
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 09 October 2018
Sec. Alzheimer's Disease and Related Dementias
Volume 10 - 2018 | https://doi.org/10.3389/fnagi.2018.00312
This article is part of the Research TopicInterphase Between Aging and Neurodegenerative DiseasesView all 10 articles
 Aarti Mishra1,2
Aarti Mishra1,2 Roberta D. Brinton2,3,4*
Roberta D. Brinton2,3,4*Neuro-inflammatory processes that contribute to development of Alzheimer’s are evident early in the latent prodromal phase and worsen during the course of the disease. Despite substantial mechanistic and clinical evidence of inflammation, therapeutic approaches targeting inflammation have failed to alter the course of the disease. Disparate results from epidemiological and clinical trials targeting inflammation, highlight the complexity of the inflammatory process. Herein we review the dynamics of the inflammatory process across aging, midlife endocrine transitions, and the APOEε4 genotype and their contribution to progression of Alzheimer’s disease (AD). We discuss the chronic inflammatory processes that are activated during midlife chronological and endocrine aging, which ultimately limit the clearance capacity of microglia and lead to immune senescence. Aging, menopause, and APOEε4 combine the three hits of a compromised bioenergetic system of menopause with the chronic low grade innate inflammation of aging with the APOEε4 dyslipidemia and adaptive immune response. The inflammatory immune response is the unifying factor that bridges across each of the risk factors for AD. Immune system regulators that are specific to stage of disease and inflammatory phenotype would provide a therapeutic strategy to disconnect the bridge that drives disease. Outcomes of this analysis provide plausible mechanisms underlying failed clinical trials of anti-inflammatory agents in Alzheimer’s patients. Further, they highlight the need for stratifying AD clinical trial cohorts based on inflammatory phenotype. Combination therapies that include targeted use of anti-inflammatory agent’s specific to the immune phenotype are considered.
Alzheimer’s disease (AD) is characterized by an extended prodromal phase of typically 10–20 years duration prior to clinical manifestation of cognitive decline (Amieva et al., 2008). The prodromal phase of AD consists of both- pre-stage symptoms and mild cognitive impairment (MCI) (Wilson et al., 2011). Clinical studies have shown that the prodromal phase is characterized by metabolic dysfunction, amyloid-β (Aβ) deposition in the brain, mild to moderate cognitive dysfunction, and chronic low-grade inflammation (Habeck et al., 2012; Olsson et al., 2013; Brinkmalm et al., 2014; Wirz et al., 2014; Rajan et al., 2015; Mosconi et al., 2017a,b). These hallmark pathologies have aided in the development of biomarkers predictive of disease pathogenesis. In some populations, Aβ42 is one of the first biomarkers to appear in the cerebrospinal fluid (CSF) (Jack et al., 2013; Brinkmalm et al., 2014; Calsolaro and Edison, 2016). Despite the recent and widespread recognition of neuroinflammation in the pathogenesis of AD, there has been sparse expansion of inflammation-based biomarkers and preventive strategies. Understanding the dynamic interplay between inflammation and the risk factors such as age, APOE genotype, and endocrine transition states, can aid in this process. This review addresses the interaction between inflammation and risk factors key to the pathogenesis of AD. Strategies to specifically target these processes are also considered.
Substantial evidence documents reactive microgliosis around plaque deposition and is now a hallmark of AD pathology (McGeer et al., 1988; Mattiace et al., 1990; Xiang et al., 2006). Reactive microgliosis and neuroinflammation in AD patients is considered a consequence of Aβ plaque deposition (McGeer et al., 1987). Microgliosis in AD is evidenced both microscopically and biochemically with increased levels of the proinflammatory cytokines including tumor necrosis factor-α (TNFα), IL-6, and IL-1β (Itagaki et al., 1989; Dickson et al., 1993; Ferretti and Cuello, 2011; Eikelenboom et al., 2012; Latta et al., 2014). While the inflammatory response to Aβ plaque deposition is irrefutable, it is a late stage response in the inflammatory cascade. Indicators of earlier inflammatory responses are apparent in multiple conditions that are risk factors for later development of AD.
