 Celia G. Fernandez
Celia G. Fernandez Mary E. Hamby
Mary E. Hamby Morgan L. McReynolds
Morgan L. McReynolds William J. Ray
William J. Ray- The Neurodegeneration Consortium, Institute of Applied Cancer Science (IACS), The University of Texas MD Anderson Cancer Center, Houston, TX, United States
APOE4 is the greatest genetic risk factor for late-onset Alzheimer’s disease (AD), increasing the risk of developing the disease by 3-fold in the 14% of the population that are carriers. Despite 25 years of research, the exact mechanisms underlying how APOE4 contributes to AD pathogenesis remain incompletely defined. APOE in the brain is primarily expressed by astrocytes and microglia, cell types that are now widely appreciated to play key roles in the pathogenesis of AD; thus, a picture is emerging wherein APOE4 disrupts normal glial cell biology, intersecting with changes that occur during normal aging to ultimately cause neurodegeneration and cognitive dysfunction. This review article will summarize how APOE4 alters specific pathways in astrocytes and microglia in the context of AD and the aging brain. APOE itself, as a secreted lipoprotein without enzymatic activity, may prove challenging to directly target therapeutically in the classical sense. Therefore, a deeper understanding of the underlying pathways responsible for APOE4 toxicity is needed so that more tractable pathways and drug targets can be identified to reduce APOE4-mediated disease risk.
Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disease of aging, the incidence of which is expected to increase exponentially as the proportion of the population over the age of 65 increases. Research in AD drug discovery has historically focused on the Amyloid Hypothesis, based primarily on findings from early-onset AD, which is caused by mutations in amyloid-β (Aβ) pathway proteins and which accounts for <2% of all AD cases. While the Amyloid Hypothesis predicts that enhanced production and diminished clearance of Aβ causes AD, therapeutics aimed at modulating Aβ levels have largely failed, although they have not yet been tested at presymptomatic stages of disease (Doig et al., 2017).
After aging, the ε4 allele of the APOE gene is the next greatest risk factor for AD, while the relatively rare ε2 allele confers AD protection (Corder et al., 1993; Saunders et al., 1993; Strittmatter et al., 1993). Although 25 years have passed since it was identified, there are still no approved drugs directly targeting APOE4, due partly to the inherent “undruggability” of lipoproteins. However, the atherosclerosis field has demonstrated that indirectly modulating the effect of lipoproteins can be a successful alternative strategy. For example, statins affect lipoprotein composition and disease risk by targeting a metabolic pathway (cholesterol synthesis); similarly, understanding the downstream pathways that mediate APOE4 disease risk might identify more tractable therapeutic targets for treating APOE4-mediated AD.
APOE in the brain is primarily expressed by astrocytes and microglia, and APOE4 expression alters the normal function of both of these glial cell types, potentially contributing to AD risk. Although the toxicity associated with APOE4 likely involves the impaired ability of APOE4-expressing glia to efficiently clear Aβ, it is also apparent that there are Aβ-independent effects on normal glial physiology. The role of APOE in mediating Aβ levels has been discussed in depth elsewhere (Ries and Sastre, 2016), and will only be briefly touched upon below. This review will instead focus on more recent findings that specifically describe the role of APOE in glial biology, in addition to and independent of Aβ modulation, particularly during aging, and will describe pathways in each glial cell type that may link APOE to disease pathogenesis.
Although astrocytes and microglia are the primary producers of APOE, whether an interaction between these cells exists in terms of APOE biology has not been carefully examined. Cross-talk between astrocytes and microglia in neurodegeneration is well-known (Jha et al., 2018); for example, astrocytes can secrete complement factor C3 in response to Aβ, which can then activate microglia via the C3a receptor (Lian et al., 2016). On the other hand, lipopolysaccharide-stimulated microglia can induce neurotoxic “A1” reactive astrocytes, as opposed to neurotrophic “A2” reactive astrocytes (Liddelow et al., 2017). The same group found that A1-type astrocytes are present in aging (Clarke et al., 2018) and AD brain (Liddelow et al., 2017), and that A1 astrocytes not only lose the neurotrophic capacity of A2 astrocytes, but also actively produce a neurotoxin to kill neurons and oligodendrocytes. Importantly, a recent study demonstrated that blocking this microglial-dependent induction of A1 astrocytes is protective in mouse models of Parkinson’s disease (Yun et al., 2018). Whether blockade of such microglia/astrocyte cross-talk can help ameliorate neurodegeneration in humans and in AD has yet to be demonstrated. Furthermore, whether APOE is one such secreted factor that mediates interactions between astrocytes and microglia has not been reported, nor has a synergistic effect of APOE from each cell type been clearly defined. Even so, since both astrocytes and microglia express APOE, this review article will separately consider specific aspects of each cell type’s normal physiology that might be impacted by APOE4 expression in aging and AD.
Overview of APOE Isoforms
APOE is a lipoprotein that normally facilitates lipid transport between cells (Mahley, 1988). APOE transcription is activated by liver X receptor (LXR) and peroxisome proliferator-activated receptor γ (PPARγ), transcription factors that regulate lipid homeostasis and inflammation (Laffitte et al., 2001; Akiyama et al., 2002; Liang et al., 2004; Mandrekar-Colucci et al., 2012; Moutinho et al., 2019). In the lipid-rich brain, APOE is predominantly expressed by astrocytes and microglia, and perhaps in limited circumstances by neurons (Boyles et al., 1985; Pitas et al., 1987; Uchihara et al., 1995; Nakai et al., 1996; Xu et al., 1998, 2006).
The human APOE gene exists as three different alleles, ε2, ε3, and ε4, which are present at ~7%, 79%, and ~14%, respectively, in the entire population (Bertram et al., 2007), and which exhibit differences in lipid and receptor binding efficiency. The presence of one ε4 allele increases the risk of AD by threefold, while carriers with two ε4 alleles are eight times as likely to develop AD compared to those without any ε4 allele; and ε4 is associated with an earlier age of disease onset, from about 85 years without any ε4, to 75 years with one and 68 years with two ε4 alleles (Corder et al., 1993). These statistics make APOE ε4 the greatest known genetic risk factor for AD, more than any other gene to date. In contrast to the human gene, mouse ApoE exists as only one isoform, and the structure of the mouse APOE protein more closely matches human APOE3 (Raffai et al., 2001); targeted-replacement mice, in which the endogenous mouse ApoE gene has been replaced with either of the human APOE isoforms, have therefore been created to study differences in human APOE isoform function, and will be referred to throughout this review article (Sullivan et al., 1997; Knouff et al., 1999).
The three human APOE isoforms differ from one another in the protein sequence at amino acid positions 112 and 158 (Figure 1; Mahley, 1988; Raffai et al., 2001; Hatters et al., 2006). These single amino acid differences are enough to change the lipid and receptor binding ability of APOE (Weisgraber et al., 1982; Dong and Weisgraber, 1996; Gong et al., 2002). Specifically, R112 in APOE4 creates a domain interaction between the N-terminal receptor binding domain and the C-terminal lipid binding domain, preventing efficient binding to HDL compared to APOE2 and APOE3, with preferential binding to VLDL (Dong et al., 1994; Dong and Weisgraber, 1996). While APOE2 is protective against AD, the ε2 allele is also associated with hyperlipoproteinemia III, which is characterized by accumulated lipoproteins in the plasma and development of atherosclerosis (Giau et al., 2015). This is thought to be caused by impairment in the receptor binding region of APOE2, leading to delayed lipoprotein clearance and increased triglyceride and cholesterol levels (Havel and Kane, 1973; Weisgraber et al., 1982; Mahley and Rall, 2000). In the context of AD, APOE2 has been relatively understudied, although some research is ongoing (Wu and Zhao, 2016). It should be noted that APOE2 in many experimental settings is similar to APOE3 or performs qualitatively better (such as in amyloid clearance). For clarity, and because there is much less in the literature to explain the mechanism of action of APOE2, the present review will focus on different phenotypes conferred by APOE3 vs. APOE4.
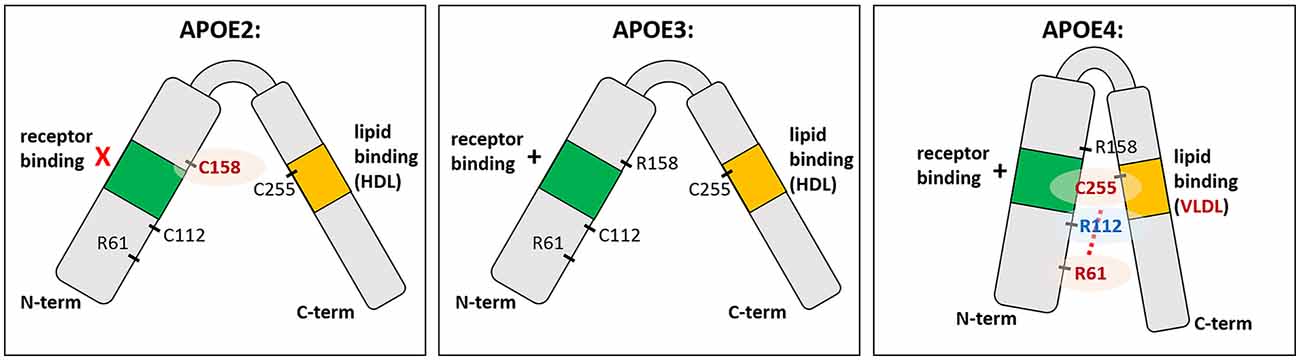
Figure 1. The structure of APOE isoforms. APOE is a soluble secreted protein, with N-terminal and C-terminal domains linked by a central hinge region. The N-terminal domain contains the receptor binding domain (indicated in green), and the C-terminal domain contains the lipid binding region (indicated in orange). Each isoform differs from one another at amino acid position 112 and 158. Cysteine at position 158 (C158) in APOE2 is thought to cause deficient receptor binding, while arginine at position 112 (R112) in APOE4 changes the conformation of the entire domain such that R61 is exposed and interacts with C255 in the C-terminal domain (red dotted line). This “domain interaction” is thought to be the biophysical basis for differences in APOE4 function compared to the other isoforms; e.g., preference for VLDL over HDL. In APOE3 and APOE2, which have C112 instead of R112, the R61 is not exposed and there is no such domain interaction.
APOE Isoforms and Amyloid Clearance
Both astrocytes and microglia clear Aβ (Paresce et al., 1996; Wyss-Coray et al., 2003; Ries and Sastre, 2016) and although there is some evidence that APOE4 may enhance Aβ production (Ye et al., 2005), it is widely thought that APOE4 confers AD risk through deficient Aβ clearance compared to APOE3 and APOE2 (Koistinaho et al., 2004; Deane et al., 2008; Simonovitch et al., 2016), although not necessarily via direct binding (Verghese et al., 2013). APOE isoforms differ not only in lipid binding ability, but also in affinity for specific APOE receptors (Ruiz et al., 2005; Holtzman et al., 2012). LRP1, a major receptor for APOE, mediates Aβ clearance in astrocytes and pericytes (Liu et al., 2017a; Ma et al., 2018), and astrocytes expressing APOE4 have reduced LRP1 surface expression, which could explain impaired amyloid clearance in vivo (Prasad and Rao, 2018). However, astrocytes also utilize other APOE receptors such as LDLR for Aβ clearance, but in an APOE-independent manner (Basak et al., 2012); furthermore, Aβ is cleared by transcytosis across the blood brain barrier, glymphatic and interstitial fluid bulk flow, and by extracellular degrading enzymes, highlighting the complexity around understanding how APOE4 contributes to amyloid accumulation.
APOE Isoforms and Tau Pathology
In addition to modulating Aβ, APOE also affects tau pathology, another hallmark of AD, in an isoform-specific manner. APOE4 worsens tau pathology in the P301S tau mouse model, and APOE4 genotype is associated with exacerbated neurodegeneration in human primary tauopathies (Shi et al., 2017). APOE4 status is associated with tau pathology particularly in instances when amyloid pathology is also present (Farfel et al., 2016). The relationship between APOE4 carrier status and CSF tau levels is more robustly correlated in women than in men (Hohman et al., 2018), suggesting a possible sex effect in APOE4-mediated toxicity. Neurons expressing P301S tau are less viable when co-cultured with APOE4-expressing glia compared to APOE2- or APOE3-expressing glia, while co-culture with APOE−/− glia leads to the greatest neuronal viability, supporting the idea that APOE4 represents a toxic gain-of-function (Shi et al., 2017). Higher CSF tau levels are associated with faster disease progression and reduced cortical plasticity in patients, but only in APOE4 carriers (Koch et al., 2017), further cementing a role for APOE4 in exacerbating tau pathology. Since some evidence suggests that APOE can be expressed by neurons under stress (Xu et al., 1998, 2006; Harris et al., 2004), it is possible that neuron-derived APOE4 directly mediates tau toxicity in neurons, but the above data suggests that glia-derived APOE4 is likely contributing as well.
Amyloid- and Tau-Independent Effects of APOE4: Glial Cell Biology
The role of APOE4 in neurological disease is certainly broader than the clearance or response to misfolded proteins, including Aβ and tau; for example, APOE receptors play diverse roles in brain physiology independent of Aβ (Holtzman et al., 2012) and APOE4 carriers may be susceptible to disorders that do not involve proteinopathy, such as chemotherapy-induced cognitive dysfunction (Mandelblatt et al., 2018; Speidell et al., 2019). In addition to the role of APOE4 derived from astrocytes and microglia, a growing body of literature also supports a role for APOE4 and pericytes at the blood brain barrier in neurovascular unit dysfunction and AD pathogenesis (Casey et al., 2015; Soto et al., 2015; Halliday et al., 2016; Ma et al., 2018). APOE has even been proposed to be proteolytically cleaved to form either cytotoxic or neuroprotective fragments, in a cell type- and isoform-specific manner (Brecht et al., 2004; Muñoz et al., 2018). Thus, the neurotoxicity conferred by APOE4 in AD may not be solely due to its effects on amyloid or tau pathology, but also to its effects on normal glial functions. How these processes fit into the current understanding of APOE function and neurodegeneration will be important for drug discovery efforts targeting APOE biology to treat AD.
