 Toshiki Mizuno
Toshiki Mizuno Ikuko Mizuta
Ikuko Mizuta Takashi Koizumi
Takashi Koizumi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 07 May 2020
Sec. Neuroinflammation and Neuropathy
Volume 12 - 2020 | https://doi.org/10.3389/fnagi.2020.00091
This article is part of the Research TopicIntramural Vascular Cells: Key Therapeutic Targets for Vascular Cognitive ImpairmentView all 15 articles
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a hereditary cerebral small vessel disease caused by mutations in NOTCH3, is characterized by recurrent stroke without vascular risk factors, mood disturbances, and dementia. MRI imaging shows cerebral white matter (WM) hyperintensity, particularly in the external capsule and temporal pole. Missense mutations related to a cysteine residue in the 34 EGFr on the NOTCH3 extracellular domain (N3ECD) are a typical mutation of CADASIL. On the other hand, atypical mutations including cysteine sparing mutation, null mutation, homozygous mutation, and other associate genes are also reported. From the viewpoint of gain of function apart from Notch signaling or loss of function of Notch signaling, we review the research article about CADASIL and summarized the pathogenesis of small vessel, stroke, and dementia in this disease.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is one of the most common hereditary cerebral small vessel diseases caused by mutations in NOTCH3. C was first recognized as a clinical and genetic hereditary small-vessel disease entity that induces cerebral infarction, white matter (WM) disease, microbleeds, and finally, vascular dementia. Understanding the impact of this disease is important for analyzing small-vessel diseases as well as vascular dementia. This review article is summarized in terms of whether CADASIL is caused by the gain of NOTCH3 function or by loss of NOTCH signaling function. Supporting data for a gain of function is pro-aggregatory property of cysteine related or cysteine-sparing NOTCH3 mutations as well as homozygous NOTCH3 mutation. Also, pathological findings of the accumulation of extracellular matrix and granular osmiophilic material (GOM) supported the hypothesis that Notch extracellular domain (NECD) can be a core of aggregation. Looking from the other side, nonsense NOTCH3 mutation or constitutive activation of NOTCH3 signaling can be related to the pathogenesis of different cerebral small vessel disease from CADASIL (Figure 1).
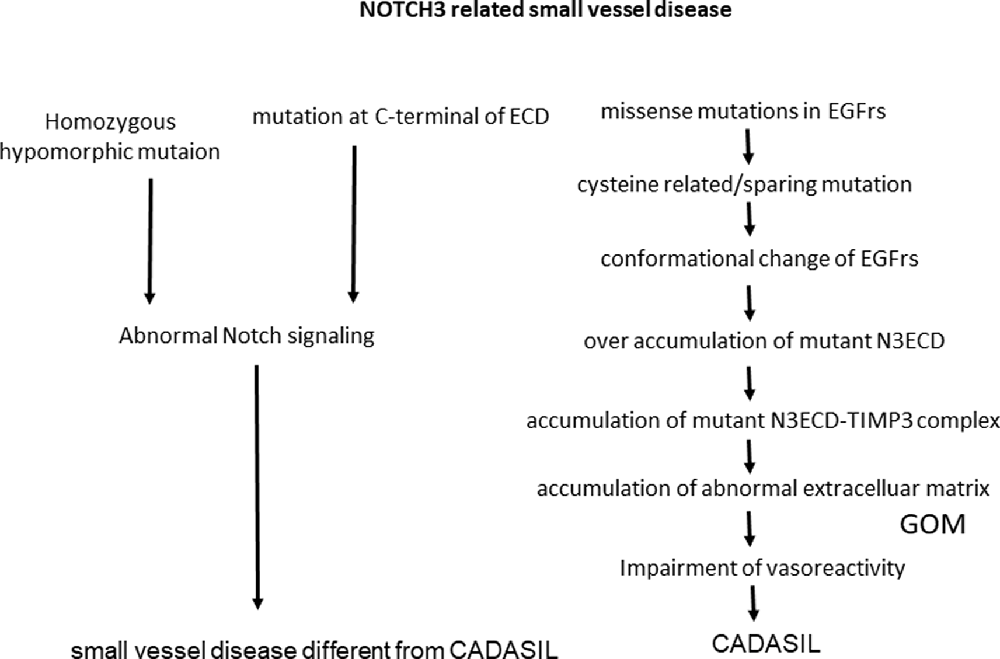
Figure 1. NOTCH3 is related to small vessel disease. ECD, extracellular domain; N3ECD, Notch3 extracellular domain; EGFrs, Epidermal Growth Factor repeats; GOM, granular osmiophilic material; TIMP3, tissue inhibitor of metalloproteinases 3.
Notch signaling is evolutionarily conserved, and four members of the Notch receptor family: NOTCH1-4 exist in humans. NOTCH3 encodes a single-pass transmembrane receptor, NOTCH3, which consists of a NECD including 34 epidermal growth factor-like repeats (EGFrs) and an Notch intracellular domain (NICD). The Notch protein is thought to undergo complex proteolytic processing events. The first cleavage, named as S1 cleavage of the receptor occurs on the Golgi for the maturation of the receptor (i.e., the formation of the NECD-NICD heterodimer). The second proteolysis, named as S2 cleavage by A disintegrin and metalloprotease 10 (ADAM10), occurs on the cell surface when the receptor interacts with NOTCH ligand, Jagged or Delta/Serrate/LAG-2 (DSL), on neighboring cells (Brou et al., 2000). The third cleavage, named as S3 cleavage: γ-secretase–dependent intra-membrane proteolysis, NICD is moved into the cytoplasm and shuttles to the nucleus. Finally, NICD activated target genes with transcriptional cofactors of the CBF1-Su(H)-Lag1 (CSL) family (Bray, 2006). While NOTCH3 mainly expressed in the central nervous system in the fetus, NOTCH3 is expressed predominantly in vascular smooth muscle cells (VSMCs) to maintain vascular contractility in adults (Joutel et al., 2000).
To date, more than 200 cysteine-related mutations, most of which are single nucleotide changes, have been reported (Supplementary Table S1, Joutel et al., 1997; Rutten et al., 2014; Koizumi et al., 2019; Leiden Open Variation Database, or references therein). A few exceptions include in-frame insertion/deletion mutations and splicing-site mutations (Tikka et al., 2009). Also, a rare mutation of in-frame 15 bp duplication in exon 7 is reported (Lee et al., 2011). CADASIL-associated mutations are localized from exon2 to 24, which encode EGFrs. Each EGFr contains six cysteine residues that likely participate in forming three pairs of disulfide bonds to maintain the normal NOTCH3 protein conformation. Most of the mutations are of the missense type, resulting in an even number to an odd number of cysteine residues (Joutel et al., 1997; Mizuta et al., 2017). The resulting unpaired cysteine is predicted to cause abnormal disulfide bridge formation that leads to aggregation of NECD (Duering et al., 2011).