Associations between the occurrence of systemic infections and chronic inflammatory conditions with Alzheimer’s disease, suggests an active participation of inflammation in early stages of disease development. Patients with higher erythrocyte sedimentation rate (ESR), which is a clinical indicator of non-specific inflammation, are at greater risk of developing AD (Li et al., 2012). This is further corroborated by epidemiological studies that show that patients who suffer from chronic periodontal infection (Van Den Heuvel et al., 2007) and HIV have a higher risk of developing AD (Stanley et al., 1994; Alisky, 2007; Xu and Ikezu, 2009; Chakradhar, 2018). Recapitulating the clinical effect, Krstic et al. (2012) established an animal model that displayed AD like neuropathology by inducing chronic inflammation prenatally using a viral antigen, thereby showing that chronic inflammation potentiates the development of AD. Traumatic brain injury (TBI) also increases the risk of developing AD. Lesions that develop during TBI lead to an acute inflammatory response that includes microglial activation to facilitate debris removal and neuroprotection (Van Den Heuvel et al., 2007; Breunig et al., 2013; Habib et al., 2014). Incomplete resolution of the acute inflammatory response in TBI, however, is often followed by hypoxia and oxidative stress, which leads to the chronic activation of microglia and the release of neurotoxic proinflammatory cytokines (Van Den Heuvel et al., 2007; Breunig et al., 2013; Habib et al., 2014).
Chronic inflammatory conditions such as autoimmune disorders alter the risk of development of dementia. A recent study found patients admitted to the hospital for an autoimmune disorder have greater risk for subsequent hospitalization due to dementia (Wotton and Goldacre, 2017). This association was particularly significant for multiple sclerosis and systemic lupus erythematosus for AD. While patients with rheumatoid arthritis (RA) had a reduced risk of developing Alzheimer’s disease, they had an increased risk of vascular dementia (Wotton and Goldacre, 2017). Multiple studies indicate that AD incidence is lower in persons with RA (Policicchio et al., 2017). Some attribute this reduction in incidence to the regular use of non-steroidal anti-inflammatory drugs (NSAIDs) (McGeer et al., 1996; Etminan et al., 2003). An alternative mechanism involves upregulation of granulocyte macrophage-colony stimulating factor (GM-CSF) with a probable gain of function in myeloid cells, thus enabling effective debris clearance is also hypothesized to reduce the incidence of AD in RA patients (McGeer et al., 1996; Boyd et al., 2010). In mice, increased levels of GM-CSF (both intrahippocampal and subcutaneous administration) significantly reduced amyloidosis and reversed cognitive impairment (McGeer et al., 1996; Boyd et al., 2010). More recent findings indicate that RA and risk of AD can be stratified based on treatment. Case-controlled study conducted on electronic medical records from 8.5 million commercially insured adults, indicate that RA patients treated with an anti-TNFα therapy, etanercept, had a lower risk of AD whereas those on other anti-inflammatory agents had increased risk of AD (Chou et al., 2016).
Disparate results from epidemiological studies and randomized clinical trials highlight the complexity of response to anti-inflammatory agents (Thal et al., 2005). Epidemiological analyses indicated that long-term NSAIDs users have a lower risk of developing AD (McGeer et al., 1996; Vlad et al., 2008). Based on epidemiological findings, a clinical study – ADAPT (Alzheimer’s Disease Anti-Inflammatory Prevention Trial) was conducted in cognitively intact elderly individuals with a family history of AD. In this trial, the selective cyclooxygenase-2 (COX-2) inhibitor Celecoxib and non-selective COX inhibitor Naproxen, were used as preventive therapies. The trial was discontinued 15 months after randomization, due to the increased cardiovascular risk of these therapies. On extended follow-up after 7 years, treatment with celecoxib or naproxen for 1–3 years did not prevent cognitive decline (Group, 2007, 2008; Breitner et al., 2011; Alzheimer’s Disease Anti-inflammatory Prevention Trial Research Group, 2013). Collectively, these findings indicate that timing of NSAID treatment for chronic inflammatory conditions are a critical factor impacting the efficacy of NSAID therapy to prevent or delay progression of Alzheimer’s disease (McGeer et al., 1996; Vlad et al., 2008).