Astrocytes play critical roles in brain lipid and energy metabolism, and both microglia and astrocytes have important immune functions in the brain. APOE4 expression in each of these cell types likely disrupts these pathways, ultimately leading to brain dysfunction in addition to any Aβ- and tau-mediated effects. The role of APOE4 and aging in each of these cell types and pathways will now be examined individually.
APOE and Astrocyte Bioenergetics
The idea that APOE isoforms differentially mediate astrocyte bioenergetics has gained increasing support in recent years and implies that APOE4-expressing astrocytes have deficient lipid and glucose metabolism, impairing their ability to support energy-demanding neurons, particularly during aging. In the following sections, we will describe different aspects of lipid homeostasis and glucose metabolism in astrocytes, and how APOE may be involved in these processes.
Astrocytes and Lipid Homeostasis in the Aging and AD Brain
The most well-studied aspect of APOE biology in AD is lipid transport, which neurons rely upon for their proper function. Lipid homeostasis is clearly altered in AD: in his first description of the disease, Alois Alzheimer noted that “many glial cells show adipose saccules” (Alzheimer et al., 1995), and lipid accumulations are present in both human AD brain and in an AD mouse model (Hamilton et al., 2015), as well as in the aging mouse brain (Shimabukuro et al., 2016). Given that the brain is the most lipid-rich organ outside of adipose tissue (O’Brien and Sampson, 1965), it is therefore not surprising that lipoproteins, cholesterol and lipid homeostasis are critical for normal brain function, including neuronal repair, membrane remodeling, and plasticity (Mahley, 2016). For example, disrupting lipid homeostasis in mice by knocking out both the α and β isoforms of LXR, which are required for cholesterol and lipid efflux from astrocytes, leads to widespread abnormalities in the brain, including an age-dependent accumulation of lipid vacuoles in perivascular astrocytes (Wang et al., 2002). When SREBP2, a major positive regulator of cholesterol and lipid synthesis, is specifically knocked out in astrocytes, mice exhibit reduced brain weight and deficits in social behavior, learning and memory, and coordinated movement, as well as elevated glucose oxidation (Ferris et al., 2017). Interestingly, the neurons in these mice show elevated SREBP2, possibly to compensate for the lack of SREBP2 in astrocytes; yet this neuron-specific SREBP2 elevation was not enough to rescue the pathological changes associated with astrocyte-specific knock-out, underscoring the dependence of neurons on astrocytic lipids.
APOE4 from primary astrocytes is poorly lipidated compared to APOE3 (Gong et al., 2002); deficient lipid binding and transport by APOE4 might therefore be expected to result in the same type of widespread brain abnormalities described above, ultimately leading to increased risk for AD (Figure 2). But despite the poor lipid transport capabilities of APOE4 and the reliance of neurons on astrocyte-supplied lipid, APOE4 carriers have generally normal brain function throughout life. How then does aging uncover the deficits conferred by APOE4? The young brain may have mechanisms in place to cope with inefficient APOE4 lipid transport; but aging leads to decreased cholesterol synthesis in astrocytes (Boisvert et al., 2018), which, when combined with lower efflux from APOE4, could tip the balance and culminate in neuronal lipid deficits. Furthermore, Aβ inhibits SREBP2 in primary cultured cells from mouse cortex (Mohamed et al., 2018), suggesting that amyloid deposition could make neurons even more dependent on astrocytic lipids, which would be lacking in ε4 carriers. Although cholesterol has been the most extensively studied, changes in other lipid classes are also observed in serum samples from AD patients, including sterols, sphingomyelin, phosphatidylcholine, glycerophosphoethanolamine, lysophosphatidylcholine, diacylglycerols, and triacylglycerols (Anand et al., 2017); therefore, APOE4 status could exacerbate other age-related changes in lipid homeostasis. While it is unclear whether these changes in lipid metabolism are a cause or an effect of AD, aging- and APOE-related perturbations may be expected to exacerbate amyloid pathology, and vice versa, culminating in widespread neurodegeneration.
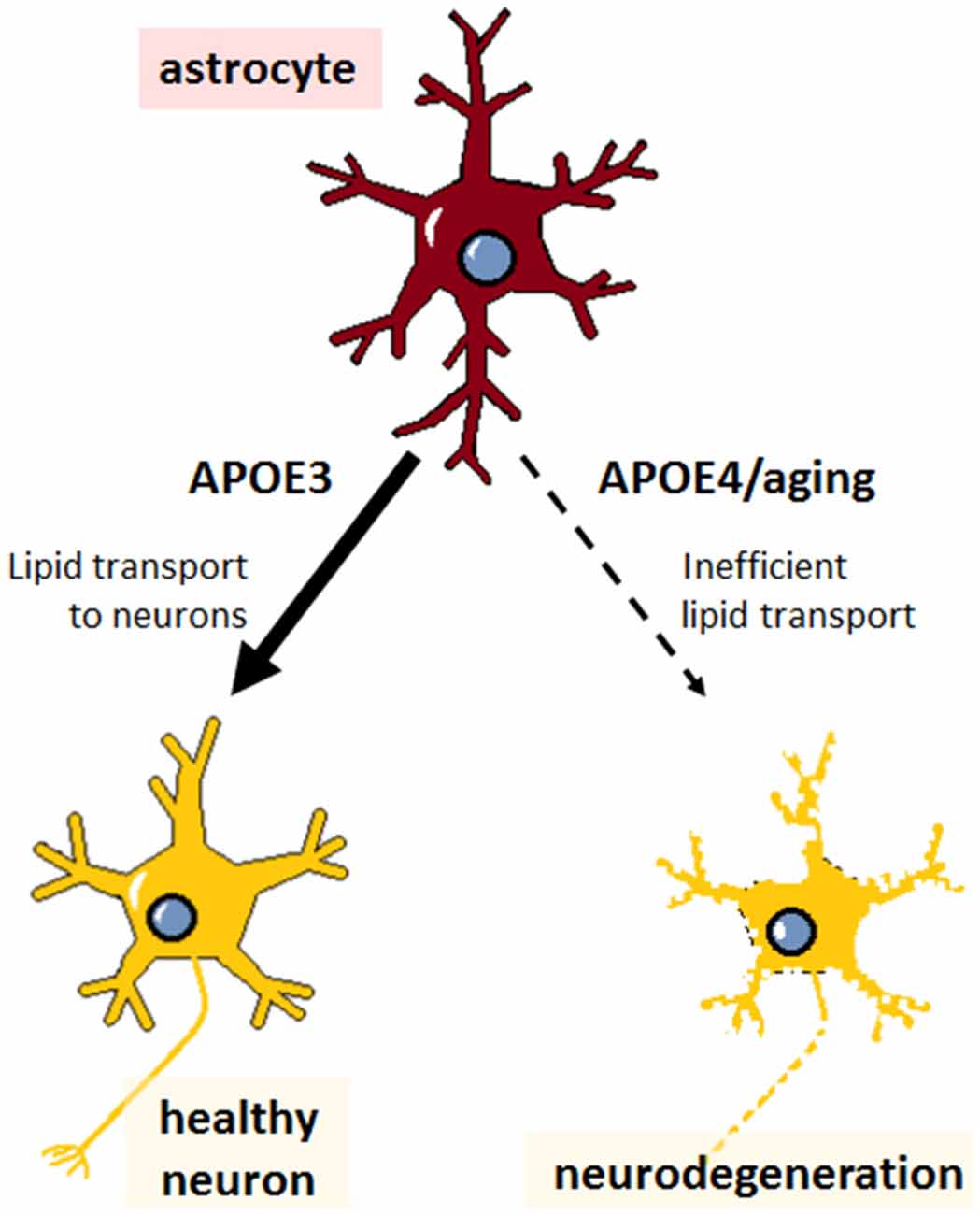
Figure 2. Impaired lipid transport capacity of astrocytic APOE4 sensitizes neurons to degeneration during aging. Astrocytes (red) expressing APOE3 supply normal levels of cholesterol and other lipids to the cells of the brain, particularly to neurons (yellow), maintaining healthy neuronal function and cognition. Astrocytes expressing APOE4 are less inefficient at lipid transport, which, compounded with aging-associated lipid dysregulation, leads to neurodegeneration (neuron with rough edges) and is expected to predispose APOE4 carriers to Alzheimer’s disease (AD).
Paradoxical Effects of APOE4 on Cholesterol Synthesis
Given that APOE4-containing lipoproteins are lipid-deficient, one might expect lipid secretion to be impaired. Surprisingly, human iPSC-derived astrocytes expressing APOE4 reportedly secrete significantly more cholesterol than their APOE3+ counterparts (Lin et al., 2018). Notably, this enhanced cholesterol secretion was accompanied by higher, not lower, intracellular cholesterol. Accumulated intracellular cholesterol is consistent with the reduced ability of APOE4 to export cholesterol, which was confirmed in an independent study showing that APOE4 iPSC-derived astrocytes produce APOE-lipoprotein particles with less cholesterol than APOE3-expressing cells (Zhao et al., 2017a). But the increased cholesterol secretion is more difficult to explain; how could APOE4 promote both the intracellular accumulation and enhanced extracellular secretion of cholesterol in vitro? One explanation to unite these seemingly contradictory findings is that these cells are unable to properly sense that intracellular cholesterol levels are high. Normally, negative feedback loops ensure that cells laden with lipids reduce synthesis and uptake while increasing efflux—consistent with this signaling mechanism, SREBP2 was in fact downregulated in APOE4+ iPSC-derived astrocytes, as would be expected from cells with excessive lipids (Lin et al., 2018). APOE4 might therefore reduce the clearance of cholesterol via enzymatic oxidation. Consistent with this idea, APOE−/− mice have reduced 24-OH-, 7α, and 7β-hydroxycholesterol in their brains (Nunes et al., 2018). Furthermore, APOE mRNA levels are reduced in the APOE4+ iPSCs, and APOE is a major target gene of LXRs, which are activated by hydroxycholesterol. In contrast to the above finding, astrocytes from targeted-replacement mice expressing APOE4 were previously found to secrete less cholesterol than astrocytes from APOE3 mice (Gong et al., 2002; Riddell et al., 2008). While it is possible that species differences in cholesterol handling between mice and humans could explain these disparate findings (Dietschy and Turley, 2002), more research is needed to clarify exactly how APOE genotype affects astrocyte cholesterol metabolism.
APOE4 Disrupts Lipid Droplet Homeostasis
Recent observations indicate that APOE regulates intracellular lipid storage. A consequence of SREBP2 inhibition, as might occur during aging or amyloid deposition, is the reduction of autophagic lipid mobilization from structures known as lipid droplets (LDs; Seo et al., 2011; Kim et al., 2016). LDs are intracellular accumulations of neutral lipids and are central to cellular lipid homeostasis, particularly in astrocytes, where they play a dual role in managing lipids from neurons and in maintaining astrocytic energy demands. Elevated reactive oxygen species (ROS) in neurons induces lipid peroxidation and triggers subsequent efflux of lipids that accumulate as LDs in neighboring astrocytes, a process that is neuroprotective and dependent on APOE (Figure 3; Liu et al., 2015, 2017b). An increase in peroxidated lipids is associated with disrupted lipid homeostasis, decreased phosphatidylcholine synthesis, decreased mitochondrial metabolism, and ultimately cognitive decline (McDougall et al., 2017), and APOE mitigates this toxicity in neurons by transferring the burden of lipid accumulation and subsequent clearance to astrocytes. In a Drosophila model of neurodegeneration, APOE4 is a complete loss of function in terms of the neuroprotective formation of LDs in glial cells, leading to neuronal cell death (Figure 3; Liu et al., 2015, 2017b). These data indicate that APOE might play an important role not only in astrocyte-mediated synthesis and transfer of lipids to neurons, but in reverse as well, as an acceptor of neuronal-derived peroxidated lipids.
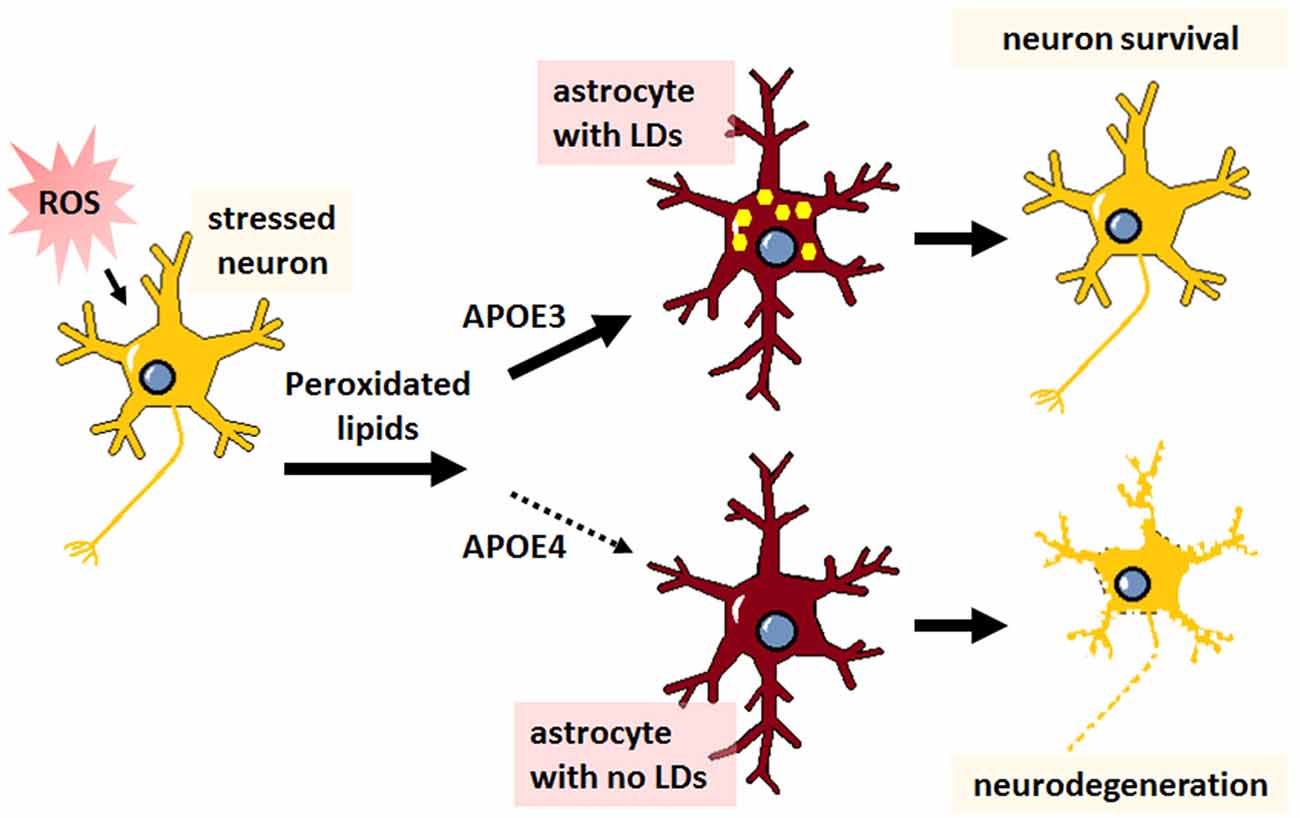
Figure 3. The neuroprotective transfer of toxic lipids from neurons to astrocytes results in lipid droplet formation, which is abrogated in APOE4-expressing cells. Agents or stressors that induce reactive oxygen species (ROS) formation in neurons leads to increased levels of toxic peroxidated lipids. Neurons transfer these lipids to astrocytes via APOE. With APOE3 expression, this transfer results in the formation of lipid droplets (LDs; yellow dots) in astrocytes and neuroprotection. Conversely, APOE4 expression is thought to prevent the transfer of peroxidated lipids to astrocytes, resulting in no LD formation in astrocytes and subsequent neurodegeneration.