The mutations accumulate in EGFrs 1–6, apart from the ligand-binding domain, EGFrs 10 and 11. Cellular experiments showed that most of the mutations do not affect Notch signaling, suggesting that CADASIL is not caused by signaling dysfunction. However, NOTCH3 harboring a p.Cys428Ser mutation in EGFr 10 and p.Cys455Arg in EGFr 11 exhibits attenuated ligand-binding activity, resulting in a significant reduction of NOTCH3 signaling (Joutel et al., 2004; Peters et al., 2004b). A previous report showed that mutations in EGFrs 10 and 11 are associated with milder cognitive deficits and a trend toward a lower volume of lacunar infarcts compared with the common mutations in EGFrs 2–5 (Monet-Leprêtre et al., 2009). This association remains to be elucidated, but it is possible that Notch signaling may affect the clinical symptoms.
Cases with duplication (Lee et al., 2011) or deletion of NOTCH3 (Dichgans et al., 2000) are reported. These mutations also change the number of Cysteine residue as well as point mutation and GOM was detected in the cases with these mutations (Lee et al., 2011).
A popular hypothesis holds that the NOTCH3 mutations causing CADASIL are gain-of-function rather than loss-of-function mutations (Carare et al., 2013). One of the evidence supporting this hypothesis was the identification of hypomorphic mutations in individuals without the CADASIL phenotype. Rutten et al. (2013) reported two NOTCH3 nonsense mutations, c.307C>T, p.Arg103*, in two brothers aged in their 50s; brain MRI and skin biopsy results showed incompatible with CADASIL. Also, they reported a CADASIL patient with compound heterozygous for a pathogenic NOTCH3 mutation, p.Tyr710Cys, and an intragenic frameshift deletion. In that patient’s family, p.Tyr710Cys segregated with the affected parent, whereas the intragenic frameshift deletion was also identified in the normal parent of the patient. They concluded that these hypomorphic NOTCH3 alleles do not cause CADASIL (Rutten et al., 2013). According to previous case reports, complete loss of, and also constitutive activation of NOTCH3 signaling are thought to cause arteriopathy. Pippucci et al. (2015) reported a 24 years-old man with childhood-onset arteriopathy and cavitating leukoencephalopathy. Exome analysis of the patient and his consanguineous parents identified homozygous NOTCH3 null mutation c.C2898A (p.C966*) in the patient. Fouillade et al. (2008) reported a 53-year-old woman with 35-years-onset stroke and MRI finding of WM hyperintensity. They identified NOTCH3 c.4544T>C resulting in p.L1515P mutation which localizes in the C-terminal end of NOTCH3 extracellular domain (N3ECD). Although the precise mechanism remains unknown, cellular experiments suggested increased NOTCH3 signaling of this mutation in a ligand-independent manner (Pippucci et al., 2015). It is of note that the pathological hallmark of CADASIL, GOM, was not detected in either of these patients (Fouillade et al., 2008; Pippucci et al., 2015), suggesting that arteriopathy related to abnormal NOTCH3 signaling is different from CADASIL. In a study using a transgenic mouse model of CADASIL, Joutel described the toxic gain-of-function properties of mutant NOTCH3. Mice harboring human NOTCH3 with p.Arg90Cys, p.Cys428Ser, or p.Arg169Cys mutations exhibited CADASIL-like pathologic changes, N3ECD accumulation, and deposition of GOM (see below). Notably, transgenic mice with p.Cys428Ser, which cannot mediate Notch signaling (see above), also showed GOM deposition, suggestive of toxic gain-of-function mutations leading to aggregation of mutated N3ECD (Joutel, 2011).
Muino et al. (2017) recently reviewed cysteine-sparing NOTCH3 missense mutations and proposed criteria for their pathogenicity: (a) typical clinical CADASIL syndrome; (b) diffuse white matter hyperintensity (WMH); (c) 33 NOTCH3 exons analyzed; (d) mutations that were not polymorphisms; and (e) GOM deposits noted in skin biopsy. Of the 25 mutations reviewed, they concluded that p.Arg61Trp, p.Arg75Pro, p.Asp80Gly, and p.Arg213Lys fulfilled these criteria. To uncover the pathogenicity of cysteine-sparing mutations, analysis of pro-aggregatory property may be important. Some cysteine-involving mutations (Duering et al., 2011) and also p.Arg75Pro and p.Asp80Gly (Wollenweber et al., 2015) was reported to be prone to aggregation by using a single-particle aggregation assay. In the very recent report of the patient with p.Gly73Ala, unfortunately, skin biopsy was declined, cellular and in vitro experiments showed pro-aggregatory property of the mutation (Huang et al., 2020).
Of the pathogenic cysteine-sparing mutations, p.Arg75Pro is probably the most frequent, and this mutation is primarily reported in eastern Asians (Kim et al., 2006; Mizuno et al., 2008; Ueda et al., 2015). In addition to positive skin biopsy findings, we also noted co-segregation in a p.Arg75Pro family and demonstrated the predicted conformational change in the EGFr harboring this mutation (Mizuno et al., 2008). Japanese CADASIL patients with p.Arg75Cys exhibit atypical and mild phenotypes, including a lower frequency of stroke/TIA and temporal pole lesions, with a tendency toward an older age at onset (Ueda et al., 2015; Koizumi et al., 2019).
Although CADASIL is generally caused by heterozygous mutations in NOTCH3, several CADASIL cases involving homozygous mutations have been reported (Mukai et al., 2018). Interestingly, some cases involving homozygous mutations showed a more-severe clinical phenotype than cases involving heterozygous mutations, but other cases were within the spectrum of the heterozygous phenotype.
The most frequent homozygous mutation is p.Arg544Cys. Mukai et al. (2018) reported a 63-year-old male case presenting first stroke attack with only mild weakness of the left leg and recovered well, involving a homozygous p.Arg544Cys mutation; GOM around the basement membrane of VSMCs on skin biopsy were detected in this case. No other stroke patients were detected in his parents and siblings indicated the tendency toward a mild phenotype with the p.Arg544Cys mutation in agreement with previous reports (Liao et al., 2015; Lee et al., 2016). p.Arg544Cys locates not in EGFr, but between the 13th and 14th EGFrs. Therefore, we hypothesize that p.Arg544Cys may contribute to a milder effect to a conformational change of EGFr resulting in a milder phenotype. The p.Arg544Cys mutation is also notable because of its geographic accumulation. The frequency of this mutation in CADASIL patients is 93.6% in Jeju Island, Korea, and 70.5% in Taiwan (Liao et al., 2015; Lee et al., 2016).
Opherk et al. (2014) performed a genome-wide association study to identify genetic modifiers of WMH volume in CADASIL. They analyzed SNP array data for 466 patients and found no SNPs reaching genome-wide significance. However, polygenic score analyses which included SNPs with weak p-values, indicated significant association with WMH volume when 10,574 SNPs (each p-value < 0.1) or 52,125 SNPs (each p-value < 0.5) were included. They suggested that multiple variants exert small effects on the WMH burden in CADASIL.
We recently reported ring finger protein 213 (RNF213)-related susceptibility to intracranial arterial stenosis (ICAS) in CADASIL patients (Yeung et al., 2018). The frequency of RNF213 variants was 23.5% in CADASIL patients with ICAS, compared with 1.9% in those without ICAS. CADASIL is recognized as a small-vessel disease, but intracranial major artery stenosis was reported in some CADASIL patients (Choi et al., 2005). The susceptibility variant rs112735431, c.14576G>A (p.R4859K) or c.14429G>A (p.R4110K) in RNF213, was originally identified as prominently associated with moyamoya disease, mainly reported in eastern Asia. Miyawaki et al. (2012) found the variant was linked to susceptibility to ICAS even in sporadic cases. These associated genes may contribute to the clinical phenotypes of CADASIL.