Discrepancies between the epidemiological and clinical trial findings indicate the need for greater refinement in considering patient populations and anti-inflammatory therapies. Elucidating the inflammatory phenotype which emerges during the progression of AD requires consideration of the triggers that initiate chronic inflammation. In the sections below, we address how age, endocrine status, and APOE genotype impact inflammatory processes across AD progression from risk to late stage disease.
Aging has a broad systems-level effect on human biology, which is evidenced by alterations in physiological function, metabolism, cognition, and inflammation (Smith et al., 2005). The effects of aging on immune responses are extensive and complex. In some individuals, adaptive immune responses will decline with age, whereas, others will experience aberrant immune responses leading to autoimmune disorders (Goronzy and Weyand, 2012; Vadasz et al., 2013; Fougère et al., 2017). Aging is associated with accumulation of oxidative stress and DNA damage and chronic low-grade inflammation (Cui et al., 2012). Though the effect of aging on cognitive function is variable, age remains the greatest risk factor for Alzheimer’s in which inflammation is an early and persistent hallmark of the disease (von Bernhardi et al., 2015).
Central nervous system microglia plays a prominent role in innate immunity. Microglia constantly conducts surveillance of brain parenchyma to detect foreign pathogens and clear debris (Streit et al., 2004; von Bernhardi et al., 2015). Microglia detects and responds to a broad range of triggers including TBI, infections and damage associated molecular patterns (DAMPs). Reactive oxygen species (ROS), extracellular DNA and ATP all act as DAMPs (von Bernhardi et al., 2015; Gulke et al., 2018).