Although these studies show that APOE4 expression leads to a decrease in LD formation when neurons are the lipid donor and astrocytes are the recipient, APOE4 can also induce LDs in a cell autonomous manner. LD formation results from interactions between the endoplasmic reticulum (ER) and mitochondria, at structures known as mitochondria-associated ER membranes, and APOE may regulate LD formation by mediating ER-mitochondria communication at these sites (Tambini et al., 2016). Fibroblasts treated with APOE4 astrocyte conditioned medium (ACM) exhibit increased LDs, and blocking ER-mitochondria tethering returns lipid levels to normal (Tambini et al., 2016). These data indicate that either the APOE4 ACM contained a factor that signaled to the cells to induce LDs, or alternatively, that the APOE4 ACM is somehow nutrient-deprived compared to APOE3 media, since glucose deprivation also induces LD formation in astrocytes. Nutrient deprivation-induced LDs are used for β-oxidation of fatty acids to generate acetyl-CoA to meet cellular energy demands (Cabodevilla et al., 2013). Thus, it is possible that APOE regulation of LDs in astrocytes is context-dependent: nutrient deprivation induces formation of LDs and subsequent breakdown by autophagy to fulfill energy requirements, which APOE4 can stimulate; whereas increased neuronal oxidative stress leads to accumulation of toxic lipids, which are transferred to astrocytes, an activity that is lacking in APOE4 cells. In either case, APOE4-dependent deficiency in autophagy would also impair LD breakdown, causing toxic accumulation in either cell type (Simonovitch et al., 2016). Further study delineating the impact APOE4 has on LD homeostasis could identify points of therapeutic intervention.
Astrocyte Glucose Metabolism in the Aging Brain
The brain is a highly energy-demanding organ, and declines in brain glucose utilization and mitochondrial function during aging may interact with AD risk factors, including APOE, to negatively impact neuronal homeostasis. The data supporting this concept range from model organisms to epidemiology. For example, yeast genes that enhance or suppress Aβ toxicity exert their effect depending on the level of mitochondrial respiration (Treusch et al., 2011), suggesting that cellular energetics determines resiliency to amyloid. Energetics also impacts AD risk profile in humans: postmenopausal women characterized as having a poor metabolic profile, which includes elevated glucose and increased insulin resistance, exhibit worse cognitive performance compared to healthy metabolic subjects, and cognitive decline in this group is exacerbated by APOE4 carrier status (Karim et al., 2019). However, this relationship is likely complex; a study including both aged women and men found no difference in glucose levels in AD and APOE4 carriers vs. healthy and non-APOE4 carriers; there were marginal reductions in insulin and insulin resistance in APOE4 carriers, which was somewhat increased in individuals with AD (Morris et al., 2017).
To clarify the underlying relationship between APOE4 and energy homeostasis, APOE4 targeted-replacement mice have been studied. Aged (22 months) mice expressing APOE4 exhibit decreased insulin signaling in cortex and hippocampus (Zhao et al., 2017b) and middle-aged (6 months) female APOE4 mice are deficient in the uptake and utilization of glucose in the brain, with compromised respiratory capacity and decreased PPARγ signaling (Wu et al., 2018). In addition to downregulated PPARγ, another study found that insulin-degrading enzyme (IDE) was also downregulated in the hippocampus of the same aged APOE4 mice (Keeney et al., 2015). Reduction of PPARγ would be expected to trigger lipid dysregulation by decreasing lipid synthesis (as described above), as well as dampen anti-inflammatory signaling; and while lower levels of IDE would be expected to decrease Aβ clearance, lower IDE should also increase insulin and affect glucose and glycogen levels, perhaps leading over time to insulin resistance, although this would need to be determined experimentally. In the same study, aged mice expressing either APOE4 or APOE3 compared to the neuroprotective APOE2 were also found to have downregulated insulin signaling proteins IGF1, IRS1, and GLUT4 (Keeney et al., 2015), in agreement with the idea that aging itself causes deficient glucose metabolism independently of APOE genotype. As discussed above, energy deficiencies might be exacerbated in APOE4 carriers as lipid β-oxidation and lipid droplet autophagy become increasingly important for cell function.
Aerobic Glycolysis
Deficits in energy metabolism associated with APOE4 might also exacerbate aging-associated declines in aerobic glycolysis (Goyal et al., 2017; Figure 4). Aerobic glycolysis is the preferential conversion of glucose to lactate rather than pyruvate, even in the presence of oxygen, and is typically associated with cancer cells, although non-cancerous cells also engage in this process (Jones and Bianchi, 2015). In fact, astrocytes in mice are capable of surviving solely by aerobic glycolysis for at least as long as 1 year without any signs of pathology or neurodegeneration (Supplie et al., 2017). Certain brain regions tend to preferentially use aerobic glycolysis (Vaishnavi et al., 2010), and as the brain ages, there is a shift towards oxidative phosphorylation (OxPhos) to meet energy requirements (Figure 4; Goyal et al., 2017). This aging-related increased reliance on OxPhos has been proposed to lead to elevated ROS and peroxidated lipids (Harris et al., 2014), a situation likely made worse in APOE4 carriers, given the reduced ability of APOE4 to traffic neuronal peroxidated lipids to astrocytes for elimination. Thus, the switch to OxPhos could be an age-dependent trigger for APOE4 pathophysiology.

Figure 4. Different modes of glucose metabolism are preferred in the young vs. aging brain. In the young brain, aerobic glycolysis is generally favored, resulting in increased lactate production, presumably by astrocytes, which should be supportive for increased neuronal activity. With aging, there is a shift toward oxidative phosphorylation (OxPhos) instead, resulting in increased electron transport chain (ETC) activity, increased ROS production, and more peroxidated lipids. Healthy cultured cells that are resistant to Aβ toxicity happen to exhibit a preference for aerobic glycolysis, and aerobic exercise, which is known to confer neuroprotection, elevates aerobic glycolysis. In contrast, cells from familial AD patients (PSEN1ΔE9) exhibit elevated OxPhos. While young APOE4 carriers may exhibit increased glycolytic activity, particularly in brain regions associated with AD [e.g., default mode network (DMN), entorhinal cortex], aged APOE4 carriers may exhibit elevated OxPhos instead, although more evidence to demonstrate whether and how such a metabolic shift occurs is warranted.
In line with the concept that aerobic glycolysis is beneficial or protective, and OxPhos is not, hiPSC-derived astrocytes from AD patients harboring the PSEN1 ΔE9 mutation are more oxidative than isogenic controls, with increased ROS production and decreased lactate secretion (Oksanen et al., 2017). On the other hand, PC12 and B12 cells that are resistant to Aβ toxicity exhibit upregulated aerobic glycolysis (Newington et al., 2011). Furthermore, aerobic exercise, which improves cognitive scores in aging and AD patients (Panza et al., 2018), increases aerobic glycolysis and lactate production in the brain (Matsui et al., 2017). Interestingly, in the entorhinal cortex of APOE4 targeted-replacement mice, genes involved in OxPhos are upregulated, suggesting an APOE4-dependent increase in OxPhos and decrease in aerobic glycolysis and lactate (Figure 4; Nuriel et al., 2017b). Therapeutic strategies aimed at improving aerobic glycolysis may therefore help ameliorate APOE4-mediated toxicity.
Glycogen in Astrocytes
Despite its high energy demands, there are few energy stores in the brain compared to the rest of the body. In addition to storing lipids in the form of LDs, astrocytes are also the primary cell type in the brain to store glycogen. Astrocytic glycogen is important for maintaining healthy neurons and overall brain function, providing an energetic buffer during periods of low glucose availability (Bak et al., 2018). Primary astrocytes cultured in high (25 mM) vs. low glucose (5.5 mM) have elevated rates of glycolysis and glycogen content (Li et al., 2018). Elevated glycogen stores in co-cultured astrocytes are neuroprotective during glucose deprivation (Swanson and Choi, 1993). Significant evidence connects glycogen with memory formation: mice lacking glycogen synthase in the brain have impairments in learning- and memory-associated synaptic plasticity (Duran et al., 2013); glycogenolysis is important for memory consolidation (Gibbs et al., 2006); glycogen is a precursor to glutamate for learning (Gibbs et al., 2007); and glycogen content changes with early memory consolidation in 1-day-old chick (Hertz et al., 2003). Activated glycogen synthase kinase 3 has long been associated with the hallmarks of AD, including Aβ deposition, tau hyperphosphorylation, and brain inflammation, and would furthermore be expected to inhibit glycogen synthesis and thus decrease glycogen stores (Rayasam et al., 2009).
Energy metabolism in the brain may change during aging in a cell type-dependent manner. Inhibition of glycogen breakdown, termed “glycogenolysis,” disrupts long-term potentiation in young, but not old, rat hippocampus (Drulis-Fajdasz et al., 2015). In a follow-up proteomics study, the same group found that, while glycogen phosphorylase (PYGB), the rate-limiting enzyme in glycogen degradation, is predominantly expressed in astrocytes in young animals, its distribution switches to being present in both neurons and astrocytes in old animals (Drulis-Fajdasz et al., 2018). As glycogen accumulation in neurons normally triggers apoptosis (Vilchez et al., 2007; Duran et al., 2012), the authors speculate that upregulation of PYGB in neurons may be a protective mechanism to keep neuronal glycogen stores low. However, total depletion of glycogen in neurons may not be desirable in all circumstances, as low levels of neuronal glycogen may be protective during hypoxia (Saez et al., 2014).
The importance of glycogen to enhanced memory is not necessarily ascribed to elevated pyruvate for mitochondrial OxPhos, since aged wild-type and adult APP/PS1 mice fed a diet supplemented with pyruvate still exhibit impairments in a passive avoidance task for fear memory, despite a preservation of glycogen stores and enhanced exploratory behavior (Koivisto et al., 2016). Rather than supplying pyruvate for OxPhos, glycogen may instead supply lactate to mediate its beneficial effects, as described in the following section.
Lactate, Glycogen and the Astrocyte-Neuron Lactate Shuttle Hypothesis
The Astrocyte-Neuron Lactate Shuttle (ANLS) hypothesis was first formulated in 1994 (Pellerin and Magistretti, 1994, 2012), and describes a process in which astrocytes metabolize glucose to export lactate for neurons during periods of high neuronal activity, during learning and memory, for example. The existence of an astrocyte-to-neuron transport of lactate would necessitate a lower basal concentration of lactate in neurons compared to astrocytes, and a recent study has indeed demonstrated such a gradient in vivo, using a genetically-encoded lactate sensor (Machler et al., 2016).
Despite findings in support of the ANLS hypothesis, there has been some disagreement in the field as to whether astrocytic lactate is really used by active neurons in the brain, if neurons are able to produce their own alternative energy substrates, or if astrocytes produce lactate in response to their own energetic demands (Dienel, 2012). For example, computer simulations of neuron/astrocyte energetics, based on fMRS data, support a model in which neurons readily metabolize glucose and export lactate, which is taken up by astrocytes, and not the other way around (Simpson et al., 2007; Mangia et al., 2009).
Although neurons are certainly capable of taking up glucose and secreting lactate themselves, there is compelling evidence that lactate secretion from astrocytes, derived from glycogen stores specifically, is important in contexts of neuronal high energy demand. For example, the transport of lactate specifically from astrocytes to neurons is necessary for long-term memory formation (Suzuki et al., 2011) and spatial working memory (Newman et al., 2011). Neuronal activity can upregulate astrocytic genes involved in lactate production and export (Hasel et al., 2017), ensuring that astrocytes are able to supply neurons with the necessary lactate during periods of intense energetic demands. Lactate derived from astrocytic glycogen can sustain neuronal activity in the absence of other forms of energy, and blocking the transfer of lactate from astrocytes to neurons in the absence of any other energy source leads to axonal/neuronal failure (Ransom and Fern, 1997; Wender et al., 2000; Brown et al., 2003, 2005; Suh et al., 2007; Walls et al., 2008). Blocking glycogen degradation or lactate transfer reduces glutamate release from neurons (Sickmann et al., 2009). Furthermore, exhaustive exercise decreases brain glycogen and elevates astrocyte-derived lactate (Matsui et al., 2017). While the original ANLS hypothesis may undergo revision and refinement, astrocytic glycogen-derived lactate certainly appears to be an important component of healthy neuronal function, particularly during times of nutrient deficiency.
