 Eriola Hoxha1,2
Eriola Hoxha1,2 Pellegrino Lippiello3
Pellegrino Lippiello3 Fabio Zurlo3
Fabio Zurlo3 Ilaria Balbo1,2
Ilaria Balbo1,2 Rita Santamaria3
Rita Santamaria3 Filippo Tempia1,2,4*
Filippo Tempia1,2,4* Maria Concetta Miniaci3
Maria Concetta Miniaci3- 1Neuroscience Institute Cavalieri Ottolenghi (NICO), Turin, Italy
- 2Department of Neuroscience, University of Torino, Turin, Italy
- 3Department of Pharmacy, School of Medicine, University of Naples Federico II, Naples, Italy
- 4National Institute of Neuroscience (INN), Turin, Italy
The role of the cerebellum in Alzheimer’s disease (AD) has been neglected for a long time. Recent studies carried out using transgenic mouse models have demonstrated that amyloid-β (Aβ) is deposited in the cerebellum and affects synaptic transmission and plasticity, sometimes before plaque formation. A wide variability of motor phenotype has been observed in the different murine models of AD, without a consistent correlation with the extent of cerebellar histopathological changes or with cognitive deficits. The loss of noradrenergic drive may contribute to the impairment of cerebellar synaptic function and motor learning observed in these mice. Furthermore, cerebellar neurons, particularly granule cells, have been used as in vitro model of Aβ-induced neuronal damage. An unexpected conclusion is that the cerebellum, for a long time thought to be somehow protected from AD pathology, is actually considered as a region vulnerable to Aβ toxic damage, even at the early stage of the disease, with consequences on motor performance.
Introduction
Accumulating evidence indicates that Alzheimer’s disease (AD) is caused by the toxicity of amyloid-β (Aβ) peptide (Haass and Selkoe, 2007; Shankar et al., 2008; Gouras et al., 2010). Aβ peptides are generated from β-amyloid precursor protein (APP) via proteolytic cleavage by the β- and γ-secretases; depending on the site of cleavage, different sized Aβ peptides are generated, with Aβ1–40 and Aβ1–42 being the most toxic Aβ isoforms (Selkoe, 1998; O’Brien and Wong, 2011). The soluble monomers can assemble into microaggregates, also termed “soluble Aβ oligomers,” protofibrils and fibrils that accumulate in the brains of AD subjects, forming insoluble amyloid plaques. It is now believed that soluble Aβ oligomers are the critical pathogenic molecules in AD leading to synaptic and cognitive dysfunctions (Selkoe, 2008; Ferreira et al., 2015). Accumulation of Aβ peptides and insoluble plaque formation trigger a number of detrimental processes, including hyperphosphorylation of the microtubule-associated protein tau, which leads to the formation of neurofibrillary tangles and neuronal death (Kopeikina et al., 2012; Lasagna-Reeves et al., 2012). In accordance with this hypothesis, recent evidence shows that, in transgenic mice expressing the human tau protein, injection of Aβ1–42 monomers can induce tau hyperphosphorylation (Manassero et al., 2016). However, the relevance of this mechanism in the cerebellum is not clear yet. Indeed, the cerebellum exhibits a lower expression of tau compared to the cerebral cortex or to the hippocampus (Hu et al., 2017).
Most of the research in the AD field has been focused on characterizing the toxicity of Aβ on the medial temporal lobe structures, especially the hippocampus, since their dysfunction is considered the main cause of memory loss in AD, the earliest cognitive deficit in this type of dementia (Hyman et al., 1984; West et al., 1994; Scheff et al., 2007; Miniaci and De Leonibus, 2018). However, patients with AD can also develop language, visual or motor symptoms, suggesting that Aβ pathology extends beyond the hippocampus (Caine and Hodges, 2001; Lambon Ralph et al., 2003; Albers et al., 2015).
Studies in both familial (FAD) and sporadic cases of AD have demonstrated that the cerebellum is a vulnerable region in AD patients (Schmahmann, 2016; Jacobs et al., 2018). Elevated levels of Aβ oligomers have been observed in the cerebellar cortex of AD patients (Larner, 1997; Sepulveda-Falla et al., 2011). Furthermore, cases of early-onset FAD, caused by mutation of presenilin-1 (PSEN1; the catalytic subunits of γ-secretase), present Purkinje cell (PC) loss as well as cerebellar deposition of Aβ and high levels of hyperphosphorylated tau (Sepulveda-Falla et al., 2011, 2014). Cerebellar atrophy is another characteristic feature of sporadic AD which affects initially the portions of the cerebellar regions connected to the default mode network, a set of highly interacting brain regions (including angular gyrus, middle temporal gyrus, precuneus and dorsal medial prefrontal cortex) involved in cognition and extensively affected by neurodegeneration such as in AD (Thomann et al., 2008; Mevel et al., 2011; Guo et al., 2016). As the atrophy extends to the cerebellar anterior lobe, patients may display motor dysfunctions, i.e., impairments in gait and limb coordination (Aggarwal et al., 2006).
In the last decades, mouse models of AD expressing mutant human APP, sometimes together with mutant presenilins, have been widely used for investigating the AD pathophysiology and to establish new therapeutic strategies (Ashe and Zahs, 2010; Puzzo et al., 2015; Drummond and Wisniewski, 2017; Sasaguri et al., 2017). Most of the studies on these mouse models have investigated the relationship between Aβ, memory impairment and hippocampal synaptic plasticity, which is considered the cellular correlate of learning and memory (Selkoe, 2002; Haass and Selkoe, 2007; Shankar and Walsh, 2009). Less is known about the effects of Aβ on cerebellar synaptic functions and motor learning. Here, we provide for the first time a comprehensive review of the literature regarding the cerebellar cellular and molecular characterization in mouse models of AD, that can help researchers to better understand cerebellar contribution in neurodegenerative disorders and eventually provide the basis for future research on this field.
Cerebellar Motor Dysfunctions in Mouse Model of AD
Motor deficits, initially classified as late-onset symptoms in AD patients, have been considered for a long time as irrelevant given the severe deterioration of cognitive functions (Ala and Frey, 1995). However, there is increasing evidence that motor performance is impaired even at the preclinical stage of the disease (Pettersson et al., 2002; Buchman and Bennett, 2011) and a link between motor skills deficits and AD onset probability has also been proposed (Aggarwal et al., 2006).