CADASIL affects small vessels in the brain WM and deep gray matter, resulting in thickening of vascular walls and luminal stenosis. In the tunica media, degeneration of VSMCs, positive PAS staining, and granular deposits of N3ECD immunoreactivity are observed (Baudrimont et al., 1993; Joutel et al., 2000). In the tunica adventitia, accumulation of various fibrous extracellular matrices is observed, including collagen, laminin, and clusterin. However, vessel occlusion or thrombosis is rarely found (Ruchoux et al., 1995). Therefore, the direct pathogenic mechanism leading to lacunar infarction in CADASIL remains to be elucidated. A popular hypothetical pathomechanism involves hemodynamic disturbance of lesion-affected arterioles and loss of compliance and autoregulation (Tikka et al., 2014).
The first ultrastructural descriptions of perivascular deposits surrounding small, penetrating arteries in the brain, designated GOM, were reported by Baudrimont et al. (1993). This material has been examined mainly in the brain, but GOM is also found surrounding VSMCs in other tissues, including muscle and skin (Ruchoux et al., 1995). Tikka et al. (2009) investigated the GOM in CADASIL patients and concluded that COM is specific to CADASIL. Immunohistochemistry and immunogold electron microscopy studies revealed the distribution of GOM and N3ECD protein in the microvasculature of brain gray matter and WM. Immunogold electron microscopy using an antibody to N3ECD revealed abundant particles in the GOM within microvessels, VSMC membranes, and perivascular cells (Yamamoto et al., 2013). These results suggest that NOTCH3 fragments are major components of GOM deposits.
Cisendocytosis of either the DSL ligands or the Notch receptor itself into the cytoplasm has been recognized as playing an important role in regulating Notch signaling (Bray, 2006; Fortini and Bilder, 2009; Pratt et al., 2011). Also, genetic and cellular biological studies have shown that Notch is endocytosed into neighboring ligand-expressing cells in Drosophila (Klueg and Muskavitch, 1999) and that endocytosis of NECD promotes Notch proteolysis and downstream signaling in mammals (Nichols et al., 2007). Therefore, endocytosis of NECD into ligand-expressing cells, known as trans-endocytosis, is believed to be more critical for Notch activation than proteolytic events (Nichols et al., 2007). Because CADASIL causing mutations localize in N3ECD, we hypothesized that impairment of N3ECD trans-endocytosis may be a pathological mechanism of CADASIL. We addressed this issue by using HEK293 cells harboring a single copy of mutant or wildtype human NOTCH3 cDNA cocultured with Jagged1-expressing cells (Watanabe-Hosomi et al., 2012). In this co-culture system, Notch signaling quantified by HES1 expression was similar between mutant and wildtype N3ECD, in agreement with previous reports. However, we found that C185R mutant N3ECD on the cell surface is degraded significantly more slowly than wild-type N3ECD in NOTCH3 cells. While vesicles containing N3ECD were observed in Jag1-expressing cells co-cultured with wild-type NOTCH3, vesicles with mutant N3ECD within the Jag1-expressing cells were significantly fewer in number. These results indicate that the process of degradation of mutant N3ECD on the cell surface is disturbed due to the impairment of trans-endocytosis (Watanabe-Hosomi et al., 2012). It can also explain the abnormal accumulation of N3ECD in vascular walls without accumulation of the intracellular domain of Notch3 intracellular domain (N3ICD) or full-length protein, and signaling is activated normally. Further approach is necessary to uncover the process between impaired trans-endocytosis and abnormal accumulation of N3ECD and GOM deposit.
Using cultured cells, Monet-Leprêtre et al. (2013) provided evidence that excess levels of or multimerization of mutant Notch3 ECD facilitate interactions with key components of the vascular extracellular matrix, including tissue inhibitor of metalloproteinases 3 (TIMP3) and VTN. Brain vessels from transgenic mice and patients with CADASIL exhibit elevated levels of both insoluble cross-linked and soluble TIMP3 species (Monet-Leprêtre et al., 2013). Later, Capone et al. (2016) showed that reducing TIMP3 or VTN ameliorated CADASIL phenotype using transgenic mice, suggesting TIMP3 or VTN may be a novel therapeutic target of CADASIL.
It is well known that transforming growth factor-β (TGF-β) signaling is important in the regulation of fibrotic events in vessels and other various tissues. Increased TGF-β signaling was reported as the pathophysiology of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL; Hara et al., 2009), suggesting TGF-β signaling as a key pathway to cerebral small vessel diseases. Kast et al. (2014) examined molecules involving in the regulation of TGF-β bioavailability, fibronectin, fibrillin-1, and latent TGF-β–binding protein 1 (LTBP-1), in post-mortem brain tissue from CADASIL patients. All the three molecules were enriched in the CADASIL vessel. However, fibronectin and fibrillin-1 did not colocalize with N3ECD deposits, whereas, LTBP-1 showed a striking co-localization with N3ECD deposits, suggesting specific recruitment of LTBP-1 into aggregates. Also, increased levels of the TGF-β prodomain indicate dysregulation of the TGF-β pathway in CADASIL development. In vitro co-aggregation assay showed a direct interaction between LTBP-1 and mutant N3-ECD but no interaction between LTBP-1 and wildtype N3-ECD. These suggested a specific co-aggregation of LTBP-1 with mutant NOTCH3 and possible TGF-β signaling impairment in CADASIL (Kast et al., 2014).
To examine the degree and extent of the pathologic changes, Okeda et al. (2002) analyzed the entire length of vessels by reconstructing 1,000 serial sections of the 11 cerebral medullary arteries in an autopsy of a CADASIL patient who was 75 years old. The predominant findings were loss of VSMCs in the tunica media and fibrosis in the tunica adventitia. Most arteries exhibited continuous complete loss of VSMCs in the WM. Severe adventitial fibrosis was found in all arteries but restricted to WM. However, no stenosis or occlusion was found in the arteries studied. Considering their results collectively, they used a “so-called earthen pipe state” to describe the state of lesioned arteries and proposed failure of autoregulation of cerebral blood flow due to the earthen pipe state as underlying the pathogenesis of CADASIL (Okeda et al., 2002).
As the receptor protein encoded by the NOTCH3 gene is expressed not only on VSMCs but also on pericytes, pericytes and capillary vessels can be damaged by CADASIL (Dziewulska and Lewandowska, 2012). Degeneration and loss of pericytes in capillary vessels were detected in the microvessels in the autopsy of the brain and skin-muscle biopsy in CADASIL patients (Dziewulska and Lewandowska, 2012). GOM was usually seen near pericyte cell membranes or within infoldings (Dziewulska and Lewandowska, 2012). These findings suggested increased permeability of the capillary vessels and disturbances in cerebral microcirculation; this degeneration can also cause defective vasomotor reactivity in CADASIL (Okeda et al., 2002; Qin et al., 2019).
Because NOTCH3 is expressed ubiquitously in VSMCs, it is not surprising that pathological changes including GOM can be detected not only in the brain but also in other organs. It is reported that peripheral vascular function, as well as in the brain, was also impaired in patients with CADASIL and also a transgenic mice model of CADASIL (Fujiwara et al., 2012).
