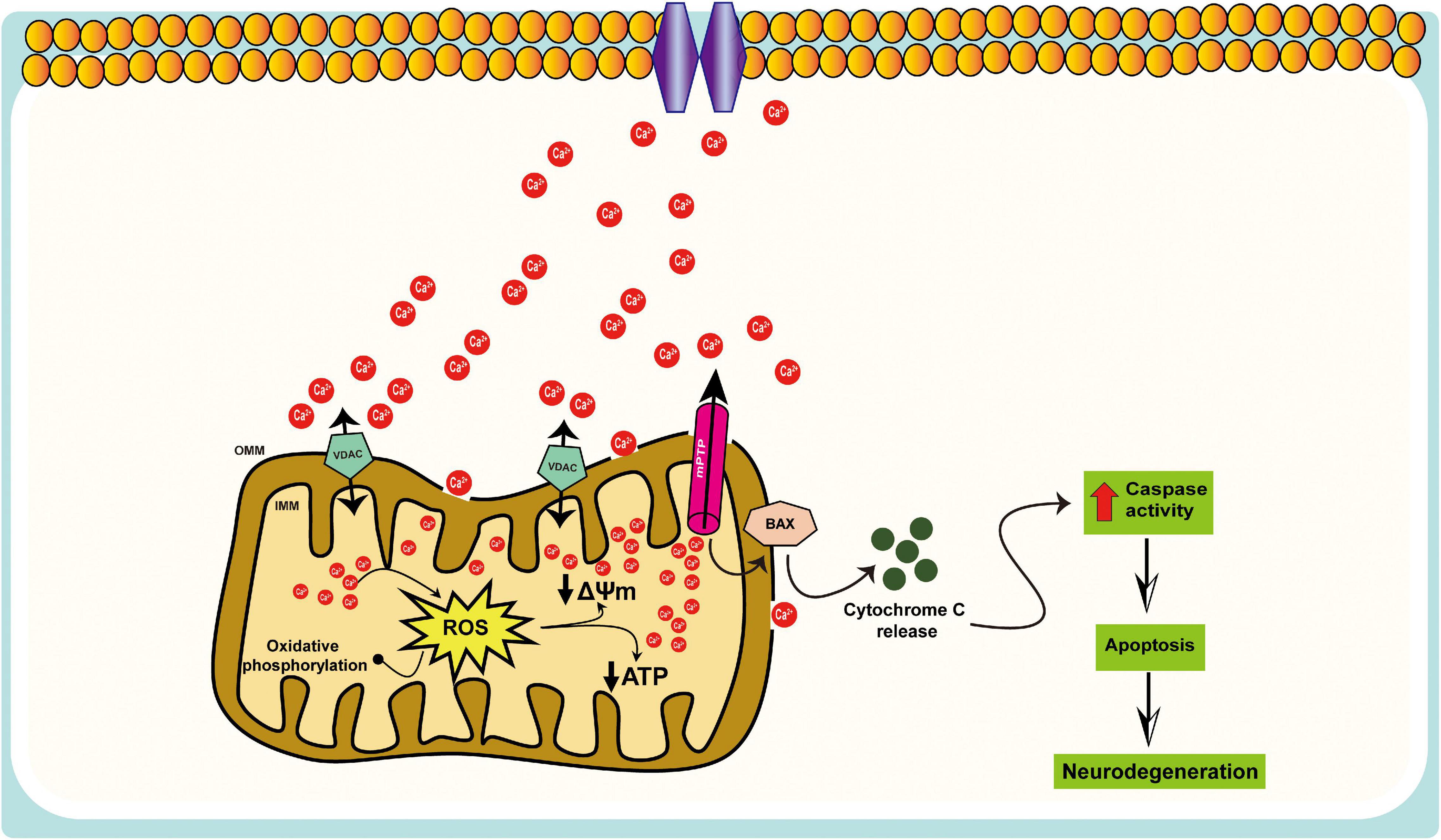
Figure 2. Schematic representation of the mitochondrial Ca2+ dysregulation in AD. Mitochondria participate in intracellular Ca2+ signaling as modulators, buffers, and sensors; excessive Ca2+ taken up by mitochondria can lead to cell death, i.e., mitochondrial Ca2+ overload, results in increased ROS production, ATP synthesis inhibition, mitochondrial permeability transition pore (mPTP) opening, the release of cytochrome c, activation of caspases, and apoptosis.
On the one hand, intracellular Ca2+ regulates multiple neuronal functions; its dyshomeostasis can trigger neuronal injury and death. Indeed, mitochondrial Ca2+ overload and subsequent dysfunction are possibly the most significant injury processes caused by excessive concentrations of cytosolic Ca2+. The fundamental role of mitochondrial dysfunction in AD, however, is clear, if many of the pathways underlying it are not. Thus, a significant aim of future studies should be to develop a clearer understanding of how, in the first place, mitochondria come to be at risk, and how this risk can be minimized. Much work needs to be done to form a better picture of the involvement of mitochondrial Ca2+ dysregulation in AD pathogenesis.
Mitochondria are dynamic, and undergo frequent changes in shape, size, number, and location. The various shapes result from the ability of mitochondria to divide, join together, and move throughout the cytoplasm. These processes are collectively referred to as mitochondrial dynamics and largely comprise two unique, closely controlled adverse processes, i.e., fission (division) and fusion (Youle and van Der Bliek, 2012; Palikaras et al., 2018; Chu, 2019; Reddy and Oliver, 2019), both of which are fundamental aspects of mitochondrial biology and quality control (Detmer and Chan, 2007; Youle and van Der Bliek, 2012; Fu et al., 2019). The equilibrium between fission and fusion is important not only for mitochondrial morphology, but also for the viability of cell and synaptic activity. Perturbations in mitochondrial fission, fusion, motility, and turnover can lead to defects in neurons. Previous studies indicate that mitochondrial fusion is neuron protective, leading to the exchange of mitochondrial DNA, reorganization of mitochondrial cristae, and protect cells from apoptosis, whereas mitochondrial fission seems a sign of apoptosis and fragmentation. It is important to note that the fission/fusion process is closely related to mitochondrial mobility and positioning. Likewise, abnormalities in mitochondrial fission and fusion, and consequent changes in mitochondrial morphology, influence mitochondrial mobility and distribution (Chen and Chan, 2009). Additionally, fission and fusion modulate mitochondrial shape, membrane topology, and intramitochondrial protein distribution, which further influence the apoptotic permeability of the mitochondrial OMM (Weaver et al., 2014; Renault et al., 2015). Disruption of fission and/or fusion processes is found in various neurodegenerative disorders including AD, PD and HD (Itoh et al., 2013; Van Laar and Berman, 2013; Hroudova et al., 2014; Reddy, 2014; Bertholet et al., 2016; Stanga et al., 2020). In this section, we discuss in detail the defects in fission/fusion and mitophagy in AD (Figure 3).
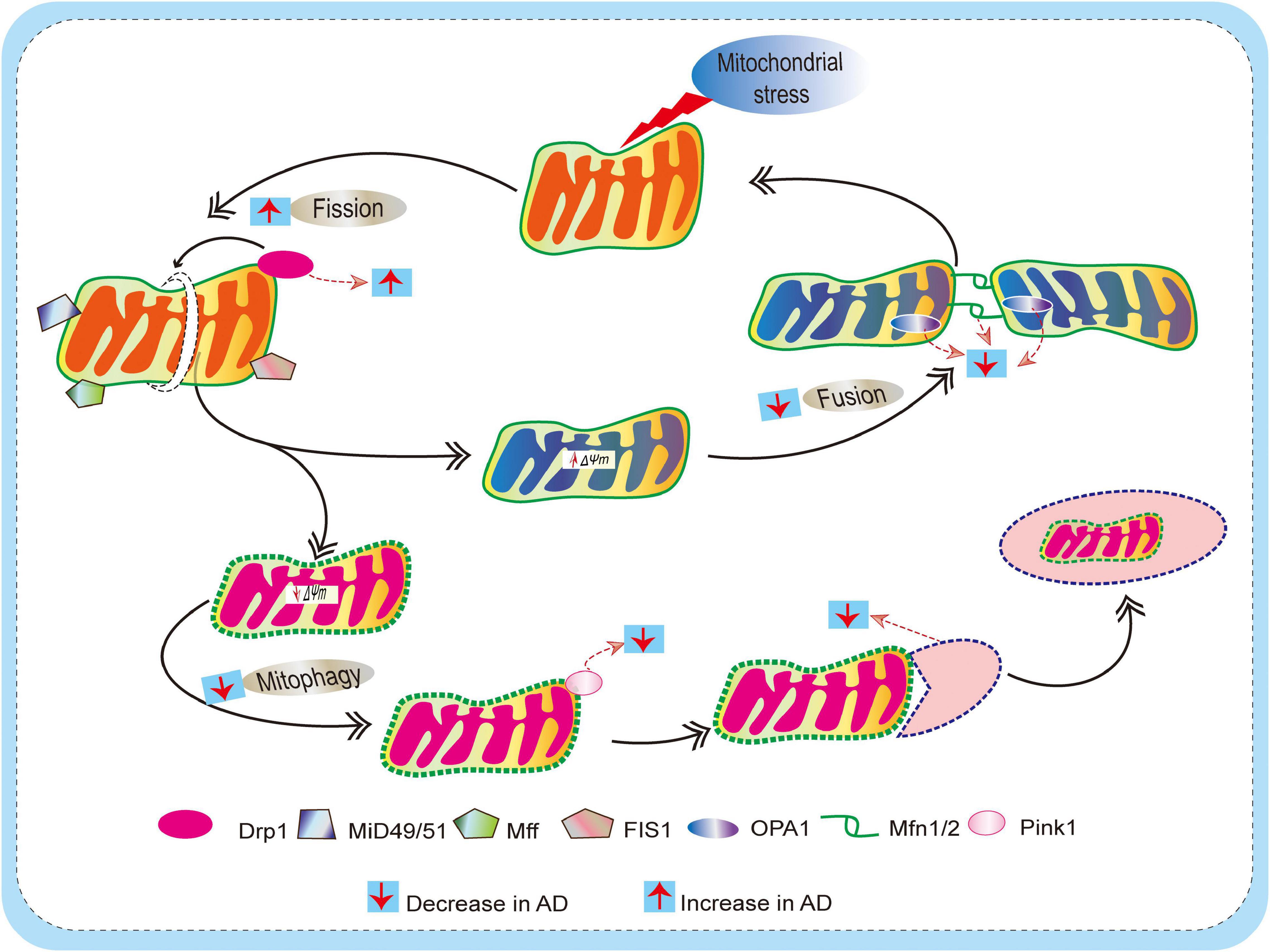
Figure 3. Schematic diagram depicting defects of mitochondrial dynamics (fission and fusion) and mitophagy mechanism in AD. Mitochondria are dynamic, and they undergo frequent changes in shape, size, number, and location to maintain mitochondrial biology and quality control. Actions of outer membrane Drp1 control mitochondrial fission. Drp1 is recruited by mitochondrial fission factor (Mff), mitochondrial fission 1 protein (Fis1), mitochondrial dynamics protein 49/51 (MiD49/51) to promote the mitochondrial fission process. On the other hand, Mitochondrial fusion is regulated by mitofusin (Mfn) 1 and 2 and optical atrophy protein 1 (OPA1). This allows for the exchange of material (matrix components and damaged mitochondrial DNA), as well as promoting a balance in bioenergetic properties (e.g., mitochondrial membrane potential ΔΨm). These fission/fusion processes also involve mitophagy mechanisms (removal of damaged mitochondria) to maintain quality control. When mitochondria become damaged due to cellular stress, sustained depolarization of their inner membrane occurs, resulting in the loss of ΔΨm, which stabilizes PINK1 at the outer membrane to initiate mitophagy. In AD, due to excessive ROS burden on neurons, impaired fission/fusion balance occurs, resulting in defective mitophagy.
Mitochondrial fission mainly involves the action of GTPase-related dynamin-related protein 1 (Drp1), which is conserved from yeast to mammals (Bleazard et al., 1999; Smirnova et al., 2001). Drp1 is recruited to the OMM by resident protein receptors, including mitochondrial fission factor (Mff), mitochondrial fission 1 protein (Fis1), and mitochondrial dynamics protein 49/51 (MiD49/51) to promote mitochondrial fission (Loson et al., 2013). In general, Drp1 is important in neurons for mitochondrial division, size, shape, and distribution, from the cell body to the axons, dendrites, and nerve terminals. In addition, it is proposed that Drp1 protein is crucial in preserving equilibrium in mitochondrial dynamics, managing fission, mitophagy, and even motility. It mainly acts as a mitochondrial fission factor, triggering mitochondrial fragmentation, and thus when it is downregulated, fusion is partially facilitated (Wenger et al., 2013). Mitochondrial dysfunction that can be attributed to an imbalance in Drp1 activity characterizes the pathology of various disorders including AD, Down syndrome (DS), multiple sclerosis (MS), ALS, PD, and triple-repeat diseases such as HD, spinobulbar muscular atrophy (SMA), spinocerebellar atrophy 1 (SCA1), and others (Wang et al., 2008, 2009b; Manczak et al., 2011; Reddy et al., 2011; Hu et al., 2017). Similarly, elevated Drp1 levels are seen in disease states that cause excessive mitochondria fragmentation, leading to mitochondrial dysfunction and neuronal damage. More specifically, prolonged mitochondrial fission and its deleterious effects are clearly illustrated in in vitro AD models: overexpression of amyloid precursor protein (APP) or Aβ treatment causes profound fragmentation and altered distribution of mitochondria, which likely trigger Aβ-induced synaptic defects in neuronal cultures (Wang et al., 2008, 2009a; Manczak et al., 2011). Likewise, experiments in Drosophila have reported Aβ-induced defects in mitochondrial dynamics and distribution as early events in vivo (Iijima-Ando et al., 2009; Zhao et al., 2010). Indeed, enhanced mitochondrial fission is well documented in AD patients and model organisms with a bias toward increased mitochondrial fragmentation (Calkins et al., 2011; Manczak et al., 2011; Kandimalla et al., 2016; Manczak et al., 2016). Similarly, increased mitochondrial ROS production, compromised mitochondrial function, and apoptosis has been associated with excessive mitochondrial fission and mitochondrial structural abnormalities (Pena-Blanco et al., 2020). Given the critical role of mitochondrial dynamics, any defects in the fidelity of the fission machinery may have a devastating impact on redox homeostasis, energy generation, and mitochondrial function.
