 Caroline L. Benn1*
Caroline L. Benn1* Lee A. Dawson2
Lee A. Dawson2- 1LoQus23 Therapeutics, Cambridge, United Kingdom
- 2Cerevance Ltd., Cambridge, United Kingdom
Kinases are an intensively studied drug target class in current pharmacological research as evidenced by the large number of kinase inhibitors being assessed in clinical trials. Kinase-targeted therapies have potential for treatment of a broad array of indications including central nervous system (CNS) disorders. In addition to the many variables which contribute to identification of a successful therapeutic molecule, drug discovery for CNS-related disorders also requires significant consideration of access to the target organ and specifically crossing the blood-brain barrier (BBB). To date, only a small number of kinase inhibitors have been reported that are specifically designed to be BBB permeable, which nonetheless demonstrates the potential for success. This review considers the potential for kinase inhibitors in the context of unmet medical need for neurodegenerative disease. A subset of kinases that have been the focus of clinical investigations over a 10-year period have been identified and discussed individually. For each kinase target, the data underpinning the validity of each in the context of neurodegenerative disease is critically evaluated. Selected molecules for each kinase are identified with information on modality, binding site and CNS penetrance, if known. Current clinical development in neurodegenerative disease are summarized. Collectively, the review indicates that kinase targets with sufficient rationale warrant careful design approaches with an emphasis on improving brain penetrance and selectivity.
Introduction
The protein kinase gene family comprises over 500 members and constitutes approximately 2% of all human genes. Protein kinases catalyze the transfer of the terminal phosphate group of adenosine triphosphate (ATP) onto an amino acid residue (typically serine, threonine or tyrosine) within a polypeptide chain. Up to 20% of all human proteins may be modified by kinase activity which results in a functional change of the target protein by altering cellular activity, location or association with other proteins. Consequently, protein kinases regulate a range of cellular functions through the orchestrated propagation and amplification of cellular stimuli into distinct biological responses through coordinated signal transduction cascades (Chico et al., 2009). Aberrant regulation of protein kinase activity has been linked to a diverse set of disease states including cancer, inflammation, metabolic, autoimmune, and neurological disorders. For example, large scale sequencing efforts have highlighted protein kinases as one of the most frequently mutated proteins across cancer subtypes. This in turn has stimulated the pursuit of these enzymes as potential drug targets for therapeutic intervention.
The catalytic subunits of protein kinases are highly conserved and over 200 structures have been solved. Protein kinases share a common bilobal-fold comprising a predominantly beta-sheet N-terminal and a helical C-terminal domain linked by a hinge polypeptide with a canonical catalytic (and ATP binding) site residing in the cleft between these two domains (Mortenson et al., 2014). Inhibitors of protein kinases can be broadly partitioned based on their binding modes (Figure 1). Type I inhibitors bind in the ATP pocket and interact directly with the kinase “hinge motif” and are thus competitive with ATP. Type II binders are also ATP competitive and generally contain a hinge binding moiety but occupy a hydrophobic pocket beyond the “gatekeeper” residue; this pocket exists in a subset of protein kinases. Type III ligands bind in a hydrophobic pocket proximal to the ATP-binding site and inhibit via an allosteric, ATP-uncompetitive mechanism. Type IV inhibitors bind to surface pockets distinct from the ATP binding site and are thus also ATP-uncompetitive. Structural analysis of selective inhibitor complexes has elucidated deeper understanding of mechanisms and accordingly, structure-guided approaches for target-specific inhibitors have been developed for a subset of kinase targets (Murray et al., 2015; Roskoski, 2016; Heightman et al., 2018).
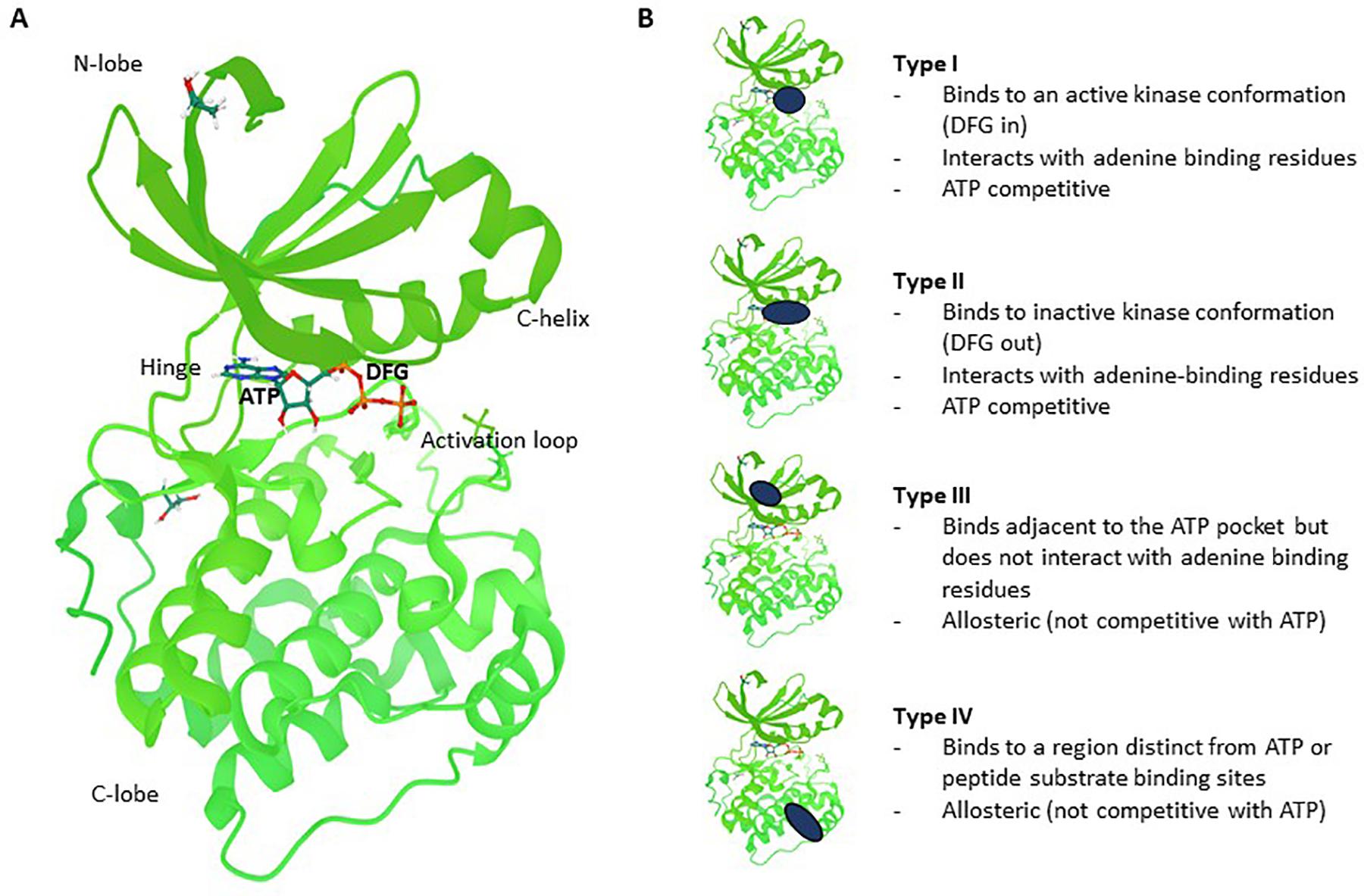
Figure 1. Kinase structure and different types of reversible small molecule kinase inhibitors. (A) Co-crystal structure of PDK1 (green) with ATP (shown in ball and sticks). Regions indicated are the N-lobe (N-terminal domain), C-lobe (C-terminal domain), C-helix, Hinge, Activation loop and the DFG regulatory motif which sits just behind part of the ATP molecule colored in orange (4RRV, PDB). (B) Type I inhibitors bind to the active conformation of the kinase, interact with the hinge motif and are ATP competitive. Type II binders are also ATP competitive but occupy a “back pocket” which exists in a subset of protein kinases. Type III ligands bind in a distinct hydrophobic pocket near the ATP binding site and are uncompetitive with ATP. Type IV inhibitors bind to distinct surface pockets away from the ATP binding site and are also uncompetitive with ATP. The PKD1 structure is used here to illustrate the different types of inhibitors.
It can thus be appreciated that kinases are eminently “druggable” using small molecule approaches but still present several challenges including ATP competition (for type 1 inhibitors), selectivity, physicochemical properties, and intellectual property. Most kinase-targeted drugs developed have been for non-CNS indications such as cancer and inflammation and have not been optimized with respect to CNS exposure. Small molecule drugs have a proven track record with respect to achieving CNS penetration, but they need to be designed with certain properties to be permeable to traverse the endothelial membranes while avoiding the efflux transporters (Figure 2). Reduced CNS penetration through the BBB can result in comparatively high systemic exposure levels in order to achieve the concurrent CNS concentrations required for efficacy – which can often lead to side effects (Figure 2). A significant proportion of the CNS-targeted kinase drugs appear to be related to neuro-oncology (Heffron, 2016). Indeed, clinical candidates with proven impact on key pathways in oncology have been leveraged with respect to systemic approaches to treating brain metastases (Franchino et al., 2018). Nevertheless, there are grounds for optimism: most kinase inhibitors have not been designed to be brain penetrant, and many screening campaigns have used compound collections loaded with “legacy” kinase inhibitor structures with suboptimal properties for CNS penetration. This situation is changing rapidly, with numerous examples now showing that it is possible to generate inhibitors that fall within a more favorable CNS property space.
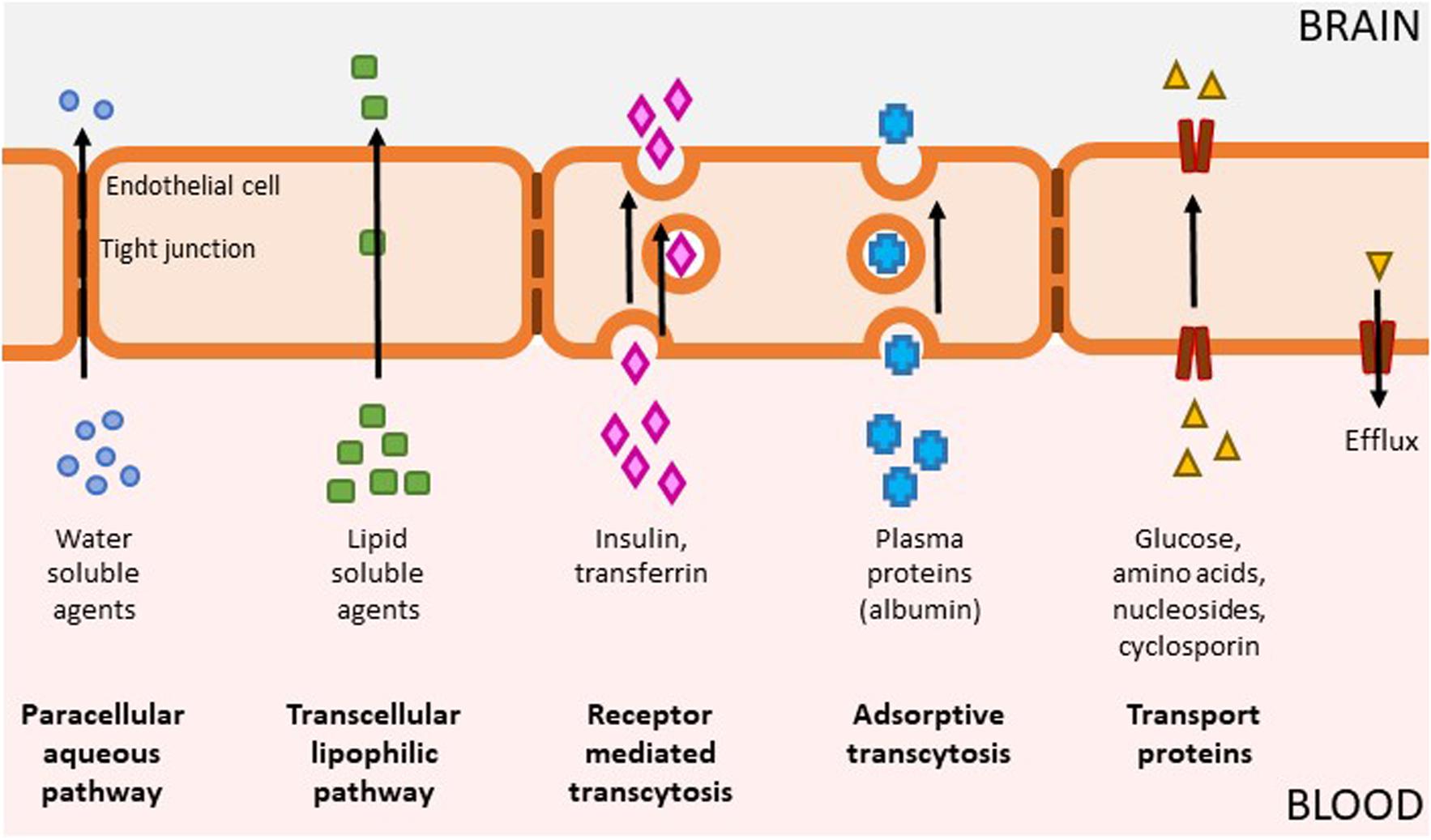
Figure 2. Mechanisms of BBB penetration. The brain is separated from the rest of the body by the blood-brain barrier (BBB) which is composed of capillary endothelium containing tight junctions and specific transporter systems to exclude pathogens and toxins, which unfortunately can also include therapeutic drugs. The BBB allows the passage of some molecules by several routes (1) Passive diffusion of water soluble agents through the paracellular aqueous pathway is negligible (2) Small, lipid soluble agents (e.g., nicotine, alcohol, antidepressants) can diffuse across a concentration gradient through endothelial cells via the transcellular lipophilic pathway which represents the main entry route into the brain for many currently approved therapeutics; (3) Receptor mediated transcytosis enable delivery of proteins such as insulin, transferrin, leptin, interleukins as well as nutrients such as iron and LDL; (4) Adsorptive transcytosis modulates transport of other proteins such as albumin; (5) Specialized transport proteins such as solute carriers can transport a range of substrates including glucose, amino acids, and drugs such as cyclosporine and azidothymidine. This category also includes efflux transporters, which is a key element of the BBB that can actively transport a variety of lipophilic drugs out of the endothelial cells. Brain penetration leading to exposure at the site of action, binding to the pharmacological target and expression of pharmacological activity are required for compound efficacy. Many drugs, including kinase inhibitors, have not been optimized for BBB penetration and as a result, they have reduced partitioning to the CNS. In order to achieve sufficient levels of exposure for compounds with suboptimal levels of CNS penetration, this means the level of systemic exposure will be significantly higher, which can lead to undesirable on- and off- target pharmacology.
Kinase Targets Clinically Assessed for Neurodegenerative Disease Indications
This review focuses on the challenges of targeting protein kinases for CNS disease indications with in-depth analyses on selected case studies for the underlying biological rationale. Here, we discuss in depth eleven protein kinase targets that have been progressed into clinical assessment for neurodegenerative disease indications from 2008 to 2018. These comprise ABL1, CSNK1D, CSF1R, MAP3K12, GSK3B, MAPK10, LRRK2, MAPK14, MTOR, RIPK1 and ROCK, discussed in the following sections and summarised in Table 1. We consider the strength of the biological rationale (although an in-depth review of the target literature will not be undertaken here but can be found in associated reviews), small molecule inhibitors and preclinical/clinical translational efforts. Of note, very few of the molecules in the clinic at the time of writing have been rationally designed with CNS penetration in mind.
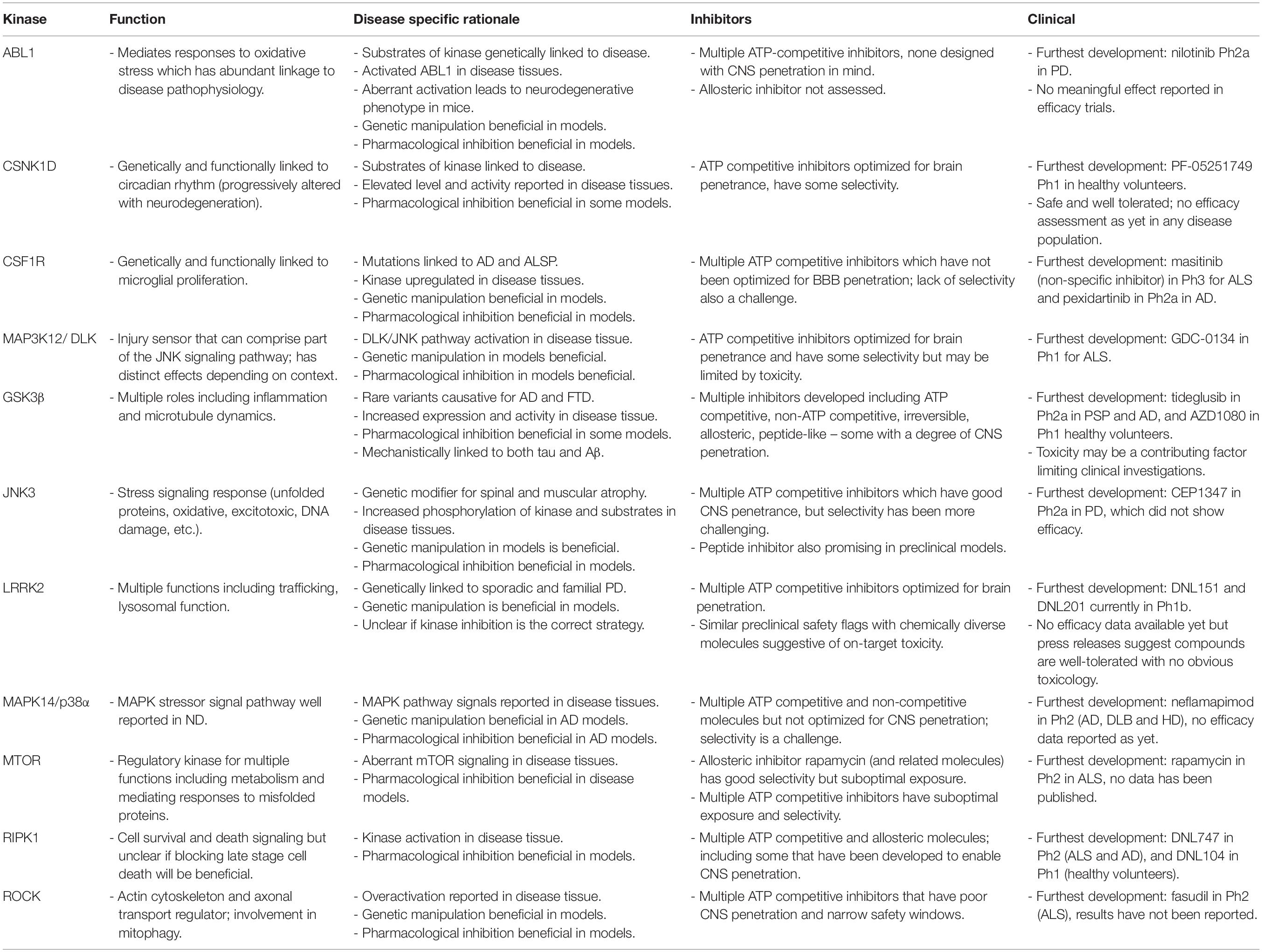
Table 1. Summary of kinases covered in manuscript with respect to function, disease association, inhibitors, and clinical development.
ABL1 (Abelson Murine Leukemia Viral Oncogene Homolog 1, c-Abl)
Biological Rationale
Oxidative stress, protein aggregate accumulation, and damaged mitochondria are common hallmarks of neurodegenerative disease and ABL1 sits at the nexus of the associated signaling pathways with roles in the regulation of receptor endocytosis, DNA repair, autophagy, cytoskeleton dynamics, cell survival, growth, and motility (Gonfloni et al., 2012). ABL1 activity is regulated by its subcellular localization, intramolecular bonds and post-translational status (in particular, its autophosphorylation at Y412 by upstream kinases) (Kharbanda et al., 1995; Shafman et al., 1997; Yuan et al., 1997; Brasher and Van Etten, 2000; Dorey et al., 2001; Ko et al., 2010; Imam et al., 2011). In adult mouse models, aberrant ABL1 activation causes neurodegeneration and neuroinflammation in forebrain neurons (Schlatterer et al., 2011). ABL1 is present in its activated form in Parkinson’s disease (PD) patients as well as in preclinical models of the disease; furthermore, ABL1 has been reported to phosphorylate α-synuclein at Y39 and the E3 ligase Parkin, both of which are genetic risk factors for PD (Ko et al., 2010; Imam et al., 2011; Schlatterer et al., 2011; Brahmachari et al., 2016; Karim et al., 2020). Deletion of the ABL1 gene reduced α-synuclein aggregation, neuropathology, and neurobehavioral deficits and protects against MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a prodrug for the neurotoxin MPP+/1-methyl-4-phenylpyridinum) challenges (Ko et al., 2010; Hebron et al., 2013; Mahul-Mellier et al., 2014; Brahmachari et al., 2016). Pharmacological inhibition of ABL1 has protective effects in mouse models of PD, although these data need to be treated with caution given the likely low CNS exposures and known promiscuity of these molecules (Hebron et al., 2013; Imam et al., 2013; Karuppagounder et al., 2014; Tanabe et al., 2014; Lee et al., 2018). ABL1 hyperactivation has been shown in other neurodegenerative diseases with inhibitor treatment generally reported as beneficial for Alzheimer’s disease (AD), Niemann-Pick Type C (NPC), and amyotrophic lateral sclerosis (ALS) (Derkinderen et al., 2005; Jing et al., 2009; Klein et al., 2011; Schlatterer et al., 2011; Imamura et al., 2017; Osaki et al., 2018; Riancho et al., 2018).
Small Molecule Inhibitors
ABL1 oncoproteins have been a prime molecular target for cancer therapy using ATP-competitive inhibitors such as imatinib which directly challenged the viewpoint at the time that kinases were not good drug targets (Druker, 2008). Subsequent related ATP-competitive inhibitors such as nilotinib, dasatinib, bosutinib, and ponatinib were developed to address the prevalence of mutations in the ABL1 kinase domains; but all exhibit notable off-target activity including c-KIT, CSF1R, and PDGFRA/B (Shah et al., 2004; Weisberg et al., 2005; Dubreuil et al., 2009; Anastassiadis et al., 2011; Cortes et al., 2011; Ashman and Griffith, 2013; Rask-Andersen et al., 2014). Co-crystal structures of the kinase domain in complex with imatinib and other inhibitors have been critical in revealing the mechanism of action of point mutations causing drug resistance in certain cancers (Schindler et al., 2000; Nagar et al., 2002). In addition to ATP-competitive molecules, allosteric targeting of the myristate pocket and the SH2-kinase interface (a major autoinhibitory mechanism) has yielded additional compounds such as GNF-2 and asciminib (Wylie et al., 2017; Schoepfer et al., 2018). GNF-2 induces myristoylated ABL1 translocation to the endoplasmic reticulum, leading to competition with the intramolecular engagement of the NH2-terminal myristate for binding to the ABL1 kinase myristate-binding pocket.
Clinical Development
Initial development for ABL1 inhibitors was for indications requiring peripheral exposure and thus molecules have not been optimized for CNS penetrance. For example, it was shown preclinically that nilotinib has some penetration with 310 ng/mg of compound in brain tissue following 10 or 20 mg/kg (I.P.; 3–4 h post- dose) (Hebron et al., 2013). Recently, nilotinib (150 and 300 mg once daily for 24 weeks) was employed in an open-label clinical trial for PD and Dementia with Lewy Body (DLB; Pagan et al., 2016, 2019). The efficacy and tolerability results were promising, albeit preliminary, and constrained by the small sample size and the low doses employed (vs. oncology doses) possibly questioning the level of target engagement attained, particularly given the free brain ratio, i.e., Kpu,u of <0.1 (Lindholm et al., 2016). Recently, it was reported that while nilotinib was safe and tolerable in a PD population, it did not appear to exert a clinically meaningful effect (MJF foundation press release1). Bosutinib is also in the clinic for “degenerative dementias” (NCT02921477), but no brain or plasma quantification data have been reported. Arguably, bafetinib (INNO-406) may be a better molecule to test the hypothesis that c-Abl mediates PD pathogenesis given the potentially improved CNS exposure (Imam et al., 2013). K-0706 from Sun Pharma appears to represent a series of Abl inhibitors that have been assessed for CNS penetrance with a reported brain: plasma ratio, i.e., Kp of 0.4 at 1 h following a 30 mg/kg dose for a representative molecule (patent WO2017/208267). This molecule was also assessed for efficacy in a mouse model of PD and is currently being evaluated in a PD population (NCT02970019). Selectivity data on these molecules were not available.
Synopsis
The biological rationale for ABL1 is reasonable, particularly in PD with multiple biological substrates of the kinase being genetically linked and mechanistically connected to pathways of relevance to the disease. However, the molecules developed to date have suboptimal levels of CNS penetration and limited selectivity which may ultimately impact efficacy and tolerability since systemic exposures are likely to be in excess of those needed for CNS target engagement. Thus, more careful evaluation with suitably brain penetrant, potent, selective, and potentially better tolerated molecules may be warranted before the long-term therapeutic utility of selective ABL1 inhibitors in neurodegenerative patient populations can be fully ascertained.