Connecting APOE4 and Brain Energy Metabolism: Future Directions
How could aging glycogen metabolism interface with APOE4 genotype to exacerbate neurodegeneration? Young adult APOE4 carriers have altered expression of proteins involved in glucose metabolism in the posterior cingulate cortex (PCC), a central component of the DMN (Perkins et al., 2016). Subregions of the PCC are proposed to be involved in internally directed cognition, including memory retrieval and planning, as well as controlling attentional focus (Leech and Sharp, 2014). The DMN is highly metabolically active and is one of the earliest regions to deteriorate in AD and in normal aging (Leech and Sharp, 2014), and young APOE4 carriers exhibit increased activity in the DMN before any signs of disease (Filippini et al., 2009). In agreement with this increased activity, hiPSC-derived neurons from APOE4 patients are hyperactive (Lin et al., 2018), and APOE4 targeted-replacement mice exhibit a hyperactive entorhinal cortex compared to APOE3-expressing mice (Nuriel et al., 2017a). While young APOE4 carriers were found to express higher levels of glycolysis enzymes (GLUT1, GLUT3, HEX1, MCT2, SCOT, AACS) and complexes I, II, and IV of the electron transport chain (ETC), there were lower levels of MCT4, an important transporter for astrocytic lactate secretion (Perkins et al., 2016). Disruption of MCT4 impairs long-term memory, which is rescued by lactate injection, while memory impairment caused by disruption of the neuronal lactate transporter MCT2, is not rescued by lactate, strongly supporting the notion that astrocytic export of lactate is critical for long-term memory formation (Suzuki et al., 2011). Thus, the decreased MCT4 in young APOE4 carriers might be expected to cause a deficit in lactate secretion by astrocytes, despite higher glycolytic activity. Interestingly, the DMN is a region that relies on aerobic glycolysis in young, healthy brain (Vaishnavi et al., 2010), and so should be a region that relies on elevated lactate production to support neuronal activity; the increased neuronal activity and decreased capacity of astrocytes to keep up with such activity in APOE4 carriers might then be expected to burn out glycogen stores early, effectively accelerating an aging-associated metabolic phenotype reliant on OxPhos. Further work is necessary to determine whether this pathway could be induced by diet, exercise, or pharmacological intervention to preserve cognitive function in presymptomatic APOE4 carriers.
In summary, APOE performs a complex set of interrelated functions in astrocytes, ranging from its long-appreciated lipid transport function to regulation of lipid storage and utilization to cellular energetics. In the next section we will review emerging concepts around APOE function in the other major producer of APOE in the brain, microglia.
Microglia-Derived APOE in Aging and AD
A large body of evidence implicates microglia in APOE-mediated AD pathogenesis, particularly in relation to aging. Microglial APOE production is strongly induced during injury and disease, including in AD (Olah et al., 2018; Ping et al., 2018; Rangaraju et al., 2018a). In 5XFAD transgenic mice, which harbor five different human familial AD-causing mutations and exhibit accelerated amyloid pathology (Oakley et al., 2006), microglial APOE mRNA is significantly increased (Wang et al., 2015). A similar increase in microglial APOE mRNA was also found in a separate but similar transgenic mouse model of accelerated amyloid pathology, APP/PS1 (Orre et al., 2014), as well as in aged (isolated from 24 month old mice) vs. younger (5 month old) mouse microglia (Hickman et al., 2013), and in the Ercc1 mutant mouse model of accelerated aging (Raj et al., 2014a; Holtman et al., 2015). The upregulation of APOE mRNA in these mouse models of AD and aging reflect concordant increases at the protein level and in human AD brain. A recent proteomics study of microglia isolated from 5XFAD mice identified APOE as one of the top upregulated proteins (Rangaraju et al., 2018a). Interestingly, immunohistochemical analysis in this same study indicated that the microglia with elevated APOE were those surrounding amyloid plaques, demonstrating that a distinct subset of microglia increase APOE expression, rather than all microglia. Furthermore, the aged mouse microglial proteome also shows an enrichment in APOE protein compared to non-aged mice (Rangaraju et al., 2018a). In agreement with these findings in mice, an analysis of frontal cortex human postmortem brain tissue found elevated APOE protein in AD patients vs. healthy controls (Ping et al., 2018). Another study performing a post-mortem human brain proteomics analysis also found APOE to be higher in the aged microglia (Olah et al., 2018). Thus aging alone, and not only disease pathogenesis, is sufficient to induce microglial APOE expression at both the mRNA and protein level.
The expression of APOE in subsets of disease- and aging-associated microglia raises an important question: what role does APOE play in the microglial response to disease and aging, and how is this impacted by APOE4 genotype? Both mouse and human studies indicate that key microglial functions are affected by APOE genotype, including transcriptomic changes towards the disease-associated phenotype, the percent of microglia coverage around plaques, increased cytokine production, as well as chemotaxis, phagocytosis and, perhaps, synaptic pruning (Figure 5). Each of these functions are discussed in the following sections.
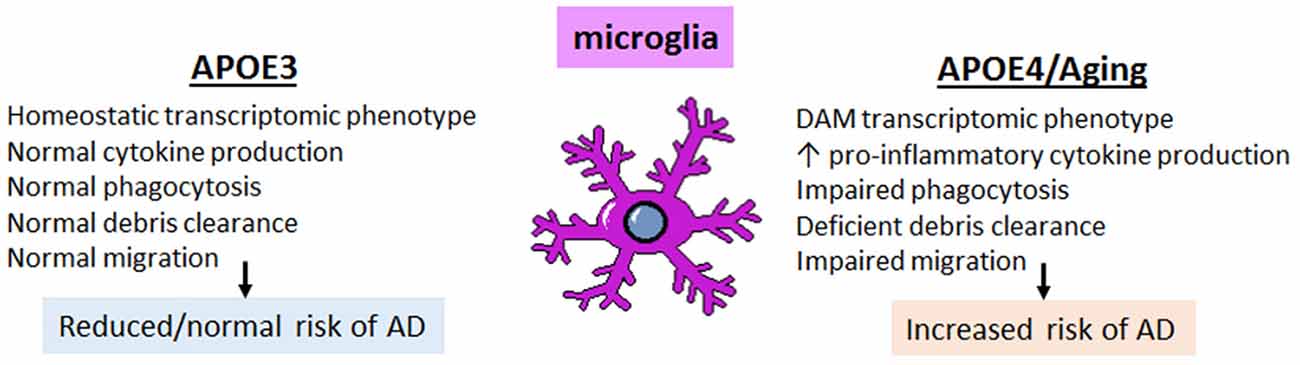
Figure 5. The effect of APOE4 expression on microglia is cell autonomous and triggers a DAM, pro-inflammatory phenotype with impaired homeostatic functions. Microglia expressing APOE4 vs. APOE3 tend to exhibit a disease-associated microglia (DAM)-like phenotype; this includes increased pro-inflammatory cytokine production with impaired phagocytic ability, deficient clearance of debris (including amyloid plaques), and impaired migratory ability. These changes, compounded with the pro-inflammatory phenotype associated with normal aging, result in an increased risk of developing AD.
APOE and the Microglial Phenotype in AD
The transcriptional profile of microglia is altered in AD, switching from a homeostatic phenotype to a molecular profile often referred to as the disease-associated microglial (DAM) phenotype (Zhang et al., 2013; Keren-Shaul et al., 2017; Sarlus and Heneka, 2017; Rangaraju et al., 2018b). Genome-wide association studies (GWAS), including large-scale meta-analyses, have indicated that the majority of genetic variants conferring risk for late onset sporadic AD are immune-related and enriched in microglia, implicating DAM microglia in AD pathogenesis (Guerreiro et al., 2013; Lambert et al., 2013; Dos Santos et al., 2017; Huang et al., 2017). The affected genes include myeloid receptors TREM2 and CD33, transcriptional regulators SPI1 (Pu.1) and MEF2C, complement pathway (CR1), antigen presentation (HLA-DRB5), the MS4A family locus, and ABCA7, amongst several others (Lambert et al., 2013). Some single nucleotide polymorphisms (SNPs) are present in non-coding regions and alter expression of microglial genes (e.g., SPI1; CD33), whereas other SNPs result in a gain or loss of function in several microglial genes related to immune function (e.g., TREM2) (Raj et al., 2014b; Malik et al., 2015; Huang et al., 2017). A large-scale weighted gene coexpression network analysis (WGCNA) combined with pathological assessment of 1647 post-mortem brain tissues from late-onset AD patients and non-demented controls pointed to immune/microglial gene networks as having the most significant functional enrichment of all modules (Rangaraju et al., 2018b). Moreover, this microglial module was significantly associated with the greatest number of AD-relevant pathological traits, including the extent of brain atrophy, and represented immune pathways consisting of complement, Fc-receptors, major histocompatibility complex (MHC), cytokines/chemokines and toll-like receptors (Zhang et al., 2013). Since then, several groups have characterized this DAM phenotype/immune network, albeit with varying nomenclature (Gjoneska et al., 2015; Keren-Shaul et al., 2017; Rangaraju et al., 2018b), and attempts to identify key regulators of this transcriptomic phenotype have been underway (Gjoneska et al., 2015). A common theme on which these genetic risk factors converge is that they alter key microglia activities, including phagocytosis, cytokine production, and microglial encapsulation of amyloid plaques (Figure 5). Altogether, this work has repositioned the thinking in the field, emphasizing microglia as a potential source of attractive therapeutic targets for AD.
Interestingly, APOE is a key regulator of the microglial transcriptional signature, as demonstrated in post-mortem human brain studies, human cellular models as well as in AD mouse models and cultured microglia in vitro (Keren-Shaul et al., 2017; Krasemann et al., 2017; Pimenova et al., 2017; Lin et al., 2018; Olah et al., 2018). Studies performing single cell RNA sequencing of CD45+ microglia from 5XFAD mice, paired with in situ hybridization of DAM signature genes, indicate that both the morphology and molecular identity of microglia around plaques, as a population, are different from microglia distal to plaques (Keren-Shaul et al., 2017). These DAMs have increased APOE expression that is triggered following a downregulation in homeostatic genes such as CX3CR1 and P2Y12 (Keren-Shaul et al., 2017; Krasemann et al., 2017). Furthermore, APOE mediates the switch from homeostatic to the DAM phenotype; notably, knocking out APOE specifically in microglia in 5XFAD mice prevents the transition to the DAM phenotype and partially rescues neuronal cell death in an axotomized facial motor nucleus model (Krasemann et al., 2017). A full knockout of APOE conferred no additional protection over that of microglia-selective APOE deletion, highlighting the importance of microglia-specific APOE to this process (as opposed to astrocytic APOE, for example; Krasemann et al., 2017). However, an astrocyte-selective APOE model was not directly compared, so it remains possible that it is not the cellular source of APOE that matters, but rather a reduction in total APOE levels that underlies this finding.
While elucidating the role of mouse APOE is informative, it is also critical to understand whether different phenotypes ensue with human APOE variants. Microglia isolated from aged vs. non-aged human postmortem brain for RNA sequencing analysis display an immune-enriched signature that is significantly associated with key traits, including APOE genotype (Olah et al., 2018). Although statistical significance was not reached for APOE4, the neuroprotective APOE2 was associated with a reduction in this aged microglial phenotype (Olah et al., 2018). In human cellular models, isogenic conversion of human iPSC-derived microglia from APOE3/E3 AD patients to APOE4/E4 is sufficient to transform the microglia transcriptome to a DAM-like phenotype (Lin et al., 2018). Notably, this APOE4 gene expression signature significantly overlapped with the transcriptional profile seen in human brain (Lin et al., 2018), in support of the notion that APOE4 may impact the DAM phenotype in human AD. A WGCNA transcriptomic analysis of brain from APOE3 or APOE4 targeted-replacement mice subjected to traumatic brain injury identified that the network most significantly associated with APOE genotype was the “innate immune response,” which included complement activation; in this network, the genes were shifted toward increased expression along with APOE4 compared to APOE3 (Castranio et al., 2017), again supporting a model in which APOE4 confers a pro-inflammatory phenotype relative to APOE3.
APOE Regulation of Microglial Plaque Association
APOE immunoreactivity in human brain is enriched in congophilic, dense-core plaques (Navarro et al., 2003), as opposed to diffuse plaques, which are heterogeneous with respect to APOE immunoreactivity (Gearing et al., 1995). Notably, microglial activation around diffuse plaques is minimal (Maat-Schieman et al., 1994; Stalder et al., 1999; Mrak, 2012), begging the question as to whether the presence of APOE in plaques is the trigger that differentially activates microglia at specific plaque types, or whether the presence of APOE in the plaques is simply due to the upregulation of APOE upon transition from homeostatic microglia to DAMs (Ulrich et al., 2014; Krasemann et al., 2017).
APOE4-expressing immune cells are less efficient at plaque engulfment compared to APOE3-expressing cells. When GFP+ bone marrow cells from human APOE3 or APOE4 donor mice were transplanted into lethally-irradiated 5 month old APPswe/PS1ΔE9 [i.e., bone marrow transplanted (BMT)-APP/PS1] mice, donor GFP+ macrophages are found in the brain 8 months later, with APOE3 exhibiting greater numbers of plaque-associated GFP+ Iba1+ cells (Yang et al., 2013). Interestingly, APOE4 was associated with reduced microglia coverage around Aβ plaques (Yang et al., 2013). Proper microglial encapsulation of plaques is thought to be protective, sequestering damage from surrounding cells, and decreased microglial coverage is associated with higher Aβ levels and increased neuronal dystrophy (Yeh et al., 2016). Indeed, the percentage of Aβ per area was significantly higher in the hippocampus and cortex of mice with APOE4 vs. APOE3 transplant (Yang et al., 2013). Furthermore, APOE4 BMT-APP/PS1 mice had significantly higher brain expression levels of the pro-inflammatory genes TNFα and macrophage migration inhibitory factor (MIF; which are upregulated in AD patients), lower levels of the anti-inflammatory gene IL-10, and impaired spatial working memory in the Barnes maze, compared with APOE3 BMT-APP/PS1 mice (Yang et al., 2013).