In mammals, the fusion of the OMM requires the action of dynamin-related GTPase proteins mitofusin-1 (Mfn1) and mitofusin-2 (Mfn2), which bind the outer membranes of two mitochondria, while inner membrane fusion is facilitated by the optic atrophy type 1 (OPA1) protein. It has been shown that the heptad repeat region of Mfn1 contains an antiparallel coil that is likely to be involved in tethering mitochondria throughout the fusion process (Koshiba et al., 2004). Apart from tethering mitochondrial membranes, Mfn2 serves additional cellular functions, such as mitochondrion-ER contact site development and stabilization, mitochondrion-lipid droplet interaction, cell proliferation, metabolic signaling, and mitophagy (Chen et al., 2014; Zorzano et al., 2015; Naon et al., 2016; Boutant et al., 2017; McLelland et al., 2018). Maintenance of the membrane potential (ΔΨm) of the IMM is necessary for mitochondrial fusion. The mechanistic relationship between ΔΨm and fusion remains to be resolved, but one consideration seems to be the dependency on the ΔΨm of post-translational OPA1 processing (Ishihara et al., 2006). In addition to its function in membrane fusion, OPA1 is also essential for preserving the organization and structure of the IMM (Frezza et al., 2006). Defects in Mfn1, Mfn2, and OPA1 have been reported in various neurodegenerative disorders, including AD. Thus, modulating the activity of these fusion proteins might have an effect on the interaction of the mitochondrial network and IMM structure, both of which affect mitochondrial functions critical for cell health and viability, such as OXPHOS and permeabilization of the membrane during apoptotic cell death.
Recent studies suggest that there is an intrinsic link between hyperphosphorylated Tau and mitochondrial alterations. Mitochondrial dysfunction is detected in P301L tau transgenic mice (David et al., 2005). Abnormal mitochondrial fusion was associated with the overexpression of Tau protein (Li et al., 2016; Kandimalla et al., 2018). Tau ablation resulted in decreased ROS, increased fusion but decreased fission, inhibited mPTP and cyclophilin D (Cyp-D), and enhanced ATP production in mice. The fact that tau indirectly resulted in elongated mitochondria (citation) could be due to tau binding and stabilizing the actin cytoskeletal, disrupting the physical association of mitochondria and Drp1, thus preventing Drp1 dependant fission. The mechanism may involve decreased fission and increased fusion, suggesting that fusion beyond its physiological limit may be detrimental to mitochondrial function. Therefore, preventing tau modifications could enhance mitochondrial health and reduce neurodegeneration.
Mitochondrial biogenesis is the process by which mitochondria increase in number and size. A constant renewal of mitochondria is central to maintaining the number of healthy mitochondria. One of the important mitochondrial biogenesis factors is peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1), a transcriptional coactivator that controls specific transcription action factors, sequentially, coordinating the expression of key nuclear-encoded mitochondrial genes that are required for the proper functioning of the organelle. PGC-1α and estrogen-related receptor-α (ERRα) together stimulate the function of Mfn2 to facilitate the fusion process (Soriano et al., 2006). Repression of PGC-1α and Mfn2 causes a decrease in oxygen consumption, glucose oxidation, and ΔΨm, and an increase in the expression of oxidative phosphorylation proteins (Chen et al., 2005). Moreover, PGC-1α activity is responsive to multiple stimuli, including but not limited to nutrient availability, Ca2+, ROS, insulin, estrogen hormone, hypoxia, ATP demand, and cytokines (Huang et al., 2020). Besides PGC-1α, other members of the PGC-1 family of coactivators, namely PGC-1β and PGC-related coactivator (PRC), are also implicated in modulating mitochondrial function, but their exact role is not well understood (Scarpulla, 2008). The capacity of mitochondrial biogenesis declines with aging and in neurodegenerative disease. Notably, decreased levels of PGC-1α and Mfn2 have been reported in AD (Qin et al., 2009; Wang et al., 2009a). The activity of PGC-1α can be modulated by posttranslational signalings such as AMP-activated kinase (AMPK), Akt, p38 MAPK, and the sirtuin 1 (Sirt1). Direct phosphorylation by AMPK activates PGC-1α and promotes PGC-1α dependent induction at the PGC-1α promoter (Canto and Auwerx, 2009). Moreover, activation of Sirt1 through caloric restriction induces PGC-1α activity and enhances mitochondrial function (Planavila et al., 2011; Tang, 2016). Importantly, impaired AMPK, Sirt1, and PGC-1α signaling have been implicated in AD pathology, drugs that activate this signaling would provide hope in alleviating AD.
The proper distribution of mitochondria throughout the cell is achieved by the mitochondrial transport mechanism. Mitochondrial transport relies on proteins that exist in the membranes of mitochondria and transport molecules and other factors such as ions into or out of the organelles (Hansen and Herrmann, 2019; Ruprecht et al., 2019). Mitochondrial transport mainly depends on the actin cytoskeleton in budding yeast (Fehrenbacher et al., 2004) and on both actin and microtubules in mammalian cells (Morris and Hollenbeck, 1995; Ligon and Steward, 2000). These transport mechanisms can ensure the proper inheritance and recruitment of mitochondria. Neuronal mitochondrial transport is essential for providing ATP to the sites of synapses and promoting axonal growth, as well asCa2+ buffering, mitochondrial repair, and degradation (Lin and Sheng, 2015). Studies with the membrane-potential indicator dye JC-1 indicate that mitochondria with high ΔΨm favorably travel to the anterograde direction, whereas mitochondria with low ΔΨm move in the retrograde direction (Miller and Sheetz, 2004). These migration patterns suggest that active mitochondria are recruited to distal regions with high energy requirements, whereas impaired mitochondria are returned to the cell soma, perhaps for destruction or repair. Multiple kinesin family members and cytoplasmic dynein have been implicated in anterograde and retrograde mitochondrial transport, respectively (Hollenbeck and Saxton, 2005). Moreover, axonal anterograde transport of mitochondria require actions of Mfn2 (fusion protein), and Milton/Miro complex (members of the molecular complex that links mitochondria to kinesin motors) (Stowers et al., 2002; Guo et al., 2005; Misko et al., 2010). In neurons, cellular signaling cues, such as Ca2+, ROS, oxygen level, nutrients, and ATP, act to regulate these Milton/Miro proteins and determine mitochondrial movement and position. Although Milton/Miro proteins have been identified as mammalian adaptors responsible for transporting mitochondria by kinesin, additional motor and adaptor proteins also participate in axonal trafficking of mitochondria transport, ensuring proper mitochondrial distribution in the cell (Melkov and Abdu, 2018). Impaired mitochondrial axonal transport contributes to several human neurodegenerative conditions, including spastic paraplegia, Charcot–Marie–Tooth, ALS, HD, PD, and AD (Charrin et al., 2005; Goldstein, 2012; Lamberts and Brundin, 2017; Flannery and Trushina, 2019). In AD, impairment of mitochondrial axonal transport precedes the accumulation of toxic protein aggregates which is linked to disturbed axonal integrity and synaptic function (Stokin et al., 2005; Calkins et al., 2011). While the precise molecular mechanisms underlying abnormal mitochondrial transport in AD remain elucidated, a disturbance in mitochondrial motility is tightly linked with an unbalanced fission/fusion mechanism, increased levels of both Aβ and pTau, and ROS.
Mitophagy, a selective type of autophagy, is a crucial pathway for mitochondrial quality control where faulty mitochondria are sequestrated into autophagosomes for subsequent lysosomal degradation (Youle and Narendra, 2011; Kerr et al., 2017). Mitophagy dysfunction has been implicated in aging and multiple neurodegenerative diseases, such as AD, PD, ALS, and HD (Chu, 2019; Cai and Jeong, 2020). In this section, we offer a detailed and timely description of the molecular mechanisms of mitophagy and discuss current therapeutic approaches that target mitophagy and improve mitochondrial function in AD. In studies of yeast, worms (Caenorhabditis elegans), fruit flies (Drosophila melanogaster), zebrafish (Danio rerio), and mammals such as human (Homo sapiens), the molecular machinery that mediates the targeting of mitochondria to lysosomes has been elucidated (Lazarou et al., 2015). Importantly, PTEN-induced putative kinase protein 1 (PINK1)-parkin-mediated mitophagy is the most widely studied mitophagy pathway (Gautier et al., 2008). When mitochondria become damaged due to cellular stress, continued depolarization of their inner membrane occurs, leading to loss of mitochondrial ΔΨm, and this stabilizes PINK1 in the OMM. There, PINK1 phosphorylates Mfn2 and then stimulates the ubiquitin-proteasome system (UPS), which, in turn, recruits parkin to the OMM (Ziviani et al., 2010; Chan et al., 2011). This further promotes the engulfment of damaged mitochondria by the phagophore or isolation membranes and hence the formation of mitophagosomes destined for removal via the lysosomal system. Numerous studies have confirmed the roles of PINK1 and Parkin in mitochondrial quality control and mitophagy (Sung et al., 2016; Lee et al., 2018).
Mitochondrial quality control mechanisms that effectively sense and eradicate damaged mitochondria are weakened due to usage, aging, or disease, and this is likely to have a marked impact on neuronal health. A growing body of evidence indicates that inhibition of the clearance of damaged mitochondria and the concomitant increase in ROS results in an accumulation of impaired neurons in AD. One important aspect is that mitophagy could be compromised in AD due to unstable fusion of lysosomes and autophagosomes. Thus, disrupted lysosomal activity in healthy cells results in neuronal phenotypes resembling those in AD (Nixon et al., 2008). In addition, autophagosome aggregation develops after oxidative stress in mouse cortical neurons, which shows similarities with AD (Boland et al., 2008), while mutations in PS1 can impair autophagy/mitophagy (Lee et al., 2010). Together, these results indicate that impaired mitophagy is implicated in neuronal degeneration in AD (Figure 3).
The above analysis makes it clear that strategies capable of targeting mitochondrial function are needed to slow the progression of AD. The focus of the research effort should be to develop a therapeutic intervention that can target ROS and excessive mitochondrial fragmentation, thereby minimizing mitochondrial dysfunction and consequent synaptic injury during AD progression (summarized in Table 1). One fascinating approach to reducing the burden of ROS and improving mitochondrial health is exercise. In the following section, we discuss in detail the impact of physical activity on mitochondrial function.
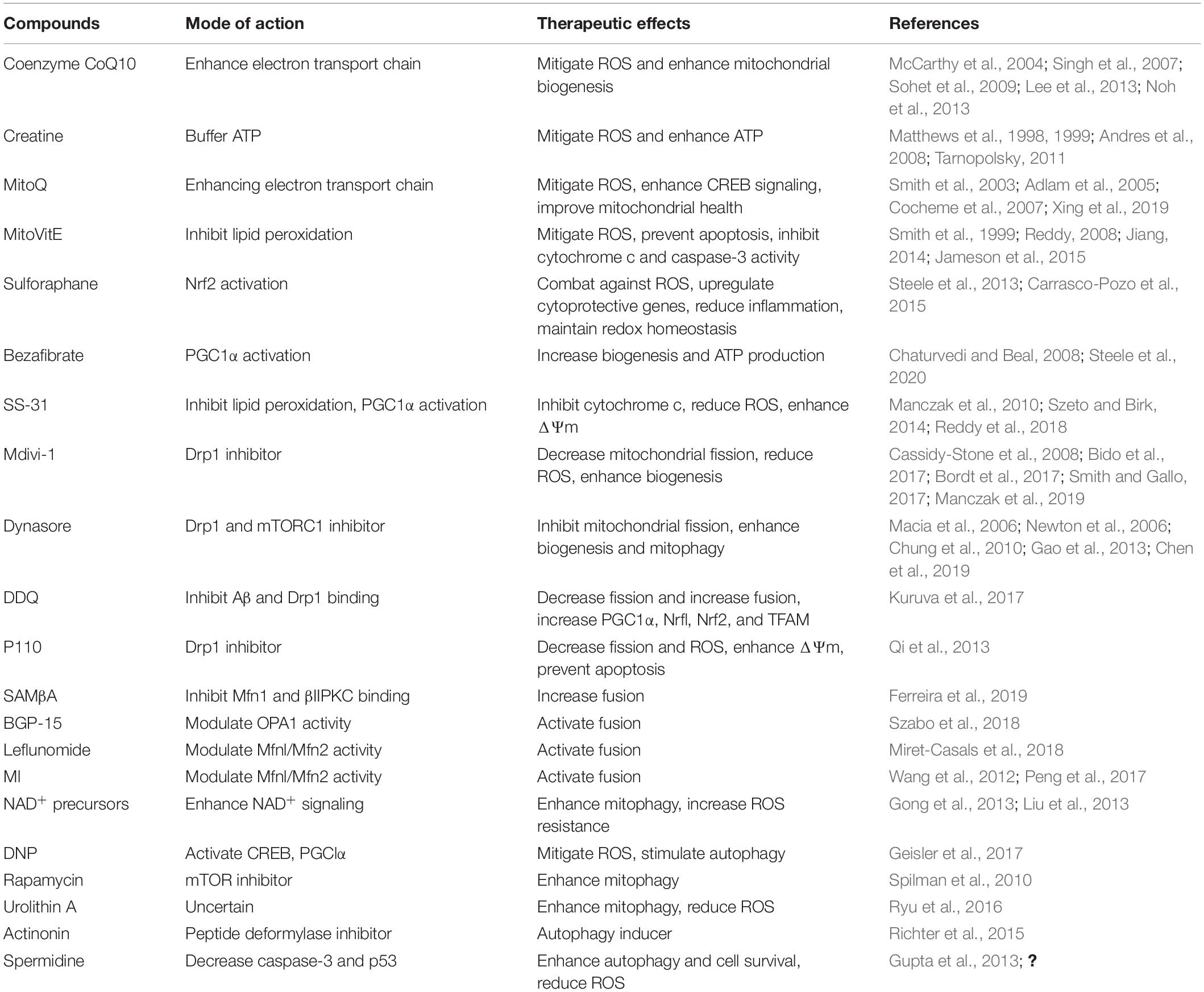
Table 1. Summary of drugs aiming to improve mitochondrial dysfunction in AD.