CSNK1D (Casein Kinase 1δ)
Biological Rationale
The casein kinase 1 family comprises seven members of constitutively active serine/threonine kinases and was first identified as a set of proteins that could readily phosphorylate casein in vitro (reviewed in Cozza and Pinna, 2016). Across the family there is high conservation in the kinase domain, with 98% homology between delta and epsilon but isoforms vary in the length and primary structure of the regulatory domains. Casein kinases are negatively regulated via dimer formation and phosphorylation (Knippschild et al., 2005). The casein kinase 1 family phosphorylate over 140 additional targets associated with membrane transport processes, trafficking and microtubule associated dynamics, cell cycle progression and apoptosis (Yang Y. et al., 2017; Xu et al., 2019). Mutations in CSNK1D have been linked to familial advanced sleep phase syndrome and acute inhibition or genetic disruption results in alterations in circadian rhythms which correlates with the observation that core clock proteins are substrates for the enzyme (Xu et al., 2005; Etchegaray et al., 2009; Isojima et al., 2009; Meng et al., 2010; Brennan et al., 2013; Kennaway et al., 2015). Furthermore, it is possible that dysregulation of circadian rhythms in AD is aligned with alterations in CSNK1D function. Elevated levels and activity of CSNK1D has been reported in AD post-mortem brain tissue and ALS (Ghoshal et al., 1999; Yasojima et al., 2000; Thal et al., 2011; Salado et al., 2014). In addition, the δ isoform has been shown to phosphorylate a range of proteins which have been genetically linked to various neurodegenerative diseases in vitro. However, this correlation has not consistently been corroborated in vivo, e.g., PS2, BACE1, Parkin, TDP43, α-synuclein, LRRK2, and tau (Walter, 2000; Walter et al., 2001; de la Torre et al., 2009; Kosten et al., 2014; Alquezar et al., 2016; Nonaka et al., 2016; Morales-Garcia et al., 2017; De Wit et al., 2018). Pharmacological modulation of CSNK1δ/ε has been reported to have beneficial impacts in ALS, FTD, and PD neurodegenerative disease phenotypes in vivo (Salado et al., 2014; Alquezar et al., 2016; Morales-Garcia et al., 2017).
Small Molecule Inhibitors
Several ATP-competitive inhibitors have been identified with a range of preferences for the δ and ε isoforms, such as the set of molecules from Pfizer which have been demonstrated to exhibit central exposure and target engagement: PF-4800567, PF-0670462, and the PET ligand PF-5236216 (Meng et al., 2010; Mente et al., 2013; Wager et al., 2014, 2017). However, true selectivity has proven to be a challenge due to the high conservation within casein kinase 1 family members and related kinases such as p38α or TTBK1/2 (Halekotte et al., 2017).
Clinical Development
Following preclinical development using circadian rhythm assessment as a measure of central target engagement, PF-05251749 was assessed in Phase 1 for safety and tolerability with single and multiple ascending dose studies (NCT02443740; NCT02691702). Pfizer has reported no further development since October 2018 but Biogen has recently acquired the molecule with a $75 million upfront payment presumably to pursue this further in AD populations (January 2020)2.
Synopsis
The biological rationale for this kinase in ND disorders would be significantly augmented via additional investigation in relevant in vivo models; particularly in TDP43-related disorders which has seen the most effort to date. Pfizer has developed molecules rationally for selectivity, potency and brain penetrance which appear to be well tolerated in the clinic as far as we can currently ascertain. Thus, it would seem further assessment of these molecules in ND populations may be warranted.
CSF1R (Colony Stimulating Factor 1 Receptor)
Biological Rationale
Microglial cells have been described as the master regulators of the neuroinflammatory response associated with neurodegenerative disease. In numerous preclinical models of neurodegenerative disease, and in post-mortem brain samples from AD patients, an increase in microglia number and their proliferation rate has been observed (Perry et al., 2010; Gómez-Nicola et al., 2013; Walker et al., 2017). This proliferative activity is regulated by the tyrosine kinase, CSF1R; indeed, CSF1R-deficient mice and rats have no microglia (Ginhoux et al., 2010; Erblich et al., 2011; Gómez-Nicola et al., 2013; Pridans et al., 2018). Furthermore, CSF1R itself is upregulated in preclinical neurodegeneration models and human post-mortem samples (Akiyama et al., 1994; Murphy et al., 2000). Genetic and pharmacological strategies to eliminate or reduce CSF1R kinase activity, and thus decrease microglial proliferation, have been shown to promote beneficial effects in preclinical models of AD, ALS, tauopathies, and MS (Asai et al., 2015; Bruttger et al., 2015; Dagher et al., 2015; Borjini et al., 2016; Martínez-Muriana et al., 2016; Olmos-Alonso et al., 2016; Spangenberg et al., 2016, 2019; El-Gamal et al., 2018; Elmore et al., 2018; Sosna et al., 2018; Spiller et al., 2018; Mancuso et al., 2019; Zhong et al., 2019). Of note, a number of PET tracers have recently been developed to monitor response to CSF1R inhibitors which we believe reflects the broad level of interest in this mechanism (Tanzey et al., 2018; Horti et al., 2019; Janssen and Mach, 2019; Mason et al., 2019).
Colony stimulating factor 1 receptor has two known ligands, CSF1 (colony stimulating factor 1) which is also expressed in microglia and IL-34 predominantly expressed in neurons (Lin et al., 2008; Wei et al., 2010; Mizuno et al., 2011; Nandi et al., 2012; Wang et al., 2012; Chitu et al., 2016). Ligand binding triggers dimerization and transphosphorylation to activate the receptor and subsequently activate a downstream cascade of cytoplasmic mediators that enable development, survival or proliferation phenotypes (Akiyama et al., 1994; Xu et al., 2015; Askew et al., 2017). Mutations in CSF1R have been associated with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) (Stabile et al., 2016; Lynch et al., 2017). Interestingly this condition can often be misdiagnosed as AD, corticobasal syndrome, DLB or atypical PD (Puschmann, 2013; Adams et al., 2018; Sharma et al., 2019). More recently, it has been reported that CSF1R may have variants associated with AD pathogenesis (Giau et al., 2019a, b).
Small Molecule Inhibitors
Structure-based drug development has been leveraged in some cases to rationally design CSF1R inhibitors including pexidartinib (PLX3397), GW2580, KI20227, AZD7507; JNJ-40346527, BLZ945/buparlisib or a set of DFG-out inhibitors (Ohno et al., 2006; Meegalla et al., 2008; Meyers et al., 2010; Uitdehaag et al., 2011; Pyonteck et al., 2013; Scott et al., 2013; Dagher et al., 2015). These molecules have largely been intended for indications such as rheumatoid arthritis and cancer related disorders where systemic exposure is required, thus these molecules have not been optimized for BBB penetration. Lack of selectivity is also a challenge with these molecules in CNS disorders; CSF1R is a class III receptor tyrosine kinase (RTK) which also includes cKIT, FLT3, and PDGFs in its family. The majority of CSF1R inhibitors also exhibit activity against these family members (as well as other kinases) and the converse is true, i.e., class III RTK inhibitors such as masitinib and sunitinib also inhibit CSF1R (Uitdehaag et al., 2011; Gandin et al., 2015). More recently, fragment-based drug design has been leveraged to identify novel starting points for CSF1R inhibition (Machiraju et al., 2019), presumably to better optimize selectivity and brain penetration.
Clinical Development
The broad-spectrum class III RTK inhibitor, masitinib, is currently in phase III for ALS (NCT02588677; NCT03127267) (Petrov et al., 2017; Mora et al., 2019) and has also been assessed in AD populations (NCT00976118), although detailed publications of these data are not yet available. Plexxikon initiated a Ph2a trial in AD populations in 2016 (Eudra 2016-000429-38) but no results have been published / reported to date.
Synopsis
While multiple data streams have indicated neuroinflammation and microglial activation as pathophysiological features of neurodegenerative disorders, it is not clear at what stage in the disease neuroinflammatory process would be the most appropriate point for intervention to lead to clinical benefit. There remains a question of when this detrimental effect of microglia may become relevant and consequently when would be an appropriate time to treat with this mechanism. The molecules developed and tested to date have suboptimal levels of CNS penetration and/or limited selectivity which again may impact the interpretation of the preclinical efficacy and ultimately clinical tolerability. That said there are preclinical data demonstrating a clear effect of CSF1R inhibitors (Olmos-Alonso et al., 2016; Mancuso et al., 2019; Spangenberg et al., 2019). The strong structural understanding of this kinase and significant interest in the target should prime the repurposing of existing molecules and the next generation of ligands designed for CNS indications.
MAP3K12 (DLK, Dual Leucine Zipper Kinase)
Biological Rationale
Dual leucine zipper kinase, a mitogen-activated protein kinase kinase kinase, functions as an injury sensor that initiates the c-Jun N-terminal kinase (JNK)-dependent stress response in neurons to mediate context-dependent axon re- and degeneration (Gallo and Johnson, 2002; Hirai et al., 2006). DLK activation can lead to rapid neuronal degeneration while both the presence and absence of DLK has been reported to promote axon regeneration following injury (Chen et al., 2008; Hammarlund et al., 2009; Miller et al., 2009; Xiong et al., 2010; Ghosh et al., 2011; Itoh et al., 2011; Shin et al., 2012, 2019; Watkins et al., 2013; Welsbie et al., 2013; Wlaschin et al., 2018). The seemingly contradictory responses in neurons appear context dependent and the substrates for JNK (see section 3.6) phosphorylation differ accordingly (reviewed in Tedeschi and Bradke (2013) and Siu et al. (2018)]. In the context of chronic neurodegenerative disease, the experimental evidence (mostly from Genentech investigators), supports the notion that DLK is a conserved regulator of neuronal degeneration following injury. Aberrant activation of the DLK-JNK pathway has been reported in transgenic mouse models and spinal cord lysates from ALS patients (Le Pichon et al., 2017). Equivalent findings have been reported in two AD transgenic mouse models and in post-mortem CNS tissues from patients with disease (Le Pichon et al., 2017). Genetic manipulation or pharmacological inhibition of DLK, with compounds developed by Genentech, was reported to attenuate synaptic loss, neuronal degeneration and functional decline in models of AD, PD, and ALS in addition to acute neuronal injury models (Miller et al., 2009; Itoh et al., 2011; Pozniak et al., 2013; Watkins et al., 2013; Patel et al., 2015a, b, 2017; Le Pichon et al., 2017; Yin et al., 2017). It should be borne in mind that the closely related family member MAP3K13 (LZK, leucine zipper kinase) also regulates axonal responses to injury via JNK signaling, and notably, co-overexpression of both kinases did not have synergistic effects (Chen et al., 2016). The potential for complex functional interaction and cross-regulation should be considered in the context of therapeutic development.
Small Molecule Inhibitors
Small molecule DLK inhibitors that reduced c-Jun phosphorylation in mouse models were initially identified following high throughput screening approaches and refined using a shape-based scaffold hopping approach (Patel et al., 2015a, b). Structure-based design was leveraged to further optimize two series with respect to pharmacokinetics (PK, particularly brain penetrance), selectivity and tolerability profiles with potential for prolonged administration exemplified by GDC-0134 (Patel et al., 2017). The selectivity profile indicates 23-fold selectivity against the closely related family member MAP3K13; which may functionally compensate in some contexts. Other molecules such as tozasertib or sunitinib have been reported to have DLK inhibitory activity, based on high content screening using retinal ganglion cells, but have not been optimized for selectivity or PK parameters commensurate with CNS exposure (Welsbie et al., 2013, 2017). More recently, a platform capable of identifying inhibitors of DLK palmitoylation in order to modulate kinase localization has been reported (Martin et al., 2019).
Clinical Development
Genentech’s DLK inhibitor molecule is currently in PhI for ALS (NCT02655614).
Synopsis
This case study represents a clear example of how structural and medicinal chemistry know how has been applied to actively develop a kinase inhibitor with CNS penetrance. It remains to be seen whether the preclinical assessment was sufficient to mitigate the risks associated with apparent context-dependent effects of DLK inhibition as the published data may suggest a narrow therapeutic index with potential toxicology concerns.
GSK3B (Glycogen Synthase Kinase 3β, GSK3β)
Biological Rationale
GSK3B codes for two isoforms of a constitutively active serine/threonine kinase which phosphorylates over 40 substrates with key roles in Wnt signaling, insulin signaling, glycogen metabolism, development, mitotic regulation, inflammation (particularly the innate immune response), neurotransmitter and neurotrophic factor signaling and microtubule dynamics (Rayasam et al., 2009; Patel and Woodgett, 2017). GSK3β activity is tightly regulated by several mechanisms including cellular localization, protein interactions and phosphorylation, leading to increasing (Y216 in the activation loop) or decreasing (S9) substrate binding ability (Hoshi et al., 1996; Haar et al., 2001; Bijur and Jope, 2003; Dajani et al., 2003). Increased expression and activity of GSK3β has been noted in AD, PD, and ALS post-mortem tissue while decreased expression and activity was reported in Huntington’s disease (HD) patient brains (Pajak et al., 2009; Lim et al., 2014; Credle et al., 2015; Nicolia et al., 2017). Furthermore, rare variants have been identified as causative in AD and FTD (frontotemporal dementia) (Schaffer et al., 2008; Kettunen et al., 2015). Treatment with GSK3β inhibitors has been shown to be impactful in a number of models, but there have also been challenges given the toxicities associated with some compounds (Selenica et al., 2007; Peineau et al., 2009; Petit-Paitel et al., 2009; Serenó et al., 2009; Eldar-Finkelman and Martinez, 2011; Ahn et al., 2012; Kramer et al., 2012; Fuchs et al., 2015). GSK3β was identified as a tau kinase in addition to directly promoting Aβ production in AD pathophysiology (Hanger et al., 1992; Sperbera et al., 1995; Spittaels et al., 2000; Phiel et al., 2003; Terwel et al., 2008). Accordingly, some inhibitors have been shown to reverse tau hyperphosphorylation and subsequently resulted in some beneficial outcomes in models (Martinez et al., 2002; Serenó et al., 2009; Onishi et al., 2011; Berg et al., 2012; Kramer et al., 2012; Xiong et al., 2013).
Small Molecule Inhibitors
Phosphorylation of target proteins by GSK3β generally inhibits their activity; inhibitor treatment thus disinhibits the pathway(s) in question. Analysis of the GSK3β structure as an unphosphorylated apo-enzyme together with Y216 monophosphorylated enzyme complexed with a peptide provided insight into both its regulation of the kinase activity and its preference for pre-phosphorylated substrates (Bax et al., 2001; Dajani et al., 2001, 2003; Haar et al., 2001). Diverse GSK3β inhibitors have been developed and can be grouped according to their mechanism of action and binding affinity: (1) ATP-competitive inhibitors which constitute the largest pool with a diverse range of scaffolds such as SB-216763 and AZD1080 (Georgievska et al., 2013; Li et al., 2015). These and other ATP-competitive inhibitors have been shown to exhibit BBB penetrance including pyrazines or oxadiazoles (Saitoh et al., 2009; Onishi et al., 2011; Berg et al., 2012; Yngve et al., 2012; Pandey and DeGrado, 2016; Prabhakaran et al., 2017). (2) non-ATP competitive inhibitors which either inhibit by binding at a catalytic triad and resisting the correct substrate orientation, such as tideglusib (also known as NP-12) and a thiadiazolidinone derivative (which has shown evidence of brain penetration) (Luna-Medina et al., 2007; Pandey and DeGrado, 2016), or by directly competing with the substrate; (3) irreversible inhibitors (Perez et al., 2009); (4) peptide-like inhibitors that act as pseudo-substrates; (5) allosteric inhibitors which bind in non-ATP and non-substrate competitive fashion and thus have potential to enable a more selective and subtle modulation of the enzyme (Martinez et al., 2002; Palomo et al., 2011, 2017) and (6) metal ions such as lithium chloride which has not been included in this review, given that it is neither specific nor potent (Maqbool et al., 2016). Structural analyses of chemically diverse inhibitors in complex with GSK3β have illuminated how the enzyme interacts with selective and non-selective inhibitor scaffolds and have provided insight and opportunity to guide further rational design approaches (Bertrand et al., 2003; Kramer et al., 2012; Sivaprakasam et al., 2015).
Clinical Development
Despite extensive promising preclinical data, GSK3β inhibitors have rarely reached clinical trials (Pandey and DeGrado, 2016). Molecules that have reached human testing for neurodegenerative indications are tideglusib (NCT00948259; NCT01049399; NCT01350362) and AZD1080. No clinical pharmacokinetic data for tideglusib has been reported despite a radiolabeled analog being reported (Pandey and DeGrado, 2016). Early studies in AD patients suggested decreased brain atrophy, reduction of phospho-tau and BACE-1 levels in cerebrospinal fluid and improved cognitive function (del Ser et al., 2013). Further assessment of tideglusib suggested no clinical benefit although it was well tolerated (up to a 1000 mg qd for 24 weeks) (Lovestone et al., 2015). Tideglusib treatment was additionally reported to decrease brain atrophy in progressive supranuclear palsy (PSP) following 1 year of treatment with 600 or 800 mg daily but with no other clinical benefits (Höglinger et al., 2014; Tolosa et al., 2014). In Phase I trials, AZD1080 was apparently safe and well tolerated with reasonable PK profiles and evidence of peripheral target engagement (Georgievska et al., 2013). No further development has been published since. ALS was also being evaluated as a potential indication (MR/K015273/1) but again no clinical progression has been reported.
Synopsis
Published data suggests GSK3β inhibition may have therapeutic benefit across a range of neurodegenerative diseases. Many small molecule inhibitors have been generated, of which some appear to have a reasonable degree of CNS penetration. Toxicity may be one potential issue, likely due in part to the essential role of the enzyme in many fundamental physiological pathways, which has limited or prevented further exploration. Nonetheless, two molecules have been identified as being sufficiently safe for clinical investigation but have not yielded positive data for CNS indications.
c-Jun N-Terminal Kinases (JNKs, Particularly JNK3/MAPK10)
Biological Rationale
c-Jun N-terminal kinases are members of the mitogen-activated protein kinase (MAPK) family and regulate serine/threonine phosphorylation of several transcription factors in response to various stimuli, e.g., endoplasmic reticulum stress / unfolded protein response signaling, oxidative and excitotoxic stress, DNA damage, neurotoxins, cytokines, and fatty acids (Haeusgen et al., 2009; Bogoyevitch et al., 2010). Activation of the JNK pathway, through phosphorylation in the activation loop by MAP2Ks, relies on the coordinated interaction of scaffold proteins enabling the assembly of signaling complexes and recruit JNK-phosphorylated transcription factors (Mooney and Whitmarsh, 2004; Guo and Whitmarsh, 2008; Engström et al., 2010; Antoniou et al., 2011; Zhan et al., 2015, 2016). JNKs have been implicated in a range of neurodegenerative disorders including PD and AD (Chambers and LoGrasso, 2011; Crocker et al., 2011; Mehan et al., 2011; Yarza et al., 2016). Increased JNK phosphorylation has been reported in human post-mortem AD patient brains; together with reports of tau and amyloid precursor protein (APP) phosphorylation resulting in a stimulation of Aβ42 peptide production (Yoshida et al., 2004; Kimberly et al., 2005; Lagalwar et al., 2006; Pearson et al., 2006; Vogel et al., 2009; Killick et al., 2014; Gourmaud et al., 2015). Genetic depletion and pharmacological inhibition of JNK have been found impactful in AD in vivo model phenotypes (Bogoyevitch et al., 2004; Bowers et al., 2011; Probst et al., 2011; Ramin et al., 2011; Hong et al., 2012; Zhou et al., 2015; Petrov et al., 2016; Gourmaud et al., 2018). Increased nuclear staining of c-Jun (JNK substrate) as a surrogate marker for JNK activity has been observed in patients with PD but not in dementia with Lewy bodies (DLB) (Ferrer et al., 2001; Hunot et al., 2004). JNK3, the predominant CNS expressed isoform with roles in brain development as well as learning and memory function (Kuan et al., 1999; Bevilaqua et al., 2003; Brecht et al., 2005; Waetzig et al., 2006; Eminel et al., 2008). Kuan et al. (1999), Bevilaqua et al. (2003), Brecht et al. (2005), Waetzig et al. (2006) and Eminel et al. (2008) is robustly activated in toxin models of PD while genetic deletion of JNK3 and pan-kinase inhibition are both protective in these models. However, protective effects of JNK3 inhibition / deletion in genetic models of PD are less clear (Choi et al., 1999, 2015; Saporito et al., 1999, 2000; Hunot et al., 2004; Wang et al., 2009; Chambers et al., 2011, 2013). Complementary data exists for other neurodegenerative disease models with respect to genetic and pharmacological interrogation (Morfini et al., 2009; Perrin et al., 2009; Suzuki and Matsuoka, 2013; Wityak et al., 2015). Finally, JNK3 has also been identified as a genetic modifier for spinal and muscular atrophy (Genabai et al., 2015).
The JNK-interacting protein 1 (JIP1) derived peptide inhibitor (D-JNK1-1, also known as XG-102, AM-111 or brimapitide) has the advantage of comparative specificity for JNK and is fused to the Tat peptide sequence to enable cellular delivery across the BBB (Barr et al., 2002; Davoli et al., 2014). Peptide treatment has been shown to be efficacious in multiple in vivo models (Repici et al., 2008; Colombo et al., 2009; Sclip et al., 2011, 2014; Tran et al., 2012). More recent preclinical data suggests that peptide inhibitor treatment in the 5xFAD model improves cognitive performance and decreases amyloid load, mirroring the effect of genetic deletion of JNK3 (Gourmaud et al., 2018). In addition, several preclinical studies revealed a protective effect of the peptide inhibitors in hearing loss following acoustic trauma; this led to subsequent clinical development for acoustic trauma and acute sensorineural hearing loss resulting in statistically significant positive effects (Grindal et al., 2010; Omotehara et al., 2011; Suckfuell et al., 2014; Eshraghi et al., 2018).
Inhibitors
Several ATP-competitive JNK inhibitors have been developed with a range of characteristics including good CNS penetration; only three are mentioned here as illustrations of their class. AS602801 (bentamapimod) is a pan-JNK inhibitor with good bioavailability and able to cross the BBB (Ferrandi et al., 2011). SP600125 is a CNS penetrant and reversible anthrapyrazolone ATP competitive inhibitor, 300-fold more selective for JNK versus ERK and p38 MAPK (Bennett et al., 2001; Vogel et al., 2009; Davies and Tournier, 2012). CC-930 (tanzisertib) is a potent, selective, orally active JNK inhibitor selective against ERK1 and p38α (Krenitsky et al., 2012). More recently, J30-8 was reported to have over 2500-fold isoform selectivity for JNK3, which could make it a good tool molecule for further mechanistic studies in ND models (Dou et al., 2019).
A structural class analysis of over 40 JNK structures, both apo and in complex with ligands or peptides has suggested the possibility for allosteric interactions between the catalytic and peptide binding sites and the A-loop (Ansideri et al., 2018). This has revealed two potential distinct autoinhibitory mechanisms leading to blocking of the formation of the active catalytic site in the inactive kinase (Laughlin et al., 2012; Mishra and Günther, 2018). Furthermore, structural assessments of peptide binding modes indicate the basis for selectivity of different docking motifs.
Clinical Development
A few small molecule JNK inhibitors have entered clinical trials including tanzisertib and bentamapimod but none for the treatment of neurodegenerative disorders. While tanzisertib appeared safe and well-tolerated, observations of dose-responsive decreases in MMP7 plasma levels halted clinical development. Clinical trials with molecules further upstream of the cascade have also been reported, such as CEP-1347, a mixed lineage kinase inhibitor, which failed to show benefit in PD patients (Shoulson and Parkinson’s Study Group and Precept Investigators, 2006; Parkinson Study Group, 2007). It is worth noting that DLK (discussed above) is also upstream in the JNK cascade so may ultimately provide evidence to indicate JNK utility in ALS if the Genentech molecule continues to progress.
Synopsis
The JNK cascade appears to be a relevant axis with rationale for inhibitor development in both AD and PD. While there has been progress in inhibitor design, the specificity across JNK isoforms, in addition to other kinases such as MAPK14, with a brain penetrant small molecule is still a challenge that has not yet been appropriately overcome. Improved selectivity as a key strategy is likely only achievable via targeting interactions such as JIP1 outside of the ATP pocket.
LRRK2 (Leucine Rich Repeat Kinase 2)
Biological Rationale
Leucine rich repeat kinase 2 is a large, multi-domain enzyme with dual GTPase and kinase activities that has been genetically linked to sporadic and familial PD (Zimprich et al., 2004; Khan et al., 2005; Ross et al., 2011; Paisán-Ruiz et al., 2013). Mutations are distributed throughout the protein, predominantly in the kinase and ROC/GTPase domain and have been shown to modulate ATP or GTP hydrolysis rates in vitro (Smith et al., 2005; West et al., 2005; Gloeckner et al., 2006; Greggio et al., 2006; Nichols et al., 2010; Dusonchet et al., 2011; Cookson, 2015; Manzoni et al., 2015). Some of these data implicate increased kinase activity (and resultant phosphorylation of downstream Rab proteins) with specific mutations such as G2019S, which has spurred development of LRRK2 kinase site inhibitors. It is noteworthy that other mutations have not recapitulated these observations (Taylor and Alessi, 2020). A computational model of the full length homodimeric protein supports the notion of cross-regulation between the GTPase and kinase domains and suggest that downstream biochemistry may be more complicated than a simple enhancement of kinase activity (Mata et al., 2006; Greggio et al., 2008; Weiss, 2008; Berger et al., 2010; Guaitoli et al., 2016; Rosenbusch and Kortholt, 2016). In particular, modeling approaches have also enabled predictions of repeat domain structure and impact of mutations using other organism homologs and the overall view is one of a compact architecture with tight, multidomain organization and intramolecular regulation of the enzymatic activities (Vancraenenbroeck et al., 2012; Mills et al., 2014; Guaitoli et al., 2016). Crystal structures for the LRRK2 ROC (Ras of complex proteins) domain in complex with GDP-Mg2+ further support the notion that LRRK2 may actually function as a dimeric GTPase (Deng et al., 2008; Deyaert et al., 2017; Nguyen and Moore, 2017; Mills et al., 2018).