Significant motor impairment has been also observed in transgenic mice modeling AD (Table 1). For example, APP23 transgenic mice overexpressing APP751 isoform with the Swedish mutation (KM670/671NL) show motor coordination deficit on the rotarod by 3 months of age, before Aβ accumulation (Van Dam et al., 2003). Rotarod motor coordination deficits preceding Aβ accumulation has been also revealed in APP/PSEN1 mice, carrying APP695 isoform with Swedish mutation and mutant presenilin 1 genes (APPswe/PSEN1dE9; Kuwabara et al., 2014). Moreover, APPswe/PSEN1dE9 mice show motor learning deficits on the ErasmusLadder Task (Sepulveda-Falla et al., 2014). Interestingly, in addition to motor coordination deficits on the beam test, TgCRND8 mice, containing human APP695 with the Swedish and Indiana (V717F) mutations, exhibit gait impairment on the footprinting test (Russo et al., 2018).
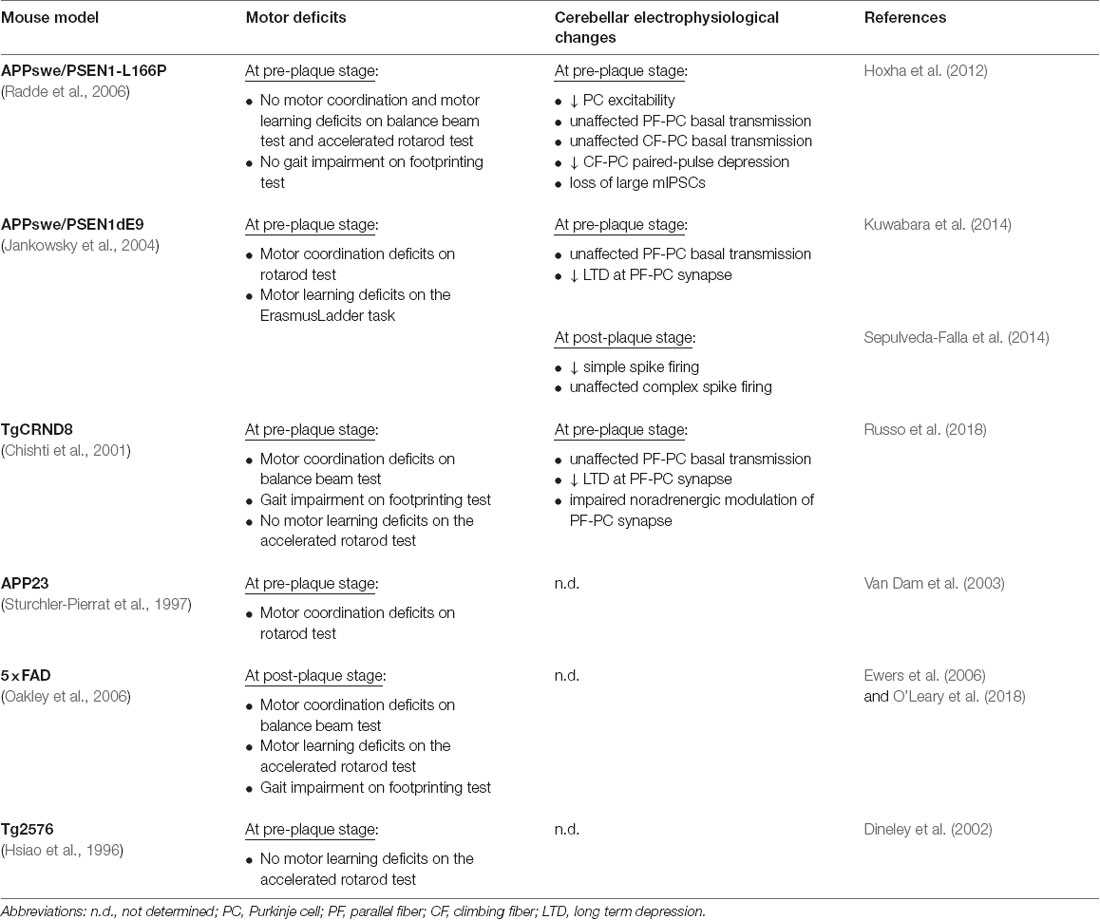
Table 1. Motor behavior and cerebellar electrophysiological phenotypes in murine models of Alzheimer’s disease (AD).
On the other hand, several studies report motor deficits only at later stages, when Aβ deposition is already present. For example, 5xFAD mice, carrying human APP695 with Swedish, Florida (APP I716V) and London (V717I) mutations plus PSEN1 with M146L and L286V mutations, exhibit a severe motor impairment at 11–13 months of age consisting of motor coordination and motor learning deficits on the balance beam test and accelerating rotarod, respectively (Ewers et al., 2006). Moreover, 5xFAD transgenic mice show also changes in gait parameters with a shorter stride length respect to wild type (WT) mice on the footprinting test (O’Leary et al., 2018).
Nevertheless, there are other transgenic (Tg) lines that not show motor dysfunctions even in the presence of cerebellar Aβ accumulation, PC death and/or cerebellar electrophysiological changes. These models include Tg2576 mice, which overexpress a mutant form of APP695 with the Swedish mutation (Dineley et al., 2002), and APP/PS1 mice overexpressing APP695 Swedish mutant form plus PSEN1 with the L166P mutation (APPswe/PSEN1-L166P; Hoxha et al., 2012).
These results, taken together, suggest that there is no clear relationship between cerebellar Aβ deposits and motor deficits. It is possible to hypothesize that soluble forms of Aβ, like monomers or oligomers, are more directly related to motor symptoms. This might explain the wide variability of the motor phenotype observed in the different murine models of AD.
Motor deficits are also not correlated to the onset of cognitive impairment. In fact, in TgCRND8 and APPswe/PSdeltaE9 mice, motor deficits precede cognitive symptoms (Chishti et al., 2001; Reiserer et al., 2007). In 5xFAD mice, the cognitive impairment becomes apparent before motor deficits (Oakley et al., 2006). In APP23 mice motor and cognitive symptoms start at the same age (Van Dam et al., 2003).
Given these behavioral outcomes, it is possible that the brain regions engaged in motor and cognitive performances are differentially affected in AD animal models.
Cerebellar Histopathological Changes
Several studies in AD patients have reported diffuse amyloid plaques in the cerebellum particularly in those with the pre-senile onset of dementia (Larner, 1997). Aβ deposits were found in the molecular layer of the cerebellar cortex extending to the PC layer, and to a lower extent in the granule cell layer (Braak et al., 1989; Sepulveda-Falla et al., 2011). The cerebellar histopathological alterations appear earlier and are more severe in FAD compared to the sporadic form of AD and are characterized by PC loss, neuritic plaques and reactive astrocytosis (Fukutani et al., 1996).