Clear phenotypes are restricted to the brain, but several studies suggested cardiovascular (van den Boom et al., 2003; Rufa et al., 2007) and renal (Kusaba et al., 2007; Guerrot et al., 2008; Ragno et al., 2012) involvement, though their causal relationship is still unclear. A recent article by Kelleher et al. (2019) showed that CADASIL iPSC-derived mural cells, including VSMCs and pericytes were susceptible to apoptotic stress. This may explain why major symptoms are restricted to the brain, because the blood-brain barrier, the crucial structure to maintain brain homeostasis, consists of astrocytes and pericytes (Ihara and Yamamoto, 2016).
To date, thousands of families with CADASIL have been diagnosed worldwide in many different ethnic groups. The disorder is often overlooked and misdiagnosed. Its minimum prevalence has been estimated at between 2 and 5 in 100,000 but may vary between populations (Razvi et al., 2005; Narayan et al., 2012; Moreton et al., 2014; Bianchi et al., 2015).
There have been a limited number of studies involving genotype-phenotype analyses, some of which reported negative results (Adib-Samii et al., 2010). On the other hand, a German study including 371 CADASIL patients showed positive findings in genotype-phenotype analyses of eight of the most frequent genotypes: p.Arg90Cys, p.Cys117Phe, p.Arg133Cys, p.Arg14.
1Cys, p.Arg153Cys, p.Arg169Cys, p.Cys174Tyr, and p.Arg182Cys (Opherk et al., 2004). Both p.Cys174Tyr and p.Cys117Phe was significantly associated with a lower median age at death, and p.Cys117Phe alone was significantly associated with a lower median age at onset of stroke and immobilization (Opherk et al., 2004). Genotype-phenotype correlation analyses of each mutation should be carefully conducted due to the wide phenotype distribution, even in the same family harboring the same mutation.
Rutten et al. (2019) recently demonstrated the effect of mutation location on the severity of the disease. By comparing CADASIL patients with mutations in EGFr 1–6 and EGFr 7–34, those in the EGFr 1–6 group had a 12-year earlier onset of stroke, lower survival, and higher WMH volume than those in the EGFr 7–34 group. As described above, NOTCH3 mutations in CADASIL patients accumulate in EGFr 1–6, at 71.8% in Europeans (Rutten et al., 2019). It is noteworthy that NOTCH3 pathogenic variants were identified in a recent analysis of a large-scale genome variation database of a general population (Rutten et al., 2016). NOTCH3 pathogenic variants in a general population accumulated in EGFr 7–34, at 97.5% in the general European population, based on the Genome Aggregation Database (Rutten et al., 2016). Taken together, the results of Rutten et al. (2019) suggest a predisposition toward EGFr 1–6 in the classical, more-severe CADASIL phenotype. They also suggest a broad disease spectrum involving EGFr 7–34, from a mild phenotype to possible non-penetrance, and these results highlight the significant role of NOTCH3 pathogenic variants in general populations.
The difference between clinical symptoms and course in the same family harboring the same NOTCH3 mutation indicates the importance of environmental factors. Of the two twin studies of CADASIL, the first reported one found apparent different phenotypes, including 14 years difference of the age at onset between the twins, suggesting an effect of environmental factors on the disease (Mykkänen et al., 2009). The second reported one found similar phenotypes, including the age at onset of 74–75 years old and parkinsonism in the twin (Ragno et al., 2016). A similar phenotype indicates similar lifestyle-related factors, and Ragno’s cases did not contradict the possible effect of environmental factors on the CADASIL phenotype (Ragno et al., 2016). Conventional vascular risk factors may influence the severity of the disease. Adib-Samii et al. (2010) analyzed 200 symptomatic CADASIL patients in the UK; they found that hypertension (odds ratio 2.57) and pack-years of smoking (odds ratio 1.07) were associated with an increased risk of stroke. Ciolli also assessed the influence of vascular risk factors and revealed that hypertension was related to both disability assessment for dementia (DAD) score and disability (Ciolli et al., 2014). In our cohort, 62.9% of CADASIL patients had vascular risk factors, including hypertension, diabetes mellitus, hyperlipidemia, smoking, or alcohol consumption, and they were more prone to stroke (Mizuta et al., 2017). Management of these factors are essential in CADASIL as well as sporadic cases.
Original criteria for CADASIL diagnosis were proposed by Davous in 1997 (Davous, 1998). This precious work contributed to the core concept of CADASIL when CADASIL was not well recognized by physicians yet. However, genetic tests of NOTCH3 revealed atypical cases, and the clinical phenotype of CADASIL was diverse, ranging from asymptomatic to severe. The Davous’s criteria cannot be applied to atypical CADASIL cases because the criteria are strict and have low sensitivity. In particular, cases involving elderly onset, no family history, or positive cardiovascular risk factors might be overlooked using these criteria. To avoid missing suspected CADASIL patients before genetic testing, we proposed more-sensitive criteria from Japanese CADASIL cases (Mizuta et al., 2017; Supplementary Table S2). The sensitivity of our new criteria is 97%, sufficient to screen candidates for CADASIL and to aid genetic testing (Mizuta et al., 2017).
To prioritize access to genetic testing for suspected CADASIL patients, a quantitative evaluation of CADASIL-specific features possessed by each patient is necessary. Markus proposed a diagnostic procedure based on skin biopsy and involvement of the anterior temporal lobe on MRI (Markus et al., 2002). As pre-genetic screening approaches are desirable, Pescini et al. (2012) developed the CADASIL scale, a screening tool applied in the clinical setting due to the high cost and time-consuming nature of genetic testing. The weighted scores to common disease features based on frequencies obtained in a pooled analysis of selected international CADASIL series. The cut-off score of the definitive CADASIL scale had a sensitivity of 96.7% and specificity of 74.2%. Unfortunately, the sensitivity of the CADASIL scale was 52.1% in our Japanese cohort because of some clinical differences about the CADASIL group as well as NOTCH3-negative CADASIL-like patients (non-NOTCH3) group between the European population and the Japanese population (Koizumi et al., 2019). Several studies revealed a low prevalence rate of migraine in Japanese CADASIL compared to European CADASIL patients (Uchino et al., 2002). Although WM lesion at the temporal pole is specific CADASIL patients in both cohort, WM lesion at the external capsule was different in the non-NOTCH3 group in each cohort. These differences decreased the sensitivity and specificity of the original CADASIL scale when applied to the Japanese cohort. Therefore, we modified the CADASIL scale based on the clinical features of 126 Japanese CADASIL patients and 53 Non-NOTCH3 to develop CADASIL scale-J (Supplementary Table S3; Koizumi et al., 2019). In CADASIL scale-J, the score ranged from 0 to 25 and a cut-off value of 16, using eight items. The sensitivity and specificity of the CADASIL scale-J were enough quality for prioritizing tool before genetic testing.