Leucine rich repeat kinase 2 is regulated by autophosphorylation and phosphorylation by other kinases and interactions with kinase-specific chaperones (Chia et al., 2014; Muda et al., 2014; Shu et al., 2016). Unbiased protein-protein interaction and other approaches have provided insights into the functional roles of LRRK2 including the regulation of vesicular and lysosomal trafficking, lysosomal maturation and autophagy regulation (Shin et al., 2008; Tong et al., 2012; Manzoni et al., 2013, 2016; Beilina et al., 2014; Cirnaru et al., 2014; Dodson et al., 2014; Gómez-Suaga et al., 2014; Parisiadou et al., 2014; Piccoli et al., 2014; Saez-Atienzar et al., 2014; Schapansky et al., 2014; Stafa et al., 2014; Waschbüsch et al., 2014; Yun et al., 2015; Bang et al., 2016; Islam et al., 2016; Kuwahara et al., 2016; Roosen and Cookson, 2016; Steger et al., 2016; Hur et al., 2019; Lewis, 2019). In support of this genetic ablation of LRRK2 leads to reduced autophagic clearance and accordingly, α-synuclein accumulation in the absence of neurodegeneration (Biskup et al., 2006; Giasson et al., 2006; Higashi et al., 2007; Alegre-Abarrategui et al., 2009; Tong et al., 2012; Baptista et al., 2013; Ness et al., 2013; Longo et al., 2017). It is noteworthy that although LRRK2 is expressed in various brain regions it also has broad systemic expression across several other organs such as lung, kidney, and heart.
Inhibitors
Several molecules were assessed and/or developed to inhibit LRRK2 kinase function based on the observations of increased kinase activity and associated neurotoxicity with the most common mutation, G2019S [reviewed in Zhao and Dzamko (2019)]. Of note, compounds have been developed specifically with the aims of combining selectivity and brain penetrance. An HTS and subsequent lead optimization provided the low nM reversible ATP competitive inhibitor LRRK2-IN-1 but PK studies revealed limited BBB permeation (Deng et al., 2011). The anthracene and phenanthrene derivatives LDN-73794 and LDN-22684 inhibited LRRK2-G2019S and wild-type LRRK2 in the low micromolar range but with different mechanisms of inhibition. LDN-73794 was confirmed to be ATP competitive, whereas LDN-22684 was found to be a non-ATP-competitive inhibitor and furthermore was not GTP- nor substrate-competitive and was thus deduced to be an allosteric inhibitor (Liu et al., 2010a, b). GSK2578215A was another highly potent and selective LRRK2 inhibitor with high brain exposures but unfortunately failed to inhibit LRRK2 in mice, potentially due to species differences (Reith et al., 2012). The initial HTS output GNE-7915 was further developed to identify the analogs GNE-0877 and GNE-9605; all of which were potent LRRK2 inhibitors with enhanced brain penetration and robust, dose-dependent inhibition in mice and non-human primates (NHP) (Estrada et al., 2014; Fuji et al., 2015). PF-06447475 is another second-generation molecule with high potency, selectivity and good BBB permeability properties but poor oral PK profiles (Daher et al., 2015; Henderson et al., 2015). Finally, an indazole termed Mli-2 was reported to exhibit a marked improvement in potency for G2019S LRRK2, orally available with good brain penetrance/PK profiles and inhibitory activities (Fell et al., 2015; Scott et al., 2017). For completeness, we also acknowledge here the development of GTP binding inhibitors which have the potential of impacting on kinase function through interaction of the two domains, although these molecules are considerably behind the plethora of kinase domain inhibitors (Sen et al., 2009; Li et al., 2014, 2015). Alternatively, the structural interface between LRRK2 and the 14-3-3 protein, a key interaction partner for LRRK2, has been reported (Stevers et al., 2017, 2018). Many pathogenic mutations in LRRK2 disrupt or weaken the dimeric or the 14-3-3 interactions; suggesting a stabilization approach as a potential alternative therapeutic strategy (Nichols et al., 2010; Fraser et al., 2013; Melrose, 2015; Zhao et al., 2015; Lavalley et al., 2016).
Clinical Development
Preclinical safety testing for LRRK2 inhibitors has been challenging, partially due to robust expression of LRRK2 in the lung, kidney and some peripheral immune cells. Knockout mice have a normal life span, but pathology has been noted in peripheral organs, particularly the kidney (Herzig et al., 2011; Tong et al., 2012; Ness et al., 2013). Kidney phenotypes have not been observed in rodents or NHP treated with LRRK2 inhibitors (Fell et al., 2015). However, phospholipidosis and various pathologies have been observed in lungs of NHP with several structurally diverse inhibitors, which appeared to be reversible upon withdrawal of the compounds, together suggesting on-target toxicity (Herzig et al., 2011; Baptista et al., 2013, 2020; Ness et al., 2013; Shayman and Abe, 2013; Daher et al., 2015; Fuji et al., 2015). Interestingly, chronic pharmacological inhibition of LRRK2 appears to result in reduced expression of the protein by promoting its ubiquitination and consequent proteasomal degradation (Herzig et al., 2011; Fuji et al., 2015; Zhao et al., 2015; Lobbestael et al., 2016; Perera et al., 2016). In this context, it will be interesting to ascertain effects of antisense oligonucleotides such as BIIB094 against specific mutations in LRRK2 (NCT03976349). The Genentech compounds have been licensed to Denali and are currently in the clinic for Phase 1b trials as DNL151 and DNL201 (NCT04056689 and NCT03710707). Denali press releases have since reported both DNL151 and 201 are well-tolerated with no obvious functional evidence of lung toxicology, and additionally have high levels of target and pathway engagement with modulation of lysosomal biomarkers3.
Synopsis
The genetic linkage in LRRK2 to PD susceptibility together with altered protein activity provides the rationale for inhibitor development in PD. There are now several chemically diverse, potent, selective and brain penetrant inhibitors. However, the apparent on-target toxicity seen with some of these inhibitors in NHP raises concerns around the therapeutic index and safety of chronic dosing which have impeded clinical investigations with many of these molecules. The ongoing trials are seeking to leverage both safety and efficacy biomarkers with a view to impacting Parkinson’s onset/progression in a genetically stratifiable cohort.
MAPK14 (p38α Mitogen Activated Protein Kinase)
Biological Rationale
P38α is a ubiquitously expressed serine/threonine kinase activated by upstream MAP2Ks in response to environmental and inflammatory/oxidative/metabolic/ER stress. In the CNS, p38α (like JNK3) has been proposed to mediate synaptic function, proteostasis and endolysosomal function in response to stress signals and indeed, it is likely that there is cross-regulation between the p38 and JNK pathways [reviewed in Borders et al. (2008)]. Accordingly, the MAPK stressor signal cascade has broad literature representation in neurodegenerative disease, and p38α is no exception with specific dysregulation observed at early stages in AD, ALS, PD, FTD, and PSP (Hensley et al., 1999; Atzori et al., 2001; Hartzler et al., 2002; Raoul et al., 2002; Sun et al., 2003). P38α, like GSK3β or CDK5-p25, has been hypothesized to link Aβ and tau or α-synuclein (Puig et al., 2004; Grassi et al., 2019). Genetic suppression of p38α has been reported to be beneficial in AD models and pharmacological inhibition of p38α has been claimed to confer benefits in vivo (Alam, 2015; Roy et al., 2015; Maphis et al., 2016; Schnöder et al., 2016; Colié et al., 2017; Lee and Kim, 2017; Zhou et al., 2017).
Small Molecule Inhibitors
While potency, selectivity and SAR data is available for several individual p38α inhibitors; Astolfi and colleagues were the first to systematically assess these from a structural perspective [reviewed in Astolfi et al. (2018)]. Both ATP-competitive and non-competitive molecules have been identified such as VX-745 (neflamapimod), MW150 and the related MW181, PH-797804, BMS-582949, PF-03715455 and an allosteric binder. Notably, multiple structures have been generated with both ATP- and non-ATP site binders, often through an attempt to improve on selectivity profiles (Fitzgerald et al., 2003; Swahn et al., 2005; Sack et al., 2008; Scior et al., 2011; Arai et al., 2012; Getlik et al., 2012; Astolfi et al., 2019) and reviewed in Astolfi et al. (2018).
Clinical Development
Neflamapimod (VX745) was discovered through structure-based design at Vertex and previously assessed in rheumatoid arthritis in 2001. The molecule appears to be tolerated up to 125 mg twice daily for 12 weeks (Alam et al., 2017). As a result of these data and the preclinical profile, a Phase IIa study in patients with early stage AD (6–12 weeks in patients with MMSE 20-28 and biomarker positive) was performed and demonstrated that neflamapimod treatment led to significant improvement in episodic memory (Alam et al., 2017). A blinded, placebo-controlled Phase 2 study of 152 people with CSF biomarker-confirmed mild AD was initiated in December 2017, with clinical output expected late 2019 (NCT03402659). Additional clinical studies of neflamapimod in DLB (NCT04001517) and HD (NCT03980938) have since been initiated.
Synopsis
The notion of MAPK pathway involvement in neurodegenerative disease is broadly supported in the literature. However, targeting these pathways are challenging due to (1) “crossover” between parallel pathways and (2) achieving selectivity and sufficient CNS exposure without notable systemic levels for a small molecule against any kinase in this pathway, including p38α. No drugs targeting this kinase are on the market yet for any indication, including chronic obstructive pulmonary disease, rheumatoid arthritis and corticosteroid-resistant asthma. Indeed, more than a dozen distinct molecules have failed in the clinic at least partly due to hepatotoxicity and development of tolerance. However, the reported favorable profile of neflamapimod and multiple ongoing clinical studies will hopefully inform whether this mechanism has therapeutic benefit.
MTOR (Mechanistic Target of Rapamycin Complex 1, mTOR/mTORC1)
Biological Rationale
The serine/threonine kinase MTOR is a regulatory nexus for cell growth and metabolism in response to nutrients, growth factors and cellular energy conditions modulating autophagy, glucose metabolism, protein synthesis, and mitochondrial functions. MTOR is the catalytic subunit of two structurally and functionally distinct complexes with respect to protein composition: the rapamycin sensitive mTORC1 which functions as a nutrient sensor activated by insulin, growth factors and amino acids; and mTORC2 which has a role in cytoskeleton organization. Under nutrient-replete conditions, mTORC1 as an autophagy repressor via phosphorylation of ULK1, ATG13, ATG14L, and TFEB, key positive regulators of autophagy; and a positive regulator of protein synthesis via phosphorylation of S6K and eIF4E BP1-4 thus promoting global and cap-end translation (Rabanal-Ruiz et al., 2017; Walters and Cox, 2018). More recently, cryoEM data have refined our thinking as to how MTOR interacts with and is regulated by mTORC1 complex members such as LST8, LAMTOR (“Ragulator”), Raptor, Rheb, PRAS40, and chaperonins (Yang et al., 2016; Yang H. et al., 2017; de Araujo et al., 2017; Cuéllar et al., 2019).
A consistent observation across neurodegenerative disease pathophysiology is the occurrence of specific misfolded proteins. These proteins ultimately appear as aggregates/inclusions that are proposed to infringe on normal cellular function including impaired protein clearance either by the ubiquitin-proteasome pathway or the lysosomal degradation pathway (including autophagy) [reviewed in Frake et al. (2015) and Menzies et al. (2017)]. Several studies have implicated impaired autophagy and aberrant mTOR signaling in models and/or post-mortem brain samples of various neurodegenerative diseases (Webb et al., 2003; Li et al., 2005; Nixon et al., 2005; Yu et al., 2005; Boland et al., 2008; Pickford et al., 2008; Jaeger and Wyss-Coray, 2009; Chu, 2010; Dehay et al., 2010; Heng et al., 2010; Spilman et al., 2010; Winslow et al., 2010; Zheng et al., 2010; Bové et al., 2011; Caccamo et al., 2011; Barmada et al., 2014; Manzoni et al., 2016). A relevant therapeutic approach could thus include facilitating the clearance of aberrantly folded proteins by increasing autophagy through repressing mTORC1; consistent with this notion, rapamycin treatment restores mTOR activity and rescues neurodegenerative disease model phenotypes (Webb et al., 2003; Ravikumar et al., 2004; Sarkar et al., 2009; Dehay et al., 2010; Menzies et al., 2010; Spilman et al., 2010; Bové et al., 2011; Cortes et al., 2012; Ozcelik et al., 2013; Jiang et al., 2014). It is also noteworthy that this approach will have effects beyond the CNS as illustrated in SOD1 mouse models for ALS, where both detrimental and beneficial immunomodulatory effects have been reported (Zhang et al., 2011; Staats et al., 2013).
Small Molecule Inhibitors
The allosteric mTORC1 inhibitor rapamycin (also known as sirolimus) was the first drug to be identified as an autophagy inducer through its interaction with FKBP12 which then binds to and inhibits the kinase activity of mTORC1 (Vézina et al., 1975; Heitman et al., 1991; Van Duyne et al., 1993; Choi et al., 1996). Crystal and cryoEM structures have illustrated how the FKBP12-rapamycin binding domain appears to regulate access to the active site and thus blocks substrate recruitment preventing downstream phosphorylation of regulatory proteins such as 4E-BP1 and S6K1 (Choi et al., 1996; Yip et al., 2010; Yang et al., 2013, 2016). Rapamycin together with other related natural product “rapalogs” such as temsirolimus (CCI-779), everolimus (RAD001), and ridaforolimus (AP23573) have good selectivity for mTORC1 versus mTORC2 but suboptimal exposure levels in the brain (O’Donnell et al., 2008; Benjamin et al., 2011). ATP-competitive inhibitors of mTOR inhibitors such as Torin 1, WYE-125132, and AZD-8055 have lower selectivity profiles for mTORC1 versus mTORC2 due to its mode of action and accordingly are able to block phosphorylation of all substrates of the two complexes as opposed to a subset of substrates. These ATP-competitive inhibitors are viewed as being more promising in oncology for tumors that are addicted to the mTOR signaling pathway and in line with this, suboptimal levels of BBB penetration in addition to toxicity issues with chronic dosing have been reported (Thoreen et al., 2009; Yu et al., 2010; Jhanwar-Uniyal et al., 2019).
Clinical Development
Rapamycin and its analogs have been used for many years in organ transplant patients (for review see Waldner et al., 2016) but its suggested utility in CNS disorders has been a more recent development driven by preclinical evidence of efficacy (reviewed above). These preclinical data together with the good safety profile led to the initiation of a phase II clinical assessment of rapamycin in an ALS population (NCT03359538). Unfortunately, no clinical outcome data has been published to date and no other trials of rapamycin or its analogs have been reported. Of note, everolimus appears to have better oral bioavailability and partitioning to the CNS and thus may be an alternative option for clinical development although none has been instigated to date for neurodegenerative indications (Frost and Hulbert, 2015; Palavra et al., 2017).
Synopsis
The notion of augmenting autophagic flux for neurodegenerative disease is cogent and warrants detailed assessment. Indeed, many organizations are pursuing approaches to do just that. Consistent with the hypothesis, inhibition of mTORC1 has consistently been beneficial in multiple neurodegenerative disease models but clinical confirmation is still awaited. This is likely because current ATP competitive inhibitors are suboptimal with respect to selectivity profile, tolerability and CNS exposure levels. Allosteric inhibitors with improved selectivity for mTORC1 may have benefit but we suggest that further improvement of these molecules or alternative approaches to impacting mTORC1 complex such as via regulators of the complex may have greater therapeutic potential.
RIPK1 (Receptor-Interacting Serine/Threonine-Protein Kinase 1)
Biological Rationale
Receptor-interacting serine/threonine-protein kinase 1 functions in a variety of pathways related to both cell survival and death. It has been linked to lysosome function (via cystatin F), inflammation (via p65 and cFos) and cell death pathways (apoptosis and necroptosis but not necrosis) (Wegner et al., 2017). For necroptosis, RIPK1’s kinase function appears essential, and it is a key part of a multiprotein complex termed the “necrosome” which triggers pathways such as the activation, phosphorylation and oligomerization of MLKL (Vandenabeele et al., 2010; Oberst, 2016). Inhibition of necroptosis has been suggested as a therapeutic strategy in diseases such as ALS and AD where RIPK1 activation has been reported (Li et al., 2012; Kaiser et al., 2014; Re et al., 2014; Vanden Berghe et al., 2015; Ito et al., 2016; Caccamo et al., 2017; Ofengeim et al., 2017; Degterev et al., 2019). However, it is not yet clear whether necroptosis is one of the key mechanisms by which neurons die (Chi et al., 2018; Heckmann et al., 2019) or if blocking it would be beneficial in the long run. Studies with optineurin knockout mice, and TBK1-TAK1 heterozygous KO mice, demonstrated RIPK1 activation [reviewed in Markovinovic et al. (2017)]. Targeting RIPK1 kinase function mitigated the axon-myelin phenotype and axonal loss observed in these models (Re et al., 2014; Ito et al., 2016; Markovinovic et al., 2017; Xu et al., 2018). Additionally, the RIPK1 inhibitor, necrostatin (Nec-1s), has been reported to be protective in the R6/2 transgenic mouse model of HD in response to ischemic insults in vivo (Zhu et al., 2011). Similarly, treatment of with Nec-1s has been reported to exert a protective effect in ALS and PD models (Re et al., 2014; Wu et al., 2015; Lin et al., 2019; Machado et al., 2019).
Small Molecule Inhibitors
Multiple small molecule inhibitors for RIPK1 inhibition have been developed with a range of binding modes. Allosteric binders such as Nec-1s are precedented in addition to ATP competitive molecules such as GSK2982772 (Najjar et al., 2015; Harris et al., 2017; Hamilton et al., 2019). The structural basis for kinase-domain based inhibition of RIPK1 by Nec-1s is reasonably well understood whereby the molecule stabilizes the protein in an inactive conformation through interactions between key amino acids in the activation loop and the surrounding structural elements; echoing the mechanism seen with ABL1 inhibitors (Xie et al., 2013; Najjar et al., 2015; Degterev et al., 2019). More recently, the trend has been to leverage structural information to guide inhibitor discovery toward type 1 and type 2 binding modes (Najjar et al., 2015; Harris et al., 2017; Hofmans et al., 2018). Interestingly, a number of kinase inhibitors defined against other kinase targets including tozasertib/VX-680, sorafenib, ponatinib and the PERK inhibitors GSK2606414/GSK2656157 have all been shown to have notable activity against RIPK1 (Fauster et al., 2015; Martens et al., 2017; Rojas-Rivera et al., 2017; Hofmans et al., 2018). There is little information available about DNL747 although DNL104, an earlier generation RIPK1 inhibitor licensed from the Yuan lab by Denali, may be related to necrostatins (Degterev et al., 2008; Wu et al., 2013).
Clinical Development
Denali has announced that they have completed recruitment for assessment of DNL747, a claimed potent, oral dosed brain penetrant RIPK1 inhibitor, in ALS (NCT03757351) and AD (NCT03757325) populations. These studies should read out in mid-2020. Another Denali molecule, DNL104, a BBB-penetrant RIPK1 inhibitor has been reported to modulate pharmacodynamic markers in a healthy volunteer trial (Grievink et al., 2019). GSK2982772 has been shown to be safe, tolerated, with good target engagement and is currently in Ph2a for chronic inflammatory diseases such as ulcerative colitis and psoriasis; but CNS penetrance has not been reported (NCT02776033, NCT02858492) (Weisel et al., 2017).
Synopsis
Although the rationale for this mechanism in necroptosis pathways seems reasonable, the clear weakness is the lack of clarity around the role this process plays in ongoing neurodegeneration; and whether blocking what is presumably quite a late stage cell death process will have real beneficial therapeutic consequences to the patient.
ROCK (Rho-Associated Protein Kinase)
Biological Rationale
Actin is one of three key components of the neuronal cytoskeleton and contributes to maintaining the axonal and somatodendritic compartments: the others being intermediate filaments and microtubules. A clear connection between cytoskeleton damage and neurodegeneration has emerged over the past few years such as actin cytoskeleton dysregulation and axonal transport defects [microtubule dependent and independent; reviewed in Eira et al. (2016) and Kounakis and Tavernarakis (2019)]. The actin cytoskeleton is regulated by Rho GTPase family signaling pathways (which are themselves dysregulated in PD, HD, AD, and ALS) via ROCK (Minamide et al., 2000; Maloney et al., 2005; Fulga et al., 2007; Mendoza-Naranjo et al., 2007; Petratos et al., 2008; Munsie and Truant, 2012; Tourette et al., 2014). ROCK1 and 2 have overlapping substrates outside of cytoskeletal proteins including those related to autophagy and vesicle dynamics (reviewed in Koch et al., 2018). ROCK1 and 2 are normally present in an autoinhibited configuration but over-activation has been implicated in AD, PD, HD, and ALS (Petratos et al., 2008; Barcia et al., 2012; Villar-Cheda et al., 2012; Herskowitz et al., 2013; Conti et al., 2014; Fujita and Yamashita, 2014; Tönges et al., 2014; Eira et al., 2016; Gentry et al., 2016; Henderson et al., 2016; Narayanan et al., 2016; Saal et al., 2017). More recently, it has been reported that ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway (Moskal et al., 2020). Genetic or pharmacological approaches using ROCK inhibitors such as fasudil and/or Y-27632 were beneficial in multiple disease models including ALS models which may be linked with the profilin genetic association in some forms of familial ALS (Zhou et al., 2003; Shao et al., 2008; Bauer et al., 2009; Deyts et al., 2009; Li et al., 2012; Tönges et al., 2012, 2014; Takata et al., 2013; Günther et al., 2014; Tatenhorst et al., 2014; Gentry et al., 2016; Saal et al., 2017). In addition to canonical ROCK signaling, it has been reported that ROCK activity plays a role in microglial phenotypes whereby activity is required to maintain proinflammatory (“M1”) phenotypes and ROCK inhibition enables a switch to an anti-inflammatory (“M2”) behavior (Zhang et al., 2013; Roser et al., 2017; Scheiblich and Bicker, 2017).
Small Molecule Inhibitors
Several ROCK inhibitors have been developed in the past few decades that are predominantly ATP-competitive type 1 inhibitors exemplified by fasudil, the closely related ripasudil/K-115 and the chemically divergent Y-27632 (Yamamoto et al., 2014 and reviewed in Abbhi and Piplani, 2018). However, selectivity is a challenge for these and other molecules. Structures of the kinase domain, which show high homology to other family members, in complex with fasudil, Y-27632 and other molecules have been reported (Li et al., 2012; Julian and Olson, 2014; Boland et al., 2015; Homan and Tesmer, 2015). Interestingly, a novel class of type I ROCK inhibitors was identified using fragment-based screening assisted by structure-guided design (Li et al., 2012). The ROCK2-preferring KD025/SLx-2119 or LYC-30937 may be interesting molecules given the predominant ROCK 2 expression in the CNS; and the hypothesis that ROCK1 may have a stronger relationship with vasodilation insensitivity and mortality (reviewed in Feng et al., 2016; Koch et al., 2018). BBB penetration optimization for ROCK inhibitors has not been disclosed though fasudil has been reported to alleviate EAE-dependent damage by decreasing BBB permeability (Huang et al., 2011).
Clinical Development
Fasudil and ripasudil are already in use or in clinical trials for pathological conditions including glaucoma, cerebral vasospasm, hypertension, atherosclerosis, and aortic stiffness (Julian and Olson, 2014). Despite beneficial effects, there are clinical limitations including poor oral bioavailability, blood pressure fluctuations and a relatively narrow safety window which makes them likely unsuitable for chronic dosing paradigms. Regardless, phase II clinical trials with fasudil have been initiated in ALS patients but results have not yet been reported (NCT01935518 and NCT03792490) (Koch et al., 2018; Lingor et al., 2019).
Synopsis
As for many other kinases, systemic inhibition of ROCK is expected to result in significant side effects. A molecule that avoids the blood pressure liabilities would be of great interest for clinical assessment in neurodegenerative disorders, however, to date this has proved very challenging.
Summary
The rationale behind protein kinases as relevant targets for CNS drug discovery is aligned with the appreciation that many cellular processes are tightly regulated by reversible phosphorylation processes; and that at least a number of these may become aberrant in disease (Figure 3). Interest in the potential of brain penetrant kinase inhibitors for neurodegenerative disease has grown in the last decade despite the dogma that brain penetrant, selective kinase inhibitors were not achievable.
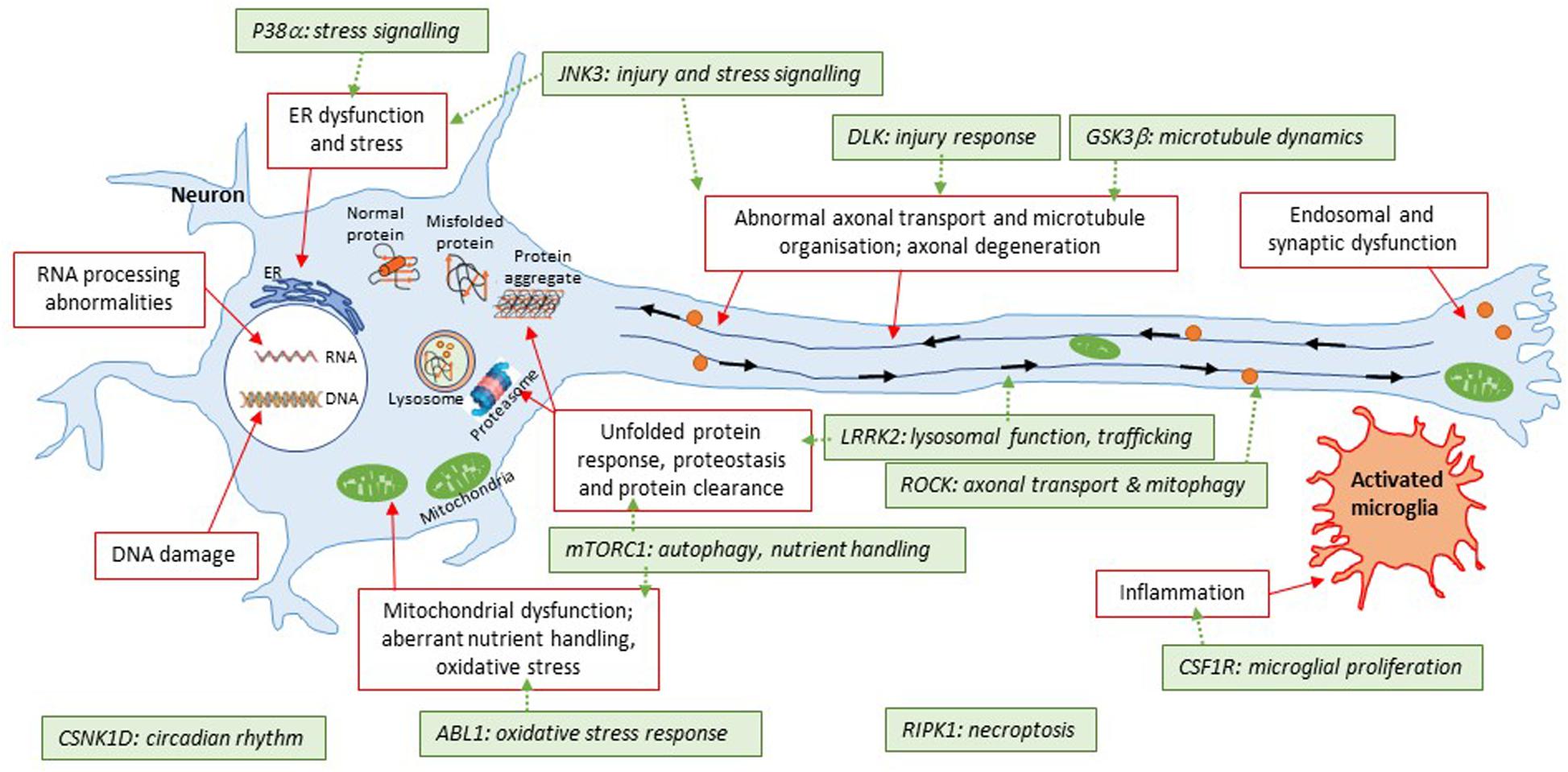
Figure 3. Illustration of kinase targets covered in this review with respect to converging pathways in neurodegeneration. This simplified schematic summarizes some of the common areas of pathophysiology in neurodegenerative diseases. The general theme is that the disease protein(s) are misfolded; which then triggers a number of downstream events such as protein aggregation, energy imbalances, altered proteostasis, stress responses to the unfolded proteins themselves or downstream consequences of events triggered by the unfolded proteins. Some of these pathways are further connected (e.g., diseased mitochondria can be removed by a specialized form of autophagy, known as mitophagy; furthermore, trafficking defects can apply to specific organelles illustrated here such as autolysosomes). The 11 kinases covered in this review have roles to play with respect to some of these pathways that have been shown to be altered. A key question for many targets is how closely related they are to initial triggers of pathophysiology; and whether inhibition of these targets will beneficially impact on multiple aspects of disease pathophysiology. LRRK2 is an exception here given that it is a bona fide genetic cause of Parkinson’s disease for a subset of patients.