In another study using 5XFAD mice crossed to APOE3 or APOE4 targeted-replacement mice, mice expressing APOE4 exhibited significantly larger and more numerous amyloid plaques, as well as increased microglial dystrophy; but in contrast to the Yang et al. (2013) study, more microglia were found surrounding plaques in APOE4 vs. APOE3 and APOE2 (Rodriguez et al., 2014). It is difficult to distinguish whether the change in microglia phenotype in relation to plaque type is indirect, in response to worsened pathology or if it is also partly due to a cell autonomous effect of APOE4 on microglia, irrespective of plaque type. While these in vivo studies are informative, other recent studies indicate cell-intrinsic APOE4 effects on microglia. More specifically, human iPSC-derived microglia from APOE4 carriers have different morphology compared to isogenic APOE3 controls, and have a reduced capacity to phagocytose Aβ (Lin et al., 2018), in agreement with a change towards the DAM phenotype. Thus, APOE4 expression impairs the ability of microglia to efficiently clear amyloid pathology, although the precise mechanisms underlying microglial recruitment to specific amyloid plaques require further characterization.
APOE Genotype and Cytokine Production
An overwhelming body of evidence supports that the presence of APOE4, either recombinantly applied or endogenously expressed, confers an increase in pro-inflammatory cytokine production across rodent and human species, in blood, brain, and microglia. In support, rat primary glial cultures comprised of astrocytes and microglia produce higher levels of IL-1β when exposed to recombinant APOE4, purified from APOE-expressing HEK293 cell culture medium, than APOE3 (Guo et al., 2004). Cultured mouse microglia derived from APOE4 targeted-replacement mice have an activated morphology, produce higher levels of pro-inflammatory cytokines including TNFα, IL-6, and IL12p40, and nitric oxide (NO) along with lower levels of anti-inflammatory cytokines than their APOE3-derived counterpart when exposed to various pro-inflammatory mediators including LPS, IFNγ, or LPS+ IFNγ (Brown et al., 2002; Colton et al., 2005; Vitek et al., 2009). Notably, some of these effects (e.g., NO production) are APOE4 gene dosage-dependent (Vitek et al., 2009).
Similar to that seen in cultured microglia, APOE4 mice immune-challenged with a peripheral injection of LPS exhibit higher brain mRNA expression levels of TNFα and IL12p40 than in that from APOE3 TR mice (Vitek et al., 2009). A similar increase in pro-inflammatory cytokines, namely TNFα and IL-6, in APOE4 mouse serum is seen following a peripheral injection with LPS compared to that in APOE3 mice (Lynch et al., 2003). Finally, when LPS is administered by intracerebroventricular injection, APOE4 mice have higher brain levels of IL-1β, IL-6, and TNFα than APOE3 mice (Zhu et al., 2012).
While this increase has been consistently observed by independent groups in vivo in APOE targeted-replacement mice in AD models (Tai et al., 2011), due to perhaps independent roles of APOE genotype on other aspects of the disease (e.g., Aβ plaque levels), it is not clear whether the increase in cytokines by APOE4 is due to the increase in pathology, or due to a direct effect of APOE4 on cytokine production, which could contribute to the increase in pathological changes. Cell culture experiments shed some light on the former, in that the effect of APOE4 seems to be a cell-autonomous effect on microglia as when stimulated in culture, they produce more pro-inflammatory cytokines such as IL-1β (Guo et al., 2004), which suggests the differential extent of pathology (e.g., amyloid plaque deposition) as not being the sole driver of the differential increase in cytokines due to APOE4 vs. APOE3 genotype. Since these APOE targeted-replacement mouse studies assess the effect of human APOE in a mouse context, it remains plausible that this toxic pro-inflammatory effect attributed to APOE4 could be specific to mouse; however, human data indicates otherwise and suggests this phenomenon is intrinsic to the human APOE isoform irrespective of species by which it is produced/acting upon. Indeed, over the past few years, studies using advanced human cellular models parallel the pro-inflammatory findings seen in mice (Lin et al., 2018). Further, human clinical data suggests something similar. In two Chinese populations with AD, APOE4 carriers, carrying either one or two copies, had elevated plasma levels of the pro-inflammatory cytokines TNF-α, IL-6, and IL-1β compared to that of APOE2 and APOE3 carriers (Fan et al., 2017). Also, APOE genotype modulates cytokine production in human peripheral blood when stimulated with pro-inflammatory mediators ex vivo as well as in vivo. More specifically, ex vivo stimulation of peripheral blood collected from healthy volunteers with TLR2 and TLR4 ligands demonstrated that TNFα, IL-1β, IL-6, IL-17, IFNγ, G-CSF, IL-8, MCP-1, MIP-1a, and IP-10 levels were robustly increased in that from APOE3/E4 compared to APOE3/E3 carriers (Gale et al., 2014). Similarly, healthy human subjects intravenously administered the TLR4 ligand LPS exhibited higher plasma TNFα levels in APOE3/E4 vs. E3/E3 (Gale et al., 2014). In recent years, more advanced human cellular models make the picture clearer and indicate the mouse findings are not species specific and extend to human microglia.
Are these effects good or bad? Notably, recent studies have been controversial as to whether the best therapeutic approach for AD with respect to targeting APOE would be to lower APOE levels or augment them. As in astrocytes, the effects of APOE4 in microglia are often confounded by reports that APOE4 production and/or protein stability is lower compared to APOE3 (Bertrand et al., 1995; Raffai et al., 2001; Glockner et al., 2002). Thus, it remains plausible that this inflammatory response could be due to a decrease in APOE levels, irrespective of genotype. APOE3 can dampen cytokine production, and removing APOE can lead to a more pro-inflammatory phenotype. So, would elevating APOE4 protein levels help ameliorate the pro-inflammatory phenotype, or worsen it? Microglial APOE is neuroprotective in rat microglia neuronal co-cultures (Polazzi et al., 2015) and this release of APOE and the resulting neuroprotective effect is lost when microglia are exposed to inflammatory stimuli, thus lowering APOE. Therefore, it is interesting to hypothesize that, in the context of AD, when microglia are exposed to pro-inflammatory stimuli, APOE synthesis and secretion is stunted (Saura et al., 2003; Polazzi et al., 2015), effectively decreasing any neuroprotective effects of the microglia. A study examining the effect of APOE genotype comparing WT neurons cultured with either APOE3 vs. APOE4 mouse-derived astrocytes or microglia found that only APOE4 microglia led to greater neurotoxicity (Maezawa et al., 2006). Interestingly, the greater toxicity of APOE4 correlated with higher pro-inflammatory cytokine levels (TNFα, IL-6, IL-1β). Finally, it should be noted that the APOE4 effects can be sex-specific in certain contexts (Colton et al., 2005).
APOE Effect on Phagocytosis, Synaptic Pruning, and Chemotaxis
While the effect of APOE genotype on synaptic pruning and phagocytosis has been not been studied in microglia, astrocytic phagocytosis has been evaluated using APOE2, APOE3, and APOE4 targeted-replacement mice crossed to mice expressing EGFP driven by the astrocyte-specific promoter Aldh1l1 (Chung et al., 2016). Fluorophore-conjugated cholera toxin-β subunit (CTB-594) was used to label axonal projections of retinal ganglion cells and the dorsal lateral geniculate nucleus, an area with a high degree of synaptic pruning during development. In agreement with in vitro phagocytic assessments, astrocytes in APOE2 mice showed significantly enhanced phagocytic capacity compared with APOE3, whereas astrocytes in APOE4 mice demonstrated a significant decrease (Chung et al., 2016). Although this study focused on astrocytes, microglia are key players in synaptic pruning (Paolicelli et al., 2011; Schafer et al., 2012); thus, it would be informative to determine whether the effect of APOE genotype on synaptic pruning is cell type-specific or not. In vitro, ApoE−/− mouse-derived peritoneal macrophages demonstrated a decreased uptake of apoptotic cells, but no change in ability to uptake latex beads, compared to WT (Grainger et al., 2004). Given that APOE4 from human CSF was found to form smaller complexes than APOE2 and APOE3, it has been proposed that APOE4 may be deficient in lipid debris clearance, in accordance with phagocytic studies conducted on APOE4 human iPSC-derived microglia (Lin et al., 2018). Finally, microglial migration has also been found to be linked to APOE genotype (Cudaback et al., 2011). Mouse ApoE−/− microglia show reduced ATP- and C5a-triggered migration; likewise, in targeted-replacement mice, APOE2 and APOE4 have reduced ATP- and C5a-triggered migration compared to APOE3 (Cudaback et al., 2011).
APOE and Other Microglial AD Risk Factors
Several studies have looked at potential interactions between APOE and TREM2, another genetic risk factor for AD (Guerreiro et al., 2013). APOE can bind TREM2 (Atagi et al., 2015; Yeh et al., 2016), and, either directly or indirectly, APOE can alter TREM2 signaling or function (Jendresen et al., 2017). Both APOE and TREM2 are implicated in key steps in the homeostatic to DAM phenotype (Krasemann et al., 2017). It is still unclear whether there is and to what extent there is an interaction in APOE and TREM2, genetically or functionally, warranting further investigation.
Other risk factors for AD have clear functional overlap with APOE. Of note, ABCA7, also expressed in microglia, has been associated with age of onset of AD in a similar manner as APOE. More specifically, the minor allele at rs3764650 in ABCA7 is associated with a delayed onset and shorter disease duration (Kim et al., 2006). While its function in regulating the homeostasis of phospholipids and cholesterol has been the most well studied function of ABCA7 in relation to APOE, it also plays a role in phagocytosis (Tomioka et al., 2017), which has been observed in vitro and in vivo and been reviewed more extensively elsewhere (Abe-Dohmae and Yokoyama, 2012; Aikawa et al., 2018).
Future Directions for Understanding Microglial APOE in Immunosenescence
Is microglial APOE upregulation in aging and AD helpful or harmful? Is it a compensatory mechanism, or does it contribute to accelerated aging and neurodegenerative disease pathogenesis? It is interesting to speculate that the two greatest risk factors for late-onset AD, aging and APOE, interact with respect to inflammation, with APOE4 promoting an enhanced inflammatory tone over the course of a lifetime (Olarte et al., 2006; Sando et al., 2008). Microglia undergo senescence with aging, a process termed immunosenescence (Costantini et al., 2018), consistent with a DAM phenotype, and accumulating evidence suggests that APOE4 genotype may aggravate this process to promote neuroinflammation and neurodegeneration in AD. The change in cellular source of APOE from predominantly astrocyte-derived, to astrocyte- and microglia-derived during disease or aging, raises questions as to whether the cellular source of APOE subserves differential functions. It should be noted however, that although APOE immunoreactivity has been demonstrated around plaques in post-mortem human AD brain, co-labeling of APOE with microglial markers has not been investigated. Thus, a careful evaluation of this is warranted given the recent advances in understanding APOE expression in AD models. Finally, although there are clearly centrally mediated and cell-autonomous effects of APOE4, several peripheral effects of APOE4 on immune cells have been observed, and as such, it is unclear as to what extent the peripheral component of APOE4 status has on the risk to AD.
Conclusion
The role of APOE4 in mediating AD risk is complex and multifactorial, involving a diverse array of cell types and functions that need to be taken into consideration for APOE-directed drug development. Studies from the last decade have made significant progress in defining what those functions are, and how aging might factor into the progression of APOE4-mediated AD. Cholesterol metabolism, LD formation and lipid transfer from neurons to glia, and glucose/glycogen/lactate metabolism from glia to neurons all appear to be important pathways in maintaining brain health, particularly during aging; and the pro-inflammatory nature of APOE4 and decreased phagocytic capacity of APOE4-expressing glia likely contributes to neurodegeneration as well (Figure 6). These pathways may yield viable therapeutic targets for treating AD, but the precise mechanisms and connections with APOE4 still remain poorly defined. It is also unclear how APOE4-mediated disrupted function in astrocytes and microglia separately could synergize to increase AD risk, warranting further investigation.
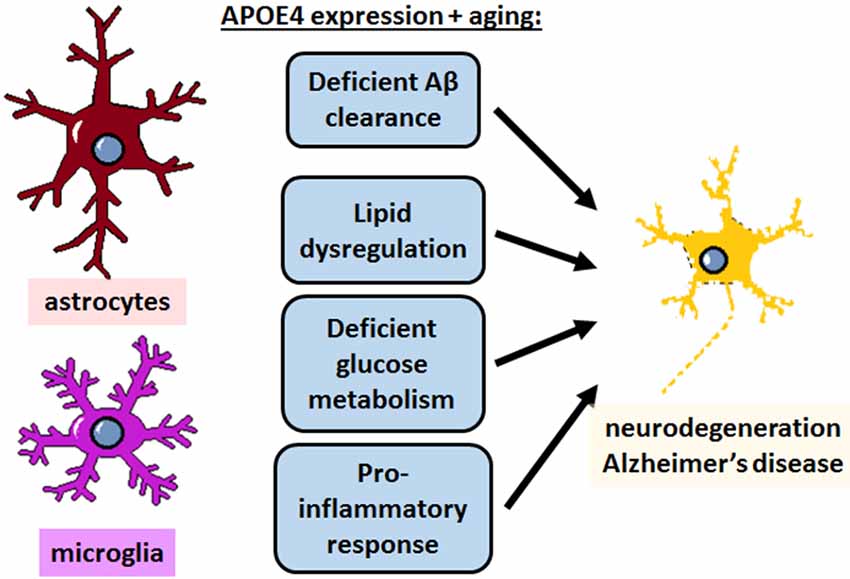
Figure 6. APOE4 disrupts homeostatic pathways in astrocytes and microglia to cause neurodegeneration and AD. APOE4 expression and the normal aging process itself impair astrocyte and microglia physiology in specific pathways, which could theoretically be targeted to treat AD. In addition to deficient clearance of Aβ, emerging evidence specifically highlights lipid dysregulation and deficient glucose metabolism in astrocytes, and a neurodegenerative pro-inflammatory response in microglia, and to some extent in astrocytes as well. All of these pathways converge with similar deficits that occur in normal aging to ultimately lead to neurodegeneration.
Author Contributions
CF wrote the first draft of the manuscript. CF, MH and WR wrote sections of the manuscript. MM and CF created the figures. All authors contributed intellectually and to manuscript revision, and read and approved the submitted version.