Exercise is one of the most effective strategies for maintaining a healthy body and normal brain activity. Moreover, exercise and a healthy diet can specifically boost several aspects of mitochondrial function. In this context, the beneficial effects of caloric restriction and exercise in slowing the aging process and enhancing mitochondrial function have been shown in humans and rodent models (Barbieri et al., 2015; Lundby and Jacobs, 2016; Fiuza-Luces et al., 2019). Significantly, exercise not only enhances mitochondrial activity in the peripheral organs, but also completely blocks brain atrophy in mouse models. The beneficial effects of physical activity are now widely accepted in humans as a way to not only improve fitness, but also treat patients with neurodegenerative diseases, including AD (Bernardo et al., 2016). Additional studies have reported numerous advantages of exercise in AD patients, including better blood flow to the brain, enhanced hippocampal thickness, enhanced neurogenesis, cognitive performance, reduced neuropsychiatric symptoms, and slower disorder (Brown et al., 2013; Cass, 2017).
Worldwide, around 30% of adults are insufficiently active (Hallal et al., 2012), which is a greater risk of ROS-induced anomalies. It is well established that a sedentary lifestyle contributes to increased ROS and neuroinflammation seen in neurodegenerative disorders. On the other hand, physical exercise can mitigate inflammation and oxidative stress (Gleeson et al., 2011; Mazzola et al., 2011). This attenuation might be one of the mechanisms responsible for improving several clinical aspects, for instance, attenuating cellular aging (Puterman et al., 2010) and increasing insulin sensitivity (Gordon et al., 2014). Moreover, physical exercise prevented ROS and normalized its various components, including thiobarbituric acid reactive substances (TBA-RS), superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) in rats (Mazzola et al., 2011). Lack of exercise leads to an overall reduction in mitochondrial ETC activity in healthy individuals. Simultaneously, endurance training can improve ETC activity, and resistance training can stimulate the integration of satellite cells into existing muscle fibers. Furthermore, exercise can stimulate mitochondrial proliferation through enhancing PGC-1α and AMPK signaling (Kang and Li Ji, 2012), and causes a reduction in the levels of systemic inflammation (McGee et al., 2003). This increase in PGC-1α and AMPK further promotes mitochondrial biogenesis.
Adequate consumption of vitamins and minerals and the use of natural foods rich in antioxidants (fruits, vegetables, etc.) could represent the ideal approach to maintaining the optimal antioxidant status. Foods that are rich in vitamin C can alleviate ROS. Vitamin C at various dosages, administered alone or in conjugation with other antioxidants, acutely or chronically, is the most commonly used antioxidant in clinical and laboratory research (Carr and Maggini, 2017; Spoelstra-de Man et al., 2018). Vitamin C attenuates ROS and maintains mitochondrial health in cells (Kc et al., 2005; Peng et al., 2019) and animal models (Singh and Rana, 2010). Similarly, Vitamin C consumption reduces Aβ plaque, preserves mitochondrial morphology, and ameliorates AD pathology in 5XFAD mice of AD (Kook et al., 2014). Moreover, the beneficial effect of caloric restriction on mitochondrial health is well documented. For example, a 15–25% continuous reduction in calorie intake in healthy adults for 24 months resulted in improved quality of life and a significant decrease in ROS levels (Redman et al., 2018). The caloric restriction mechanisms comprise activation of autophagy/mitophagy via AMPK-dependent inhibition of mTOR signaling, and the activation of Sirt1, which are important modulators of resistance to cellular stress, aging, and cell death. Moreover, caloric restriction has been shown to increase mitochondrial biogenesis and turnover, leading to a lesser accumulation of dysfunctional organelles, improved mitochondrial dynamics, morphology, and decreased mitochondrial permeability to Ca2+ retention capacity, ultimately leading to protection against excitotoxicity, a major mechanism involved in AD pathogenesis (Amigo et al., 2017). In addition to caloric restrictions, the ketogenic diet can slow down the development of cognitive dysfunction in patients with mild cognitive impairment (MCI) and AD. The neuroprotective effect of the ketogenic diet in AD is attributed to ketone bodies’ ability to provide a more efficient energy fuel source for mitochondria under conditions where glucose uptake is altered. Furthermore, ketone bodies have been shown to improve mitochondrial respiration, reduce the ROS production, improve antioxidant defense mechanism, and inhibit mPTP opening, thus ultimately protect mitochondrial and neuronal function (Masino and Rho, 2012). Overall, these studies suggest that physical exercise and diet have beneficial effects on mitochondrial health, redox homeostasis, and neuronal function, supporting the adoption of a healthy lifestyle as an invaluable tool against AD.
Antioxidant therapies, innovative pharmacological strategies designed to boost mitochondrial function, and mitigate local ROS production in mitochondria competing to reduce global levels of ROS. These compounds include coenzyme CoQ10, idebenone, creatine, MitoQ, MitoVitE, MitoTEMPOL, sulforaphane, bezafibrate, latrepirdine, methylene blue, triterpenoids, a series of Szeto-Schiller (SS) peptides such as SS-31, curcumin, Ginkgo biloba, omega-3 polyunsaturated fatty acids (Murphy and Hartley, 2018) and resveratrol which indirectly activates PGC-1α and induces mitochondrial biogenesis (Lagouge et al., 2006; Chuang et al., 2019). Numerous laboratories have extensively evaluated these mitochondrial-targeted compounds using in vivo and in vitro models of AD. Advantages of these compounds include improving bioenergetics, reducing ROS, maintaining mitochondrial dynamics.
CoQ10 is an essential cofactor of the ETC, functions by maintaining the mitochondrial ΔΨm, supporting ATP synthesis, and inhibiting ROS generation, thus protecting neuronal cells from oxidative stress and neurodegenerative diseases (McCarthy et al., 2004). Furthermore, it protects the membrane phospholipids, and mitochondrial membrane proteins against the damage of free radicals, increases mitochondrial mass and bioenergetic function (Singh et al., 2007; Noh et al., 2013). Other studies established that daily administration of CoQ10 significantly increased antioxidant enzyme activities and reduced inflammation (Sohet et al., 2009; Lee et al., 2013). Idebenone is an analog of CoQ10 that has better potency and a more promising pharmacokinetic profile. Idebenone can protect vision loss by enhancing mitochondrial ETC, in individuals with discordant visual acuities (Klopstock et al., 2011). CoQ10 has been utilized in many disease states to reduce ROS and improve mitochondrial health; this warrants CoQ10 use in preclinical and controlled human trials.
Creatine in mitochondria combines with phosphate to form phosphocreatine, which functions as a source of high-energy phosphate released during anaerobic metabolism. Thus, creatine serves as an intracellular buffer for ATP and as an energy shuttle for the movement of high energy phosphates from mitochondrial sites of production to cytoplasmic sites of utilization. Creatine is present in the highest concentration in tissues with high energy demands, such as muscle and brain (Parikh et al., 2009). Studies suggest reduced phosphocreatine levels in muscle tissue were shown in individuals with mitochondrial dysfunction (Tarnopolsky and Parise, 1999), and administration with creatine monohydrate can enhance exercise capacity in some individuals with mitochondrial dysfunction (Tarnopolsky, 2011). Similarly, beneficial effects of creatine supplementation have been shown in neurodegenerative and neurological diseases linked with mitochondrial dysfunction, such as PD, HD, and ALS (Andres et al., 2008). Findings from rodent research suggest that creatine exerts neuroprotective effects by buffering ATP levels to counter neurotoxic assaults by mPTP opening, and malonate (Matthews et al., 1998, 1999). These data implicate that oral creatine may serve as a potential therapy against ROS and subsequent reduction of bioenergetics, which occurred in AD.
MitoQ is a mitochondria-targeted compound that enhances the mitochondrial protection against oxidative damage (Cocheme et al., 2007). MitoQ consists of a lipophilic cation moiety that enables mitochondria-specific accumulation and ubiquinone converted to the antioxidant ubiquinol by the activity of complex II of the ETC (Smith et al., 2003). MitoQ, water-soluble that can be administered orally through the drinking water, and can cross the blood–brain barrier (Rodriguez-Cuenca et al., 2010), have protective effects against mitochondrial alterations induced by oxidative stress in animal models (Adlam et al., 2005). Further, MitoQ prevented cognitive decline and neuropathology in a mouse model of AD (McManus et al., 2011). Treatment with MitoQ causes activation of cAMP response element-binding protein (CREB), thus improve mitochondrial health (Xing et al., 2019). Overall, these studies suggest the antioxidant and mitochondria protecting role of MitoQ in many pathological conditions, including AD.
Vitamin E belongs to a group of compounds that includes both tocopherols and tocotrienols (Jiang, 2014). Tocopherol can protect cell membranes from oxidation, reacting with lipid radicals produced formed during lipid peroxidation (Jiang, 2014). MitoVitE is basically the chromanol moiety of vitamin E that bounds to a triphenyl phosphonium (TPP) cation and accumulates within mitochondria due to the large negative charge of the IMM. MitoVitE has been shown to accumulate in all major organs of mice and rats after oral, intraperitoneal, or intravenous administration and exerts a potent antioxidant activity (Jameson et al., 2015). Trolox, a synthetic, water-soluble, and cell-permeable derivative of vitamin E, often serves as a potent antioxidant in several model organisms (Wu et al., 1990; Guo et al., 2012). MitoVitE was more effective in vitro and in vivo than trolox (Jameson et al., 2015). MitoVitE can protect mitochondria from oxidative damage by reducing H2O2, inhibiting caspase activation, and blocking apoptosis (Reddy, 2008). Another study demonstrates that MitoVitE can prevent the release of cytochrome c, and staving off apoptosis by inhibiting caspase-3 activation, thus, rejuvenating ΔΨm for effective bioenergetics (Smith et al., 1999). Importantly, antioxidants which specifically accumulate within the mitochondrial matrix are suggested to offer better protection against oxidative stress.
Sulforaphane, a natural isothiocyanate-derived from a glucosinolate found in cruciferous vegetables, particularly broccoli, which is considered to be a common activator of Nrf2, can combat oxidative damage in mitochondria (Carrasco-Pozo et al., 2015). The activation of the Nrf2 pathway leads to upregulation of many downstream products involved in protection against oxidative stress, including NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), glutathione peroxidase 1 (GPx1), and gamma-glutamylcysteine synthetase (γGCS) (Steele et al., 2013). Sulforaphane has powerful antioxidant and anti-inflammatory properties, which allow it to reduce cytotoxicity and ROS dramatically. Animal studies suggest that sulforaphane supplementation could be disease-modifying for many common, devastating neurological conditions, such as AD, PD, epilepsy, stroke, etc. Collectively, these results indicate that Nrf2 activators can have antioxidant effects by retaining mitochondrial redox homeostasis. Sulforaphane is a potential neuroprotective phytochemical that needs further human trials to determine its effectiveness in preventing and reducing the burden of multiple neurological diseases, including AD.
Through mitochondrial biogenesis, cells increase their mitochondrial population in response to increased energy demand. This is driven by the PGC-1α activation, which is a transcriptional coactivator that controls mitochondrial biogenesis. Bezafibrate, a peroxisome proliferator-activated receptor (PPAR) agonist widely used to treat dyslipidemia. Bezafibrate supplementation resulted in the initiation of mitochondrial biogenesis, leading to an increase in mitochondrial mass, oxidative phosphorylation capacity, and energy generation (Chaturvedi and Beal, 2008; Steele et al., 2020). These findings imply that bezafibrate might be a promising therapeutic agent for treating any neurodegenerative diseases associated with mitochondrial dysfunction.
SS-31 is a small molecule that has been shown to exert potent antioxidant effects against ROS to protect mitochondrial function. SS-31 can prevent the peroxidase activity of cytochrome c in mitochondria, reduce ROS production, and aid reversal of mitochondrial dysfunction (Szeto and Birk, 2014). Moreover, SS-31 was proven to inhibit lipid peroxidation and hydrogen peroxide scavenging (Reddy et al., 2018). SS-31 has the benefit of being localized to the mitochondrion, explicitly targeting the IMM rather than the mitochondrial matrix. Treatment with SS-31 prevented mitochondrial dysfunction and enhanced ΔΨm, and increased neuroprotective gene PGC-1α in neuroblastoma N2a cells grown with mutant APP (Manczak et al., 2010), suggesting beneficial effects of the drug on mitochondrial alterations in AD. It is worth noting that mitochondria-targeted small molecules such as SS-31, have been tested in cell culture and animal models of neurodegenerative diseases, such as AD, HD, PD, ALS, multiple sclerosis, and other human diseases. Therefore, it is important to consider using small molecules for preclinical models and human clinical trials.