The greatest challenge facing any CNS-targeted drug is effective penetration of the BBB and this is certainly not restricted to kinase inhibitors (Figure 2). At least some kinase inhibitors currently in the clinic appear to be opportunistic as opposed to the output of focused discovery and development workflows. Accordingly, the molecules in question have properties that do not align with those of CNS-penetrant compounds which raises concerns around the outcome of pending clinical trials with drugs that were developed and approved for indications requiring systemic exposure (reviewed in Shi and Mader, 2018). Indeed, a common concern when considering the current set of kinase inhibitors in clinical assessment is toxicity, which may be due at least in part to the suboptimal partitioning of the molecules in the human body, i.e., requirement of high systemic exposure to achieve therapeutically significant brain concentrations. With increased focused development of CNS-intended kinase inhibitors, we anticipate increased reporting of relevant pharmacokinetic properties commensurate with assessing appropriate compartmental exposure. This could include information such as unbound partition coefficient (Kpu,u) data together with confirmation of target engagement with clinical relevance such as PET (Watterson et al., 2013; Prabhakaran et al., 2017; Wager et al., 2017). Indeed, intentionally designed kinase inhibitor candidates for CNS indications exemplified by the LRRK2 and DLK molecules have been reported in the context of detailed DMPK and safety assessments.
Another consistent theme in this review is the cross-reactivity seen with a range of kinase inhibitors although this is improving with kinase panel screening activities to assess selectivity (Uitdehaag et al., 2012). Caution still must be leveraged as the use of different ATP concentrations used during the functional assay, the format of the assay and the selection of representative kinases for assessment does not fully indicate the true extent of the selectivity profile.
Structurally guided approaches and/or fragment-based drug discovery are key approaches that can be leveraged which address many of these challenges. Kinases are eminently amenable to x-ray crystallography to generated detailed structural data that, together with biophysical and bioinformatic approaches, can be effectively leveraged to understand the three-dimensional binding mode of ligands and inform the rational optimization into potent and selective lead compounds with CNS drug-like properties (Chessari and Woodhead, 2009; Brooijmans et al., 2010; Mortenson et al., 2014). Structurally guided approaches are further augmented when performed in conjunction with unbiased fragment screening which has potential to identify novel chemotypes/scaffolds or allosteric sites. In addition, with the rapid development of cryoEM and related technologies we may be able to gain much greater insight into the role kinases play in regulating multiprotein complexes and subsequently how we may be able to drug these enzymes in a more precise manner.
Author Contributions
CB and LD collected and reviewed the literature, wrote, and critically reviewed. Both authors contributed to the article and approved the submitted version.
Conflict of Interest
CB is a paid employee of Dementia Discovery Fund Ltd., as the CSO of LoQus23 Therapeutics. LD is a paid employee of Cerevance Ltd.
Acknowledgments
The authors would like to thank David Reynolds for critical reading of the manuscript.
Abbreviations
AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; ALSP, adult onset leukoencephalopathy with axonal spheroids and pigmented glia; ATP, adenosine triphosphate; BBB, blood brain barrier; CNS, central nervous system; DLB, dementia with Lewy bodies; DMPK, drug metabolism and pharmacokinetics; FTD, frontotemporal dementia; GTP, guanosine triphosphate; HD, Huntington’s disease; HTS, high throughput screen; Kp/Kpu,u, Partition coefficient/unbound partition coefficient; MPTP/MPP+, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/1-methyl-4-phenylpyridinium; MS, multiple sclerosis; ND, neurodegenerative disease; NHP, non-human primate; NPC, Niemann-Pick disease type C; PD, Parkinson’s disease; PET, positron emission tomography; PK, pharmacokinetics; PSP, progressive supranuclear palsy; RTK, receptor tyrosine kinase.
Footnotes
- ^ https://www.cureparkinsons.org.uk/mjff-press-release-nilo-pd
- ^ https://www.fiercebiotech.com/biotech/biogen-makes-another-alzheimer-s-bet-paying-pfizer-75m-for-early-phase-drug
- ^ https://www.denalitherapeutics.com/investors/press-release?id=7361
References
Abbhi, V., and Piplani, P. (2018). Rho-kinase (ROCK) inhibitors- a neuroprotective therapeutic paradigm with a focus on ocular utility. Curr. Med. Chem. 27, 2222–2256. doi: 10.2174/0929867325666181031102829
Adams, S. J., Kirk, A., and Auer, R. N. (2018). Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): integrating the literature on hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). J. Clin. Neurosci. Off. J. Neurosurg. Soc. Aust. 48, 42–49. doi: 10.1016/j.jocn.2017.10.060
Ahn, S.-W., Kim, J.-E., Park, K. S., Choi, W.-J., Hong, Y.-H., Kim, S.-M., et al. (2012). The neuroprotective effect of the GSK-3β inhibitor and influence on the extrinsic apoptosis in the ALS transgenic mice. J. Neurol. Sci. 320, 1–5. doi: 10.1016/j.jns.2012.05.038
Akiyama, H., Nishimura, T., Kondo, H., Ikeda, K., Hayashi, Y., and McGeer, P. L. (1994). Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res. 639, 171–174.
Alam, J., Blackburn, K., and Patrick, D. (2017). Neflamapimod: clinical phase 2b-ready oral small molecule inhibitor of p38α to reverse synaptic dysfunction in early Alzheimer’s disease. J. Prev. Alzheimers Dis. 4, 273–278. doi: 10.14283/jpad.2017.41
Alam, J. J. (2015). Selective brain-targeted antagonism of p38 MAPKα reduces Hippocampal IL-1β levels and improves morris water maze performance in aged rats. J. Alzheimers Dis. 48, 219–227. doi: 10.3233/JAD-150277
Alegre-Abarrategui, J., Christian, H., Lufino, M. M. P., Mutihac, R., Venda, L. L., Ansorge, O., et al. (2009). LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 18, 4022–4034. doi: 10.1093/hmg/ddp346
Alquezar, C., Salado, I. G., de la Encarnación, A., Pérez, D. I, Moreno, F., Gil, C., et al. (2016). Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: a novel strategy for the treatment of frontotemporal dementia. Mol. Neurodegener. 11:36. doi: 10.1186/s13024-016-0102-7
Anastassiadis, T., Deacon, S. W., Devarajan, K., Ma, H., and Peterson, J. R. (2011). Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045. doi: 10.1038/nbt.2017
Ansideri, F., Macedo, J. T., Eitel, M., El-Gokha, A., Zinad, D. S., Scarpellini, C., et al. (2018). Structural optimization of a pyridinylimidazole scaffold: shifting the selectivity from p38α mitogen-activated protein kinase to c-Jun N-Terminal Kinase 3. ACS Omega 3, 7809–7831. doi: 10.1021/acsomega.8b00668
Antoniou, X., Falconi, M., Di Marino, D., and Borsello, T. (2011). JNK3 as a therapeutic target for neurodegenerative diseases. J. Alzheimers Dis. 24, 633–642. doi: 10.3233/JAD-2011-091567
Arai, T., Ohno, M., Inoue, H., Hayashi, S., Aoki, T., Hirokawa, H., et al. (2012). Design and synthesis of novel p38α MAP kinase inhibitors: discovery of pyrazole-benzyl ureas bearing 2-molpholinopyrimidine moiety. Bioorg. Med. Chem. Lett. 22, 5118–5122. doi: 10.1016/j.bmcl.2012.05.095
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Ashman, L. K., and Griffith, R. (2013). Therapeutic targeting of c-KIT in cancer. Expert Opin. Investig. Drugs 22, 103–115. doi: 10.1517/13543784.2013.740010
Askew, K., Li, K., Olmos-Alonso, A., Garcia-Moreno, F., Liang, Y., Richardson, P., et al. (2017). Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 18, 391–405. doi: 10.1016/j.celrep.2016.12.041
Astolfi, A., Kudolo, M., Brea, J., Manni, G., Manfroni, G., Palazzotti, D., et al. (2019). Discovery of potent p38α MAPK inhibitors through a funnel like workflow combining in silico screening and in vitro validation. Eur. J. Med. Chem. 182:111624. doi: 10.1016/j.ejmech.2019.111624
Astolfi, A., Manfroni, G., Cecchetti, V., and Barreca, M. L. (2018). A comprehensive structural overview of p38α mitogen-activated protein kinase in complex with ATP-Site and Non-ATP-Site binders. ChemMedChem 13, 7–14. doi: 10.1002/cmdc.201700636
Atzori, C., Ghetti, B., Piva, R., Srinivasan, A. N., Zolo, P., Delisle, M. B., et al. (2001). Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J. Neuropathol. Exp. Neurol. 60, 1190–1197. doi: 10.1093/jnen/60.12.1190
Bang, Y., Kim, K.-S., Seol, W., and Choi, H. J. (2016). LRRK2 interferes with aggresome formation for autophagic clearance. Mol. Cell. Neurosci. 75, 71–80. doi: 10.1016/j.mcn.2016.06.007
Baptista, M. A. S., Dave, K. D., Frasier, M. A., Sherer, T. B., Greeley, M., Beck, M. J., et al. (2013). Loss of Leucine-Rich Repeat Kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One 8:e80705. doi: 10.1371/journal.pone.0080705
Baptista, M. A. S., Merchant, K., Barrett, T., Bhargava, S., Bryce, D. K., Ellis, J. M., et al. (2020). LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci. Transl. Med. 12:eaav0820. doi: 10.1126/scitranslmed.aav0820
Barcia, C., Ros, C. M., Annese, V., Carrillo-de Sauvage, M. A., Ros-Bernal, F., Gómez, A., et al. (2012). ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci. Rep. 2:809. doi: 10.1038/srep00809
Barmada, S. J., Serio, A., Arjun, A., Bilican, B., Daub, A., Ando, D. M., et al. (2014). Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 10, 677–685. doi: 10.1038/nchembio.1563
Barr, R. K., Kendrick, T. S., and Bogoyevitch, M. A. (2002). Identification of the critical features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 277, 10987–10997. doi: 10.1074/jbc.M107565200
Bauer, P. O., Wong, H. K., Oyama, F., Goswami, A., Okuno, M., Kino, Y., et al. (2009). Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J. Biol. Chem. 284, 13153–13164. doi: 10.1074/jbc.M809229200
Bax, B., Carter, P. S., Lewis, C., Guy, A. R., Bridges, A., Tanner, R., et al. (2001). The structure of phosphorylated GSK-3β complexed with a peptide, FRATtide, that Inhibits β-Catenin phosphorylation. Structure 9, 1143–1152. doi: 10.1016/S0969-2126(01)00679-7
Beilina, A., Rudenko, I. N., Kaganovich, A., Civiero, L., Chau, H., Kalia, S. K., et al. (2014). Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 111, 2626–2631. doi: 10.1073/pnas.1318306111
Benjamin, D., Colombi, M., Moroni, C., and Hall, M. N. (2011). Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 10, 868–880. doi: 10.1038/nrd3531
Bennett, B. L., Sasaki, D. T., Murray, B. W., O’Leary, E. C., Sakata, S. T., Xu, W., et al. (2001). SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U.S.A. 98, 13681–13686. doi: 10.1073/pnas.251194298
Berg, S., Bergh, M., Hellberg, S., Högdin, K., Lo-Alfredsson, Y., Söderman, P., et al. (2012). Discovery of novel potent and highly selective glycogen synthase kinase-3β (GSK3β) inhibitors for Alzheimer’s disease: design, synthesis, and characterization of pyrazines. J. Med. Chem. 55, 9107–9119. doi: 10.1021/jm201724m
Berger, Z., Smith, K. A., and LaVoie, M. J. (2010). Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49, 5511–5523. doi: 10.1021/bi100157u
Bertrand, J. A., Thieffine, S., Vulpetti, A., Cristiani, C., Valsasina, B., Knapp, S., et al. (2003). Structural characterization of the GSK-3β active site using selective and non-selective ATP-mimetic inhibitors. J. Mol. Biol. 333, 393–407. doi: 10.1016/j.jmb.2003.08.031
Bevilaqua, L. R. M., Kerr, D. S., Medina, J. H., Izquierdo, I., and Cammarota, M. (2003). Inhibition of hippocampal Jun N-terminal kinase enhances short-term memory but blocks long-term memory formation and retrieval of an inhibitory avoidance task. Eur. J. Neurosci. 17, 897–902. doi: 10.1046/j.1460-9568.2003.02524.x
Bijur, G., and Jope, R. (2003). Glycogen synthase kinase-3β is highly activated in nuclei and mitochondria. Neuroreport 14, 2415–2419.
Biskup, S., Moore, D. J., Celsi, F., Higashi, S., West, A. B., Andrabi, S. A., et al. (2006). Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 60, 557–569. doi: 10.1002/ana.21019
Bogoyevitch, M. A., Boehm, I., Oakley, A., Ketterman, A. J., and Barr, R. K. (2004). Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential. Biochim. Biophys. Acta BBA Proteins Proteomics 1697, 89–101. doi: 10.1016/j.bbapap.2003.11.016
Bogoyevitch, M. A., Ngoei, K. R. W., Zhao, T. T., Yeap, Y. Y. C., and Ng, D. C. H. (2010). c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim. Biophys. Acta BBA Proteins Proteomics 1804, 463–475. doi: 10.1016/j.bbapap.2009.11.002
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008
Boland, S., Bourin, A., Alen, J., Geraets, J., Schroeders, P., Castermans, K., et al. (2015). Design, synthesis, and biological evaluation of novel, highly active soft ROCK inhibitors. J. Med. Chem. 58, 4309–4324. doi: 10.1021/acs.jmedchem.5b00308
Borders, A. S., de Almeida, L., Van Eldik, L. J., and Watterson, D. M. (2008). The p38alpha mitogen-activated protein kinase as a central nervous system drug discovery target. BMC Neurosci. 9(Suppl. 2):S12. doi: 10.1186/1471-2202-9-S2-S12
Borjini, N., Fernández, M., Giardino, L., and Calzà, L. (2016). Cytokine and chemokine alterations in tissue, CSF, and plasma in early presymptomatic phase of experimental allergic encephalomyelitis (EAE), in a rat model of multiple sclerosis. J. Neuroinflamm. 13:291. doi: 10.1186/s12974-016-0757-6
Bové, J., Martínez-Vicente, M., and Vila, M. (2011). Fighting neurodegeneration with rapamycin: mechanistic insights. Nat. Rev. Neurosci. 12, 437–452. doi: 10.1038/nrn3068
Bowers, S., Truong, A. P., Neitz, R. J., Neitzel, M., Probst, G. D., Hom, R. K., et al. (2011). Design and synthesis of a novel, orally active, brain penetrant, tri-substituted thiophene based JNK inhibitor. Bioorg. Med. Chem. Lett. 21, 1838–1843. doi: 10.1016/j.bmcl.2011.01.046
Brahmachari, S., Ge, P., Lee, S. H., Kim, D., Karuppagounder, S. S., Kumar, M., et al. (2016). Activation of tyrosine kinase c-Abl contributes to α-synuclein-induced neurodegeneration. J. Clin. Invest. 126, 2970–2988. doi: 10.1172/JCI85456
Brasher, B. B., and Van Etten, R. A. (2000). C-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 275, 35631–35637. doi: 10.1074/jbc.M005401200
Brecht, S., Kirchhof, R., Chromik, A., Willesen, M., Nicolaus, T., Raivich, G., et al. (2005). Specific pathophysiological functions of JNK isoforms in the brain. Eur. J. Neurosci. 21, 363–377. doi: 10.1111/j.1460-9568.2005.03857.x
Brennan, K. C., Bates, E. A., Shapiro, R. E., Zyuzin, J., Hallows, W. C., Huang, Y., et al. (2013). Casein kinase iδ mutations in familial migraine and advanced sleep phase. Sci. Transl. Med. 5:183ra56. doi: 10.1126/scitranslmed.3005784
Brooijmans, N., Chang, Y.-W., Mobilio, D., Denny, R. A., and Humblet, C. (2010). An enriched structural kinase database to enable kinome-wide structure-based analyses and drug discovery. Protein Sci. Publ. Protein Soc. 19, 763–774. doi: 10.1002/pro.355
Bruttger, J., Karram, K., Wörtge, S., Regen, T., Marini, F., Hoppmann, N., et al. (2015). Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 43, 92–106. doi: 10.1016/j.immuni.2015.06.012
Caccamo, A., Branca, C., Piras, I. S., Ferreira, E., Huentelman, M. J., Liang, W. S., et al. (2017). Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 20, 1236–1246. doi: 10.1038/nn.4608
Caccamo, A., Maldonado, M. A., Majumder, S., Medina, D. X., Holbein, W., Magrí, A., et al. (2011). Naturally secreted Amyloid-β increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J. Biol. Chem. 286, 8924–8932. doi: 10.1074/jbc.M110.180638
Chambers, J. W., and LoGrasso, P. V. (2011). Mitochondrial c-Jun N-terminal Kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 286, 16052–16062. doi: 10.1074/jbc.M111.223602
Chambers, J. W., Pachori, A., Howard, S., Ganno, M., Hansen, D., Kamenecka, T., et al. (2011). Small molecule c-jun-N-Terminal kinase inhibitors protect dopaminergic neurons in a model of Parkinson’s disease. ACS Chem. Neurosci. 2, 198–206. doi: 10.1021/cn100109k
Chambers, J. W., Pachori, A., Howard, S., Iqbal, S., and LoGrasso, P. V. (2013). Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. J. Biol. Chem. 288, 4000–4011. doi: 10.1074/jbc.M112.406777
Chen, M., Geoffroy, C. G., Wong, H. N., Tress, O., Nguyen, M. T., Holzman, L. B., et al. (2016). Leucine Zipper-bearing kinase promotes axon growth in mammalian central nervous system neurons. Sci. Rep. 6:31482. doi: 10.1038/srep31482
Chen, X., Rzhetskaya, M., Kareva, T., Bland, R., During, M. J., Tank, A. W., et al. (2008). Antiapoptotic and trophic effects of dominant-negative forms of dual leucine zipper kinase in dopamine neurons of the substantia nigra in vivo. J. Neurosci. 28, 672–680. doi: 10.1523/JNEUROSCI.2132-07.2008
Chessari, G., and Woodhead, A. J. (2009). From fragment to clinical candidate–a historical perspective. Drug Discov. Today 14, 668–675. doi: 10.1016/j.drudis.2009.04.007
Chi, H., Chang, H.-Y., and Sang, T.-K. (2018). Neuronal cell death mechanisms in major neurodegenerative diseases. Int. J. Mol. Sci. 19:3082. doi: 10.3390/ijms19103082
Chia, R., Haddock, S., Beilina, A., Rudenko, I. N., Mamais, A., Kaganovich, A., et al. (2014). Phosphorylation of LRRK2 by casein kinase 1α regulates trans-Golgi clustering via differential interaction with ARHGEF7. Nat. Commun. 5:5827. doi: 10.1038/ncomms6827
Chico, L. K., Van Eldik, L. J., and Watterson, D. M. (2009). Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discov. 8, 892–909. doi: 10.1038/nrd2999
Chitu, V., Gokhan, Ş, Nandi, S., Mehler, M. F., and Stanley, E. R. (2016). Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. 39, 378–393. doi: 10.1016/j.tins.2016.03.005
Choi, J., Chen, J., Schreiber, S. L., and Clardy, J. (1996). Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239–242. doi: 10.1126/science.273.5272.239
Choi, W.-S., Kim, H.-W., and Xia, Z. (2015). JNK inhibition of VMAT2 contributes to rotenone-induced oxidative stress and dopamine neuron death. Toxicology 328, 75–81. doi: 10.1016/j.tox.2014.12.005
Choi, W.-S., Yoon, S.-Y., Oh, T. H., Choi, E.-J., O’Malley, K. L., and Oh, Y. J. (1999). Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J. Neurosci. Res. 57, 86–94. doi: 10.1002/(SICI)1097-4547(19990701)57:1<86::AID-JNR9<3.0.CO;2-E
Chu, C. T. (2010). Tickled PINK1: mitochondrial homeostasis and autophagy in recessive Parkinsonism. Biochim. Biophys. Acta 1802, 20–28. doi: 10.1016/j.bbadis.2009.06.012
Cirnaru, M. D., Marte, A., Belluzzi, E., Russo, I., Gabrielli, M., Longo, F., et al. (2014). LRRK2 kinase activity regulates synaptic vesicle trafficking and neurotransmitter release through modulation of LRRK2 macro-molecular complex. Front. Mol. Neurosci. 7:49. doi: 10.3389/fnmol.2014.00049
Colié, S., Sarroca, S., Palenzuela, R., Garcia, I., Matheu, A., Corpas, R., et al. (2017). Neuronal p38α mediates synaptic and cognitive dysfunction in an Alzheimer’s mouse model by controlling β-amyloid production. Sci. Rep. 7:45306. doi: 10.1038/srep45306
Colombo, A., Bastone, A., Ploia, C., Sclip, A., Salmona, M., Forloni, G., et al. (2009). JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol. Dis. 33, 518–525. doi: 10.1016/j.nbd.2008.12.014
Conti, A., Riva, N., Pesca, M., Iannaccone, S., Cannistraci, C. V., Corbo, M., et al. (2014). Increased expression of Myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim. Biophys. Acta 1842, 99–106. doi: 10.1016/j.bbadis.2013.10.013
Cookson, M. R. (2015). LRRK2 pathways leading to neurodegeneration. Curr. Neurol. Neurosci. Rep. 15:42. doi: 10.1007/s11910-015-0564-y
Cortes, C. J., Qin, K., Cook, J., Solanki, A., and Mastrianni, J. A. (2012). Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of gerstmann–sträussler–scheinker disease. J. Neurosci. 32, 12396–12405. doi: 10.1523/JNEUROSCI.6189-11.2012
Cortes, J. E., Kantarjian, H. M., Brümmendorf, T. H., Kim, D.-W., Turkina, A. G., Shen, Z.-X., et al. (2011). Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 118, 4567–4576. doi: 10.1182/blood-2011-05-355594
Cozza, G., and Pinna, L. A. (2016). Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 20, 319–340. doi: 10.1517/14728222.2016.1091883
Credle, J. J., George, J. L., Wills, J., Duka, V., Shah, K., Lee, Y.-C., et al. (2015). GSK-3β dysregulation contributes to parkinson’s-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and α-synuclein. Cell Death Differ. 22, 838–851. doi: 10.1038/cdd.2014.179
Crocker, C. E., Khan, S., Cameron, M. D., Robertson, H. A., Robertson, G. S., and LoGrasso, P. (2011). JNK inhibition protects dopamine neurons and provides behavioral improvement in a Rat 6-Hydroxydopamine model of Parkinson’s disease. ACS Chem. Neurosci. 2, 207–212. doi: 10.1021/cn1001107
Cuéllar, J., Ludlam, W. G., Tensmeyer, N. C., Aoba, T., Dhavale, M., Santiago, C., et al. (2019). Structural and functional analysis of the role of the chaperonin CCT in mTOR complex assembly. Nat. Commun. 10:2865. doi: 10.1038/s41467-019-10781-1
Dagher, N. N., Najafi, A. R., Kayala, K. M. N., Elmore, M. R. P., White, T. E., Medeiros, R., et al. (2015). Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J. Neuroinflammation 12:139. doi: 10.1186/s12974-015-0366-9
Daher, J. P. L., Abdelmotilib, H. A., Hu, X., Volpicelli-Daley, L. A., Moehle, M. S., Fraser, K. B., et al. (2015). Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J. Biol. Chem. 290, 19433–19444. doi: 10.1074/jbc.M115.660001
Dajani, R., Fraser, E., Roe, S. M., Yeo, M., Good, V. M., Thompson, V., et al. (2003). Structural basis for recruitment of glycogen synthase kinase 3β to the axin–APC scaffold complex. Embo J. 22, 494–501. doi: 10.1093/emboj/cdg068