Funding
The Neurodegeneration Consortium is supported by the Robert A. and Renee E. Belfer Family Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abe-Dohmae, S., and Yokoyama, S. (2012). ABCA7: a potential mediator between cholesterol homeostasis and the host defense system. Clin. Lipidol. 7, 677–687. doi: 10.2217/clp.12.67
Aikawa, T., Holm, M. L., and Kanekiyo, T. (2018). ABCA7 and pathogenic pathways of Alzheimer’s disease. Brain Sci. 8:E27. doi: 10.3390/brainsci8020027
Akiyama, T. E., Sakai, S., Lambert, G., Nicol, C. J., Matsusue, K., Pimprale, S., et al. (2002). Conditional disruption of the peroxisome proliferator-activated receptor γ gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol. Cell. Biol. 22, 2607–2619. doi: 10.1128/mcb.22.8.2607-2619.2002
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Anand, S., Barnes, J. M., Young, S. A., Garcia, D. M., Tolley, H. D., Kauwe, J. S. K., et al. (2017). Discovery and confirmation of diagnostic serum lipid biomarkers for Alzheimer’s disease using direct infusion mass spectrometry. J. Alzheimers Dis. 59, 277–290. doi: 10.3233/JAD-170035
Atagi, Y., Liu, C. C., Painter, M. M., Chen, X. F., Verbeeck, C., Zheng, H., et al. (2015). Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050. doi: 10.1074/jbc.M115.679043
Bak, L. K., Walls, A. B., Schousboe, A., and Waagepetersen, H. S. (2018). Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 293, 7108–7116. doi: 10.1074/jbc.r117.803239
Basak, J. M., Verghese, P. B., Yoon, H., Kim, J., and Holtzman, D. M. (2012). Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J. Biol. Chem. 287, 13959–13971. doi: 10.1074/jbc.M111.288746
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23. doi: 10.1038/ng1934
Bertrand, P., Poirier, J., Oda, T., Finch, C. E., and Pasinetti, G. M. (1995). Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer disease. Mol. Brain Res. 33, 174–178. doi: 10.1016/0169-328x(95)00097-c
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N., and Allen, N. J. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22, 269–285. doi: 10.1016/j.celrep.2017.12.039
Boyles, J. K., Pitas, R. E., Wilson, E., Mahley, R. W., and Taylor, J. M. (1985). Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Invest. 76, 1501–1513. doi: 10.1172/jci112130
Brecht, W. J., Harris, F. M., Chang, S., Tesseur, I., Yu, G. Q., Xu, Q., et al. (2004). Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 24, 2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004
Brown, A. M., Sickmann, H. M., Fosgerau, K., Lund, T. M., Schousboe, A., Waagepetersen, H. S., et al. (2005). Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J. Neurosci. Res. 79, 74–80. doi: 10.1002/jnr.20335
Brown, A. M., Tekkök, S. B., and Ransom, B. R. (2003). Glycogen regulation and functional role in mouse white matter. J. Physiol. 549, 501–512. doi: 10.1113/jphysiol.2003.042416
Brown, C. M., Wright, E., Colton, C. A., Sullivan, P. M., Laskowitz, D. T., and Vitek, M. P. (2002). Apolipoprotein E isoform mediated regulation of nitric oxide release. Free Radic. Biol. Med. 32, 1071–1075. doi: 10.1016/S0891-5849(02)00803-1
Cabodevilla, A. G., Sánchez-Caballero, L., Nintou, E., Boiadjieva, V. G., Picatoste, F., Gubern, A., et al. (2013). Cell survival during complete nutrient deprivation depends on lipid droplet-fueled β-oxidation of fatty acids. J. Biol. Chem. 288, 27777–27788. doi: 10.1074/jbc.m113.466656
Casey, C. S., Atagi, Y., Yamazaki, Y., Shinohara, M., Tachibana, M., Fu, Y., et al. (2015). Apolipoprotein E inhibits cerebrovascular pericyte mobility through a RhoA protein-mediated pathway. J. Biol. Chem. 290, 14208–14217. doi: 10.1074/jbc.m114.625251
Castranio, E. L., Mounier, A., Wolfe, C. M., Nam, K. N., Fitz, N. F., Letronne, F., et al. (2017). Gene co-expression networks identify Trem2 and Tyrobp as major hubs in human APOE expressing mice following traumatic brain injury. Neurobiol. Dis. 105, 1–14. doi: 10.1016/j.nbd.2017.05.006
Chung, W. S., Verghese, P. B., Chakraborty, C., Joung, J., Hyman, B. T., Ulrich, J. D., et al. (2016). Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. U S A 113, 10186–10191. doi: 10.1073/pnas.1609896113
Clarke, L. E., Liddelow, S. A., Chakraborty, C., Munch, A. E., Heiman, M., and Barres, B. A. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U S A 115, E1896–e1905. doi: 10.1073/pnas.1800165115
Colton, C. A., Brown, C. M., and Vitek, M. P. (2005). Sex steroids, APOE genotype and the innate immune system. Neurobiol. Aging 26, 363–372. doi: 10.1016/j.neurobiolaging.2004.08.001
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Costantini, E., D’Angelo, C., and Reale, M. (2018). The role of immunosenescence in neurodegenerative diseases. Mediators Inflamm. 2018:6039171. doi: 10.1155/2018/6039171
Cudaback, E., Li, X., Montine, K. S., Montine, T. J., and Keene, C. D. (2011). Apolipoprotein E isoform-dependent microglia migration. FASEB J. 25, 2082–2091. doi: 10.1096/fj.10-176891
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. doi: 10.1172/jci36663
Dienel, G. A. (2012). Brain lactate metabolism: the discoveries and the controversies. J. Cereb. Blood Flow Metab. 32, 1107–1138. doi: 10.1038/jcbfm.2011.175
Dietschy, J. M., and Turley, S. D. (2002). Control of cholesterol turnover in the mouse. J. Biol. Chem. 277, 3801–3804. doi: 10.1074/jbc.r100057200
Doig, A. J., Del Castillo-Frias, M. P., Berthoumieu, O., Tarus, B., Nasica-Labouze, J., Sterpone, F., et al. (2017). Why is research on amyloid-β failing to give new drugs for Alzheimer’s disease? ACS Chem. Neurosci. 8, 1435–1437. doi: 10.1021/acschemneuro.7b00188
Dong, L. M., and Weisgraber, K. H. (1996). Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 271, 19053–19057. doi: 10.1074/jbc.271.32.19053
Dong, L. M., Wilson, C., Wardell, M. R., Simmons, T., Mahley, R. W., Weisgraber, K. H., et al. (1994). Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J. Biol. Chem. 269, 22358–22365.
Dos Santos, L. R., Pimassoni, L. H. S., Sena, G. G. S., Camporez, D., Belcavello, L., Trancozo, M., et al. (2017). Validating GWAS variants from microglial genes implicated in Alzheimer’s disease. J. Mol. Neurosci. 62, 215–221. doi: 10.1007/s12031-017-0928-7
Drulis-Fajdasz, D., Gizak, A., Wojtowicz, T., Wisniewski, J. R., and Rakus, D. (2018). Aging-associated changes in hippocampal glycogen metabolism in mice. Evidence for and against astrocyte-to-neuron lactate shuttle. Glia 66, 1481–1495. doi: 10.1002/glia.23319
Drulis-Fajdasz, D., Wójtowicz, T., Wawrzyniak, M., Wlodarczyk, J., Mozrzymas, J. W., and Rakus, D. (2015). Involvement of cellular metabolism in age-related LTP modifications in rat hippocampal slices. Oncotarget 6, 14065–14081. doi: 10.18632/oncotarget.4188
Duran, J., Saez, I., Gruart, A., Guinovart, J. J., and Delgado-García, J. M. (2013). Impairment in long-term memory formation and learning-dependent synaptic plasticity in mice lacking glycogen synthase in the brain. J. Cereb. Blood Flow Metab. 33, 550–556. doi: 10.1038/jcbfm.2012.200
Duran, J., Tevy, M. F., Garcia-Rocha, M., Calbó, J., Milán, M., and Guinovart, J. J. (2012). Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO Mol. Med. 4, 719–729. doi: 10.1002/emmm.201200241
Fan, Y. Y., Cai, Q. L., Gao, Z. Y., Lin, X., Huang, Q., Tang, W., et al. (2017). APOE ε4 allele elevates the expressions of inflammatory factors and promotes Alzheimer’s disease progression: a comparative study based on Han and She populations in the Wenzhou area. Brain Res. Bull. 132, 39–43. doi: 10.1016/j.brainresbull.2017.04.017
Farfel, J. M., Yu, L., De Jager, P. L., Schneider, J. A., and Bennett, D. A. (2016). Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol. Aging 37, 19–25. doi: 10.1016/j.neurobiolaging.2015.09.011
Ferris, H. A., Perry, R. J., Moreira, G. V., Shulman, G. I., Horton, J. D., and Kahn, C. R. (2017). Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc. Natl. Acad. Sci. U S A 114, 1189–1194. doi: 10.1073/pnas.1620506114
Filippini, N., MacIntosh, B. J., Hough, M. G., Goodwin, G. M., Frisoni, G. B., Smith, S. M., et al. (2009). Distinct patterns of brain activity in young carriers of the APOE-ε4 allele. Proc. Natl. Acad. Sci. U S A 106, 7209–7214. doi: 10.1073/pnas.0811879106
Gale, S. C., Gao, L., Mikacenic, C., Coyle, S. M., Rafaels, N., Murray Dudenkov, T., et al. (2014). APOε4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 134, 127–134. doi: 10.1016/j.jaci.2014.01.032
Gearing, M., Schneider, J. A., Robbins, R. S., Hollister, R. D., Mori, H., Games, D., et al. (1995). Regional variation in the distribution of apolipoprotein E and A β in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 54, 833–841. doi: 10.1097/00005072-199511000-00010
Giau, V. V., Bagyinszky, E., An, S. S., and Kim, S. Y. (2015). Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 11, 1723–1737. doi: 10.2147/ndt.s84266
Gibbs, M. E., Anderson, D. G., and Hertz, L. (2006). Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 54, 214–222. doi: 10.1002/glia.20377
Gibbs, M. E., Lloyd, H. G., Santa, T., and Hertz, L. (2007). Glycogen is a preferred glutamate precursor during learning in 1-day-old chick: biochemical and behavioral evidence. J. Neurosci. Res. 85, 3326–3333. doi: 10.1002/jnr.21307
Gjoneska, E., Pfenning, A. R., Mathys, H., Quon, G., Kundaje, A., Tsai, L. H., et al. (2015). Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369. doi: 10.1038/nature14252
Glockner, F., Meske, V., and Ohm, T. G. (2002). Genotype-related differences of hippocampal apolipoprotein E levels only in early stages of neuropathological changes in Alzheimer’s disease. Neuroscience 114, 1103–1114. doi: 10.1016/s0306-4522(02)00178-1
Gong, J. S., Kobayashi, M., Hayashi, H., Zou, K., Sawamura, N., Fujita, S. C., et al. (2002). Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. J. Biol. Chem. 277, 29919–29926. doi: 10.1074/jbc.m203934200
Goyal, M. S., Vlassenko, A. G., Blazey, T. M., Su, Y., Couture, L. E., Durbin, T. J., et al. (2017). Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 26, 353.e3–360.e3. doi: 10.1016/j.cmet.2017.07.010
Grainger, D. J., Reckless, J., and McKilligin, E. (2004). Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J. Immunol. 173, 6366–6375. doi: 10.4049/jimmunol.173.10.6366
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guo, L., LaDu, M. J., and Van Eldik, L. J. (2004). A dual role for apolipoprotein e in neuroinflammation: anti- and pro-inflammatory activity. J. Mol. Neurosci. 23, 205–212. doi: 10.1385/jmn:23:3:205
Halliday, M. R., Rege, S. V., Ma, Q., Zhao, Z., Miller, C. A., Winkler, E. A., et al. (2016). Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 36, 216–227. doi: 10.1038/jcbfm.2015.44
Hamilton, L. K., Dufresne, M., Joppé, S. E., Petryszyn, S., Aumont, A., Calon, F., et al. (2015). Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer’s disease. Cell Stem Cell 17, 397–411. doi: 10.1016/j.stem.2015.08.001
Harris, F. M., Tesseur, I., Brecht, W. J., Xu, Q., Mullendorff, K., Chang, S., et al. (2004). Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer’s disease. J. Biol. Chem. 279, 3862–3868. doi: 10.1074/jbc.m309475200
Harris, R. A., Tindale, L., and Cumming, R. C. (2014). Age-dependent metabolic dysregulation in cancer and Alzheimer’s disease. Biogerontology 15, 559–577. doi: 10.1007/s10522-014-9534-z
Hasel, P., Dando, O., Jiwaji, Z., Baxter, P., Todd, A. C., Heron, S., et al. (2017). Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 8:15132. doi: 10.1038/ncomms15132
Hatters, D. M., Peters-Libeu, C. A., and Weisgraber, K. H. (2006). Apolipoprotein E structure: insights into function. Trends Biochem. Sci. 31, 445–454. doi: 10.1038/npg.els.0005909
