 Aaron Song Chuan Foo
Aaron Song Chuan Foo Tuck Wah Soong
Tuck Wah Soong Tseng Tsai Yeo2
Tseng Tsai Yeo2 Kah-Leong Lim
Kah-Leong Lim
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 03 April 2020
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 12 - 2020 | https://doi.org/10.3389/fnagi.2020.00089
This article is part of the Research Topic Progress in Risk Factors and Development of Parkinson’s Disease View all 28 articles
As the main driver of energy production in eukaryotes, mitochondria are invariably implicated in disorders of cellular bioenergetics. Given that dopaminergic neurons affected in Parkinson’s disease (PD) are particularly susceptible to energy fluctuations by their high basal energy demand, it is not surprising to note that mitochondrial dysfunction has emerged as a compelling candidate underlying PD. A recent approach towards forestalling dopaminergic neurodegeneration in PD involves near-infrared (NIR) photobiomodulation (PBM), which is thought to enhance mitochondrial function of stimulated cells through augmenting the activity of cytochrome C oxidase. Notwithstanding this, our understanding of the neuroprotective mechanism of PBM remains far from complete. For example, studies focusing on the effects of PBM on gene transcription are limited, and the mechanism through which PBM exerts its effects on distant sites (i.e., its “abscopal effect”) remains unclear. Also, the clinical application of NIR in PD proves to be challenging. Efficacious delivery of NIR light to the substantia nigra pars compacta (SNpc), the primary site of disease pathology in PD, is fraught with technical challenges. Concerted efforts focused on understanding the biological effects of PBM and improving the efficiency of intracranial NIR delivery are therefore essential for its successful clinical translation. Nonetheless, PBM represents a potential novel therapy for PD. In this review, we provide an update on the role of mitochondrial dysfunction in PD and how PBM may help mitigate the neurodegenerative process. We also discussed clinical translation aspects of this treatment modality using intracranially implanted NIR delivery devices.
Parkinson’s disease (PD) is one of the most prevalent neurodegenerative disorders worldwide. Its incidence rises steeply above the age of 65, with one in every 50 people above the age of 80 being diagnosed with PD (Titova and Chaudhuri, 2018). From 1990 to 2016, the number of PD patients across the globe more than doubled from 2.5 million to 6.1 million. At the current pace of growth, this number is projected to hit 12 million by 2040 (Dorsey et al., 2018), making PD the most rapidly accruing neurological disease worldwide. Epidemiological data from developed countries (United Kingdom, United States, Singapore, Brazil) estimates the cost of each PD patient to be between USD 5,835–10,349 annually. As the natural course of PD is slow and progressive, PD patients continue to live for many years in a state of dependence (Hoehn and Yahr, 1998). Productivity losses, the need for a permanent carer, nursing home costs and hospitalization fees constitute the main expenses for upkeeping this group of patients (Findley, 2007; Zhao et al., 2013; Bovolenta et al., 2017). In response to the projected surge of PD, several groups have advocated urgent action to address this impending “pandemic” (Dorsey et al., 2018).
The reason for progressive disability in PD patients is the dysregulation of neural circuits in the basal ganglia that are responsible for movement control. One of the characteristic features of PD is the selective degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), which exerts a profound influence on muscle tone, ambulation, and limb movement through its effects on the basal ganglia motor loop. A hypo-dopaminergic state, as exemplified in PD, manifests clinically with “lead-pipe” rigidity of the affected limbs, “bradykinesia” i.e., generalized slowing of movement, and a distinctive 4–6 Hz “pill-rolling” tremor which characteristically affects the distal part of one’s upper extremity and spreads proximally before involving the contralateral side. Histologically, affected neurons exhibit an exuberance of intracytoplasmic inclusions containing misfolded α-synuclein proteinaceous fibrils—a neuropathological hallmark of PD known as “Lewy bodies” (Spillantini et al., 1997). Post-mortem analysis of PD brains has led to the proposal that Lewy body pathology is propagative, with the lower brainstem, upper brainstem and cerebral hemispheres being involved at successive stages of the disease (Del Tredici and Braak, 2016). In recent times there has also been an increasing body of evidence implicating the gut-brain axis in the spread of Lewy pathology, with alterations in the gut microbiome being the proposed mechanism for initiation of α-synucleinopathy (Braak et al., 2003a,b; Phillips et al., 2008; Fasano et al., 2013; Tan et al., 2014; Hasegawa et al., 2015; Kim et al., 2019; Yang et al., 2019). Intriguingly, not all neurons that demonstrate Lewy body accumulation undergo significant degeneration. Similarly, at any one stage, only a select group of neurons in each diseased region demonstrates the predilection for α-synuclein fibril aggregation (Surmeier and Schumacker, 2013). These peculiar observations suggest that the most severely affected neuronal groups ought to possess traits that confer increased vulnerability to α-synucleinopathy-mediated insults.
Neurons consume much larger amounts of energy compared to other cell types as the maintenance of resting membrane potential is an extremely energy-intensive process (Purves et al., 2018). Not surprisingly, neuronal groups with increased vulnerability in PD possess characteristics that contribute to a greater energy burden (Figure 1). Neurons with their cell bodies in the dorsal motor nucleus of the vagus (DMV), the medullary reticular nuclei, the pontine raphe nuclei, the locus coeruleus (LC), the pedunculopontine nucleus (PPN) and the SNpc share similar morphological features—long, thin and poorly myelinated axons with extensive terminal branching (Sulzer and Surmeier, 2013). These structural attributes impose substantial challenges on neuronal metabolism. First, the excessively arborized axonal tree implies a multi-fold increase in the number of synaptic terminals, which places a strain on vesicular recycling and mitochondrial trafficking machinery (Liang et al., 2007). Second, the lack of myelination gives rise to multiple leak points along the axon, increasing the burden of resting membrane potential maintenance. Atop the inefficiencies of their morphological characteristics, several of these neuronal groups also possess burdensome physiological phenotypes. Unlike the majority of neurons whose activity is triggered by the opening of voltage-gated ion channels in response to membrane depolarization, these neurons fire action potentials even in the absence of external stimulation due to their innate pace-making abilities (Williams et al., 1984; Chan and Chan, 1989; Travagli et al., 1991; Zhang et al., 2012; Cooper et al., 2015). Autonomous pace-maker dependent firing prolongs the amount of time these neurons spend in the depolarized state, which commensurately increases the amount of energy needed by adenosine triphosphate (ATP)-dependent ion pumps to maintain resting membrane potential. Finally, some of these neuronal groups (e.g., SNpc dopaminergic neurons, DMV cholinergic neurons) rely on Cav1.3 L-type calcium channel-mediated pacemaker activity for sustained autonomous firing. Cav1.3 generated currents create broad action potentials, implying slow opening/closing kinetics which lead to larger amounts of Ca2+ influx. In comparison with Na+ currents, Ca2+ currents are far more energy-intensive to maintain as the gradient of [intracellular Ca2+]:[extracellular Ca2+] is 2,000 times that of [intracellular Na+]:[extracellular Na+] (Surmeier and Schumacker, 2013). Ca2+ dependent firing also strains intracellular Ca2+ buffering mechanisms and predisposes the neuron to excitotoxicity. Cumulatively, the energy-intensive properties of the above neuronal groups lower their capacity to cope with an energy crisis. This translates into increased neuronal vulnerability when exposed to further stresses, such as α-synucleinopathy in PD.
