 Maria Antonietta Casu1
Maria Antonietta Casu1 Ignazia Mocci1
Ignazia Mocci1 Raffaella Isola2
Raffaella Isola2 Augusta Pisanu3
Augusta Pisanu3 Laura Boi2
Laura Boi2 Giovanna Mulas4
Giovanna Mulas4 Nigel H. Greig5
Nigel H. Greig5 Maria Dolores Setzu2*†
Maria Dolores Setzu2*† Anna R. Carta2*†
Anna R. Carta2*†- 1CNR Institute of Translational Pharmacology, Cagliari, Italy
- 2Department of Biomedical Sciences, University of Cagliari, Cagliari, Italy
- 3CNR Institute of Neuroscience, Cagliari, Italy
- 4Department of Life and Environmental Sciences, University of Cagliari, Cagliari, Italy
- 5National Institute of Aging (NIA), Drug Design & Development Section, Translational Gerontology Branch, Baltimore, MD, United States
The search for new disease-modifying drugs for Parkinson’s disease (PD) is a slow and highly expensive process, and the repurposing of drugs already approved for different medical indications is becoming a compelling alternative option for researchers. Genetic variables represent a predisposing factor to the disease and mutations in leucine-rich repeat kinase 2 (LRRK2) locus have been correlated to late-onset autosomal-dominant PD. The common fruit fly Drosophila melanogaster carrying the mutation LRRK2 loss-of-function in the WD40 domain (LRRK2WD40), is a simple in vivo model of PD and is a valid tool to first evaluate novel therapeutic approaches to the disease. Recent studies have suggested a neuroprotective activity of immunomodulatory agents in PD models. Here the immunomodulatory drug Pomalidomide (POM), a Thalidomide derivative, was examined in the Drosophila LRRK2WD40 genetic model of PD. Mutant and wild type flies received increasing POM doses (1, 0.5, 0.25 mM) through their diet from day 1 post eclosion, until postnatal day (PN) 7 or 14, when POM’s actions were evaluated by quantifying changes in climbing behavior as a measure of motor performance, the number of brain dopaminergic neurons and T-bars, mitochondria integrity. LRRK2WD40 flies displayed a spontaneous age-related impairment of climbing activity, and POM significantly and dose-dependently improved climbing performance both at PN 7 and PN 14. LRRK2WD40 fly motor disability was underpinned by a progressive loss of dopaminergic neurons in posterior clusters of the protocerebrum, which are involved in the control of locomotion, by a low number of T-bars density in the presynaptic bouton active zones. POM treatment fully rescued the cell loss in all posterior clusters at PN 7 and PN 14 and significantly increased the T-bars density. Moreover, several damaged mitochondria with dilated cristae were observed in LRRK2WD40 flies treated with vehicle but not following POM. This study demonstrates the neuroprotective activity of the immunomodulatory agent POM in a genetic model of PD. POM is an FDA-approved clinically available and well-tolerated drug used for the treatment of multiple myeloma. If further validated in mammalian models of PD, POM could rapidly be clinically tested in humans.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder whose pathology primarily targets dopaminergic neurons of the substantia nigra pars compacta, together with non-motor areas of the CNS. This leads to typical PD-associated motor symptoms, which includes bradykinesia, tremor, and rigidity, as well as a range of non-motor symptoms, typified by postural instability, cognitive impairment and olfactory deficits (Parkinson, 2002; Dauer and Przedborski, 2003; Lees et al., 2009; Chaudhuri and Odin, 2010; Erkkinen et al., 2018). The degeneration of dopamine neurons in the substantia nigra is progressive and motor symptoms appear predictably when cell loss exceeds 50 percent.
The development of new drugs for the treatment of PD has been a slow and highly expensive process and, to date, has led to the approval of drugs that have proved to be symptomatic in their efficacy, but ineffective in slowing disease progression. As a response to these limitations and in the light of increasing knowledge of PD neuropathology, the pre-clinical and clinical testing of drugs with regulatory-approved clinical safety data, namely drug-repurposing, has become a compelling option for the pharmaceutical industry and researchers in the search for new PD therapeutics. Drug repurposing or repositioning is a strategy aimed at identifying new uses for already approved or investigational drugs, that fall outside their original medical indication (Brundin et al., 2013; Pushpakom et al., 2019).
Neuroinflammatory mechanisms are recognized as contributors to the neuropathology of PD and are classically characterized by reactive microgliosis with excessive production of proinflammatory molecules within degenerating mesencephalic areas (Troncoso-Escudero et al., 2018). Under physiological conditions, neuroinflammatory responses are finely orchestrated via a regulated production of cytokines, growth factors and toxic free radicals. On the contrary, in the case of PD, deregulation of neuroinflammatory responses occurs, and the chronic release of inflammatory cytokines, such as tumor necrosis factor (TNF)-α, is regarded as a principal pathological contributor to the ensuing progressive neurodegeneration (Joers et al., 2017). In this light, pharmacologically targeting the mechanisms underpinning cytokine production or actions may provide a compelling disease-modifying strategy for PD (Martinez and Peplow, 2018).
Based on the recognized role of neuroinflammation in PD neuropathology, evaluating commercially available immunomodulatory drugs for repositioning in PD has been considered an auspicious approach (Martinez and Peplow, 2018). Different classes of clinically available drugs active on the immune system have been investigated across various experimental models of PD, suggesting a benefit in slowing the disease progression and the development of motor symptoms (Van der Perren et al., 2015; Ren et al., 2017; Zhao et al., 2017). In recent years, immunomodulatory drugs, such as Thalidomide and its derivatives Lenalidomide and Pomalidomide (POM), have been appraised for the treatment of neurological disorders with a neuroinflammatory component; however, their potential utility in PD models has, to date, been poorly investigated (Tweedie et al., 2011). Thalidomide and derivatives display a potent biological effect on cytokine-mediated responses, acting primarily through the inhibition of TNF-α production via posttranslational mechanisms, with consequent dampening of the inflammatory cascade (Sampaio et al., 1991; Moreira et al., 1993; Tweedie et al., 2011; Chanan-Khan et al., 2013; Terpos et al., 2013). Among Thalidomide-derived immunomodulatory compounds, POM holds particular interest for its potent anti-TNF-α activity at significantly lower concentrations than the parent compound, as described in embryos and in vitro assays (Mahony et al., 2013). Moreover, POM displayed less adverse effects than Thalidomide and Lenalidomide, in relation to its teratogenic, anti-angiogenic and neurotoxic activity (Mahony et al., 2013; Vargesson et al., 2013).
Although the majority of human PD cases are idiopathic, genetic variables may represent a predisposing factor to the disease, and mutations in several specific genes have been linked to familial forms of PD. Among them, multiple mutations in the leucine-rich repeat kinase 2 (LRRK2) gene have been correlated to late-onset autosomal dominant PD (Kumari and Tan, 2009; Hernandez et al., 2016), accounting for up to 13% of familial PD cases and have been detected in 1–2% of idiopathic PD cases, making LRRK2 the most commonly linked PD gene. LRRK2 holds a dual enzymatic activity with two domains involved, namely the N-terminal and the C-terminal WD40 domain (Mills et al., 2012). In particular, the missense substitution G2385R within the WD40 domain leads to a partial loss-of-function of LRRK2, and is pathologically relevant for PD, being associated with an increased risk of developing idiopathic PD in Chinese and Korean ethnicity (Tan et al., 2009; Carrion et al., 2017).
The common fruit fly Drosophila melanogaster (Dm) is a useful organism for modeling neurodegenerative diseases with a translational value, carrying 75% homology with human disease genes (Bilen and Bonini, 2005). Dm carrying the LRRK2 loss-of-function mutation in the WD40 domain (LRRKWD40) is a simple in vivo model of PD that recapitulates key features of the disease, including motor impairment and mitochondrial abnormalities (Lee et al., 2012; De Rose et al., 2016; Hewitt and Whitworth, 2017). Of note, signaling pathways that regulate immune responses are highly conserved throughout evolution (Buchmann, 2014) and cytokines are important mediators of the innate immune system used by Drosophila to fight pathogen invasion (Vanha-Aho et al., 2016). Specifically, Drosophila has the ortholog of the tumor necrosis factor (TNF) Eiger as well as of TNF receptor (TNFR) Wengen (Moreno et al., 2002; Igaki and Miura, 2014). Genetic studies have identified a high homology of Eiger with the human gene, and evolutionarily conserved functions (Moreno et al., 2002; Salazar-Jaramillo et al., 2014). Therefore, the Drosophila is a useful and simple model for targeting cytokines with therapeutic agents.
