 Patricia Molina-Martínez1†
Patricia Molina-Martínez1† Rubén Corpas1,2†
Rubén Corpas1,2† Elisa García-Lara1,2
Elisa García-Lara1,2 Marta Cosín-Tomás1
Marta Cosín-Tomás1 Rosa Cristòfol1
Rosa Cristòfol1 Perla Kaliman1,3
Perla Kaliman1,3 Carme Solà1,2José Luis Molinuevo2,4,5,6
Carme Solà1,2José Luis Molinuevo2,4,5,6 Raquel Sánchez-Valle2,4,5Anna Antonell2,4,5
Raquel Sánchez-Valle2,4,5Anna Antonell2,4,5 Albert Lladó2,4,5*‡
Albert Lladó2,4,5*‡ Coral Sanfeliu1,2*‡
Coral Sanfeliu1,2*‡- 1Institut d’Investigacions Biomèdiques de Barcelona (IIBB), Consejo Superior de Investigaciones Científicas (CSIC), Barcelona, Spain
- 2Institut d’Investigació Biomèdica August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 3Faculty of Health Sciences, Universitat Oberta de Catalunya, Barcelona, Spain
- 4Alzheimer’s Disease and Other Cognitive Disorders Unit, Department of Neurology, Hospital Clínic, Barcelona, Spain
- 5Fundació Clínic per a la Recerca Biomèdica, Universitat de Barcelona, Barcelona, Spain
- 6Centro de Investigación Biomédica en Red de Fragilidad y Envejecimiento Saludable (CIBERFES), Madrid, Spain
Neuroinflammation is a risk factor for Alzheimer’s disease (AD). We sought to study the glial derangement in AD using diverse experimental models and human brain tissue. Besides classical pro-inflammatory cytokines, we analyzed chitinase 3 like 1 (CHI3L1 or YKL40) and triggering receptor expressed on myeloid cells 2 (TREM2) that are increasingly being associated with astrogliosis and microgliosis in AD, respectively. The SAMP8 mouse model of accelerated aging and AD traits showed elevated pro-inflammatory cytokines and activated microglia phenotype. Furthermore, 6-month-old SAMP8 showed an exacerbated inflammatory response to peripheral lipopolysaccharide in the hippocampus and null responsiveness at the advanced age (for this strain) of 12 months. Gene expression of TREM2 was increased in the hippocampus of transgenic 5XFAD mice and in the cingulate cortex of autosomal dominant AD patients, and to a lesser extent in aged SAMP8 mice and sporadic early-onset AD patients. However, gene expression of CHI3L1 was increased in mice but not in human AD brain samples. The results support the relevance of microglia activation in the pathways leading to neurodegeneration and suggest diverse neuroinflammatory responses according to the AD process. Therefore, the SAMP8 mouse model with marked alterations in the dynamics of microglia activation and senescence may provide a complementary approach to transgenic mouse models for the study of the neuroinflammatory mechanisms underlying AD risk and progression.
Introduction
Neuroinflammation and peripheral inflammatory conditions associated with aging or some pathological conditions are known risk factors for triggering sporadic Alzheimer’s disease (AD) (Bolós et al., 2017; Tejera et al., 2019). Furthermore, lower peripheral inflammation is one of the markers associated with preserved memory at middle age (Corpas et al., 2019). Accordingly, experimental studies have shown neuroprotection by diverse anti-inflammatory agents against memory loss and AD-like pathology (Corpas et al., 2017; Ettcheto et al., 2017). Furthermore, some observational studies have shown the potential of non-steroidal anti-inflammatory drugs (NSAIDs) for prevention of AD (Wang et al., 2015a). However, a number of clinical trials of NSAIDs and other anti-inflammatory drugs reported to date have shown either their lack of efficacy against AD or serious side effects (Elmaleh et al., 2019). The recent failure of the NSAID naproxen to reduce the progression of presymptomatic AD (Meyer et al., 2019) has prompted the redesign of clinical studies (Hershey and Lipton, 2019) and efforts to broaden the pathways of study in the search for new inflammatory targets (Sharman et al., 2019). The relevance of neuroinflammation in the development of AD is further confirmed by the fact that inflammation-related proteins, including the astroglia-based YKL40 [also known as chitinase 3 like 1 (CHI3L1)] and the microglia-based triggering receptor expressed on myeloid cells 2 (TREM2), are currently considered to be potential AD biomarkers in CSF or blood (Molinuevo et al., 2018). The corresponding genes CHI3L1 and TREM2 are widely expressed in cerebral tissue, including the hippocampus and cortical areas sensitive to AD pathology (Guerreiro et al., 2013; Sanfilippo et al., 2019).
In experimental studies, mouse models of AD exhibit a diverse degree of neuroinflammation that generally correlates with the amyloid burden. For instance, the 5XFAD transgenic AD mouse model develops abundant amyloid plaque deposits surrounded by activated astrocyte and microglia (Oakley et al., 2006) in parallel with increased brain levels of pro-inflammatory cytokines (Griñán-Ferré et al., 2016b). However, the neuroinflammatory mechanism may differ between AD mouse models and humans (Hemonnot et al., 2019), which may have contributed to the failure of neuroinflammation targeted drugs in clinical trials of AD (Kodamullil et al., 2017). The senescence-accelerated mouse prone 8 (SAMP8) strain is a spontaneous mouse model of accelerated aging that has been proposed as a model of late-onset sporadic AD (Cheng et al., 2014; Liu et al., 2020). It exhibits neuroinflammation, memory loss and mild AD-like pathology (Álvarez-López et al., 2014; Griñán-Ferré et al., 2016a; Ito et al., 2020). At the systemic level, SAMP8 shows increased concentrations of circulating pro-inflammatory factors and increased activation of inflammatory pathways in several organs in comparison with the control strain senescence-resistant mouse 1 (SAMR1) (i.e., Miró et al., 2017; Fernández-García et al., 2019). Furthermore, we have previously demonstrated that cell cultures of SAMP8 embryonic or neonatal brain maintain the pathogenic dysfunctions found in the adult mouse brain (García-Matas et al., 2008; Díez-Vives et al., 2009; Cristòfol et al., 2012). Therefore, SAMP8 may be a good model in which to study the mechanisms underlying the risk of sporadic AD associated with age-related inflammation. Moreover, systemic inflammation may induce or aggravate neuroinflammation. In this regard, peripheral administration of lipopolysaccharide (LPS) from bacterial endotoxin is widely used to induce inflammatory responses in mice for modeling neurodegeneration processes (for a review see: Catorce and Gevorkian, 2016). LPS is the major component of the outer membrane leaflet of Gram-negative bacteria. A structural motif of LPS is recognized by the innate immune cells as one of the pathogen-associated molecular patterns (PAMPs) that stimulate Toll-like receptors (TLRs); LPS mainly stimulates TLR4. The LPS/TLR4 signaling pathway may trigger potent immune responses (Lu et al., 2008). In brief, LPS-activated peritoneal macrophages and dendritic cells of the innate immune system produce Interleukin 1 (IL1α and IL1β), a first-line mediator in the signaling cascade that would help to fight the infection; this includes vagal stimulation to induce sickness behavior, and further synthesis of IL1β in cells lying outside the blood–brain barrier that would reach brain target cells (mainly microglia, but also endothelial cells and macrophage-like cells among others) to induce prostaglandins and pro-inflammatory cytokines (Konsman et al., 2002). A comparable response of transient sickness behavior and microglial activation is also elicited in humans after a single peripheral administration of LPS (Schedlowski et al., 2014; Sandiego et al., 2015), thus giving value to the LPS challenge in experimental models. Besides changes in the classical pro-inflammatory cytokines and nitric oxide pathways, analysis of the proposed AD biomarkers CHI3L1 and TREM2 may help us to understand the mechanisms involved in neuroinflammatory changes.
We aimed to analyze the reactive gliosis of SAMP8 mouse brain and those of astrocytes and microglia in vitro, either in basal conditions or after an acute injury with LPS. Using SAMR1 and SAMP8 mice in adulthood and advanced age we aimed to discern age-related changes. Next, we analyzed CHI3L1 and TREM2 gene expression in several scenarios, including the transgenic mouse 5XFAD and post-mortem human samples with advanced AD pathology. A study of these genes in the hippocampus of SAMP8 and 5XFAD mice and in the posterior cingulate cortex of human patients with sporadic early-onset AD (sEOAD) and autosomal dominant AD (ADAD) may help reveal the neuroinflammatory mechanisms underlying AD risk and progression.
Materials and Methods
Animals
The senescence-prone SAMP8 mouse strain and the control senescence-resistant SAMR1 strain were developed by selective breeding at Kyoto University (Takeda et al., 1994). SAMP8 mice show accelerated brain senescence with cognitive impairment, neuroinflammation, oxidative stress, traits of amyloid and tau pathologies, and epigenetic alterations (López-Ramos et al., 2012; Cosín-Tomás et al., 2014; Griñán-Ferré et al., 2018). Male SAMP8 and SAMR1 mice aged 6 and 12 months were used for this study. Six-month-old SAMP8 mice show the full pathological phenotype, whereas 12-month-old mice are near the mean survival age and have been poorly analyzed (Porquet et al., 2013). Heterozygous transgenic AD mice of the strain 5XFAD (Oakley et al., 2006) and their wild type siblings (WT), 9-month-old males, were used for selected analysis. All mice were bred at the Animal House of the University of Barcelona (UB, Barcelona, Spain). First progenitors of the SAMP8/SAMR1 strains and 5XFAD were obtained from Harlan (Envigo, Barcelona, Spain) and from Jackson Laboratory (Bar Harbor, ME, United States), respectively. Animals were maintained under standard laboratory conditions of food and water ad libitum, 22 ± 2°C, and 12 h:12 h light-dark cycle. All experimental protocols and procedures were approved by the local Ethics Committee for Animal Experimentation (CEEA, UB; DAAM 7136 and DAAM 9323), in accordance with the Decree 214/97 of the Generalitat de Catalunya, Spanish legislation and the European Union Directive 2010/63/EU for animal experiments.
Glial Cell Cultures
Astrocytes and microglia cultured from neonatal SAMP8 neocortex show senescent and pathological traits that might greatly contribute to pathological brain aging in these mice (García-Matas et al., 2008, 2015; Díez-Vives et al., 2009). Here we used mixed glial cultures enriched in astrocytes and almost pure microglia cultures to discern inflammation mechanisms at the cellular level.
Mixed glial cultures enriched in astrocytes were prepared from the cerebral cortices of 2-day-old SAMP8 and SAMR1 mice as previously described (Sola et al., 2011). Briefly, brains were dissected free of the meninges, diced into small cubes and dissociated by incubation with a 0.5% trypsin-EDTA solution (Gibco) for 25 min. Cells were seeded at 5 × 104 cells/cm2 in multi-well plates or on glass coverslips in DMEM supplemented with 2.5 mM glutamine, 100 μg/mL gentamycin and 20% fetal bovine serum (FBS) at 37°C in a humidified incubator with 5% CO2. The culture medium was changed every 3–4 days and FBS was progressively lowered to 10% during the first 2 weeks of culture. Experiments were routinely carried out at 21 days in vitro. Established mixed glia cultures of both SAMR1 and SAMP8 consisted of 85–90% astrocytes, 10–15% microglia, and 0.1–1% oligodendroglia.
Microglial cultures were obtained from mature mixed glial cultures prepared as described above, followed by mild trypsinization to detach an upper layer of mixed glia while maintaining a bottom layer of microglia attached to the plate (Saura et al., 2003). Cultures were used immediately. Established microglia cultures of both SAMR1 and SAMP8 consisted of >98% microglial cells.