Innate immune responses by microglia are phenotypically typified by enlargement of the cell body, and molecularly by the upregulation of CD68, major histocompatibility complex-II (MHC-II) along with costimulatory molecules and secretion of pro and anti-inflammatory cytokines (Kim and Joh, 2006). The onset of innate immune responses leads to activation of the adaptive immune response. The innate activation of the adaptive immune response results in infiltration of peripheral immune cells, particularly T cell invasion of the brain (Kim and Joh, 2006). Together the innate and adaptive immune responses create the chronic low-grade inflammation typical of aging (Kim and Joh, 2006; see Figures 1, 2).
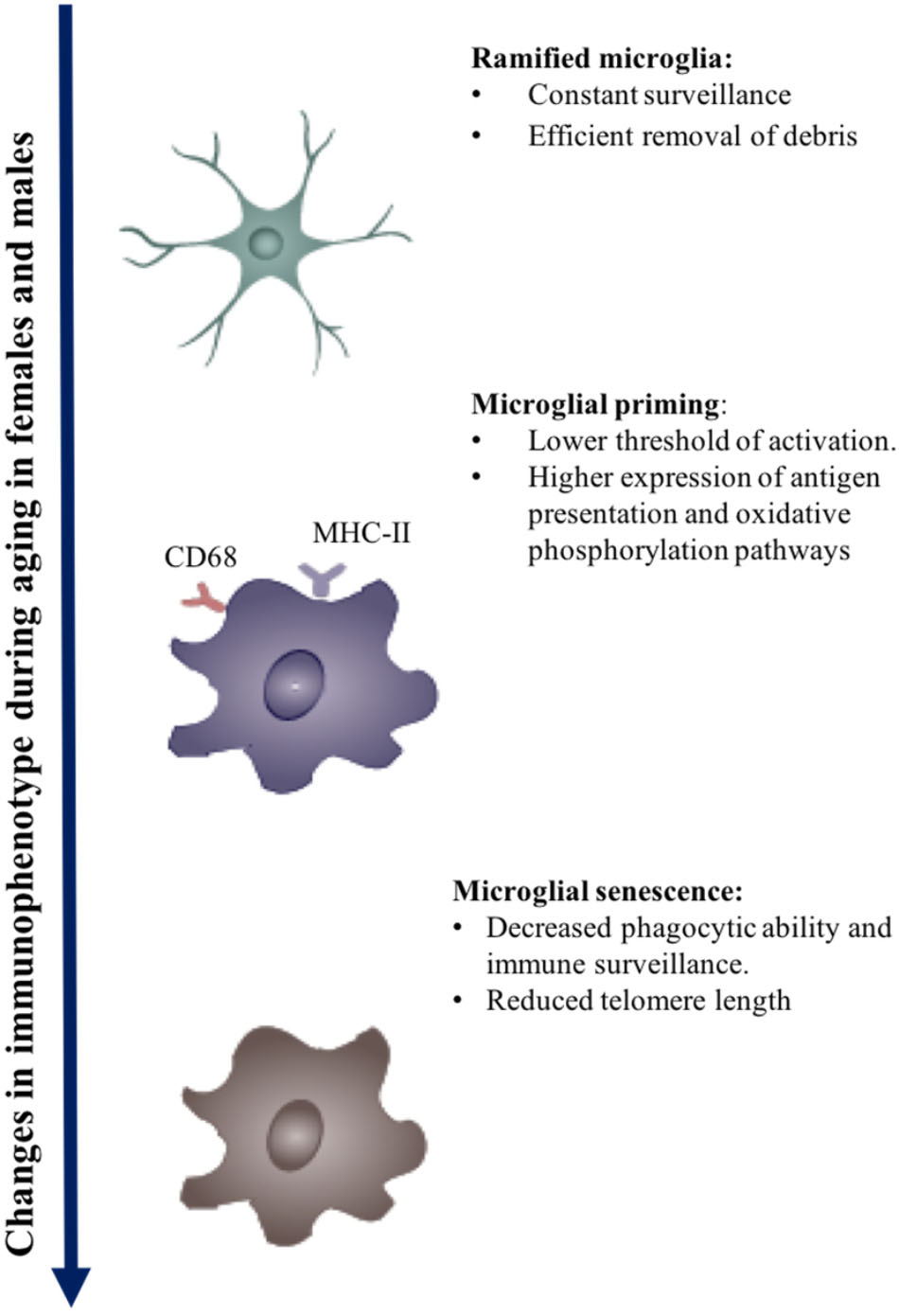
FIGURE 1. Effect of aging on microglial immunophenotype. Changes in the immunophenotype of aging. Early to midlife aging can induced microglial priming, which is evident in the upregulation of MHC-II molecules, Fcg receptors, and microglial receptors. Chronic activation may eventually lead to immune senescence.
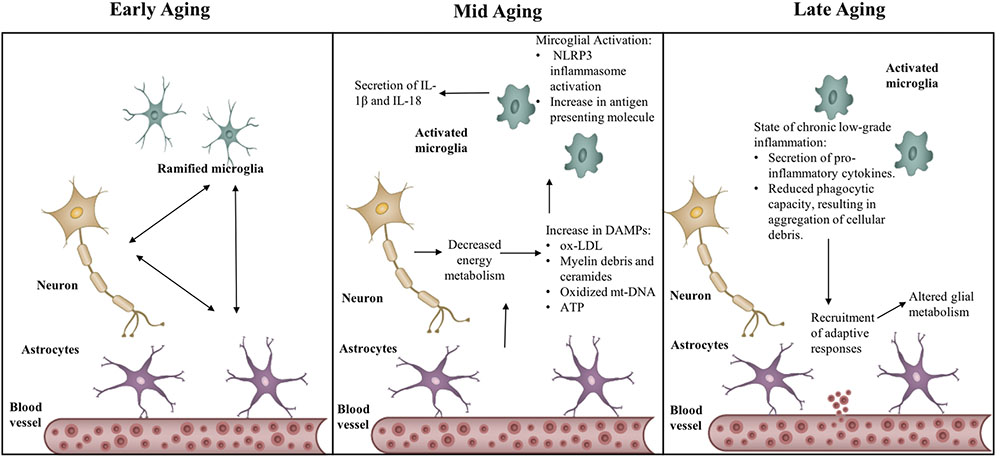
FIGURE 2. Transitions in the inflammatory phenotype across aging. Aging causes activation of neuro-inflammatory mechanisms – NLRP3 inflammasome and increased MHC-II expression, through production of sterile triggers, such as, oxidized low-density lipoprotein (Ox-LDL), myelin debris, oxidized mitochondrial DNA (mt-DNA), ATP. State of chronic low-grade inflammation leads to recruitment of adaptive responses, in turn affecting glial metabolism.