Here, we first characterized the progressive dopaminergic neuronal loss in the Drosophila LRRK2WD40 genetic model of PD. Thereafter, we investigated the neuroprotective outcome of the immunomodulatory agent POM in this model. Specifically, POM activity was evaluated across ages in fly motor performance and loss of dopamine neurons in anatomically identified cell clusters within the brain. Ultrastructural analysis of mitochondrial integrity, and of T-bars as the essential part of the molecular machinery mediating neurotransmitter release in Drosophila, was also performed (Wagh et al., 2006).
Materials and Methods
Adult wild type (WT; Oregon-R) and LRRKWD40 mutants (LRRKex1, #34750, from Bloomington Stock Center) Dm males were used. After emergence from pupae, WT or LRRKWD40 mutant males were separated from females, reared and tested at ages 1–7–14 days (respectively PN 1, PN 7, PN 14). WT and mutant flies were reared on a standard cornmeal-yeast-agar medium in controlled environmental conditions (24–25°C; 60% relative humidity; light/dark = 12/12 h). In addition, groups of mutant and WT flies were reared on the standard medium supplemented with increasing concentrations of POM (0, 25–0, 5–1–2 mM) for 1 and 2 weeks.
Climbing Assay
The climbing assay (negative geotaxis assay) was used to assess locomotor ability (Liu et al., 2008; De Rose et al., 2016). Climbing data were obtained from different age groups (PN 1, PN 7 and PN 14) of untreated and treated WT and LRRK2WD40 mutants. Cohorts of 60 flies from each experimental group were subjected to the assay. Flies were placed individually in a vertically-positioned plastic tube (length 10 cm; diameter 1.5 cm) and tapped to the bottom. Climbing time (s) was recorded upon crossing a line drawn at 6 cm from the bottom. The number of flies that could climb onto, or above this line within 10 s was recorded and expressed as a percentage of total flies evaluated. Data were expressed as the average ± standard error of the mean (SEM) from three different experiments.
Immunohistochemistry
Free-floating fluorescent immunostaining for Tyrosine Hydroxylase (TH) was performed on the whole dissected adult brain from 6 to 8 flies from each experimental group.
LRRK2WD40 and WT Drosophila were anesthetized on ice, and brains were rapidly dissected and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS). After fixation and washes in PBS, whole brains were incubated with the TH primary antibody (1:100; AB 152 Millipore) and 10% normal donkey serum in PBS + 0.3% Tween 20 (PBST), at 4°C for 72 h. After washing, the brains were incubated with a donkey anti-rabbit Alexa Fluor 594 secondary antibody (1:200 Jackson ImmunoResearch, West Grove, PA, USA) and 10% normal donkey serum in PBST at 4°C for another 72 h. Afterward, the brains were mounted on glass slides and coverslipped with Vectashield.
Fluorescent images were captured with a fluorescence spinning disk confocal microscope (Crisel Instruments). The brains were scanned through Z-stacks (63× objective) and the number of TH-positive neurons was calculated in identified cell clusters, including paired posterior lateral clusters PPL1, PPL2, paired posterior medial clusters PPM1/2, PPM3, paired anterior lateral and medial clusters PAL and PAM. Cells were counted manually in both hemispheres for each stack via NIH ImageJ software, by an observer blinded to the treatments.
Electron Microscopy Analysis
LRRK2WD40 mutants untreated or treated with POM 0.5 mM (n = 3) were anesthetized on ice, and brains were rapidly dissected and fixed in a mixture of 1% paraformaldehyde and 1.25% glutaraldehyde in 0.15 M cacodylate buffer. After fixation and rinsing in the same buffer, brains were post-fixed with 1% osmium tetroxide in distilled water for 2 h and then stained overnight with 0.5% uranyl acetate at 4°C. Brains were then dehydrated in a graded acetone series, and embedded in EPON resin. To identify the protocerebrum, semi-thin coronal sections of the whole brains, cut with a Reichert Supernova ultramicrotome, were stained with toluidine blue. Ultrathin sections (90 nm) were observed under a JEOL JEM 1400 Plus electron microscope, equipped with a CCD camera, at an acceleration voltage of 80 kV.
Morphometry: the total number of mitochondria, the percentage of mitochondria with swollen cristae (expressed as the percentage of mitochondria displaying swollen cristae out of total mitochondria with discernible cristae in the sampled area) and the T-bars density was evaluated in the unitary area (25 μm2) within dorsal or ventral protocerebrum, in each experimental group. Forty to fifty unitary area fields for each area were considered for each brain. More than 10,970 mitochondria and 1,400 T-bars were randomly sampled on 504 non-overlapping micrographs at a final magnification of 10,000×. Swollen cristae were recognized when the individual cristae size (i.e., the distance between two contiguous membranes of one crista) doubled the average cristae size. T-bars were unambiguously identified at presynaptic active zones by the presence of T-shaped electron-dense projections.
Statistics
Statistically significant differences between groups in the behavioral and immunohistochemical analysis were evaluated by one-way or two-way ANOVA followed by Tukey’s multiple comparisons test when appropriate. TEM analysis was performed Mann–Whitney Rank Sum Test as data did not follow a normal distribution. A P-value ≤ 0.05 was considered significant.
Results
LRRK2WD40 Mutants Display Cardinal Features of PD
We first evaluated LRRK2WD40 mutants, as compared to WT Oregon-R flies of increasing ages (PN 1–7–14), for motor impairment and loss of dopaminergic neurons as these are two cardinal features of PD.
The climbing assay (negative geotaxis assay) was used to assess locomotor ability. The analysis of climbing activity showed that LRRK2WD40 mutants developed an age-related motor impairment measured as the time spent to reach the target. At PN 1 LRRK2WD40 displayed a climbing activity similar to WT flies, while at PN 7 and PN 14 LRRK2WD40 mutants displayed a significant motor impairment as compared to WT flies, as shown by the strain effect (F(1,472) = 59.64, P < 0.0001), age effects (F(2,472) = 24.80, P < 0.0001) and strain × age interaction (F(2,472) = 5.271, P < 0.01; Figure 1A). Moreover, the motor impairment was significantly greater at PN 14 than PN 7, suggesting a progressive deterioration of motor abilities (P < 0.05 as compared to PN14 by Tukey’s post hoc test; Figure 1A).
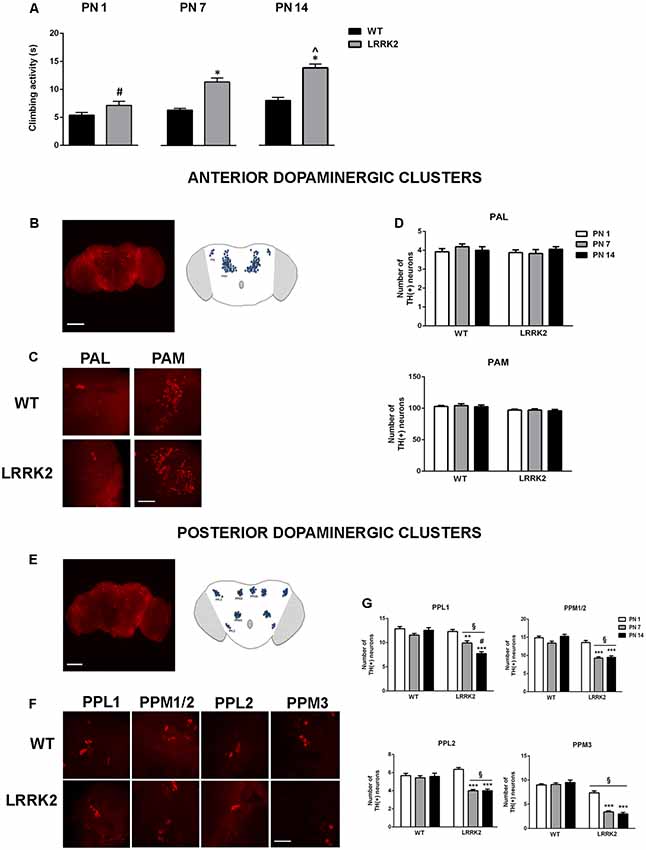
Figure 1. (A) Climbing behavior of wild type (WT) and leucine-rich repeat kinase 2 (LRRK2)WT40 (LRRK2) flies at postnatal days (PN) 1, 7, 14. Bars represent the mean ± SEM of 60 flies per group. *p < 0.001 vs. WT, #p < 0.001 vs. LRRK2 at PN 7 and PN 14 days, respectively; ∧p < 0.05 vs. LRRK2 PN 7, by two-way ANOVA followed by Tukey’s post hoc. (B,C) Representative images (10×) of the anterior brain (B) and higher magnification (63×) of anterior dopaminergic clusters PAM and PAL (C) of the Drosophila brain. (D) Number of dopaminergic neurons in the anterior dopaminergic clusters PAM and PAL in WT and LRRK2 mutants at PN 1, 7, 14. (E–F) Representative images (10×) of the posterior brain (E) and higher magnification (63×) of posterior dopaminergic clusters PPL1, PPL2, PPM1/2, PPM3 (F) of the Drosophila brain. (G) Number of dopaminergic neurons in the posterior dopaminergic clusters in WT and LRRK2 mutants at PN 1, 7, 14. §p < 0.05 vs. the corresponding WT; **,***p < 0.05 and 0.01 vs. LRRK2 PN 1; #p < 0.05 vs. PN7, by two-way ANOVA followed by Tukey’s post hoc.