Lipopolysaccharide Treatment
In vivo, SAMP8 and SAMR1 mice were subjected to a pro-inflammatory injury by administration of LPS from Escherichia coli (serotype 026:B6; Sigma, Saint Louis, MO, United States). LPS was dissolved in sterile physiological saline solution to obtain 100 μg in 0.1 mL for intraperitoneal injection. Mice received a dose of 3 mg per kg body weight or the equivalent volume of vehicle. The experiment was terminated after 3 h of treatment with LPS or vehicle. The experimental groups were as follows: SAMR1-Control 6 months (n = 10); SAMR1-Control 12 months (n = 7); SAMP8-Control 6 months (n = 6); SAMP8-Control 12 months (n = 3); SAMR1-LPS 6 months (n = 10); SAMR1-LPS 12 months (n = 7); SAMP8-LPS 6 months (n = 5); and SAMP8-LPS 12 months (n = 4). SAMP8 mice have a reduced lifespan with median and maximum life expectancies of 10 and 16 months, respectively (Porquet et al., 2013). Therefore, older SAMP8 mice were visually inspected for overall health appearance prior to their inclusion in the study.
In vitro, pro-inflammatory injury was induced in glial cell cultures by adding LPS at the final concentration of 100 ng/mL in the culture medium. Interferon-γ (IFN; Sigma) at the final concentration of 0.1 ng/mL was simultaneously added to potentiate the effects of LPS (Straccia et al., 2011). Experiments were terminated after 24 h of exposure to LPS + IFN or vehicle (saline). Conditioned culture media and/or cells were immediately collected for further assays. All experiments were performed in cells from at least n = 3 independent primary cultures.
Sickness Behavior
Decreased motivation to engage in social exploratory behavior is used to assess sickness behavior induced by an infection (Dantzer, 2001). Sickness behavior shows the adaptive reorganization of an animal’s priorities. SAMP8 and SAMR1 mice were subjected to the social behavior test immediately before administration of LPS or vehicle and again 3 h later. A juvenile conspecific male mouse was introduced into the test subject’s home cage for a 5-min period. The social interaction between the subject and the juvenile intruder was video-recorded and the length of time spent engaged in social investigation was determined from the video records. Social behavior was determined as the amount of time that the experimental subject spent investigating (e.g., anogenital sniffing and trailing) the intruder (Fishkin and Winslow, 1997). The results were expressed as the ratio between the time devoted to social behavior after the treatment, and the respective baseline response.
Mouse Blood and Brain Tissue Samples
After completion of the behavioral test, 6- and 12-month-old SAMP8 and SAMR1 mice were decapitated to collect trunk blood. Blood was allowed to clot at 4°C for 30 min and centrifuged to obtain serum. The cerebral cortex and hippocampus were immediately dissected on a cold plate. Samples of blood serum and cerebral tissues were stored at −80°C for further analysis. All samples were obtained 3 h after LPS or vehicle injection. Brain tissue of 9-month-old 5XFAD (n = 4) and WT mice (n = 4) was also obtained and stored at −80°C until analysis.
Human Tissue Samples
Brain tissue samples of sEOAD, ADAD and neurologically healthy controls (NHC) were obtained from the Neurological Tissue Biobank of Hospital Clínic – IDIBAPS and the Neuropathology Institute of the Hospital Universitari de Bellvitge. All subjects or their legal representatives provided written consent for the use of the brain samples and the study was approved by the Ethics Committee of the Hospital Clínic of Barcelona. All procedures were conducted in accordance with the 1964 Declaration of Helsinki and its later amendments. Postmortem brain tissue was evaluated following standardized pathological procedures and each patient’s disease was classified according to international consensus criteria (Hyman et al., 2012; Montine et al., 2012). Following these criteria, AD patients included in the study were classified as A3, B3, and C3. All patients were previously screened for the APOE genotype and for mutations in the PSEN1, PSEN2, and APP genes (Antonell et al., 2013). Eight samples per group were used for this study. Characteristics of the subjects are displayed in Table 1. ADAD subjects bore PSEN1 mutations (L286P, V89L, M139T and E120G). The three groups were balanced for sex, postmortem delay and APOE genotype. ADAD and NHC groups were also matched by age, but sEOAD was significantly older than the other two groups [one-way ANOVA, F(2,21) = 12.92, p < 0.001; sEOAD vs. ADAD p < 0.01 and sEOAD vs. NHC, p < 0.001].
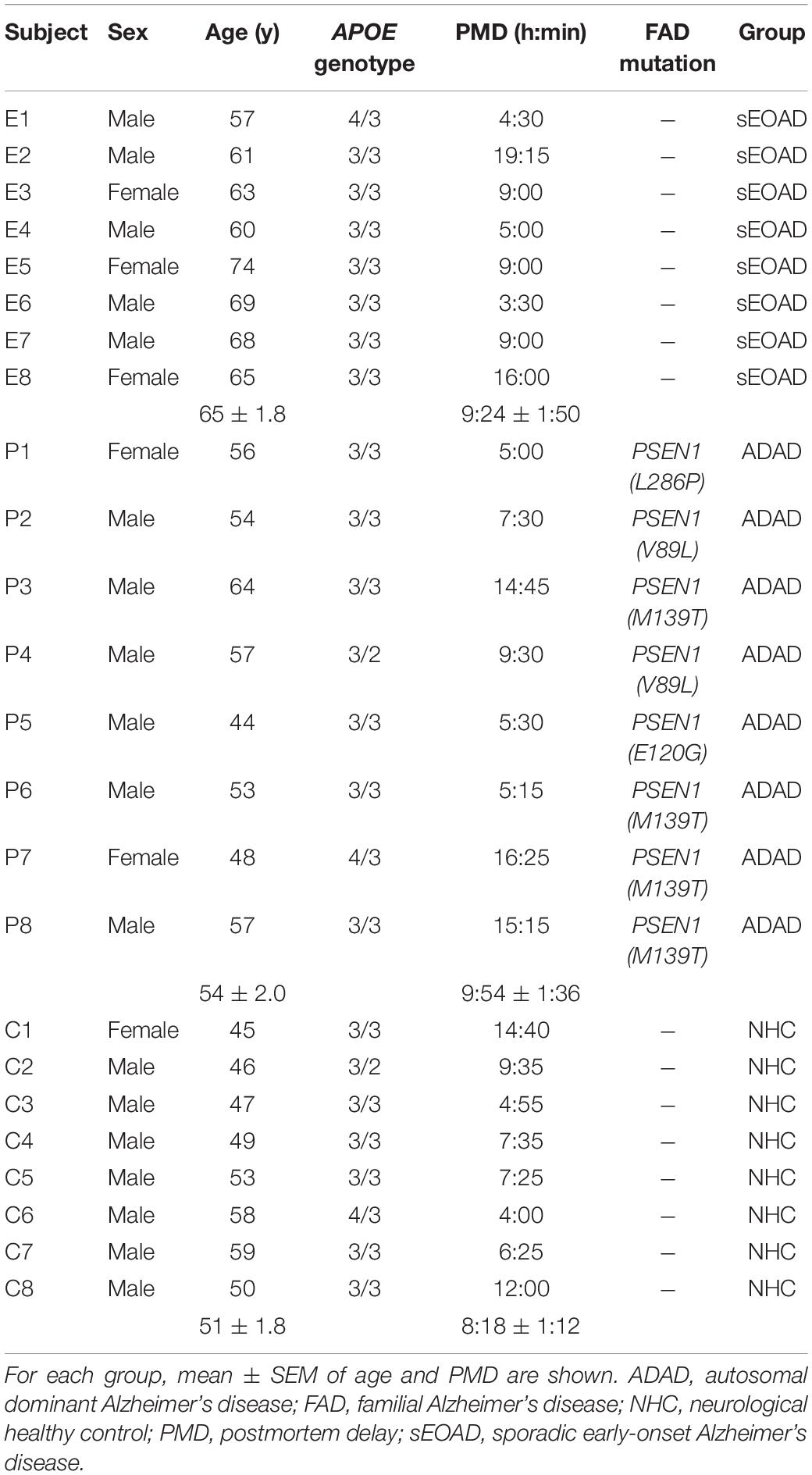
Table 1. Characteristics of the human subjects included in the study.
RNA Extraction and qPCR Analysis
Total RNA was isolated from mouse tissue samples using the mirVanaTM miRNA Isolation Kit with phenol (Applied Biosystems, Foster City, CA, United States) in accordance with the manufacturer’s instructions for obtaining total RNA, including small RNA. Isolation of total RNA from frozen brain tissue of the posterior cingulate area at the thalamus level was performed using an RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, Germany). RNA yield, purity, and quality were determined using a NanoDropTM ND1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). RNAs with a 260/280 ratio of >1.9 were selected. Random-primed cDNA synthesis was performed using the High-Capacity cDNA Archive kit (Applied Biosystems). Gene expression was measured in a CFX96 Real-Time qPCR Detection System (Bio-Rad, Hercules, CA, United States), using specific TaqMan FAM-labeled probes (Applied Biosystems). The expression of the genes Il1b, Il6 and Tnf, coding for the common pro-inflammatory cytokines IL1β, Interleukin 6 (IL6), and tumor necrosis factor α (TNFα), respectively, were analyzed in SAMP8 and SAMR1 cortex and hippocampus. The expression of the mouse genes Chil1 and Trem2 that code for CHI3L1 and TREM2, respectively, were analyzed in the hippocampus of SAMP8, SAMR1, 5XFAD, and WT mice. The human counterpart genes CHI3L1 and TREM2 were analyzed in human samples of the posterior cingulate area at the thalamus level. Mouse data were normalized to TBP gene expression and human data to PGK1 and B2M. mRNA levels were expressed as fold change of the control group (6-month-old SAMR1 treated with vehicle, WT mice or NHC, as appropriate). A list of primers utilized is presented in Table 2.
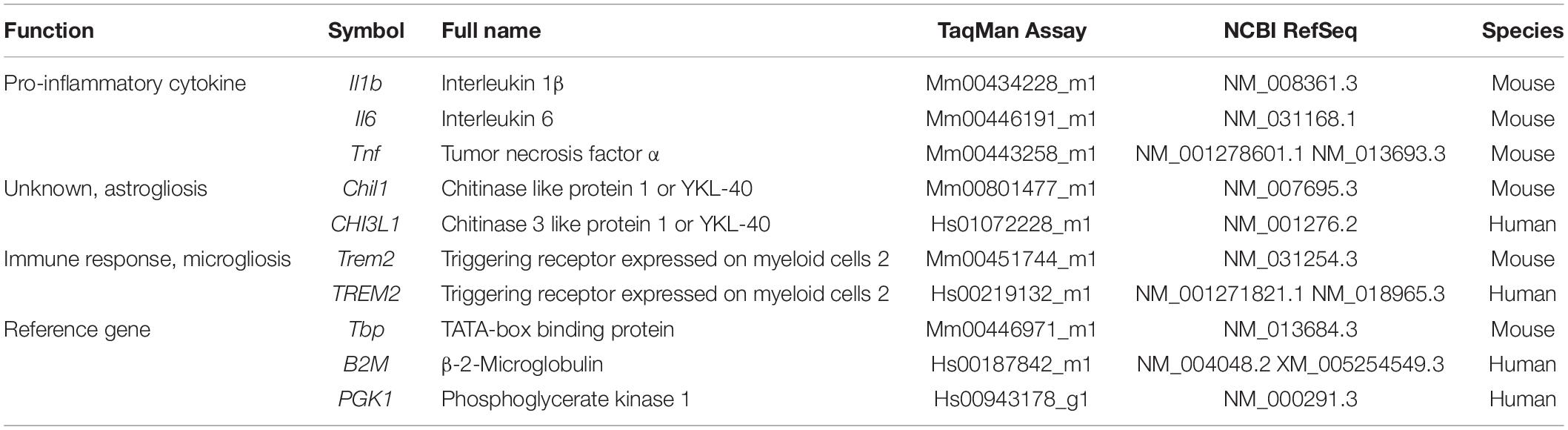
Table 2. Genes analyzed and TaqMan FAM-labeled probes for real-time qPCR.