MRI can visualize the characterization of WM lesions and lacunar infarction in stroke syndromes. In CADASIL, T2-weighted hyperintensity was noted in the deep WM, internal and external capsules, and the temporal pole. Temporal pole hyperintensity on T2-weighted and FLAIR MR sequences can be detected even in the early 20s of CADASIL patients (Chabriat et al., 1998; Mizuno, 2012). Dilated perivascular spaces (PVS) in CADASIL patients are located in the lentiform nuclei (94%) and subcortical WM of the temporal lobes (66%; Chabriat et al., 1998). PVS around small perforating arteries are pial-lined, interstitial fluid-filled spaces, readily seen to be enlarged in the WM of elderly subjects. Yamamoto et al. (2009) performed a postmortem study to quantify the degree and extent of PVS and arteriopathic changes within the temporal pole WM of CADASIL subjects. They concluded that the MRI hyperintensity in the temporal pole of CADASIL patients could be explained by enlarged PVS and degeneration of myelin rather than lacunar infarcts.
Viswanathan analyzed clinical data from 147 consecutive patients and revealed a significant independent association between age, volume of lacunar lesions, and global cognitive function scales, although WMH and microbleeds had no independent influence on cognitive function. Disability was associated with the volume of lacunar lesions, microbleeds, systolic blood pressure, and age but not with WMH (Viswanathan et al., 2007). Liem et al. (2007) analyzed 62 symptomatic and 15 asymptomatic members of CADASIL and revealed that the severity of cognitive dysfunction in mutation carriers is independently associated with MRI infarct lesion load. In contrast, WMH lesion load and microbleeds were not associated with cognitive dysfunction after correcting for age. These data indicate the importance of lacunar infarction in the progression of CADASIL.
Viswanathan examined the relative impact of lesion burden and location of these MRI markers on cognitive impairment and disability combined with whole-brain mean apparent diffusion coefficient (mean-ADC) and brain parenchymal fraction (BPF). In multivariate models accounting for lesion burden and location, the volume of lacunar lesion, mean-ADC, and BPF each had an independent influence on global cognitive function and disability (Viswanathan et al., 2010). Particularly, brain atrophy was shown to have the strongest independent influence on clinical impairment in CADASIL when all MRI markers in the disease are considered together. These results suggest that the clinical impact of cerebral cortical loss is important for CADASIL dementia (Viswanathan et al., 2010).
Jouvent et al. (2011b) used high-resolution postmortem 7-T MRI to examine infarcts of the cerebral cortex in a CADASIL patient with pathology examination. These lesions were not visible on the in vivo MRI obtained at 1.5 T. They examined cortex morphology and clinical worsening in 190 CADASIL patients and showed that reduction of sulcal depth is independently associated with increased time to complete trail making test A and B and that of cortical thickness to increased disability. They also showed that the impact of volume of lacunar lesions on cortical changes is greater than that of the volume of WMH and that cortical changes related to lacunar lesions evolve parallel to clinical worsening. These results support the hypothesis that cortical changes in CADASIL play a role in disease pathophysiology (Jouvent et al., 2011a).
Few prospective studies have examined the natural history of CADASIL. Davous summarized the natural history of 134 documented cases and showed a mean age at onset of 40.3 years. The mean duration of the disease was 13.6 years, and the mean age at death was 56.7 years (Davous, 1999). Opherk summarized the natural history of a larger CADASIL cohort and reported a median age at onset of stroke of 50.7 years in men and 52.5 years in women, the median age at death of 64.6 years in men and 70.7 years in women (Opherk et al., 2004). In our cohort including 200 CADASIL patients, the mean age at onset of stroke was 48.3 years in men and 52.2 years in women (Koizumi et al., 2019). These results indicated a similar clinical course in any ethics, but more studies need to clarify a difference in each ethics before starting a disease-modifying-therapy on CADASIL.
Peters et al. (2004a) reported that their cohort deteriorated for all clinical scales over 2 years. There were 18 strokes within 173 person-years, giving an average incidence rate of stroke of 10.4 per 100 person-years. Age at baseline was found to be a predictor of clinical progression (Peters et al., 2004a). These data indicate a younger age of onset of stroke and death compared with the general population. CADASIL researchers should thus develop new approaches to improve this natural history.
TM and IM conceived the original idea. TM wrote the manuscript with support from IM, AW-H, MM, and TK.
Grant-in-aid for Research on Intractable Disease from the Japanese Ministry of Health, Labour, and Welfare H28-Nanchitou(Nan)-Ippan-029, H30-Nanchitou(Nan)-Ippan-006.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ADAM10, A disintegrin and metalloprotease 10; ADC, apparent diffusion coefficient; BPF, brain parenchymal fraction; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; CM, cerebral microbleeds; CSL, CBF1-Su(H)-Lag1; DAD, Disability Assessment for Dementia; DSL, Delta/Serrate/LAG-2; ExAC, exome aggregation consortium; EGFrs, epidermal growth factor-like repeats; ICAS, intracranial arterial stenosis; Jag1, Jagged1; LTBP-1, latent TGF-β–binding protein 1; mean-ADC, mean apparent diffusion coefficient; NECD, Notch extracellular domain; NICD, Notch intracellular domain; N3ECD, Notch3 extracellular domain; N3ICD, Notch3 intracellular domain; Non-NOTCH3, NOTCH3-negative CADASIL–like patients; GOM, granular osmiophilic material; PVS, perivascular spaces; RNF213, ring finger protein 213; TGF-β, transforming growth factor-β; TIMP3, tissue inhibitor of metalloproteinases 3; VSMC, vascular smooth muscle cell; VTN, vitronectin; WM, white matter; WMH, white matter hyperintensity; WML, white matter lesion.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2020.00091/full#supplementary-material.