Havel, R. J., and Kane, J. P. (1973). Primary dysβlipoproteinemia: predominance of a specific apoprotein species in triglyceride-rich lipoproteins. Proc. Natl. Acad. Sci. U S A 70, 2015–2019. doi: 10.1073/pnas.70.7.2015
Hertz, L., O’Dowd, B. S., Ng, K. T., and Gibbs, M. E. (2003). Reciprocal changes in forebrain contents of glycogen and of glutamate/glutamine during early memory consolidation in the day-old chick. Brain Res. 994, 226–233. doi: 10.1016/j.brainres.2003.09.043
Hickman, S. E., Kingery, N. D., Ohsumi, T. K., Borowsky, M. L., Wang, L. C., Means, T. K., et al. (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. doi: 10.1038/nn.3554
Hohman, T. J., Dumitrescu, L., Barnes, L. L., Thambisetty, M., Beecham, G., Kunkle, B., et al. (2018). Sex-specific association of apolipoprotein E with cerebrospinal fluid levels of Tau. JAMA Neurol. 75, 989–998. doi: 10.1001/jamaneurol.2018.0821
Holtman, I. R., Noback, M., Bijlsma, M., Duong, K. N., van der Geest, M. A., Ketelaars, P. T., et al. (2015). Glia open access database (GOAD): a comprehensive gene expression encyclopedia of glia cells in health and disease. Glia 63, 1495–1506. doi: 10.1002/glia.22810
Holtzman, D. M., Herz, J., and Bu, G. (2012). Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006312. doi: 10.1101/cshperspect.a006312
Huang, K. L., Marcora, E., Pimenova, A. A., Di Narzo, A. F., Kapoor, M., Jin, S. C., et al. (2017). A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 20, 1052–1061. doi: 10.1038/nn.4587
Jendresen, C., Arskog, V., Daws, M. R., and Nilsson, L. N. (2017). The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J. Neuroinflammation 14:59. doi: 10.1186/s12974-017-0835-4
Jha, M. K., Jo, M., Kim, J. H., and Suk, K. (2018). Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist 1:1073858418783959. doi: 10.1177/1073858418783959
Jones, W., and Bianchi, K. (2015). Aerobic glycolysis: beyond proliferation. Front. Immunol. 6:227. doi: 10.3389/fimmu.2015.00227
Karim, R., Koc, M., Rettberg, J. R., Hodis, H. N., Henderson, V. W., St John, J. A., et al. (2019). Apolipoprotein E4 genotype in combination with poor metabolic profile is associated with reduced cognitive performance in healthy postmenopausal women: implications for late onset alzheimer’s disease. Menopause 26, 7–15. doi: 10.1097/GME.0000000000001160
Keeney, J. T., Ibrahimi, S., and Zhao, L. (2015). Human ApoE isoforms differentially modulate glucose and amyloid metabolic pathways in female brain: evidence of the mechanism of neuroprotection by ApoE2 and implications for Alzheimer’s disease prevention and early intervention. J. Alzheimers Dis. 48, 411–424. doi: 10.3233/jad-150348
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276.e17–1290.e17. doi: 10.1016/j.cell.2017.05.018
Kim, K. Y., Jang, H. J., Yang, Y. R., Park, K. I., Seo, J., Shin, I. W., et al. (2016). SREBP-2/PNPLA8 axis improves non-alcoholic fatty liver disease through activation of autophagy. Sci. Rep. 6:35732. doi: 10.1038/srep35732
Kim, W. S., Guillemin, G. J., Glaros, E. N., Lim, C. K., and Garner, B. (2006). Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport 17, 891–896. doi: 10.1097/01.wnr.0000221833.41340.cd
Knouff, C., Hinsdale, M. E., Mezdour, H., Altenburg, M. K., Watanabe, M., Quarfordt, S. H., et al. (1999). Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J. Clin. Invest. 103, 1579–1586. doi: 10.1172/jci6172
Koch, G., Di Lorenzo, F., Loizzo, S., Motta, C., Travaglione, S., Baiula, M., et al. (2017). CSF tau is associated with impaired cortical plasticity, cognitive decline and astrocyte survival only in APOE4-positive Alzheimer’s disease. Sci. Rep. 7:13728. doi: 10.1038/s41598-017-14204-3
Koistinaho, M., Lin, S., Wu, X., Esterman, M., Koger, D., Hanson, J., et al. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 10, 719–726. doi: 10.1038/nm1058
Koivisto, H., Leinonen, H., Puurula, M., Hafez, H. S., Barrera, G. A., Stridh, M. H., et al. (2016). Chronic pyruvate supplementation increases exploratory activity and brain energy reserves in young and middle-aged mice. Front. Aging Neurosci. 8:41. doi: 10.3389/fnagi.2016.00041
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566.e9–581.e9. doi: 10.1016/j.immuni.2017.08.008
Laffitte, B. A., Repa, J. J., Joseph, S. B., Wilpitz, D. C., Kast, H. R., Mangelsdorf, D. J., et al. (2001). LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl. Acad. Sci. U S A 98, 507–512. doi: 10.1073/pnas.021488798
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Leech, R., and Sharp, D. J. (2014). The role of the posterior cingulate cortex in cognition and disease. Brain 137, 12–32. doi: 10.1093/brain/awt162
Li, W., Roy Choudhury, G., Winters, A., Prah, J., Lin, W., Liu, R., et al. (2018). Hyperglycemia alters astrocyte metabolism and inhibits astrocyte proliferation. Aging Dis. 9, 674–684. doi: 10.14336/ad.2017.1208
Lian, H., Litvinchuk, A., Chiang, A. C., Aithmitti, N., Jankowsky, J. L., and Zheng, H. (2016). Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 36, 577–589. doi: 10.1523/JNEUROSCI.2117-15.2016
Liang, Y., Lin, S., Beyer, T. P., Zhang, Y., Wu, X., Bales, K. R., et al. (2004). A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J. Neurochem. 88, 623–634. doi: 10.1111/j.1471-4159.2004.02183.x
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141.e7–1154.e7. doi: 10.1016/j.neuron.2018.05.008
Liu, C. C., Hu, J., Zhao, N., Wang, J., Wang, N., Cirrito, J. R., et al. (2017a). Astrocytic LRP1 mediates brain aβ clearance and impacts amyloid deposition. J. Neurosci. 37, 4023–4031. doi: 10.1523/JNEUROSCI.3442-16.2017
Liu, L., MacKenzie, K. R., Putluri, N., Maletić-Savatić, M., and Bellen, H. J. (2017b). The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 26, 719.e6–737.e6. doi: 10.1016/j.cmet.2017.08.024
Liu, L., Zhang, K., Sandoval, H., Yamamoto, S., Jaiswal, M., Sanz, E., et al. (2015). Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190. doi: 10.1016/j.cell.2014.12.019
Lynch, J. R., Tang, W., Wang, H., Vitek, M. P., Bennett, E. R., Sullivan, P. M., et al. (2003). APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 278, 48529–48533. doi: 10.1074/jbc.m306923200
Ma, Q., Zhao, Z., Sagare, A. P., Wu, Y., Wang, M., Owens, N. C., et al. (2018). Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 13:57. doi: 10.1186/s13024-018-0286-0
Maat-Schieman, M. L., Rozemuller, A. J., van Duinen, S. G., Haan, J., Eikelenboom, P., and Roos, R. A. (1994). Microglia in diffuse plaques in hereditary cerebral hemorrhage with amyloidosis (Dutch). An immunohistochemical study. J. Neuropathol. Exp. Neurol. 53, 483–491. doi: 10.1097/00005072-199409000-00007
Machler, P., Wyss, M. T., Elsayed, M., Stobart, J., Gutierrez, R., von Faber-Castell, A., et al. (2016). In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 23, 94–102. doi: 10.1016/j.cmet.2015.10.010
Maezawa, I., Nivison, M., Montine, K. S., Maeda, N., and Montine, T. J. (2006). Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 20, 797–799. doi: 10.1096/fj.05-5423fje
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630. doi: 10.1126/science.3283935
Mahley, R. W. (2016). Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler. Thromb. Vasc. Biol. 36, 1305–1315. doi: 10.1161/atvbaha.116.307023
Mahley, R. W., and Rall, S. C. Jr. (2000). Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1, 507–537. doi: 10.1146/annurev.genom.1.1.507
Malik, M., Chiles, J. III., Xi, H. S., Medway, C., Simpson, J., Potluri, S., et al. (2015). Genetics of CD33 in Alzheimer’s disease and acute myeloid leukemia. Hum. Mol. Genet. 24, 3557–3570. doi: 10.1093/hmg/ddv092
Mandelblatt, J. S., Small, B. J., Luta, G., Hurria, A., Jim, H., McDonald, B. C., et al. (2018). Cancer-related cognitive outcomes among older breast cancer survivors in the thinking and living with cancer study. J. Clin. Oncol. doi: 10.1200/JCO.18.00140 [Epub ahead of print].
Mandrekar-Colucci, S., Karlo, J. C., and Landreth, G. E. (2012). Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 32, 10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012
Mangia, S., Simpson, I. A., Vannucci, S. J., and Carruthers, A. (2009). The in vivo neuron-to-astrocyte lactate shuttle in human brain: evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 109, 55–62. doi: 10.1111/j.1471-4159.2009.06003.x
Matsui, T., Omuro, H., Liu, Y. F., Soya, M., Shima, T., McEwen, B. S., et al. (2017). Astrocytic glycogen-derived lactate fuels the brain during exhaustive exercise to maintain endurance capacity. Proc. Natl. Acad. Sci. U S A 114, 6358–6363. doi: 10.1073/pnas.1702739114
McDougall, M., Choi, J., Magnusson, K., Truong, L., Tanguay, R., and Traber, M. G. (2017). Chronic vitamin E deficiency impairs cognitive function in adult zebrafish via dysregulation of brain lipids and energy metabolism. Free Radic. Biol. Med. 112, 308–317. doi: 10.1016/j.freeradbiomed.2017.08.002
Mohamed, A., Viveiros, A., Williams, K., and Posse de Chaves, E. (2018). Aβ inhibits SREBP-2 activation through Akt inhibition. J. Lipid Res. 59, 1–13. doi: 10.1194/jlr.m076703
Morris, J. K., Uy, R. A. Z., Vidoni, E. D., Wilkins, H. M., Archer, A. E., Thyfault, J. P., et al. (2017). Effect of APOE ε4 genotype on metabolic biomarkers in aging and Alzheimer’s disease. J. Alzheimers Dis. 58, 1129–1135. doi: 10.3233/jad-170148
Moutinho, M., Codocedo, J. F., Puntambekar, S. S., and Landreth, G. E. (2019). Nuclear receptors as therapeutic targets for neurodegenerative diseases: lost in translation. Annu. Rev. Pharmacol. Toxicol. 59, 237–261. doi: 10.1146/annurev-pharmtox-010818-021807
Mrak, R. E. (2012). Microglia in Alzheimer brain: a neuropathological perspective. Int. J. Alzheimers Dis. 2012:165021. doi: 10.1155/2012/165021
Muñoz, S. S., Garner, B., and Ooi, L. (2018). Understanding the role of ApoE fragments in Alzheimer’s disease. Neurochem. Res. doi: 10.1007/s11064-018-2629-1 [Epub ahead of print].
Nakai, M., Kawamata, T., Taniguchi, T., Maeda, K., and Tanaka, C. (1996). Expression of apolipoprotein E mRNA in rat microglia. Neurosci. Lett. 211, 41–44. doi: 10.1016/0304-3940(96)12716-6
Navarro, A., Del Valle, E., Astudillo, A., González del Rey, C., and Tolivia, J. (2003). Immunohistochemical study of distribution of apolipoproteins E and D in human cerebral β amyloid deposits. Exp. Neurol. 184, 697–704. doi: 10.1016/s0014-4886(03)00315-7
Newington, J. T., Pitts, A., Chien, A., Arseneault, R., Schubert, D., and Cumming, R. C. (2011). Amyloid β resistance in nerve cell lines is mediated by the Warburg effect. PLoS One 6:e19191. doi: 10.1371/journal.pone.0019191
Newman, L. A., Korol, D. L., and Gold, P. E. (2011). Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6:e28427. doi: 10.1371/journal.pone.0028427
Nunes, V. S., Cazita, P. M., Catanozi, S., Nakandakare, E. R., and Quintão, E. C. R. (2018). Decreased content, rate of synthesis and export of cholesterol in the brain of apoE knockout mice. J. Bioenerg. Biomembr. 50, 283–287. doi: 10.1007/s10863-018-9757-9
Nuriel, T., Angulo, S. L., Khan, U., Ashok, A., Chen, Q., Figueroa, H. Y., et al. (2017a). Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun. 8:1464. doi: 10.1038/s41467-017-01444-0
Nuriel, T., Peng, K. Y., Ashok, A., Dillman, A. A., Figueroa, H. Y., Apuzzo, J., et al. (2017b). The endosomal-lysosomal pathway is dysregulated by APOE4 expression in vivo. Front. Neurosci. 11:702. doi: 10.3389/fnins.2017.00702
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal β-amyloid aggregates, neurodegeneration and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
O’Brien, J. S., and Sampson, E. L. (1965). Lipid composition of the normal human brain: gray matter, white matter, and myelin. J. Lipid Res. 6, 537–544.