Morphology of Cultured Glial Cells
The activated phenotype of glial cells was confirmed by microscopic examination of stained cultures. Glial cells grown on glass coverslips were fixed with 4% paraformaldehyde and astrocytes and microglia were specifically stained using standard histological procedures. Astrocytes were immunostained with the primary antibody to Glial fibrillary acidic protein (GFAP) (1:500; Dako Z0334; Agilent, Santa Clara, CA, United States) followed by the fluorescent Alexa Fluor 546 species-specific conjugated secondary antibody (1:1000; Thermo Fisher Scientific, Waltham, MA, United States). Microglia were stained with lectin from Bandeiraea simplicifolia conjugated with fluorescein (1:400; Sigma, L2895). Coverslips were mounted upside down on glass slides with Mowiol 4-88 (Sigma 81381) media. Astrocyte cultures from SAMR1 and SAMP8 showed similar basal morphology and similar morphological transformation in response to LPS + IFN. Microglia proliferation and relative number of cells with morphological changes were analyzed by cell count in microphotographs using Cell F software (Olympus, Shinjuku, Tokyo, Japan).
Nitrite Assay
Nitric oxide generation by activated glia in culture was measured by the colorimetric Griess reaction that detects nitrite (NO2–), a stable reaction product of nitric oxide and molecular oxygen. Briefly, 100 μL of conditioned medium were incubated with 100 μL of Griess reagent for 10 min at room temperature. The optical density was measured at 540 nm using a microplate reader (iEMS Reader MF; Labsystems, Vantaa, Finland). The nitrite μM concentration was determined from a sodium nitrite standard curve.
Cytokine Determinations
The levels of the pro-inflammatory cytokines IL1β, IL6, and TNFα were determined in homogenates of SAMP8 and SAMR1 cerebral cortical tissue and in conditioned culture media. The protein concentration of tissue homogenates was determined using the Bradford assay. IL1β in the serum of the mice was also determined to identify the peripheral response to LPS. Cytokines were determined using commercial ELISA kits, following the manufacturer’s instructions. Mouse IL1β Quantikine ELISA Kit (MLB00C) was purchased from R&D Systems (Minneapolis, MI, United States), Murine IL6 ELISA Set (861.020.005) from Diaclone (Besançon, France) and Mouse TNFα ELISA Ready-SET-Go (88-7324-22) from Thermo Fisher. Samples were measured at 450 nm using a plate reader (iEMS Reader MF; Labsystems, Vantaa, Finland). Data were expressed as pg/ml of blood serum or pg/mg of protein, as appropriate.
Statistical Analysis
The results are expressed as mean ± SEM. The distribution of the data was checked with the Shapiro–Wilk test, and data were log-transformed into normality where required. Data from the SAMP8 and SAMR1 mice with two factors (strain and treatment in the in vitro experiments) or three factors (strain, treatment and age in the in vivo experiments) were analyzed by ANOVA to obtain the respective main effects and interaction effects. All ANOVA factors have two levels and therefore no further analysis was performed in the absence of statistically significant interaction. Experimental groups were compared using Fisher’s least significant difference (LSD) test when there was an interaction between factors. Student’s t-test was used for comparison between 5XFAD and WT mice. The human tissue results were analyzed by one-way ANOVA followed by LSD post hoc test. Statistical analyses were performed using IBM SPSS Statistics v22.
Results
Inflammatory Phenotype of SAMP8 Mice
Lipopolysaccharide induced a reduction in social exploration indicative of sickness behavior as expected, showing the effectiveness of the treatment in 6- and 12-month-old SAMR1 and SAMP8 mice. The results 3 h after the injection are depicted in Figure 1A. SAMR1 and SAMP8 mice showed similar responses to LPS injection. Furthermore, both SAMR1 and SAMP8 showed lower social exploration with age [three-way ANOVA, main effect of treatment: F(1,44) = 55.086, p < 0.001; main effect of age: F(1,44) = 4.323, p = 0.043].
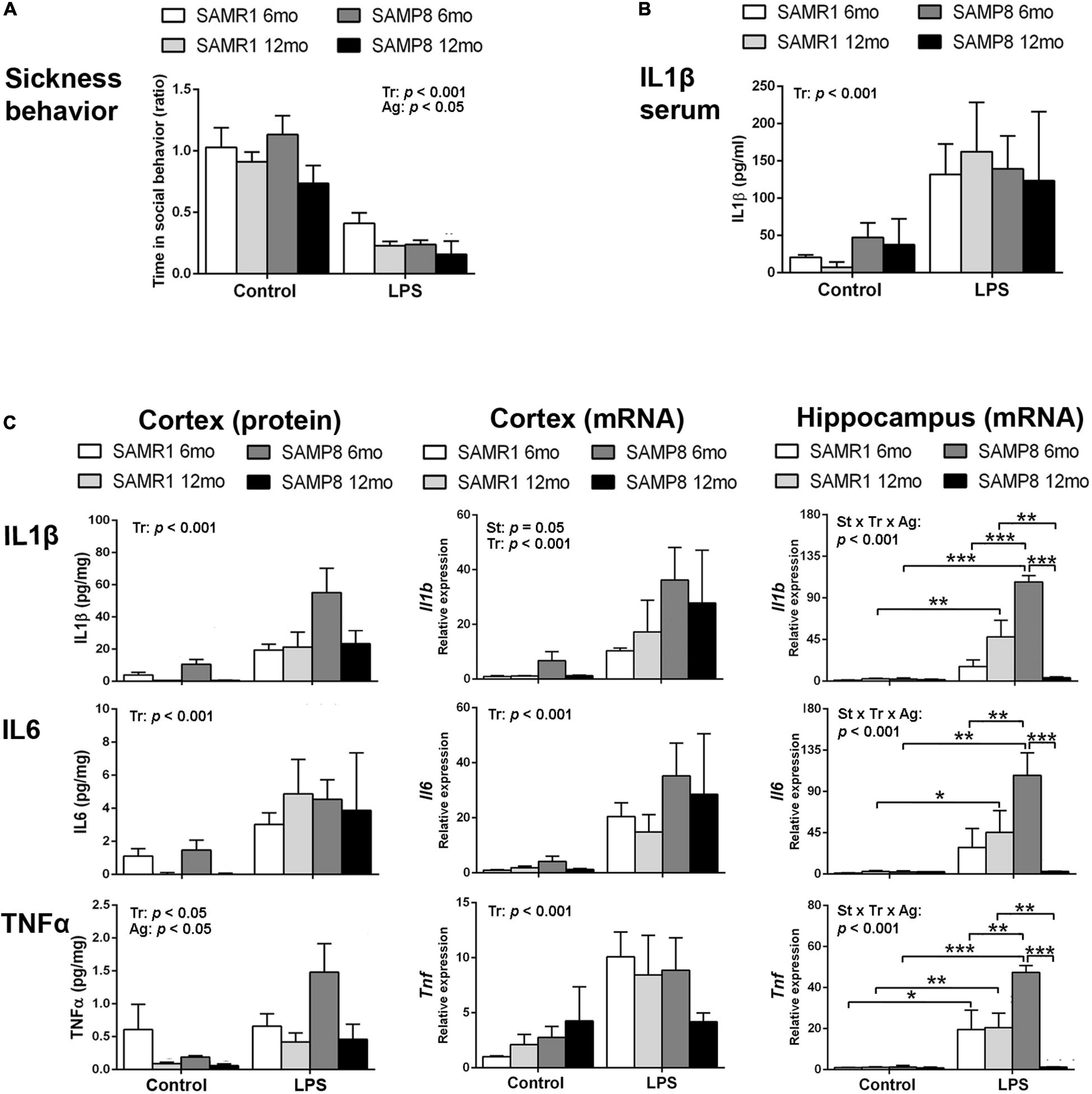
Figure 1. Hippocampus of SAMP8 mice revealed an exacerbated acute inflammatory response to lipopolysaccharide (LPS) at young age and null responsiveness at old age. Sickness behavior 3 h after 3 mg/kg i.p. of LPS showed the effect of treatment and a lowering effect of age in the response of both SAMR1 and SAMP8 mice (A). Blood serum levels of Interleukin 1β (IL1β) were increased in all groups of mice treated with LPS for 3 h regardless of strain and age (B). Cerebral cortical levels of protein and mRNA of pro-inflammatory cytokines Interleukin 1β (IL1β/Il1b, protein/gene), Interleukin 6 (IL6/Il6, protein/gene) and Tumor necrosis factor α (TNFα/Tnf, protein/gene) were generally elevated by LPS in all mice, with lower protein levels of TNFα in all aged mice and higher mRNA levels of Il1b in SAMP8 mice; mRNA levels of all cytokines in the hippocampus showed exacerbated levels in 6-month-old SAMP8 compared to SAMR1 mice and low levels of cytokines indicative of null responsiveness to a pro-inflammatory injury in 12 month-old SAMP8 mice 3 h after LPS injection (C). P-values for two-way ANOVA analysis are indicated at the top area of the graph: St, strain main effect; Tr, treatment main effect; Ag, age main effect; and St × Tr × Ag, interaction effect. P-values for Fisher’s LSD post hoc tests are indicated as: *p < 0.05; **p < 0.01, ***p < 0.001 compared to the corresponding SAMR1 group, the corresponding control treatment group, or the corresponding younger mice group, as indicated. N = 3–10 mice/group.
Protein levels of the first-line pro-inflammatory cytokine IL1β in the blood serum of these animals are shown in Figure 1B. There was a significant increase in response to LPS stimulus in all groups, without effect of strain or age [three-way ANOVA, main effect of treatment: F(1,18) = 18,371, p < 0.001].
Protein and mRNA levels of the pro-inflammatory cytokines IL6, IL1β, and TNFα for these mice 3 h after the injection of LPS or vehicle are shown in Figure 1C. Protein levels of IL1β, IL6, and TNFα generally increased in the cerebral cortex of SAMR1 and SAMP8 mice in response to LPS systemic injury as indicated by statistical significance of treatment in ANOVA [three-way ANOVA, main effect of treatment: F(1,51) = 15.639, p < 0.001 for IL1β; F(1,42) = 15,475, p < 0.001 for IL6; and F(1,48) = 5.179, p = 0.027 for TNFα]. However, the results of 6-month SAMR1 protein levels and 12-month SAMP8 mRNA levels showed no tendency to increase those of TNFα after treatment with LPS. Furthermore, TNFα levels generally decreased with age [three-way ANOVA, main effect of age: F(1,48) = 7.165, p = 0.010 for TNFα]. A visible trend for lower IL1β and IL6 levels in the control treatment in 12-month-old mice compared to 6-month-old mice for both strains did reach significance when vehicle-injected mice were analyzed separately from LPS groups (not indicated in the figure) [two-way ANOVA, main effect of age: F(1,24) = 7.723, p = 0.010 for IL1β; F(1,22) = 10.257, p = 0.004 for IL6].