In the last two decades, some inhibitors of Drp1, including, in particular, mitochondrial division inhibitor 1 (Mdivi-1), dynasore, diethyl (3,4-dihydroxyphenethylamino) (quinolin-4-yl) methylphosphonate (DDQ), and P110, have been developed, and their beneficial effects have been studied in cell cultures and mouse models. The quinazolinone derivative, Mdivi-1, identified initially as a selective inhibitor of mitochondrial fission protein DRP1, induces neuroprotection in AD and PD models, as well as other neurodegenerative disorders, by improving mitochondrial fusion, and increasing mitochondrial biogenesis and synaptic protein levels (Bido et al., 2017; Smith and Gallo, 2017; Manczak et al., 2019). Recently, Mdivi-1 was shown to serve as a reversible mitochondrial complex I inhibitor that decreases mitochondrial fission and ROS production and further enhances mitochondrial function (Bordt et al., 2017). In addition, recent detailed studies of neuronal N2a cells support the theory that Mdivi-1 inhibits mitochondrial heterogeneity and increases energy efficiency (Manczak et al., 2019). On the other hand, the capacity of Mdivi-1 to suppress Drp1 and trigger mitochondrial fission has recently been questioned (Bordt et al., 2017). The authors did not notice any treatment effect with Mdivi-1 on mitochondrial morphology in mammalian cells in this study. However, they confirmed the impact of Mdivi-1 on yeast Drp1, consistent with a previous report (Cassidy-Stone et al., 2008). As Mdivi-1 is being considered for clinical trials, it may be appropriate to carry out more thorough investigations into its molecular targets to ensure its safety and effectiveness in humans.
Another cell-permeable small molecule that inhibits Drp1 activity is dynasore (Macia et al., 2006). A low dose of dynasore is sufficient to inhibit mitochondrial fission caused by ROS in cultured cells (Gao et al., 2013), which, in turn, inhibits endocytosis in neuronal cells and phagocytosis (Newton et al., 2006; Chung et al., 2010). Furthermore, dynasore inhibits mTORC1, which leads to nuclear translocation of TFEB and TFE3, the master regulators of autophagy and lysosomal biogenesis, thereby increasing autophagic flux. Dynasore therapy greatly improves the clearance by autophagy of protein aggregates of mutant HD in cells (Chen et al., 2019). In this context, a recent study suggests dynasore treatment decreases Aβ internalization and processing to the secretory pathway. However, there is currently a lack of data on the impact of dynasore treatment on fission-fusion and mitophagy in early and late-onset AD. Nevertheless, pharmacological interventions that inhibit the actions of Drp1 provide hope that excessive mitochondrial fragmentation can be abrogated in AD. Finally, the inhibition of the fission mechanism will benefit mitophagy, and energy production.
Recently, the role of mitochondrial dysfunction in AD has been studied using the pharmacological compound diethyl (3,4 dihydroxyphenethylamino) (quinolin-4-yl) methylphosphonate (DDQ). DDQ has shown promising effects on mRNA and protein levels associated with mitochondrial dysfunction and AD-related synaptic dysregulation. In addition, DDQ decreases mitochondrial fission proteins (Drp1 and Fis1), increases fusion proteins (Mfn1 and 2), and inhibit Aβ interactions. A novel property of DDQ is that it binds at the active binding sites of Aβ and Drp1, inhibiting the formation of complexes between of Aβ and Drp1 (Kuruva et al., 2017). This research indicates that DDQ can decrease the levels of Drp1 and Aβ, inhibit irregular Drp1-Aβ interactions, further decrease excessive mitochondrial fragmentation, and maintain mitochondrial function and synaptic activity in AD neurons. The effect of DDQ either before or after Aβ treatment on levels of various mRNAs and proteins important for mitochondrial function (PGC-1α, Nrf1, Nrf2, TFAM, DRP1, Fis1, Mfn1, and Mfn2), as well as those involved in synaptic activation (synaptophysin, PSD95, synapsin1 and 2, synaptobrevin1 and 2, synaptopodin, and GAP43), has also been examined. The mRNA and protein levels of mitochondrial-enhancing molecules such as PGC-1α, Nrf1, Nrf2, and TFAM were substantially increased following incubation with Aβ, followed by DDQ treatment. In addition, after DDQ therapy, a decrease in levels of mitochondrial fission proteins (DRP1 and Fis1) and an increase in levels of mitochondrial fusion proteins (Mfn1 and 2) was observed. This led to the conclusion that in the presence of Aβ, pretreatment with DDQ decreases fission activity (DRP1 and Fis1) and increases fusion activity (Mfn1 and 2). In order to evaluate its protective effects against Aβ-induced neuronal toxicity, more preclinical research using AD mouse models and clinical trials using AD patients treated with DDQ are needed.
P110 is another inhibitor of Drp1, which acts by blocking the interaction of Drp1 and Fis1. P110 was first used to protect neuronal cells: in cultured neurons, it reduces mitochondrial fragmentation and generation of ROS, restores mitochondrial integrity and ΔΨm, and protects cells against apoptosis caused by ROS (Qi et al., 2013). Other studies have used P110 to inhibit mitochondrial fission, thereby protecting the cell from death caused by stress or damage, especially in heart disease models. Moreover, treatment with P110 increases acute infarction-induced cell death and reduces heart failure both in vitro and in vivo (Disatnik et al., 2013). Therefore, P110 treatment should be assessed in early- and late-onset AD, at least in animal models, to confirm the therapeutic effects of this compound on mitochondrial fragmentation.
Therapeutic interventions directed at the fusion machinery (Mfn1, Mfn2, and OPA1) will enhance mitochondrial health by optimizing and rebalancing the regulation and control of fusion. Drugs that improve mitochondrial fusion function have recently been documented to suppress the death of apoptotic cardiac cells in vivo. When the association between Mfn1 and βIIPKC is blocked by the novel agent SAMβA, mitochondrial fusion and cardiac function are enhanced in rats (Ferreira et al., 2019). Another promising compound, which modulates the activity of OPA1 in lung epithelial cells, is the small molecule BGP-15 (Szabo et al., 2018). Leflunomide, a new compound launched in 2018, is another small molecule that activates fusion, as recognized by Mfn1/Mfn2-dependent mitochondrial elongation. Leflunomide-treated HeLa cells display an elongated mitochondrial network and enhanced expression of Mfn1/2. Mechanistically, leflunomide inhibits pyrimidine synthesis; this minimizes the activity of doxorubicin-induced PARP and cleaved caspase in3 in embryonic mouse fibroblast (MEF) cells and shields PC12 cells from apoptosis (Miret-Casals et al., 2018). In 2012, another mitochondrial fusion activator hydrazone M1 was introduced. Mitochondrial fragmentation is common in SH-SY5Y cells treated with 1-methyl-4-phenyl-pyridinium-(MPP+-), which models the death of neurons in PD. Treatment with the fusion promoter M1 in this model reduces the release of cytochrome c and prevents cell death (Wang et al., 2012). Similarly, M1 also protects mitochondrial function in a rotenone-induced PD model (Peng et al., 2017). These findings indicate that maintaining mitochondrial fusion is a promising approach to the treatment of a number of human diseases, including AD. Thus, further research is warranted, at least in animal models of AD, to test the effects of these drugs on impaired fusion machinery in AD.
Pharmacological agents and lifestyle interventions targeted at enhancing mitophagy are a promising approach for achieving a significant therapeutic benefit (Kerr et al., 2017; Lou et al., 2020). Caloric restriction, prolonged fasting, and physical exercise are bioenergetic challenges that can enhance neuroplasticity (i.e., they promote synapse formation and hippocampal neurogenesis, reduce ROS, stimulate mitochondrial biogenesis, and enhance autophagy) (Longo and Mattson, 2014). For example, in mice, fasting and exercise result in elevated numbers of autophagosomes in cerebral cortical neurons, increased expression of SIRT3, and activation of mitochondrial biogenesis by a pathway involving BDNF signaling and PGC-1α upregulation (Alirezaei et al., 2010; Cheng et al., 2012, 2016). Thus, exercise and fasting can augment the numbers of healthy mitochondria in neurons by promoting the pathways that enhance mitophagy. A variety of mitophagy-enhancing compounds in AD and other neurodegenerative diseases have been investigated recently, including nicotinamide adenine dinucleotide (NAD+) precursors, urolithin A (Ryu et al., 2016), the antibiotic actinonin (Richter et al., 2015), and spermidine (Gupta et al., 2013). NAD+ levels are reduced in animal models of AD, and elevation of cellular NAD+ levels by treatment with NAD+ precursors such as nicotinamide, nicotinamide mononucleotide, and nicotinamide riboside attenuates Aβ and tau pathologies, improves SIRT3 function, increases mitochondrial resistance to ROS, enhances mitophagy and prevents cognitive dysfunction, likely by upregulating the activity of the CREB transcription factor (Gong et al., 2013; Liu et al., 2013). In addition, mitochondrial uncoupling agents, such as 2,4-dinitrophenol (DNP), can induce autophagy and are useful in maintaining neuronal activity in animal AD models (Geisler et al., 2017). The mTOR inhibitor, rapamycin, is another mitophagy-inducing drug that can prevent cognitive defects and reduce Aβ pathology in an APP-mutant AD mouse model (Spilman et al., 2010). Collectively, these results provide encouragement for controlled human trials to explore the therapeutic potential of the above compounds.
One approach that might accelerate the translation of these mitochondrial agents into the clinic is to screen compounds in animal models at the prodromal stage and after neurological symptoms. If the drug impedes disease development in these models, there would be a firm basis for moving to human trials. We remain enthusiastic about the prospects for the treatment of neurodegenerative diseases using mitochondrial therapies; specifically, those designed to prevent mitochondrial damage, stimulate organelle biogenesis, and improve mitochondrial quality control (Figure 4). However, these advances require improvements in early diagnosis, the development of clinically appropriate biomarkers, and better trial design to allow for more rapid identification of compounds for the clinic.
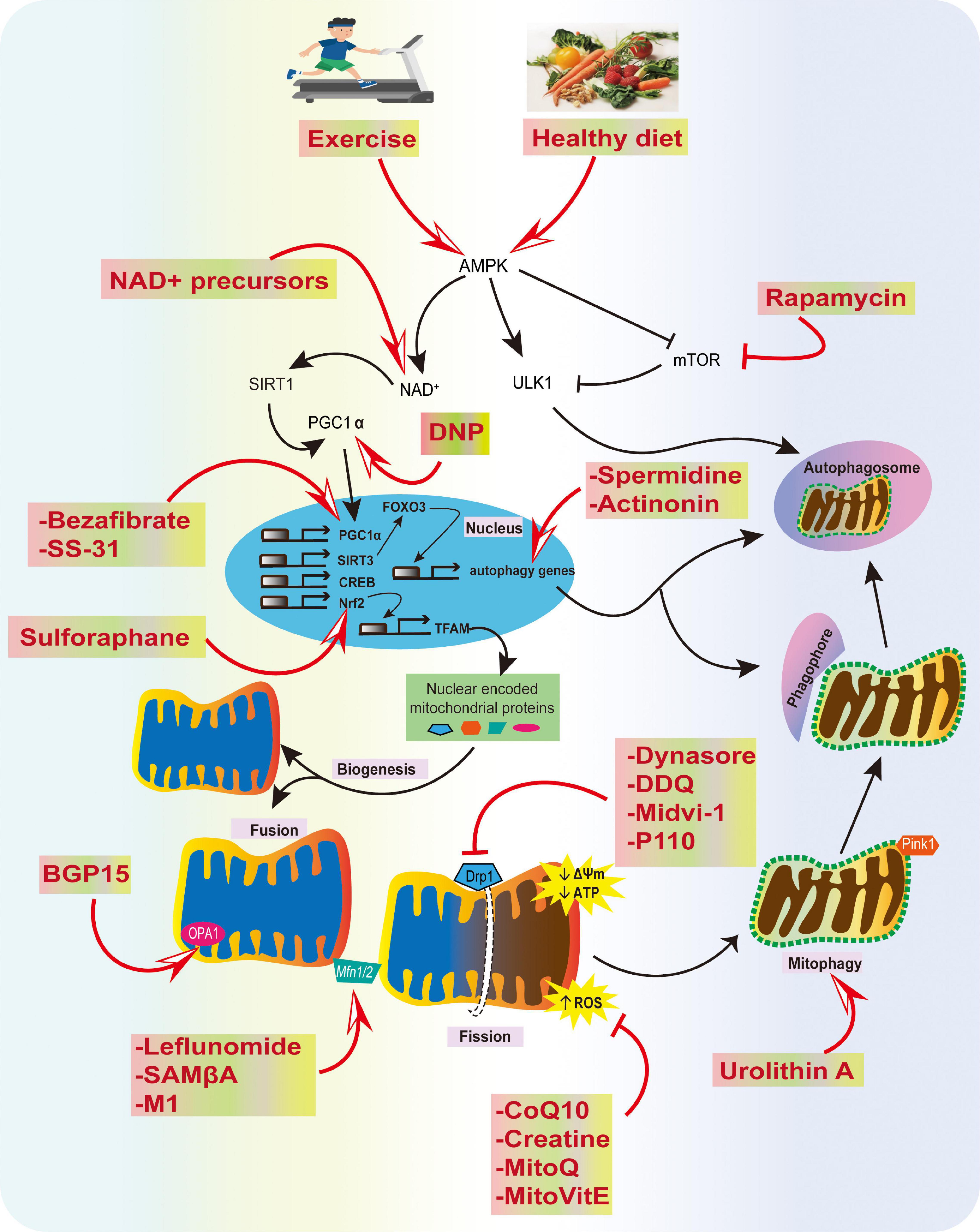
Figure 4. Summary of physiological and pharmacological interventions and molecular targets to improve mitochondrial function in AD. We highlight several strategies that can contribute to mitochondrial health, such as exercise and a healthy diet, inhibition of excessive mitochondrial fragmentation and ROS, and improving fusion, biogenesis, transport, and mitophagy using various compounds, as potential strategies for AD prevention.