Microglial phenotype is dynamic. It changes with age and is typified by both homeostatic and disease phenotypes (Butovsky et al., 2014; Keren-Shaul et al., 2017; Krasemann et al., 2017). The telomerase deficient accelerated aging mouse model exhibits reduced microglial numbers and deficient morphological and cellular processes (Khan et al., 2015). Further, microglial response to activation is stage of development dependent. Production of cytokines and trophic factors by microglia increases linearly with age (Lai et al., 2013). Microglia isolated from younger mice (2–4 months) exhibit a lower expression of proinflammatory cytokines: TNFα, IL-6, and IL-1β than older mice (Helenius et al., 1996; Crain et al., 2013; Latta et al., 2015). On activation by ATP, microglia isolated from neonatal rats and 13–15 month old adult rats have a more robust inflammatory response exhibited by upregulation of nitric oxide, TNFα, and brain derived neurotrophic factor (BDNF) in comparison to microglia derived from younger animals (2–11 months) (Lai et al., 2013). Aging distinctly affects migratory function of microglia; younger microglia on encountering activating signals exhibit an increase in motility and rapid extension of ramifications, whereas older microglia are less dynamic. Transcriptomic studies corroborate a reduction in migratory ability of microglia with age as age affects actin cytoskeleton reogranization which is vital in both phagocytosis and migration (Damani et al., 2011; Orre et al., 2014).
Comparison of the transcriptomic profiles of young and aged microglia revealed that microglial receptors (Trem2c, P2yr12, P2yr13, and Adora) involved in recognizing DAMPs such as, oxidized low-density lipoprotein, mitochondrial DNA, extracellular ATP decreased with age (Orre et al., 2014). In contrast, the expression of receptors that recognize pathogens and microbes (Tlr2, CD74, Ltf, Clec7a, Cxcl16, and Ifitm6) increases with age (Orre et al., 2014). Age-related changes in microglial transcriptome are not ubiquitous as the expression of phagocytic receptors (Cd14, Cd68, Cd11b, and ICAM) remained unaltered in aging (Hickman et al., 2013; Smith and Dragunow, 2014). However, activation of microglial phagocytosis is diminished in aging. Studies characterizing microglial phagocytic capacity, report a reduction in phagocytosis with age which is especially evident following activation (Li M.D. et al., 2015; Ritzel et al., 2015). These findings indicate that despite stable expression of phagocytic receptors, the functional capacity of microglia decreases with age (Li M.D. et al., 2015; Ritzel et al., 2015). For example, the ability of microglia to phagocytose Aβ is affected by age, with microglia isolated from postnatal animals effectively phagocytosing Aβ fibrils, whereas adult microglia lose their capacity to do so (Floden and Combs, 2011). Other contributing factors to microglial senescence are age-related myelin degeneration and lysosomal storage in microglia, which in turn burden microglial clearance function (Holtman et al., 2015; Safaiyan et al., 2016).
Systemic inflammation and aging cause microglial priming. Primed microglia have a lower threshold for activation, are hypersensitive, develop an exaggerated immune response on activation, and have a distinct molecular signature from the M1–M2 phenotype (Perry and Teeling, 2013; Holtman et al., 2015; Ojo et al., 2015). The molecular signatures of primed microglia include the overexpression of antigen presentation, redox pathways, oxidative phosphorylation, and lysosomes.
In summary, during aging primed microglia generate a pro-inflammatory cascade due to their lower threshold of activation, enhanced reactivity, and limited functional capacity on encountering secondary triggers. The chronic activation of microglia coupled with age-related microglial priming hastens the process of senescence to cause loss of function over time (Franceschi et al., 2000; Streit and Xue, 2014). Senescent microglia appear to have lesser ramifications and stouter cell bodies, often referred to as dystrophic microglia.
The inflammasome complex is a sensor of DAMPs. DAMPs act as an inflammatory challenge to the host defense mechanism and lead to the activation of the NLRP3 (Nod-like receptor pyrin domain 3) inflammasome complex (Youm et al., 2013; Zhang et al., 2013). Within the family of innate inflammasome sensors, the NLRP3 inflammasome has the unique ability to detect sterile inflammatory triggers. It can detect a wide range of metabolic and aging-related DAMPS, such as ROS production, glucose tolerance, and insulin resistance (Vandanmagsar et al., 2011; Salminen et al., 2012; Walsh et al., 2014), lipotoxic fatty acids, ceramides, free cholesterol, uric acid, and ATP, and it releases IL-1β and IL-18 (Youm et al., 2012, 2013; Zhang et al., 2013; see Figure 2).