LPS induced increased mRNA levels of Il1b, Il6, and Tnf in the cerebral cortex of both strains of mice [three-way ANOVA, main effect of treatment: F(1,30) = 40.574, p < 0.001 for Il1b, F(1,33) = 28.382, p < 0.001 for Il6, and F(1,30) = 19.163, p < 0.001 for Tnf]. Furthermore, SAMP8 showed higher levels of Il1b mRNA [main effect of strain: F(1,30) = 4.159, p = 0.050 for Il1b].
In the hippocampus, LPS also induced an increase in the mRNA of the three cytokines [three-way ANOVA, main effect of treatment: F(1,23) = 27.187, p < 0.001 for Il1b, F(1,22) = 30.821, p < 0.001 for Il6, F(1,23) = 34.536, p < 0.001 for Tnf]. However, the levels attained were very high in the 6-month-old SAMP8 mice compared to SAMR1 and to the corresponding levels in SAMP8 cerebral cortical tissue. Furthermore, 12-month-old SAMP8 mice showed significantly lower mRNA levels of the three cytokines than in 6-month-old SAMP8 mice after LPS injection [three-way ANOVA, main effect of age: F(1,23) = 4.839, p = 0.038 for Il1b and F(1,23) = 10.082, p = 0.004 for Tnf; effect of treatment × strain × age interaction: F(1,23) = 17.257, p < 0.001 for Il1b, F(1,22) = 4.326, p = 0.049 for Il6, F(1,23) = 10.542, p = 0.004 for Tnf].
Overall, SAMP8 mice were similarly responsive than SAMR1 mice against LPS injection at the peripheral level. However, there was a differential response in the brain tissue. Specifically, SAMP8 mice showed a general increase of Il1b expression in the cerebral cortex and, most noticeably, an extreme activation of the gene expression of Il1b, Il6 and Tnf induced by LPS in the hippocampus at young age followed by null responsiveness to infection-like stimulus at old age. Therefore, there was an exacerbated immune response in young SAMP8 and immunosuppression in aged SAMP8 hippocampi.
Inflammatory Phenotype of SAMP8 Glial Cultures
Phenotypic images of astrocytes and microglia in mixed glial cultures from SAMR1 and SAMP8 mice are shown in Figure 2A. SAMP8 astrocytes were visualized by immunostaining of the GFAP marker in mixed glial cultures highly enriched in these cells. In control conditions, SAMP8 astrocytes had a similar morphology to SAMR1 astrocytes. Both astrocyte strains acquired a typical activated morphology with filiform processes after pro-inflammatory injury induced by LPS + IFN treatment. However, SAMP8 microglia stained with lectin showed a globular morphology indicative of the activated state in control conditions similar to that of SAMR1 microglia after LPS + IFN injury. Pro-inflammatory treatment also induced proliferation in SAMP8 microglia, but not in SAMR1 microglia, as confirmed by cell count (upper histogram, Figure 2A) [two-way ANOVA, main effect of strain: F(1,29) = 11.42, p = 0.0021, main effect of treatment: F(1,29) = 15.26, p = 0.0005, and effect of strain × treatment: F(1,29) = 14.72, p = 0.0006]. Furthermore, the morphological transformation appeared more intense in SAMP8 microglia treated with LPS + IFN than in the SAMR1 counterparts. Cell counts revealed a higher percentage of activated microglia in SAMP8 under both control and pro-inflammatory conditions (lower histogram, Figure 2A) [two-way ANOVA, main effect of strain: F(1,29) = 39.19, p < 0.0001, main effect of treatment: (1, 29) = 17.97, p = 0.0002].
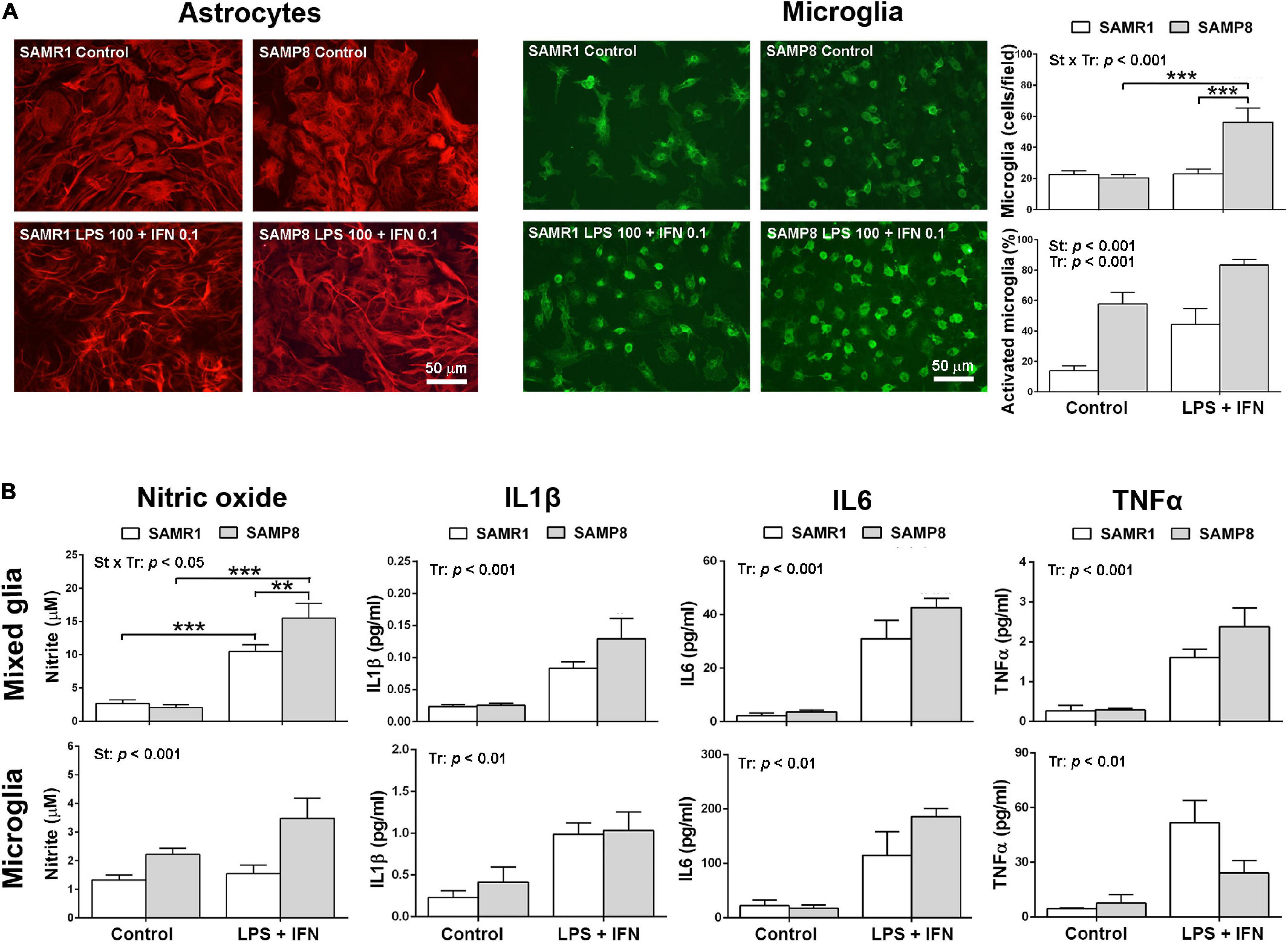
Figure 2. Mixed glial cultures of SAMP8 senescent mice had a pro-inflammatory phenotype mainly driven by microglia. Representative images of astrocytes stained with glial fibrillary acidic protein (GFAP) and microglia cultures stained with lectin in mixed glial cultures in control conditions or submitted to a 24-h treatment with lipopolysaccharide (LPS) 100 ng/ml + interferon γ (IFN) 0.1 ng/ml; histograms of the average cell counts of microglia per microscopic field and the percentage of microglia with reactive phenotype shown by globular morphology, as indicated (A). Histograms of the nitric oxide generation, and the levels of pro-inflammatory cytokines Interleukin 1β (IL1β), Interleukin 6 (IL6) and Tumor necrosis factor α (TNFα) released into the culture media of mixed cultures and microglia cultures, in control conditions or treated with LPS 100 ng/ml + IFN 0.1 ng/ml, as indicated (B). P-values for two-way ANOVA analysis are indicated at the top area of the graph: St, strain main effect; Tr, treatment main effect; and St × Tr, interaction effect. P-values for Fisher’s LSD post hoc tests between the groups indicated in the graph are as follows: **p < 0.01, ***p < 0.001. N = 6–10 (microglia micrographs) from 3 independent cultures per group for analysis of microglia number and phenotype in mixed glial cultures, N = 18–24 (mixed glia)/15–26 (microglia) from 4 to 6 independent cultures per group for nitrite determination and N = 4–8 (mixed glia)/3–6 (microglia) independent cultures per group for cytokine analysis. Scale bar = 50 μm.
Pro-inflammatory physiological changes in SAMR1 and SAMP8 cultures were evaluated by the levels of nitric oxide and cytokine release. The results are shown in Figure 2B. Mixed glial cultures increased nitric oxide generation following treatment with LPS + IFN, as determined by nitrite accumulation in the culture media. This response was higher in SAMP8 cultures than SAMR1 cultures [two-way ANOVA, main effect of treatment: F(1,74) = 89.93, p < 0.0001; main effect of strain: F(1,74) = 3.992, p = 0.0494; effect of strain × treatment interaction: F(1,74) = 6.255, p = 0.0146]. SAMP8 pure microglia cultures also showed higher nitric oxide generation than SAMR1 microglia, although the effect of treatment or strain × treatment interaction did not reach significance [two-way ANOVA, main effect of strain: F(1,75) = 12.61, p = 0.0007]. However, there was a tendency for a difference between nitric oxide levels after LPS + IFN injury in SAMR1 and SAMP8 microglia that was significant by a direct Student’s t–test comparison (not indicated in the figure) [t(44) = 2.706, p = 0.010].
As a whole, glial cell cultures obtained from newborn brain of SAMP8 mice showed higher responsiveness to a pro-inflammatory LPS + IFN injury than those of SAMR1 mice. This effect was primarily driven by microglia, as demonstrated by nitric oxide generation in control and stimulated conditions in pure microglia cultures. Furthermore, the morphological analysis confirmed a state of basal activation, as shown by a higher number of cells with globular morphology, and a hyperreactivity to LPS + IFN, as shown by an increase in the total number and in the percentage of activated SAMP8 microglia.
Alzheimer’s Disease Neuroinflammatory Markers
Phenotypic gene expression of Chil1 and Trem2 in SAMR1 and SAMP8 hippocampus 3 h after acute LPS injury or control conditions is shown in Figure 3A. Both Chil1 and Trem2 mRNA increased with age, although the effect was almost null in the vehicle-injected SAMR1 mice [three-way ANOVA, main effect of age: F(1,25) = 7.602, p = 0. 011 for Chil1, and F(1,24) = 7.174, p = 0.013 for Trem2]. Meanwhile, LPS treatment induced Chil1 expression in 12-month-old SAMR1 mice but not in the younger counterparts, whereas LPS-treated SAMP8 mice at both ages showed similar expression than control 12-month-old SAMP8 [three-way ANOVA, effect of treatment × strain × age interaction: F(1,25) = 4.386, p = 0. 047 for Chil1]. The variability of the Trem2 data did not allow obtaining further ANOVA significant results in addition to the main effect of age; however, there was a tendency for a difference between 12-month-old SAMR1 in control conditions and LPS treatment that was significant by a direct Student’s t–test comparison (not indicated in the figure) [t(11) = 2.669, p = 0.0218].