Apart from offering a cure, most therapeutic interventions can have numerous adverse side effects, which can also be the case with the use of some mitochondria-targeted therapeutics. Targetting mitochondria is a novel strategy; nevertheless, despite very encouraging results in using these mitochondrial-targeted therapeutics, it is challenging to target the population of damaged mitochondria selectively. Notably, the targeting of compounds to the subcellular compartments represents one of the modern molecular pharmacology trends. The drug substance’s target is located inside an intracellular compartment such as a mitochondrion, and the drug molecule has to penetrate several membranes to reach the final destination. The molecule would require exact physicochemical properties to cross the different barriers. For example, to suppress ROS, antioxidants administered orally or intraventricularly, or intra-muscularly, must travel through the blood and finally reach the targeted organ. However, in this case, healthy tissues other than targeted organs that have not undergone oxidative damage, could be unavoidably targeted by frequent use of these antioxidants. As a result, upon increasing antioxidant doses to permit the repair of pathological mitochondria (especially toxic hyperpolarized mitochondria), normal mitochondria may also be adversely affected, as their ROS levels may fall below their physiologically acceptable limit. Undoubtedly, these compounds protected mitochondrial health and delayed aging in various animal trials; however, clinical evidence has not fully supported these preliminary findings. Thus, we have to be mindful of dosage, timing, and modes of exposure for different pathologies, with all the apparent benefits of mitochondrial-targeted therapeutics.
Significant progress has been made in discovering the causes of the debilitating neurodegenerative disorder AD. Accumulating data suggest that mitochondria play a vital role in the pathogenesis of AD by causing increased ROS production and oxidative damage, disturbance of Ca2+ homeostasis, activation of the mPTP, and alterations in dynamics, and mitophagy. The exploration of treatments that target mitochondria in AD is in progress, but there is an immediate need to develop novel therapeutic approaches that block or slow down the progression of this incurable disease. Here, we reviewed novel strategies for targeting altered pathways based on an aggregation of misfolded protein, defects in mitochondrial dynamics, OXPHOS dysfunction, oxidative stress, and compromised mitophagy in AD pathology. It remains a significant challenge to develop mitochondrially targeted AD therapeutics using innovative drug delivery systems and transfer them from the lab bench to the hospital bed. Nevertheless, in our opinion, focusing on mitochondria and expanding the area of mitochondrial pharmacology has enormous potential for modern, reliable mitochondrial therapy in AD.
AM, and LY designed the theme of the manuscript. AM contributed writing the following sections “Introduction,” “Oxidative Stress and Mitochondrial Defects in AD,” “Impaired Mitochondrial Dynamics and Mitophagy in AD,” “Strategies to Improve Mitochondrial Function in AD,” and created Figures 1, 4. ST wrote “Shortage of Neuronal ATP in AD” and “Mitophagy Defects in AD” and made Figures 2, 3. LY conducted a critical revision of the manuscript. All authors contributed to the article and approved the submitted version.
Work on this manuscript was supported by grants from the National Natural Science Foundation of China (31771219, 31871170, and 31970915) and the Guangdong Natural Science Foundation for Major Cultivation Project (2018B030336001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adlam, V. J., Harrison, J. C., Porteous, C. M., James, A. M., Smith, R. A., Murphy, M. P., et al. (2005). Targeting an antioxidant to mitochondria decreases cardiac ischemia–reperfusion injury. FASEB J. 19, 1088–1095. doi: 10.1096/fj.05-3718com
Alirezaei, M., Kemball, C. C., Flynn, C. T., Wood, M. R., Whitton, J. L., and Kiosses, W. B. (2010). Short–term fasting induces profound neuronal autophagy. Autophagy 6, 702–710. doi: 10.4161/auto.6.6.12376
Alzheimer’s Association (2020). Alzheimer’s disease facts and figures. Physician 16, 391–460. doi: 10.1002/alz.12068
Amigo, I., Menezes–Filho, S. L., Luevano–Martinez, L. A., Chausse, B., and Kowaltowski, A. J. (2017). Caloric restriction increases brain mitochondrial calcium retention capacity and protects against excitotoxicity. Aging Cell 16, 73–81. doi: 10.1111/acel.12527
Andres, R. H., Ducray, A. D., Schlattner, U., Wallimann, T., and Widmer, H. R. (2008). Functions and effects of creatine in the central nervous system. Brain Res. Bull. 76, 329–343. doi: 10.1016/j.brainresbull.2008.02.035
Arimon, M., Takeda, S., Post, K. L., Svirsky, S., Hyman, B. T., and Berezovska, O. (2015). Oxidative stress and lipid peroxidation are upstream of amyloid pathology. Neurobiol. Dis. 84, 109–119. doi: 10.1016/j.nbd.2015.06.013
Assaly, R., De Tassigny, A., Paradis, S., Jacquin, S., Berdeaux, A., and Morin, D. (2012). Oxidative stress, mitochondrial permeability transition pore opening and cell death during hypoxia–reoxygenation in adult cardiomyocytes. Eur. J. Pharmacol. 675, 6–14. doi: 10.1016/j.ejphar.2011.11.036
Barbieri, E., Agostini, D., Polidori, E., Potenza, L., Guescini, M., Lucertini, F., et al. (2015). The pleiotropic effect of physical exercise on mitochondrial dynamics in aging skeletal muscle. Oxid. Med. Cell Longev. 2015:917085. doi: 10.1155/2015/917085
Beck, S. J., Guo, L., Phensy, A., Tian, J., Wang, L., Tandon, N., et al. (2016). Deregulation of mitochondrial F1FO–ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 7:11483. doi: 10.1038/ncomms11483
Bermejo, P., Gomez–Serranillos, P., Santos, J., Pastor, E., Gil, P., and Martin–Aragon, S. (1997). Determination of malonaldehyde in Alzheimer’s disease: a comparative study of high–performance liquid chromatography and thiobarbituric acid test. Gerontology 43, 218–222. doi: 10.1159/000213853
Bernardi, P., Krauskopf, A., Basso, E., Petronilli, V., Blachly–Dyson, E., Di Lisa, F., et al. (2006). The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x
Bernardo, T. C., Marques–Aleixo, I., Beleza, J., Oliveira, P. J., Ascensao, A., and Magalhaes, J. (2016). Physical Exercise and Brain Mitochondrial Fitness: The Possible Role Against Alzheimer’s Disease. Brain Pathol. 26, 648–663. doi: 10.1111/bpa.12403
Bertholet, A. M., Delerue, T., Millet, A. M., Moulis, M. F., David, C., Daloyau, M., et al. (2016). Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 90, 3–19. doi: 10.1016/j.nbd.2015.10.011
Bido, S., Soria, F. N., Fan, R. Z., Bezard, E., and Tieu, K. (2017). Mitochondrial division inhibitor–1 is neuroprotective in the A53T–alpha–synuclein rat model of Parkinson’s disease. Sci. Rep. 7:7495. doi: 10.1038/s41598-017-07181-0
Bleazard, W., Mccaffery, J. M., King, E. J., Bale, S., Mozdy, A., Tieu, Q., et al. (1999). The dynamin–related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1, 298–304. doi: 10.1038/13014
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008
Bordt, E. A., Clerc, P., Roelofs, B. A., Saladino, A. J., Tretter, L., Adam–Vizi, V., et al. (2017). The Putative Drp1 Inhibitor mdivi–1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell. 40, 583–594 e586. doi: 10.1016/j.devcel.2017.02.020
Boutant, M., Kulkarni, S. S., Joffraud, M., Ratajczak, J., Valera–Alberni, M., Combe, R., et al. (2017). Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 36, 1543–1558. doi: 10.15252/embj.201694914
Boyman, L., Coleman, A. K., Zhao, G., Wescott, A. P., Joca, H. C., Greiser, B. M., et al. (2019). Dynamics of the mitochondrial permeability transition pore: Transient and permanent opening events. Arch. Biochem. Biophys. 666, 31–39. doi: 10.1016/j.abb.2019.03.016
Broniowska, K. A., and Hogg, N. (2012). The chemical biology of S–nitrosothiols. Antioxid. Redox. Signal 17, 969–980. doi: 10.1089/ars.2012.4590
Brown, B. M., Peiffer, J. J., and Martins, R. N. (2013). Multiple effects of physical activity on molecular and cognitive signs of brain aging: can exercise slow neurodegeneration and delay Alzheimer’s disease? Mol. Psych. 18, 864–874. doi: 10.1038/mp.2012.162
Butterfield, D. A. (2002). Amyloid beta–peptide (1–42)–induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. Free Radic. Res. 36, 1307–1313. doi: 10.1080/1071576021000049890
Butterfield, D. A., and Bader Lange, M. L., and Sultana, R. (2010). Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 1801, 924–929. doi: 10.1016/j.bbalip.2010.02.005
Butterfield, D. A., Drake, J., Pocernich, C., and Castegna, A. (2001). Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta–peptide. Trends Mol. Med. 7, 548–554. doi: 10.1016/s1471-4914(01)02173-6
Butterfield, D. A., Reed, T., Newman, S. F., and Sultana, R. (2007a). Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 43, 658–677. doi: 10.1016/j.freeradbiomed.2007.05.037
Butterfield, D. A., Reed, T. T., Perluigi, M., De Marco, C., Coccia, R., Keller, J. N., et al. (2007b). Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 1148, 243–248. doi: 10.1016/j.brainres.2007.02.084
Butterfield, D. A., Reed, T., and Sultana, R. (2011). Roles of 3–nitrotyrosine– and 4–hydroxynonenal–modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic. Res. 45, 59–72. doi: 10.3109/10715762.2010.520014
Cai, Q., and Jeong, Y. Y. (2020). Mitophagy in Alzheimer’s Disease and Other Age–Related Neurodegenerative Diseases. Cells 9:150. doi: 10.3390/cells9010150
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., and Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 20, 4515–4529. doi: 10.1093/hmg/ddr381
Calvo-Rodriguez, M., Hou, S. S., Snyder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 11:2146. doi: 10.1038/s41467-020-16074-2
Canto, C., and Auwerx, J. (2009). PGC–1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 20, 98–105. doi: 10.1097/MOL.0b013e328328d0a4
Carr, A. C., and Maggini, S. (2017). Vitamin C and Immune Function. Nutrients 9:1211. doi: 10.3390/nu9111211
Carraro, M., and Bernardi, P. (2020). Measurement of membrane permeability and the mitochondrial permeability transition. Methods Cell. Biol. 155, 369–379. doi: 10.1016/bs.mcb.2019.10.004
Carrasco-Pozo, C., Tan, K. N., and Borges, K. (2015). Sulforaphane is anticonvulsant and improves mitochondrial function. J. Neurochem. 135, 932–942. doi: 10.1111/jnc.13361
Cass, S. P. (2017). Alzheimer’s Disease and Exercise: A Literature Review. Curr. Sports Med. Rep. 16, 19–22. doi: 10.1249/JSR.0000000000000332
Cassidy, L., Fernandez, F., Johnson, J. B., Naiker, M., Owoola, A. G., and Broszczak, D. A. (2020). Oxidative stress in alzheimer’s disease: A review on emergent natural polyphenolic therapeutics. Compl. Ther. Med. 49:102294. doi: 10.1016/j.ctim.2019.102294
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak–dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204. doi: 10.1016/j.devcel.2007.11.019
Castegna, A., Thongboonkerd, V., Klein, J. B., Lynn, B., Markesbery, W. R., and Butterfield, D. A. (2003). Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J. Neurochem. 85, 1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L., et al. (2011). Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Charrin, B. C., Saudou, F., and Humbert, S. (2005). Axonal transport failure in neurodegenerative disorders: the case of Huntington’s disease. Pathol. Biol. 53, 189–192. doi: 10.1016/j.patbio.2004.12.008
Chaturvedi, R. K., and Beal, M. F. (2008). Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci. 1147, 395–412. doi: 10.1196/annals.1427.027