Similar anatomical and temporal patterns of alterations develop in the cerebellum of transgenic mice models of FAD. For example, APPswe/PSEN1dE9 mice show few diffuse Aβ deposits in the molecular layer at 6 months of age; the number of cerebellar deposits increases with disease progression and at higher rate in females vs. males (Lomoio et al., 2012; Kuwabara et al., 2014). At 18/20 months of age, the Aβ plaques appear surrounded by dystrophic neurites, microglia and astrocytes; focal loss of PCs and degeneration of parallel fibers (PFs) have been also observed. Significant loss of PCs (about 60%) has been also revealed in Tg2576 mice (Kozuki et al., 2011); PC loss progresses with animal aging and becomes much more prominent in older transgenic mice. Deep cerebellar nuclei (DCN) neurons appear vulnerable, even at the early stage of disease, in 3xTg-AD mouse model that contains three mutant genes, APPSwe, PS1M146V and tauP301L, leading to both plaque and tangle pathology (Esquerda-Canals et al., 2013). In these mice, loss of DCN is more pronounced at the level of the fastigial nucleus than the interpositus and dentate nuclei.
Taken together these studies suggest that the involvement of the cerebellum is quite variable depending on the mutation, on the region and cell type analyzed. More experiments are needed to understand the role of specific mutations and the involvement of tau in the pathology of animal models of AD. A deeper level of understanding might be obtained by studies of the alterations in cellular function by electrophysiologicaltechniques.
Alteration of PC Activity and Synaptic Plasticity in Mouse Models of AD
In the cerebellar cortex, elevated levels of soluble Aβ1–42 and Aβ oligomers are associated with the reduction of PC intrinsic excitability. Patch-clamp recordings from PCs of 8-month-old APPswe/PSEN1-L166P mice revealed a larger afterhyperpolarization following the first action potential and longer second and third interspike intervals compared to control PCs, which are indicative of higher frequency adaptation (Table 1; Hoxha et al., 2012). No changes of PC membrane excitability were found in 2-month-old APPswe/PSEN1-L166P mice in which the Aβ levels are scarcely detectable. Since PCs are the sole output of the cerebellar cortex, changes in their firing properties can alter the activity of target neurons in the deep cerebellar and vestibular nuclei (Hoxha et al., 2018).
According to different studies, Aβ1–42 does not affect the PF-PC basal synaptic transmission (paired-pulse facilitation and/or input-output relationship) whether it is autonomously produced in AD mouse models, including APP/PS1, or administered to cerebellar slices of WT mice, at a concentration of 500 nM (Hoxha et al., 2012; Kuwabara et al., 2014; Russo et al., 2018). Conversely, application of 1 μM Aβ1–42 peptides determines a significant decrease in PF-PC synapse activity in cerebellar slices of C57/Bl6 mice (Arbez et al., 2007). Such effect is associated with the decrease of miniature excitatory postsynaptic currents (mEPSCs) amplitude and frequency suggesting an acute effect of Aβ on both presynaptic and postsynaptic compartments. According to the authors, the PF-PC synaptic alterations are caused by the Aβ-induced activation of pro-apoptotic MAP kinases JNK and p38 since their inhibition reversed the negative effect of Aβ on mEPSC amplitude and frequency.
Single-unit extracellular recordings of PCs in vivo in the cerebellar lobules (I–V) of 22-week old APPswe/PSEN1dE9 mice revealed a firing frequency reduction of simple spikes, generated by intrinsic membrane properties and PF inputs to PCs, whereas the firing frequency of complex spikes, driven by climbing fiber (CF) inputs, was not affected (Sepulveda-Falla et al., 2014). The CF-PC basal synaptic transmission is also unaffected in cerebellar slices of 8-month-old APPswe/PSEN1-L166P mice, but the paired-pulse depression at the CF-PC synapse is reduced suggesting an altered release of glutamate from CFs (Hoxha et al., 2012).
Interestingly, the PCs of APPswe/PSEN1-L166P mice also showed a selective loss of large amplitude miniature inhibitory postsynaptic currents mediated by gamma-aminobutyric acid (GABA) receptors pointing to a reduction of the GABAergic inhibitory drive on PCs (Hoxha et al., 2012). The decrease of GABA inputs might be considered a compensatory mechanism for the impairment of intrinsic excitability aimed at re-establishing the physiological rate of PC firing. Alternatively, the reduction in membrane excitability might be a homeostatic compensation for the impairment of GABAergic transmission. Dysfunction in the GABAergic signaling system has been also documented in different brain regions of AD patients, including the cerebellum (Mohanakrishnan et al., 1997; Seidl et al., 2001). In particular, a significant loss of GABA content has been found in post-mortem cerebellum samples from AD patients associated with a significant decrease of glutamic acid decarboxylase (GAD), the synthesizing enzyme of GABA (Burbaeva et al., 2014). The results from animal models, together with the immunohistochemical studies on human AD brains, indicate that a GABAergic dysfunction is present in the AD cerebellum, with possible consequences on the complex excitatory/inhibitory balance, thus contributing to the disease pathophysiology (McCormick, 1989).
Although basal synaptic transmission is intact at the PF-PC synapse, cerebellar cortex synaptic plasticity deficits have been observed in AD mice models in correlation with motor learning impairment (Kuwabara et al., 2014). In most cases, the PF- long-term depression (LTD) is impaired by either exogenous synthetic Aβ1–42 application or by the abnormally high levels of endogenous Aβ present in AD Tg mouse models. Russo et al. (2018) have recently demonstrated that cerebellar synaptic plasticity is affected early in the time course of the Alzheimer’s pathology. Indeed, electrophysiological recordings on cerebellar slices from 2-month-old TgCRND8 mice revealed a significant alteration of LTD at the PF-PC synapse at the pre-plaque stage. Indeed, TgCRND8 mice exhibit no Aβ plaques before 3-months, but in several brain areas including the cerebellum small amounts of Aβ peptides, which are likely to interfere with synaptic plasticity and learning, have been detected (Chishti et al., 2001; Yu et al., 2014).
These data suggest that, in the cerebellum, deficits of neuronal signaling are present in early stages of AD, and they are paralleled by the onset of motor symptoms. However, the results are sparse and have been obtained in different animal models of AD, so that currently it is difficult to draw conclusions. Future studies are necessary to throw light on the cerebellar neuronal dysfunctions that are involved in motor symptoms of patients with AD. In addition, the activity of cerebellar neuronal networks is under continuous control by diffuse-projection neuromodulatory systems, some of which are severely affected in AD. In the cerebellum, the noradrenergic system is one of the most important neuromodulatory systems.
Alteration of Noradrenergic Neurotransmission in the Cerebellar Cortex
Noradrenaline (NA) is a well-known neuromodulator involved in a broad variety of brain processes, including attention, arousal, decision making and memory. The cerebellar cortex receives a widespread noradrenergic projection, from the locus coeruleus (LC), which is consistent with the demonstration that the NA is involved in the modulation of cerebellar function including motor learning (Pompeiano, 1998; Cartford et al., 2004; Schweighofer et al., 2004).