Dajani, R., Fraser, E., Roe, S. M., Young, N., Good, V., Dale, T. C., et al. (2001). Crystal structure of glycogen synthase kinase 3β: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105, 721–732. doi: 10.1016/S0092-8674(01)00374-9
Davies, C., and Tournier, C. (2012). Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem. Soc. Trans. 40, 85–89. doi: 10.1042/BST20110641
Davoli, E., Sclip, A., Cecchi, M., Cimini, S., Carrà, A., Salmona, M., et al. (2014). Determination of tissue levels of a neuroprotectant drug: the cell permeable JNK inhibitor peptide. J. Pharmacol. Toxicol. Methods 70, 55–61. doi: 10.1016/j.vascn.2014.04.001
de Araujo, M. E. G., Naschberger, A., Fürnrohr, B. G., Stasyk, T., Dunzendorfer-Matt, T., Lechner, S., et al. (2017). Crystal structure of the human lysosomal mTORC1 scaffold complex and its impact on signaling. Science 358, 377–381. doi: 10.1126/science.aao1583
de la Torre, E. R., Luzón-Toro, B., Forte-Lago, I., Minguez-Castellanos, A., Ferrer, I., and Hilfiker, S. (2009). Combined kinase inhibition modulates parkin inactivation. Hum. Mol. Genet. 18, 809–823. doi: 10.1093/hmg/ddn407
De Wit, T., Baekelandt, V., and Lobbestael, E. (2018). Inhibition of LRRK2 or casein kinase 1 results in LRRK2 protein destabilization. Mol. Neurobiol. 56, 5273–5286. doi: 10.1007/s12035-018-1449-2
Degterev, A., Hitomi, J., Germscheid, M., Ch’en, I. L., Korkina, O., Teng, X., et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321. doi: 10.1038/nchembio.83
Degterev, A., Ofengeim, D., and Yuan, J. (2019). Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. U.S.A. 116, 9714–9722. doi: 10.1073/pnas.1901179116
Dehay, B., Bové, J., Rodríguez-Muela, N., Perier, C., Recasens, A., Boya, P., et al. (2010). Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010
del Ser, T., Steinwachs, K. C., Gertz, H. J., Andrés, M. V., Gómez-Carrillo, B., Medina, M., et al. (2013). Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J. Alzheimers Dis. 33, 205–215. doi: 10.3233/JAD-2012-120805
Deng, J., Lewis, P. A., Greggio, E., Sluch, E., Beilina, A., and Cookson, M. R. (2008). Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 1499–1504. doi: 10.1073/pnas.0709098105
Deng, X., Dzamko, N., Prescott, A., Davies, P., Liu, Q., Yang, Q., et al. (2011). Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 7, 203–205. doi: 10.1038/nchembio.538
Derkinderen, P., Scales, T. M. E., Hanger, D. P., Leung, K.-Y., Byers, H. L., Ward, M. A., et al. (2005). Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J. Neurosci. Off. J. Soc. Neurosci. 25, 6584–6593. doi: 10.1523/JNEUROSCI.1487-05.2005
Deyaert, E., Kortholt, A., and Versées, W. (2017). The LRR-Roc-COR module of the Chlorobium tepidum Roco protein: crystallization and X-ray crystallographic analysis. Acta Crystallogr. Sect. F Struct. Biol. Commun. 73, 520–524. doi: 10.1107/S2053230X17011955
Deyts, C., Galan-Rodriguez, B., Martin, E., Bouveyron, N., Roze, E., Charvin, D., et al. (2009). Dopamine D2 receptor stimulation potentiates PolyQ-Huntingtin-induced mouse striatal neuron dysfunctions via Rho/ROCK-II activation. PLoS One 4:e8287. doi: 10.1371/journal.pone.0008287
Dodson, M. W., Leung, L. K., Lone, M., Lizzio, M. A., and Guo, M. (2014). Novel ethyl methanesulfonate (EMS)-induced null alleles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis. Model. Mech. 7, 1351–1363. doi: 10.1242/dmm.017020
Dorey, K., Engen, J. R., Kretzschmar, J., Wilm, M., Neubauer, G., Schindler, T., et al. (2001). Phosphorylation and structure-based functional studies reveal a positive and a negative role for the activation loop of the c-Abl tyrosine kinase. Oncogene 20, 8075–8084. doi: 10.1038/sj.onc.1205017
Dou, X., Huang, H., Li, Y., Jiang, L., Wang, Y., Jin, H., et al. (2019). Multistage screening reveals 3-substituted indolin-2-one derivatives as novel and isoform-selective c-Jun N-terminal Kinase 3 (JNK3) inhibitors: implications to drug discovery for potential treatment of neurodegenerative diseases. J. Med. Chem. 62, 6645–6664. doi: 10.1021/acs.jmedchem.9b00537
Druker, B. J. (2008). Translation of the Philadelphia chromosome into therapy for CML. Blood 112, 4808–4817. doi: 10.1182/blood-2008-07-077958
Dubreuil, P., Letard, S., Ciufolini, M., Gros, L., Humbert, M., Castéran, N., et al. (2009). Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One 4:e7258. doi: 10.1371/journal.pone.0007258
Dusonchet, J., Kochubey, O., Stafa, K., Young, S. M., Zufferey, R., Moore, D. J., et al. (2011). A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J. Neurosci. 31, 907–912. doi: 10.1523/JNEUROSCI.5092-10.2011
Eira, J., Silva, C. S., Sousa, M. M., and Liz, M. A. (2016). The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog. Neurobiol. 141, 61–82. doi: 10.1016/j.pneurobio.2016.04.007
Eldar-Finkelman, H., and Martinez, A. (2011). GSK-3 inhibitors: preclinical and clinical focus on CNS. Front. Mol. Neurosci. 4:32. doi: 10.3389/fnmol.2011.00032
El-Gamal, M. I., Al-Ameen, S. K., Al-Koumi, D. M., Hamad, M. G., Jalal, N. A., and Oh, C.-H. (2018). Recent advances of colony-stimulating factor-1 receptor (CSF-1R) kinase and its inhibitors. J. Med. Chem. 61, 5450–5466. doi: 10.1021/acs.jmedchem.7b00873
Elmore, M. R. P., Hohsfield, L. A., Kramár, E. A., Soreq, L., Lee, R. J., Pham, S. T., et al. (2018). Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell 17:e12832. doi: 10.1111/acel.12832
Eminel, S., Roemer, L., Waetzig, V., and Herdegen, T. (2008). c-Jun N-terminal kinases trigger both degeneration and neurite outgrowth in primary hippocampal and cortical neurons. J. Neurochem. 104, 957–969. doi: 10.1111/j.1471-4159.2007.05101.x
Engström, W., Ward, A., and Moorwood, K. (2010). The role of scaffold proteins in JNK signalling. Cell Prolif. 43, 56–66. doi: 10.1111/j.1365-2184.2009.00654.x
Erblich, B., Zhu, L., Etgen, A. M., Dobrenis, K., and Pollard, J. W. (2011). Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 6:e26317. doi: 10.1371/journal.pone.0026317
Eshraghi, A. A., Aranke, M., Salvi, R., Ding, D., Coleman, J. K. M., Ocak, E., et al. (2018). Preclinical and clinical otoprotective applications of cell-penetrating peptide D-JNKI-1 (AM-111). Hear. Res. 368, 86–91. doi: 10.1016/j.heares.2018.03.003
Estrada, A. A., Chan, B. K., Baker-Glenn, C., Beresford, A., Burdick, D. J., Chambers, M., et al. (2014). Discovery of highly potent, selective, and brain-penetrant aminopyrazole leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J. Med. Chem. 57, 921–936. doi: 10.1021/jm401654j
Etchegaray, J.-P., Machida, K. K., Noton, E., Constance, C. M., Dallmann, R., Di Napoli, M. N., et al. (2009). Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol. Cell. Biol. 29, 3853–3866. doi: 10.1128/MCB.00338-09
Fauster, A., Rebsamen, M., Huber, K. V. M., Bigenzahn, J. W., Stukalov, A., Lardeau, C.-H., et al. (2015). A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 6:e1767. doi: 10.1038/cddis.2015.130
Fell, M. J., Mirescu, C., Basu, K., Cheewatrakoolpong, B., DeMong, D. E., Ellis, J. M., et al. (2015). MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J. Pharmacol. Exp. Ther. 355, 397–409. doi: 10.1124/jpet.115.227587
Feng, Y., LoGrasso, P. V., Defert, O., and Li, R. (2016). Rho Kinase (ROCK) inhibitors and their therapeutic potential. J. Med. Chem. 59, 2269–2300. doi: 10.1021/acs.jmedchem.5b00683
Ferrandi, C., Richard, F., Tavano, P., Hauben, E., Barbié, V., Gotteland, J.-P., et al. (2011). Characterization of immune cell subsets during the active phase of multiple sclerosis reveals disease and c-Jun N-terminal kinase pathway biomarkers. Mult. Scler. J. 17, 43–56. doi: 10.1177/1352458510381258
Ferrer, I., Blanco, R., Carmona, M., Puig, B., Barrachina, M., Gómez, C., et al. (2001). Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson’s disease and Dementia with Lewy bodies. J. Neural Transm. 108, 1383–1396. doi: 10.1007/s007020100015
Fitzgerald, C. E., Patel, S. B., Becker, J. W., Cameron, P. M., Zaller, D., Pikounis, V. B., et al. (2003). Structural basis for p38alpha MAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat. Struct. Biol. 10, 764–769. doi: 10.1038/nsb949
Frake, R. A., Ricketts, T., Menzies, F. M., and Rubinsztein, D. C. (2015). Autophagy and neurodegeneration. J. Clin. Invest. 125, 65–74. doi: 10.1172/JCI73944
Franchino, F., Rudà, R., and Soffietti, R. (2018). Mechanisms and therapy for cancer metastasis to the brain. Front. Oncol. 8:161. doi: 10.3389/fonc.2018.00161
Fraser, K. B., Moehle, M. S., Daher, J. P. L., Webber, P. J., Williams, J. Y., Stewart, C. A., et al. (2013). LRRK2 secretion in exosomes is regulated by 14-3-3. Hum. Mol. Genet. 22, 4988–5000. doi: 10.1093/hmg/ddt346
Frost, M., and Hulbert, J. (2015). Clinical management of tuberous sclerosis complex over the lifetime of a patient. Pediatr. Health Med. Ther. 6, 139–146. doi: 10.2147/PHMT.S67342
Fuchs, C., Rimondini, R., Viggiano, R., Trazzi, S., De Franceschi, M., Bartesaghi, R., et al. (2015). Inhibition of GSK3β rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol. Dis. 82, 298–310. doi: 10.1016/j.nbd.2015.06.018
Fuji, R. N., Flagella, M., Baca, M., Baptista, M. A. S., Brodbeck, J., Chan, B. K., et al. (2015). Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 7:273ra15. doi: 10.1126/scitranslmed.aaa3634
Fujita, Y., and Yamashita, T. (2014). Axon growth inhibition by RhoA/ROCK in the central nervous system. Front. Neurosci. 8:338. doi: 10.3389/fnins.2014.00338
Fulga, T. A., Elson-Schwab, I., Khurana, V., Steinhilb, M. L., Spires, T. L., Hyman, B. T., et al. (2007). Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 9, 139–148. doi: 10.1038/ncb1528
Gallo, K. A., and Johnson, G. L. (2002). Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol. 3, 663–672. doi: 10.1038/nrm906
Gandin, V., Ferrarese, A., Via, M. D., Marzano, C., Chilin, A., and Marzaro, G. (2015). Targeting kinases with anilinopyrimidines: discovery of N-phenyl-N’-[4-(pyrimidin-4-ylamino)phenyl]urea derivatives as selective inhibitors of class III receptor tyrosine kinase subfamily. Sci. Rep. 5:16750. doi: 10.1038/srep16750
Genabai, N. K., Ahmad, S., Zhang, Z., Jiang, X., Gabaldon, C. A., and Gangwani, L. (2015). Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum. Mol. Genet. 24, 6986–7004. doi: 10.1093/hmg/ddv401
Gentry, E. G., Henderson, B. W., Arrant, A. E., Gearing, M., Feng, Y., Riddle, N. C., et al. (2016). Rho Kinase inhibition as a therapeutic for progressive supranuclear palsy and corticobasal degeneration. J. Neurosci. Off. J. Soc. Neurosci. 36, 1316–1323. doi: 10.1523/JNEUROSCI.2336-15.2016
Georgievska, B., Sandin, J., Doherty, J., Mörtberg, A., Neelissen, J., Andersson, A., et al. (2013). AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J. Neurochem. 125, 446–456. doi: 10.1111/jnc.12203
Getlik, M., Grütter, C., Simard, J. R., Nguyen, H. D., Robubi, A., Aust, B., et al. (2012). Structure-based design, synthesis and biological evaluation of N-pyrazole, N’-thiazole urea inhibitors of MAP kinase p38α. Eur. J. Med. Chem. 48, 1–15. doi: 10.1016/j.ejmech.2011.11.019
Ghosh, A. S., Wang, B., Pozniak, C. D., Chen, M., Watts, R. J., and Lewcock, J. W. (2011). DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol. 194, 751–764. doi: 10.1083/jcb.201103153
Ghoshal, N., Smiley, J. F., DeMaggio, A. J., Hoekstra, M. F., Cochran, E. J., Binder, L. I., et al. (1999). A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer’s disease. Am. J. Pathol. 155, 1163–1172. doi: 10.1016/s0002-9440(10)65219-4
Giasson, B. I., Covy, J. P., Bonini, N. M., Hurtig, H. I., Farrer, M. J., Trojanowski, J. Q., et al. (2006). Biochemical and pathological characterization of Lrrk2. Ann. Neurol. 59, 315–322. doi: 10.1002/ana.20791
Giau, V. V., Bagyinszky, E., Yang, Y. S., Youn, Y. C., An, S. S. A., and Kim, S. Y. (2019a). Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 9:8368. doi: 10.1038/s41598-019-44848-2
Giau, V. V., Senanarong, V., Bagyinszky, E., An, S. S. A., and Kim, S. (2019b). Analysis of 50 neurodegenerative genes in clinically diagnosed early-onset Alzheimer’s disease. Int. J. Mol. Sci. 20:1514. doi: 10.3390/ijms20061514
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Gloeckner, C. J., Kinkl, N., Schumacher, A., Braun, R. J., O’Neill, E., Meitinger, T., et al. (2006). The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 15, 223–232. doi: 10.1093/hmg/ddi439
Gómez-Nicola, D., Fransen, N. L., Suzzi, S., and Perry, V. H. (2013). Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 33, 2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013
Gómez-Suaga, P., Rivero-Ríos, P., Fdez, E., Blanca Ramírez, M., Ferrer, I., Aiastui, A., et al. (2014). LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 23, 6779–6796. doi: 10.1093/hmg/ddu395
Gonfloni, S., Maiani, E., Di Bartolomeo, C., Diederich, M., and Cesareni, G. (2012). Oxidative stress, DNA damage, and c-Abl signaling: at the crossroad in neurodegenerative diseases? Int. J. Cell Biol. 2012:683097. doi: 10.1155/2012/683097
Gourmaud, S., Paquet, C., Dumurgier, J., Pace, C., Bouras, C., Gray, F., et al. (2015). Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: links to cognitive decline. J. Psychiatry Neurosci. 40, 151–161. doi: 10.1503/jpn.140062
Gourmaud, S., Thomas, P., Thomasseau, S., Tible, M., Abadie, C., Paquet, C., et al. (2018). Brimapitide reduced neuronal stress markers and cognitive deficits in 5XFAD transgenic mice. J. Alzheimers Dis. 63, 665–674. doi: 10.3233/JAD-171099
Grassi, D., Diaz-Perez, N., Volpicelli-Daley, L. A., and Lasmézas, C. I. (2019). Pα-syn∗ mitotoxicity is linked to MAPK activation and involves tau phosphorylation and aggregation at the mitochondria. Neurobiol. Dis. 124, 248–262. doi: 10.1016/j.nbd.2018.11.015
Greggio, E., Jain, S., Kingsbury, A., Bandopadhyay, R., Lewis, P., Kaganovich, A., et al. (2006). Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341. doi: 10.1016/j.nbd.2006.04.001
Greggio, E., Zambrano, I., Kaganovich, A., Beilina, A., Taymans, J.-M., Daniëls, V., et al. (2008). The parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 283, 16906–16914. doi: 10.1074/jbc.M708718200
Grievink, H. W., Heuberger, J. A. A. C., Huang, F., Chaudhary, R., Birkhoff, W. A. J., Tonn, G. R., et al. (2019). DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin. Pharmacol. Ther. 107, 406–414. doi: 10.1002/cpt.1615
Grindal, T. C., Sampson, E. M., and Antonelli, P. J. (2010). AM-111 prevents hearing loss from semicircular canal injury in otitis media. Laryngoscope 120, 178–182. doi: 10.1002/lary.20759
Guaitoli, G., Raimondi, F., Gilsbach, B. K., Gómez-Llorente, Y., Deyaert, E., Renzi, F., et al. (2016). Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. U.S.A. 113, E4357–E4366. doi: 10.1073/pnas.1523708113
Günther, R., Saal, K.-A., Suhr, M., Scheer, D., Koch, J. C., Bähr, M., et al. (2014). The rho kinase inhibitor Y-27632 improves motor performance in male SOD1(G93A) mice. Front. Neurosci. 8:304. doi: 10.3389/fnins.2014.00304
Guo, C., and Whitmarsh, A. J. (2008). The β-Arrestin-2 scaffold protein promotes c-Jun N-terminal Kinase-3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 283:15903. doi: 10.1074/jbc.M710006200
Haar, E. t., Coll, J. T., Austen, D. A., Hsiao, H.-M., Swenson, L., and Jain, J. (2001). Structure of GSK3β reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 8, 593–596. doi: 10.1038/89624
Haeusgen, W., Boehm, R., Zhao, Y., Herdegen, T., and Waetzig, V. (2009). Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience 161, 951–959. doi: 10.1016/j.neuroscience.2009.04.014
Halekotte, J., Witt, L., Ianes, C., Krüger, M., Bührmann, M., Rauh, D., et al. (2017). Optimized 4,5-diarylimidazoles as potent/selective inhibitors of protein kinase CK1δ and their structural relation to p38α MAPK. Molecules 22:522. doi: 10.3390/molecules22040522
Hamilton, G. L., Chen, H., Deshmukh, G., Eigenbrot, C., Fong, R., Johnson, A., et al. (2019). Potent and selective inhibitors of receptor-interacting protein kinase 1 that lack an aromatic back pocket group. Bioorg. Med. Chem. Lett. 29, 1497–1501. doi: 10.1016/j.bmcl.2019.04.014
Hammarlund, M., Nix, P., Hauth, L., Jorgensen, E. M., and Bastiani, M. (2009). Axon regeneration requires a conserved MAP kinase pathway. Science 323:802. doi: 10.1126/science.1165527
Hanger, D. P., Hughes, K., Woodgett, J. R., Brion, J.-P., and Anderton, B. H. (1992). Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62. doi: 10.1016/0304-3940(92)90774-2
Harris, P. A., Berger, S. B., Jeong, J. U., Nagilla, R., Bandyopadhyay, D., Campobasso, N., et al. (2017). Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60, 1247–1261. doi: 10.1021/acs.jmedchem.6b01751
Hartzler, A. W., Zhu, X., Siedlak, S. L., Castellani, R. J., Avilá, J., Perry, G., et al. (2002). The p38 pathway is activated in Pick disease and progressive supranuclear palsy: a mechanistic link between mitogenic pathways, oxidative stress, and tau. Neurobiol. Aging 23, 855–859. doi: 10.1016/s0197-4580(02)00029-5
Hebron, M. L., Lonskaya, I., and Moussa, C. E.-H. (2013). Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 22, 3315–3328. doi: 10.1093/hmg/ddt192
Heckmann, B. L., Tummers, B., and Green, D. R. (2019). Crashing the computer: Apoptosis vs. necroptosis in neuroinflammation. Cell Death Differ. 26, 41–52. doi: 10.1038/s41418-018-0195-3
Heffron, T. P. (2016). Small molecule kinase inhibitors for the treatment of brain cancer. J. Med. Chem. 59, 10030–10066. doi: 10.1021/acs.jmedchem.6b00618
Heightman, T. D., Berdini, V., Braithwaite, H., Buck, I. M., Cassidy, M., Castro, J., et al. (2018). Fragment-based discovery of a potent, orally bioavailable inhibitor that modulates the phosphorylation and catalytic activity of ERK1/2. J. Med. Chem. 61, 4978–4992. doi: 10.1021/acs.jmedchem.8b00421
Heitman, J., Movva, N. R., Hiestand, P. C., and Hall, M. N. (1991). FK 506-binding protein proline rotamase is a target for the immunosuppressive agent FK 506 in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 88, 1948–1952. doi: 10.1073/pnas.88.5.1948
Henderson, B. W., Gentry, E. G., Rush, T., Troncoso, J. C., Thambisetty, M., Montine, T. J., et al. (2016). Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer’s disease and ROCK1 depletion reduces amyloid-β levels in brain. J. Neurochem. 138, 525–531. doi: 10.1111/jnc.13688
Henderson, J. L., Kormos, B. L., Hayward, M. M., Coffman, K. J., Jasti, J., Kurumbail, R. G., et al. (2015). Discovery and preclinical profiling of 3-[4-(Morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 Kinase inhibitor. J. Med. Chem. 58, 419–432. doi: 10.1021/jm5014055
Heng, M. Y., Duong, D. K., Albin, R. L., Tallaksen-Greene, S. J., Hunter, J. M., Lesort, M. J., et al. (2010). Early autophagic response in a novel knock-in model of Huntington disease. Hum. Mol. Genet. 19, 3702–3720. doi: 10.1093/hmg/ddq285
Hensley, K., Floyd, R. A., Zheng, N. Y., Nael, R., Robinson, K. A., Nguyen, X., et al. (1999). p38 kinase is activated in the Alzheimer’s disease brain. J. Neurochem. 72, 2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x
Herskowitz, J. H., Feng, Y., Mattheyses, A. L., Hales, C. M., Higginbotham, L. A., Duong, D. M., et al. (2013). Pharmacologic inhibition of ROCK2 suppresses amyloid-β production in an Alzheimer’s disease mouse model. J. Neurosci. Off. J. Soc. Neurosci. 33, 19086–19098. doi: 10.1523/JNEUROSCI.2508-13.2013
Herzig, M. C., Kolly, C., Persohn, E., Theil, D., Schweizer, T., Hafner, T., et al. (2011). LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. 20, 4209–4223. doi: 10.1093/hmg/ddr348
Higashi, S., Biskup, S., West, A. B., Trinkaus, D., Dawson, V. L., Faull, R. L. M., et al. (2007). Localization of Parkinson’s disease-associated LRRK2 in normal and pathological human brain. Brain Res. 1155, 208–219. doi: 10.1016/j.brainres.2007.04.034
Hirai, S., Cui, D. F., Miyata, T., Ogawa, M., Kiyonari, H., Suda, Y., et al. (2006). The c-Jun N-Terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J. Neurosci. 26:11992. doi: 10.1523/JNEUROSCI.2272-06.2006
Hofmans, S., Devisscher, L., Martens, S., Van Rompaey, D., Goossens, K., Divert, T., et al. (2018). Tozasertib analogues as inhibitors of necroptotic cell death. J. Med. Chem. 61, 1895–1920. doi: 10.1021/acs.jmedchem.7b01449
Höglinger, G. U., Huppertz, H.-J., Wagenpfeil, S., Andrés, M. V., Belloch, V., León, T., et al. (2014). Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov. Disord. 29, 479–487. doi: 10.1002/mds.25815
Homan, K. T., and Tesmer, J. J. G. (2015). Molecular basis for small molecule inhibition of G protein-coupled receptor kinases. ACS Chem. Biol. 10, 246–256. doi: 10.1021/cb5003976
Hong, Y. K., Lee, S., Park, S. H., Lee, J. H., Han, S. Y., Kim, S. T., et al. (2012). Inhibition of JNK/dFOXO pathway and caspases rescues neurological impairments in Drosophila Alzheimer’s disease model. Biochem. Biophys. Res. Commun. 419, 49–53. doi: 10.1016/j.bbrc.2012.01.122
Horti, A. G., Naik, R., Foss, C. A., Minn, I., Misheneva, V., Du, Y., et al. (2019). PET imaging of microglia by targeting macrophage colony-stimulating factor 1 receptor (CSF1R). Proc. Natl. Acad. Sci. U.S.A. 116, 1686–1691. doi: 10.1073/pnas.1812155116
Hoshi, M., Takashima, A., Noguchi, K., Murayama, M., Sato, M., Kondo, S., et al. (1996). Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci. U.S.A. 93, 2719–2723. doi: 10.1073/pnas.93.7.2719
Huang, X. N., Fu, J., and Wang, W. Z. (2011). The effects of fasudil on the permeability of the rat blood-brain barrier and blood-spinal cord barrier following experimental autoimmune encephalomyelitis. J. Neuroimmunol. 239, 61–67. doi: 10.1016/j.jneuroim.2011.08.015
Hunot, S., Vila, M., Teismann, P., Davis, R. J., Hirsch, E. C., Przedborski, S., et al. (2004). JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 665–670. doi: 10.1073/pnas.0307453101
Hur, E.-M., Jang, E.-H., Jeong, G. R., and Lee, B. D. (2019). LRRK2 and membrane trafficking: nexus of Parkinson’s disease. BMB Rep. 52, 533–539.