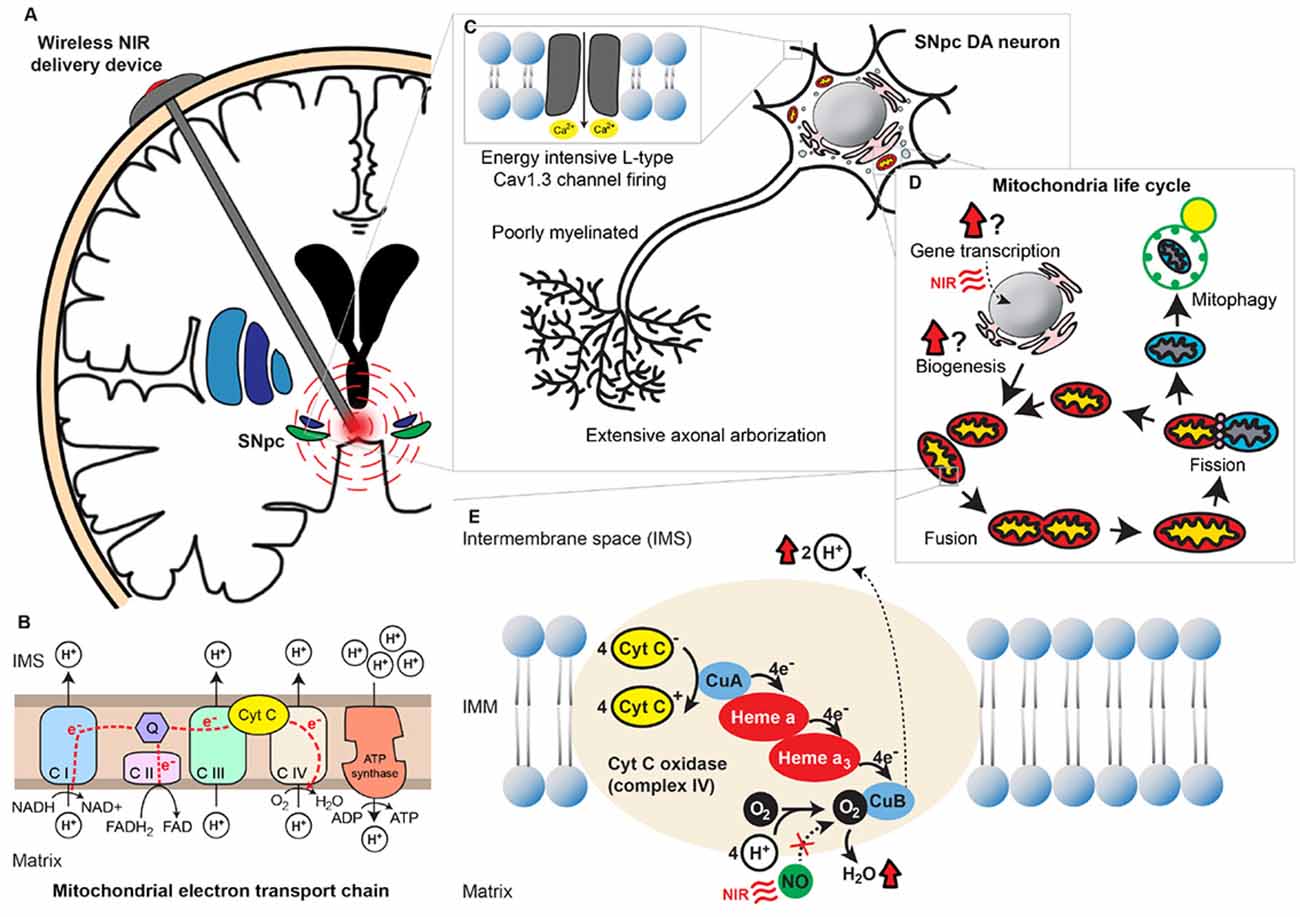
Figure 1. Near-infrared (NIR) stimulation and its potential neuroprotective role in Parkinson’s disease (PD). (A) A wirelessly powered intracranial device implanted in the human brain for delivery of NIR PBM to the SNpc. (B) Mitochondrial oxidative phosphorylation chain consisting of 5 different transmembrane protein complexes on the IMM that comprise the electron transport chain (ETC). The ETC pumps electrons derived from the Krebs cycle across the IMM into the intermembranous space through the actions of NADH dehydrogenase (complex I) and succinate dehydrogenase (complex II). The electrons are subsequently transferred to ubiquinone, which is reduced to ubiquinol upon electron receival. Cytochrome bc1 complex (complex III) then transfers these ubiquinol bound electrons to cytochrome C, which in turn binds to cytochrome C oxidase (complex IV) and facilitates the final electron transfer to oxygen. This transfer of electrons is coupled with the outward movement of protons across the IMM, establishing a transmembrane electrochemical gradient. Potential energy stored in the transmembrane gradient [proton motive force ΔPm] is utilized by adenosine triphosphate (ATP) synthetase (complex V) to drive the production of ATP from ADP and phosphate, which is coupled to the inward flow of protons across the IMM. (C) Energy-demanding features of SNpc dopaminergic neurons. These neurons possess extensive axonal arborization, long and poorly myelinated axonal projections, as well as L-type Cav1.3 channel-mediated pacemaker-type firing. Cumulatively these properties deplete the reserve capacity of these neurons to respond to an energy crisis. (D) The mitochondria life cycle depicting the processes of biogenesis, fusion, fission, and degradation. The effects of NIR on these processes remain uncertain. (E) An illustration of mitochondrial complex IV, COX, with its heme and copper centers. Electrons are transferred from cytochrome C to O2 via a series of redox reactions involving these centers. Binding of endogenous NO to O2’s binding site on CuB and prevents the final electron transfer step in oxidative phosphorylation. NIR PBM is purported to act via photodissociation of NO from O2’s binding site, releasing the NO’s inhibition on COX and thereby enhancing mitochondrial energy production. Abbreviations: ATP, Adenosine triphosphate; C I, Complex I; C II, Complex II; C III, Complex III; C IV, Complex IV; C V, Complex V; CuA, Copper A; CuB, Copper B; Cyt C, Cytochrome C; DA, Dopaminergic; IMM, Inner mitochondrial membrane; IMS, Intermembrane space; NIR, Near-infrared; NO, Nitric oxide; O2, Oxygen; Q, Coenzyme Q; PBM, Photobiomodulation; SNpc, Substantia nigra pars compacta.
The fact that energy-intensive neurons have increased vulnerability in PD suggests that some of the underlying disease processes may be linked to impairments in cellular energy production. This lends further weight to the hypothesis that disruptions in mitochondrial function constitute some of the key pathogenic processes in PD.
Mitochondria are believed to have originated from the fusion of an oxidative α-propeobacterium with a pre-eukaryotic cell 1.5–2 billion years ago (Margulis, 1975; Zimmer, 2009; Richards and Archibald, 2011). This union achieved a synergistic effect, with the resultant organism being capable of harnessing oxidative phosphorylation—a mechanism of energy production 15 times more efficient than the process of anerobic glycolysis utilized by unicellular organisms (Kadenbach, 2012). The ability to cope with higher energy demands enabled these hybrids to grow to larger dimensions and paved the way for their evolution into pluricellular organisms—the present-day eukaryotes. The oxidative phosphorylation system (Figure 1) comprises of the mitochondrial electron transport chain (ETC) and ATP synthase, all of which are transmembrane proteins located on the inner mitochondrial membrane (IMM). Complex IV of the ETC, cytochrome C oxidase (COX), is thought to be the enzyme that catalyzes the rate-limiting step in the chain of electron transfers (Villani and Attardi, 1997). This property is unique to mammalian cells and is not observed in isolated mitochondria. It is believed that COX has a regulatory role in cellular energy production, which varies with tissue-type and metabolic demands. The presence of tissue-specific isoforms of cytochrome C and COX in mammals lends strength to this theory (Pierron et al., 2012). Neuronal energy levels fluctuate in sync with their frequency of action potential firing. Unsurprisingly, it has been demonstrated in vitro that COX expression closely mirrors neuronal activity (Liang et al., 2006b). Being a bi-genomically encoded enzyme with a total of 13 subunits, transcription of both nuclear and mitochondrial DNA (mtDNA) is necessary for COX expression (Kadenbach et al., 2000). This process is mediated via transcription factors nuclear respiratory factors 1 and 2 (NRF1 and NRF2), which have been identified as promoters of COX expression in times of increased cellular energy demand (Ongwijitwat and Wong-Riley, 2005).
In addition to the regulation of oxidative phosphorylation, maintaining healthy mitochondrial volumes is also essential for efficient energy production in cells. The intracellular mitochondrial network is constantly pruned by the processes of biogenesis, fusion, fission, and degradation. These processes constitute the mitochondrial life cycle, which takes place every 5–20 min (Ferree and Shirihai, 2012). Under physiological conditions, the events in the mitochondrial life cycle are in dynamic equilibrium, maintaining a stable mass of intracellular mitochondria. If cellular metabolic requirements are altered, the mitochondrial network undergoes remodeling to accommodate the changes in energy demand (Ferree and Shirihai, 2012). The various stages in the mitochondrial life cycle are illustrated in Figure 1. Excellent reviews on the various aspects of the mitochondrial life cycle have been written elsewhere and we will therefore not elaborate on this topic in the present article (Hyde et al., 2010; Youle and Narendra, 2011; Ferree and Shirihai, 2012).
The mitochondrial theory of PD is based on observations in which disease processes impairing oxidative phosphorylation, mitochondrial biogenesis or mitophagy manifest as phenotypes that share common Parkinsonian features. The first link between PD and mitochondrial dysfunction was conceived by Langston (2017) after observing rapidly developing Parkinsonian features in intravenous drug abusers who were exposed to 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP), a toxin whose metabolite 1-methyl-4-phenylpyridinium (MPP+) inhibits mitochondrial complex I (Ramsay et al., 1986, 1987; Langston, 2017). Once in the central nervous system, MPP+ is taken up by the dopamine active transporter and induces the selective degeneration of SNpc dopaminergic neurons. This phenomenon has been successfully reproduced in MPTP-treated rodents and nonhuman primates, popularizing these animal models for PD research (Dauer and Przedborski, 2003; Konnova and Swanberg, 2018). Complementing the findings in MPTP, rotenone, a pesticide that inhibits mitochondrial complex I, produced similar PD-like locomotor deficits in treated animals. Despite it having widespread uptake in the central nervous system, the toxic effects of rotenone are most pronounced in SNpc dopaminergic neurons, reinforcing the belief that neurons with greater susceptibility to energy perturbances would be affected the most (Betarbet et al., 2000). Further evidence is gleaned from epidemiological studies, which identified rotenone-exposed agrarian populations to be at greater risk of developing PD (Dhillon et al., 2008; Tanner et al., 2011).
Genetic mutations in PD patients further cement the link between mitochondrial dysfunction and PD. Mutations in the PINK1 and PRKN (formerly PARK2) genes, which encode the mitophagy-related proteins PINK1 and Parkin, respectively, result in autosomal recessively inherited, early-onset forms of PD (Kitada et al., 1998; Valente et al., 2004). Phenotypes of Parkin-type and PINK1-type early-onset PD are strikingly similar, implicating not just individual gene mutations but the entire mitochondrial quality control machinery in the pathogenesis of PD (Bruggemann and Klein, 1993; Schneider and Klein, 1993). Concomitantly, mutations in mtDNA have been identified in sporadic PD patients. The number of these somatic mutations accumulate with age, mirroring the increased incidence of PD in older subjects (Corral-Debrinski et al., 1992). As several ETC proteins require bi-genomic transcription involving both nuclear and mtDNA (Hermann et al., 2012), an accumulation of mtDNA mutations would likely impair mitochondrial oxidative phosphorylation capacity. Interestingly, post-mortem studies in aged patients depict a higher prevalence of mtDNA mutations in the SNpc compared to other parts of the brain, regardless of whether these patients had PD or not (Soong et al., 1992; Bender et al., 2006, 2008). Whilst the reasons underlying why mtDNA mutations have a predilection for accumulating in SNpc neurons remain elusive, these findings align aging, PD and mitochondrial bioenergetic failure in a unifying pathogenic process beginning with strained mitochondrial function due to age-related genomic instability, subsequent bioenergetic failure of the most vulnerable neuronal groups, and finally, neuronal loss leading to the disease phenotype. This belief is further substantiated by age-related Parkinsonism observed in the MitoPark mouse, a transgenic mouse model with conditional knockout of mitochondrial transcription factor A (Tfam) in dopaminergic neurons. MitoPark mice are born healthy but develop progressive degeneration of SNpc dopaminergic neurons and motor retardation as they grow older, mimicking the natural course of PD in humans (Sterky et al., 2011). On the whole, evidence supporting mitochondrial dysfunction as one of the key pathogenic processes in PD is compelling and promotes acceptance of PD as a disease of bioenergetic failure.