Degeneration of LC and loss of noradrenergic innervation in various brain regions have been revealed in AD in correlation with decreased cognitive functions (Bondareff et al., 1987; Bickford, 1993; Grudzien et al., 2007). Studies of post-mortem tissue have demonstrated a significant reduction of total LC cell number in MCI and AD as well as decrease in norepinephrine transporter (NET), responsible for the reuptake of NA, in several brain areas of AD patients including the cerebellum (Tejani-Butt et al., 1993; Gulyás et al., 2010; Kelly et al., 2017). Interestingly, AD patients with aggressive behavior show higher levels of α2- and β-adrenergic receptors (ARs) in the cerebellar cortex compared to non-agitated AD subjects, whereas no significant changes in ARs levels were found in prefrontal cortex and hypothalamus (Russo-Neustadt and Cotman, 1997). According to the authors, alterations of cerebellar ARs expression may affect the PC activity and as a consequence, the cerebellar output to the cerebral cortical areas encoding affective and defensive/aggressive behavior.
Depletion of NA has also been reported in the cerebellum of TgCRND8 mice starting at 4–5 weeks of age (Francis et al., 2012). The loss of noradrenergic innervation is likely to contribute to the impairment of cerebellar synaptic function and eventually, motor learning observed in these mice (Russo et al., 2018). Under normal conditions, activation of α-ARs by NA or the α2-AR agonist UK 14,304, produces synaptic depression at the PF-PC synapse whereas β2-AR activation by isoproterenol facilitates the PF-PC synaptic transmission (Lippiello et al., 2015; Hoxha et al., 2016). On the contrary, application of α- or β-ARs agonist does not induce any effect on PF-EPSCs in the cerebellar slices of 2-month-old TgCRND8 (Figure 1). Furthermore, unlike controls, 2-month-old TgCRND8 mice exhibit a significant impairment of isoproterenol-induced plasticity at the PF-PC in correlation with significant motor coordination and balance deficits (Russo et al., 2018).
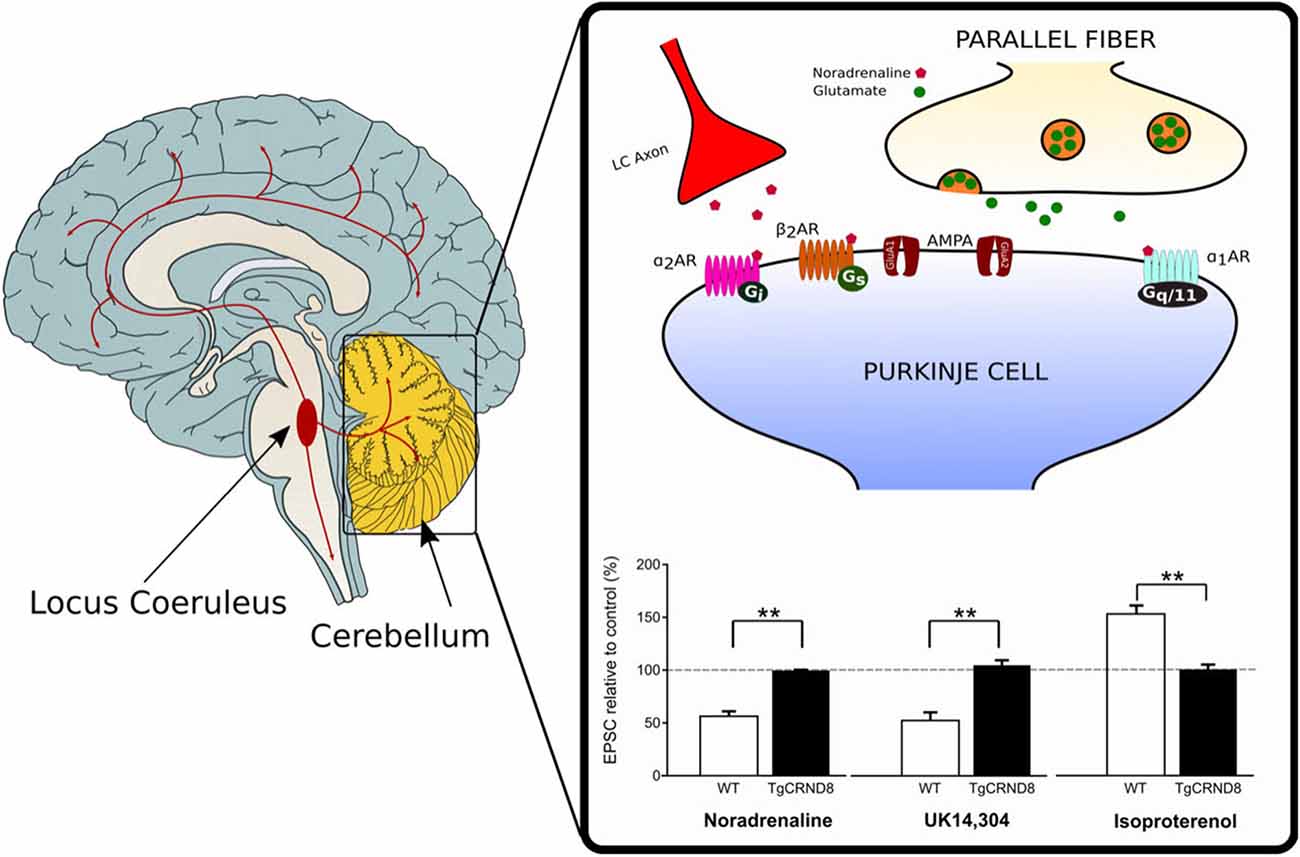
Figure 1. Loss of noradrenergic modulation of the parallel fiber-Purkinje cell (PF-PC) synapse in TgCRND8 mice. In wild-type (WT) mice, the PF-PC excitatory synaptic transmission is depressed by the endogenous agonist noradrenaline (NA) as well as by the α2-adrenergic receptor (AR) agonist UK 14,304, but it is potentiated by the β-AR agonist isoproterenol. The same agonists do not induce any effect on PF-excitatory postsynaptic currents (EPSCs) in the cerebellar slices of 2-month-old TgCRND8. Bar graph shows the mean (±SEM) percentage of change of EPSC recorded in PCs of WT and TgCRND8 (Tg) mice. **p < 0.01 Tg vs. WT (modified with permission from Russo et al., 2018).