We then evaluated the integrity of dopaminergic neurons in anterior and posterior neuronal clusters of the Drosophila brain via analysis of TH IR (Figures 1B–G). The number of anterior dopamine cell clusters proved to be stable across both WT flies and LRRK2WD40 mutants across all ages analyzed (Figures 1B–D). In contrast, dopaminergic neurons of posterior clusters underwent an age-related reduction in LRRK2WD40 but not in WT flies (P < 0.01 as compared to PN 1 by Tukey’s post hoc test), in accord with the role played by these nuclei in Drosophila motor activity (Figures 1E–G). Of note, the loss of dopaminergic neurons in PPL1 was gradual, with a lower number of neurons at PN 14 as compared with PN 7 (P < 0.05 vs. PN 7), in line with the progressive deterioration of motor ability (Figure 1E). Interestingly, LRRK2WD40 flies showed a significantly lower number of dopaminergic neurons within PPM3 as compared with WT flies, already at PN 1 (Figure 1G).
POM Prevented the Age-Dependent Motor Impairment in LRRK2WD40 Drosophila
LRRK2WD40 and WT flies were exposed to increasing doses of POM in their diet (0.25–0.5–1 mM) starting from PN 1, to evaluate the effect on locomotor ability parameters. The motor test was performed at PN 7 and PN 14, when the climbing activity and the percentage of flies reaching the target within 10 s was measured. The exposure to 0.5 and 1 mM POM for 7 days (PN 7) reduced the climbing time in LRRK2WD40 as compared to vehicle-exposed mutants (strain effect: F(1,628) = 19.97 P < 0.0001; treatment effect: F(3,628) = 2.681 P < 0.05; strain × treatment interaction: F(3,628) = 5.623, P < 0.001; Figure 2A), whereas the lowest POM concentration (0.25 mM) failed to produce any effect at this age. After 14 days of treatment (PN 14) a recovery of motor disability in the LRRK2WD40 was observed with all POM concentrations, as shown by the reduction of climbing time as compared to vehicle-exposed flies (strain: F(1,560) = 14.93, P < 0.001; treatment: F(3,560) = 2.892, P < 0.05; strain × treatment interaction: F(3,560) = 7.783, P < 0.0001). The motor activity of WT flies was unaffected by POM at all tested concentrations (Figures 2A,B).
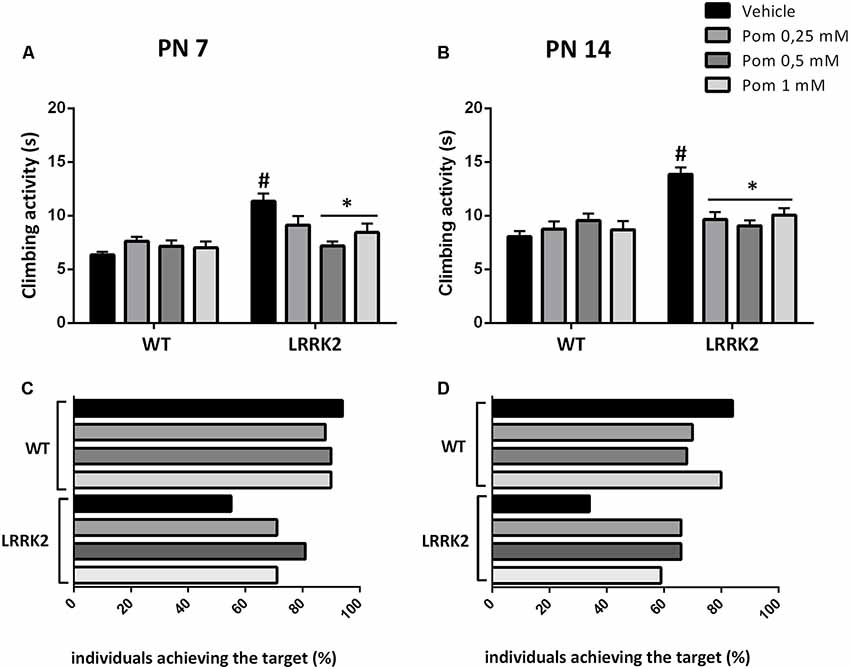
Figure 2. (A,B) Climbing activity of WT and LRRK2 mutant flies treated with increasing concentrations of pomalidomide (POM) in their diet at PN 7 (A) and 14 (B). (C,D) Percentage of WT and LRRK2 individuals reaching the target within 10 s. #p < 0.01 vs. the corresponding WT; *p < 0.01 vs. vehicle, by two-way ANOVA followed by Tukey’s post hoc.
As an additional parameter of motor ability, we counted the percentage of WT and LRRK2WD40 flies completing the climbing test within 10 s (Figures 2C,D). We found that at PN 7, and even more so at PN 14, the percent of flies reaching the target was lower as compared to WT flies of the same age (respectively, 55% and 34% as compared to 94% in WT; Figures 2C,D). Moreover, exposure to all concentrations of POM for 7 or 14 days increased the number of LRRK2WD40 flies that reached the target at PN 7 and PN 14 (Figures 2C,D).
POM Prevented the Neuronal Loss in LRRK2WD40Drosophila
Flies were sacrificed after the climbing test to quantitatively evaluate POM’s actions on TH-IR in anterior and posterior brain clusters of dopaminergic neurons (Figures 3, 4). Since POM displayed a maximal behavioral effect at the concentration of 0.5 mM, with no further improvement at 1 mM, only brains from flies exposed to 0.25 and 0.5 mM were processed for TH immunohistochemistry.
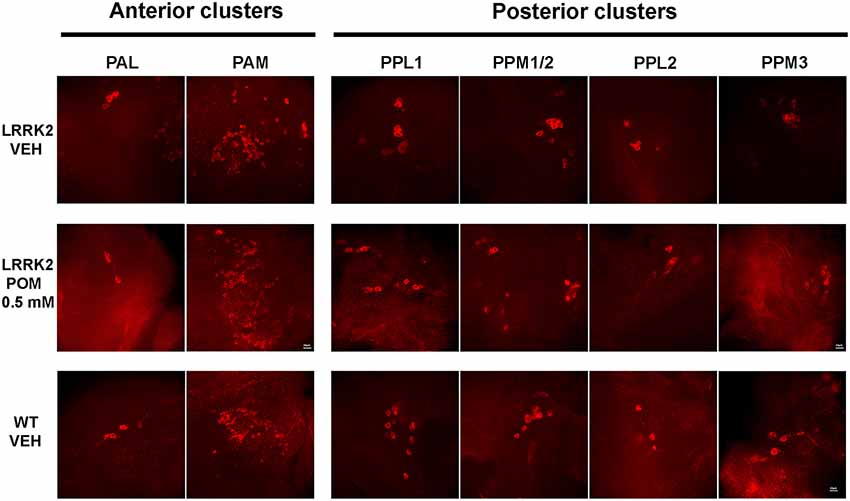
Figure 3. Representative images (63×) of tyrosine hydroxylase (TH)-positive neurons in anterior (PAL, PAM) and posterior (PPLs, PPMs) dopaminergic clusters, in WT and LRRK2 flies treated with vehicle or with POM 0.5 mM for 14 days (PN 14).