As the mitochondrial theory of PD gains prominence, therapeutics aimed at mitochondrial modulation are also garnering interest in the PD community. Near-infrared (NIR) light therapy, also known as “photobiomodulation” (PBM), is a treatment modality that has been under investigation for many ailments since the 1960 s. Discovery of the therapeutic properties of NIR light was rather accidental—in an experiment where photodynamic therapy was delivered to tumor cells in rodents, the intensity of laser administered was mistakenly reduced, and whilst this failed to achieve the intended tumoricidal effects of the experiment, the attenuated laser therapy successfully promoted superficial wound healing in the treated animals (Mester et al., 1971). This accidental finding revealed the therapeutic effects of low-intensity laser therapy and prompted further investigation of its mechanism of action.
PBM works on the principle that light-sensitive molecules in the body known as chromophores are excited by photonic stimulation. It is now known that hemoglobin, myoglobin, and COX are the only 3 chromophores in mammalian tissue capable of absorbing light in the NIR range (600–900 nm wavelength). Passarella and Karu (2014) were the first to discover that one of the main NIR-sensitive chromophores was associated with mitochondria. In the early experiments conducted by Passarella et al. (1984, 1988), irradiation of rat liver mitochondria with low dose Helium-Neon laser light of 632.8 nm wavelength enhanced oxygen consumption, increased membrane potential, promoted ATP generation and encouraged protein synthesis. In their attempts to identify the molecule responsible for these effects, Karu et al. (1996) experimented with NIR PBM on HeLa cells, believing that by identifying the wavelength(s) at which delivered light produced the greatest cellular response they could determine the peak absorption spectrum of the target molecule and hence uncover its identity. Interestingly, their study revealed peak DNA/RNA synthesis and cellular adhesive levels at multiple wavelengths within the NIR range—620 nm, 680 nm, 760 nm and 820 nm (Karu et al., 1996; Karu, 1999). These wavelengths corresponded with excitation energies required for metal-ligand and d-d electron transfers in copper (Cu) complexes and prompted the search for a photo-acceptor with multiple Cu centers. As mitochondrial complex IV COX possessed two redox-active Cu sites (CuA and CuB) within its core, it emerged as the apparent candidate responsible for the effects of NIR.
Mitochondrial cytochrome C undergoes oxidation by COX and in turn facilitates the reduction of O2 to water as per the chemical equation below (Sarti et al., 2012).
The transfer of electrons from cytochrome C to O2 is coupled with the expulsion of 4 protons from the mitochondrial matrix across the IMM, contributing to the transmembrane electrochemical gradient that drives ATP synthesis. Nitric oxide (NO), a molecule known for its vasodilatory properties in vivo, competes with O2 for its binding site on the CuB center of COX (Hamblin, 2018). By binding noncovalently to CUB, NO inhibits the activity of COX and hence impairs the process of oxidative phosphorylation. It is surmised that under physiological conditions, the inhibitory effect of NO may have a role in the regulation of energy production. To maximize the efficiency of resource utilization, the body reduces O2 consumption in tissues whose metabolism is low (e.g., keratinocytes) to ensure that demand in energy-intensive tissues (e.g., neurons, hepatocytes and cardiomyocytes) can be met (Lane, 2006). The prevailing theory underpinning PBM is that NIR enhances mitochondrial energy production by causing photodissociation of NO from its binding site on COX, which facilitates O2 binding and electron transfer (Hamblin, 2018). In a series of noteworthy experiments, Wong-Riley et al. (2001, 2005) investigated the mechanism through which PBM modulated energy production in rodent visual cortex neuronal cultures. Being post-mitotic cells with energy demands that are largely reflective of their spiking activity, neurons are ideal candidates for bioenergetic studies. Wong-Riley et al. (2001, 2005) demonstrated that while PBM with 670 nm wavelength light was capable of fully reversing the effects of tetrodotoxin (TTX) induced sodium (Na+) channel blockade on neuronal COX activity (Wong-Riley et al., 2001, 2005), its effect was attenuated significantly when TTX was substituted with potassium cyanide (KCN), a potent COX inhibitor (Wong-Riley et al., 2005). These results strongly suggested that the beneficial effects of PBM were at least partially mediated through enhancing mitochondrial COX activity. Further supplementation of the above results by findings demonstrating that PBM augmented neuronal ATP levels in the presence of toxin treatment further supported its role as an enhancer of mitochondrial energy production (Wong-Riley et al., 2005; Ying et al., 2008).
Notwithstanding these claims, there have been several alternative hypotheses refuting the mitochondrial COX theory of PBM in recent years. For example, Lima et al. (2019) demonstrated increased cellular proliferation after the administration of NIR PBM in cell lines and animal models with deficient COX expression. An alternative mechanism to augmenting COX activity has also been suggested by another study showing that enhanced mitochondrial energy production is the effect of a reduction in intra-mitochondrial water viscosity induced by NIR (Sommer, 2019). It is believed that the viscosity of the thin layer of water molecules on the inner side of the IMM, termed the interfacial water layer (IWL), influences the driving rate of ATP synthase—the enzyme that catalyzes ATP production in the mitochondria. ATP generation is facilitated by the rotatory motion of the ATP synthase machinery, which is in turn propelled by the diffusion of protons across IMM as a result of an established transmembrane electrochemical gradient. NIR-mediated reduction in IWL viscosity decreases the friction opposing ATP synthase rotation and results in “smoother” turning of the ATP synthase machinery. This theory is supported by the fact that cellular ATP level increases are immediate after PBM stimulation (Quirk et al., 2016), suggesting that the intracellular changes mediating elevated ATP production are alterations in physical properties rather than chemical processes, as the latter tends to occur with a slight time lag (Sommer, 2019). Yet another group suggested that the photo-absorbent metabolite pyropheophorbide-a (P-a) of dietary chlorophyl could facilitate light-driven energy production processes in animals (Xu et al., 2014). Xu et al. (2014) first examined whether P-a associates with the mitochondria after chlorophyl ingestion by looking at fluorescence emission levels of P-a from mammalian mitochondrial fragments. Elevated levels of 675 nm fluorescence emission characteristics of P-a were noticed after a certain concentration of mitochondrial fragments was amassed, suggesting the possibility of P-a being bound to mitochondrial proteins. This finding was followed up with experiments measuring ATP levels in isolated mammalian mitochondria treated with P-a and/or 670 nm wavelength NIR light. In the subsequent experiment, ATP levels only increased in groups where P-a and NIR light were co-administered, and not in those in which P-a or NIR were given in isolation. This observation was further supported by in vivo demonstration in a Caenorhabditis elegans model that lifespan, respiration rate, and ATP levels increased significantly when both P-a and NIR were administered concomitantly (Xu et al., 2014). Given the multiplicity of these competing theories, it may be that NIR exerts its modulatory effects through several mechanisms instead of one.
Intriguingly, the beneficial effects of NIR PBM follow a biphasic dose-response curve which adheres closely to the Arndt-Schulz principle (Huang et al., 2009; Sharma et al., 2011; Xuan et al., 2016). At very low doses of stimulation, the effects of PBM are negligible. As the dose of PBM is increased its effects are correspondingly amplified until a peak is observed, oftentimes at an intensity that is still considered relatively low. Further increments of intensity beyond this level only serve to elicit inhibitory or harmful effects in treated specimens. This property of NIR has led to the belief that the PBM delivers its benefits by acting as a gentle stressor of cells, which triggers the activation of intrinsic cellular stress response machinery. Part of this response involves the activation of transcription factors and secondary signaling pathways, which can explain the sustained effects of PBM even after a single treatment session (Liang et al., 2006a). The nuclear transcription factor NF-kB is upregulated in response to PBM-induced elevation of intracellular reactive oxygen species (ROS) and leads to activation of several anti-apoptotic, antioxidant and pro-proliferation downstream processes (de Freitas and Hamblin, 2016). Whilst low-stress levels achieve beneficial outcomes for cells, overzealous NIR stimulation leads to pathological levels of ROS production, which initiates a self-propagating vicious cycle that begets more ROS release, cellular damage, and activation of intracellular apoptotic pathways (Zorov et al., 2006). It seems like cellular tolerance for ROS depends on the capacity of mitochondrial superoxide dismutase (MnSOD) to cope with the partitioning of superoxide (O−) radicals, and once a critical threshold is breached any benefits associated with NIR are nullified (Stone and Yang, 2006; Sies, 2017).
There are some reports that NIR PBM also promotes neurogenesis and synaptogenesis atop its neuroprotective properties. An increase in neuro-progenitor proliferation and synaptogenesis markers have been observed in the dentate gyrus and subventricular zone after PBM stimulation in rodents (Xuan et al., 2013, 2014, 2015). Furthermore, Johnstone et al. (2014) proposed that in addition to its local cellular effects PBM also elicits a systemic type of response that is immunomodulatory (Rochkind et al., 1989; Johnstone et al., 2015). This arises from clinical observations in which the local application of NIR can elicit positive effects at distant sites in the body. The exact mechanisms underpinning this phenomenon are still unknown, but is believed to involve the release of trophic factors (e.g., brain-derived neurotrophic factor), downregulation of pro-inflammatory cytokines and upregulation of anti-inflammatory cytokines in the body (Braverman et al., 1989; Johnstone et al., 2014). One such effector molecule is PPARγ, a nuclear receptor involved in the production of anti-inflammatory heat shock protein 70 (HSP 70), which is increased after NIR treatment (de Lima et al., 2013; Croasdell et al., 2015). The distant effects of NIR has been likened to the “abscopal effect” observed in radiotherapy of metastatic cancer, where local treatment of a tumor not only leads to its shrinkage but also a diminution of metastases at distant sites (Johnstone et al., 2015).