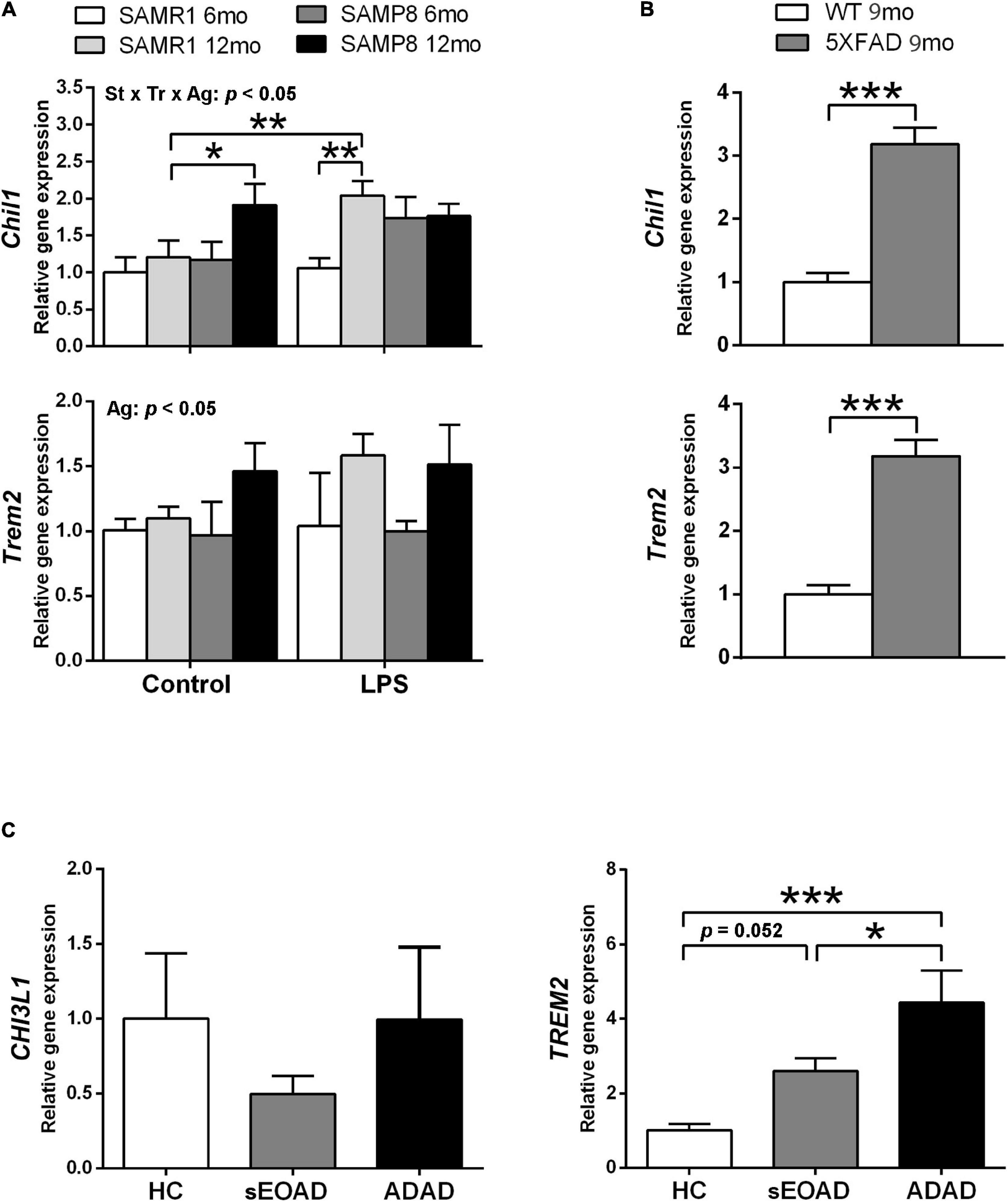
Figure 3. Gene expression of Alzheimer’s disease (AD)-associated markers YKL40, also known as Chitinase 3-Like 1 (CHI3L1/Chil1, human/mouse gene), and Triggering receptor expressed on myeloid cells (TREM2/Trem2, human/mouse gene) showed differential patterns in the hippocampus of SAMP8 mice in comparison with AD samples. Trem2 expression showed a general increase with age in SAMP8 and SAMR1 mice, whereas Chil1 showed a greater increase with age in SAMP8 under control treatment and in SAMR1 3 h after 3 mg/kg i.p. of lipopolysaccharide (LPS) (A). Both Chil1 and Trem2 genes were highly expressed in the hippocampus of the AD mouse model 5XFAD (B). The respective human homologous gene CHI3L1 showed no changes of expression in the posterior cingulate cortex of early onset AD (sEOAD) or autosomal dominant AD (ADAD) as compared to neurologically healthy controls (NHC), whereas TREM2 showed increased expression in AD (C). P-values for three-way ANOVA analysis are indicated at the top area of the graph (Ag, age main effect; and St × Tr × Ag, strain × treatment × age interaction effect) and p-values for Fisher’s LSD post hoc tests between the groups indicated in the graph are indicated as follows: *p < 0.05 and **p < 0.01, N = 3–7, in (A). P-values for t-test are indicated as: ***p < 0.001, N = 4, in (B). P-values for LSD tests after one-way ANOVA are indicated as: *p < 0.05 and ***p < 0.001, N = 8, in (C).
The AD mouse model 5XFAD showed greater hippocampus expression of both Chil1 and Trem2 than WT mouse siblings, as shown in Figure 3B [t(6) = 7.39, p < 0.001, for Chil1 and t(6) = 7.39, p < 0.001 for Trem2].
The mRNA levels of the corresponding human genes CHI3L1 and TREM2 in posterior cingulate tissue samples are shown in Figure 3C. No significant differences were obtained between the NHC and AD groups for CHI3L1. However, TREM2 mRNA levels were higher in the AD groups than the NHC group. This increase was of borderline significance for sEOAD but highly significant for ADAD. Furthermore, ADAD showed higher TREM2 expression than sEOAD [one-way ANOVA, F(2,21) = 9.839, p = 0.001].
Here we found a differential expression in the respective markers for astrogliosis (Chil1/CHI3L1) and microgliosis (Trem2/TREM2) in the several brain tissues analyzed from mouse models and AD patients. SAMP8 hippocampus showed increased expression of Chil1 at old age, although the increase of Trem2 did not reach statistical significance compared to SAMR1. sEOAD patients showed also a borderline significance in the increase of TREM2. However, ADAD patients and 5XFAD mice showed a distinct increased expression of Trem2/TREM2. 5XFAD mice also showed high levels of Chil1 expression.
Discussion
SAMP8 Mice Showed Dysregulation of the Neuroimmune Response to an Acute Stimulus in the Hippocampus
The SAMP8 mouse model of accelerated brain aging and sporadic AD traits showed a higher neuroinflammatory profile compared to SAMR1, as expected from previous reports (Tha et al., 2000; Álvarez-López et al., 2014; Griñán-Ferré et al., 2016a; Rancán et al., 2017). However, we found a major dysregulation of the neuroimmune response to an acute stimulus in the SAMP8 hippocampus that ranged from hyperresponsiveness at young age to null responsiveness at older age. It is known that the main pro-inflammatory cytokines IL1β, IL6, and TNFα are transiently overexpressed in various regions of mouse brain after an LPS injury (Layé et al., 1994; Catorce and Gevorkian, 2016). Accordingly, both SAMP8 and SAMR1 mice were responsive to LPS by showing an increase in the protein and mRNA levels of these cytokines in the cerebral cortex. A similar response to LPS was also found at the peripheral level. Interestingly, the hippocampus analysis showed a much higher inflammatory response in 6-month-old SAMP8 mice than in the SAMR1 mice of the same age. Furthermore, 12-month-old SAMP8 hippocampus showed no responsiveness to LPS challenge, whereas SAMR1 mice conserved a response of increased cytokine gene expression. Two previous studies performed in young mice reported either no strain differences after a lower dose of 20 μg/kg body weight LPS (Tha et al., 2000) or higher protein levels of the NLRP3 inflammasome in whole brain tissue and IL6 in blood serum of SAMP8 than SAMR1 after 0.33 mg/kg body weight LPS (Ito et al., 2020). Therefore, both young age and a strong LPS challenge may be required to cause an exacerbated generation of pro-inflammatory mediators in SAMP8. Likewise, an insufficient resolution response to age-related inflammation has been reported in SAMP8 hippocampus (Wang et al., 2015b). In fact the hippocampus is highly vulnerable to deterioration by neurotoxic activation of glial cells (Ojo et al., 2015). Accordingly, genome-wide association studies have shown that immune response and related pathways are highly activated in AD brain, suggesting that an increasingly deregulated response with age and infections may result in progressive neurodegeneration and AD (Liu and Yu, 2019). We speculate that chronic neuroinflammation in the SAMP8 hippocampus causes a deregulation of the local neuroimmune response as shown by hyperreactivity to an infectious-like stimulus in young adulthood age and immunosuppression in old age; these processes would contribute to memory loss and neurodegeneration.
SAMP8 Microglia From Neonatal Mice Showed a Pro-inflammatory State in vitro
The in vitro analysis showed a higher contribution of microglia than astrocytes in the SAMP8 neuroinflammatory processes. SAMP8 microglia generally showed the amoeboid morphology that is associated with an activated state (Aldana, 2019). However, SAMP8 astrocytes did not show overt reactive phenotype in control conditions, although they have shown some functional deficiencies in vitro (García-Matas et al., 2008, 2015). Experiments in cell cultures have shown that both microglia and astrocytes are able to synthesize IL1β, IL6 and TNFα (Lam et al., 2017; Rodgers et al., 2020). Here there was a similar response to LPS + IFN injury in the release of pro-inflammatory cytokines in mixed glial cultures enriched in astrocytes and in microglia cultures. However, we found higher nitric oxide production in LPS-challenged SAMP8 mixed glia and more crucially, in SAMP8 pure microglia regardless of treatment conditions. These effects were paralleled by increased proliferation and morphological changes of SAMP8 microglia. Excessive nitric oxide generated by microglia through sustained expression of inducible nitric oxide synthase (iNOS) is believed to contribute to age-associated neurodegeneration (Yuste et al., 2015). Accordingly, higher nitric oxide content (Wang et al., 2013) and iNOS expression (Griñán-Ferré et al., 2016a) have been reported in SAMP8 brain tissue than in SAMR1. Neuroinflammatory priming has also been reported in microglia isolated from the brains of adult SAMP8 as compared to SAMR1 mice (Ito et al., 2020). Exacerbated inflammatory responses in neurodegenerative disorders are mainly attributed to microglia, in a proposed vicious cycle of sustained microglial activation, neuroinflammation and neurodegeneration (Aldana, 2019; Ahmad et al., 2019). Nevertheless, there is a current consent on the complex dynamics and heterogeneity of microglia activation beyond the initially proposed pro-inflammatory M1 (classical activation) and immunosuppressive M2 (alternative activation) states (Ransohoff, 2016; Song and Suk, 2017; García-Revilla et al., 2019). Microglia polarization can also evolve to alternative activated phenotypes involved in repair functions (Colton et al., 2006; Masuda et al., 2019). Specifically, a subset of microglia, the “disease-associated microglia” (DAM), may be able to sense neuronal damage signals from AD and other conditions and promote phagocytosis, barrier formation and activation of protective pathways (Deczkowska et al., 2018). Furthermore, the presence of microglia with a senescent phenotype in the hippocampus of 12-month-old SAMP8 would be consistent with the senescent dystrophic microglia that have been described in the human aging brain (Streit et al., 2004). Senescent microglia no longer perform their functions of immune surveillance and response or other active neuronal supportive functions including activity-dependent remodeling of synaptic connections (Fields et al., 2014).