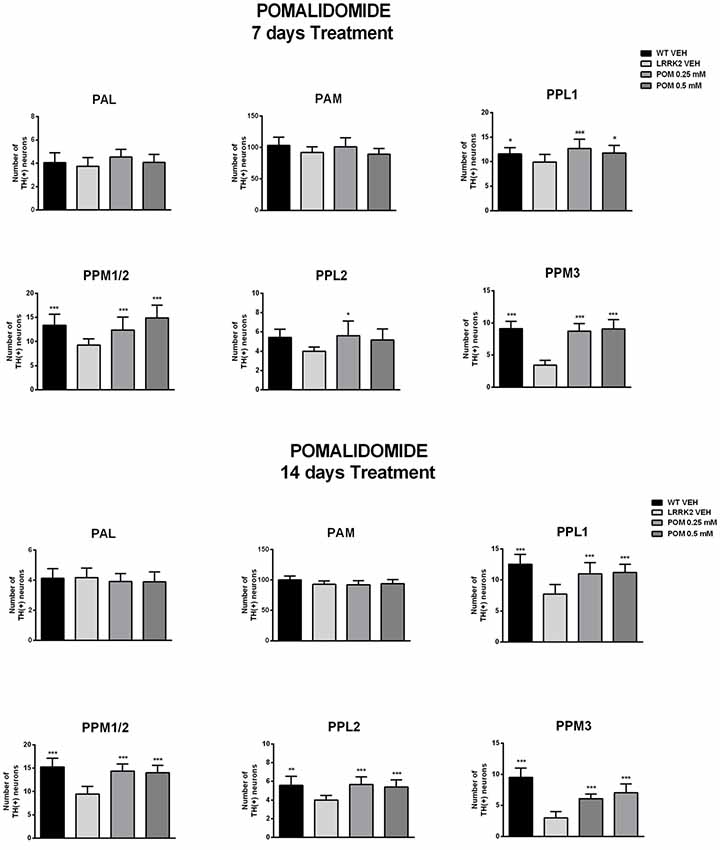
Figure 4. POM effect on the number of TH-positive neurons in anterior (PAM, PAM) and posterior (PPLs and PPMs) dopaminergic cluster of the Drosophila brain, from flies treated with vehicle, 0.25, 0.5 mM POM, after 7 and 14 days of treatment (PN 7 and PN 14). *,***p < 0.05 or 0.01 vs. LRRK2 VEH, by Two-way ANOVA followed by Tukey’s post hoc.
The analysis of TH-IR confirmed that neuronal loss was confined to posterior clusters in LRRK2WD40 mutants and showed that treatment with POM prevented it. Specifically, exposure to POM 0.25 and 0.5 mM similarly increased the number of TH-positive neurons within the posterior clusters of the brain in LRRK2WD40 flies, namely PPL1, PPL2, PPM1/2 and PPM3 (Figure 4). Importantly, POM fully prevented the loss of dopamine neurons at PN 7 as well as at PN 14, when the neuronal loss in LRRK2WD40 was greater, in accord with the complete prevention of motor deficits. Exposure to POM did not produce any effect on TH-IR in WT flies (data not shown).
POM Prevented the Loss of T-bars and Mitochondrial Damage in LRRK2WD40 Drosophila
Transmission electron microscopy (TEM) analysis was conducted on LRRK2WD40 flies treated with vehicle or with 0.5 mM POM for 14 days. Ultrastructural morphology of Drosophila LRRK2WD40 brain was investigated both in the dorsal and ventral protocerebrum and showed longitudinally cut axons and terminal boutons with numerous synaptic vesicles and T-bar shaped presynaptic densities (Figures 5A,B). The morphometric analysis did not reveal any difference in the number of total mitochondria between control and treated LRRK2WD40 (Figure 5C). Inside axons and terminal boutons of vehicle-treated LRRK2WD40 flies several pleomorphic mitochondria were present, some displaying swollen cristae (Figure 5A). POM treatment clearly decreased the occurrence of mitochondria with swollen cristae as compared with vehicle treatment, both in dorsal and ventral protocerebrum (p < 0.01; Figures 5B,C). Moreover, POM significantly increased the number of T-bar in the presynaptic bouton active zones of the dorsal protocerebrum as compared to vehicle treatment (p < 0.01; Figure 5C). Exposure of WT flies to POM did not produce any effect on mitochondria number and morphology, nor in the number of T-bars (data not shown).
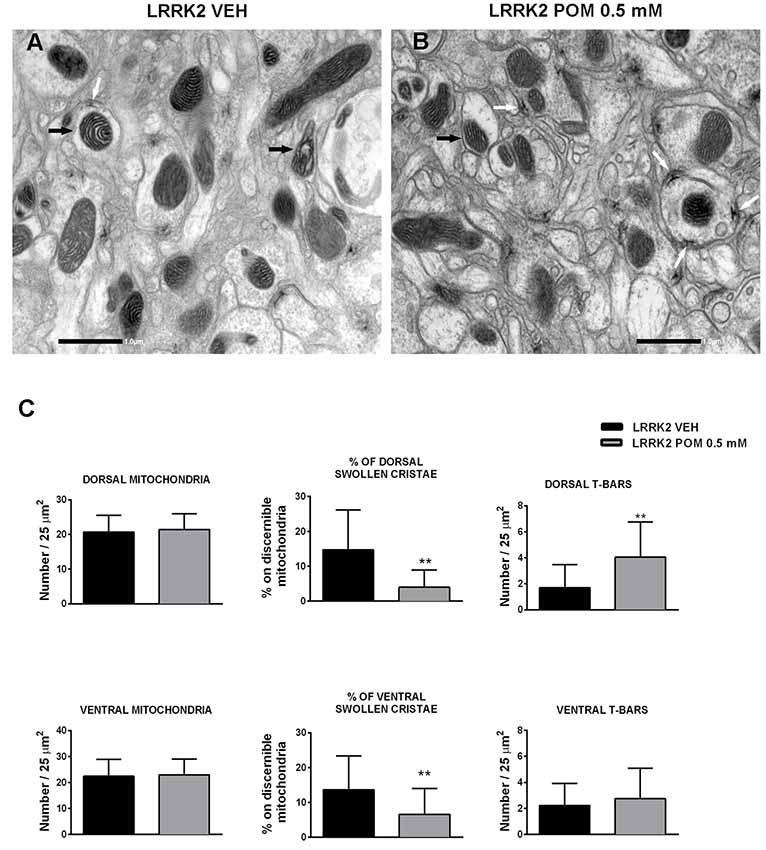
Figure 5. Representative images of mitochondria (10,000×) acquired from the protocerebrum of LRRK2 Drosophila treated with vehicle (A) or with POM 0.5 mM (B). Black arrows indicate mitochondria with swollen cristae in (A) and with normal cristae in (B); white arrows indicate T-Bars. (C) POM effect on the number of total mitochondria, percentage of swollen cristae and T-Bars in the dorsal and ventral protocerebrum. **p < 0.01 vs. LRRK2 VEH, by Mann–Whitney test.
Discussion
In the present study, we investigated the neuroprotective activity of the immunomodulatory drug POM in a transgenic Drosophila model of PD. We demonstrate that POM effectively prevented motor deficits, protected against the progressive loss of dopaminergic neurons, increased the number of T-bars and reduced mitochondrial damage. To our knowledge, this is the first report evaluating POM as a neuroprotective agent in a PD model. Moreover, this is the first report on the efficacy of an immunomodulatory drug in a transgenic model of PD, which complement studies investigating the neuroprotective efficacy of these agents in toxin-based or alpha-synuclein-based models (Martinez and Peplow, 2018).