Imam, S. Z., Trickler, W., Kimura, S., Binienda, Z. K., Paule, M. G., Slikker, W., et al. (2013). Neuroprotective efficacy of a new brain-penetrating C-Abl inhibitor in a murine Parkinson’s disease model. PLoS One 8:e65129. doi: 10.1371/journal.pone.0065129
Imam, S. Z., Zhou, Q., Yamamoto, A., Valente, A. J., Ali, S. F., Bains, M., et al. (2011). Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 31, 157–163. doi: 10.1523/JNEUROSCI.1833-10.2011
Imamura, K., Izumi, Y., Watanabe, A., Tsukita, K., Woltjen, K., Yamamoto, T., et al. (2017). The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 9:eaaf3962. doi: 10.1126/scitranslmed.aaf3962
Islam, M. S., Nolte, H., Jacob, W., Ziegler, A. B., Pütz, S., Grosjean, Y., et al. (2016). Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 25, 5365–5382. doi: 10.1093/hmg/ddw352
Isojima, Y., Nakajima, M., Ukai, H., Fujishima, H., Yamada, R. G., Masumoto, K., et al. (2009). CKIepsilon/delta-dependent phosphorylation is a temperature-insensitive, period-determining process in the mammalian circadian clock. Proc. Natl. Acad. Sci. U.S.A. 106, 15744–15749. doi: 10.1073/pnas.0908733106
Ito, Y., Ofengeim, D., Najafov, A., Das, S., Saberi, S., Li, Y., et al. (2016). RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608. doi: 10.1126/science.aaf6803
Itoh, A., Horiuchi, M., Wakayama, K., Xu, J., Bannerman, P., Pleasure, D., et al. (2011). ZPK/DLK, a mitogen-activated protein kinase kinase kinase, is a critical mediator of programmed cell death of motoneurons. J. Neurosci. 31, 7223–7228. doi: 10.1523/JNEUROSCI.5947-10.2011
Jaeger, P. A., and Wyss-Coray, T. (2009). All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Mol. Neurodegener. 4:16. doi: 10.1186/1750-1326-4-16
Janssen, B., and Mach, R. H. (2019). “Chapter 7 - Development of brain PET imaging agents: strategies for imaging neuroinflammation in Alzheimer’s disease,” in Progress in Molecular Biology and Translational Science Brain Imaging, eds J. T. Becker and A. D. Cohen (Cambridge, MA: Academic Press), 371–399. doi: 10.1016/bs.pmbts.2019.04.005
Jhanwar-Uniyal, M., Wainwright, J. V., Mohan, A. L., Tobias, M. E., Murali, R., Gandhi, C. D., et al. (2019). Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 72, 51–62. doi: 10.1016/j.jbior.2019.03.003
Jiang, T., Yu, J.-T., Zhu, X.-C., Zhang, Q.-Q., Cao, L., Wang, H.-F., et al. (2014). Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 85, 121–130. doi: 10.1016/j.neuropharm.2014.05.032
Jing, Z., Caltagarone, J., and Bowser, R. (2009). Altered subcellular distribution of c-Abl in Alzheimer’s disease. J. Alzheimers Dis. 17, 409–422.
Julian, L., and Olson, M. F. (2014). Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases 5:e29846. doi: 10.4161/sgtp.29846
Kaiser, W. J., Daley-Bauer, L. P., Thapa, R. J., Mandal, P., Berger, S. B., Huang, C., et al. (2014). RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. U.S.A. 111, 7753–7758. doi: 10.1073/pnas.1401857111
Karim, R., Liao, E. E., Kim, J., Meints, J., Martinez, H. M., Pletnikova, O., et al. (2020). α-Synucleinopathy associated c-Abl activation causes p53-dependent autophagy impairment. Mol. Neurodegener. 15:27. doi: 10.1186/s13024-020-00364-w
Karuppagounder, S. S., Brahmachari, S., Lee, Y., Dawson, V. L., Dawson, T. M., and Ko, H. S. (2014). The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep. 4:4874. doi: 10.1038/srep04874
Kennaway, D. J., Varcoe, T. J., Voultsios, A., Salkeld, M. D., Rattanatray, L., and Boden, M. J. (2015). Acute inhibition of casein kinase 1δ/ε rapidly delays peripheral clock gene rhythms. Mol. Cell. Biochem. 398, 195–206. doi: 10.1007/s11010-014-2219-8
Kettunen, P., Larsson, S., Holmgren, S., Olsson, S., Minthon, L., Zetterberg, H., et al. (2015). Genetic variants of GSK3B are associated with biomarkers for Alzheimer’s disease and cognitive function. J. Alzheimers Dis. 44, 1313–1322. doi: 10.3233/JAD-142025
Khan, N. L., Jain, S., Lynch, J. M., Pavese, N., Abou-Sleiman, P., Holton, J. L., et al. (2005). Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128, 2786–2796. doi: 10.1093/brain/awh667
Kharbanda, S., Ren, R., Pandey, P., Shafman, T. D., Feller, S. M., Weichselbaum, R. R., et al. (1995). Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 376, 785–788. doi: 10.1038/376785a0
Killick, R., Ribe, E. M., Al-Shawi, R., Malik, B., Hooper, C., Fernandes, C., et al. (2014). Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt–PCP–JNK pathway. Mol. Psychiatry 19, 88–98. doi: 10.1038/mp.2012.163
Kimberly, W. T., Zheng, J. B., Town, T., Flavell, R. A., and Selkoe, D. J. (2005). Physiological regulation of the β-Amyloid precursor protein signaling domain by c-Jun N-Terminal Kinase JNK3 during neuronal differentiation. J. Neurosci. 25, 5533–5543. doi: 10.1523/JNEUROSCI.4883-04.2005
Klein, A., Maldonado, C., Vargas, L. M., Gonzalez, M., Robledo, F., Perez de Arce, K., et al. (2011). Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiol. Dis. 41, 209–218. doi: 10.1016/j.nbd.2010.09.008
Knippschild, U., Gocht, A., Wolff, S., Huber, N., Löhler, J., and Stöter, M. (2005). The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 17, 675–689. doi: 10.1016/j.cellsig.2004.12.011
Ko, H. S., Lee, Y., Shin, J.-H., Karuppagounder, S. S., Gadad, B. S., Koleske, A. J., et al. (2010). Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc. Natl. Acad. Sci. U.S.A. 107, 16691–16696. doi: 10.1073/pnas.1006083107
Koch, J. C., Tatenhorst, L., Roser, A.-E., Saal, K.-A., Tönges, L., and Lingor, P. (2018). ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 189, 1–21. doi: 10.1016/j.pharmthera.2018.03.008
Kosten, J., Binolfi, A., Stuiver, M., Verzini, S., Theillet, F.-X., Bekei, B., et al. (2014). Efficient modification of alpha-synuclein serine 129 by protein kinase CK1 requires phosphorylation of tyrosine 125 as a priming event. ACS Chem. Neurosci. 5, 1203–1208. doi: 10.1021/cn5002254
Kounakis, K., and Tavernarakis, N. (2019). The cytoskeleton as a modulator of aging and neurodegeneration. Adv. Exp. Med. Biol. 1178, 227–245. doi: 10.1007/978-3-030-25650-0_12
Kramer, T., Schmidt, B., and Monte, F. L. (2012). Small-molecule inhibitors of GSK-3: structural insights and their application to Alzheimer’s disease models. Int. J. Alzheimers Dis. 2012:381029. doi: 10.1155/2012/381029
Krenitsky, V. P., Delgado, M., Nadolny, L., Sahasrabudhe, K., Ayala, L., Clareen, S. S., et al. (2012). Aminopurine based JNK inhibitors for the prevention of ischemia reperfusion injury. Bioorg. Med. Chem. Lett. 22, 1427–1432. doi: 10.1016/j.bmcl.2011.12.028
Kuan, C.-Y., Yang, D. D., Roy, D. R. S., Davis, R. J., Rakic, P., and Flavell, R. A. (1999). The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22, 667–676. doi: 10.1016/S0896-6273(00)80727-8
Kuwahara, T., Inoue, K., D’Agati, V. D., Fujimoto, T., Eguchi, T., Saha, S., et al. (2016). LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci. Rep. 6:29945. doi: 10.1038/srep29945
Lagalwar, S., Guillozet-Bongaarts, A. L., Berry, R. W., and Binder, L. I. (2006). Formation of Phospho-SAPK/JNK granules in the hippocampus is an early event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 65, 455–464. doi: 10.1097/01.jnen.0000229236.98124.d8
Laughlin, J. D., Nwachukwu, J. C., Figuera-Losada, M., Cherry, L., Nettles, K. W., and LoGrasso, P. V. (2012). Structural mechanisms of allostery and autoinhibition in JNK family kinases. Struct. Lond. Engl. 20, 2174–2184. doi: 10.1016/j.str.2012.09.021
Lavalley, N. J., Slone, S. R., Ding, H., West, A. B., and Yacoubian, T. A. (2016). 14-3-3 Proteins regulate mutant LRRK2 kinase activity and neurite shortening. Hum. Mol. Genet. 25, 109–122. doi: 10.1093/hmg/ddv453
Le Pichon, C. E., Meilandt, W. J., Dominguez, S., Solanoy, H., Lin, H., Ngu, H., et al. (2017). Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med. 9:eaag0394. doi: 10.1126/scitranslmed.aag0394
Lee, J. K., and Kim, N.-J. (2017). Recent advances in the inhibition of P38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 22:1287. doi: 10.3390/molecules22081287
Lee, S., Kim, S., Park, Y. J., Yun, S. P., Kwon, S.-H., Kim, D., et al. (2018). The c-Abl inhibitor, Radotinib HCl, is neuroprotective in a preclinical Parkinson’s disease mouse model. Hum. Mol. Genet. 27, 2344–2356. doi: 10.1093/hmg/ddy143
Lewis, P. A. (2019). Leucine rich repeat kinase 2: a paradigm for pleiotropy. J. Physiol. 597, 3511–3521. doi: 10.1113/JP276163
Li, L., Shao, X., Cole, E. L., Ohnmacht, S. A., Ferrari, V., Hong, Y. T., et al. (2015). Synthesis and initial in vivo studies with [11C]SB-216763: the first radiolabeled brain penetrative inhibitor of GSK-3. ACS Med. Chem. Lett. 6, 548–552. doi: 10.1021/acsmedchemlett.5b00044
Li, X., Alafuzoff, I., Soininen, H., Winblad, B., and Pei, J.-J. (2005). Levels of MTOR and Its downstream targets 4E-BP1, EEF2, and EEF2 kinase in relationships with Tau in Alzheimer’s disease brain. FEBS J. 272, 4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x
Li, R., Martin, M. P., Liu, Y., Wang, B., Patel, R. A., Zhu, J.-Y., et al. (2012). Fragment-based and structure-guided discovery and optimization of Rho kinase inhibitors. J. Med. Chem. 55, 2474–2478. doi: 10.1021/jm201289r
Li, T., Yang, D., Zhong, S., Thomas, J. M., Xue, F., Liu, J., et al. (2014). Novel LRRK2 GTP-binding inhibitors reduced degeneration in Parkinson’s disease cell and mouse models. Hum. Mol. Genet. 23, 6212–6222. doi: 10.1093/hmg/ddu341
Lin, Q.-S., Chen, P., Wang, W.-X., Lin, C.-C., Zhou, Y., Yu, L.-H., et al. (2019). RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab. Invest. 100, 503–511. doi: 10.1038/s41374-019-0319-5
Lim, N. K. H., Hung, L. W., Pang, T. Y., Mclean, C. A., Liddell, J. R., Hilton, J. B., et al. (2014). Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington’s disease affected brain. Hum. Mol. Genet. 23, 4051–4063. doi: 10.1093/hmg/ddu119
Lin, H., Lee, E., Hestir, K., Leo, C., Huang, M., Bosch, E., et al. (2008). Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 320, 807–811. doi: 10.1126/science.1154370
Lindholm, D., Pham, D. D., Cascone, A., Eriksson, O., Wennerberg, K., and Saarma, M. (2016). c-Abl inhibitors enable insights into the pathophysiology and neuroprotection in Parkinson’s disease. Front. Aging Neurosci. 8:254. doi: 10.3389/fnagi.2016.00254
Lingor, P., Weber, M., Camu, W., Friede, T., Hilgers, R., Leha, A., et al. (2019). ROCK-ALS: protocol for a randomized, placebo-controlled, double-blind phase IIa Trial of safety, tolerability and efficacy of the Rho Kinase (ROCK) inhibitor fasudil in amyotrophic lateral sclerosis. Front. Neurol. 10:293. doi: 10.3389/fneur.2019.00293
Liu, M., Dobson, B., Glicksman, M. A., Yue, Z., and Stein, R. L. (2010a). Kinetic mechanistic studies of WT Leucine-Rich Repeat Kinase2: characterization of the kinase and GTPase activities. Biochemistry 49, 2008–2017. doi: 10.1021/bi901851y
Liu, M., Poulose, S., Schuman, E., Zaitsev, A. D., Dobson, B., Auerbach, K., et al. (2010b). Development of a mechanism-based HTS Assay for Leucine Rich Repeat Kinase2—discovery of LRRK2 inhibitors. Anal. Biochem. 404, 186–192. doi: 10.1016/j.ab.2010.05.033
Lobbestael, E., Civiero, L., De Wit, T., Taymans, J.-M., Greggio, E., and Baekelandt, V. (2016). Pharmacological LRRK2 kinase inhibition induces LRRK2 protein destabilization and proteasomal degradation. Sci. Rep. 6:33897. doi: 10.1038/srep33897
Longo, F., Mercatelli, D., Novello, S., Arcuri, L., Brugnoli, A., Vincenzi, F., et al. (2017). Age-dependent dopamine transporter dysfunction and Serine129 phospho-α-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol. Commun. 5:22. doi: 10.1186/s40478-017-0426-8
Lovestone, S., Boada, M., Dubois, B., Hüll, M., Rinne, J. O., Huppertz, H.-J., et al. (2015). A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 45, 75–88. doi: 10.3233/JAD-141959
Luna-Medina, R., Cortes-Canteli, M., Sanchez-Galiano, S., Morales-Garcia, J. A., Martinez, A., Santos, A., et al. (2007). NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: potential therapeutic role in brain disorders. J. Neurosci. 27:5766. doi: 10.1523/JNEUROSCI.1004-07.2007
Lynch, D. S., Rodrigues Brandão de Paiva, A., Zhang, W. J., Bugiardini, E., Freua, F., Tavares Lucato, L., et al. (2017). Clinical and genetic characterization of leukoencephalopathies in adults. Brain J. Neurol. 140, 1204–1211. doi: 10.1093/brain/awx045
Machado, C. B., Pluchon, P., Harley, P., Rigby, M., Gonzalez Sabater, V., Stevenson, D. C., et al. (2019). In vitro modelling of nerve-muscle connectivity in a compartmentalised tissue culture device. Adv. Biosyst. 3:1800307. doi: 10.1002/adbi.201800307
Machiraju, P. K., Yedla, P., Gubbala, S. P., Bohari, T., Abdul, J. K. V., Xu, S., et al. (2019). Identification, synthesis and evaluation of CSF1R inhibitors using fragment based drug design. Comput. Biol. Chem. 80, 374–383. doi: 10.1016/j.compbiolchem.2019.04.015
Mahul-Mellier, A.-L., Fauvet, B., Gysbers, A., Dikiy, I., Oueslati, A., Georgeon, S., et al. (2014). c-Abl phosphorylates α-synuclein and regulates its degradation: implication for α-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 23, 2858–2879. doi: 10.1093/hmg/ddt674
Maloney, M. T., Minamide, L. S., Kinley, A. W., Boyle, J. A., and Bamburg, J. R. (2005). Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: a feedforward mechanism for Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 25, 11313–11321. doi: 10.1523/JNEUROSCI.3711-05.2005
Mancuso, R., Fryatt, G., Cleal, M., Obst, J., Pipi, E., Monzón-Sandoval, J., et al. (2019). CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 142, 3243–3264. doi: 10.1093/brain/awz241
Manzoni, C., Denny, P., Lovering, R. C., and Lewis, P. A. (2015). Computational analysis of the LRRK2 interactome. PeerJ 3;e778. doi: 10.7717/peerj.778
Manzoni, C., Mamais, A., Dihanich, S., Abeti, R., Soutar, M. P. M., Plun-Favreau, H., et al. (2013). Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim. Biophys. Acta 1833, 2900–2910. doi: 10.1016/j.bbamcr.2013.07.020
Manzoni, C., Mamais, A., Roosen, D. A., Dihanich, S., Soutar, M. P. M., Plun-Favreau, H., et al. (2016). mTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin-1. Sci. Rep. 6:35106. doi: 10.1038/srep35106
Maphis, N., Jiang, S., Xu, G., Kokiko-Cochran, O. N., Roy, S. M., Van Eldik, L. J., et al. (2016). Selective suppression of the α isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res. Ther. 8:54. doi: 10.1186/s13195-016-0221-y
Maqbool, M., Mobashir, M., and Hoda, N. (2016). Pivotal role of glycogen synthase kinase-3: a therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 107, 63–81. doi: 10.1016/j.ejmech.2015.10.018
Markovinovic, A., Cimbro, R., Ljutic, T., Kriz, J., Rogelj, B., and Munitic, I. (2017). Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog. Neurobiol. 154, 1–20. doi: 10.1016/j.pneurobio.2017.04.005
Martens, S., Jeong, M., Tonnus, W., Feldmann, F., Hofmans, S., Goossens, V., et al. (2017). Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell Death Dis. 8:e2904. doi: 10.1038/cddis.2017.298
Martin, D. D. O., Kanuparthi, P. S., Holland, S. M., Sanders, S. S., Jeong, H.-K., Einarson, M. B., et al. (2019). Identification of novel inhibitors of DLK palmitoylation and signaling by high content screening. Sci. Rep. 9:3632. doi: 10.1038/s41598-019-39968-8
Martinez, A., Alonso, M., Castro, A., Pérez, C., and Moreno, F. J. (2002). First Non-ATP competitive glycogen synthase kinase 3 β (GSK-3β) inhibitors:? thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 45, 1292–1299. doi: 10.1021/jm011020u
Martínez-Muriana, A., Mancuso, R., Francos-Quijorna, I., Olmos-Alonso, A., Osta, R., Perry, V. H., et al. (2016). CSF1R blockade slows the progression of amyotrophic lateral sclerosis by reducing microgliosis and invasion of macrophages into peripheral nerves. Sci. Rep. 6:25663. doi: 10.1038/srep25663
Mason, C., Kossatz, S., Carter, L., Pirovano, G., Brand, C., Guru, N., et al. (2019). A 89Zr-HDL PET tracer monitors response to a CSF1R inhibitor. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 61, 433–436. doi: 10.2967/jnumed.119.230466
Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P., and Gallo, K. A. (2006). LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci. 29, 286–293. doi: 10.1016/j.tins.2006.03.006
Meegalla, S. K., Wall, M. J., Chen, J., Wilson, K. J., Ballentine, S. K., DesJarlais, R. L., et al. (2008). Structure-based optimization of a potent class of arylamide FMS inhibitors. Bioorg. Med. Chem. Lett. 18, 3632–3637. doi: 10.1016/j.bmcl.2008.04.059
Mehan, S., Meena, H., Sharma, D., and Sankhla, R. (2011). JNK: a stress-activated protein kinase therapeutic strategies and involvement in Alzheimer’s and various neurodegenerative abnormalities. J. Mol. Neurosci. 43, 376–390. doi: 10.1007/s12031-010-9454-6
Melrose, H. L. (2015). LRRK2 and ubiquitination: implications for kinase inhibitor therapy. Biochem. J. 470, e21–e24. doi: 10.1042/BJ20150785
Mendoza-Naranjo, A., Gonzalez-Billault, C., and Maccioni, R. B. (2007). Abeta1-42 stimulates actin polymerization in hippocampal neurons through Rac1 and Cdc42 Rho GTPases. J. Cell Sci. 120, 279–288. doi: 10.1242/jcs.03323
Meng, Q.-J., Maywood, E. S., Bechtold, D. A., Lu, W.-Q., Li, J., Gibbs, J. E., et al. (2010). Entrainment of disrupted circadian behavior through inhibition of casein kinase 1 (CK1) enzymes. Proc. Natl. Acad. Sci. U.S.A. 107, 15240–15245. doi: 10.1073/pnas.1005101107
Mente, S., Arnold, E., Butler, T., Chakrapani, S., Chandrasekaran, R., Cherry, K., et al. (2013). Ligand-protein interactions of selective casein kinase 1δ inhibitors. J. Med. Chem. 56, 6819–6828. doi: 10.1021/jm4006324
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi: 10.1016/j.neuron.2017.01.022
Menzies, F. M., Huebener, J., Renna, M., Bonin, M., Riess, O., and Rubinsztein, D. C. (2010). Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 133, 93–104. doi: 10.1093/brain/awp292
Meyers, M. J., Pelc, M., Kamtekar, S., Day, J., Poda, G. I., Hall, M. K., et al. (2010). Structure-based drug design enables conversion of a DFG-in binding CSF-1R kinase inhibitor to a DFG-out binding mode. Bioorg. Med. Chem. Lett. 20, 1543–1547. doi: 10.1016/j.bmcl.2010.01.078
Miller, B. R., Press, C., Daniels, R. W., Sasaki, Y., Milbrandt, J., and DiAntonio, A. (2009). A DLK-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci. 12:387. doi: 10.1038/nn.2290
Mills, R. D., Liang, L.-Y., Lio, D. S.-S., Mok, Y.-F., Mulhern, T. D., Cao, G., et al. (2018). The Roc-COR tandem domain of leucine-rich repeat kinase 2 forms dimers and exhibits conventional Ras-like GTPase properties. J. Neurochem. 147, 409–428. doi: 10.1111/jnc.14566
Mills, R. D., Mulhern, T. D., Liu, F., Culvenor, J. G., and Cheng, H.-C. (2014). Prediction of the repeat domain structures and impact of parkinsonism-associated variations on structure and function of all functional domains of Leucine-Rich Repeat Kinase 2 (LRRK2). Hum. Mutat. 35, 395–412. doi: 10.1002/humu.22515
Minamide, L. S., Striegl, A. M., Boyle, J. A., Meberg, P. J., and Bamburg, J. R. (2000). Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2, 628–636. doi: 10.1038/35023579
Mishra, P., and Günther, S. (2018). New insights into the structural dynamics of the kinase JNK3. Sci. Rep. 8:9435. doi: 10.1038/s41598-018-27867-3
Mizuno, T., Doi, Y., Mizoguchi, H., Jin, S., Noda, M., Sonobe, Y., et al. (2011). Interleukin-34 selectively enhances the neuroprotective effects of microglia to attenuate oligomeric amyloid-β neurotoxicity. Am. J. Pathol. 179, 2016–2027. doi: 10.1016/j.ajpath.2011.06.011
Mooney, L. M., and Whitmarsh, A. J. (2004). Docking interactions in the c-Jun N-terminal kinase pathway. J. Biol. Chem. 279, 11843–11852. doi: 10.1074/jbc.M311841200
Mora, J. S., Genge, A., Chio, A., Estol, C. J., Chaverri, D., Hernández, M., et al. (2019). Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 21, 5–14. doi: 10.1080/21678421.2019.1632346
Morales-Garcia, J. A., Salado, I. G., Sanz-San Cristobal, M., Gil, C., Pérez-Castillo, A., Martínez, A., et al. (2017). Biological and pharmacological characterization of benzothiazole-based CK-1δ inhibitors in models of Parkinson’s disease. ACS Omega 2, 5215–5220. doi: 10.1021/acsomega.7b00869
Morfini, G. A., You, Y.-M., Pollema, S. L., Kaminska, A., Liu, K., Yoshioka, K., et al. (2009). Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 12, 864–871. doi: 10.1038/nn.2346
Mortenson, P. N., Berdini, V., and O’Reilly, M. (2014). Fragment-based approaches to the discovery of kinase inhibitors. Methods Enzymol. 548, 69–92. doi: 10.1016/B978-0-12-397918-6.00003-3
Moskal, N., Riccio, V., Bashkurov, M., Taddese, R., Datti, A., Lewis, P. N., et al. (2020). ROCK inhibitors upregulate the neuroprotective parkin-mediated mitophagy pathway. Nat. Commun. 11:88. doi: 10.1038/s41467-019-13781-3
Muda, K., Bertinetti, D., Gesellchen, F., Hermann, J. S., von Zweydorf, F., Geerlof, A., et al. (2014). Parkinson-related LRRK2 mutation R1441C/G/H impairs PKA phosphorylation of LRRK2 and disrupts its interaction with 14-3-3. Proc. Natl. Acad. Sci. U.S.A. 111, E34–E43. doi: 10.1073/pnas.1312701111
Munsie, L. N., and Truant, R. (2012). The role of the cofilin-actin rod stress response in neurodegenerative diseases uncovers potential new drug targets. Bioarchitecture 2, 204–208. doi: 10.4161/bioa.22549
Murphy, G. M., Zhao, F., Yang, L., and Cordell, B. (2000). Expression of macrophage colony-stimulating factor receptor is increased in the AbetaPP(V717F) transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 157, 895–904.
Murray, C. W., Berdini, V., Buck, I. M., Carr, M. E., Cleasby, A., Coyle, J. E., et al. (2015). Fragment-based discovery of potent and selective DDR1/2 inhibitors. ACS Med. Chem. Lett. 6, 798–803. doi: 10.1021/acsmedchemlett.5b00143
Nagar, B., Bornmann, W. G., Pellicena, P., Schindler, T., Veach, D. R., Miller, W. T., et al. (2002). Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62, 4236–4243.
Najjar, M., Suebsuwong, C., Ray, S. S., Thapa, R. J., Maki, J. L., Nogusa, S., et al. (2015). Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 10, 1850–1860. doi: 10.1016/j.celrep.2015.02.052
Nandi, S., Gokhan, S., Dai, X.-M., Wei, S., Enikolopov, G., Lin, H., et al. (2012). The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev. Biol. 367, 100–113. doi: 10.1016/j.ydbio.2012.03.026
Narayanan, K. L., Chopra, V., Rosas, H. D., Malarick, K., and Hersch, S. (2016). Rho Kinase pathway alterations in the brain and leukocytes in Huntington’s disease. Mol. Neurobiol. 53, 2132–2140. doi: 10.1007/s12035-015-9147-9
Ness, D., Ren, Z., Gardai, S., Sharpnack, D., Johnson, V. J., Brennan, R. J., et al. (2013). Leucine-Rich Repeat Kinase 2 (LRRK2)-deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS One 8:e66164. doi: 10.1371/journal.pone.0066164
Nguyen, A. P. T., and Moore, D. J. (2017). Understanding the GTPase Activity of LRRK2: regulation, function, and neurotoxicity. Adv. Neurobiol. 14, 71–88. doi: 10.1007/978-3-319-49969-7_4
Nichols, R. J., Dzamko, N., Morrice, N. A., Campbell, D. G., Deak, M., Ordureau, A., et al. (2010). 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 430, 393–404. doi: 10.1042/BJ20100483
Nicolia, V., Ciraci, V., Cavallaro, R. A., Ferrer, I., Scarpa, S., and Fuso, S. S. (2017). GSK3β 5’-flanking DNA methylation and expression in Alzheimer’s disease patients. Curr. Alzheimer Res. 14, 753–759.