Gene Expression of the Astrogliosis Marker CHI3L1 Was Increased in SAMP8 and 5XFAD Mice but Not in Alzheimer’s Disease Brains
In the brain, CHI3L1 is expressed mainly by astrocytes and it increases in regions of neuroinflammation in AD and tauopathies (Querol-Vilaseca et al., 2017), and also in other neuroinflammatory conditions (Bonneh-Barkay et al., 2010). We found increased expression of Chil1 gene in the hippocampus of 12-month-old SAMP8 and 9 month-old 5XFAD mice, in agreement with the progression of age-related astrogliosis (Girard et al., 2014; Corpas et al., 2017) and the advanced stage of neurodegeneration in both mouse models. Noticeably, Chil1 expression increased in 12-month-old hippocampus of control SAMR1 mice after LPS challenge. Therefore, the level of neuroinflammation needed to upregulate Chil1 may require the interaction of advancing age and systemic infection in these mice. Also, increased expression of Chil1 has been shown in other AD transgenic mouse models at advancing age (Colton et al., 2006; Xiao et al., 2016). In human brain, increased expression of the CHI3L1 gene has been reported in postmortem tissue samples of sporadic AD patients in their 70s and 80s (Colton et al., 2006; Llorens et al., 2017) and further potentiated by systemic infection (Rakic et al., 2018). However, we found no significant changes in expression in the mRNA of the younger AD cohorts tested despite its severe pathology. Intriguingly, it may be required an older age and/or the presence of infection for detecting increased expression of CHI3L1 despite the increased protein levels in the CSF of AD patients (Schindler et al., 2019; Antonell et al., 2020).
The function of CHI3L1 protein remains unknown. In vitro, soluble inflammatory mediators released by macrophages or microglia have been reported to induce CHI3L1 transcription and morphological changes in astrocytes (Bonneh-Barkay et al., 2012). The results of an in vivo study of traumatic brain injury have suggested that CHI3L1 is protective (Wiley et al., 2015), whereas the results from a study with chronic brain infusion of amyloid beta suggested otherwise (Choi et al., 2018). It is possible that expression of CHI3L1 with advancing neurodegeneration is deleterious, as shown for instance by its inverse correlation with survival in amyotrophic lateral sclerosis (Sanfilippo et al., 2019).
Gene Expression of the Microgliosis Marker TREM2 Was Highly Increased in Mice and Alzheimer’s Brain With Familial Disease Type
Upregulation of the Trem2 gene in SAMP8 hippocampus was detected in aged mice, consistent with a previously reported age-related increase in whole brain protein levels in these mice (Jiang et al., 2014b). Remarkably, the gene upregulation was driven by aging and was statistically unrelated to peripheral challenge with LPS in both SAMP8 and SAMR1. Furthermore, middle-aged 5XFAD mice with advanced AD-like pathology showed robust upregulation of Trem2 in agreement with previous results reported in this and other transgenic AD mouse models (Jiang et al., 2014a; Brendel et al., 2017; Hüttenrauch et al., 2018). Our results in human AD brain showed increased TREM2 mRNA in the posterior cingulate brain area of AD patients, although the increase of the sEOAD group did not reach significance compared to NHC. We cannot rule out a contribution of age factor on the small increase of TREM2 mRNA levels in sEOAD patients, who were slightly older than NHC and ADAD groups. However, previous authors have also shown a trend to increase the overall TREM2 expression levels in sporadic AD cases in comparison to controls (Roussos et al., 2015; Del-Aguila et al., 2019). Remarkably, we found a differential increase between sEOAD and ADAD patients, namely, gene-driven AD increased TREM2 expression levels by almost twice as much as sporadic AD, a similar fold difference as in the transgenic 5XFAD mouse model compared to aged SAMP8 mice. Therefore, the increased expression of TREM2 in ADAD brain could indicate a differential microglia response depending on the AD etiology. What is more, the differential level of expression of TREM2 does not seem driven by the degree of pathology, since sEOAD and ADAD had a similar high level of neuropathological changes. Gene expression of TREM2 has not been previously analyzed in ADAD brain, to our knowledge. However, higher gene expression of TREM2 in the superior temporal gyrus has been reported in AD carriers of a missense mutation of this gene (Roussos et al., 2015). Also, the analysis of the differential expression of gene transcripts for the three TREM2 isoforms (Zhong and Chen, 2019) by RNA-Seq has shown an association between AD cases and higher levels of the shortest transcript that lacks the transmembrane domain, in the parietal lobe of sporadic AD brain and TREM2 mutation carriers (Del-Aguila et al., 2019). Unfortunately, our qPCR analysis did not discern the different TREM2 transcripts.
The transmembrane glycoprotein TREM2 is a crucial component of a receptor-signaling complex that regulates the immune response and phagocytic activity of microglia, macrophages, dendritic cells and osteoclasts (Sudom et al., 2018). Furthermore, it is involved in neuroprotection mediated by the DAM phenotype (Deczkowska et al., 2018). TREM2 gene variants with loss-of-function effects are a known risk factor for late-onset AD (Sudom et al., 2018). However, higher levels of TREM2 protein have been reported in the middle temporal cortices of late-onset AD patients in their 80s than in age-matched control tissue, and this increase was correlated with that of phosphorylated tau and apoptotic markers (Lue et al., 2015). This is consistent with the proposed deleterious effect of TREM2 at advanced stages of AD when it would aggravate neuroinflammation (Karanfilian et al., 2020). Animal studies have shown that brain levels of soluble TREM2 increase during aging in parallel with amyloidosis and microglia activation (Brendel et al., 2017). At middle age, Trem2 studies of gene modulation showed a protective effect in transgenic AD mice (Jiang et al., 2014a) and SAMP8 mice (Jiang et al., 2014b). TREM2 is emerging as a critical factor in the regulation of complex microglia activation states (García-Revilla et al., 2019).
Conclusion
Pathological changes of microglia activation in the postmortem AD brain have been described in the pioneering studies of the McGeer’s group in the late 1980s and 1990s (McGeer et al., 1987). Later studies overwhelmingly suggest that reactive microglia play an important role in the neuroinflammatory processes of AD. Age-related microglia changes showing a moderately activated phenotype were subsequently described in the normal human brain (Sheng et al., 1998). There is also current agreement on the AD risk induced by low-grade chronic neuroinflammation in the aging brain.
Here we confirmed the presence of increased expression of TREM2 in AD microglia and unveiled a differential expression between sporadic and gene-driven AD, despite a similarly severe pathology in human (sEOAD and ADAD) and similarly severe cognitive loss and neuroinflammation in mouse models (SAMP8 and 5XFAD). Unexpectedly, we found no changes in the gene expression of the astrocytic protein CHI3L1 (also known as YKL-40) in the human brain of our cohorts of sEOAD or ADAD, while it is known to increase in sporadic AD at older ages. However, the mouse counterpart Chil1 did increase its gene expression. Furthermore, we found that the SAMP8 mice show early microglia-exacerbated reactivity to an infectious-like stimulus in vitro and in vivo, followed by immunosuppression at old age in the hippocampus. Therefore, SAMP8 mice open a new scenario in the relationship between the progressive impairment of the innate immune system and the expression of AD-related gliosis markers. However, microglia phenotypic changes and derangement of their innate immune function driven by age-related neuroinflammation and AD pathology are complex and warrant further study using diverse experimental systems, including genetic and non-genetic AD models and human samples.
Data Availability Statement
The original contributions generated for this study are included in the article results, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Hospital Clínic of Barcelona, Barcelona, Spain. Written informed consent was not provided because brain samples were obtained from the Neurological Tissue Biobank of Hospital Clínic - IDIBAPS and the Neuropathology Institute of the Hospital Universitari de Bellvitge. Tissue donations follow the legal procedures for their use in research studies. The animal study was reviewed and approved by the Ethics Committee for Animal Experimentation (CEAA) of the University of Barcelona, Spain.
Author Contributions
RCr, PK, CSo, JM, RS-V, AA, AL, and CSa contributed to the conception and design of the study. AL and CSa jointly supervised the study. PM-M, RCo, EG-L, MC-T, RCr, and CSa did the experimental analysis and data processing. RCo and CSa wrote the manuscript draft. All authors read and approved the final manuscript.
Funding
This research was funded by Spanish MINECO and European Regional Development Fund, grant number SAF2016-77703; Spanish MCINN, grant number PID2019-106285RB; Catalan Autonomous Government AGAUR, grant number 2017-SGR-106; Competitiveness Operational Programme 2014-2020, C-Reactive protein therapy for stroke-associated dementia, ID P_37_674, MySMIS code: 103432, contract 51/05.09.2016; and the CERCA Programme/Generalitat de Catalunya. RCo was supported by a post-doctoral research contract of the Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Instituto de Salud Carlos III, Madrid, Spain. AL (PERIS SLT008/18/00061) received funding from Departament de Salut de la Generalitat de Catalunya. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Isidre Ferrer (Bellvitge University Hospital and Bellvitge Biomedical Research Institute-IDIBELL, L’Hospitalet de Llobregat, Spain) for his contribution to the selection and provision of postmortem human tissue samples.