The NLRP3 inflammasome complex activation is a two-step process. Molecular pathogens like lipopolysaccharides (LPS) have been shown to prime cells, leading to the activation of pattern recognition receptors (PRRs), the release of IL-1β, and increased expression of NLRP3. When followed by a secondary trigger such as ATP, this process causes the inflammasome to assemble and causes further activation. NLRP3 is also activated by the accumulation of damaged mitochondria due to the inhibition of autophagy, resulting in excessive production of ROS. oxidized mitochondrial DNA is also implicated in the activation of NLRP3 (Dixit, 2013). The activation of NLRP3 by ROS is mediated by thioredoxin interacting protein (TXNIIP) (Youm et al., 2012, 2013; Zhang et al., 2013).
The activation patterns of NLRP3 are similar in both macrophages and microglia. NLRP3 activation leads to the priming of microglia and reducing the threshold for activation (Halle et al., 2008; Heneka et al., 2013). Increase in caspase-1 activity in postmortem MCI and AD brains indicates the possible participation of the NLRP3 inflammasome in AD pathogenesis (Heneka et al., 2013). Targeting NLRP3 and NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) has been associated with a reduction in pathology of AD (Tang et al., 2015). In animal models carrying AD pathology, NLRP3 knockout and caspase-1 knockout caused spatial memory to remain intact. Moreover, the microglial phenotype in the NLRP3 knockout model shifted to the M2 anti-inflammatory phenotype with greater neuroprotection improved clearance of the plaque burden (Halle et al., 2008; Heneka et al., 2013).
With several studies linking NLRP3 inflammasome activation to chronic low-grade inflammation observed in aging, therapeutics targeting this sensor has also emerged. In a recent study, the ketone body – β-hydroxybutyrate (BHB) was found to suppress NLRP3 activation caused by urate crystals, lipotoxic fatty acids, and ATP. The levels of BHB increase with starvation, caloric restriction, and high-intensity exercise. Aging also marks a shift in the fuel usage and dependence on different fuel mechanisms. The inhibition of NLRP3 by BHB resulted in a decrease of IL-1β and IL-18 production by monocytes. BHB also reduced caspase-1 activation and IL-1β secretion in mouse models of NLRP3-mediated chronic inflammatory diseases like Muckle-Wells syndrome (Youm et al., 2015).
Another molecule, MCC950, blocked canonical and non-canonical activation of NLRP3 and attenuated experimental autoimmune encephalomyelitis (EAE). Both, MCC950 and BHB, were used in a mouse model of Muckle-Wells syndrome, which is characterized by chronic inflammation mediated by NLRP3. Thus, targeting NLRP3 in aging and aging-related disorders could be an important therapeutic strategy (Coll et al., 2015).
The endocrine transition of the perimenopause to the post-menopause, while associated with loss of reproductive function (Brinton et al., 2015), is also associated with rise in chronic low-grade inflammation (Yin et al., 2015). Chronic systemic inflammation accelerates ovarian failure (Ağaçayak et al., 2016). Conversely, depleting proinflammatory cytokines IL-1α and IL-1β extends ovarian function and lifespan (Uri-Belapolsky et al., 2014). Concurrent with the chronic low-grade inflammation, the perimenopausal transition is typified by decline in brain glucose metabolism and mitochondrial respiration (Yao et al., 2010; Ding et al., 2013; Brinton et al., 2015; Yin et al., 2015; Mosconi et al., 2017a,b), myelin catabolism (Klosinski et al., 2015) and loss of white matter volume (Mosconi et al., 2017a,b), beta-amyloid deposition in brain (Mosconi et al., 2017a,b) and changes in neurological function (Brinton et al., 2015). Later age at natural and surgical menopause is associated with better verbal memory (Kuh et al., 2018). Surgically induced menopause prior to natural menopause is associated with rapid cognitive decline and earlier onset of AD (Rocca et al., 2008; Bove et al., 2014). Further, studies have shown that post-menopausal women with higher estradiol levels have a reduced risk of developing AD (Manly et al., 2000).