The concept of using PBM as a therapeutic approach in PD is particularly enticing for several reasons: first and foremostly, the mechanisms underpinning both disease and intervention converge on the common subject of mitochondrial bioenergetics. That PD is gaining recognition as a disease of bioenergetic failure makes PBM, which is focused on enhancing cellular energy production, a befitting remedy for this disease. This matchup is further complemented with neuroinflammation being another proposed pathogenic process in PD and NIR purported to have both local and systemic immunomodulatory effects (Johnstone et al., 2015). Long term PBM administration has been shown to limit astrogliosis, a consequence of neuroinflammation that recruits exuberant infiltrates of astrocytes and microglia to form glial scars (El Massri et al., 2016, 2018; O’Brien and Austin, 2019). Further supporting this claim are observed reductions of immunogenic cytokine levels after PBM administration (Byrnes et al., 2005; Muili et al., 2012). Being able to address two of the key pathogenic processes in PD makes NIR an attractive candidate for clinical translation amidst the list of experimental therapeutics targeted at neurodegeneration. Further promoting the feasibility of NIR is the absence of adverse events associated with its usage, which suggests a robust safety profile. To date, there have been no acute adverse effects reported with NIR PBM delivered in doses within the determined therapeutic ranges [4–30 J/cm2; 5–50 mW/cm2], both in short and long term animal and patient studies (Desmet et al., 2006; Ilic et al., 2006; Zivin et al., 2009; McCarthy et al., 2010; Rojas and Gonzalez-Lima, 2011; Quirk et al., 2012a; Moro et al., 2017; Hamilton et al., 2019). With negligible side effects and the potential for immense benefits, the favorable risk: reward ratio of NIR light therapy significantly raises its feasibility for clinical application. Lastly, NIR PBM has demonstrated the potential to be neuroprotective, which is unprecedented in PD therapeutics. At present, there are no treatments that can forestall the neurodegenerative process in PD. The principle modality of PD treatment is drug replacement therapy, which is targeted at artificially restoring central nervous system dopamine levels either by inhibition of dopamine metabolizing enzymes or replacement by levodopa. The effects of drug treatment eventually wear off as the disease progresses into its later stages and the non-physiological pattern of dopamine replacement in the central nervous system triggers untoward dyskinetic movements in PD patients which can prove to be extremely debilitating. Once the undesirable side effects of medical therapy become intolerable, advanced PD treatments such as deep brain stimulation, continuous subcutaneous apomorphine infusions, and intra-jejunal levodopa-carbidopa intestinal gel (LCIG) are offered to patients. Although being able to effectively address medication failure and levodopa-induced dyskinesia, these treatments remain extremely costly and are unable to confer protection against the neurodegenerative process in PD. The anticipated “pandemic” of PD patients in the coming years and its associated escalation in healthcare costs herald the need for new therapies focusing on neuroprotection in the disease’s early stages.
A few groups have examined the neuroprotective effects of NIR PBM in in vitro PD models (Table 1). For example, Ying et al. (2008) and Liang et al. (2008) have demonstrated reduced cell death, increased ATP production and elevated COX activity after delivery of pulsed 670 nm wavelength light in PD neurotoxin-treated rodent primary neuronal cultures, A30P over-expressing neuroblastoma cell lines and PD cybrid cells with damaged mtDNA (Zorov et al., 2006; Trimmer et al., 2009; Quirk et al., 2012b). Several principles guiding optimal delivery of NIR stimulation were derived from such in vitro studies. First, NIR seems to work best with regimes requiring pre-treatment. The neuroprotective effects of PBM are most accentuated when its administration starts before toxin treatment of neurons and continues throughout toxin exposure (Wong-Riley et al., 2005; Liang et al., 2006b; Ying et al., 2008). Nonetheless, this phenomenon was not necessarily observed in in vivo models, as evidenced by the absence of benefit in NIR pre-treated vis-á-vis NIR post-treated mice which received MPTP (Reinhart et al., 2016). The benefits of pre-treatment with NIR can allude to ischemic preconditioning in which episodes of tolerable ischemia mitigates damage from a serious insult that follows, such as in stroke or peripheral arterial disease. Second, the optimal dose of NIR for neuronal stimulation is fluence and radiance values of 4–30 J/cm2 and 50 mW/cm2, respectively (Wong-Riley et al., 2005; Liang et al., 2006b; Ying et al., 2008). Keeping to these parameters would avert light toxicity from NIR, in keeping with the biphasic dose-response of NIR stimulation as per the Arndt–Schultz Law. Lastly, lights in the NIR spectrum that produce the greatest stimulatory effects in neurons are those at 670 nm and 830 nm wavelengths (Wong-Riley et al., 2005); 670 nm being relatively close to the peak absorption spectrum of oxidized CuB (Karu, 1999). The successful optimization studies performed in neuronal cultures paved the way for PBM testing in animal models, in which subsequent rodent and non-human primate experiments often used the above treatment paradigms as their benchmark.
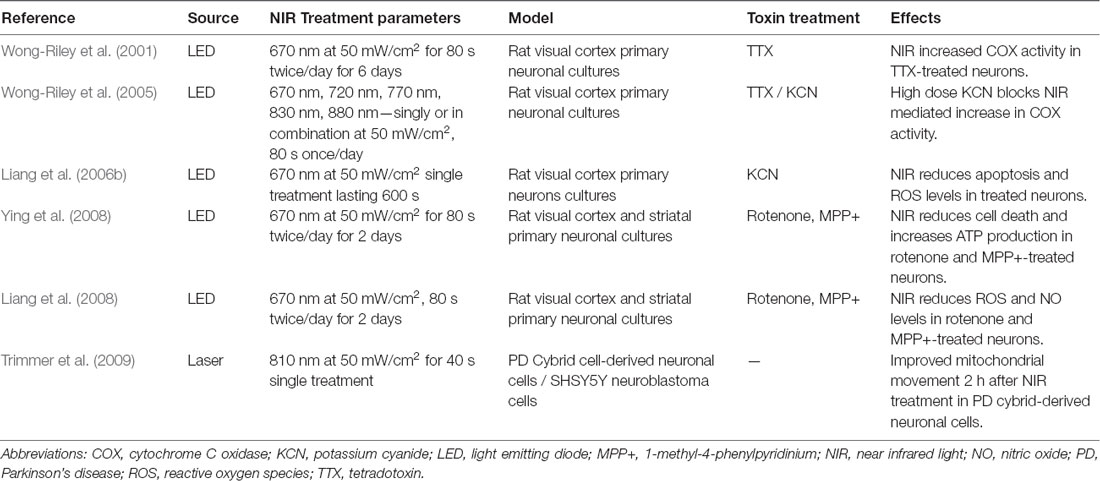
Table 1. In vitro studies in PD models.
Several in vivo studies validated the efficacy of NIR PBM in PD animal models (Table 2). Oueslati et al. (2015) using an adeno-associated virus (AAV) rat model of PD, managed to demonstrate retardation of α-synuclein toxicity and alleviation of synucleinopathy-related motor deficits with PBM. Several studies also showed similar rescue of SNpc dopaminergic neurons in MPTP–treated mice following PBM treatment (Shaw et al., 2010; Johnstone et al., 2014; Reinhart et al., 2015). PBM using a combination of lights with varying wavelengths (670 nm and 810 nm) in the NIR range delivered synchronously or sequentially were also tested in rodents, with positive results (Reinhart et al., 2017). Encouraging results from these studies prompted experiments investigating the feasibility of delivering NIR from an intracranial source. This was first demonstrated successfully in two rodent studies by Benabid and colleagues (Moro et al., 2014; Reinhart et al., 2016). In the study conducted by Reinhart et al. (2016), an intracranial NIR delivery device was implanted in the vicinity of the SNpc of 6-hydroxydopamine (6-OHDA) lesioned Wistar rats, demonstrating the feasibility of stereotaxic device implantation into the ventral midbrain. Interestingly, in the experiments conducted by Moro et al. (2014) and Reinhart et al. (2016), pulsed delivery of NIR achieved better results than continuous delivery for the same dose of NIR treatment. Following these accomplishments, Darlot et al. (2016) were the first to report successful PBM treatment via an intracranially implanted NIR device in a monkey model of PD. This was followed by further experiments in nonhuman primate models exploring different dosage protocols, the extent of astrogliosis and long term safety of NIR using implanted devices (El Massri et al., 2016, 2017; Moro et al., 2016). With regards to non-invasive PBM treatments for PD, there are a few groups that have examined its efficacy in human subjects. Hamilton et al. (2019) and Santos et al. (2019) have recently reported slight improvements in motor and non-motor symptoms in PD patients after the administration of PBM using extracranial sources. In their trial, Santos et al. (2019) delivered PBM by alternating a 670 nm LED between the subjects’ left and right temples in 6 1-min blocks per session. Subjects were treated twice a week for a total of 9 weeks and demonstrated significant improvements in motor scores (Santos et al., 2019). In parallel, Hamilton et al. (2019) performed a study involving 6 PD subjects who adopted the practice of wearing of “PBM helmets” –buckets lined on the inside with NIR lights—regularly for 24 months and reported that treated subjects experienced substantial alleviation of both motor as well as non-motor symptoms. One postulation is that PBM targeted at cortical regions in PD can modulate dysfunctional neural circuits caused by a lack of dopamine, even if there is no direct action on SNpc dopaminergic neurons. If this is true, PBM delivered to cortical regions may be capable of symptom relief, but is unlikely to achieve any neuroprotective effect (Hamilton et al., 2019).