Cheignon, C., Tomas, M., Bonnefont–Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics—-fusion, fission, movement, and mitophagy—-in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi: 10.1093/hmg/ddp326
Chen, H., Chomyn, A., and Chan, D. C. (2005). Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 280, 26185–26192. doi: 10.1074/jbc.M503062200
Chen, K. H., Dasgupta, A., Ding, J., Indig, F. E., Ghosh, P., and Longo, D. L. (2014). Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. FASEB J. 28, 382–394. doi: 10.1096/fj.13-230037
Chen, Y., Xu, S., Wang, N., Ma, Q., Peng, P., Yu, Y., et al. (2019). Dynasore Suppresses mTORC1 Activity and Induces Autophagy to Regulate the Clearance of Protein Aggregates in Neurodegenerative Diseases. Neurotox. Res. 36, 108–116. doi: 10.1007/s12640-019-00027-9
Cheng, A., Wan, R., Yang, J. L., Kamimura, N., Son, T. G., Ouyang, X., et al. (2012). Involvement of PGC–1alpha in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 3:1250. doi: 10.1038/ncomms2238
Cheng, A., Yang, Y., Zhou, Y., Maharana, C., Lu, D., Peng, W., et al. (2016). Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell. Metab. 23, 128–142. doi: 10.1016/j.cmet.2015.10.013
Chu, C. T. (2019). Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 122, 23–34. doi: 10.1016/j.nbd.2018.07.015
Chuang, Y. C., Chen, S. D., Hsu, C. Y., Chen, S. F., Chen, N. C., and Jou, S. B. (2019). Resveratrol Promotes Mitochondrial Biogenesis and Protects against Seizure–Induced Neuronal Cell Damage in the Hippocampus Following Status Epilepticus by Activation of the PGC–1alpha Signaling Pathway. Int. J. Mol. Sci. 20:998. doi: 10.3390/ijms20040998
Chung, C., Barylko, B., Leitz, J., Liu, X., and Kavalali, E. T. (2010). Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J. Neurosci. 30, 1363–1376. doi: 10.1523/JNEUROSCI.3427-09.2010
Cocheme, H. M., Kelso, G. F., James, A. M., Ross, M. F., Trnka, J., Mahendiran, T., et al. (2007). Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion 7, S94–S102. doi: 10.1016/j.mito.2007.02.007
Cortes, L., Malva, J., Rego, A. C., and Pereira, C. F. (2020). Calcium Signaling in Aging and Neurodegenerative Diseases 2019. Int. J. Mol. Sci. 21:1125. doi: 10.3390/ijms21031125
Cunnane, S. C., Trushina, E., Morland, C., Prigione, A., Casadesus, G., Andrews, Z. B., et al. (2020). Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 19, 609–633. doi: 10.1038/s41573-020-0072-x
David, D. C., Hauptmann, S., Scherping, I., Schuessel, K., Keil, U., Rizzu, P., et al. (2005). Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 280, 23802–23814. doi: 10.1074/jbc.M500356200
Dei, R., Takeda, A., Niwa, H., Li, M., Nakagomi, Y., Watanabe, M., et al. (2002). Lipid peroxidation and advanced glycation end products in the brain in normal aging and in Alzheimer’s disease. Acta Neuropathol. 104, 113–122. doi: 10.1007/s00401-002-0523-y
Detmer, S. A., and Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol, 8, 870–879. doi: 10.1038/nrm2275
Disatnik, M. H., Ferreira, J. C., Campos, J. C., Gomes, K. S., Dourado, P. M., Qi, X., et al. (2013). Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long–term cardiac dysfunction. J. Am. Heart Assoc. 2:e000461. doi: 10.1161/JAHA.113.000461
Du, H., Guo, L., and Yan, S. S. (2012). Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid. Redox. Signal 16, 1467–1475. doi: 10.1089/ars.2011.4277
Elgenaidi, I. S., and Spiers, J. P. (2019). Regulation of the phosphoprotein phosphatase 2A system and its modulation during oxidative stress: A potential therapeutic target? Pharmacol. Ther. 198, 68–89. doi: 10.1016/j.pharmthera.2019.02.011
Fehrenbacher, K. L., Yang, H. C., Gay, A. C., Huckaba, T. M., and Pon, L. A. (2004). Live cell imaging of mitochondrial movement along actin cables in budding yeast. Curr. Biol. 14, 1996–2004. doi: 10.1016/j.cub.2004.11.004
Ferreira, J. C. B., Campos, J. C., Qvit, N., Qi, X., Bozi, L. H. M., Bechara, L. R. G., et al. (2019). A selective inhibitor of mitofusin 1–betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 10:329. doi: 10.1038/s41467-018-08276-6
Ferrer, M. D., Sureda, A., Mestre, A., Tur, J. A., and Pons, A. (2010). The double edge of reactive oxygen species as damaging and signaling molecules in HL60 cell culture. Cell Physiol. Biochem. 25, 241–252. doi: 10.1159/000276558
Fiuza-Luces, C., Valenzuela, P. L., Laine-Menendez, S., Fernandez-De La Torre, M., Bermejo-Gomez, V., Rufian-Vazquez, L., et al. (2019). Physical Exercise and Mitochondrial Disease: Insights From a Mouse Model. Front. Neurol. 10, 790. doi: 10.3389/fneur.2019.00790
Flannery, P. J., and Trushina, E. (2019). Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell Neurosci. 98, 109–120. doi: 10.1016/j.mcn.2019.06.009
Frezza, C., Cipolat, S., Martins De Brito, O., Micaroni, M., Beznoussenko, G. V., Rudka, T., et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. doi: 10.1016/j.cell.2006.06.025
Fu, W., Liu, Y., and Yin, H. (2019). Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019:9757201. doi: 10.1155/2019/9757201
Galbusera, C., Facheris, M., Magni, F., Galimberti, G., Sala, G., Tremolada, L., et al. (2004). Increased susceptibility to plasma lipid peroxidation in Alzheimer disease patients. Curr. Alzheimer Res. 1, 103–109. doi: 10.2174/1567205043332171
Gamba, P., Staurenghi, E., Testa, G., Giannelli, S., Sottero, B., and Leonarduzzi, G. (2019). A Crosstalk Between Brain Cholesterol Oxidation and Glucose Metabolism in Alzheimer’s Disease. Front. Neurosci. 13:556. doi: 10.3389/fnins.2019.00556
Gao, D., Zhang, L., Dhillon, R., Hong, T. T., Shaw, R. M., and Zhu, J. (2013). Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS One 8:e60967. doi: 10.1371/journal.pone.0060967
Gauba, E., Chen, H., Guo, L., and Du, H. (2019). Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis. 121, 138–147. doi: 10.1016/j.nbd.2018.09.020
Gautier, C. A., Kitada, T., and Shen, J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U. S. A 105, 11364–11369. doi: 10.1073/pnas.0802076105
Geisler, J. G., Marosi, K., Halpern, J., and Mattson, M.P. (2017). DNP, mitochondrial uncoupling, and neuroprotection: A little dab’ll do ya. Alzheimers Dement 13, 582–591. doi: 10.1016/j.jalz.2016.08.001
Gibson, G. E., Starkov, A., Blass, J. P., Ratan, R. R., and Beal, M.F. (2010). Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age–associated neurodegenerative diseases. Biochim. Biophys. Acta 1802, 122–134. doi: 10.1016/j.bbadis.2009.08.010
Giorgi, C., Marchi, S., and Pinton, P. (2018). The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730. doi: 10.1038/s41580-018-0052-8
Gleeson, M., Bishop, N. C., Stensel, D. J., Lindley, M. R., Mastana, S. S., and Nimmo, M. A. (2011). The anti–inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat. Rev. Immunol. 11, 607–615. doi: 10.1038/nri3041
Goldstein, L. S. (2012). Axonal transport and neurodegenerative disease: can we see the elephant? Prog. Neurobiol. 99, 186–190. doi: 10.1016/j.pneurobio.2012.03.006
Gong, B., Pan, Y., Vempati, P., Zhao, W., Knable, L., Ho, L., et al. (2013). Nicotinamide riboside restores cognition through an upregulation of proliferator–activated receptor–gamma coactivator 1alpha regulated beta–secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging. 34, 1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005
Gordon, B., Chen, S., and Durstine, J. L. (2014). The effects of exercise training on the traditional lipid profile and beyond. Curr. Sports Med. Rep. 13, 253–259. doi: 10.1249/JSR.0000000000000073
Guo, C., He, Z., Wen, L., Zhu, L., Lu, Y., Deng, S., et al. (2012). Cytoprotective effect of trolox against oxidative damage and apoptosis in the NRK–52e cells induced by melamine. Cell. Biol. Int. 36, 183–188. doi: 10.1042/CBI20110036
Guo, X., Macleod, G. T., Wellington, A., Hu, F., Panchumarthi, S., Schoenfield, M., et al. (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. doi: 10.1016/j.neuron.2005.06.027
Gupta, V. K., Scheunemann, L., Eisenberg, T., Mertel, S., Bhukel, A., Koemans, T. S., et al. (2013). Restoring polyamines protects from age–induced memory impairment in an autophagy–dependent manner. Nat. Neurosci. 16, 1453–1460. doi: 10.1038/nn.3512
Hallal, P. C., Andersen, L. B., Bull, F. C., Guthold, R., Haskell, W., Ekelund, U., et al. (2012). Global physical activity levels: surveillance progress, pitfalls, and prospects. Lancet 380, 247–257. doi: 10.1016/S0140-6736(12)60646-1
Hansen, K. G., and Herrmann, J. M. (2019). Transport of Proteins into Mitochondria. Protein J. 38, 330–342. doi: 10.1007/s10930-019-09819-6
Hollenbeck, P. J., and Saxton, W. M. (2005). The axonal transport of mitochondria. J. Cell Sci. 118, 5411–5419. doi: 10.1242/jcs.02745
Hroudova, J., Singh, N., and Fisar, Z. (2014). Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer’s disease. Biomed. Res. Int. 2014:175062. doi: 10.1155/2014/175062
Hu, C., Huang, Y., and Li, L. (2017). Drp1–Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 18:144. doi: 10.3390/ijms18010144
Huang, J. B., Hsu, S. P., Pan, H. Y., Chen, S. D., Chen, S. F., Lin, T. K., et al. (2020). Peroxisome Proliferator–Activated Receptor gamma Coactivator 1alpha Activates Vascular Endothelial Growth Factor That Protects Against Neuronal Cell Death Following Status Epilepticus through PI3K/AKT and MEK/ERK Signaling. Int. J. Mol. Sci. 21:247. doi: 10.3390/ijms21197247
Iijima-Ando, K., Hearn, S. A., Shenton, C., Gatt, A., Zhao, L., and Iijima, K. (2009). Mitochondrial mislocalization underlies Abeta42–induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS One 4:e8310. doi: 10.1371/journal.pone.0008310
Ishihara, N., Fujita, Y., Oka, T., and Mihara, K. (2006). Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977. doi: 10.1038/sj.emboj.7601184
Itoh, K., Nakamura, K., Iijima, M., and Sesaki, H. (2013). Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 23, 64–71. doi: 10.1016/j.tcb.2012.10.006
Jameson, V. J., Cocheme, H. M., Logan, A., Hanton, L. R., Smith, R. A., and Murphy, M. P. (2015). Synthesis of triphenylphosphonium vitamin E derivatives as mitochondria–targeted antioxidants. Tetrahedron 71, 8444–8453. doi: 10.1016/j.tet.2015.09.014
Jiang, Q. (2014). Natural forms of vitamin E: metabolism, antioxidant, and anti–inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 72, 76–90. doi: 10.1016/j.freeradbiomed.2014.03.035
Jing, Y.-H., Yan, J.-L., Wang, Q.-J., Chen, H.-C., Ma, X.-Z., Yin, J., et al. (2018). Spermidine ameliorates the neuronal aging by improving the mitochondrial function in vitro. Exp. Gerontol. 108, 77–86. doi: 10.1016/j.exger.2018.04.005
Kandimalla, R., Manczak, M., Fry, D., Suneetha, Y., Sesaki, H., and Reddy, P. H. (2016). Reduced dynamin–related protein 1 protects against phosphorylated Tau–induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 25, 4881–4897. doi: 10.1093/hmg/ddw312
Kandimalla, R., Manczak, M., Yin, X., Wang, R., and Reddy, P. H. (2018). Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 27, 30–40. doi: 10.1093/hmg/ddx381
Kang, C., and Li Ji, L. (2012). Role of PGC–1alpha signaling in skeletal muscle health and disease. Ann. N. Y. Acad. Sci. 1271, 110–117. doi: 10.1111/j.1749-6632.2012.06738.x
Kann, O., and Kovacs, R. (2007). Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 292, C641–C657. doi: 10.1152/ajpcell.00222.2006
Kc, S., Carcamo, J. M., and Golde, D. W. (2005). Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 19, 1657–1667. doi: 10.1096/fj.05-4107com
Kerr, J. S., Adriaanse, B. A., Greig, N. H., Mattson, M. P., Cader, M. Z., Bohr, V. A., et al. (2017). Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 40, 151–166. doi: 10.1016/j.tins.2017.01.002
Khatri, N., and Man, H. Y. (2013). Synaptic activity and bioenergy homeostasis: implications in brain trauma and neurodegenerative diseases. Front. Neurol. 4:199. doi: 10.3389/fneur.2013.00199