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Nonaka, T., Suzuki, G., Tanaka, Y., Kametani, F., Hirai, S., Okado, H., et al. (2016). Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by truncated casein kinase 1δ triggers mislocalization and accumulation of TDP-43. J. Biol. Chem. 291, 5473–5483. doi: 10.1074/jbc.M115.695379
Oberst, A. (2016). Death in the fast lane: what’s next for necroptosis? FEBS J. 283, 2616–2625. doi: 10.1111/febs.13520
O’Donnell, A., Faivre, S., Burris, H. A., Rea, D., Papadimitrakopoulou, V., Shand, N., et al. (2008). Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 26, 1588–1595. doi: 10.1200/JCO.2007.14.0988
Ofengeim, D., Mazzitelli, S., Ito, Y., DeWitt, J. P., Mifflin, L., Zou, C., et al. (2017). RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, E8788–E8797. doi: 10.1073/pnas.1714175114
Ohno, H., Kubo, K., Murooka, H., Kobayashi, Y., Nishitoba, T., Shibuya, M., et al. (2006). A c-fms tyrosine kinase inhibitor, Ki20227, suppresses osteoclast differentiation and osteolytic bone destruction in a bone metastasis model. Mol. Cancer Ther. 5, 2634–2643.
Olmos-Alonso, A., Schetters, S. T. T., Sri, S., Askew, K., Mancuso, R., Vargas-Caballero, M., et al. (2016). Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain J. Neurol. 139, 891–907. doi: 10.1093/brain/awv379
Omotehara, Y., Hakuba, N., Hato, N., Okada, M., and Gyo, K. (2011). Protection against ischemic cochlear damage by intratympanic administration of AM-111. Otol. Neurotol. 32, 1422–1427. doi: 10.1097/MAO.0b013e3182355658
Onishi, T., Iwashita, H., Uno, Y., Kunitomo, J., Saitoh, M., Kimura, E., et al. (2011). A novel glycogen synthase kinase-3 inhibitor 2-methyl-5-(3-4-[(S)-methylsulfinyl]phenyl-1-benzofuran-5-yl)-1,3,4-oxadiazole decreases tau phosphorylation and ameliorates cognitive deficits in a transgenic model of Alzheimer’s disease. J. Neurochem. 119, 1330–1340. doi: 10.1111/j.1471-4159.2011.07532.x
Osaki, T., Uzel, S. G. M., and Kamm, R. D. (2018). Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 4:eaat5847. doi: 10.1126/sciadv.aat5847
Ozcelik, S., Fraser, G., Castets, P., Schaeffer, V., Skachokova, Z., Breu, K., et al. (2013). Rapamycin Attenuates the progression of Tau Pathology in P301S Tau Transgenic mice. PLoS One 8:e62459. doi: 10.1371/journal.pone.0062459
Pagan, F., Hebron, M., Valadez, E. H., Torres-Yaghi, Y., Huang, X., Mills, R. R., et al. (2016). Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Park. Dis. 6, 503–517. doi: 10.3233/JPD-160867
Pagan, F. L., Hebron, M. L., Wilmarth, B., Torres-Yaghi, Y., Lawler, A., Mundel, E. E., et al. (2019). Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharmacol. Res. Perspect. 7:e00470. doi: 10.1002/prp2.470
Paisán-Ruiz, C., Lewis, P. A., and Singleton, A. B. (2013). LRRK2: cause, risk, and mechanism. J. Park. Dis. 3, 85–103. doi: 10.3233/JPD-130192
Pajak, B., Songin, M., Strosznajder, J. B., and Gajkowska, B. (2009). Alzheimer’s disease genetic mutation evokes ultrastructural alterations: correlation to an intracellular Aβ deposition and the level of GSK-3β-P(Y216) phosphorylated form. NeuroToxicology 30, 581–588. doi: 10.1016/j.neuro.2009.05.008
Palavra, F., Robalo, C., and Reis, F. (2017). Recent advances and challenges of mTOR inhibitors use in the treatment of patients with tuberous sclerosis complex. Oxid. Med. Cell. Longev. 2017:9820181. doi: 10.1155/2017/9820181
Palomo, V., Perez, D. I., Roca, C., Anderson, C., Rodríguez-Muela, N., Perez, C., et al. (2017). Subtly modulating glycogen synthase Kinase 3 β: allosteric inhibitor development and their potential for the treatment of chronic diseases. J. Med. Chem. 60, 4983–5001. doi: 10.1021/acs.jmedchem.7b00395
Palomo, V., Soteras, I., Perez, D. I., Perez, C., Gil, C., Campillo, N. E., et al. (2011). Exploring the binding sites of glycogen synthase Kinase 3. Identification and characterization of allosteric modulation cavities. J. Med. Chem. 54, 8461–8470. doi: 10.1021/jm200996g
Pandey, M. K., and DeGrado, T. R. (2016). Glycogen synthase Kinase-3 (GSK-3)-targeted therapy and imaging. Theranostics 6, 571–593. doi: 10.7150/thno.14334
Parisiadou, L., Yu, J., Sgobio, C., Xie, C., Liu, G., Sun, L., et al. (2014). LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat. Neurosci. 17, 367–376. doi: 10.1038/nn.3636
Parkinson Study Group. (2007). Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 69, 1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0
Patel, P., and Woodgett, J. R. (2017). “Chapter Eight - Glycogen synthase Kinase 3: a kinase for all pathways?,” in Current Topics in Developmental Biology Protein Kinases in Development and Disease, ed. A. Jenny (Cambridge, MA: Academic Press), 277–302. doi: 10.1016/bs.ctdb.2016.11.011
Patel, S., Cohen, F., Dean, B. J., De La Torre, K., Deshmukh, G., Estrada, A. A., et al. (2015a). Discovery of dual Leucine Zipper Kinase (DLK, MAP3K12) inhibitors with activity in neurodegeneration models. J. Med. Chem. 58, 401–418. doi: 10.1021/jm5013984
Patel, S., Harris, S. F., Gibbons, P., Deshmukh, G., Gustafson, A., Kellar, T., et al. (2015b). Scaffold-Hopping and structure-based discovery of potent, selective, and brain Penetrant N-(1H-Pyrazol-3-yl)pyridin-2-amine inhibitors of dual Leucine Zipper Kinase (DLK, MAP3K12). J. Med. Chem. 58, 8182–8199. doi: 10.1021/acs.jmedchem.5b01072
Patel, S., Meilandt, W. J., Erickson, R. I., Chen, J., Deshmukh, G., Estrada, A. A., et al. (2017). Selective inhibitors of dual Leucine Zipper Kinase (DLK, MAP3K12) with activity in a model of Alzheimer’s disease. J. Med. Chem. 60, 8083–8102. doi: 10.1021/acs.jmedchem.7b00843
Pearson, A. G., Byrne, U. T. E., MacGibbon, G. A., Faull, R. L. M., and Dragunow, M. (2006). Activated c-Jun is present in neurofibrillary tangles in Alzheimer’s disease brains. Neurosci. Lett. 398, 246–250. doi: 10.1016/j.neulet.2006.01.031
Peineau, S., Nicolas, C. S., Bortolotto, Z. A., Bhat, R. V., Ryves, W. J., Harwood, A. J., et al. (2009). A systematic investigation of the protein kinases involved in NMDA receptor-dependent LTD: evidence for a role of GSK-3 but not other serine/threonine kinases. Mol. Brain 2:22. doi: 10.1186/1756-6606-2-22
Perera, G., Ranola, M., Rowe, D. B., Halliday, G. M., and Dzamko, N. (2016). Inhibitor treatment of peripheral mononuclear cells from Parkinson’s disease patients further validates LRRK2 dephosphorylation as a pharmacodynamic biomarker. Sci. Rep. 6:31391. doi: 10.1038/srep31391
Perez, D. I., Conde, S., Pérez, C., Gil, C., Simon, D., Wandosell, F., et al. (2009). Thienylhalomethylketones: irreversible glycogen synthase kinase 3 inhibitors as useful pharmacological tools. Bioorg. Med. Chem. 17, 6914–6925. doi: 10.1016/j.bmc.2009.08.042
Perrin, V., Dufour, N., Raoul, C., Hassig, R., Brouillet, E., Aebischer, P., et al. (2009). Implication of the JNK pathway in a rat model of Huntington’s disease. Exp. Neurol. 215, 191–200. doi: 10.1016/j.expneurol.2008.10.008
Perry, V. H., Nicoll, J. A. R., and Holmes, C. (2010). Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201. doi: 10.1038/nrneurol.2010.17
Petit-Paitel, A., Brau, F., Cazareth, J., and Chabry, J. (2009). Involvment of cytosolic and mitochondrial GSK-3β in mitochondrial dysfunction and neuronal cell death of MPTP/MPP+-treated neurons. PLoS One 4:e5491. doi: 10.1371/journal.pone.0005491
Petratos, S., Li, Q.-X., George, A. J., Hou, X., Kerr, M. L., Unabia, S. E., et al. (2008). The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain J. Neurol. 131, 90–108. doi: 10.1093/brain/awm260
Petrov, D., Luque, M., Pedrós, I., Ettcheto, M., Abad, S., Pallàs, M., et al. (2016). Evaluation of the role of JNK1 in the Hippocampus in an experimental model of familial Alzheimer’s disease. Mol. Neurobiol. 53, 6183–6193. doi: 10.1007/s12035-015-9522-6
Petrov, D., Mansfield, C., Moussy, A., and Hermine, O. (2017). ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 9:68. doi: 10.3389/fnagi.2017.00068
Phiel, C. J., Wilson, C. A., Lee, V. M.-Y., and Klein, P. S. (2003). GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature 423, 435–439. doi: 10.1038/nature01640
Piccoli, G., Onofri, F., Cirnaru, M. D., Kaiser, C. J. O., Jagtap, P., Kastenmüller, A., et al. (2014). Leucine-rich repeat kinase 2 binds to neuronal vesicles through protein interactions mediated by its C-Terminal WD40 domain. Mol. Cell. Biol. 34, 2147–2161. doi: 10.1128/MCB.00914-13
Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P. A., et al. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Invest. 118, 2190–2199. doi: 10.1172/JCI33585
Pozniak, C. D., Sengupta Ghosh, A., Gogineni, A., Hanson, J. E., Lee, S.-H., Larson, J. L., et al. (2013). Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J. Exp. Med. 210, 2553–2567. doi: 10.1084/jem.20122832
Prabhakaran, J., Zanderigo, F., Sai, K. K. S., Rubin-Falcone, H., Jorgensen, M. J., Kaplan, J. R., et al. (2017). Radiosynthesis and in vivo evaluation of [11C]A1070722, a high affinity GSK-3 PET tracer in primate brain. ACS Chem. Neurosci. 8, 1697–1703. doi: 10.1021/acschemneuro.6b00376
Pridans, C., Raper, A., Davis, G. M., Alves, J., Sauter, K. A., Lefevre, L., et al. (2018). Pleiotropic impacts of macrophage and microglial deficiency on development in rats with targeted mutation of the Csf1r locus. J. Immunol. 201, 2683–2699. doi: 10.4049/jimmunol.1701783
Probst, G. D., Bowers, S., Sealy, J. M., Truong, A. P., Hom, R. K., Galemmo, R. A., et al. (2011). Highly selective c-Jun N-terminal kinase (JNK) 2 and 3 inhibitors with in vitro CNS-like pharmacokinetic properties prevent neurodegeneration. Bioorg. Med. Chem. Lett. 21, 315–319. doi: 10.1016/j.bmcl.2010.11.010
Puig, B., Gómez-Isla, T., Ribé, E., Cuadrado, M., Torrejón-Escribano, B., Dalfó, E., et al. (2004). Expression of stress-activated kinases c-Jun N-terminal kinase (SAPK/JNK-P) and p38 kinase (p38-P), and tau hyperphosphorylation in neurites surrounding betaA plaques in APP Tg2576 mice. Neuropathol. Appl. Neurobiol. 30, 491–502. doi: 10.1111/j.1365-2990.2004.00569.x
Puschmann, A. (2013). Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkins. Relat. Disord. 19, 407–415. doi: 10.1016/j.parkreldis.2013.01.020
Pyonteck, S. M., Akkari, L., Schuhmacher, A. J., Bowman, R. L., Sevenich, L., Quail, D. F., et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19:1264. doi: 10.1038/nm.3337
Rabanal-Ruiz, Y., Otten, E. G., and Korolchuk, V. I. (2017). mTORC1 as the main gateway to autophagy. Essays Biochem. 61, 565–584. doi: 10.1042/EBC20170027
Ramin, M., Azizi, P., Motamedi, F., Haghparast, A., and Khodagholi, F. (2011). Inhibition of JNK phosphorylation reverses memory deficit induced by β-amyloid (1–42) associated with decrease of apoptotic factors. Behav. Brain Res. 217, 424–431. doi: 10.1016/j.bbr.2010.11.017
Raoul, C., Estévez, A. G., Nishimune, H., Cleveland, D. W., deLapeyrière, O., Henderson, C. E., et al. (2002). Motoneuron death triggered by a specific pathway downstream of Fas. Potentiation by ALS-linked SOD1 mutations. Neuron 35, 1067–1083. doi: 10.1016/s0896-6273(02)00905-4
Rask-Andersen, M., Zhang, J., Fabbro, D., and Schiöth, H. B. (2014). Advances in kinase targeting: current clinical use and clinical trials. Trends Pharmacol. Sci. 35, 604–620. doi: 10.1016/j.tips.2014.09.007
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Rayasam, G. V., Tulasi, V. K., Sodhi, R., Davis, J. A., and Ray, A. (2009). Glycogen synthase kinase 3: more than a namesake. Br. J. Pharmacol. 156, 885–898. doi: 10.1111/j.1476-5381.2008.00085.x
Re, D. B., Le Verche, V., Yu, C., Amoroso, M. W., Politi, K. A., Phani, S., et al. (2014). Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81, 1001–1008. doi: 10.1016/j.neuron.2014.01.011
Reith, A. D., Bamborough, P., Jandu, K., Andreotti, D., Mensah, L., Dossang, P., et al. (2012). GSK2578215A; a potent and highly selective 2-arylmethyloxy-5-substitutent-N-arylbenzamide LRRK2 kinase inhibitor. Bioorg. Med. Chem. Lett. 22, 5625–5629. doi: 10.1016/j.bmcl.2012.06.104
Repici, M., Zanjani, H. S., Gautheron, V., Borsello, T., Dusart, I., and Mariani, J. (2008). Specific JNK inhibition by D-jnki1 protects purkinje cells from cell death in lurcher mutant mouse. Cerebellum 7, 534–538. doi: 10.1007/s12311-008-0070-8
Riancho, J., Gil-Bea, F. J., Castanedo-Vazquez, D., Sedano, M. J., Zufiría, M., de Eulate, G. F. G., et al. (2018). Clinical evidences supporting the Src/c-Abl pathway as potential therapeutic target in amyotrophic lateral sclerosis. J. Neurol. Sci. 393, 80–82. doi: 10.1016/j.jns.2018.08.013
Rojas-Rivera, D., Delvaeye, T., Roelandt, R., Nerinckx, W., Augustyns, K., Vandenabeele, P., et al. (2017). When PERK inhibitors turn out to be new potent RIPK1 inhibitors: critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 24, 1100–1110. doi: 10.1038/cdd.2017.58
Roosen, D. A., and Cookson, M. R. (2016). LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 11:73. doi: 10.1186/s13024-016-0140-1
Rosenbusch, K. E., and Kortholt, A. (2016). Activation mechanism of LRRK2 and its cellular functions in Parkinson’s disease. Park. Dis. 2016:7351985. doi: 10.1155/2016/7351985
Roser, A.-E., Tönges, L., and Lingor, P. (2017). Modulation of microglial activity by Rho-Kinase (ROCK) inhibition as therapeutic strategy in Parkinson’s disease and amyotrophic lateral sclerosis. Front. Aging Neurosci. 9:94. doi: 10.3389/fnagi.2017.00094
Roskoski, R. (2016). Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 103, 26–48. doi: 10.1016/j.phrs.2015.10.021
Ross, O. A., Soto-Ortolaza, A. I., Heckman, M. G., Aasly, J. O., Abahuni, N., Annesi, G., et al. (2011). LRRK2 exonic variants and susceptibility to Parkinson’s disease. Lancet Neurol. 10, 898–908. doi: 10.1016/S1474-4422(11)70175-2
Roy, S. M., Grum-Tokars, V. L., Schavocky, J. P., Saeed, F., Staniszewski, A., Teich, A. F., et al. (2015). Targeting human central nervous system protein kinases: an isoform selective p38αMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem. Neurosci. 6, 666–680. doi: 10.1021/acschemneuro.5b00002
Saal, K.-A., Galter, D., Roeber, S., Bähr, M., Tönges, L., and Lingor, P. (2017). Altered expression of growth associated protein-43 and Rho Kinase in human patients with Parkinson’s disease. Brain Pathol. Zurich Switz. 27, 13–25. doi: 10.1111/bpa.12346
Sack, J. S., Kish, K. F., Pokross, M., Xie, D., Duke, G. J., Tredup, J. A., et al. (2008). Structural basis for the high-affinity binding of pyrrolotriazine inhibitors of p38 MAP kinase. Acta Crystallogr. D Biol. Crystallogr. D 64, 705–710. doi: 10.1107/S0907444908010032
Saez-Atienzar, S., Bonet-Ponce, L., Blesa, J. R., Romero, F. J., Murphy, M. P., Jordan, J., et al. (2014). The LRRK2 inhibitor GSK2578215A induces protective autophagy in SH-SY5Y cells: involvement of Drp-1-mediated mitochondrial fission and mitochondrial-derived ROS signaling. Cell Death Dis. 5:e1368. doi: 10.1038/cddis.2014.320
Saitoh, M., Kunitomo, J., Kimura, E., Iwashita, H., Uno, Y., Onishi, T., et al. (2009). 2-{3-[4-(Alkylsulfinyl)phenyl]-1-benzofuran-5-yl}-5-methyl-1,3,4-oxadiazole derivatives as novel inhibitors of glycogen synthase kinase-3β with good brain permeability. J. Med. Chem. 52, 6270–6286. doi: 10.1021/jm900647e
Salado, I. G., Redondo, M., Bello, M. L., Perez, C., Liachko, N. F., Kraemer, B. C., et al. (2014). Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 57, 2755–2772. doi: 10.1021/jm500065f
Saporito, M. S., Brown, E. M., Miller, M. S., and Carswell, S. (1999). CEP-1347/KT-7515, an inhibitor of c-jun N-Terminal kinase activation, attenuates the 1-Methyl-4-Phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J. Pharmacol. Exp. Ther. 288, 421–427.
Saporito, M. S., Thomas, B. A., and Scott, R. W. (2000). MPTP activates c-Jun NH2-Terminal Kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J. Neurochem. 75, 1200–1208. doi: 10.1046/j.1471-4159.2000.0751200.x
Sarkar, S., Ravikumar, B., Floto, R. A., and Rubinsztein, D. C. (2009). Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 16, 46–56. doi: 10.1038/cdd.2008.110
Schaffer, B. A. J., Bertram, L., Miller, B. L., Mullin, K., Weintraub, S., Johnson, N., et al. (2008). Association of GSK3B With Alzheimer disease and frontotemporal dementia. Arch. Neurol. 65:1368. doi: 10.1001/archneur.65.10.1368
Schapansky, J., Nardozzi, J. D., Felizia, F., and LaVoie, M. J. (2014). Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum. Mol. Genet. 23, 4201–4214. doi: 10.1093/hmg/ddu138
Scheiblich, H., and Bicker, G. (2017). Regulation of microglial phagocytosis by RhoA/ROCK-inhibiting drugs. Cell. Mol. Neurobiol. 37, 461–473. doi: 10.1007/s10571-016-0379-7
Schindler, T., Bornmann, W., Pellicena, P., Miller, W. T., Clarkson, B., and Kuriyan, J. (2000). Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289, 1938–1942.