Adib-Samii, P., Brice, G., Martin, R. J., and Markus, H. S. (2010). Clinical spectrum of CADASIL and the effect of cardiovascular risk factors on phenotype: study in 200 consecutively recruited individuals. Stroke 41, 630–634. doi: 10.1161/strokeaha.109.568402
Baudrimont, M., Dubas, F., Joutel, A., Tournier-Lasserve, E., and Bousser, M. G. (1993). Autosomal dominant leukoencephalopathy and subcortical ischemic stroke. A clinicopathological study. Stroke 24, 122–125. doi: 10.1161/01.str.24.1.122
Bianchi, S., Zicari, E., Carluccio, A., Di Donato, I., Pescini, F., Nannucci, S., et al. (2015). CADASIL in central Italy: a retrospective clinical and genetic study in 229 patients. J. Neurol. 1, 134–141. doi: 10.1007/s00415-014-7533-2
Bray, S. J. (2006). Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689. doi: 10.1038/nrm2009
Brou, C., Logeat, F., Gupta, N., Bessia, C., LeBail, O., Doedens, J. R., et al. (2000). A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol. Cell 2, 207–216. doi: 10.1016/s1097-2765(00)80417-7
Capone, C., Cognat, E., Ghezali, L., Baron-Menguy, C., Aubin, D., Mesnard, L., et al. (2016). Reducing Timp3 or vitronectin ameliorates disease manifestations in CADASIL mice. Ann. Neurol. 3, 387–403. doi: 10.1002/ana.24573
Carare, R. O., Hawkes, C. A., Jeffrey, M., Kalaria, R. N., and Weller, R. O. (2013). Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol. Appl. Neurobiol. 39, 593–611. doi: 10.1111/nan.12042
Chabriat, H., Levy, C., Taillia, H., Iba-Zizen, M. T., Vahedi, K., Joutel, A., et al. (1998). Patterns of MRI lesions in CADASIL. Neurology 2, 452–457. doi: 10.1212/wnl.51.2.452
Choi, E. J., Choi, C. G., and Kim, J. S. (2005). Large cerebral artery involvement in CADASIL. Neurology 65, 1322–1324. doi: 10.1212/01.wnl.0000180965.79209.50
Ciolli, L., Pescini, F., Salvadori, E., Del Bene, A., Pracucci, G., Poggesi, A., et al. (2014). Influence of vascular risk factors and neuropsychological profile on functional performances in CADASIL: results from the MIcrovascular LEukoencephalopathy Study (MILES). Eur. J. Neurol. 1, 65–71. doi: 10.1111/ene.12241
Davous, P. (1998). CADASIL: a review with proposed diagnostic criteria. Eur. J. Neurol. 5, 219–233. doi: 10.1046/j.1468-1331.1998.530219.x
Dichgans, M., Ludwig, H., Müller-Höcker, J., Messerschmidt, A., and Gasser, T. (2000). Small in-frame deletions and missense mutations in CADASIL: 3D models predict misfolding of Notch3 EGF-like repeat domains. Eur. J. Hum. Genet. 8, 280–285. doi: 10.1038/sj.ejhg.5200460
Duering, M., Karpinska, A., Rosner, S., Hopfner, F., Zechmeister, M., Peters, N., et al. (2011). Co-aggregate formation of CADASIL-mutant NOTCH3: a single-particle analysis. Hum. Mol. Genet. 16, 3256–3265. doi: 10.1093/hmg/ddr237
Dziewulska, D., and Lewandowska, E. (2012). Pericytes as a new target for pathological processes in CADASIL. Neuropathology 32, 515–521. doi: 10.1111/j.1440-1789.2011.01290.x
Fortini, M. E., and Bilder, D. (2009). Endocytic regulation of Notch signaling. Curr. Opin. Genet. Dev. 19, 323–328. doi: 10.1016/j.gde.2009.04.005
Fouillade, C., Chabriat, H., Riant, F., Mine, M., Arnoud, M., Magy, L., et al. (2008). Activating NOTCH3 mutation in a patient with small-vessel-disease of the brain. Hum. Mutat. 3:452. doi: 10.1002/humu.9527
Fujiwara, Y., Mizuno, T., Okuyama, C., Nagakane, Y., Watanabe-Hosomi, A., Kondo, M., et al. (2012). Simultaneous impairment of intracranial and peripheral artery vasoreactivity in CADASIL patients. Cerebrovasc. Dis. 2, 128–134. doi: 10.1159/000334185
Guerrot, D., François, A., Boffa, J. J., Boulos, N., Hanoy, M., Legallicier, B., et al. (2008). Nephroangiosclerosis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: is NOTCH3 mutation the common culprit? Am. J. Kidney Dis. 52, 340–345. doi: 10.1053/j.ajkd.2008.04.017
Hara, K., Shiga, A., Fukutake, T., Nozaki, H., Miyashita, A., Yokoseki, A., et al. (2009). Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N. Engl. J. Med. 17, 1729–1739. doi: 10.1056/NEJMoa0801560
Huang, L., Li, W., Li, Y., Song, C., Wang, P., Wang, H., et al. (2020). A novel cysteine-sparing G73A mutation of NOTCH3 in a Chinese CADASIL family. Neurogenetics 1, 39–49. doi: 10.1007/s10048-019-00592-3
Ihara, M., and Yamamoto, Y. (2016). Emerging evidence for pathogenesis of sporadic cerebral small vessel disease. Stroke 47, 554–560. doi: 10.1161/strokeaha.115.009627
Joutel, A. (2011). Pathogenesis of CADASIL: transgenic and knock-out mice to probe function and dysfunction of the mutated gene, Notch3, in the cerebrovasculature. Bioessays 33, 73–80. doi: 10.1002/bies.201000093
Joutel, A., Andreux, F., Gaulis, S., Domenga, V., Cecillon, M., Battail, N., et al. (2000). The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Invest. 5, 597–605. doi: 10.1172/jci8047
Joutel, A., Monet, M., Domenga, V., Riant, F., and Tournier-Lasserve, E. (2004). Pathogenic mutations associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy differently affect Jagged1 binding and Notch3 activity via the RBP/JK signaling Pathway. Am. J. Hum. Genet. 74, 338–347. doi: 10.1086/381506
Joutel, A., Vahedi, K., Corpechot, C., Troesch, A., Chabriat, H., Vayssière, C., et al. (1997). Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 9090, 1511–1515. doi: 10.1016/s0140-6736(97)08083-5
Jouvent, E., Mangin, J. F., Duchesnay, E., Porcher, R., Düring, M., Mewald, Y., et al. (2011a). Longitudinal changes of cortical morphology in CADASIL. Neurobiol. Aging 33, 1002.e29–1002.e36. doi: 10.1016/j.neurobiolaging.2011.09.013
Jouvent, E., Poupon, C., Gray, F., Paquet, C., Mangin, J. F., Le Bihan, D., et al. (2011b). Intracortical infarcts in small vessel disease: a combined 7-T postmortem MRI and neuropathological case study in cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 42, e27–e30. doi: 10.1161/strokeaha.110.594218
Kast, J., Hanecker, P., Beaufort, N., Giese, A., Joutel, A., Dichgans, M., et al. (2014). Sequestration of latent TGF-β binding protein 1 into CADASIL-related Notch3-ECD deposits. Acta neuropathol. Commun. 1:96. doi: 10.1186/s40478-014-0096-8
Kelleher, J., Dickinson, A., Cain, S., Hu, Y., Bates, N., Harvey, A., et al. (2019). Patient-specific iPSC model of a genetic vascular dementia syndrome reveals failure of mural cells to stabilize capillary structures. Stem Cell Reports 5, 817–831. doi: 10.1016/j.stemcr.2019.10.004
Kim, Y., Choi, E. J., Choi, C. G., Kim, G., Choi, J. H., Yoo, H. W., et al. (2006). Characteristics of CADASIL in Korea: a novel cysteine-sparing Notch3 mutation. Neurology 10, 1511–1516. doi: 10.1212/01.wnl.0000216259.99811.50
Klueg, K. M., and Muskavitch, M. A. (1999). Ligand-receptor interactions and trans-endocytosis of Delta, Serrate and Notch: members of the Notch signalling pathway in Drosophila. J. Cell Sci. 112, 3289–3297.