Kim, S. H., Vlkolinsky, R., Cairns, N., and Lubec, G. (2000). Decreased levels of complex III core protein 1 and complex V beta chain in brains from patients with Alzheimer’s disease and Down syndrome. Cell Mol. Life Sci. 57, 1810–1816. doi: 10.1007/pl00000661
Kim, S. H., Vlkolinsky, R., Cairns, N., Fountoulakis, M., and Lubec, G. (2001). The reduction of NADH ubiquinone oxidoreductase 24– and 75–kDa subunits in brains of patients with Down syndrome and Alzheimer’s disease. Life Sci. 68, 2741–2750. doi: 10.1016/s0024-3205(01)01074-8
Klopstock, T., Yu–Wai–Man, P., Dimitriadis, K., Rouleau, J., Heck, S., Bailie, M., et al. (2011). A randomized bo–controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 134, 2677–2686. doi: 10.1093/brain/awr170
Kook, S. Y., Lee, K. M., Kim, Y., Cha, M. Y., Kang, S., Baik, S. H., et al. (2014). High–dose of vitamin C supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell Death Dis. 5:e1083. doi: 10.1038/cddis.2014.26
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., Mccaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi: 10.1126/science.1099793
Kuruva, C. S., Manczak, M., Yin, X., Ogunmokun, G., Reddy, A. P., and Reddy, P. H. (2017). Aqua–soluble DDQ reduces the levels of Drp1 and Abeta and inhibits abnormal interactions between Abeta and Drp1 and protects Alzheimer’s disease neurons from Abeta– and Drp1–induced mitochondrial and synaptic toxicities. Hum. Mol. Genet. 26, 3375–3395. doi: 10.1093/hmg/ddx226
Lagouge, M., Argmann, C., Gerhart–Hines, Z., Meziane, H., Lerin, C., Daussin, F., et al. (2006). Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC–1alpha. Cell 127, 1109–1122. doi: 10.1016/j.cell.2006.11.013
Lamberts, J. T., and Brundin, P. (2017). Axonal transport dysfunction in neurodegenerative diseases: the “holy grail” for developing disease modifying therapies? Neurobiol. Dis. 105, 271–272. doi: 10.1016/j.nbd.2017.06.018
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, B. J., Tseng, Y. F., Yen, C. H., and Lin, P. T. (2013). Effects of coenzyme Q10 supplementation (300 mg/day) on antioxidation and anti–inflammation in coronary artery disease patients during statins therapy: a randomized, bo–controlled trial. Nutr. J. 12, 142. doi: 10.1186/1475-2891-12-142
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer–related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Lee, J. J., Sanchez–Martinez, A., Zarate, A. M., Beninca, C., Mayor, U., Clague, M. J., et al. (2018). Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell. Biol. 217, 1613–1622. doi: 10.1083/jcb.201801044
Li, X. C., Hu, Y., Wang, Z. H., Luo, Y., Zhang, Y., Liu, X. P., et al. (2016). Human wild–type full–length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 6:24756. doi: 10.1038/srep24756
Liang, W. S., Reiman, E. M., Valla, J., Dunckley, T., Beach, T. G., Grover, A., et al. (2008). Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. U. S. A. 105, 4441–4446. doi: 10.1073/pnas.0709259105
Ligon, L. A., and Steward, O. (2000). Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 427, 351–361. doi: 10.1002/1096-9861(20001120)427
Lin, M. Y., and Sheng, Z. H. (2015). Regulation of mitochondrial transport in neurons. Exp. Cell Res. 334, 35–44. doi: 10.1016/j.yexcr.2015.01.004
Liu, D., Pitta, M., Jiang, H., Lee, J. H., Zhang, G., Chen, X., et al. (2013). Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 34, 1564–1580. doi: 10.1016/j.neurobiolaging.2012.11.020
Llanos-Gonzalez, E., Henares-Chavarino, A. A., Pedrero-Prieto, C. M., Garcia-Carpintero, S., Frontinan-Rubio, J., Sancho-Bielsa, F. J., et al. (2019). Interplay Between Mitochondrial Oxidative Disorders and Proteostasis in Alzheimer’s Disease. Front. Neurosci. 13:1444. doi: 10.3389/fnins.2019.01444
Longo, V. D., and Mattson, M. P. (2014). Fasting: molecular mechanisms and clinical applications. Cell Metab. 19, 181–192. doi: 10.1016/j.cmet.2013.12.008
Loson, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell. 24, 659–667. doi: 10.1091/mbc.E12-10-0721
Lou, G., Palikaras, K., Lautrup, S., Scheibye–Knudsen, M., Tavernarakis, N., and Fang, E. F. (2020). Mitophagy and Neuroprotection. Trends Mol. Med. 26, 8–20. doi: 10.1016/j.molmed.2019.07.002
Lovell, M. A., Ehmann, W. D., Butler, S. M., and Markesbery, W. R. (1995). Elevated thiobarbituric acid–reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 45, 1594–1601. doi: 10.1212/wnl.45.8.1594
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., Chan, J., et al. (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. doi: 10.1038/nature02661
Lundby, C., and Jacobs, R. A. (2016). Adaptations of skeletal muscle mitochondria to exercise training. Exp. Physiol. 101, 17–22. doi: 10.1113/EP085319
Macia, E., Ehrlich, M., Massol, R., Boucrot, E., Brunner, C., and Kirchhausen, T. (2006). Dynasore, a cell–permeable inhibitor of dynamin. Dev. Cell 10, 839–850. doi: 10.1016/j.devcel.2006.04.002
Manczak, M., Calkins, M. J., and Reddy, P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509. doi: 10.1093/hmg/ddr139
Manczak, M., Kandimalla, R., Fry, D., Sesaki, H., and Reddy, P. H. (2016). Protective effects of reduced dynamin–related protein 1 against amyloid beta–induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 25, 5148–5166. doi: 10.1093/hmg/ddw330
Manczak, M., Kandimalla, R., Yin, X., and Reddy, P. H. (2019). Mitochondrial division inhibitor 1 reduces dynamin–related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 28, 177–199. doi: 10.1093/hmg/ddy335
Manczak, M., Mao, P., Calkins, M. J., Cornea, A., Reddy, A. P., Murphy, M. P., et al. (2010). Mitochondria–targeted antioxidants protect against amyloid–beta toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 20, S609–S631. doi: 10.3233/JAD-2010-100564
Masino, S. A., Rho, J. M. (2012). “Mechanisms of Ketogenic Diet Action,” in Jasper’s Basic Mechanisms of the Epilepsies, eds Th, J.L., Noebels, M., Avoli, M.A., and Rogawski, R.W. Olsen & A.V. Delgado–Escueta. (Bethesda, MD: Springer).
Matthews, R. T., Ferrante, R. J., Klivenyi, P., Yang, L., Klein, A. M., Mueller, G., et al. (1999). Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp. Neurol. 157, 142–149. doi: 10.1006/exnr.1999.7049
Matthews, R. T., Yang, L., Jenkins, B. G., Ferrante, R. J., Rosen, B. R., Kaddurah–Daouk, R., et al. (1998). Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J. Neurosci. 18, 156–163.
Mazzola, P. N., Terra, M., Rosa, A. P., Mescka, C. P., Moraes, T. B., Piccoli, B., et al. (2011). Regular exercise prevents oxidative stress in the brain of hyperphenylalaninemic rats. Metab Brain Dis. 26, 291–297. doi: 10.1007/s11011-011-9264-8
McCarthy, S., Somayajulu, M., Sikorska, M., Borowy-Borowski, H., and Pandey, S. (2004). Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water–soluble Coenzyme Q10. Toxicol. Appl. Pharmacol. 201, 21–31. doi: 10.1016/j.taap.2004.04.019
McGee, S. L., Howlett, K. F., Starkie, R. L., Cameron-Smith, D., Kemp, B. E., and Hargreaves, M. (2003). Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes 52, 926–928. doi: 10.2337/diabetes.52.4.926
McLelland, G. L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 ubiquitination by PINK1/parkin gates the p97–dependent release of ER from mitochondria to drive mitophagy. Elife 7:2866. doi: 10.7554/eLife.32866
McManus, M. J., Murphy, M. P., and Franklin, J. L. (2011). The mitochondria–targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 31, 15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011
Melkov, A., and Abdu, U. (2018). Regulation of long–distance transport of mitochondria along microtubules. Cell Mol. Life Sci. 75, 163–176. doi: 10.1007/s00018-017-2590-1
Miller, K. E., and Sheetz, M. P. (2004). Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 117, 2791–2804. doi: 10.1242/jcs.01130
Miret-Casals, L., Sebastian, D., Brea, J., Rico-Leo, E. M., Palacin, M., Fernandez-Salguero, P. M., et al. (2018). Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell. Chem. Biol. 25, 268–278 e264. doi: 10.1016/j.chembiol.2017.12.001
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J., and Baloh, R. H. (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 30, 4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010
Morris, R. L., and Hollenbeck, P. J. (1995). Axonal transport of mitochondria along microtubules and F–actin in living vertebrate neurons. J. Cell. Biol. 131, 1315–1326. doi: 10.1083/jcb.131.5.1315
Murphy, M. P., and Hartley, R. C. (2018). Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 17, 865–886. doi: 10.1038/nrd.2018.174
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. U. S. A. 113, 11249–11254. doi: 10.1073/pnas.1606786113
Nesci, S. (2020). The mitochondrial permeability transition pore in cell death: A promising drug binding bioarchitecture. Med. Res. Rev. 40, 811–817. doi: 10.1002/med.21635
Newton, A. J., Kirchhausen, T., and Murthy, V. N. (2006). Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc. Natl. Acad. Sci. U. S. A. 103, 17955–17960. doi: 10.1073/pnas.0606212103
Nichols, M., Pavlov, E. V., and Robertson, G. S. (2018). Tamoxifen–induced knockdown of the mitochondrial calcium uniporter in Thy1–expressing neurons protects mice from hypoxic/ischemic brain injury. Cell Death Dis. 9:606. doi: 10.1038/s41419-018-0607-9
Nixon, R. A., Yang, D. S., and Lee, J. H. (2008). Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4, 590–599. doi: 10.4161/auto.6259
Noh, Y. H., Kim, K. Y., Shim, M. S., Choi, S. H., Choi, S., Ellisman, M. H., et al. (2013). Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 4:e820. doi: 10.1038/cddis.2013.341
Novorolsky, R. J., Nichols, M., Kim, J. S., Pavlov, E. V., and Wilson, J. J. (2020). The cell–permeable mitochondrial calcium uniporter inhibitor Ru265 preserves cortical neuron respiration after lethal oxygen glucose deprivation and reduces hypoxic/ischemic brain injury. J. Cereb. Blood Flow Metab. 40, 1172–1181. doi: 10.1177/0271678X20908523
Ohta, S., and Ohsawa, I. (2006). Dysfunction of mitochondria and oxidative stress in the pathogenesis of Alzheimer’s disease: on defects in the cytochrome c oxidase complex and aldehyde detoxification. J. Alzheimers Dis. 9, 155–166. doi: 10.3233/jad-2006-9208
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Parikh, S., Saneto, R., Falk, M. J., Anselm, I., Cohen, B. H., Haas, R., et al. (2009). A modern approach to the treatment of mitochondrial disease. Curr. Treat Options. Neurol. 11, 414–430. doi: 10.1007/s11940-009-0046-0
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell. Calcium. 70, 87–94. doi: 10.1016/j.ceca.2017.06.008
Pena-Blanco, A., Haschka, M. D., Jenner, A., Zuleger, T., Proikas–Cezanne, T., Villunger, A., et al. (2020). Drp1 modulates mitochondrial stress responses to mitotic arrest. Cell Death Differ. 27, 2620–2634. doi: 10.1038/s41418-020-0527-y
Peng, K., Yang, L., Wang, J., Ye, F., Dan, G., Zhao, Y., et al. (2017). The Interaction of Mitochondrial Biogenesis and Fission/Fusion Mediated by PGC–1alpha Regulates Rotenone–Induced Dopaminergic Neurotoxicity. Mol. Neurobiol. 54, 3783–3797. doi: 10.1007/s12035-016-9944-9
Peng, W., Xu, S., Zhang, J., and Zhang, Y. (2019). Vitamin C Attenuates Sodium Fluoride–Induced Mitochondrial Oxidative Stress and Apoptosis via Sirt1–SOD2 Pathway in F9 Cells. Biol. Trace Elem. Res. 191, 189–198. doi: 10.1007/s12011-018-1599-0
Planavila, A., Iglesias, R., Giralt, M., and Villarroya, F. (2011). Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 90, 276–284. doi: 10.1093/cvr/cvq376
Pratico, D., Uryu, K., Leight, S., Trojanoswki, J. Q., and Lee, V. M. (2001). Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 21, 4183–4187.