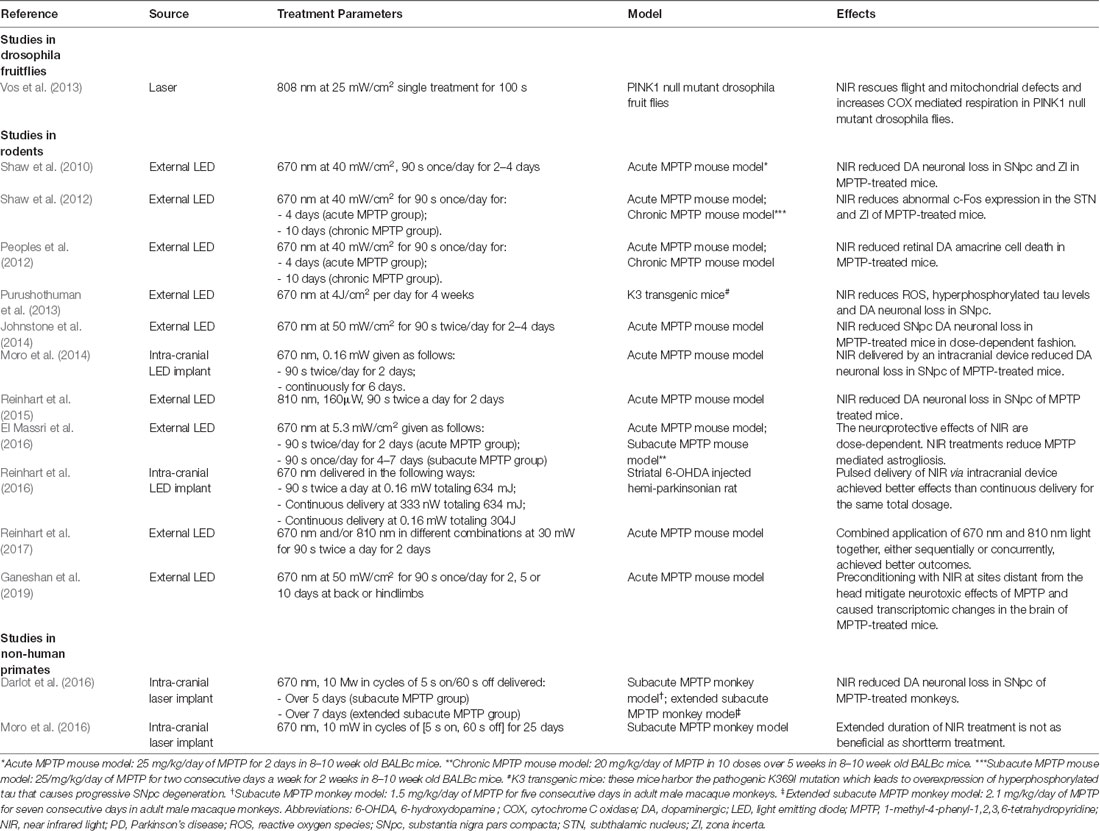
Table 2. In vivo studies in PD models.
The development of an intracranially implanted NIR delivery device for PD is challenging for several reasons. First, based on their studies in mice brains, Moro et al. (2014) realized that the signal intensity of NIR decays at a rate of 65% per millimeter of brain tissue. In comparison with the brains of mice and small monkeys, the human brain is much larger and shielded by a thick bony calvarium. These barriers to light penetration, compounded by the fact that the midbrain SNpc is one of the most deep-seated parts of the human brain, present significant challenges for NIR delivery to the target region. Ultimately, this endeavor would involve the insertion of an LED via a safe trajectory deep into the ventral midbrain to effect NIR stimulation. Being our “seat of consciousness,” the midbrain is an exquisitely sensitive region of the human brain, with zero tolerance for any surgical mishap. Operating in such an important region of the brain inadvertently translates into greater risks for surgical candidates. Also, for neuroprotection to be effective, it has to be administered early in the course of the disease. Given the lack of effective early screening modalities, PD is largely diagnosed based on its clinical features which only manifest after 70%–80% of SNpc dopaminergic neurons have undergone degeneration. This implies that only 20%–30% of SNpc dopaminergic neurons remain available for salvage even if neuroprotective measures were implemented immediately at the time of diagnosis. Conversely, even if pre-clinical PD were able to be effectively screened for and diagnosed, the significant risks of surgical implantation may deter patients who are “seemingly well” from undertaking such a procedure, culminating in a poor uptake rate for this treatment modality. Perhaps safer regions for intracranial implantation of the LED could be explored, such as placement of the NIR LED just rostral to the midbrain tegmentum within the body of the third ventricle. This can be achieved safely under direct vision with the guidance of a neuro-endoscope and a carefully planned trajectory (Figure 2). Another site that could be considered for implantation is within the sphenoid sinus (SS) located in the posterior nasal space given its proximity to the ventral midbrain (Figure 2). Nevertheless, although near the upper brainstem, the SS remains separated from it by the bony clivus and the dura mater, which impedes the transmission of the NIR signal. Hence, simulation studies looking at the penetration of NIR through these layers of tissue would be required before commissioning the SS as a satisfactory location for LED placement. Second, given that PD runs its course over years, another challenge for such a device is the sustained delivery of NIR stimulation over a prolonged period. The presence of an efficient power source is imperative for such an undertaking. Existing batteries for intracranial implantable devices often require implantation in a subcutaneous pocket over the chest, with wires tunneled under the skin to connect it with the device’s intracranial portion. These device components are subject to the risks of breakage and microbial infection, which significantly increase device-related morbidity. The advent of miniaturized wirelessly powered devices may be able to overcome this technological limitation (Montgomery et al., 2015). Recent evidence advocating early DBS for PD (EARLYSTIM Trial, 2013) may be used to promote the use of NIR devices as co-implantation of NIR and DBS devices could be performed in the same setting (Schuepbach et al., 2013).
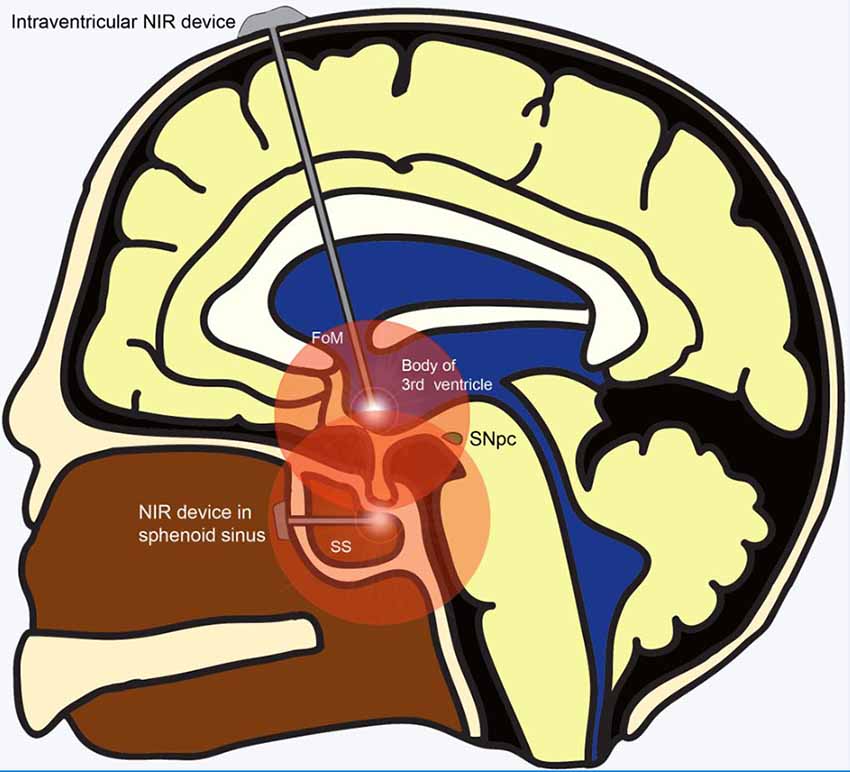
Figure 2. Alternative sites for implantation of NIR delivery device apart from the ventral midbrain. Wirelessly powered NIR delivery devices in the 3rd ventricle and the sphenoid sinus (SS), both of which are close to the ventral midbrain. Insertion of the device into the 3rd ventricle can be achieved safely via a straight trajectory traversing the Foramen of Monroe. Insertion of the NIR device within the SS in the posterior part of the nasal cavity can be achieved using an endoscopic endonasal approach. Abbreviations: FoM, foramen of Monroe; NIR, near-infrared; SNpc, substantia nigra pars compacta; SS, sphenoid sinus.
The idea of matchmaking NIR PBM—a mitochondrial modulatory instrument, with PD—a disease of bioenergetic failure, remains an attractive proposition. However, before getting mired in the race towards clinical translation, a step back to examine the gaps in our understanding of PBM seems worthy of our efforts. It seems that the mechanisms through which PBM augments mitochondrial function remain unclear. Information on its effects on gene transcription, mitochondrial dynamics and immunomodulation are also scant and limited. These shortfalls are areas that we believe future research efforts can be directed at. A further concern of intracranial NIR delivery surrounds the need for device implantation into the midbrain. Given that manipulation in this treacherous region of the brain could potentially lead to devastating clinical deficits it would not be unreasonable to anticipate that such a treatment modality would be met with resistance from patients with early PD. Alternative sites for implantation of the NIR delivery device, such as the intranasally or intraventricularly, could potentially be explored. We hope that technological advancements in the future can overcome these shortcomings and produce a device optimized for use in human subjects.
AF wrote the first draft in consultation with all other co-authors, who provided inputs. K-LL proof-read and completed the final draft. TS and TY edited the manuscript and provided expert advice and intellectual inputs.
This work was supported by grants from the National Medical Research Council—Translational Clinical Research Program in Parkinson’s disease (K-LL) and Lee Kong Chian School of Medicine (K-LL). Dr. AF is supported by the Department of Surgery, National University Hospital and a graduate scholarship from the National University of Singapore Research Scholars Programme.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Liting Hang for help with illustrations, and Ms. Hui-Ying Chan for critical reading of the manuscript.
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Bender, A., Schwarzkopf, R. M., McMillan, A., Krishnan, K. J., Rieder, G., Neumann, M., et al. (2008). Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J. Neurol. 255, 1231–1235. doi: 10.1007/s00415-008-0892-9
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
Bovolenta, T. M., de Azevedo Silva, S. M. C., Saba, R. A., Borges, V., Ferraz, H. B., and Felicio, A. C. (2017). Average annual cost of Parkinson’s disease in São Paulo, Brazil, with a focus on disease-related motor symptoms. Clin. Interv. Aging 12, 2095–2108. doi: 10.2147/cia.s151919
Braak, H., Del Tredici, K., Rüb, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003a). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Braak, H., Rüb, U., Gai, W. P., and Del Tredici, K. (2003b). Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 110, 517–536. doi: 10.1007/s00702-002-0808-2
Braverman, B., McCarthy, R. J., Ivankovich, A. D., Forde, D. E., Overfield, M., and Bapna, M. S. (1989). Effect of helium-neon and infrared laser irradiation on wound healing in rabbits. Lasers Surg. Med. 9, 50–58. doi: 10.1002/lsm.1900090111
Bruggemann, N., and Klein, C. (1993). “Parkin type of early-onset Parkinson disease,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens and A. Amemiya (Seattle, WA: University of Washington).