Oksanen, M., Petersen, A. J., Naumenko, N., Puttonen, K., Lehtonen, Š., Gubert Olive, M., et al. (2017). PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Reports 9, 1885–1897. doi: 10.1016/j.stemcr.2017.10.016
Olah, M., Patrick, E., Villani, A. C., Xu, J., White, C. C., Ryan, K. J., et al. (2018). A transcriptomic atlas of aged human microglia. Nat. Commun. 9:539. doi: 10.1038/s41467-018-02926-5
Olarte, L., Schupf, N., Lee, J. H., Tang, M. X., Santana, V., Williamson, J., et al. (2006). Apolipoprotein E ε4 and age at onset of sporadic and familial Alzheimer disease in Caribbean Hispanics. Arch. Neurol. 63, 1586–1590. doi: 10.1001/archneur.63.11.1586
Orre, M., Kamphuis, W., Osborn, L. M., Jansen, A. H. P., Kooijman, L., Bossers, K., et al. (2014). Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760. doi: 10.1016/j.neurobiolaging.2014.06.004
Panza, G. A., Taylor, B. A., MacDonald, H. V., Johnson, B. T., Zaleski, A. L., Livingston, J., et al. (2018). Can exercise improve cognitive symptoms of Alzheimer’s disease? J. Am. Geriatr. Soc. 66, 487–495. doi: 10.1111/jgs.15241
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Paresce, D. M., Ghosh, R. N., and Maxfield, F. R. (1996). Microglial cells internalize aggregates of the Alzheimer’s disease amyloid β-protein via a scavenger receptor. Neuron 17, 553–565. doi: 10.1016/s0896-6273(00)80187-7
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U S A 91, 10625–10629. doi: 10.1073/pnas.91.22.10625
Pellerin, L., and Magistretti, P. J. (2012). Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 32, 1152–1166. doi: 10.1038/jcbfm.2011.149
Perkins, M., Wolf, A. B., Chavira, B., Shonebarger, D., Meckel, J. P., Leung, L., et al. (2016). Altered energy metabolism pathways in the posterior cingulate in young adult apolipoprotein E ε4 carriers. J. Alzheimers Dis. 53, 95–106. doi: 10.3233/jad-151205
Pimenova, A. A., Marcora, E., and Goate, A. M. (2017). A tale of two genes: microglial apoe and trem2. Immunity 47, 398–400. doi: 10.1016/j.immuni.2017.08.015
Ping, L., Duong, D. M., Yin, L., Gearing, M., Lah, J. J., Levey, A. I., et al. (2018). Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s disease. Sci. Data 5:180036. doi: 10.1038/sdata.2018.36
Pitas, R. E., Boyles, J. K., Lee, S. H., Foss, D., and Mahley, R. W. (1987). Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta 917, 148–161. doi: 10.1016/0005-2760(87)90295-5
Polazzi, E., Mengoni, I., Pena-Altamira, E., Massenzio, F., Virgili, M., Petralla, S., et al. (2015). Neuronal regulation of neuroprotective microglial apolipoprotein E secretion in rat in vitro models of brain pathophysiology. J. Neuropathol. Exp. Neurol. 74, 818–834. doi: 10.1097/nen.0000000000000222
Prasad, H., and Rao, R. (2018). Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc. Natl. Acad. Sci. U S A 115, E6640–E6649. doi: 10.1073/pnas.1801612115
Raffai, R. L., Dong, L. M., Farese, R. V. Jr., and Weisgraber, K. H. (2001). Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc. Natl. Acad. Sci. U S A 98, 11587–11591. doi: 10.1073/pnas.201279298
Raj, D. D., Jaarsma, D., Holtman, I. R., Olah, M., Ferreira, F. M., Schaafsma, W., et al. (2014a). Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol. Aging 35, 2147–2160. doi: 10.1016/j.neurobiolaging.2014.03.025
Raj, T., Ryan, K. J., Replogle, J. M., Chibnik, L. B., Rosenkrantz, L., Tang, A., et al. (2014b). CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet. 23, 2729–2736. doi: 10.1093/hmg/ddt666
Rangaraju, S., Dammer, E. B., Raza, S. A., Gao, T., Xiao, H., βrbet, R., et al. (2018a). Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer’s disease-related proteins. Mol. Neurodegener. 13:34. doi: 10.1186/s13024-018-0266-4
Rangaraju, S., Dammer, E. B., Raza, S. A., Rathakrishnan, P., Xiao, H., Gao, T., et al. (2018b). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 13:24. doi: 10.1186/s13024-018-0254-8
Ransom, B. R., and Fern, R. (1997). Does astrocytic glycogen benefit axon function and survival in CNS white matter during glucose deprivation? Glia 21, 134–141. doi: 10.1002/(sici)1098-1136(199709)21:1<134::aid-glia15>3.0.co;2-t
Rayasam, G. V., Tulasi, V. K., Sodhi, R., Davis, J. A., and Ray, A. (2009). Glycogen synthase kinase 3: more than a namesake. Br. J. Pharmacol. 156, 885–898. doi: 10.1111/j.1476-5381.2008.00085.x
Riddell, D. R., Zhou, H., Atchison, K., Warwick, H. K., Atkinson, P. J., Jefferson, J., et al. (2008). Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci. 28, 11445–11453. doi: 10.1523/JNEUROSCI.1972-08.2008
Ries, M., and Sastre, M. (2016). Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. 8:160. doi: 10.3389/fnagi.2016.00160
Rodriguez, G. A., Tai, L. M., LaDu, M. J., and Rebeck, G. W. (2014). Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflammation 11:111. doi: 10.1186/1742-2094-11-111
Ruiz, J., Kouiavskaia, D., Migliorini, M., Robinson, S., Saenko, E. L., Gorlatova, N., et al. (2005). The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 46, 1721–1731. doi: 10.1194/jlr.m500114-jlr200
Saez, I., Duran, J., Sinadinos, C., Beltran, A., Yanes, O., Tevy, M. F., et al. (2014). Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J. Cereb. Blood Flow Metab. 34, 945–955. doi: 10.1038/jcbfm.2014.33
Sando, S. B., Melquist, S., Cannon, A., Hutton, M. L., Sletvold, O., Saltvedt, I., et al. (2008). APOE ε 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol. 8:9. doi: 10.1186/1471-2377-8-9
Sarlus, H., and Heneka, M. T. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249. doi: 10.1172/JCI90606
Saunders, A. M., Strittmatter, W. J., Schmechel, D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., et al. (1993). Association of apolipoprotein E allele ε 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472. doi: 10.1212/WNL.43.8.1467
Saura, J., Petegnief, V., Wu, X., Liang, Y., and Paul, S. M. (2003). Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: opposite effects of lipopolysaccharide. J. Neurochem. 85, 1455–1467. doi: 10.1046/j.1471-4159.2003.01788.x
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Seo, Y. K., Jeon, T. I., Chong, H. K., Biesinger, J., Xie, X., and Osborne, T. F. (2011). Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. 13, 367–375. doi: 10.1016/j.cmet.2011.03.005
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Shimabukuro, M. K., Langhi, L. G., Cordeiro, I., Brito, J. M., Batista, C. M., Mattson, M. P., et al. (2016). Lipid-laden cells differentially distributed in the aging brain are functionally active and correspond to distinct phenotypes. Sci. Rep. 6:23795. doi: 10.1038/srep23795
Sickmann, H. M., Walls, A. B., Schousboe, A., Bouman, S. D., and Waagepetersen, H. S. (2009). Functional significance of brain glycogen in sustaining glutamatergic neurotransmission. J. Neurochem. 109, 80–86. doi: 10.1111/j.1471-4159.2009.05915.x
Simonovitch, S., Schmukler, E., Bespalko, A., Iram, T., Frenkel, D., Holtzman, D. M., et al. (2016). Impaired autophagy in APOE4 astrocytes. J. Alzheimers Dis. 51, 915–927. doi: 10.3233/jad-151101
Simpson, I. A., Carruthers, A., and Vannucci, S. J. (2007). Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J. Cereb. Blood Flow Metab. 27, 1766–1791. doi: 10.1038/sj.jcbfm.9600521
Soto, I., Graham, L. C., Richter, H. J., Simeone, S. N., Radell, J. E., Grabowska, W., et al. (2015). APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biol. 13:e1002279. doi: 10.1371/journal.pbio.1002279
Speidell, A. P., Demby, T., Lee, Y., Rodriguez, O., Albanese, C., Mandelblatt, J., et al. (2019). Development of a human APOE knock-in mouse model for study of cognitive function after cancer chemotherapy. Neurotox. Res. 35, 291–303. doi: 10.1007/s12640-018-9954-7
Stalder, M., Phinney, A., Probst, A., Sommer, B., Staufenbiel, M., and Jucker, M. (1999). Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol. 154, 1673–1684. doi: 10.1016/s0002-9440(10)65423-5
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993). Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U S A 90, 1977–1981. doi: 10.1073/pnas.90.5.1977
Suh, S. W., Bergher, J. P., Anderson, C. M., Treadway, J. L., Fosgerau, K., and Swanson, R. A. (2007). Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)pro pyl]-1H-indole-2-carboxamide). J. Pharmacol. Exp. Ther. 321, 45–50. doi: 10.1124/jpet.106.115550
Sullivan, P. M., Mezdour, H., Aratani, Y., Knouff, C., Najib, J., Reddick, R. L., et al. (1997). Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 272, 17972–17980. doi: 10.1074/jbc.272.29.17972
Supplie, L. M., Duking, T., Campbell, G., Diaz, F., Moraes, C. T., Götz, M., et al. (2017). Respiration-deficient astrocytes survive as glycolytic cells in vivo. J. Neurosci. 37, 4231–4242. doi: 10.1523/JNEUROSCI.0756-16.2017
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. doi: 10.1016/j.cell.2011.02.018
Swanson, R. A., and Choi, D. W. (1993). Glial glycogen stores affect neuronal survival during glucose deprivation in vitro. J. Cereb. Blood Flow Metab. 13, 162–169. doi: 10.1038/jcbfm.1993.19
Tai, L. M., Youmans, K. L., Jungbauer, L., Yu, C., and Ladu, M. J. (2011). Introducing human APOE into aβ transgenic mouse models. Int. J. Alzheimers Dis. 2011:810981. doi: 10.4061/2011/810981
Tambini, M. D., Pera, M., Kanter, E., Yang, H., Guardia-Laguarta, C., Holtzman, D., et al. (2016). ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 17, 27–36. doi: 10.15252/embr.201540614
Tomioka, M., Toda, Y., Mañucat, N. B., Akatsu, H., Fukumoto, M., Kono, N., et al. (2017). Lysophosphatidylcholine export by human ABCA7. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 658–665. doi: 10.1016/j.bbalip.2017.03.012
Treusch, S., Hamamichi, S., Goodman, J. L., Matlack, K. E., Chung, C. Y., Baru, V., et al. (2011). Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 334, 1241–1245. doi: 10.1126/science.1213210
Uchihara, T., Duyckaerts, C., He, Y., Kobayashi, K., Seilhean, D., Amouyel, P., et al. (1995). ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain. Neurosci. Lett. 195, 5–8. doi: 10.1016/0304-3940(95)11763-m
Ulrich, J. D., Finn, M. B., Wang, Y., Shen, A., Mahan, T. E., Jiang, H., et al. (2014). Altered microglial response to Aβ plaques in APPPS1–21 mice heterozygous for TREM2. Mol. Neurodegener. 9:20. doi: 10.1186/1750-1326-9-20
Vaishnavi, S. N., Vlassenko, A. G., Rundle, M. M., Snyder, A. Z., Mintun, M. A., and Raichle, M. E. (2010). Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. U S A 107, 17757–17762. doi: 10.1073/pnas.1010459107
Verghese, P. B., Castellano, J. M., Garai, K., Wang, Y., Jiang, H., Shah, A., et al. (2013). ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U S A 110, E1807–E1816. doi: 10.1073/pnas.1220484110
Vilchez, D., Ros, S., Cifuentes, D., Pujadas, L., Valles, J., García-Fojeda, B., et al. (2007). Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 10, 1407–1413. doi: 10.1038/nn1998
Vitek, M. P., Brown, C. M., and Colton, C. A. (2009). APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360. doi: 10.1016/j.neurobiolaging.2007.11.014
Walls, A. B., Sickmann, H. M., Brown, A., Bouman, S. D., Ransom, B., Schousboe, A., et al. (2008). Characterization of 1,4-dideoxy-1,4-imino-d-arabinitol (DAB) as an inhibitor of brain glycogen shunt activity. J. Neurochem. 105, 1462–1470. doi: 10.1111/j.1471-4159.2008.05250.x
Wang, L., Schuster, G. U., Hultenby, K., Zhang, Q., Andersson, S., and Gustafsson, J. A. (2002). Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc. Natl. Acad. Sci. U S A 99, 13878–13883. doi: 10.1073/pnas.172510899
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Weisgraber, K. H., Innerarity, T. L., and Mahley, R. W. (1982). Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J. Biol. Chem. 257, 2518–2521.
Wender, R., Brown, A. M., Fern, R., Swanson, R. A., Farrell, K., and Ransom, B. R. (2000). Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 20, 6804–6810. doi: 10.1523/JNEUROSCI.20-18-06804.2000
Wu, L., and Zhao, L. (2016). ApoE2 and Alzheimer’s disease: time to take a closer look. Neural Regen. Res. 11, 412–413. doi: 10.4103/1673-5374.179044
Wu, L., Zhang, X., and Zhao, L. (2018). Human ApoE isoforms differentially modulate brain glucose and ketone body metabolism: implications for Alzheimer’s disease risk reduction and early intervention. J. Neurosci. 38, 6665–6681. doi: 10.1523/JNEUROSCI.2262-17.2018
Wyss-Coray, T., Loike, J. D., Brionne, T. C., Lu, E., Anankov, R., Yan, F., et al. (2003). Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med. 9, 453–457. doi: 10.1038/nm838
Xu, P. T., Gilbert, J. R., Qiu, H. L., Rothrock-Christian, T., Settles, D. L., Roses, A. D., et al. (1998). Regionally specific neuronal expression of human APOE gene in transgenic mice. Neurosci. Lett. 246, 65–68. doi: 10.1016/s0304-3940(98)00247-x
Xu, Q., Bernardo, A., Walker, D., Kanegawa, T., Mahley, R. W., and Huang, Y. (2006). Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006
Yang, Y., Cudaback, E., Jorstad, N. L., Hemingway, J. F., Hagan, C. E., Melief, E. J., et al. (2013). APOE3, but not APOE4, bone marrow transplantation mitigates behavioral and pathological changes in a mouse model of Alzheimer disease. Am. J. Pathol. 183, 905–917. doi: 10.1016/j.ajpath.2013.05.009
Ye, S., Huang, Y., Mullendorff, K., Dong, L., Giedt, G., Meng, E. C., et al. (2005). Apolipoprotein (apo) E4 enhances amyloid β peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc. Natl. Acad. Sci. U S A 102, 18700–18705. doi: 10.1073/pnas.0508693102
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-β by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yun, S. P., Kam, T. I., Panicker, N., Kim, S., Oh, Y., Park, J. S., et al. (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 24, 931–938. doi: 10.1038/s41591-018-0051-5
Zhang, B., Gaiteri, C., Bodea, L. G., Wang, Z., McElwee, J., Podtelezhnikov, A. A., et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. doi: 10.1016/j.cell.2013.03.030
Zhao, J., Davis, M. D., Martens, Y. A., Shinohara, M., Graff-Radford, N. R., Younkin, S. G., et al. (2017a). APOE ε4/ε4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 26, 2690–2700. doi: 10.1093/hmg/ddx155
Zhao, N., Liu, C. C., Van Ingelgom, A. J., Martens, Y. A., Linares, C., Knight, J. A., et al. (2017b). Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron 96, 115.e5–129.e5. doi: 10.1016/j.neuron.2017.09.003
Keywords: APOE, Alzheimer’s disease, astrocytes, microglia, aging
Citation: Fernandez CG, Hamby ME, McReynolds ML and Ray WJ (2019) The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 11:14. doi: 10.3389/fnagi.2019.00014
Received: 31 October 2018; Accepted: 16 January 2019;
Published: 11 February 2019.
Edited by:
David Baglietto-Vargas, University of California, Irvine, United StatesReviewed by:
Jennifer S. Yokoyama, University of San Francisco, United StatesChristian J. Pike, University of Southern California, United States
Copyright © 2019 Fernandez, Hamby, McReynolds and Ray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Celia G. Fernandez, Y2Zlcm5hbmRlejJAbWRhbmRlcnNvbi5vcmc=