Based on the recognized role of neuroinflammation and inflammatory cytokines in the neuropathology of PD, clinically available agents that act on the central immune system via an immunosuppressive or immunomodulatory mechanism have been tested in experimental models of PD (Martinez and Peplow, 2018). Among drugs in clinical use, the immunosuppressants Fingolimod (Gilenya), Tacrolimus (Fujimycin) and Cyclosporin have been reported as effective neuroprotective treatments in rodent models of dopaminergic degeneration (Tamburrino et al., 2015; Van der Perren et al., 2015; Vidal-Martínez et al., 2016; Ren et al., 2017; Zhao et al., 2017). However, the systemic adverse effects of immunosuppressants and their narrow therapeutic window, together with their limited ability to cross the blood-brain barrier (BBB) heavily limit the use of these drugs in PD. Dimethyl Fumarate, another FDA-approved drug with a mixed mechanism of action that includes an anti-inflammatory activity, was reported effective in ameliorating PD features in an alpha-synuclein based model of PD (Lastres-Becker et al., 2016). More recently, the focus of research for the treatment of neurological disorders has moved toward the investigation of agents acting through an immunomodulatory, rather than immunosuppressive mechanism (Martinez and Peplow, 2018). Hence, given the dual beneficial and potentially harmful roles of activated immune cells within the CNS, the strategy of boosting reparative and protective functions, while suppressing pro-inflammatory activity and the detrimental effects of inflammatory cytokines, might be efficacious to achieve neuroprotection in PD (Frankola et al., 2011; Lecca et al., 2018). Thalidomide is a small molecule drug that acts by inhibiting the synthesis of the potent inflammatory cytokine TNF-α causing a consequent dampening of the inflammatory cascade. Unlike larger molecules, it is readily capable of crossing the BBB (Tweedie et al., 2007; Boi et al., 2019). Therefore, Thalidomide and its analogs are promising candidate agents for evaluation in a variety of neurological disorders underpinned by neuroinflammation (Tweedie et al., 2007, 2012; Yoon et al., 2013; Wang et al., 2016; Batsaikhan et al., 2019; Tsai et al., 2019). In PD models, Thalidomide and the analog Lenalidomide, administered at the same dose of 100 mg/kg p.o. to mice overexpressing alpha-synuclein, induced an improvement in motor performance and dopaminergic fibers loss (Valera et al., 2015). Regrettably, both drugs are associated with unwanted side effects when used in chronic regimens. Thalidomide is currently used mainly to treat multiple myeloma and erythema nodosum leprosum (Franks et al., 2004), but its long-term use at doses required to stop inflammation has been associated with peripheral neuropathy as well as teratogenic effects (Vargesson, 2009). Indeed, in a recent clinical trial of Thalidomide in Alzheimer’s disease, poor tolerability precluded the agent being administered at the perceived efficacious dose (Decourt et al., 2017). Lenalidomide is a more potent anti-inflammatory drug as compared with Thalidomide, effectively inhibiting TNF-α at lower doses than the parent compound. However, the drug has been reported to be a substrate for the p-glycoprotein efflux pump at the BBB, which may limit it achieving and maintaining therapeutic levels in the brain (Hughes et al., 2019). Furthermore, long-term use of Lenalidomide can also result in peripheral neuropathy (Christian et al., 2007; Palumbo et al., 2012; Voorhees et al., 2013) and, similar to Thalidomide, it displays teratogenic, antiangiogenic, and neurotoxic activity at anti-inflammatory doses (Mahony et al., 2013). Hence, Thalidomide derivatives with potent anti-inflammatory properties but fewer side effects are currently under investigation (Beedie et al., 2016; Luo et al., 2018). Among them, POM is an FDA-approved and clinically available immunomodulatory drug in use for the treatment of multiple myeloma that is resistant to Lenalidomide (Siegel et al., 2019). POM is a structural analog of Thalidomide, with potent anti-inflammatory activity at significantly lower concentrations than the parent compound or Lenalidomide, and with a TNF-α inhibitory action of up to 50,000-fold greater than Thalidomide (Mahony et al., 2013; Wang et al., 2016). In a recent study, POM displayed an excellent BBB permeability in mice, achieving a brain/plasma concentration ratio of 0.71 (Tsai et al., 2019). Moreover, unlike Thalidomide and Lenalidomide, POM was devoid of anti-angiogenic and teratogenic actions at potently anti-inflammatory concentrations (Mahony et al., 2013), suggesting that it may be a more effective and safer drug, which justifies its preferential use with respect to its analog compounds in cancer treatment (Shortt et al., 2013; Mo and Richardson, 2019). In neurological diseases, POM was proven effective in reducing neuronal loss in a rat model of moderate traumatic brain injury (Wang et al., 2016) and mitigated H2O2-induced death of cortical neurons in culture (Tsai et al., 2018) and neuronal loss in rats subjected to ischemic stroke (Tsai et al., 2019) via its anti-oxidative and anti-inflammatory activity; however, this drug has yet to be tested in PD.
In the present study, POM was tested in the Drosophila LRRK2 model of PD carrying the G2385R mutation within the WD40 protein domain. Several studies have demonstrated that the G2385R mutation is a pathologically relevant variant, causing a partial loss of the kinase function of LRRK2 (Rudenko et al., 2012; Carrion et al., 2017). Accordingly, an increased risk of developing idiopathic PD in Chinese and Korean ethnicity holding this mutation has been widely reported (Tan et al., 2009; Xie et al., 2014). Whereas many studies have characterized LRRK2 gain-of-function models of PD in the rodent and insect (Liu et al., 2008), only one previous study has validated the Drosophila LRRK2WD40 as a model of PD (De Rose et al., 2016), showing that LRRK2WD40 mutants displayed features of PD including motor disability, reduced life span and mitochondrial damage (De Rose et al., 2016). Here, we further validated the Drosophila LRRK2WD40 as a PD model by conducting a detailed analysis of dopaminergic neuron clusters in the fly brain in an age-dependent manner. In the Drosophila brain, DA neurons are organized in distinct bilateral symmetric clusters comprising a stereotypical number of cells, with specific projections onto target regions that represent distinct functional areas (Monastirioti, 1999; Lima and Miesenbock, 2005). While PAL neurons mostly innervate the optic tubercle, PPL1 neurons innervate distinct regions of the mushroom bodies that are implicated with learning and memory (Zars, 2000). Moreover, PPM3 neurons innervate the central complex, which is a higher center for the control of locomotion (Strauss, 2002).
We demonstrate an age-related loss of dopaminergic neurons in LRRK2WD40 mutants but not in WT flies. Of note, the dopaminergic cell loss was limited to the posterior dopaminergic clusters, while sparing the anterior clusters, in line with the prominent role played by posterior clusters in the control of locomotion (Strauss, 2002; Mao and Davis, 2009). Moreover, we show that the cell loss was absent in LRRK2WD40 mutants at birth (1–2 days old flies), with the exception of the PPM3, while a significant decrease in the number of TH-positive neurons was observed in 7 days old mutants, becoming even greater in older flies. This supports the progressive nature of dopaminergic degeneration, adding important translational value to the LRRK2WD40 fly as a model of PD. Accordingly, motor disability paralleled dopaminergic degeneration in our study, being recorded in 7 and 14 days old mutants but not at birth (1–2 days old). The age-related decline of dopamine neurons was dependent upon the presence of the G2385R mutation, since the number of cells remained unvaried in WT flies with aging, in line with previous studies showing lack of age-related degeneration in the brain of WT Drosophila (White et al., 2010). Confirming previous findings, we also observed the presence of damaged mitochondria as indicated by multiple dilated cristae (De Rose et al., 2016). Of note, the G2385R variant has been associated with reduced LRRK2 binding to synaptic proteins and reduced neurotransmission, which may underpin defective T-bars in the LRRK2 WD40 Drosophila model (Carrion et al., 2017).
This further demonstration of dopaminergic degeneration in an LRRK2-loss of function model adds a piece to a yet puzzling issue, where the pathological role of loss of function variants in PD is still debated (Blauwendraat et al., 2018). Kinase inhibitors blocking LRRK2 gain of function are currently considered as potential treatments in PD, suggesting that further studies are warranted to define the molecular mechanisms underpinning the involvement of LKKR2 variants in PD.
We report the neuroprotective effect of POM in the LRRK2WD40 model. This neuroprotective effect was established by the POM-mediated improvement of motor performance, the rescue of TH-positive neurons and mitigation of mitochondrial damage. Moreover, POM recovered the loss of T-bars, the presynaptic active zone involved in neurotransmitter release in Drosophila (Fouquet et al., 2009). Recent evidence on the role of LRRK2 suggests that the LRRK2WD40 Drosophila might represent an optimal PD model for the initial screening of drugs with a disease-modifying potential that acts on the neuroimmune system. LRRK2 is an enzymatic protein involved in multiple cellular functions including neuronal outgrowth, cytoskeletal organization, mitochondrial dynamics, autophagy, protein interactions, and vesicle trafficking (Shin et al., 2008; Cirnaru et al., 2014; Piccoli et al., 2014; Arranz et al., 2015; Roosen and Cookson, 2016). Importantly, more recent reports have shown that LRRK2 is highly expressed in both brain and peripheral immune cells, playing a pivotal role in regulating the neuroinflammatory responses via the modulation of nuclear factor (NF)-kB pathway (Gardet et al., 2010; Hakimi et al., 2011; Thevenet et al., 2011; Moehle et al., 2012; Kluss et al., 2019). Notably, NF-kB is central to the efficacious actions of POM in multiple myeloma (Offidani et al., 2014). Moreover, recent evidence points to a pivotal role of LRRK2 in microglia phagocytic function and in Drosophila phagocytes ensheathing glia (Kim et al., 2018). As mentioned previously, an unremitting proinflammatory profile together with altered microglia phagocytic function are hallmarks of PD pathogenesis, therefore LRRK2 genetic models are highly appealing for investigating immunomodulatory agents (Joers et al., 2017; Janda et al., 2018).