Further studies in APP23 transgenic mice have shown that degeneration of LC results in elevated Aβ deposition and increased neuroinflammation in brain areas usually innervated by LC (Heneka et al., 2010). This result is in accordance with the demonstration that NA positively modulates microglia phagocytosis and migration, therefore facilitating Aβ clearance and reducing the inflammatory response to Aβ.
Mechanisms Involved in Aβ-Induced Neurotoxicity
A general state of inflammation of the central nervous system has been widely documented in AD (Hensley, 2010; Maione et al., 2018). Amongst the elements contributing to the development of neuroinflammation in AD, there are certain cell types, such as astroglia and microglia, that manifest high reactivity and morphological alterations (Akiyama et al., 2000). At the cerebellar level, a severe astrocytosis has been found in the granule layer and white matter of FAD patients, in absence of amyloid plaques (Fukutani et al., 1996). Astrocytosis has been also observed in sporadic AD patients, although the number of astrocytes was significantly lower than in FAD patients.
Further studies in rats have demonstrated that cerebellar astrocytosis can be directly induced by intracerebroventricular administration of Aβ peptides. In particular, Aβ injection determines a significant increase in astrocyte number in the granular and molecular layers (Lee et al., 2014). In addition, Aβ can also activate phagocytosis by microglia in rat mixed granular/glial cell cultures causing the loss of cerebellar neurons (Neniskyte et al., 2011). In the presence of the inhibitor of microglial phagocytosis, L-leucine-methyl-ester, the administration of Aβ1–42 (250 nM) does not induce any granule cell loss. However, L-leucine-methyl-ester is not able to prevent granule cell loss at higher concentrations of Aβ1–42 (10 μM). This suggests that the loss of neurons induced by low concentrations of Aβ is directly linked to microglial activity, whereas in the presence of higher concentrations of Aβ1–42 the cellular loss occurs through a microglia-independent mechanism. According to Neniskyte et al. (2011), the administration of Aβ1–42 induces the exposure of phosphatidylserine (PS) on the neuronal surface and activates phagocytosis by microglia. Indeed, in addition to the treatment with phagocytosis inhibitors, antibodies to PS prevented Aβ1–42-induced neuronal loss.
Interestingly, an increased secretion of β-amyloid has been assessed in cerebellar granule cells undergoing apoptosis. Aβ can activate signaling cascades responsible for the neurodegeneration of nearby cells; the granular cellular death rate is reduced by anti-Aβ antibodies (Galli et al., 1998).
The apoptotic effect of Aβ25–35 on cerebellar granule cells involves the activation of different classes of caspase, including caspase-2, -3 and -6 (Allen et al., 1999). These caspases can cleave tau proteins to generate fragments with toxic activity, such as NH2-26–44 tau fragment, which may impair the oxidative phosphorylation leading to the accumulation of reactive oxygen species (ROS; Atlante et al., 2008). The excessive presence of free radicals and ROS in the nucleus determines the oxidation of nucleic acids and, consequently, harmful mutations and cellular death. An increase in 8-OH-adenine, 8-OH-guanosine and fapyadenine in nuclear DNA has been reported in the cerebellar cortex of patients in the last stage of AD (Wang et al., 2005). The oxidative stress is associated with an increase of redox-active iron at the early preclinical stage of AD. The redox metal accumulation affects PCs less than other cerebellar cell types, including glial cells, likely due to the different redox environment within the cerebellum (Smith et al., 2010).
In TgCRND8 mice, accumulation of Aβ is accompanied by an increase of oxidative stress (Yu et al., 2014). The same mice show an increased cerebellar NADPH oxidase activation at pre-plaque stage accompanied by enhanced phospholipid peroxidation compared to WT mice (Russo et al., 2018). It should be pointed out that the oxidative stress level in the cerebellum is lower than in the cortex and this may be correlated with the differences in the antioxidant activity and levels of trace metals including iron.
Conclusions
In this review article, by exploiting the knowledge derived from single and original studies, the involvement and contribution of the cerebellum in AD are outlined for the first time. According to these studies, the cerebellar involvement by AD pathology is more pronounced than previously thought. Aβ cerebellar deposits increase with disease progression in the cerebellar cortex of both patients and mouse models of AD, leading to plaque formation and loss of PCs. Further investigation on Tg mice has recently provided important insight into the effects of Aβ on cerebellar synaptic transmission and plasticity at the early stage of disease, that may lead to motor coordination and learning deficit. Such synaptic and motor learning alterations are in part mediated by deficits in NA signaling. The in vitro approach has also uncovered the role of cerebellar astrocytes and microglia phagocytosis as well as oxidative stress in the Aβ-induced neuronal damage.
Given the role of the cerebellum in motor and cognitive processes, additional studies must be undertaken to understand how neurodegenerative changes that develop progressively in this brain structure impact the progression of AD from the earliest to the final stages of the disease.
Author Contributions
MM and FT designed the outline of the article. EH, PL, FZ, IB, RS, FT and MM wrote the manuscript. IB and PL prepared the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer ET declared a shared affiliation, with no collaboration, with several of the authors, EH, IB, FT, to the handling editor at time of review.