Puterman, E., Lin, J., Blackburn, E., O’donovan, A., Adler, N., and Epel, E. (2010). The power of exercise: buffering the effect of chronic stress on telomere length. PLoS One 5:e10837. doi: 10.1371/journal.pone.0010837
Qi, X., Qvit, N., Su, Y. C., and Mochly–Rosen, D. (2013). A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 126, 789–802. doi: 10.1242/jcs.114439
Qin, W., Haroutunian, V., Katsel, P., Cardozo, C. P., Ho, L., Buxbaum, J. D., et al. (2009). PGC–1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 66, 352–361. doi: 10.1001/archneurol.2008.588
Rak, M., Benit, P., Chretien, D., Bouchereau, J., Schiff, M., El–Khoury, R., et al. (2016). Mitochondrial cytochrome c oxidase deficiency. Clin. Sci. 130, 393–407. doi: 10.1042/CS20150707
Rao, W., Zhang, L., Peng, C., Hui, H., Wang, K., Su, N., et al. (2015). Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim. Biophys. Acta 1852, 2402–2413. doi: 10.1016/j.bbadis.2015.08.014
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24, 981–990. doi: 10.1016/j.cellsig.2012.01.008
Reddy, P. H. (2006). Mitochondrial oxidative damage in aging and Alzheimer’s disease: implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006:31372. doi: 10.1155/JBB/2006/31372
Reddy, P. H. (2008). Mitochondrial medicine for aging and neurodegenerative diseases. Neuromole. Med. 10, 291–315. doi: 10.1007/s12017-008-8044-z
Reddy, P. H. (2014). Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug. Discov. Today 19, 951–955. doi: 10.1016/j.drudis.2014.03.020
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53. doi: 10.1016/j.molmed.2007.12.002
Reddy, P. H., and Oliver, D. M. (2019). Amyloid Beta and Phosphorylated Tau–Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 8:488. doi: 10.3390/cells8050488
Reddy, P. H., Manczak, M., Yin, X., and Reddy, A. P. (2018). Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria–Targeted Small Peptide SS31 in Alzheimer’s Disease. J. Alzheimers Dis. 62, 1549–1565. doi: 10.3233/JAD-170988
Reddy, P. H., Reddy, T. P., Manczak, M., Calkins, M. J., Shirendeb, U., and Mao, P. (2011). Dynamin–related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 67, 103–118. doi: 10.1016/j.brainresrev.2010.11.004
Redman, L. M., Smith, S. R., Burton, J. H., Martin, C. K., Il’yasova, D., and Ravussin, E. (2018). Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 27, 805–815 e804. doi: 10.1016/j.cmet.2018.02.019
Reed, T. T., Pierce, W. M. Jr., Turner, D. M., Markesbery, W. R., and Butterfield, D. A. (2009). Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J. Cell. Mol. Med. 13, 2019–2029. doi: 10.1111/j.1582-4934.2008.00478.x
Reed, T., Perluigi, M., Sultana, R., Pierce, W. M., Klein, J. B., Turner, D. M., et al. (2008). Redox proteomic identification of 4–hydroxy–2–nonenal–modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 30, 107–120. doi: 10.1016/j.nbd.2007.12.007
Renault, T. T., Floros, K. V., Elkholi, R., Corrigan, K. A., Kushnareva, Y., Wieder, S. Y., et al. (2015). Mitochondrial shape governs BAX–induced membrane permeabilization and apoptosis. Mol. Cell 57, 69–82. doi: 10.1016/j.molcel.2014.10.028
Richter, U., Lahtinen, T., Marttinen, P., Suomi, F., and Battersby, B. J. (2015). Quality control of mitochondrial protein synthesis is required for membrane integrity and cell fitness. J. Cell. Biol. 211, 373–389. doi: 10.1083/jcb.201504062
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Rodriguez-Cuenca, S., Cocheme, H. M., Logan, A., Abakumova, I., Prime, T. A., Rose, C., et al. (2010). Consequences of long–term oral administration of the mitochondria–targeted antioxidant MitoQ to wild–type mice. Free Radic. Biol. Med. 48, 161–172. doi: 10.1016/j.freeradbiomed.2009.10.039
Roger, A. J., Munoz-Gomez, S. A., and Kamikawa, R. (2017). The Origin and Diversification of Mitochondria. Curr. Biol. 27, R1177–R1192. doi: 10.1016/j.cub.2017.09.015
Ruprecht, J. J., King, M. S., Zogg, T., Aleksandrova, A. A., Pardon, E., Crichton, P. G., et al. (2019). The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 176, 435–447 e415. doi: 10.1016/j.cell.2018.11.025
Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba, E., Moullan, N., Nicolet-Dit-Felix, A. A., et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888. doi: 10.1038/nm.4132
Scarpulla, R. C. (2008). Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC–1–related coactivator. Ann. N. Y. Acad. Sci. 1147, 321–334. doi: 10.1196/annals.1427.006
Selfridge, J. E., Lu, J., and Swerdlow, R. H. (2013). Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis. 51, 3–12. doi: 10.1016/j.nbd.2011.12.057
Sies, H., and Jones, D. P. (2020). Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383. doi: 10.1038/s41580-020-0230-3
Singh, S., and Rana, S. V. (2010). Ascorbic acid improves mitochondrial function in liver of arsenic–treated rat. Toxicol. Ind. Health 26, 265–272. doi: 10.1177/0748233710365694
Singh, U., Devaraj, S., and Jialal, I. (2007). Coenzyme Q10 supplementation and heart failure. Nutr. Rev. 65, 286–293. doi: 10.1301/nr.2007.jun.286-293
Smirnova, E., Griparic, L., Shurland, D. L., and Van Der Bliek, A. M. (2001). Dynamin–related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256. doi: 10.1091/mbc.12.8.2245
Smith, G., and Gallo, G. (2017). To mdivi–1 or not to mdivi–1: Is that the question? Dev. Neurobiol. 77, 1260–1268. doi: 10.1002/dneu.22519
Smith, R. A., Porteous, C. M., Coulter, C. V., and Murphy, M. P. (1999). Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 263, 709–716. doi: 10.1046/j.1432-1327.1999.00543.x
Smith, R. A., Porteous, C. M., Gane, A. M., and Murphy, M. P. (2003). Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. U. S. A. 100, 5407–5412. doi: 10.1073/pnas.0931245100
Sohet, F. M., Neyrinck, A. M., Pachikian, B. D., De Backer, F. C., Bindels, L. B., Niklowitz, P., et al. (2009). Coenzyme Q10 supplementation lowers hepatic oxidative stress and inflammation associated with diet–induced obesity in mice. Biochem. Pharmacol. 78, 1391–1400. doi: 10.1016/j.bcp.2009.07.008
Soriano, F. X., Liesa, M., Bach, D., Chan, D. C., Palacin, M., and Zorzano, A. (2006). Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator–activated receptor–gamma coactivator–1 alpha, estrogen–related receptor–alpha, and mitofusin 2. Diabetes 55, 1783–1791. doi: 10.2337/db05-0509
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid–beta levels in a mouse model of Alzheimer’s disease. PLoS One 5:e9979. doi: 10.1371/journal.pone.0009979
Spoelstra-de Man, A. M. E., Elbers, P. W. G., and Oudemans-Van Straaten, H. M. (2018). Vitamin C: should we supplement? Curr. Opin. Crit. Care 24, 248–255. doi: 10.1097/MCC.0000000000000510
Stanga, S., Caretto, A., Boido, M., and Vercelli, A. (2020). Mitochondrial Dysfunctions: A Red Thread across Neurodegenerative Diseases. Int. J. Mol. Sci. 21:719. doi: 10.3390/ijms21103719
Steele, H., Gomez–Duran, A., Pyle, A., Hopton, S., Newman, J., Stefanetti, R. J., et al. (2020). Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol. Med. 12:e11589. doi: 10.15252/emmm.201911589
Steele, M. L., Fuller, S., Patel, M., Kersaitis, C., Ooi, L., and Munch, G. (2013). Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox. Biol. 1, 441–445. doi: 10.1016/j.redox.2013.08.006
Stokin, G. B., Lillo, C., Falzone, T. L., Brusch, R. G., Rockenstein, E., Mount, S. L., et al. (2005). Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307, 1282–1288. doi: 10.1126/science.1105681
Stowers, R. S., Megeath, L. J., Gorska-Andrzejak, J., Meinertzhagen, I. A., and Schwarz, T. L. (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. doi: 10.1016/s0896-6273(02)01094-2
Sultana, R., Poon, H. F., Cai, J., Pierce, W. M., Merchant, M., Klein, J. B., et al. (2006). Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 22, 76–87. doi: 10.1016/j.nbd.2005.10.004
Sung, H., Tandarich, L. C., Nguyen, K., and Hollenbeck, P. J. (2016). Compartmentalized Regulation of Parkin–Mediated Mitochondrial Quality Control in the Drosophila Nervous System In Vivo. J. Neurosci. 36, 7375–7391. doi: 10.1523/JNEUROSCI.0633-16.2016
Swerdlow, R. H. (2011). Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 1812, 1630–1639. doi: 10.1016/j.bbadis.2011.08.012
Szabo, A., Sumegi, K., Fekete, K., Hocsak, E., Debreceni, B., and Setalo, G. Jr., et al. (2018). Activation of mitochondrial fusion provides a new treatment for mitochondria–related diseases. Biochem. Pharmacol. 150, 86–96. doi: 10.1016/j.bcp.2018.01.038
Szeto, H. H., and Birk, A. V. (2014). Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 96, 672–683. doi: 10.1038/clpt.2014.174
Tang, B. L. (2016). Sirt1 and the Mitochondria. Mol. Cells 39, 87–95. doi: 10.14348/molcells.2016.2318
Tarnopolsky, M. A. (2011). Creatine as a therapeutic strategy for myopathies. Amino. Acids 40, 1397–1407. doi: 10.1007/s00726-011-0876-4
Tarnopolsky, M. A., and Parise, G. (1999). Direct measurement of high–energy phosphate compounds in patients with neuromuscular disease. Muscle Nerve 22, 1228–1233. doi: 10.1002/(sici)1097-4598(199909)22:9
Tobore, T. O. (2019). On the central role of mitochondria dysfunction and oxidative stress in Alzheimer’s disease. Neurol. Sci. 40, 1527–1540. doi: 10.1007/s10072-019-03863-x
Toral-Rios, D., Pichardo-Rojas, P. S., Alonso-Vanegas, M., and Campos-Pena, V. (2020). GSK3beta and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell Neurosci. 14:19. doi: 10.3389/fncel.2020.00019
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84. doi: 10.1016/j.biocel.2006.07.001
Van Laar, V. S., and Berman, S. B. (2013). The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson’s disease. Neurobiol. Dis. 51, 43–55. doi: 10.1016/j.nbd.2012.05.015
Vercesi, A. E., Kowaltowski, A. J., Oliveira, H. C., and Castilho, R. F. (2006). Mitochondrial Ca2+ transport, permeability transition and oxidative stress in cell death: implications in cardiotoxicity, neurodegeneration and dyslipidemias. Front. Biosci. 11, 2554–2564. doi: 10.2741/1990
Verschueren, K. H. G., Blanchet, C., Felix, J., Dansercoer, A., De Vos, D., Bloch, Y., et al. (2019). Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature 568, 571–575. doi: 10.1038/s41586-019-1095-5
Wang, D., Wang, J., Bonamy, G. M., Meeusen, S., Brusch, R. G., Turk, C., et al. (2012). A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem. Int. Ed. Engl. 51, 9302–9305. doi: 10.1002/anie.201204589
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009a). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Wang, X., Su, B., Siedlak, S. L., Moreira, P. I., Fujioka, H., Wang, Y., et al. (2008). Amyloid–beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U. S. A. 105, 19318–19323. doi: 10.1073/pnas.0804871105
Wang, X., Su, B., Zheng, L., Perry, G., Smith, M. A., and Zhu, X. (2009b). The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 1091, 153–159. doi: 10.1111/j.1471-4159.2009.05867.x
Wang, X., Wang, W., Li, L., Perry, G., Lee, H. G., and Zhu, X. (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 1842, 1240–1247. doi: 10.1016/j.bbadis.2013.10.015
Weaver, D., Eisner, V., Liu, X., Varnai, P., Hunyady, L., Gross, A., et al. (2014). Distribution and apoptotic function of outer membrane proteins depend on mitochondrial fusion. Mol. Cell 54, 870–878. doi: 10.1016/j.molcel.2014.03.048
Wenger, J., Klinglmayr, E., Frohlich, C., Eibl, C., Gimeno, A., Hessenberger, M., et al. (2013). Functional mapping of human dynamin–1–like GTPase domain based on x–ray structure analyses. PLoS One 8:e71835. doi: 10.1371/journal.pone.0071835
Wu, T. W., Hashimoto, N., Wu, J., Carey, D., Li, R. K., Mickle, D. A., et al. (1990). The cytoprotective effect of Trolox demonstrated with three types of human cells. Biochem. Cell Biol. 68, 1189–1194. doi: 10.1139/o90-176
Xing, J., Han, D., Xu, D., Li, X., and Sun, L. (2019). CREB Protects against Temporal Lobe Epilepsy Associated with Cognitive Impairment by Controlling Oxidative Neuronal Damage. Neurodegener. Dis. 19, 225–237. doi: 10.1159/000507023
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 12, 9–14. doi: 10.1038/nrm3028
Youle, R. J., and van Der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Zago, E. B., Castilho, R. F., and Vercesi, A. E. (2000). The redox state of endogenous pyridine nucleotides can determine both the degree of mitochondrial oxidative stress and the solute selectivity of the permeability transition pore. FEBS Lett. 478, 29–33. doi: 10.1016/s0014-5793(00)01815-9
Zahid, S., Khan, R., Oellerich, M., Ahmed, N., and Asif, A. R. (2014). Differential S–nitrosylation of proteins in Alzheimer’s disease. Neuroscience 256, 126–136. doi: 10.1016/j.neuroscience.2013.10.026
Zhang, X. D., Wang, Y., Wu, J. C., Lin, F., Han, R., Han, F., et al. (2009). Down–regulation of Bcl–2 enhances autophagy activation and cell death induced by mitochondrial dysfunction in rat striatum. J. Neurosci. Res. 87, 3600–3610. doi: 10.1002/jnr.22152
Zhao, X. L., Wang, W. A., Tan, J. X., Huang, J. K., Zhang, X., Zhang, B. Z., et al. (2010). Expression of beta–amyloid induced age–dependent presynaptic and axonal changes in Drosophila. J. Neurosci. 30, 1512–1522. doi: 10.1523/JNEUROSCI.3699-09.2010
Ziviani, E., Tao, R. N., and Whitworth, A. J. (2010). Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. U. S. A. 107, 5018–5023. doi: 10.1073/pnas.0913485107
Keywords: mitochondria, oxidative stress, fission, fusion, mitophagy, Alzheimer’s disease
Citation: Misrani A, Tabassum S and Yang L (2021) Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 13:617588. doi: 10.3389/fnagi.2021.617588
Received: 15 October 2020; Accepted: 28 January 2021;
Published: 18 February 2021.
Edited by:
Anne Eckert, University Psychiatric Clinics Basel, SwitzerlandReviewed by:
Amandine Grimm, University of Basel, SwitzerlandCopyright © 2021 Misrani, Tabassum and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Yang, eWFuZ19saUBnemh1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.