Byrnes, K. R., Waynant, R. W., Ilev, I. K., Wu, X., Barna, L., Smith, K., et al. (2005). Light promotes regeneration and functional recovery and alters the immune response after spinal cord injury. Lasers Surg. Med. 36, 171–185. doi: 10.1002/lsm.20143
Chan, J. Y., and Chan, S. H. (1989). Passive electrical properties of spontaneously active neurons in the nucleus reticularis gigantocellularis of the cat. Neurosci. Lett. 97, 169–174. doi: 10.1016/0304-3940(89)90158-4
Cooper, G., Lasser-Katz, E., Simchovitz, A., Sharon, R., Soreq, H., Surmeier, D. J., et al. (2015). Functional segregation of voltage-activated calcium channels in motoneurons of the dorsal motor nucleus of the vagus. J. Neurophysiol. 114, 1513–1520. doi: 10.1152/jn.00432.2014
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., Beal, M. F., and Wallace, D. C. (1992). Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 2, 324–329. doi: 10.1038/ng1292-324
Croasdell, A., Duffney, P. F., Kim, N., Lacy, S. H., Sime, P. J., and Phipps, R. P. (2015). PPARγ and the innate immune system mediate the resolution of inflammation. PPAR Res. 2015:549691. doi: 10.1155/2015/549691
Darlot, F., Moro, C., El Massri, N., Chabrol, C., Johnstone, D. M., Reinhart, F., et al. (2016). Near-infrared light is neuroprotective in a monkey model of Parkinson disease. Ann. Neurol. 79, 59–75. doi: 10.1002/ana.24542
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39, 889–909. doi: 10.1016/s0896-6273(03)00568-3
de Freitas, L. F., and Hamblin, M. R. (2016). Proposed mechanisms of photobiomodulation or low-level light therapy. IEEE J. Sel. Top. Quantum Electron. 22:700412. doi: 10.1109/JSTQE.2016.2561201
de Lima, F. M., Albertini, R., Dantas, Y., Maia-Filho, A. L., Santana Cde, L., Castro-Faria-Neto, H. C., et al. (2013). Low-level laser therapy restores the oxidative stress balance in acute lung injury induced by gut ischemia and reperfusion. Photochem. Photobiol. 89, 179–188. doi: 10.1111/j.1751-1097.2012.01214.x
Del Tredici, K., and Braak, H. (2016). Review: sporadic Parkinson’s disease: development and distribution of α-synuclein pathology. Neuropathol. Appl. Neurobiol. 42, 33–50. doi: 10.1111/nan.12298
Desmet, K. D., Paz, D. A., Corry, J. J., Eells, J. T., Wong-Riley, M. T., Henry, M. M., et al. (2006). Clinical and experimental applications of NIR-LED photobiomodulation. Photomed. Laser Surg. 24, 121–128. doi: 10.1089/pho.2006.24.121
Dhillon, A. S., Tarbutton, G. L., Levin, J. L., Plotkin, G. M., Lowry, L. K., Nalbone, J. T., et al. (2008). Pesticide/environmental exposures and Parkinson’s disease in East Texas. J. Agromedicine 13, 37–48. doi: 10.1080/10599240801986215
Dorsey, E. R., Sherer, T., Okun, M. S., and Bloem, B. R. (2018). The emerging evidence of the parkinson pandemic. J. Parkinsons Dis. 8, S3–s8. doi: 10.3233/jpd-181474
El Massri, N., Lemgruber, A. P., Rowe, I. J., Moro, C., Torres, N., Reinhart, F., et al. (2017). Photobiomodulation-induced changes in a monkey model of Parkinson’s disease: changes in tyrosine hydroxylase cells and GDNF expression in the striatum. Exp. Brain Res. 235, 1861–1874. doi: 10.1007/s00221-017-4937-0
El Massri, N., Moro, C., Torres, N., Darlot, F., Agay, D., Chabrol, C., et al. (2016). Near-infrared light treatment reduces astrogliosis in MPTP-treated monkeys. Exp. Brain Res. 234, 3225–3232. doi: 10.1007/s00221-016-4720-7
El Massri, N., Weinrich, T. W., Kam, J. H., Jeffery, G., and Mitrofanis, J. (2018). Photobiomodulation reduces gliosis in the basal ganglia of aged mice. Neurobiol. Aging 66, 131–137. doi: 10.1016/j.neurobiolaging.2018.02.019
Fasano, A., Bove, F., Gabrielli, M., Petracca, M., Zocco, M. A., Ragazzoni, E., et al. (2013). The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov. Disord. 28, 1241–1249. doi: 10.1002/mds.25522
Ferree, A., and Shirihai, O. (2012). “Mitochondrial dynamics: the intersection of form and function,” in Mitochondrial Oxidative Phosphorylation, ed. B. Kadenbach (New York, NY: Springer), 13–40.
Findley, L. J. (2007). The economic impact of Parkinson’s disease. Parkinsonism Relat. Disord. 13, S8–S12. doi: 10.1016/j.parkreldis.2007.06.003
Ganeshan, V., Skladnev, N. V., Kim, J. Y., Mitrofanis, J., Stone, J., and Johnstone, D. M. (2019). Pre-conditioning with remote photobiomodulation modulates the brain transcriptome and protects against MPTP insult in mice. Neuroscience 400, 85–97. doi: 10.1016/j.neuroscience.2018.12.050
Hamblin, M. R. (2018). Mechanisms and mitochondrial redox signaling in photobiomodulation. Photochem. Photobiol. 94, 199–212. doi: 10.1111/php.12864
Hamilton, C. L., El Khoury, H., Hamilton, D., Nicklason, F., and Mitrofanis, J. (2019). “Buckets”: early observations on the use of red and infrared light helmets in Parkinson’s disease patients. Photobiomodul. Photomed. Laser Surg. 37, 615–622. doi: 10.1089/photob.2019.4663
Hasegawa, S., Goto, S., Tsuji, H., Okuno, T., Asahara, T., Nomoto, K., et al. (2015). Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One 10:e0142164. doi: 10.1371/journal.pone.0142164
Hermann, J., Longen, S., Weckbecker, D., and Depuydt, M. (2012). “Biogenesis of mitochondrial proteins,” in Mitochondrial Oxidative Phosphorylation, (Vol. 748) ed. B. Kadenbach (New York, Heidelberg, Dordrecht, London: Springer), 41–64.
Hoehn, M. M., and Yahr, M. D. (1998). Parkinsonism: onset, progression, and mortality. 1967. Neurology 50:318 and 16 pages following. doi: 10.1212/wnl.50.2.318
Huang, Y. Y., Chen, A. C., Carroll, J. D., and Hamblin, M. R. (2009). Biphasic dose response in low level light therapy. Dose Response 7, 358–383. doi: 10.2203/dose-response.09-027.Hamblin
Hyde, B. B., Twig, G., and Shirihai, O. S. (2010). Organellar vs. cellular control of mitochondrial dynamics. Semin. Cell Dev. Biol. 21, 575–581. doi: 10.1016/j.semcdb.2010.01.003
Ilic, S., Leichliter, S., Streeter, J., Oron, A., DeTaboada, L., and Oron, U. (2006). Effects of power densities, continuous and pulse frequencies, and number of sessions of low-level laser therapy on intact rat brain. Photomed. Laser Surg. 24, 458–466. doi: 10.1089/pho.2006.24.458
Johnstone, D. M., el Massri, N., Moro, C., Spana, S., Wang, X. S., Torres, N., et al. (2014). Indirect application of near infrared light induces neuroprotection in a mouse model of parkinsonism—an abscopal neuroprotective effect. Neuroscience 274, 93–101. doi: 10.1016/j.neuroscience.2014.05.023
Johnstone, D. M., Moro, C., Stone, J., Benabid, A. L., and Mitrofanis, J. (2015). Turning on lights to stop neurodegeneration: the potential of near infrared light therapy in Alzheimer’s and Parkinson’s disease. Front. Neurosci. 9:500. doi: 10.3389/fnins.2015.00500
Kadenbach, B. (2012). “Introduction to mitochondrial oxidative phosphorylation,” in Mitochondrial Oxidative Phosphorylation, (Vol. 748) ed. B. Kadenbach (New York, Heidelberg, Dordrecht, London: Springer), 1–12.
Kadenbach, B., Hüttemann, M., Arnold, S., Lee, I., and Bender, E. (2000). Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic. Biol. Med. 29, 211–221. doi: 10.1016/s0891-5849(00)00305-1
Karu, T. I. (1999). Primary and secondary mechanisms of action of visible to near-IR radiation on cells. J. Photochem. Photobiol. B Biol. 49, 1–17. doi: 10.1016/s1011-1344(98)00219-x
Karu, T. I., Pyatibrat, L. V., Kalendo, G. S., and Esenaliev, R. O. (1996). Effects of monochromatic low-intensity light and laser irradiation on adhesion of HeLa cells in vitro. Lasers Surg. Med. 18, 171–177. doi: 10.1002/(sici)1096-9101(1996)18:2<171::aid-lsm7>3.0.co;2-p
Kim, S., Kwon, S. H., Kam, T. I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103, 627.e7–641.e7. doi: 10.1016/j.neuron.2019.05.035
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Konnova, E., and Swanberg, M. (2018). Animal models of Parkinson’s disease Parkinson’s Disease: Pathogenesis and Clinical Aspects, eds T. B. Stoker and J. C. Greenland (Brisbane, AU: Codon Publications).