Koizumi, T., Mizuta, I., Watanabe-Hosomi, A., Mukai, M., Hamano, A., Matsuura, J., et al. (2019). The CADASIL scale-J, a modified scale to prioritize access to genetic testing for japanese CADASIL-suspected patients. J. Stroke Cerebrovasc. Dis. 6, 1431–1439. doi: 10.1016/j.jstrokecerebrovasdis.2019.03.026
Kusaba, T., Hatta, T., Kimura, T., Sonomura, K., Tanda, S., Kishimoto, N., et al. (2007). Renal involvement in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Clin. Nephrol. 3, 182–187. doi: 10.5414/cnp67182
Lee, J. S., Ko, K., Oh, J. H., Park, J. H., and Lee, H. K. (2016). Phenotypic features of cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy subjects with R544C mutation. Dement. Neurocogn. Disord. 15, 15–19. doi: 10.12779/dnd.2016.15.1.15
Lee, S. J., Meng, H., Elmadhoun, O., Blaivas, M., and Wang, M. M. (2011). Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy affecting an African American man: identification of a novel 15-base pair NOTCH3 duplication. Arch. Neurol. 68, 1584–1586. doi: 10.1001/archneurol.2011.781
Liao, Y. C., Hsiao, C. T., Fuh, J. L., Chern, C. M., Lee, W. J., Guo, Y. C., et al. (2015). Characterization of CADASIL among the han chinese in taiwan: distinct genotypic and phenotypic profiles. PLoS One 10:e0136501. doi: 10.1371/journal.pone.0136501
Liem, M. K., van der Grond, J., Haan, J., van den Boom, R., Ferrari, M. D., Knaap, Y. M., et al. (2007). Lacunar infarcts are the main correlate with cognitive dysfunction in CADASIL. Stroke 3, 923–928. doi: 10.1161/01.str.0000257968.24015.bf
Markus, H. S., Martin, R. J., Simpson, M. A., Dong, Y. B., Ali, N., Crosby, A. H., et al. (2002). Diagnostic strategies in CADASIL. Neurology 8, 1134–1138. doi: 10.1212/wnl.59.8.1134
Miyawaki, S., Imai, H., Takayanagi, S., Mukasa, A., Nakatomi, H., and Saito, N. (2012). Identification of a genetic variant common to moyamoya disease and intracranial major artery stenosis/occlusion. Stroke 43, 3371–3374. doi: 10.1161/strokeaha.112.663864
Mizuno, T. (2012). Diagnosis, pathomechanism and treatment of CADASIL. Rinsho Shinkeigaku 52, 303–313. doi: 10.5692/clinicalneurol.52.303
Mizuno, T., Muranishi, M., Torugun, T., Tango, H., Nagakane, Y., Kudeken, T., et al. (2008). Two Japanese CADASIL families exhibiting Notch3 mutation R75P not involving cysteine residue. Intern. Med. 23, 2067–2072. doi: 10.2169/internalmedicine.47.1391
Mizuta, I., Watanabe-Hosomi, A., Koizumi, T., Mukai, M., Hamano, A., Tomii, Y., et al. (2017). New diagnostic criteria for cerebral autosomal dominant arteriopathy with subcortical infarcts and leukocencephalopathy in Japan. J. Neurol. Sci. 381, 62–67. doi: 10.1016/j.jns.2017.08.009
Monet-Leprêtre, M., Bardot, B., Lemaire, B., Domenga, V., Godin, O., Dichgans, M., et al. (2009). Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain. Brain 132, 1601–1612. doi: 10.1093/brain/awp049
Monet-Leprêtre, M., Haddad, I., Baron-Menguy, C., Fouillot-Panchal, M., Riani, M., Domenga-Denier, V., et al. (2013). Abnormal recruitment of extracellular matrix proteins by excess Notch3ECD: a new pathomechanism in CADASIL. Brain 136, 1830–1845. doi: 10.1093/brain/awt092
Moreton, F. C., Razvi, S. S., Davidson, R., and Muir, K. W. (2014). Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol. Scand. 130, 197–203. doi: 10.1111/ane.12266
Muino, E., Gallego-Fabrega, C., Cullell, N., Carrera, C., Torres, N., Krupinski, J., et al. (2017). Systematic review of cysteine-sparing NOTCH3 missense mutations in patients with clinical suspicion of CADASIL. Int. J. Mol. Sci. 18:E1964. doi: 10.3390/ijms18091964
Mukai, M., Mizuta, I., Ueda, A., Nakashima, D., Kushimura, Y., Noto, Y. I., et al. (2018). A Japanese CADASIL patient with homozygous NOTCH3 p.Arg544Cys mutation confirmed pathologically. J. Neurol. Sci. 394, 38–40. doi: 10.1016/j.jns.2018.08.029
Mykkänen, K., Junna, M., Amberla, K., Bronge, L., Kääriäinen, H., Poyhonen, M., et al. (2009). Different clinical phenotypes in monozygotic CADASIL twins with a novel NOTCH3 mutation. Stroke 6, 2215–2218. doi: 10.1161/strokeaha.108.528661
Narayan, S. K., Gorman, G., Kalaria, R. N., Ford, G. A., and Chinnery, P. F. (2012). The minimum prevalence of CADASIL in northeast England. Neurology 78, 1025–1027. doi: 10.1212/wnl.0b013e31824d586c
Nichols, J. T., Miyamoto, A., Olsen, S. L., D’Souza, B., Yao, C., and Weinmaster, G. (2007). DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J. Cell Biol. 176, 445–458. doi: 10.1083/jcb.200609014
Okeda, R., Arima, K., and Kawai, M. (2002). Arterial changes in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) in relation to pathogenesis of diffuse myelin loss of cerebral white matter: examination of cerebral medullary arteries by reconstruction of serial sections of an autopsy case. Stroke 33, 2565–2569. doi: 10.1161/01.str.0000032620.91848.1c
Opherk, C., Gonik, M., Duering, M., Malik, R., Jouvent, E., Hervé, D., et al. (2014). Genome-wide genotyping demonstrates a polygenic risk score associated with white matter hyperintensity volume in CADASIL. Stroke 964, 968–972. doi: 10.1161/strokeaha.113.004461
Opherk, C., Peters, N., Herzog, J., Luedtke, R., and Dichgans, M. (2004). Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain 127, 2533–2539. doi: 10.1093/brain/awh282
Pescini, F., Nannucci, S., Bertaccini, B., Salvadori, E., Bianchi, S., Ragno, M., et al. (2012). The cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) Scale: a screening tool to select patients for NOTCH3 gene analysis. Stroke 11, 2871–2876. doi: 10.1161/STROKEAHA.112.665927
Peters, N., Herzog, J., Opherk, C., and Dichgans, M. (2004a). A two-year clinical follow-up study in 80 CADASIL subjects: progression patterns and implications for clinical trials. Stroke 35, 1603–1608. doi: 10.1161/01.str.0000131546.71733.f1
Peters, N., Opherk, C., Zacherle, S., Capell, A., Gempel, P., and Dichgans, M. (2004b). CADASIL-associated Notch3 mutations have differential effects both on ligand binding and ligand-induced Notch3 receptor signaling through RBP-Jk. Exp. Cell Res. 299, 454–464. doi: 10.1016/j.yexcr.2004.06.004
Pippucci, T., Maresca, A., Magini, P., Cenacchi, G., Donadio, V., Palombo, F., et al. (2015). Homozygous NOTCH3 null mutation and impaired NOTCH3 signaling in recessive early-onset arteriopathy and cavitating leukoencephalopathy. EMBO Mol. Med. 6, 848–858. doi: 10.15252/emmm.201404399