References
Aggarwal, N. T., Wilson, R. S., Beck, T. L., Bienias, J. L., and Bennett, D. A. (2006). Motor dysfunction in mild cognitive impairment and the risk of incident Alzheimer disease. Arch. Neurol. 63, 1763–1769. doi: 10.1001/archneur.63.12.1763
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., et al. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. doi: 10.1016/S0197-4580(00)00124-X
Ala, T. A., and Frey, W. H. II. (1995). Validation of the NINCDS-ADRDA criteria regarding gait in the clinical diagnosis of Alzheimer disease. A clinicopathologic study. Alzheimer Dis. Assoc. Disord. 9, 152–159. doi: 10.1097/00002093-199500930-00006
Albers, M. W., Gilmore, G. C., Kaye, J., Murphy, C., Wingfield, A., Bennett, D. A., et al. (2015). At the interface of sensory and motor dysfunctions and Alzheimer’s disease. Alzheimers Dement. 11, 70–98. doi: 10.1016/j.jalz.2014.04.514
Allen, J. W., Eldadah, B. A., and Faden, A. I. (1999). β-amyloid-induced apoptosis of cerebellar granule cells and cortical neurons: exacerbation by selective inhibition of group I metabotropic glutamate receptors. Neuropharmacology 38, 1243–1252. doi: 10.1016/s0028-3908(99)00044-1
Arbez, N., Gautheron, V., Brugg, B., Mariani, J., and Rovira, C. (2007). β-Amyloid(1–42) induces a reduction in the parallel fiber responses of Purkinje cells: possible involvement of pro-inflammatory processes. Exp. Gerontol. 42, 951–962. doi: 10.1016/j.exger.2007.05.007
Ashe, K. H., and Zahs, K. R. (2010). Probing the biology of Alzheimer’s disease in mice. Neuron 66, 631–645. doi: 10.1016/j.neuron.2010.04.031
Atlante, A., Amadoro, G., Bobba, A., de Bari, L., Corsetti, V., Pappalardo, G., et al. (2008). A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 1777, 1289–1300. doi: 10.1016/j.bbabio.2008.07.004
Bickford, P. (1993). Motor learning deficits in aged rats are correlated with loss of cerebellar noradrenergic function. Brain Res. 620, 133–138. doi: 10.1016/0006-8993(93)90279-v
Bondareff, W., Mountjoy, C. Q., Roth, M., Rossor, M. N., Iversen, L. L., Reynolds, G. P., et al. (1987). Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1, 256–262. doi: 10.1097/00002093-198701040-00005
Braak, H., Braak, E., Bohl, J., and Lang, W. (1989). Alzheimer’s disease: amyloid plaques in the cerebellum. J. Neurol. Sci. 93, 277–287. doi: 10.1016/0022-510X(89)90197-4
Buchman, A. S., and Bennett, D. A. (2011). Loss of motor function in preclinical Alzheimer’s disease. Expert Rev. Neurother. 11, 665–676. doi: 10.1586/ern.11.57
Burbaeva, G. S., Boksha, I. S., Tereshkina, E. B., Savushkina, O. K., Prokhorova, T. A., and Vorobyeva, E. A. (2014). Glutamate and GABA-metabolizing enzymes in post-mortem cerebellum in Alzheimer’s disease: phosphate-activated glutaminase and glutamic acid decarboxylase. Cerebellum 13, 607–615. doi: 10.1007/s12311-014-0573-4
Caine, D., and Hodges, J. R. (2001). Heterogeneity of semantic and visuospatial deficits in early Alzheimer’s disease. Neuropsychology 15, 155–164. doi: 10.1037//0894-4105.15.2.155
Cartford, M. C., Gould, T., and Bickford, P. C. (2004). A central role for norepinephrine in the modulation of cerebellar learning tasks. Behav. Cogn. Neurosci. Rev. 3, 131–138. doi: 10.1177/1534582304270783
Chishti, M. A., Yang, D. S., Janus, C., Phinney, A. L., Horne, P., Pearson, J., et al. (2001). Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570. doi: 10.1074/jbc.M100710200
Dineley, K. T., Xia, X. F., Bui, D., Sweatt, J. D., and Zheng, H. (2002). Accelerated plaque accumulation, associative learning deficits, and up-regulation of α 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J. Biol. Chem. 277, 22768–22780. doi: 10.1074/jbc.m200164200
Drummond, E., and Wisniewski, T. (2017). Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 133, 155–175. doi: 10.1007/s00401-016-1662-x
Esquerda-Canals, G., Marti, J., Rivera-Hernández, G., Giménez-Llort, L., and Villegas, S. (2013). Loss of deep cerebellar nuclei neurons in the 3xTg-AD mice and protection by an anti-amyloid β antibody fragment. MAbs 5, 660–664. doi: 10.4161/mabs.25428
Ewers, M., Morgan, D. G., Gordon, M. N., and Woodruff-Pak, D. S. (2006). Associative and motor learning in 12-month-old transgenic APP + PS1 mice. Neurobiol. Aging 27, 1118–1128. doi: 10.1016/j.neurobiolaging.2005.05.019
Ferreira, S. T., Lourenco, M. V., Oliveira, M. M., and De Felice, F. G. (2015). Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 9:191. doi: 10.3389/fncel.2015.00191
Francis, B. M., Yang, J., Hajderi, E., Brown, M. E., Michalski, B., McLaurin, J., et al. (2012). Reduced tissue levels of noradrenaline are associated with behavioral phenotypes of the TgCRND8 mouse model of Alzheimer’s disease. Neuropsychopharmacology 37, 1934–1944. doi: 10.1038/npp.2012.40
Fukutani, Y., Cairns, N. J., Rossor, M. N., and Lantos, P. L. (1996). Purkinje cell loss and astrocytosis in the cerebellum in familial and sporadic Alzheimer’s disease. Neurosci. Lett. 214, 33–36. doi: 10.1016/0304-3940(96)12875-5
Galli, C., Piccini, A., Ciotti, M. T., Castellani, L., Calissano, P., Zaccheo, D., et al. (1998). Increased amyloidogenic secretion in cerebellar granule cells undergoing apoptosis. Proc. Natl. Acad. Sci. U S A 95, 1247–1252. doi: 10.1073/pnas.95.3.1247
Gouras, G. K., Tampellini, D., Takahashi, R. H., and Capetillo-Zarate, E. (2010). Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 119, 523–541. doi: 10.1007/s00401-010-0679-9
Grudzien, A., Shaw, P., Weintraub, S., Bigio, E., Mash, D. C., and Mesulam, M. M. (2007). Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging 28, 327–335. doi: 10.1016/j.neurobiolaging.2006.02.007
Gulyás, B., Brockschnieder, D., Nag, S., Pavlova, E., Kása, P., Beliczai, Z., et al. (2010). The norepinephrine transporter (NET) radioligand (S,S)-[18F]FMeNER-D2 shows significant decreases in NET density in the human brain in Alzheimer’s disease: a post-mortem autoradiographic study. Neurochem. Int. 56, 789–798. doi: 10.1016/j.neuint.2010.03.001
Guo, C. C., Tan, R., Hodges, J. R., Hu, X., Sami, S., and Hornberger, M. (2016). Network-selective vulnerability of the human cerebellum to Alzheimer’s disease and frontotemporal dementia. Brain 139, 1527–1538. doi: 10.1093/brain/aww003