References
Ahmad, M. H., Fatima, M., and Mondal, A. C. (2019). Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: rational insights for the therapeutic approaches. J. Clin. Neurosci. 59, 6–11. doi: 10.1016/j.jocn.2018.10.034
Aldana, B. I. (2019). Microglia-specific metabolic changes in neurodegeneration. J. Mol. Biol. 431, 1830–1842. doi: 10.1016/j.jmb.2019.03.006
Álvarez-López, M. J., Molina-Martínez, P., Castro-Freire, M., Cosín-Tomás, M., Cristòfol, R., Párrizas, M., et al. (2014). Rcor2 underexpression in senescent mice: a target for inflammaging? J. Neuroinflammation 11:126. doi: 10.1186/1742-2094-11-126
Antonell, A., Lladó, A., Altirriba, J., Botta-Orfila, T., Balasa, M., Fernández, M., et al. (2013). A preliminary study of the whole-genome expression profile of sporadic and monogenic early-onset Alzheimer’s disease. Neurobiol. Aging 34, 1772–1778. doi: 10.1016/j.neurobiolaging.2012.12.026
Antonell, A., Tort-Merino, A., Ríos, J., Balasa, M., Borrego-Écija, S., Auge, J. M., et al. (2020). Synaptic, axonal damage and inflammatory cerebrospinal fluid biomarkers in neurodegenerative dementias. Alzheimers Dement. 16, 262–272. doi: 10.1016/j.jalz.2019.09.001
Bolós, M., Perea, J. R., and Avila, J. (2017). Alzheimer’s disease as an inflammatory disease. Biomol. Concepts 8, 37–43. doi: 10.1515/bmc-2016-0029
Bonneh-Barkay, D., Bissel, S. J., Kofler, J., Starkey, A., Wang, G., and Wiley, C. A. (2012). Astrocyte and macrophage regulation of YKL-40 expression and cellular response in neuroinflammation. Brain Pathol. 22, 530–546. doi: 10.1111/j.1750-3639.2011.00550.x
Bonneh-Barkay, D., Wang, G., Starkey, A., Hamilton, R. L., and Wiley, C. A. (2010). In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflammation 7:34. doi: 10.1186/1742-2094-7-34
Brendel, M., Kleinberger, G., Probst, F., Jaworska, A., Overhoff, F., Blume, T., et al. (2017). Increase of TREM2 during aging of an Alzheimer’s disease mouse model is paralleled by microglial activation and amyloidosis. Front. Aging Neurosci. 9:8. doi: 10.3389/fnagi.2017.00008
Catorce, M. N., and Gevorkian, G. (2016). LPS-induced murine neuroinflammation model: main features and suitability for pre-clinical assessment of nutraceuticals. Curr. Neuropharmacol. 14, 155–164. doi: 10.2174/1570159x14666151204122017
Cheng, X. R., Zhou, W. X., and Zhang, Y. X. (2014). The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer’s disease animal model. Ageing Res. Rev. 13, 13–37. doi: 10.1016/j.arr.2013.10.002
Choi, J. Y., Yeo, I. J., Kim, K. C., Choi, W. R., Jung, J. K., Han, S. B., et al. (2018). K284-6111 prevents the amyloid beta-induced neuroinflammation and impairment of recognition memory through inhibition of NF-κB-mediated CHI3L1 expression. J. Neuroinflammation 15:224. doi: 10.1186/s12974-018-1269-3
Colton, C. A., Mott, R. T., Sharpe, H., Xu, Q., Van Nostrand, W. E., and Vitek, M. P. (2006). Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J. Neuroinflammation 3:27. doi: 10.1186/1742-2094-3-27
Corpas, R., Hernández-Pinto, A. M., Porquet, D., Hernández-Sánchez, C., Bosch, F., Ortega-Aznar, A., et al. (2017). Proinsulin protects against age-related cognitive loss through anti-inflammatory convergent pathways. Neuropharmacology 123, 221–232. doi: 10.1016/j.neuropharm.2017.06.014
Corpas, R., Solana, E., De la Rosa, A., Sarroca, S., Griñán-Ferré, C., Oriol, M., et al. (2019). Peripheral maintenance of the axis SIRT1-SIRT3 at youth level may contribute to brain resilience in middle-aged amateur rugby players. Front. Aging Neurosci. 11:352. doi: 10.3389/fnagi.2019.00352
Cosín-Tomás, M., Alvarez-López, M. J., Sanchez-Roige, S., Lalanza, J. F., Bayod, S., Sanfeliu, C., et al. (2014). Epigenetic alterations in hippocampus of SAMP8 senescent mice and modulation by voluntary physical exercise. Front. Aging Neurosci. 6:51. doi: 10.3389/fnagi.2014.00051
Cristòfol, R., Porquet, D., Corpas, R., Coto-Montes, A., Serret, J., Camins, A., et al. (2012). Neurons from senescence-accelerated SAMP8 mice are protected against frailty by the sirtuin 1 promoting agents melatonin and resveratrol. J. Pineal Res. 52, 271–281. doi: 10.1111/j.1600-079X.2011.00939.x
Dantzer, R. (2001). Cytokine-induced sickness behavior: mechanisms and implications. Ann. N. Y. Acad. Sci. 933, 222–234. doi: 10.1111/j.1749-6632.2001.tb05827.x
Deczkowska, A., Keren-Shaul, H., Weiner, A., Colonna, M., Schwartz, M., and Amit, I. (2018). Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081. doi: 10.1016/j.cell.2018.05.003
Del-Aguila, J. L., Benitez, B. A., Li, Z., Dube, U., Mihindukulasuriya, K. A., Budde, J. P., et al. (2019). TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers. Mol. Neurodegener. 14:18. doi: 10.1186/s13024-019-0319-3
Díez-Vives, C., Gay, M., García-Matas, S., Comellas, F., Carrascal, M., Abian, J., et al. (2009). Proteomic study of neuron and astrocyte cultures from senescence-accelerated mouse SAMP8 reveals degenerative changes. J. Neurochem. 111, 945–955. doi: 10.1111/j.1471-4159.2009.06374.x
Elmaleh, D. R., Farlow, M. R., Conti, P. S., Tompkins, R. G., Kundakovic, L., and Tanzi, R. E. (2019). Developing effective Alzheimer’s disease therapies: clinical experience and future directions. J. Alzheimers Dis. 71, 715–732. doi: 10.3233/JAD-190507
Ettcheto, M., Sánchez-López, E., Pons, L., Busquets, O., Olloquequi, J., Beas-Zarate, C., et al. (2017). Dexibuprofen prevents neurodegeneration and cognitive decline in APPswe/PS1dE9 through multiple signaling pathways. Redox Biol. 13, 345–352. doi: 10.1016/j.redox.2017.06.003
Fernández-García, C., Rancan, L., Paredes, S. D., Montero, C., de la Fuente, M., Vara, E., et al. (2019). Xanthohumol exerts protective effects in liver alterations associated with aging. Eur. J. Nutr. 58, 653–663. doi: 10.1007/s00394-018-1657-6
Fields, R. D., Araque, A., Johansen-Berg, H., Lim, S. S., Lynch, G., Nave, K. A., et al. (2014). Glial biology in learning and cognition. Neuroscientist 20, 426–431. doi: 10.1177/1073858413504465
Fishkin, R. J., and Winslow, J. T. (1997). Endotoxin-induced reduction of social investigation by mice: interaction with amphetamine and anti-inflammatory drugs. Psychopharmacology (Berl) 132, 335–341. doi: 10.1007/s002130050353
García-Matas, S., Gutierrez-Cuesta, J., Coto-Montes, A., Rubio-Acero, R., Díez-Vives, C., Camins, A., et al. (2008). Dysfunction of astrocytes in senescence-accelerated mice SAMP8 reduces their neuroprotective capacity. Aging Cell 7, 630–640. doi: 10.1111/j.1474-9726.2008.00410.x
García-Matas, S., Paul, R. K., Molina-Martínez, P., Palacios, H., Gutierrez, V. M., Corpas, R., et al. (2015). In vitro caloric restriction induces protective genes and functional rejuvenation in senescent SAMP8 astrocytes. Aging Cell 14, 334–344. doi: 10.1111/acel.12259
García-Revilla, J., Alonso-Bellido, I. M., Burguillos, M. A., Herrera, A. J., Espinosa-Oliva, A. M., Ruiz, R., et al. (2019). Reformulating pro-oxidant microglia in neurodegeneration. J. Clin. Med. 8:1719. doi: 10.3390/jcm8101719
Girard, S. D., Jacquet, M., Baranger, K., Migliorati, M., Escoffier, G., Bernard, A., et al. (2014). Onset of hippocampus-dependent memory impairments in 5XFAD transgenic mouse model of Alzheimer’s disease. Hippocampus 24, 762–767. doi: 10.1002/hipo.22267
Griñán-Ferré, C., Izquierdo, V., Otero, E., Puigoriol-Illamola, D., Corpas, R., Sanfeliu, C., et al. (2018). Environmental enrichment improves cognitive deficits, AD hallmarks and epigenetic alterations presented in 5xFAD mouse model. Front. Cell Neurosci. 12:224. doi: 10.3389/fncel.2018.00224
Griñán-Ferré, C., Palomera-Ávalos, V., Puigoriol-Illamola, D., Camins, A., Porquet, D., Plá, V., et al. (2016a). Behaviour and cognitive changes correlated with hippocampal neuroinflammaging and neuronal markers in female SAMP8, a model of accelerated senescence. Exp. Gerontol. 80, 57–69. doi: 10.1016/j.exger.2016.03.014
Griñán-Ferré, C., Sarroca, S., Ivanova, A., Puigoriol-Illamola, D., Aguado, F., Camins, A., et al. (2016b). Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging (Albany NY) 8, 664–684. doi: 10.18632/aging.100906
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Hemonnot, A. L., Hua, J., Ulmann, L., and Hirbec, H. (2019). Microglia in Alzheimer disease: well-known targets and new opportunities. Front. Aging Neurosci. 11:233. doi: 10.3389/fnagi.2019.00233
Hershey, L. A., and Lipton, R. B. (2019). Naproxen for presymptomatic Alzheimer disease: is this the end, or shall we try again? Neurology 92, 829–830. doi: 10.1212/WNL.0000000000007233
Hüttenrauch, M., Ogorek, I., Klafki, H., Otto, M., Stadelmann, C., Weggen, S., et al. (2018). Glycoprotein NMB: a novel Alzheimer’s disease associated marker expressed in a subset of activated microglia. Acta Neuropathol. Commun. 6:108. doi: 10.1186/s40478-018-0612-3
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13. doi: 10.1016/j.jalz.2011.10.007
Ito, N., Takemoto, H., Hasegawa, A., Sugiyama, C., Honma, K., Nagai, T., et al. (2020). Neuroinflammaging underlies emotional disturbances and circadian rhythm disruption in young male senescence-accelerated mouse prone 8 mice. Exp. Gerontol. 142:111109. doi: 10.1016/j.exger.2020.111109
Jiang, T., Tan, L., Zhu, X. C., Zhang, Q. Q., Cao, L., Tan, M. S., et al. (2014a). Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 2949–2962. doi: 10.1038/npp.2014.164
Jiang, T., Yu, J. T., Zhu, X. C., Tan, M. S., Gu, L. Z., Zhang, Y. D., et al. (2014b). Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol. Aging 35, 1243–1251. doi: 10.1016/j.neurobiolaging.2013.11.026
Karanfilian, L., Tosto, M. G., and Malki, K. (2020). The role of TREM2 in Alzheimer’s disease; evidence from transgenic mouse models. Neurobiol. Aging 86, 39–53. doi: 10.1016/j.neurobiolaging.2019.09.004
Kodamullil, A. T., Iyappan, A., Karki, R., Madan, S., Younesi, E., and Hofmann-Apitius, M. (2017). Of mice and men: comparative analysis of neuro-inflammatory mechanisms in human and mouse using cause-and-effect models. J. Alzheimers Dis. 59, 1045–1055. doi: 10.3233/JAD-170255
Konsman, J. P., Parnet, P., and Dantzer, R. (2002). Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 25, 154–159. doi: 10.1016/s0166-2236(00)02088-9
Lam, D., Lively, S., and Schlichter, L. C. (2017). Responses of rat and mouse primary microglia to pro- and anti-inflammatory stimuli: molecular profiles, K + channels and migration. J. Neuroinflammation 14:166. doi: 10.1186/s12974-017-0941-3
Layé, S., Parnet, P., Goujon, E., and Dantzer, R. (1994). Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res. Mol. Brain Res. 27, 157–162. doi: 10.1080/009841098159222
Liu, B., Liu, J., and Shi, J. S. (2020). SAMP8 mice as a model of age-related cognition decline with underlying mechanisms in Alzheimer’s disease. J. Alzheimers Dis. 75, 385–395. doi: 10.3233/JAD-200063
Liu, C., and Yu, J. (2019). Genome-wide association studies for cerebrospinal fluid soluble TREM2 in Alzheimer’s disease. Front. Aging Neurosci. 1:297. doi: 10.3389/fnagi.2019.00297
Llorens, F., Thüne, K., Tahir, W., Kanata, E., Diaz-Lucena, D., Xanthopoulos, K., et al. (2017). YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 12:83. doi: 10.1186/s13024-017-0226-4
López-Ramos, J. C., Jurado-Parras, M. T., Sanfeliu, C., Acuña-Castroviejo, D., and Delgado-García, J. M. (2012). Learning capabilities and CA1-prefrontal synaptic plasticity in a mice model of accelerated senescence. Neurobiol. Aging 33, 627.e13-26. doi: 10.1016/j.neurobiolaging.2011.04.005
Lu, Y. C., Yeh, W. C., and Ohashi, P. S. (2008). LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. doi: 10.1016/j.cyto.2008.01.006
Lue, L. F., Schmitz, C. T., Serrano, G., Sue, L. I., Beach, T. G., and Walker, D. G. (2015). TREM2 protein expression changes correlate with Alzheimer’s disease neurodegenerative pathologies in post-mortem temporal cortices. Brain Pathol. 25, 469–480. doi: 10.1111/bpa.12190
Masuda, T., Sankowski, R., Staszewski, O., Böttcher, C., Amann, L., Sagar, et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. doi: 10.1038/s41586-019-0924-x Author Correction in: Nature 568, E4.