POM possesses selective and potent TNF-α inhibitory activity, therefore suggesting that the inhibition of inflammatory pathways triggered by the ortholog of this cytokine Eiger in LRRK2WD40 flies may be the main neuroprotective mechanism of this agent. Of note, a recent study has provided a clear link between dysfunctional LRRK2 in dopamine neurons and altered glial response in Drosophila PD models, by showing that the genetic inhibition of the pro-inflammatory bone morphogenic protein (BMP) signaling in glia, was protective against PD-related neurotoxicity in aged mutant flies (Maksoud et al., 2019). Moreover, although further studies are needed to fully elucidate the functional targets of POM in PD, we suggest that modulation of phagocytosis may represent an additional mechanism of neuroprotection downstream to the anti-inflammatory activity. Hence, phagocytosis is traditionally associated with an anti-inflammatory profile of microglia. In support of this suggestion, we have recently shown in a mouse model of PD that impaired phagocytosis was restored by a neuroprotective agent acting as a modulator of microglia function (Lecca et al., 2018).
In synopsis, our study strongly supports the use of immunomodulatory drugs in PD. Importantly, POM is an FDA-approved clinically available drug, with a toxicological safer profile than the structural analog molecules. If validated in mammalian models of PD, this agent could potentially and rapidly be evaluated in humans.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
MC, IM, MS, and AC conceived, designed the study and analyzed the data. MC, IM, GM, AP, RI, LB, and MS performed the experiments. MC and MS performed the statistical analysis. NG and AC contributed reagents, materials, wrote and revised the manuscript. MC, IM, and MS contributed writing sections of the manuscript. All authors reviewed and approved the submitted version.
Funding
This research was supported in part by the Intramural Research Program, National Institute on Aging (NIA), NIH, in part by the intramural research program at the University of Cagliari (Università degli Studi di Cagliari), Italy.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AK declared a past co-authorship with one of the authors MS to the handling Editor.
Acknowledgments
We acknowledge CeSAR (intramural research service center) for providing assistance at TEM and confocal microscope facilities.
References
Arranz, A. M., Delbroek, L., Van Kolen, K., Guimarães, M. R., Mandemakers, W., Daneels, G., et al. (2015). LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J. Cell Sci. 128, 541–552. doi: 10.1242/jcs.158196
Batsaikhan, B., Wang, J. Y., Scerba, M. T., Tweedie, D., Greig, N. H., Miller, J. P., et al. (2019). Post-injury neuroprotective effects of the thalidomide analog 3,6’-dithiothalidomide on traumatic brain injury. Int. J. Mol. Sci. 20:E502. doi: 10.3390/ijms20030502
Beedie, S. L., Rore, H. M., Barnett, S., Chau, C. H., Luo, W., Greig, N. H., et al. (2016). In vivo screening and discovery of novel candidate thalidomide analogs in the zebrafish embryo and chicken embryo model systems. Oncotarget 7, 33237–33245. doi: 10.18632/oncotarget.8909
Bilen, J., and Bonini, N. M. (2005). Drosophila as a model for human neurodegenerative disease. Annu. Rev. Genet. 39, 153–171. doi: 10.1146/annurev.genet.39.110304.095804
Blauwendraat, C., Reed, X., Kia, D. A., Gan-Or, Z., Lesage, S., Pihlstrøm, L., et al. (2018). Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol. 75, 1416–1422. doi: 10.1001/jamaneurol.2018.1885
Boi, L., Pisanu, A., Greig, N. H., Scerba, M. T., Tweedie, D., Mulas, G., et al. (2019). Immunomodulatory drugs alleviate l-dopa-induced dyskinesia in a rat model of Parkinson’s disease. Mov. Disord. 34, 1818–1830. doi: 10.1002/mds.27799
Brundin, P., Barker, R. A., Conn, P. J., Dawson, T. M., Kieburtz, K., Lees, A. J., et al. (2013). Linked clinical trials—the development of new clinical learning studies in Parkinson’s disease using screening of multiple prospective new treatments. J. Parkinsons Dis. 3, 231–239. doi: 10.3233/jpd-139000
Buchmann, K. (2014). Evolution of innate immunity: clues from invertebrates via fish to mammals. Front. Immunol. 5:459. doi: 10.3389/fimmu.2014.00459
Carrion, M. D. P., Marsicano, S., Daniele, F., Marte, A., Pischedda, F., Di Cairano, E., et al. (2017). The LRRK2 G2385R variant is a partial loss-of-function mutation that affects synaptic vesicle trafficking through altered protein interactions. Sci. Rep. 7:5377. doi: 10.1038/s41598-017-05760-9
Chanan-Khan, A. A., Swaika, A., Paulus, A., Kumar, S. K., Mikhael, J. R., Rajkumar, S. V., et al. (2013). Pomalidomide: the new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. 3:e143. doi: 10.1038/bcj.2013.38
Chaudhuri, K. R., and Odin, P. (2010). The challenge of non-motor symptoms in Parkinson’s disease. Prog. Brain Res. 184, 325–341. doi: 10.1016/S0079-6123(10)84017-8
Christian, M. S., Laskin, O. L., Sharper, V., Hoberman, A., Stirling, D. I., and Latriano, L. (2007). Evaluation of the developmental toxicity of lenalidomide in rabbits. Birth Defects Res. B Dev. Reprod. Toxicol. 80, 188–207. doi: 10.1002/bdrb.20115
Cirnaru, M. D., Marte, A., Belluzzi, E., Russo, I., Gabrielli, M., Longo, F., et al. (2014). LRRK2 kinase activity regulates synaptic vesicle trafficking and neurotransmitter release through modulation of LRRK2 macro-molecular complex. Front. Mol. Neurosci. 7:49. doi: 10.3389/fnmol.2014.00049
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39, 889–909. doi: 10.1016/s0896-6273(03)00568-3
De Rose, F., Marotta, R., Poddighe, S., Talani, G., Catelani, T., Setzu, M. D., et al. (2016). Functional and morphological correlates in the Drosophila LRRK2 loss-of-function model of Parkinson’s disease: drug effects of withania somnifera (dunal) administration. PLoS One 11:e0146140. doi: 10.1371/journal.pone.0146140
Decourt, B., Drumm-Gurnee, D., Wilson, J., Jacobson, S., Belden, C., Sirrel, S., et al. (2017). Poor safety and tolerability hamper reaching a potentially therapeutic dose in the use of thalidomide for Alzheimer’s disease: results from a double-blind, placebo-controlled trial. Curr. Alzheimer Res. 14, 403–411. doi: 10.2174/1567205014666170117141330
Erkkinen, M. G., Kim, M. O., and Geschwind, M. D. (2018). Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 10:a033118. doi: 10.1101/cshperspect.a033118
Fouquet, W., Owald, D., Wichmann, C., Mertel, S., Depner, H., Dyba, M., et al. (2009). Maturation of active zone assembly by Drosophila Bruchpilot. J. Cell Biol. 186, 129–145. doi: 10.1083/jcb.200812150
Frankola, K. A., Greig, N. H., Luo, W., and Tweedie, D. (2011). Targeting TNF-α to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 10, 391–403. doi: 10.2174/187152711794653751
Franks, M. E., Macpherson, G. R., and Figg, W. D. (2004). Thalidomide. Lancet 363, 1802–1811. doi: 10.1016/S0140-6736(04)16308-3
Gardet, A., Benita, Y., Li, C., Sands, B. E., Ballester, I., Stevens, C., et al. (2010). LRRK2 is involved in the IFN-γ response and host response to pathogens. J. Immunol. 185, 5577–5585. doi: 10.4049/jimmunol.1000548
Hakimi, M., Selvanantham, T., Swinton, E., Padmore, R. F., Tong, Y., Kabbach, G., et al. (2011). Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural Transm. 118, 795–808. doi: 10.1007/s00702-011-0653-2
Hernandez, D. G., Reed, X., and Singleton, A. B. (2016). Genetics in Parkinson disease: mendelian versus non-Mendelian inheritance. J. Neurochem. 139, 59–74. doi: 10.1111/jnc.13593
Hewitt, V. L., and Whitworth, A. J. (2017). Mechanisms of Parkinson’s disease: lessons from Drosophila. Curr. Top. Dev. Biol. 121, 173–200. doi: 10.1016/bs.ctdb.2016.07.005