Schlatterer, S. D., Tremblay, M. A., Acker, C. M., and Davies, P. (2011). Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J. Alzheimers Dis. 25, 119–133. doi: 10.3233/JAD-2011-102025
Schnöder, L., Hao, W., Qin, Y., Liu, S., Tomic, I., Liu, X., et al. (2016). Deficiency of neuronal p38α MAPK attenuates amyloid pathology in Alzheimer disease mouse and cell models through facilitating lysosomal degradation of BACE1. J. Biol. Chem. 291, 2067–2079. doi: 10.1074/jbc.M115.695916
Schoepfer, J., Jahnke, W., Berellini, G., Buonamici, S., Cotesta, S., Cowan-Jacob, S. W., et al. (2018). Discovery of Asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J. Med. Chem. 61, 8120–8135. doi: 10.1021/acs.jmedchem.8b01040
Scior, T., Domeyer, D. M., Cuanalo-Contreras, K., and Laufer, S. A. (2011). Pharmacophore design of p38α MAP kinase inhibitors with either 2,4,5-trisubstituted or 1,2,4,5-tetrasubstituted imidazole scaffold. Curr. Med. Chem. 18, 1526–1539. doi: 10.2174/092986711795328409
Sclip, A., Antoniou, X., Colombo, A., Camici, G. G., Pozzi, L., Cardinetti, D., et al. (2011). c-Jun N-terminal kinase regulates soluble Aβ oligomers and cognitive impairment in AD mouse model. J. Biol. Chem. 286, 43871–43880. doi: 10.1074/jbc.M111.297515
Sclip, A., Tozzi, A., Abaza, A., Cardinetti, D., Colombo, I., Calabresi, P., et al. (2014). c-Jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death Dis. 5:e1019. doi: 10.1038/cddis.2013.559
Scott, D. A., Dakin, L. A., Daly, K., Del Valle, D. J., Diebold, R. B., Drew, L., et al. (2013). Mitigation of cardiovascular toxicity in a series of CSF-1R inhibitors, and the identification of AZD7507. Bioorg. Med. Chem. Lett. 23, 4591–4596. doi: 10.1016/j.bmcl.2013.06.031
Scott, J. D., DeMong, D. E., Greshock, T. J., Basu, K., Dai, X., Harris, J., et al. (2017). Discovery of a 3-(4-Pyrimidinyl) Indazole (MLi-2), an orally available and selective Leucine-Rich Repeat Kinase 2 (LRRK2) inhibitor that reduces brain kinase activity. J. Med. Chem. 60, 2983–2992. doi: 10.1021/acs.jmedchem.7b00045
Selenica, M.-L., Jensen, H. S., Larsen, A. K., Pedersen, M. L., Helboe, L., Leist, M., et al. (2007). Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br. J. Pharmacol. 152:959. doi: 10.1038/sj.bjp.0707471
Sen, S., Webber, P. J., and West, A. B. (2009). Dependence of Leucine-rich Repeat Kinase 2 (LRRK2) kinase activity on dimerization. J. Biol. Chem. 284, 36346–36356. doi: 10.1074/jbc.M109.025437
Serenó, L., Coma, M., Rodríguez, M., Sánchez-Ferrer, P., Sánchez, M. B., Gich, I., et al. (2009). A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367. doi: 10.1016/j.nbd.2009.05.025
Shafman, T., Khanna, K. K., Kedar, P., Spring, K., Kozlov, S., Yen, T., et al. (1997). Interaction between ATM protein and c-Abl in response to DNA damage. Nature 387, 520–523. doi: 10.1038/387520a0
Shah, N. P., Tran, C., Lee, F. Y., Chen, P., Norris, D., and Sawyers, C. L. (2004). Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305, 399–401. doi: 10.1126/science.1099480
Shao, J., Welch, W. J., Diprospero, N. A., and Diamond, M. I. (2008). Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol. Cell. Biol. 28, 5196–5208. doi: 10.1128/MCB.00079-08
Sharma, R., Graff-Radford, J., Rademakers, R., Boeve, B. F., Petersen, R. C., and Jones, D. T. (2019). CSF1R mutation presenting as dementia with Lewy bodies. Neurocase 25, 17–20. doi: 10.1080/13554794.2019.1601230
Shayman, J. A., and Abe, A. (2013). Drug induced phospholipidosis: an acquired lysosomal storage disorder. Biochim. Biophys. Acta 1831, 602–611. doi: 10.1016/j.bbalip.2012.08.013
Shi, Y., and Mader, M. (2018). Brain penetrant kinase inhibitors: learning from kinase neuroscience discovery. Bioorg. Med. Chem. Lett. 28, 1981–1991. doi: 10.1016/j.bmcl.2018.05.007
Shin, J. E., Cho, Y., Beirowski, B., Milbrandt, J., Cavalli, V., and DiAntonio, A. (2012). Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 74:1015. doi: 10.1016/j.neuron.2012.04.028
Shin, J. E., Ha, H., Kim, Y. K., Cho, Y., and DiAntonio, A. (2019). DLK regulates a distinctive transcriptional regeneration program after peripheral nerve injury. Neurobiol. Dis. 127, 178–192. doi: 10.1016/j.nbd.2019.02.001
Shin, N., Jeong, H., Kwon, J., Heo, H. Y., Kwon, J. J., Yun, H. J., et al. (2008). LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 314, 2055–2065. doi: 10.1016/j.yexcr.2008.02.015
Shoulson, I., and Parkinson’s Study Group and Precept Investigators (2006). CEP-1347 treatment fails to favorably modify the progression of Parkinson’s Disease (PRECEPT) Study: S61.003. Neurology 67, 185–185–b. doi: 10.1212/01.wnl.0000234884.42413.bc
Shu, Y., Ming, J., Zhang, P., Wang, Q., Jiao, F., and Tian, B. (2016). Parkinson-related LRRK2 mutation R1628P enables Cdk5 phosphorylation of LRRK2 and upregulates its kinase activity. PLoS One 11:e0149739. doi: 10.1371/journal.pone.0149739
Siu, M., Sengupta Ghosh, A., and Lewcock, J. W. (2018). Dual Leucine zipper kinase inhibitors for the treatment of neurodegeneration. J. Med. Chem. 61, 8078–8087. doi: 10.1021/acs.jmedchem.8b00370
Sivaprakasam, P., Han, X., Civiello, R. L., Jacutin-Porte, S., Kish, K., Pokross, M., et al. (2015). Discovery of new acylaminopyridines as GSK-3 inhibitors by a structure guided in-depth exploration of chemical space around a pyrrolopyridinone core. Bioorg. Med. Chem. Lett. 25, 1856–1863. doi: 10.1016/j.bmcl.2015.03.046
Smith, W. W., Pei, Z., Jiang, H., Moore, D. J., Liang, Y., West, A. B., et al. (2005). Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 18676–18681. doi: 10.1073/pnas.0508052102
Sosna, J., Philipp, S., Albay, R., Reyes-Ruiz, J. M., Baglietto-Vargas, D., LaFerla, F. M., et al. (2018). Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 13:11. doi: 10.1186/s13024-018-0244-x
Spangenberg, E., Severson, P. L., Hohsfield, L. A., Crapser, J., Zhang, J., Burton, E. A., et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 10:3758. doi: 10.1038/s41467-019-11674-z
Spangenberg, E. E., Lee, R. J., Najafi, A. R., Rice, R. A., Elmore, M. R. P., Blurton-Jones, M., et al. (2016). Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain J. Neurol. 139, 1265–1281. doi: 10.1093/brain/aww016
Sperbera, B. R., Leight, S., Goedert, M., and Lee, V. M.-Y. (1995). Glycogen synthase kinase-3β phosphorylates tau protein at multiple sites in intact cells. Neurosci. Lett. 197, 149–153. doi: 10.1016/0304-3940(95)11902-9
Spiller, K. J., Restrepo, C. R., Khan, T., Dominique, M. A., Fang, T. C., Canter, R. G., et al. (2018). Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 21, 329–340. doi: 10.1038/s41593-018-0083-7
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces Amyloid-β levels in a mouse model of Alzheimer’s disease. PLoS One 5:e9979. doi: 10.1371/journal.pone.0009979
Spittaels, K., den Haute, C. V., Dorpe, J. V., Geerts, H., Mercken, M., Bruynseels, K., et al. (2000). Glycogen synthase Kinase-3β phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J. Biol. Chem. 275, 41340–41349. doi: 10.1074/jbc.M006219200
Staats, K. A., Hernandez, S., Schönefeldt, S., Bento-Abreu, A., Dooley, J., Van Damme, P., et al. (2013). Rapamycin increases survival in ALS mice lacking mature lymphocytes. Mol. Neurodegener. 8:31. doi: 10.1186/1750-1326-8-31
Stabile, C., Taglia, I., Battisti, C., Bianchi, S., and Federico, A. (2016). Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): update on molecular genetics. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 37, 1565–1569. doi: 10.1007/s10072-016-2634-6
Stafa, K., Tsika, E., Moser, R., Musso, A., Glauser, L., Jones, A., et al. (2014). Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet. 23, 2055–2077. doi: 10.1093/hmg/ddt600
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 5:e12813. doi: 10.7554/eLife.12813
Stevers, L. M., de Vink, P. J., Ottmann, C., Huskens, J., and Brunsveld, L. (2018). A thermodynamic model for multivalency in 14-3-3 protein–protein interactions. J. Am. Chem. Soc. 140, 14498–14510. doi: 10.1021/jacs.8b09618
Stevers, L. M., de Vries, R. M. J. M., Doveston, R. G., Milroy, L.-G., Brunsveld, L., and Ottmann, C. (2017). Structural interface between LRRK2 and 14-3-3 protein. Biochem. J. 474, 1273–1287. doi: 10.1042/BCJ20161078
Suckfuell, M., Lisowska, G., Domka, W., Kabacinska, A., Morawski, K., Bodlaj, R., et al. (2014). Efficacy and Safety of AM-111 in the treatment of acute sensorineural hearing loss: a double-blind, randomized, placebo-controlled phase II study. Otol. Neurotol. 35, 1317–1326. doi: 10.1097/MAO.0000000000000466
Sun, A., Liu, M., Nguyen, X. V., and Bing, G. (2003). P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 183, 394–405. doi: 10.1016/s0014-4886(03)00180-8
Suzuki, H., and Matsuoka, M. (2013). The JNK/c-Jun signaling axis contributes to the TDP-43-induced cell death. Mol. Cell. Biochem. 372, 241–248. doi: 10.1007/s11010-012-1465-x
Swahn, B.-M., Huerta, F., Kallin, E., Malmström, J., Weigelt, T., Viklund, J., et al. (2005). Design and synthesis of 6-anilinoindazoles as selective inhibitors of c-Jun N-terminal kinase-3. Bioorg. Med. Chem. Lett. 15, 5095–5099. doi: 10.1016/j.bmcl.2005.06.083
Takata, M., Tanaka, H., Kimura, M., Nagahara, Y., Tanaka, K., Kawasaki, K., et al. (2013). Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br. J. Pharmacol. 170, 341–351. doi: 10.1111/bph.12277
Tanabe, A., Yamamura, Y., Kasahara, J., Morigaki, R., Kaji, R., and Goto, S. (2014). A novel tyrosine kinase inhibitor AMN107 (nilotinib) normalizes striatal motor behaviors in a mouse model of Parkinson’s disease. Front. Cell. Neurosci. 8:50. doi: 10.3389/fncel.2014.00050
Tanzey, S. S., Shao, X., Stauff, J., Arteaga, J., Sherman, P., Scott, P. J. H., et al. (2018). Synthesis and initial in vivo evaluation of [11C]AZ683-A novel PET radiotracer for colony stimulating factor 1 receptor (CSF1R). Pharm. Basel Switz. 11:136. doi: 10.3390/ph11040136
Tatenhorst, L., Tönges, L., Saal, K.-A., Koch, J. C., Szegõ, ÉM., Bähr, M., et al. (2014). Rho kinase inhibition by fasudil in the striatal 6-hydroxydopamine lesion mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 73, 770–779. doi: 10.1097/NEN.0000000000000095
Taylor, M., and Alessi, D. R. (2020). Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol. 63, 102–113. doi: 10.1016/j.ceb.2020.01.001
Tedeschi, A., and Bradke, F. (2013). The DLK signalling pathway—a double-edged sword in neural development and regeneration. EMBO Rep. 14:605. doi: 10.1038/embor.2013.64
Terwel, D., Muyllaert, D., Dewachter, I., Borghgraef, P., Croes, S., Devijver, H., et al. (2008). Amyloid activates GSK-3β to aggravate neuronal tauopathy in bigenic Mice. Am. J. Pathol. 172:786. doi: 10.2353/ajpath.2008.070904
Thal, D. R., Del Tredici, K., Ludolph, A. C., Hoozemans, J. J. M., Rozemuller, A. J., Braak, H., et al. (2011). Stages of granulovacuolar degeneration: their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol. 122, 577–589. doi: 10.1007/s00401-011-0871-6
Thoreen, C. C., Kang, S. A., Chang, J. W., Liu, Q., Zhang, J., Gao, Y., et al. (2009). An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032. doi: 10.1074/jbc.M900301200
Tolosa, E., Litvan, I., Höglinger, G. U., Burn, D., Lees, A., Andrés, M. V., et al. (2014). A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 29, 470–478. doi: 10.1002/mds.25824
Tong, Y., Giaime, E., Yamaguchi, H., Ichimura, T., Liu, Y., Si, H., et al. (2012). Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 7:2. doi: 10.1186/1750-1326-7-2
Tönges, L., Frank, T., Tatenhorst, L., Saal, K. A., Koch, J. C., Szego, ÉM., et al. (2012). Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson’s disease. Brain J. Neurol. 135, 3355–3370. doi: 10.1093/brain/aws254
Tönges, L., Günther, R., Suhr, M., Jansen, J., Balck, A., Saal, K.-A., et al. (2014). Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 62, 217–232. doi: 10.1002/glia.22601
Tourette, C., Li, B., Bell, R., O’Hare, S., Kaltenbach, L. S., Mooney, S. D., et al. (2014). A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. J. Biol. Chem. 289, 6709–6726. doi: 10.1074/jbc.M113.523696
Tran, H. T., Sanchez, L., and Brody, D. L. (2012). Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J. Neuropathol. Exp. Neurol. 71, 116–129. doi: 10.1097/NEN.0b013e3182456aed
Uitdehaag, J. C. M., Sünnen, C. M., van Doornmalen, A. M., de Rouw, N., Oubrie, A., Azevedo, R., et al. (2011). Multidimensional profiling of CSF1R screening hits and inhibitors: assessing cellular activity, target residence time, and selectivity in a higher throughput way. J. Biomol. Screen. 16, 1007–1017. doi: 10.1177/1087057111418113
Uitdehaag, J. C. M., Verkaar, F., Alwan, H., de Man, J., Buijsman, R. C., and Zaman, G. J. R. (2012). A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br. J. Pharmacol. 166, 858–876. doi: 10.1111/j.1476-5381.2012.01859.x
Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L., and Clardy, J. (1993). Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124. doi: 10.1006/jmbi.1993.1012
Vancraenenbroeck, R., Lobbestael, E., Weeks, S. D., Strelkov, S. V., Baekelandt, V., Taymans, J.-M., et al. (2012). Expression, purification and preliminary biochemical and structural characterization of the leucine rich repeat namesake domain of leucine rich repeat kinase 2. Biochim. Biophys. Acta BBA Proteins Proteomics 1824, 450–460. doi: 10.1016/j.bbapap.2011.12.009
Vanden Berghe, T., Kaiser, W. J., Bertrand, M. J., and Vandenabeele, P. (2015). Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2:e975093. doi: 10.4161/23723556.2014.975093
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi: 10.1038/nrm2970
Vézina, C., Kudelski, A., and Sehgal, S. N. (1975). Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 28, 721–726. doi: 10.7164/antibiotics.28.721
Villar-Cheda, B., Dominguez-Meijide, A., Joglar, B., Rodriguez-Perez, A. I., Guerra, M. J., and Labandeira-Garcia, J. L. (2012). Involvement of microglial RhoA/Rho-kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol. Dis. 47, 268–279. doi: 10.1016/j.nbd.2012.04.010
Vogel, J., Anand, V. S., Ludwig, B., Nawoschik, S., Dunlop, J., and Braithwaite, S. P. (2009). The JNK pathway amplifies and drives subcellular changes in tau phosphorylation. Neuropharmacology 57, 539–550. doi: 10.1016/j.neuropharm.2009.07.021
Waetzig, V., Zhao, Y., and Herdegen, T. (2006). The bright side of JNKs—Multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog. Neurobiol. 80, 84–97. doi: 10.1016/j.pneurobio.2006.08.002
Wager, T. T., Chandrasekaran, R. Y., Bradley, J., Rubitski, D., Berke, H., Mente, S., et al. (2014). Casein kinase 1δ/ε inhibitor PF-5006739 attenuates opioid drug-seeking behavior. ACS Chem. Neurosci. 5, 1253–1265. doi: 10.1021/cn500201x
Wager, T. T., Galatsis, P., Chandrasekaran, R. Y., Butler, T. W., Li, J., Zhang, L., et al. (2017). Identification and profiling of a selective and brain penetrant radioligand for in vivo target occupancy measurement of casein kinase 1 (CK1) inhibitors. ACS Chem. Neurosci. 8, 1995–2004. doi: 10.1021/acschemneuro.7b00155
Waldner, M., Fantus, D., Solari, M., and Thomson, A. W. (2016). New perspectives on MTOR inhibitors (Rapamycin, Rapalogs and TORKinibs) in transplantation. Br. J. Clin. Pharmacol. 82, 1158–1170. doi: 10.1111/bcp.12893
Walker, D. G., Tang, T. M., and Lue, L.-F. (2017). Studies on colony stimulating factor receptor-1 and ligands colony stimulating factor-1 and Interleukin-34 in Alzheimer’s disease brains and human microglia. Front. Aging Neurosci. 9:244. doi: 10.3389/fnagi.2017.00244
Walter, J. (2000). The phosphorylation of presenilin proteins. Methods Mol. Med. 32, 317–331. doi: 10.1385/1-59259-195-7
Walter, J., Fluhrer, R., Hartung, B., Willem, M., Kaether, C., Capell, A., et al. (2001). Phosphorylation regulates intracellular trafficking of β-Secretase. J. Biol. Chem. 276, 14634–14641. doi: 10.1074/jbc.M011116200
Walters, H. E., and Cox, L. S. (2018). mTORC inhibitors as broad-spectrum therapeutics for age-related diseases. Int. J. Mol. Sci. 19:2325. doi: 10.3390/ijms19082325
Wang, Y., Szretter, K. J., Vermi, W., Gilfillan, S., Rossini, C., Cella, M., et al. (2012). IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 13, 753–760. doi: 10.1038/ni.2360
Wang, Y., Zhang, Y., Wei, Z., Li, H., Zhou, H., Zhang, Z., et al. (2009). JNK inhibitor protects dopaminergic neurons by reducing COX-2 expression in the MPTP mouse model of subacute Parkinson’s disease. J. Neurol. Sci. 285, 172–177. doi: 10.1016/j.jns.2009.06.034
Waschbüsch, D., Michels, H., Strassheim, S., Ossendorf, E., Kessler, D., Gloeckner, C. J., et al. (2014). LRRK2 transport is regulated by its novel interacting partner Rab32. PLoS One 9:e111632. doi: 10.1371/journal.pone.0111632
Watkins, T. A., Wang, B., Huntwork-Rodriguez, S., Yang, J., Jiang, Z., Eastham-Anderson, J., et al. (2013). DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. U.S.A. 110, 4039–4044. doi: 10.1073/pnas.1211074110
Watterson, D. M., Grum-Tokars, V. L., Roy, S. M., Schavocky, J. P., Bradaric, B. D., Bachstetter, A. D., et al. (2013). Development of novel in vivo chemical probes to address CNS protein kinase involvement in synaptic dysfunction. PLoS One 8:e66226. doi: 10.1371/journal.pone.0066226
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.M300227200
Wegner, K. W., Saleh, D., and Degterev, A. (2017). Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol. Sci. 38, 202–225. doi: 10.1016/j.tips.2016.12.005
Wei, S., Nandi, S., Chitu, V., Yeung, Y.-G., Yu, W., Huang, M., et al. (2010). Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J. Leukoc. Biol. 88, 495–505. doi: 10.1189/jlb.1209822
Weisberg, E., Manley, P. W., Breitenstein, W., Brüggen, J., Cowan-Jacob, S. W., Ray, A., et al. (2005). Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7, 129–141. doi: 10.1016/j.ccr.2005.01.007
Weisel, K., Scott, N. E., Tompson, D. J., Votta, B. J., Madhavan, S., Povey, K., et al. (2017). Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 5:e00365. doi: 10.1002/prp2.365
Weiss, B. (2008). ROCO kinase activity is controlled by internal GTPase function. Sci. Signal. 1, e27–e27. doi: 10.1126/scisignal.123pe27
Welsbie, D. S., Mitchell, K. L., Ranganathan, V., Sluch, V. M., Yang, Z., Kim, J., et al. (2017). Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase-dependent injury signaling in neurons. Neuron 94:1142. doi: 10.1016/j.neuron.2017.06.008
Welsbie, D. S., Yang, Z., Ge, Y., Mitchell, K. L., Zhou, X., Martin, S. E., et al. (2013). Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. U.S.A. 110, 4045–4050. doi: 10.1073/pnas.1211284110
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., et al. (2005). Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847. doi: 10.1073/pnas.0507360102
Winslow, A. R., Chen, C.-W., Corrochano, S., Acevedo-Arozena, A., Gordon, D. E., Peden, A. A., et al. (2010). α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 190, 1023–1037. doi: 10.1083/jcb.201003122
Wityak, J., McGee, K. F., Conlon, M. P., Song, R. H., Duffy, B. C., Clayton, B., et al. (2015). Lead optimization toward proof-of-concept tools for Huntington’s disease within a 4-(1H-Pyrazol-4-yl)pyrimidine Class of Pan-JNK Inhibitors. J. Med. Chem. 58, 2967–2987. doi: 10.1021/jm5013598
Wlaschin, J. J., Gluski, J. M., Nguyen, E., Silberberg, H., Thompson, J. H., Chesler, A. T., et al. (2018). Dual leucine zipper kinase is required for mechanical allodynia and microgliosis after nerve injury. eLife 7:e33910. doi: 10.7554/eLife.33910
Wu, J.-R., Wang, J., Zhou, S.-K., Yang, L., Yin, J., Cao, J.-P., et al. (2015). Necrostatin-1 protection of dopaminergic neurons. Neural Regen. Res. 10, 1120–1124. doi: 10.4103/1673-5374.160108
Wu, Z., Li, Y., Cai, Y., Yuan, J., and Yuan, C. (2013). A novel necroptosis inhibitor-necrostatin-21 and its SAR study. Bioorg. Med. Chem. Lett. 23, 4903–4906. doi: 10.1016/j.bmcl.2013.06.073
Wylie, A. A., Schoepfer, J., Jahnke, W., Cowan-Jacob, S. W., Loo, A., Furet, P., et al. (2017). The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737. doi: 10.1038/nature21702
Xie, T., Peng, W., Liu, Y., Yan, C., Maki, J., Degterev, A., et al. (2013). Structural basis of RIP1 inhibition by necrostatins. Struct. Lond. Engl. 21, 493–499. doi: 10.1016/j.str.2013.01.016
Xiong, X., Wang, X., Ewanek, R., Bhat, P., DiAntonio, A., and Collins, C. A. (2010). Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J. Cell Biol. 191, 211–223. doi: 10.1083/jcb.201006039
Xiong, Y.-S., Wang, D.-L., Tan, L., Wang, X., Chen, L.-M., Gong, C.-X., et al. (2013). Inhibition of glycogen synthase Kinase-3 Reverses Tau hyperphosphorylation induced by Pin1 down-regulation. CNS Neurol. Disord. Drug Targets 12, 436–443.
Xu, P., Ianes, C., Gärtner, F., Liu, C., Burster, T., Bakulev, V., et al. (2019). Structure, regulation, and (patho-)physiological functions of the stress-induced protein kinase CK1 delta (CSNK1D). Gene 715:144005. doi: 10.1016/j.gene.2019.144005
Xu, D., Jin, T., Zhu, H., Chen, H., Ofengeim, D., Zou, C., et al. (2018). TBK1 suppresses RIPK1-driven Apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e19. doi: 10.1016/j.cell.2018.07.041
Xu, Q., Malecka, K. L., Fink, L., Jordan, E. J., Duffy, E., Kolander, S., et al. (2015). Identifying three-dimensional structures of autophosphorylation complexes in crystals of protein kinases. Sci. Signal. 8:rs13. doi: 10.1126/scisignal.aaa6711
Xu, Y., Padiath, Q. S., Shapiro, R. E., Jones, C. R., Wu, S. C., Saigoh, N., et al. (2005). Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature 434, 640–644. doi: 10.1038/nature03453
Yamamoto, K., Maruyama, K., Himori, N., Omodaka, K., Yokoyama, Y., Shiga, Y., et al. (2014). The novel Rho kinase (ROCK) inhibitor K-115: a new candidate drug for neuroprotective treatment in glaucoma. Invest. Ophthalmol. Vis. Sci. 55, 7126–7136. doi: 10.1167/iovs.13-13842
Yang, H., Jiang, X., Li, B., Yang, H. J., Miller, M., Yang, A., et al. (2017). Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373. doi: 10.1038/nature25023
Yang, Y., Xu, T., Zhang, Y., and Qin, X. (2017). Molecular basis for the regulation of the circadian clock kinases CK1δ and CK1ε. Cell. Signal. 31, 58–65. doi: 10.1016/j.cellsig.2016.12.010
Yang, H., Rudge, D. G., Koos, J. D., Vaidialingam, B., Yang, H. J., and Pavletich, N. P. (2013). mTOR kinase structure, mechanism and regulation. Nature 497, 217–223. doi: 10.1038/nature12122
Yang, H., Wang, J., Liu, M., Chen, X., Huang, M., Tan, D., et al. (2016). 4.4 Å Resolution Cryo-EM structure of human mTOR Complex 1. Protein Cell 7, 878–887. doi: 10.1007/s13238-016-0346-6
Yarza, R., Vela, S., Solas, M., and Ramirez, M. J. (2016). c-Jun N-terminal Kinase (JNK) signaling as a therapeutic target for Alzheimer’s Disease. Front. Pharmacol. 6:321. doi: 10.3389/fphar.2015.00321
Yasojima, K., Kuret, J., DeMaggio, A. J., McGeer, E., and McGeer, P. L. (2000). Casein Kinase 1 delta MRNA is upregulated in Alzheimer disease brain. Brain Res. 865, 116–120. doi: 10.1016/s0006-8993(00)02200-9
Yin, C., Huang, G., Sun, X., Guo, Z., and Zhang, J. H. (2017). DLK silencing attenuated neuron apoptosis through JIP3/MA2K7/JNK pathway in early brain injury after SAH in rats. Neurobiol. Dis. 103, 133–143. doi: 10.1016/j.nbd.2017.04.006
Yip, C. K., Murata, K., Walz, T., Sabatini, D. M., and Kang, S. A. (2010). Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 38, 768–774. doi: 10.1016/j.molcel.2010.05.017
Yngve, U., Söderman, P., Svensson, M., Rosqvist, S., and Arvidsson, P. I. (2012). Imidazopyridine-based inhibitors of glycogen synthase kinase 3: synthesis and evaluation of amide isostere replacements of the carboxamide scaffold. Chem. Biodivers. 9, 2442–2452. doi: 10.1002/cbdv.201200308
Yoshida, H., Hastie, C. J., McLauchlan, H., Cohen, P., and Goedert, M. (2004). Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J. Neurochem. 90, 352–358. doi: 10.1111/j.1471-4159.2004.02479.x
Yu, W. H., Cuervo, A. M., Kumar, A., Peterhoff, C. M., Schmidt, S. D., Lee, J.-H. et al. (2005). Macroautophagy—a novel β-Amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 171, 87–98. doi: 10.1083/jcb.200505082
Yu, K., Shi, C., Toral-Barza, L., Lucas, J., Shor, B., Kim, J. E., et al. (2010). Beyond Rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of MTORC1 and MTORC2. Cancer Res. 70, 621–631. doi: 10.1158/0008-5472.CAN-09-2340
Yuan, Z. M., Huang, Y., Ishiko, T., Kharbanda, S., Weichselbaum, R., and Kufe, D. (1997). Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 1437–1440.
Yun, H. J., Kim, H., Ga, I., Oh, H., Ho, D. H., Kim, J., et al. (2015). An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J. Biochem. 157, 485–495. doi: 10.1093/jb/mvv005
Zhan, X., Kook, S., Kaoud, T. S., Dalby, K. N., Gurevich, E. V., and Gurevich, V. V. (2015). Arrestin-3-dependent activation of c-Jun N-terminal kinases (JNKs). Curr. Protoc. Pharmacol 68, 2.12.1–2.12.26. doi: 10.1002/0471141755.ph0212s68
Zhan, X., Stoy, H., Kaoud, T. S., Perry, N. A., Chen, Q., Perez, A., et al. (2016). Peptide mini-scaffold facilitates JNK3 activation in cells. Sci. Rep. 6:21025. doi: 10.1038/srep21025
Zhang, H., Li, Y., Yu, J., Guo, M., Meng, J., Liu, C., et al. (2013). Rho kinase inhibitor fasudil regulates microglia polarization and function. Neuroimmunomodulation 20, 313–322. doi: 10.1159/000351221
Zhang, X., Li, L., Chen, S., Yang, D., Wang, Y., Zhang, X., et al. (2011). Rapamycin treatment augments motor neuron degeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Autophagy 7, 412–425. doi: 10.4161/auto.7.4.14541
Zhao, J., Molitor, T. P., Langston, J. W., and Nichols, R. J. (2015). LRRK2 dephosphorylation increases its ubiquitination. Biochem. J. 469, 107–120. doi: 10.1042/BJ20141305
Zhao, Y., and Dzamko, N. (2019). Recent developments in LRRK2-targeted therapy for Parkinson’s Disease. Drugs 79, 1037–1051. doi: 10.1007/s40265-019-01139-4
Zheng, S., Clabough, E. B. D., Sarkar, S., Futter, M., Rubinsztein, D. C., and Zeitlin, S. O. (2010). Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet. 6:e1000838. doi: 10.1371/journal.pgen.1000838
Zhong, L., Xu, Y., Zhuo, R., Wang, T., Wang, K., Huang, R., et al. (2019). Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 10:1365. doi: 10.1038/s41467-019-09118-9
Zhou, Q., Wang, M., Du, Y., Zhang, W., Bai, M., Zhang, Z., et al. (2015). Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 77, 637–654. doi: 10.1002/ana.24361
Zhou, Y., Su, Y., Li, B., Liu, F., Ryder, J. W., Wu, X., et al. (2003). Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Abeta42 by inhibiting Rho. Science 302, 1215–1217. doi: 10.1126/science.1090154
Zhou, Z., Bachstetter, A. D., Späni, C. B., Roy, S. M., Watterson, D. M., and Van Eldik, L. J. (2017). Retention of normal glia function by an isoform-selective protein kinase inhibitor drug candidate that modulates cytokine production and cognitive outcomes. J. Neuroinflamm. 14:75. doi: 10.1186/s12974-017-0845-2
Zhu, S., Zhang, Y., Bai, G., and Li, H. (2011). Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Dis. 2:e115. doi: 10.1038/cddis.2010.94
Keywords: inhibitor, central nervous system, ATP, small molecule, clinical development, blood brain barrier
Citation: Benn CL and Dawson LA (2020) Clinically Precedented Protein Kinases: Rationale for Their Use in Neurodegenerative Disease. Front. Aging Neurosci. 12:242. doi: 10.3389/fnagi.2020.00242
Received: 21 May 2020; Accepted: 13 July 2020;
Published: 02 September 2020.
Edited by:
Rubem C. A. Guedes, Federal University of Pernambuco, BrazilReviewed by:
Bingmei Fu, The City College of New York (CUNY), United StatesElisa Mitiko Kawamoto, University of São Paulo, Brazil
Copyright © 2020 Benn and Dawson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline L. Benn, Y2Jlbm5AbG9xdXMyMy5jb20=