McGeer, P. L., Itagaki, S., Tago, H., and McGeer, E. G. (1987). Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 79, 195–200. doi: 10.1016/0304-3940(87)90696-3
Meyer, P. F., Tremblay-Mercier, J., Leoutsakos, J., Madjar, C., Lafaille-Maignan, M. É, Savard, M., et al. (2019). INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 92, e2070–e2080. doi: 10.1212/WNL.0000000000007232
Miró, L., Garcia-Just, A., Amat, C., Polo, J., Moretó, M., and Pérez-Bosque, A. (2017). Dietary animal plasma proteins improve the intestinal immune response in senescent mice. Nutrients 9:12. doi: 10.3390/nu9121346
Molinuevo, J. L., Ayton, S., Batrla, R., Bednar, M. M., Bittner, T., Cummings, J., et al. (2018). Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 136, 821–853. doi: 10.1007/s00401-018-1932-x
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. doi: 10.1007/s00401-011-0910-3
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Ojo, J. O., Rezaie, P., Gabbott, P. L., and Stewart, M. G. (2015). Impact of age-related neuroglial cell responses on hippocampal deterioration. Front. Aging Neurosci. 7:57. doi: 10.3389/fnagi.2015.00057
Porquet, D., Casadesús, G., Bayod, S., Vicente, A., Canudas, A. M., Vilaplana, J., et al. (2013). Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age (Dordr) 35, 1851–1865. doi: 10.1007/s11357-012-9489-4
Querol-Vilaseca, M., Colom-Cadena, M., Pegueroles, J., San Martín-Paniello, C., Clarimon, J., Belbin, O., et al. (2017). YKL-40 (Chitinase 3-like I) is expressed in a subset of astrocytes in Alzheimer’s disease and other tauopathies. J. Neuroinflammation 14:118. doi: 10.1186/s12974-017-0893-7
Rakic, S., Hung, Y. M. A., Smith, M., So, D., Tayler, H. M., Varney, W., et al. (2018). Systemic infection modifies the neuroinflammatory response in late stage Alzheimer’s disease. Acta Neuropathol. Commun. 6:88. doi: 10.1186/s40478-018-0592-3
Rancán, L., Paredes, S. D., García, I., Muñoz, P., García, C., López de Hontanar, G., et al. (2017). Protective effect of xanthohumol against age-related brain damage. J. Nutr. Biochem. 49, 133–140. doi: 10.1016/j.jnutbio.2017.07.011
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Rodgers, K. R., Lin, Y., Langan, T. J., Iwakura, Y., and Chou, R. C. (2020). Innate immune functions of astrocytes are dependent upon tumor necrosis factor-alpha. Sci. Rep. 10:7047. doi: 10.1038/s41598-020-63766-2
Roussos, P., Katsel, P., Fam, P., Tan, W., Purohit, D. P., and Haroutunian, V. (2015). The triggering receptor expressed on myeloid cells 2 (TREM2) is associated with enhanced inflammation, neuropathological lesions and increased risk for Alzheimer’s dementia. Alzheimers Dement. 11, 1163–1170. doi: 10.1016/j.jalz.2014.10.013
Sandiego, C. M., Gallezot, J. D., Pittman, B., Nabulsi, N., Lim, K., Lin, S. F., et al. (2015). Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. U.S.A. 112, 12468–12473. doi: 10.1073/pnas.1511003112
Sanfilippo, C., Castrogiovanni, P., Imbesi, R., Kazakowa, M., Musumeci, G., Blennow, K., et al. (2019). Sex difference in CHI3L1 expression levels in human brain aging and in Alzheimer’s disease. Brain Res. 1720:146305. doi: 10.1016/j.brainres.2019.146305
Saura, J., Tusell, J. M., and Serratosa, J. (2003). High-yield isolation of murine microglia by mild trypsinization. Glia 44, 183–189. doi: 10.1002/glia.10274
Schedlowski, M., Engler, H., and Grigoleit, J. S. (2014). Endotoxin-induced experimental systemic inflammation in humans: a model to disentangle immune-to-brain communication. Brain Behav. Immun. 35, 1–8. doi: 10.1016/j.bbi.2013.09.015
Schindler, S. E., Li, Y., Todd, K. W., Herries, E. M., Henson, R. L., Gray, J. D., et al. (2019). Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer’s disease. Alzheimers Dement. 15, 655–665. doi: 10.1016/j.jalz.2018.12.019
Sharman, M. J., Verdile, G., Kirubakaran, S., Parenti, C., Singh, A., Watt, G., et al. (2019). Targeting inflammatory pathways in Alzheimer’s disease: a focus on natural products and phytomedicines. CNS Drugs 33, 457–480. doi: 10.1007/s40263-019-00619-1
Sheng, J. G., Mrak, R. E., and Griffin, W. S. (1998). Enlarged and phagocytic, but not primed, interleukin-1 alpha-immunoreactive microglia increase with age in normal human brain. Acta Neuropathol. 95, 229–234. doi: 10.1007/s004010050792
Sola, C., Cristòfol, R., Suñol, C., and Sanfeliu, C. (2011). “Primary cultures for neurotoxicity testing,” in Cell Culture Techniques, Neuromethods, Vol. 56, eds M. Aschner, C. Suñol, and A. Bal-Price (New York, NY: Humana Press), 87–103. doi: 10.1007/978-1-61779-077-5_4
Song, G. J., and Suk, K. (2017). Pharmacological modulation of functional phenotypes of microglia in neurodegenerative diseases. Front. Aging Neurosci. 9:139. doi: 10.3389/fnagi.2017.00139
Straccia, M., Gresa-Arribas, N., Dentesano, G., Ejarque-Ortiz, A., Tusell, J. M., Serratosa, J., et al. (2011). Pro-inflammatory gene expression and neurotoxic effects of activated microglia are attenuated by absence of CCAAT/enhancer binding protein β. J. Neuroinflammation 8:156. doi: 10.1186/1742-2094-8-156
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Spark, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212. doi: 10.1002/glia.10319
Sudom, A., Talreja, S., Danao, J., Bragg, E., Kegel, R., Min, X., et al. (2018). Molecular basis for the loss-of-function effects of the Alzheimer’s disease-associated R47H variant of the immune receptor TREM2. J. Biol. Chem. 293, 12634–12646. doi: 10.1074/jbc.RA118.002352
Takeda, T., Hosokawa, M., Higuchi, K., Hosono, M., Akiguchi, I., and Katoh, H. (1994). A novel murine model of aging, senescence-accelerated mouse (SAM). Arch. Gerontol. Geriatr. 19, 185–192. doi: 10.1016/0167-4943(94)90039-6
Tejera, D., Mercan, D., Sanchez-Caro, J. M., Hanan, M., Greenberg, D., Soreq, H., et al. (2019). Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 38:e101064. doi: 10.15252/embj.2018101064
Tha, K. K., Okuma, Y., Miyazaki, H., Murayama, T., Uehara, T., Hatakeyama, R., et al. (2000). Changes in expressions of proinflammatory cytokines IL-1beta, TNF-alpha and IL-6 in the brain of senescence accelerated mouse (SAM) P8. Brain Res. 885, 25–31. doi: 10.1016/s0006-8993(00)02883-3
Wang, H. M., Wang, L. W., Liu, X. M., Li, C. L., Xu, S. P., and Farooq, A. D. (2013). Neuroprotective effects of forsythiaside on learning and memory deficits in senescence-accelerated mouse prone (SAMP8) mice. Pharmacol. Biochem. Behav. 105, 134–141. doi: 10.1016/j.pbb.2012.12.016
Wang, J., Tan, L., Wang, H. F., Tan, C. C., Meng, X. F., Wang, C., et al. (2015b). Anti-inflammatory drugs and risk of Alzheimer’s disease: an updated systematic review and meta-analysis. J. Alzheimers Dis. 44, 385–396. doi: 10.3233/JAD-141506
Wang, X., Puerta, E., Cedazo-Minguez, A., Hjorth, E., and Schultzberg, M. (2015a). Insufficient resolution response in the hippocampus of a senescence-accelerated mouse model–SAMP8. J. Mol. Neurosci. 55, 396–405. doi: 10.1007/s12031-014-0346-z
Wiley, C. A., Bonneh-Barkay, D., Dixon, C. E., Lesniak, A., Wang, G., Bissel, S. J., et al. (2015). Role for mammalian chitinase 3-like protein 1 in traumatic brain injury. Neuropathology 35, 95–106. doi: 10.1111/neup.12158
Xiao, Q., Shi, R., Yang, W., Zou, Y., Du, Y., Zhang, M., et al. (2016). Time-dependent increase of chitinase1 in APP/PS1 double transgenic mice. Neurochem. Res. 41, 1604–1611. doi: 10.1007/s11064-016-1874-4
Yuste, J. E., Tarragon, E., Campuzano, C. M., and Ros-Bernal, F. (2015). Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell Neurosci. 9:322. doi: 10.3389/fncel.2015.00322
Keywords: neuroinflammation, SAMP8 mice, autosomal dominant Alzheimer’s disease (ADAD), sporadic early-onset Alzheimer’s disease (sEOAD), triggering receptor expressed on myeloid cells 2 (TREM2)
Citation: Molina-Martínez P, Corpas R, García-Lara E, Cosín-Tomás M, Cristòfol R, Kaliman P, Solà C, Molinuevo JL, Sánchez-Valle R, Antonell A, Lladó A and Sanfeliu C (2021) Microglial Hyperreactivity Evolved to Immunosuppression in the Hippocampus of a Mouse Model of Accelerated Aging and Alzheimer’s Disease Traits. Front. Aging Neurosci. 12:622360. doi: 10.3389/fnagi.2020.622360
Received: 28 October 2020; Accepted: 31 December 2020;
Published: 28 January 2021.
Edited by:
Yong-Jing Gao, Nantong University, ChinaReviewed by:
Xiaoping Tong, Shanghai Jiao Tong University, ChinaHuiming Gao, Nanjing University, China
Copyright © 2021 Molina-Martínez, Corpas, García-Lara, Cosín-Tomás, Cristòfol, Kaliman, Solà, Molinuevo, Sánchez-Valle, Antonell, Lladó and Sanfeliu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Albert Lladó, YWxsYWRvQGNsaW5pYy5jYXQ=; Coral Sanfeliu, Y29yYWwuc2FuZmVsaXVAaWliYi5jc2ljLmVz
†These authors have contributed equally to this work
‡These authors have contributed equally to this work and share senior authorship