Pratt, E. B., Wentzell, J. S., Maxson, J. E., Courter, L., Hazelett, D., and Christian, J. L. (2011). The cell giveth and the cell taketh away: an overview of Notch pathway activation by endocytic trafficking of ligands and receptors. Acta Histochem. 113, 248–255. doi: 10.1016/j.acthis.2010.01.006
Qin, W., Li, J., Zhu, R., Gao, S., Fan, J., Xia, M., et al. (2019). Melatonin protects blood-brain barrier integrity and permeability by inhibiting matrix metalloproteinase-9 via the NOTCH3/NF-κB pathway. Aging 23, 11391–11415. doi: 10.18632/aging.102537
Ragno, M., Sanguigni, S., Manca, A., Pianese, L., Paci, C., Berbellini, A., et al. (2016). Parkinsonism in a pair of monozygotic CADASIL twins sharing the R1006C mutation: a transcranial sonography study. Neurol. Sci. 6, 875–881. doi: 10.1007/s10072-016-2497-x
Ragno, M., Trojano, L., Pianese, L., Boni, M. V., Silvestri, S., Mambelli, V., et al. (2012). Renal involvement in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL): report of a case with a six-year follow-up. Histol. Histopathol. 10, 1307–1314. doi: 10.14670/HH-27.1307
Razvi, S. S., Davidson, R., Bone, I., and Muir, K. W. (2005). The prevalence of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) in the west of Scotland. J. Neurol. Neurosurg. Psychiatry 76, 739–741. doi: 10.1136/jnnp.2004.051847
Ruchoux, M. M., Guerouaou, D., Vandenhaute, B., Pruvo, J. P., Vermersch, P., and Leys, D. (1995). Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol. 89, 500–512. doi: 10.1007/bf00571504
Rufa, A., Guideri, F., Acampa, M., Cevenini, G., Bianchi, S., De Stefano, N., et al. (2007). Cardiac autonomic nervous system and risk of arrhythmias in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Stroke 2, 276–280. doi: 10.1161/01.STR.0000254530.38417.07
Rutten, J. W., Boon, E. M., Liem, M. K., Dauwerse, J. G., Pont, M. J., Vollebregt, E., et al. (2013). Hypomorphic NOTCH3 alleles do not cause CADASIL in humans. Hum. Mutat. 11, 1486–1489. doi: 10.1002/humu.22432
Rutten, J. W., Dauwerse, H. G., Gravesteijn, G., van Belzen, M. J., van der Grond, J., Polke, J. M., et al. (2016). Archetypal NOTCH3 mutations frequent in public exome: implications for CADASIL. Ann. Clin. Transl. Neurol. 11, 844–853. doi: 10.1002/acn3.344
Rutten, J. W., Haan, J., Terwindt, G. M., van Duinen, S. G., Boon, E. M., and Lesnik Oberstein, S. A. (2014). Interpretation of NOTCH3 mutations in the diagnosis of CADASIL. Expert Rev. Mol. Diagn. 14, 593–603. doi: 10.1586/14737159.2014.922880
Rutten, J. W., Van Eijsden, B. J., Duering, M., Jouvent, E., Opherk, C., Pantoni, L., et al. (2019). The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1-6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7–34 pathogenic variant. Genet. Med. 3, 676–682. doi: 10.1038/s41436-018-0088-3
Tikka, S., Baumann, M., Siitonen, M., Pasanen, P., Pöyhönen, M., Myllykangas, L., et al. (2014). CADASIL and CARASIL. Brain Pathol. 5, 525–544. doi: 10.1111/bpa.12181
Tikka, S., Mykkänen, K., Ruchoux, M. M., Bergholm, R., Junna, M., Pöyhönen, M., et al. (2009). Congruence between NOTCH3 mutations and GOM in 131 CADASIL patients. Brain 132, 933–939. doi: 10.1093/brain/awn364
Uchino, M., Hirano, T., Uyama, E., and Hashimoto, Y. (2002). Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) and CADASIL-like disorders in Japan. Ann. N Y Acad. Sci. 977, 273–278. doi: 10.1111/j.1749-6632.2002.tb04826.x
Ueda, A., Ueda, M., Nagatoshi, A., Hirano, T., Ito, T., Arai, N., et al. (2015). Genotypic and phenotypic spectrum of CADASIL in Japan: the experience at a referral center in Kumamoto University from 1997 to 2014. J. Neurol. 8, 1828–1836. doi: 10.1007/s00415-015-7782-8
van den Boom, R., Lesnik Oberstein, S. A., Spilt, A., Behloul, F., Ferrari, M. D., Haan, J., et al. (2003). Cerebral hemodynamics and white matter hyperintensities in CADASIL. J. Cereb. Blood Flow Metab. 5, 599–604. doi: 10.1097/01.WCB.0000062341.61367.D3
Viswanathan, A., Godin, O., Jouvent, E., O’Sullivan, M., Gschwendtner, A., Peters, N., et al. (2010). Impact of MRI markers in subcortical vascular dementia: a multi-modal analysis in CADASIL. Neurobiol. Aging 9, 1629–1636. doi: 10.1016/j.neurobiolaging.2008.09.001
Viswanathan, A., Gschwendtner, A., Guichard, J. P., Buffon, F., Cumurciuc, R., O’Sullivan, M., et al. (2007). Lacunar lesions are independently associated with disability and cognitive impairment in CADASIL. Neurology 2, 172–179. doi: 10.1212/01.wnl.0000265221.05610.70
Watanabe-Hosomi, A., Watanabe, Y., Tanaka, M., Nakagawa, M., and Mizuno, T. (2012). Transendocytosis is impaired in CADASIL-mutant NOTCH3. Exp. Neurol. 233, 303–311. doi: 10.1016/j.expneurol.2011.10.020
Wollenweber, F. A., Hanecker, P., Bayer-Karpinska, A., Malik, R., Bäzner, H., Moreton, F., et al. (2015). Cysteine-sparing CADASIL mutations in NOTCH3 show proaggregatory properties in vitro. Stroke 3, 786–792. doi: 10.1161/STROKEAHA.114.007472
Yamamoto, Y., Craggs, L. J., Watanabe, A., Booth, T., Attems, J., Low, R. W., et al. (2013). Brain microvascular accumulation and distribution of the NOTCH3 ectodomain and granular osmiophilic material in CADASIL. J. Neuropathol. Exp. Neurol. 5, 416–431. doi: 10.1097/nen.0b013e31829020b5
Yamamoto, Y., Ihara, M., Tham, C., Low, R. W., Slade, J. Y., Moss, T., et al. (2009). Neuropathological correlates of temporal pole white matter hyperintensities in CADASIL. Stroke 6, 2004–2011. doi: 10.1161/strokeaha.108.528299
Keywords: CADASIL, notch signaling, GOM, white matter lesion, vascular dementia
Citation: Mizuno T, Mizuta I, Watanabe-Hosomi A, Mukai M and Koizumi T (2020) Clinical and Genetic Aspects of CADASIL. Front. Aging Neurosci. 12:91. doi: 10.3389/fnagi.2020.00091
Received: 18 December 2019; Accepted: 18 March 2020;
Published: 07 May 2020.
Edited by:
Satoshi Saito, National Cerebral and Cardiovascular Center (Japan), JapanReviewed by:
Yumi Yamamoto, National Cerebral and Cardiovascular Center (Japan), JapanCopyright © 2020 Mizuno, Mizuta, Watanabe-Hosomi, Mukai and Koizumi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshiki Mizuno, bWl6dW5vQGtvdG8ua3B1LW0uYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.