Liang, H. L., Ongwijitwat, S., and Wong-Riley, M. T. (2006a). Bigenomic functional regulation of all 13 cytochrome c oxidase subunit transcripts in rat neurons in vitro and in vivo. Neuroscience 140, 177–190. doi: 10.1016/j.neuroscience.2006.01.056
Liang, H. L., Whelan, H. T., Eells, J. T., Meng, H., Buchmann, E., Lerch-Gaggl, A., et al. (2006b). Photobiomodulation partially rescues visual cortical neurons from cyanide-induced apoptosis. Neuroscience 139, 639–649. doi: 10.1016/j.neuroscience.2005.12.047
Liang, C. L., Wang, T. T., Luby-Phelps, K., and German, D. C. (2007). Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson’s disease. Exp. Neurol. 203, 370–380. doi: 10.1016/j.expneurol.2006.08.015
Liang, H. L., Whelan, H. T., Eells, J. T., and Wong-Riley, M. T. (2008). Near-infrared light via light-emitting diode treatment is therapeutic against rotenone- and 1-methyl-4-phenylpyridinium ion-induced neurotoxicity. Neuroscience 153, 963–974. doi: 10.1016/j.neuroscience.2008.03.042
Lima, P. L. V., Pereira, C. V., Nissanka, N., Arguello, T., Gavini, G., Maranduba, C., et al. (2019). Photobiomodulation enhancement of cell proliferation at 660nm does not require cytochrome c oxidase. J. Photochem. Photobiol. B 194, 71–75. doi: 10.1016/j.jphotobiol.2019.03.015
Margulis, L. (1975). The microbes’ contribution to evolution. Biosystems 7, 266–292. doi: 10.1016/0303-2647(75)90034-9
McCarthy, T. J., De Taboada, L., Hildebrandt, P. K., Ziemer, E. L., Richieri, S. P., and Streeter, J. (2010). Long-term safety of single and multiple infrared transcranial laser treatments in Sprague-Dawley rats. Photomed. Laser Surg. 28, 663–667. doi: 10.1089/pho.2009.2581
Mester, E., Spiry, T., Szende, B., and Tota, J. G. (1971). Effect of laser rays on wound healing. Am. J. Surg. 122, 532–535. doi: 10.1016/0002-9610(71)90482-x
Montgomery, K. L., Yeh, A. J., Ho, J. S., Tsao, V., Mohan Iyer, S., Grosenick, L., et al. (2015). Wirelessly powered, fully internal optogenetics for brain, spinal and peripheral circuits in mice. Nat. Methods 12, 969–974. doi: 10.1038/nmeth.3536
Moro, C., El Massri, N., Darlot, F., Torres, N., Chabrol, C., Agay, D., et al. (2016). Effects of a higher dose of near-infrared light on clinical signs and neuroprotection in a monkey model of Parkinson’s disease. Brain Res. 1648, 19–26. doi: 10.1016/j.brainres.2016.07.005
Moro, C., Massri, N. E., Torres, N., Ratel, D., De Jaeger, X., Chabrol, C., et al. (2014). Photobiomodulation inside the brain: a novel method of applying near-infrared light intracranially and its impact on dopaminergic cell survival in MPTP-treated mice. J. Neurosurg. 120, 670–683. doi: 10.3171/2013.9.jns13423
Moro, C., Torres, N., Arvanitakis, K., Cullen, K., Chabrol, C., Agay, D., et al. (2017). No evidence for toxicity after long-term photobiomodulation in normal non-human primates. Exp. Brain Res. 235, 3081–3092. doi: 10.1007/s00221-017-5048-7
Muili, K. A., Gopalakrishnan, S., Meyer, S. L., Eells, J. T., and Lyons, J. A. (2012). Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by photobiomodulation induced by 670 nm light. PLoS One 7:e30655. doi: 10.1371/journal.pone.0030655
O’Brien, J. A., and Austin, P. J. (2019). Effect of photobiomodulation in rescuing lipopolysaccharide-induced dopaminergic cell loss in the male sprague–dawley rat. Biomolecules 9:381. doi: 10.3390/biom9080381
Ongwijitwat, S., and Wong-Riley, M. T. (2005). Is nuclear respiratory factor 2 a master transcriptional coordinator for all ten nuclear-encoded cytochrome c oxidase subunits in neurons? Gene 360, 65–77. doi: 10.1016/j.gene.2005.06.015
Oueslati, A., Lovisa, B., Perrin, J., Wagnières, G., van den Bergh, H., Tardy, Y., et al. (2015). Photobiomodulation suppresses α-synuclein-induced toxicity in an AAV-based rat genetic model of Parkinson’s disease. PLoS One 10:e0140880. doi: 10.1371/journal.pone.0140880
Passarella, S., Casamassima, E., Molinari, S., Pastore, D., Quagliariello, E., Catalano, I. M., et al. (1984). Increase of proton electrochemical potential and ATP synthesis in rat liver mitochondria irradiated in vitro by helium-neon laser. FEBS Lett. 175, 95–99. doi: 10.1016/0014-5793(84)80577-3
Passarella, S., and Karu, T. (2014). Absorption of monochromatic and narrow band radiation in the visible and near IR by both mitochondrial and non-mitochondrial photoacceptors results in photobiomodulation. J. Photochem. Photobiol. B 140, 344–358. doi: 10.1016/j.jphotobiol.2014.07.021
Passarella, S., Ostuni, A., Atlante, A., and Quagliariello, E. (1988). Increase in the ADP/ATP exchange in rat liver mitochondria irradiated in vitro by helium-neon laser. Biochem. Biophys. Res. Commun. 156, 978–986. doi: 10.1016/s0006-291x(88)80940-9
Peoples, C., Shaw, V. E., Stone, J., Jeffery, G., Baker, G. E., and Mitrofanis, J. (2012). Survival of dopaminergic amacrine cells after near-infrared light treatment in MPTP-treated mice. ISRN Neurol. 2012:850150. doi: 10.5402/2012/850150
Phillips, R. J., Walter, G. C., Wilder, S. L., Baronowsky, E. A., and Powley, T. L. (2008). α-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson’s disease? Neuroscience 153, 733–750. doi: 10.1016/j.neuroscience.2008.02.074
Pierron, D., Wildman, D. E., Hüttemann, M., Letellier, T., and Grossman, L. I. (2012). “Evolution of the couple cytochrome c and cytochrome c oxidase in primates,” in Mitochondrial Oxidative Phosphorylation, ed. B. Kadenbach (New York, Heidelberg, Dordrecht, London: Springer), 185–214.
Purushothuman, S., Nandasena, C., Johnstone, D. M., Stone, J., and Mitrofanis, J. (2013). The impact of near-infrared light on dopaminergic cell survival in a transgenic mouse model of parkinsonism. Brain Res. 1535, 61–70. doi: 10.1016/j.brainres.2013.08.047
Purves, D., Augustine, G. J., Fitzpatrick, D., Hall, W. C., LaMantia, A.-S., Mooney, R. D., et al. (2018). “Electrical signals of nerve cells,” in Neuroscience, 6th Edition, eds D. Purves, G. J. Augustine, D. Fitzpatrick, W. C. Hall, A. S. LaMantia, R. D. Mooney, M. L. Platt and L. E. White (New York, NY: Oxford University Press), 31–46.
Quirk, B. J., Desmet, K. D., Henry, M., Buchmann, E., Wong-Riley, M., Eells, J. T., et al. (2012a). Therapeutic effect of near infrared (NIR) light on Parkinson’s disease models. Front. Biosci. 4, 818–823. doi: 10.2741/421
Quirk, B. J., Torbey, M., Buchmann, E., Verma, S., and Whelan, H. T. (2012b). Near-infrared photobiomodulation in an animal model of traumatic brain injury: improvements at the behavioral and biochemical levels. Photomed. Laser Surg. 30, 523–529. doi: 10.1089/pho.2012.3261
Quirk, B. J., Sannagowdara, K., Buchmann, E. V., Jensen, E. S., Gregg, D. C., and Whelan, H. T. (2016). Effect of near-infrared light on in vitro cellular ATP production of osteoblasts and fibroblasts and on fracture healing with intramedullary fixation. J. Clin. Orthop. Trauma 7, 234–241. doi: 10.1016/j.jcot.2016.02.009
Ramsay, R. R., Kowal, A. T., Johnson, M. K., Salach, J. I., and Singer, T. P. (1987). The inhibition site of MPP+, the neurotoxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine is near the Q-binding site of NADH dehydrogenase. Arch. Biochem. Biophys. 259, 645–649. doi: 10.1016/0003-9861(87)90531-5
Ramsay, R. R., Salach, J. I., and Singer, T. P. (1986). Uptake of the neurotoxin 1-methyl-4-phenylpyridine (MPP+) by mitochondria and its relation to the inhibition of the mitochondrial oxidation of NAD+-linked substrates by MPP+. Biochem. Biophys. Res. Commun. 134, 743–748. doi: 10.1016/s0006-291x(86)80483-1
Reinhart, F., El Massri, N., Chabrol, C., Cretallaz, C., Johnstone, D. M., Torres, N., et al. (2016). Intracranial application of near-infrared light in a hemi-parkinsonian rat model: the impact on behavior and cell survival. J. Neurosurg. 124, 1829–1841. doi: 10.3171/2015.5.jns15735
Reinhart, F., El Massri, N., Darlot, F., Torres, N., Johnstone, D. M., Chabrol, C., et al. (2015). 810nm near-infrared light offers neuroprotection and improves locomotor activity in MPTP-treated mice. Neurosci. Res. 92, 86–90. doi: 10.1016/j.neures.2014.11.005
Reinhart, F., El Massri, N., Torres, N., Chabrol, C., Molet, J., Johnstone, D. M., et al. (2017). The behavioural and neuroprotective outcomes when 670nm and 810nm near infrared light are applied together in MPTP-treated mice. Neurosci. Res. 117, 42–47. doi: 10.1016/j.neures.2016.11.006
Richards, T. A., and Archibald, J. M. (2011). Cell evolution: gene transfer agents and the origin of mitochondria. Curr. Biol. 21, R112–R114. doi: 10.1016/j.cub.2010.12.036
Rochkind, S., Rousso, M., Nissan, M., Villarreal, M., Barr-Nea, L., and Rees, D. G. (1989). Systemic effects of low-power laser irradiation on the peripheral and central nervous system, cutaneous wounds, and burns. Lasers Surg. Med. 9, 174–182. doi: 10.1002/lsm.1900090214
Rojas, J. C., and Gonzalez-Lima, F. (2011). Low-level light therapy of the eye and brain. Eye Brain 3, 49–67. doi: 10.2147/eb.s21391
Santos, L., Olmo-Aguado, S. D., Valenzuela, P. L., Winge, K., Iglesias-Soler, E., Arguelles-Luis, J., et al. (2019). Photobiomodulation in Parkinson’s disease: a randomized controlled trial. Brain Stimul. 12, 810–812. doi: 10.1016/j.brs.2019.02.009
Sarti, P., Forte, E., Mastronicola, D., Giuffre, A., and Arese, M. (2012). Cytochrome c oxidase and nitric oxide in action: molecular mechanisms and pathophysiological implications. Biochim. Biophys. Acta 1817, 610–619. doi: 10.1016/j.bbabio.2011.09.002
Schneider, S. A., and Klein, C. (1993). “PINK1 type of young-onset parkinson disease,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens and A. Amemiya (Seattle, WA: University of Washington).