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Heneka, M. T., O’Banion, M. K., Terwel, D., and Kummer, M. P. (2010). Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. 117, 919–947. doi: 10.1007/s00702-010-0438-z
Hensley, K. (2010). Neuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Alzheimers Dis. 21, 1–14. doi: 10.3233/JAD-2010-1414
Hoxha, E., Balbo, I., Miniaci, M. C., and Tempia, F. (2018). Purkinje cell signaling deficits in animal models of ataxia. Front. Synaptic Neurosci. 10:6. doi: 10.3389/fnsyn.2018.00006
Hoxha, E., Boda, E., Montarolo, F., Parolisi, R., and Tempia, F. (2012). Excitability and synaptic alterations in the cerebellum of APP/PS1 mice. PLoS One 7:e34726. doi: 10.1371/journal.pone.0034726
Hoxha, E., Tempia, F., Lippiello, P., and Miniaci, M. C. (2016). Modulation, plasticity and pathophysiology of the parallel fiber-purkinje cell synapse. Front. Synaptic Neurosci. 8:35. doi: 10.3389/fnsyn.2016.00035
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 274, 99–103. doi: 10.1126/science.274.5284.99
Hu, W., Wu, F., Zhang, Y., Gong, C. X., Iqbal, K., and Liu, F. (2017). Expression of tau pathology-related proteins in different brain regions: a molecular basis of tau pathogenesis. Front. Aging Neurosci. 9:311. doi: 10.3389/fnagi.2017.00311
Hyman, B. T., Van Hoesen, G. W., Damasio, A. R., and Barnes, C. L. (1984). Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science 225, 1168–1170. doi: 10.1126/science.6474172
Jacobs, H. I. L., Hopkins, D. A., Mayrhofer, H. C., Bruner, E., van Leeuwen, F. W., Raaijmakers, W., et al. (2018). The cerebellum in Alzheimer’s disease: evaluating its role in cognitive decline. Brain 141, 37–47. doi: 10.1093/brain/awx194
Jankowsky, J. L., Fadale, D. J., Anderson, J., Xu, G. M., Gonzales, V., Jenkins, N. A., et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 13, 159–170. doi: 10.1093/hmg/ddh019
Kelly, S. C., He, B., Perez, S. E., Ginsberg, S. D., Mufson, E. J., and Counts, S. E. (2017). Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 5:8. doi: 10.1186/s40478-017-0411-2
Kopeikina, K. J., Hyman, B. T., and Spires-Jones, T. L. (2012). Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci. 3, 223–233. doi: 10.2478/s13380-012-0032-y
Kozuki, M., Kurata, T., Miyazaki, K., Morimoto, N., Ohta, Y., Ikeda, Y., et al. (2011). Atorvastatin and pitavastatin protect cerebellar Purkinje cells in AD model mice and preserve the cytokines MCP-1 and TNF-α. Brain Res. 1388, 32–38. doi: 10.1016/j.brainres.2011.03.024
Kuwabara, Y., Ishizeki, M., Watamura, N., Toba, J., Yoshii, A., Inoue, T., et al. (2014). Impairments of long-term depression induction and motor coordination precede Aβ accumulation in the cerebellum of APPswe/PS1dE9 double transgenic mice. J. Neurochem. 130, 432–443. doi: 10.1111/jnc.12728
Lambon Ralph, M. A., Patterson, K., Graham, N., Dawson, K., and Hodges, J. R. (2003). Homogeneity and heterogeneity in mild cognitive impairment and Alzheimer’s disease: a cross-sectional and longitudinal study of 55 cases. Brain 126, 2350–2362. doi: 10.1093/brain/awg236
Larner, A. J. (1997). The cerebellum in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 8, 203–209. doi: 10.1159/000106632
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Guerrero-Munoz, M. J., Kiritoshi, T., Neugebauer, V., et al. (2012). Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2:700. doi: 10.1038/srep00700
Lee, J.-M., Shin, M.-S., Ji, E.-S., Kim, T.-W., Cho, H.-S., Kim, C.-J., et al. (2014). Treadmill exercise improves motor coordination through ameliorating Purkinje cell loss in amyloid β23–35-induced Alzheimer’s disease rats. J. Exerc. Rehabil. 10, 258–264. doi: 10.12965/jer.140163
Lippiello, P., Hoxha, E., Volpicelli, F., Lo Duca, G., Tempia, F., and Miniaci, M. C. (2015). Noradrenergic modulation of the parallel fiber-Purkinje cell synapse in mouse cerebellum. Neuropharmacology 89, 33–42. doi: 10.1016/j.neuropharm.2014.08.016
Lomoio, S., López-González, I., Aso, E., Carmona, M., Torrejón-Escribano, B., Scherini, E., et al. (2012). Cerebellar amyloid-β plaques: disturbed cortical circuitry in aβPP/PS1 transgenic mice as a model of familial Alzheimer’s disease. J. Alzheimers Dis. 31, 285–300. doi: 10.3233/JAD-2012-112198
Maione, F., Piccolo, M., De Vita, S., Chini, M. G., Cristiano, C., De Caro, C., et al. (2018). Down regulation of pro-inflammatory pathways by tanshinone IIA and cryptotanshinone in a non-genetic mouse model of Alzheimer’s disease. Pharmacol. Res. 129, 482–490. doi: 10.1016/j.phrs.2017.11.018
Manassero, G., Guglielmotto, M., Zamfir, R., Borghi, R., Colombo, L., Salmona, M., et al. (2016). β-amyloid 1–42 monomers, but not oligomers, produce PHF-like conformation of tau protein. Aging Cell 15, 914–923. doi: 10.1111/acel.12500
McCormick, D. A. (1989). GABA as an inhibitory neurotransmitter in human cerebral cortex. J. Neurophysiol. 62, 1018–1027. doi: 10.1152/jn.1989.62.5.1018
Mevel, K., Chételat, G., Eustache, F., and Desgranges, B. (2011). The default mode network in healthy aging and Alzheimer’s disease. Int. J. Alzheimers Dis. 2011:535816. doi: 10.4061/2011/535816
Miniaci, M. C., and De Leonibus, E. (2018). Missing the egocentric spatial reference: a blank on the map. F1000Res. 7:168. doi: 10.12688/f1000research.13675.1
Mohanakrishnan, P., Fowler, A. H., Vonsattel, J. P., Jolles, P. R., Husain, M. M., Liem, P., et al. (1997). Regional metabolic alterations in Alzheimer’s disease: an in vitro 1H NMR study of the hippocampus and cerebellum. J. Gerontol. A Biol. Sci. Med. Sci. 52A, B111–B117. doi: 10.1093/gerona/52A.2.B111
Neniskyte, U., Neher, J. J., and Brown, G. C. (2011). Neuronal death induced by nanomolar amyloid β is mediated by primary phagocytosis of neurons by microglia. J. Biol. Chem. 286, 39904–39913. doi: 10.1074/jbc.M111.267583
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
O’Leary, T. P., Robertson, A., Chipman, P. H., Rafuse, V. F., and Brown, R. E. (2018). Motor function deficits in the 12 month-old female 5xFAD mouse model of Alzheimer’s disease. Behav. Brain Res. 337, 256–263. doi: 10.1016/j.bbr.2017.09.009