Hughes, J. H., Upton, R. N., Reuter, S. E., Rozewski, D. M., Phelps, M. A., and Foster, D. J. R. (2019). Development of a physiologically based pharmacokinetic model for intravenous lenalidomide in mice. Cancer Chemother. Pharmacol. 84, 1073–1087. doi: 10.1007/s00280-019-03941-z
Igaki, T., and Miura, M. (2014). The Drosophila TNF ortholog Eiger: emerging physiological roles and evolution of the TNF system. Semin. Immunol. 26, 267–274. doi: 10.1016/j.smim.2014.05.003
Janda, E., Boi, L., and Carta, A. R. (2018). Microglial phagocytosis and its regulation: a therapeutic target in Parkinson’s disease? Front. Mol. Neurosci. 11:144. doi: 10.3389/fnmol.2018.00144
Joers, V., Tansey, M. G., Mulas, G., and Carta, A. R. (2017). Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 155, 57–75. doi: 10.1016/j.pneurobio.2016.04.006
Kim, K. S., Marcogliese, P. C., Yang, J., Callaghan, S. M., Resende, V., Abdel-Messih, E., et al. (2018). Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 115, E5164–E5173. doi: 10.1073/pnas.1718946115
Kluss, J. H., Mamais, A., and Cookson, M. R. (2019). LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 47, 651–661. doi: 10.1042/bst20180462
Kumari, U., and Tan, E. K. (2009). LRRK2 in Parkinson’s disease: genetic and clinical studies from patients. FEBS J. 276, 6455–6463. doi: 10.1111/j.1742-4658.2009.07344.x
Lastres-Becker, I., García-Yagüe, A. J., Scannevin, R. H., Casarejos, M. J., Kügler, S., Rábano, A., et al. (2016). Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 25, 61–77. doi: 10.1089/ars.2015.6549
Lecca, D., Janda, E., Mulas, G., Diana, A., Martino, C., Angius, F., et al. (2018). Boosting phagocytosis and anti-inflammatory phenotype in microglia mediates neuroprotection by PPARγ agonist MDG548 in Parkinson’s disease models. Br. J. Pharmacol. 175, 3298–3314. doi: 10.1111/bph.14214
Lees, A. J., Hardy, J., and Revesz, T. (2009). Parkinson’s disease. Lancet 373, 2055–2066. doi: 10.1016/S0140-6736(09)60492-X
Lee, S., Imai, Y., Gehrke, S., Liu, S., and Lu, B. (2012). The synaptic function of LRRK2. Biochem. Soc. Trans. 40, 1047–1051. doi: 10.1042/bst20120113
Lima, S. Q., and Miesenbock, G. (2005). Remote control of behavior through genetically targeted photostimulation of neurons. Cell 121, 141–152. doi: 10.1016/j.cell.2005.02.004
Liu, Z., Wang, X., Yu, Y., Li, X., Wang, T., Jiang, H., et al. (2008). A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. U S A 105, 2693–2698. doi: 10.1073/pnas.0708452105
Luo, W., Tweedie, D., Beedie, S. L., Vargesson, N., Figg, W. D., Greig, N. H., et al. (2018). Design, synthesis and biological assessment of N-adamantyl, substituted adamantyl and noradamantyl phthalimidines for nitrite, TNF-α and angiogenesis inhibitory activities. Bioorg. Med. Chem. 26, 1547–1559. doi: 10.1016/j.bmc.2018.01.032
Mahony, C., Erskine, L., Niven, J., Greig, N. H., Figg, W. D., and Vargesson, N. (2013). Pomalidomide is nonteratogenic in chicken and zebrafish embryos and nonneurotoxic in vitro. Proc. Natl. Acad. Sci. U S A 110, 12703–12708. doi: 10.1073/pnas.1307684110
Maksoud, E., Liao, E. H., and Haghighi, A. P. (2019). A neuron-glial trans-signaling cascade mediates LRRK2-induced neurodegeneration. Cell Rep. 26, 1774.e4–1786.e4. doi: 10.1016/j.celrep.2019.01.077
Mao, Z., and Davis, R. L. (2009). Eight different types of dopaminergic neurons innervate the Drosophila mushroom body neuropil: anatomical and physiological heterogeneity. Front Neural Circuits 3:5. doi: 10.3389/neuro.04.005.2009
Martinez, B., and Peplow, P. V. (2018). Neuroprotection by immunomodulatory agents in animal models of Parkinson’s disease. Neural Regen. Res. 13, 1493–1506. doi: 10.4103/1673-5374.237108
Mills, R. D., Mulhern, T. D., Cheng, H. C., and Culvenor, J. G. (2012). Analysis of LRRK2 accessory repeat domains: prediction of repeat length, number and sites of Parkinson’s disease mutations. Biochem. Soc. Trans. 40, 1086–1089. doi: 10.1042/bst20120088
Mo, C. C., and Richardson, P. G. (2019). Pomalidomide in lenalidomide-refractory multiple myeloma: far from futile. Br. J. Haematol. doi: 10.1111/bjh.16214 [Epub ahead of print].
Moehle, M. S., Webber, P. J., Tse, T., Sukar, N., Standaert, D. G., DeSilva, T. M., et al. (2012). LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 32, 1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012
Monastirioti, M. (1999). Biogenic amine systems in the fruit fly Drosophila melanogaster. Microsc. Res. Tech. 45, 106–121. doi: 10.1002/(sici)1097-0029(19990415)45:2<106::aid-jemt5>3.0.co;2-3
Moreira, A. L., Sampaio, E. P., Zmuidzinas, A., Frindt, P., Smith, K. A., and Kaplan, G. (1993). Thalidomide exerts its inhibitory action on tumor necrosis factor α by enhancing mRNA degradation. J. Exp. Med. 177, 1675–1680. doi: 10.1084/jem.177.6.1675
Moreno, E., Yan, M., and Basler, K. (2002). Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 12, 1263–1268. doi: 10.1016/s0960-9822(02)00954-5
Offidani, M., Corvatta, L., Caraffa, P., Leoni, P., Pautasso, C., Larocca, A., et al. (2014). Pomalidomide for the treatment of relapsed-refractory multiple myeloma: a review of biological and clinical data. Expert Rev. Anticancer Ther. 14, 499–510. doi: 10.1586/14737140.2014.906904
Palumbo, A., Freeman, J., Weiss, L., and Fenaux, P. (2012). The clinical safety of lenalidomide in multiple myeloma and myelodysplastic syndromes. Expert Opin. Drug Saf. 11, 107–120. doi: 10.1517/14740338.2011.619975
Parkinson, J. (2002). An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 14, 223–236; discussion 222. doi: 10.1176/jnp.14.2.223
Piccoli, G., Onofri, F., Cirnaru, M. D., Kaiser, C. J., Jagtap, P., Kastenmuller, A., et al. (2014). Leucine-rich repeat kinase 2 binds to neuronal vesicles through protein interactions mediated by its C-terminal WD40 domain. Mol. Cell. Biol. 34, 2147–2161. doi: 10.1128/mcb.00914-13
Pushpakom, S., Iorio, F., Eyers, P. A., Escott, K. J., Hopper, S., Wells, A., et al. (2019). Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58. doi: 10.1038/nrd.2018.168
Ren, M., Han, M., Wei, X., Guo, Y., Shi, H., Zhang, X., et al. (2017). FTY720 attenuates 6-OHDA-associated dopaminergic degeneration in cellular and mouse Parkinsonian models. Neurochem. Res. 42, 686–696. doi: 10.1007/s11064-016-2125-4
Roosen, D. A., and Cookson, M. R. (2016). LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 11:73. doi: 10.1186/s13024-016-0140-1
Rudenko, I. N., Kaganovich, A., Hauser, D. N., Beylina, A., Chia, R., Ding, J., et al. (2012). The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem. J. 446, 99–111. doi: 10.1042/bj20120637
Salazar-Jaramillo, L., Paspati, A., van de Zande, L., Vermeulen, C. J., Schwander, T., and Wertheim, B. (2014). Evolution of a cellular immune response in Drosophila: a phenotypic and genomic comparative analysis. Genome Biol. Evol. 6, 273–289. doi: 10.1093/gbe/evu012
Sampaio, E. P., Sarno, E. N., Galilly, R., Cohn, Z. A., and Kaplan, G. (1991). Thalidomide selectively inhibits tumor necrosis factor α production by stimulated human monocytes. J. Exp. Med. 173, 699–703. doi: 10.1084/jem.173.3.699
Shin, N., Jeong, H., Kwon, J., Heo, H. Y., Kwon, J. J., Yun, H. J., et al. (2008). LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 314, 2055–2065. doi: 10.1016/j.yexcr.2008.02.015
Shortt, J., Hsu, A. K., and Johnstone, R. W. (2013). Thalidomide-analogue biology: immunological, molecular and epigenetic targets in cancer therapy. Oncogene 32, 4191–4202. doi: 10.1038/onc.2012.599
Siegel, D. S., Schiller, G. J., Song, K. W., Agajanian, R., Stockerl-Goldstein, K., Kaya, H., et al. (2019). Pomalidomide plus low-dose dexamethasone in relapsed refractory multiple myeloma after lenalidomide treatment failure. Br. J. Haematol. doi: 10.1111/bjh.16213 [Epub ahead of print].