Schuepbach, W. M., Rau, J., Knudsen, K., Volkmann, J., Krack, P., Timmermann, L., et al. (2013). Neurostimulation for Parkinson’s disease with early motor complications. N. Engl. J. Med. 368, 610–622. doi: 10.1056/NEJMoa1205158
Sharma, S. K., Kharkwal, G. B., Sajo, M., Huang, Y. Y., De Taboada, L., McCarthy, T., et al. (2011). Dose response effects of 810 nm laser light on mouse primary cortical neurons. Lasers Surg. Med. 43, 851–859. doi: 10.1002/lsm.21100
Shaw, V. E., Peoples, C., Spana, S., Ashkan, K., Benabid, A. L., Stone, J., et al. (2012). Patterns of cell activity in the subthalamic region associated with the neuroprotective action of near-infrared light treatment in MPTP-treated mice. Parkinsons Dis. 2012:296875. doi: 10.1155/2012/296875
Shaw, V. E., Spana, S., Ashkan, K., Benabid, A. L., Stone, J., Baker, G. E., et al. (2010). Neuroprotection of midbrain dopaminergic cells in MPTP-treated mice after near-infrared light treatment. J. Comp. Neurol. 518, 25–40. doi: 10.1002/cne.22207
Sies, H. (2017). Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox. Biol. 11, 613–619. doi: 10.1016/j.redox.2016.12.035
Sommer, A. P. (2019). Mitochondrial cytochrome c oxidase is not the primary acceptor for near infrared light-it is mitochondrial bound water: the principles of low-level light therapy. Ann. Transl. Med. 7:S13. doi: 10.21037/atm.2019.01.43
Soong, N. W., Hinton, D. R., Cortopassi, G., and Arnheim, N. (1992). Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 2, 318–323. doi: 10.1038/ng1292-318
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Sterky, F. H., Lee, S., Wibom, R., Olson, L., and Larsson, N. G. (2011). Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. U S A 108, 12937–12942. doi: 10.1073/pnas.1103295108
Stone, J. R., and Yang, S. (2006). Hydrogen peroxide: a signaling messenger. Antioxid. Redox Signal. 8, 243–270. doi: 10.1089/ars.2006.8.243
Sulzer, D., and Surmeier, D. J. (2013). Neuronal vulnerability, pathogenesis and Parkinson’s disease. Mov. Disord. 28, 715–724. doi: 10.1002/mds.25095
Surmeier, D. J., and Schumacker, P. T. (2013). Calcium, bioenergetics and neuronal vulnerability in Parkinson’s disease. J. Biol. Chem. 288, 10736–10741. doi: 10.1074/jbc.R112.410530
Tan, A. H., Mahadeva, S., Thalha, A. M., Gibson, P. R., Kiew, C. K., Yeat, C. M., et al. (2014). Small intestinal bacterial overgrowth in Parkinson’s disease. Parkinsonism Relat. Disord. 20, 535–540. doi: 10.1016/j.parkreldis.2014.02.019
Tanner, C. M., Kamel, F., Ross, G. W., Hoppin, J. A., Goldman, S. M., Korell, M., et al. (2011). Rotenone, paraquat and Parkinson’s disease. Environ. Health Perspect. 119, 866–872. doi: 10.1289/ehp.1002839
Titova, N., and Chaudhuri, K. R. (2018). Non-motor Parkinson disease: new concepts and personalised management. Med. J. Aust. 208, 404–409. doi: 10.5694/mja17.00993
Travagli, R. A., Gillis, R. A., Rossiter, C. D., and Vicini, S. (1991). Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. Am. J. Physiol. 260, G531–G536. doi: 10.1152/ajpgi.1991.260.3.G531
Trimmer, P. A., Schwartz, K. M., Borland, M. K., De Taboada, L., Streeter, J., and Oron, U. (2009). Reduced axonal transport in Parkinson’s disease cybrid neurites is restored by light therapy. Mol. Neurodegener. 4:26. doi: 10.1186/1750-1326-4-26
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Villani, G., and Attardi, G. (1997). in vivo control of respiration by cytochrome c oxidase in wild-type and mitochondrial DNA mutation-carrying human cells. Proc. Natl. Acad. Sci. U S A 94, 1166–1171. doi: 10.1073/pnas.94.4.1166
Vos, M., Lovisa, B., Geens, A., Morais, V. A., Wagnieres, G., van den Bergh, H., et al. (2013). Near-infrared 808 nm light boosts complex IV-dependent respiration and rescues a Parkinson-related pink1 model. PLoS One 8:e78562. doi: 10.1371/journal.pone.0078562
Williams, J. T., North, R. A., Shefner, S. A., Nishi, S., and Egan, T. M. (1984). Membrane properties of rat locus coeruleus neurones. Neuroscience 13, 137–156. doi: 10.1016/0306-4522(84)90265-3
Wong-Riley, M. T., Bai, X., Buchmann, E., and Whelan, H. T. (2001). Light-emitting diode treatment reverses the effect of TTX on cytochrome oxidase in neurons. Neuroreport 12, 3033–3037. doi: 10.1097/00001756-200110080-00011
Wong-Riley, M. T., Liang, H. L., Eells, J. T., Chance, B., Henry, M. M., Buchmann, E., et al. (2005). Photobiomodulation directly benefits primary neurons functionally inactivated by toxins: role of cytochrome c oxidase. J. Biol. Chem. 280, 4761–4771. doi: 10.1074/jbc.M409650200
Xu, C., Zhang, J., Mihai, D. M., and Washington, I. (2014). Light-harvesting chlorophyll pigments enable mammalian mitochondria to capture photonic energy and produce ATP. J. Cell Sci. 127, 388–399. doi: 10.1242/jcs.134262
Xuan, W., Agrawal, T., Huang, L., Gupta, G. K., and Hamblin, M. R. (2015). Low-level laser therapy for traumatic brain injury in mice increases brain derived neurotrophic factor (BDNF) and synaptogenesis. J. Biophotonics 8, 502–511. doi: 10.1002/jbio.201400069
Xuan, W., Huang, L., and Hamblin, M. R. (2016). Repeated transcranial low-level laser therapy for traumatic brain injury in mice: biphasic dose response and long-term treatment outcome. J. Biophotonics 9, 1263–1272. doi: 10.1002/jbio.201500336
Xuan, W., Vatansever, F., Huang, L., and Hamblin, M. R. (2014). Transcranial low-level laser therapy enhances learning, memory and neuroprogenitor cells after traumatic brain injury in mice. J. Biomed. Opt. 19:108003. doi: 10.1117/1.JBO.19.10.108003
Xuan, W., Vatansever, F., Huang, L., Wu, Q., Xuan, Y., Dai, T., et al. (2013). Transcranial low-level laser therapy improves neurological performance in traumatic brain injury in mice: effect of treatment repetition regimen. PLoS One 8:e53454. doi: 10.1371/journal.pone.0053454
Yang, D., Zhao, D., Ali Shah, S. Z., Wu, W., Lai, M., Zhang, X., et al. (2019). The role of the gut microbiota in the pathogenesis of Parkinson’s disease. Front. Neurol. 10:1155. doi: 10.3389/fneur.2019.01155
Ying, R., Liang, H. L., Whelan, H. T., Eells, J. T., and Wong-Riley, M. T. (2008). Pretreatment with near-infrared light via light-emitting diode provides added benefit against rotenone- and MPP+-induced neurotoxicity. Brain Res. 1243, 167–173. doi: 10.1016/j.brainres.2008.09.057
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Zhang, M. L., Hultborn, H., and Sukiasyan, N. (2012). “The distribution of calcium channel Cav1.3 in the central nervous system and its functions in relation to motor control,” in Calcium Channels: Properties, Functions and Regulation, ed. M. R. Figgins (Hauppauge, New York: Nova Science Publishers Inc.), 1–48.
Zhao, Y. J., Tan, L. C., Au, W. L., Heng, D. M., Soh, I. A., Li, S. C., et al. (2013). Estimating the lifetime economic burden of Parkinson’s disease in Singapore. Eur. J. Neurol. 20, 368–374. doi: 10.1111/j.1468-1331.2012.03868.x
Zimmer, C. (2009). Origins. On the origin of eukaryotes. Science 325, 666–668. doi: 10.1126/science.325_666
Zivin, J. A., Albers, G. W., Bornstein, N., Chippendale, T., Dahlof, B., Devlin, T., et al. (2009). Effectiveness and safety of transcranial laser therapy for acute ischemic stroke. Stroke 40, 1359–1364. doi: 10.1161/STROKEAHA.109.547547
Keywords: Parkinson’s disease, mitochondria dysfunction, neurodegeneration, energy dysregulation, near infrared
Citation: Foo ASC, Soong TW, Yeo TT and Lim K-L (2020) Mitochondrial Dysfunction and Parkinson’s Disease—Near-Infrared Photobiomodulation as a Potential Therapeutic Strategy. Front. Aging Neurosci. 12:89. doi: 10.3389/fnagi.2020.00089
Received: 20 December 2019; Accepted: 17 March 2020;
Published: 03 April 2020.
Edited by:
Micaela Morelli, University of Cagliari, ItalyReviewed by:
John Mitrofanis, University of Sydney, AustraliaCopyright © 2020 Foo, Soong, Yeo and Lim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kah-Leong Lim, a2FobGVvbmcubGltQG50dS5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.