Pettersson, A. F., Engardt, M., and Wahlund, L. O. (2002). Activity level and balance in subjects with mild Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 13, 213–216. doi: 10.1159/000057699
Pompeiano, O. (1998). Noradrenergic influences on the cerebellar cortex: effects on vestibular reflexes under basic and adaptive conditions. Otolaryngol. Head Neck Surg. 119, 93–105. doi: 10.1016/s0194-5998(98)70178-0
Puzzo, D., Gulisano, W., Palmeri, A., and Arancio, O. (2015). Rodent models for Alzheimer’s disease drug discovery. Expert Opin. Drug Discov. 10, 703–711. doi: 10.1517/17460441.2015.1041913
Radde, R., Bolmont, T., Kaeser, S. A., Coomaraswamy, J., Lindau, D., Stoltze, L., et al. (2006). Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946. doi: 10.1038/sj.embor.7400784
Reiserer, R. S., Harrison, F. E., Syverud, D. C., and McDonald, M. P. (2007). Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer’s disease. Genes Brain Behav. 1, 54–65. doi: 10.1111/j.1601-183x.2006.00221.x
Russo, R., Cattaneo, F., Lippiello, P., Cristiano, C., Zurlo, F., Castaldo, M., et al. (2018). Motor coordination and synaptic plasticity deficits are associated with increased cerebellar activity of NADPH oxidase, CAMKII, and PKC at preplaque stage in the TgCRND8 mouse model of Alzheimer’s disease. Neurobiol. Aging 68, 123–133. doi: 10.1016/j.neurobiolaging.2018.02.025
Russo-Neustadt, A., and Cotman, C. W. (1997). Adrenergic receptors in Alzheimer’s disease brain: selective increases in the cerebella of aggressive patients. J. Neurosci. 17, 5573–5580. doi: 10.1523/JNEUROSCI.17-14-05573.1997
Sasaguri, H., Nilsson, P., Hashimoto, S., Nagata, K., Saito, T., De Strooper, B., et al. (2017). APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 36, 2473–2487. doi: 10.15252/embj.201797397
Scheff, S. W., Price, D. A., Schmitt, F. A., DeKosky, S. T., and Mufson, E. J. (2007). Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68, 1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f
Schmahmann, J. D. (2016). Cerebellum in Alzheimer’s disease and frontotemporal dementia: not a silent bystander. Brain 139, 1314–1318. doi: 10.1093/brain/aww064
Schweighofer, N., Doya, K., and Kuroda, S. (2004). Cerebellar aminergic neuromodulation: towards a functional understanding. Brain Res. Rev. 44, 103–116. doi: 10.1016/j.brainresrev.2003.10.004
Seidl, R., Cairns, N., Singewald, N., Kaehler, S. T., and Lubec, G. (2001). Differences between GABA levels in Alzheimer’s disease and down syndrome with Alzheimer-like neuropathology. Naunyn Schmiedebergs. Arch. Pharmacol. 363, 139–145. doi: 10.1007/s002100000346
Selkoe, D. J. (1998). The cell biology of β-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 8, 447–453. doi: 10.1016/s0962-8924(98)01363-4
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Selkoe, D. J. (2008). Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113. doi: 10.1016/j.bbr.2008.02.016
Sepulveda-Falla, D., Barrera-Ocampo, A., Hagel, C., Korwitz, A., Vinueza-Veloz, M. F., Zhou, K., et al. (2014). Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Invest. 124, 1552–1567. doi: 10.1172/JCI66407
Sepulveda-Falla, D., Matschke, J., Bernreuther, C., Hagel, C., Puig, B., Villegas, A., et al. (2011). Deposition of hyperphosphorylated tau in cerebellum of PS1 E280A Alzheimer’s disease. Brain Pathol. 21, 452–463. doi: 10.1111/j.1750-3639.2010.00469.x
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shankar, G. M., and Walsh, D. M. (2009). Alzheimer’s disease: synaptic dysfunction and Aβ. Mol. Neurodegener. 4:48. doi: 10.1186/1750-1326-4-48
Smith, M. A., Zhu, X., Tabaton, M., Liu, G., McKeel, D. W., Cohen, M. L., et al. (2010). Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 19, 353–372. doi: 10.3233/jad-2010-1239
Sturchler-Pierrat, C., Abramowski, D., Duke, M., Wiederhold, K. H., Mistl, C., Rothacher, S., et al. (1997). Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U S A 94, 13287–13292. doi: 10.1073/pnas.94.24.13287
Tejani-Butt, S. M., Yang, J., and Zaffar, H. (1993). Norepinephrine transporter sites are decreased in the locus coeruleus in Alzheimer’s disease. Brain Res. 631, 147–150. doi: 10.1016/0006-8993(93)91201-3
Thomann, P. A., Schläfer, C., Seidl, U., Santos, V. D., Essig, M., and Schröder, J. (2008). The cerebellum in mild cognitive impairment and Alzheimer’s disease—a structural MRI study. J. Psychiatr. Res. 42, 1198–1202. doi: 10.1016/j.jpsychires.2007.12.002
Van Dam, D., D’Hooge, R., Staufenbiel, M., Van Ginneken, C., Van Meir, F., and De Deyn, P. P. (2003). Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 17, 388–396. doi: 10.1046/j.1460-9568.2003.02444.x
Wang, J., Xiong, S., Xie, C., Markesbery, W. R., and Lovell, M. A. (2005). Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 93, 953–962. doi: 10.1111/j.1471-4159.2005.03053.x
West, M. J., Coleman, P. D., Flood, D. G., and Troncoso, J. C. (1994). Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 344, 769–772. doi: 10.1016/s0140-6736(94)92338-8
Yu, W., Bonnet, M., Farso, M., Ma, K., Chabot, J. G., Martin, E., et al. (2014). The expression of apoptosis inducing factor (AIF) is associated with aging-related cell death in the cortex but not in the hippocampus in the TgCRND8 mouse model of Alzheimer’s disease. BMC Neurosci. 15:73. doi: 10.1186/1471-2202-15-73
Keywords: cerebellum, Alzheimer’s disease, β-amyloid, purkinje cell, synaptic plasticity, noradrenaline
Citation: Hoxha E, Lippiello P, Zurlo F, Balbo I, Santamaria R, Tempia F and Miniaci MC (2018) The Emerging Role of Altered Cerebellar Synaptic Processing in Alzheimer’s Disease. Front. Aging Neurosci. 10:396. doi: 10.3389/fnagi.2018.00396
Received: 07 August 2018; Accepted: 15 November 2018;
Published: 27 November 2018.
Edited by:
Jean Mariani, Université Pierre et Marie Curie, FranceReviewed by:
Julien Rossignol, Central Michigan University, United StatesElena Tamagno, Università degli Studi di Torino, Italy
Rachel M. Sherrard, Sorbonne Universités, France
Copyright © 2018 Hoxha, Lippiello, Zurlo, Balbo, Santamaria, Tempia and Miniaci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filippo Tempia, ZmlsaXBwby50ZW1waWFAdW5pdG8uaXQ=