Strauss, R. (2002). The central complex and the genetic dissection of locomotor behaviour. Curr. Opin. Neurobiol. 12, 633–638. doi: 10.1016/s0959-4388(02)00385-9
Tamburrino, A., Churchill, M. J., Wan, O. W., Colino-Sanguino, Y., Ippolito, R., Bergstrand, S., et al. (2015). Cyclosporin promotes neurorestoration and cell replacement therapy in pre-clinical models of Parkinson’s disease. Acta Neuropathol. Commun. 3:84. doi: 10.1186/s40478-015-0263-6
Tan, E. K., Peng, R., Wu, Y. R., Wu, R. M., Wu-Chou, Y. H., Tan, L. C., et al. (2009). LRRK2 G2385R modulates age at onset in Parkinson’s disease: a multi-center pooled analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 1022–1023. doi: 10.1002/ajmg.b.30923
Terpos, E., Kanellias, N., Christoulas, D., Kastritis, E., and Dimopoulos, M. A. (2013). Pomalidomide: a novel drug to treat relapsed and refractory multiple myeloma. Onco Targets Ther. 6, 531–538. doi: 10.2147/ott.s34498
Thevenet, J., Pescini Gobert, R., Hooft van Huijsduijnen, R., Wiessner, C., and Sagot, Y. J. (2011). Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One 6:e21519. doi: 10.1371/journal.pone.0021519
Troncoso-Escudero, P., Parra, A., Nassif, M., and Vidal, R. L. (2018). Outside in: unraveling the role of neuroinflammation in the progression of Parkinson’s disease. Front. Neurol. 9:860. doi: 10.3389/fneur.2018.00860
Tsai, Y. R., Chang, C. F., Lai, J. H., Wu, J. C., Chen, Y. H., Kang, S. J., et al. (2018). Pomalidomide ameliorates H2O2-induced oxidative stress injury and cell death in rat primary cortical neuronal cultures by inducing anti-oxidative and anti-apoptosis effects. Int. J. Mol. Sci. 19:E3252. doi: 10.3390/ijms19103252
Tsai, Y. R., Tweedie, D., Navas-Enamorado, I., Scerba, M. T., Chang, C. F., Lai, J. H., et al. (2019). Pomalidomide reduces ischemic brain injury in rodents. Cell Transplant. 28, 439–450. doi: 10.1177/0963689719850078
Tweedie, D., Ferguson, R. A., Fishman, K., Frankola, K. A., Van Praag, H., Holloway, H. W., et al. (2012). Tumor necrosis factor-α synthesis inhibitor 3,6’-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflammation 9:106. doi: 10.1186/1742-2094-9-106
Tweedie, D., Frankola, K. A., Luo, W., Li, Y., and Greig, N. H. (2011). Thalidomide analogues suppress lipopolysaccharide-induced synthesis of TNF-α and nitrite, an intermediate of nitric oxide, in a cellular model of inflammation. Open Biochem. J. 5, 37–44. doi: 10.2174/1874091x01105010037
Tweedie, D., Sambamurti, K., and Greig, N. H. (2007). TNF-α inhibition as a treatment strategy for neurodegenerative disorders: new drug candidates and targets. Curr. Alzheimer Res. 4, 378–385. doi: 10.2174/156720507781788873
Valera, E., Mante, M., Anderson, S., Rockenstein, E., and Masliah, E. (2015). Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of Parkinson’s disease. J. Neuroinflammation 12:93. doi: 10.1186/s12974-015-0320-x
Van der Perren, A., Macchi, F., Toelen, J., Carlon, M. S., Maris, M., Loor de, H., et al. (2015). FK506 reduces neuroinflammation and dopaminergic neurodegeneration in an α-synuclein-based rat model for Parkinson’s disease. Neurobiol. Aging 36, 1559–1568. doi: 10.1016/j.neurobiolaging.2015.01.014
Vanha-Aho, L. M., Valanne, S., and Ramet, M. (2016). Cytokines in Drosophila immunity. Immunol. Lett. 170, 42–51. doi: 10.1016/j.imlet.2015.12.005
Vargesson, N. (2009). Thalidomide-induced limb defects: resolving a 50-year-old puzzle. Bioessays 31, 1327–1336. doi: 10.1002/bies.200900103
Vargesson, N., Mahony, C., Erskine, L., Niven, J., Greig, N. H., and Figg, W. D. (2013). Reply to D’Amato et al. and Zeldis et al.: screening of thalidomide derivatives in chicken and zebrafish embryos. Proc. Natl. Acad. Sci. U S A 110:E4820. doi: 10.1073/pnas.1318475110
Vidal-Martínez, G., Vargas-Medrano, J., Gil-Tommee, C., Medina, D., Garza, N. T., Yang, B., et al. (2016). FTY720/fingolimod reduces synucleinopathy and improves gut motility in A53T mice: contributions of pro-brain-derived neurotrophic factor (pro-BDNF) and mature BDNF. J. Biol. Chem. 291, 20811–20821. doi: 10.1074/jbc.M116.744029
Voorhees, P. M., Laubach, J., Anderson, K. C., and Richardson, P. G. (2013). Peripheral neuropathy in multiple myeloma patients receiving lenalidomide, bortezomib, and dexamethasone (RVD) therapy. Blood 121:858. doi: 10.1182/blood-2012-11-465765
Wagh, D. A., Rasse, T. M., Asan, E., Hofbauer, A., Schwenkert, I., Durrbeck, H., et al. (2006). Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron 49, 833–844. doi: 10.1016/j.neuron.2006.06.022
Wang, J. Y., Huang, Y. N., Chiu, C. C., Tweedie, D., Luo, W., Pick, C. G., et al. (2016). Pomalidomide mitigates neuronal loss, neuroinflammation and behavioral impairments induced by traumatic brain injury in rat. J. Neuroinflammation 13:168. doi: 10.1186/s12974-016-0631-6
White, K. E., Humphrey, D. M., and Hirth, F. (2010). The dopaminergic system in the aging brain of Drosophila. Front. Neurosci. 4:205. doi: 10.3389/fnins.2010.00205
Xie, C. L., Pan, J. L., Wang, W. W., Zhang, Y., Zhang, S. F., Gan, J., et al. (2014). The association between the LRRK2 G2385R variant and the risk of Parkinson’s disease: a meta-analysis based on 23 case-control studies. Neurol. Sci. 35, 1495–1504. doi: 10.1007/s10072-014-1878-2
Yoon, J. S., Lee, J. H., Tweedie, D., Mughal, M. R., Chigurupati, S., Greig, N. H., et al. (2013). 3,6’-dithiothalidomide improves experimental stroke outcome by suppressing neuroinflammation. J. Neurosci. Res. 91, 671–680. doi: 10.1002/jnr.23190
Zars, T. (2000). Behavioral functions of the insect mushroom bodies. Curr. Opin. Neurobiol. 10, 790–795. doi: 10.1016/s0959-4388(00)00147-1
Keywords: Parkinson, Drosophila, LRRK2, immunomodulation, neuroprotection, drug repurposing, repositioning
Citation: Casu MA, Mocci I, Isola R, Pisanu A, Boi L, Mulas G, Greig NH, Setzu MD and Carta AR (2020) Neuroprotection by the Immunomodulatory Drug Pomalidomide in the Drosophila LRRK2WD40 Genetic Model of Parkinson’s Disease. Front. Aging Neurosci. 12:31. doi: 10.3389/fnagi.2020.00031
Received: 20 November 2019; Accepted: 03 February 2020;
Published: 13 February 2020.
Edited by:
Simone Engelender, Technion Israel Institute of Technology, IsraelReviewed by:
Ameya Sanjay Kasture, University of Vienna, AustriaVeronica Ghiglieri, University of Perugia, Italy
Copyright © 2020 Casu, Mocci, Isola, Pisanu, Boi, Mulas, Greig, Setzu and Carta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Dolores Setzu, bWRzZXR6dUB1bmljYS5pdA==; Anna R. Carta, YWNhcnRhQHVuaWNhLml0
† These authors have contributed equally to this work