Post-menopausal women are at higher risk for developing autoimmune disorders and obesity (Doran et al., 2002; Bove, 2013). The incidence of RA is higher in peri- and post-menopausal women (Doran et al., 2002). The pathology of multiple sclerosis worsens after menopause (Tutuncu et al., 2013). Post-menopausal women are more prone to robust immune responses. The lack of ovarian steroidal hormones potentiates the inflammatory process predisposing menopausal women to immune disorders (Benedusi et al., 2012; Kireev et al., 2014; Sharma et al., 2018). Menopause and the associated lack of steroidal hormones further potentiate inflammation, which is reflected in levels of circulating cytokine levels and inflammatory responses. IL-6 and sIL-6 levels are higher in postmenopausal women (Giuliani et al., 2001). IL-4 and IL-2 levels also increase with menopause, which can be reversed by hormone therapy (Yasui et al., 2007). Serum IFN-γ levels increase during early menopause but decrease in later menopause (Goetzl et al., 2010). Peripheral blood mononuclear cells (PBMCs) isolated from postmenopausal women produced higher IL-6, IL-1β, and TNFα upon induction by LPS than PBMCs isolated from premenopausal women (Brooks-Asplund et al., 2002).
In addition to an altered cytokine profile, changes in T cell biology occur in women during this endocrine transition. Pre-menopausal women have higher CD4 counts than men and thus a more robust response to vaccination. Menopause causes a reduction in CD4 T cell numbers. This eventually causes an inversion of the CD4/CD8 T cell ratio, which is indicative of aging and can be correlated with increased oxidative stress (Larbi et al., 2008; Gameiro et al., 2010; Muller et al., 2015). The number of B2 cells (involved with antibody production) also decreases with menopause, especially during late menopause in comparison to perimenopause (Kamada et al., 2001). In mice, ovariectomy causes a reduction in the LPS-induced proliferation of leukocytes and subsequent chemotaxis, which is indicative of premature immune senescence (Baeza et al., 2011). Ovariectomized animals generally have a reduced and delayed adaptive response to vaccination, leading to decreased IgG titers in comparison to animals with intact ovaries (Haberthur et al., 2010). These changes are indicative of immune senescence occurring during menopausal transition. In the context of AD, the systemic effect of menopause on inflammation combined with effects on neurological function indicates cruciality of the menopausal transition in AD pathogensis.
Menopause is composed of three transitions; the perimenopause that precedes menopause, the cessation of reproductive capacity, menopause, and the years following menopause, post-menopause. Concomitant with this endocrine aging is chronological aging as the endocrine transition states span multiple years. Each of these endocrine stages is characterized by complex hormonal fluctuations (Brinton et al., 2015).
Decline in estradiol level during perimenopause and menopause coincides with a bioenergetic deficit in brain (Ding et al., 2013). Estradiol is a master regulator of metabolic function in the female (Rettberg et al., 2014). Clinical evidence of decline in glucose metabolism in brain and the coincident bioenergetic deficit is evidenced by reduced uptake of 18-fludeoxyglucose detected by positron emission tomography (PET) in perimenopausal and menopausal women (Mosconi et al., 2017a,b). The bioenergetic deficit precedes a shift to utilization of ketone bodies as a compensatory response to decline in brain glucose as bioenergetic fuel to generate ATP in brain. This shift to utilizing an auxiliary fuel during female endocrine aging activates catabolism of white matter as an endogenous lipid source of ketone bodies in brain (Klosinski et al., 2015) and a concomitant increase in microglial and astrocytic reactivity (Xie et al., 2013; Suenaga et al., 2015).
Analyses of microarray data obtained from the brains of postmenopausal women made available by NCBI revealed an inflammatory gene expression profile in the post central and superior frontal gyrus (Sárvári et al., 2012). In comparison to pre-menopausal women, post-menopausal women showed an upregulation in microglial markers CD14, CD18, and CD45, as well as TLR4 and MHC-II markers CD74 and C3 (Sárvári et al., 2012). These findings in the human female brain were consistent with the pattern of gene expression in the frontal cortex of ovariectomized middle-aged rats (13 months old) (Sárvári et al., 2012). Ovariectomy caused an upregulation of microglial reactivity markers CD11b, C18, CD45, and CD86, complement pathway C3, and phagocytic markers Msr2 and CD32. Together, these data are indicative of a shift in the microglial phenotype to an activated state (see Figure 3).
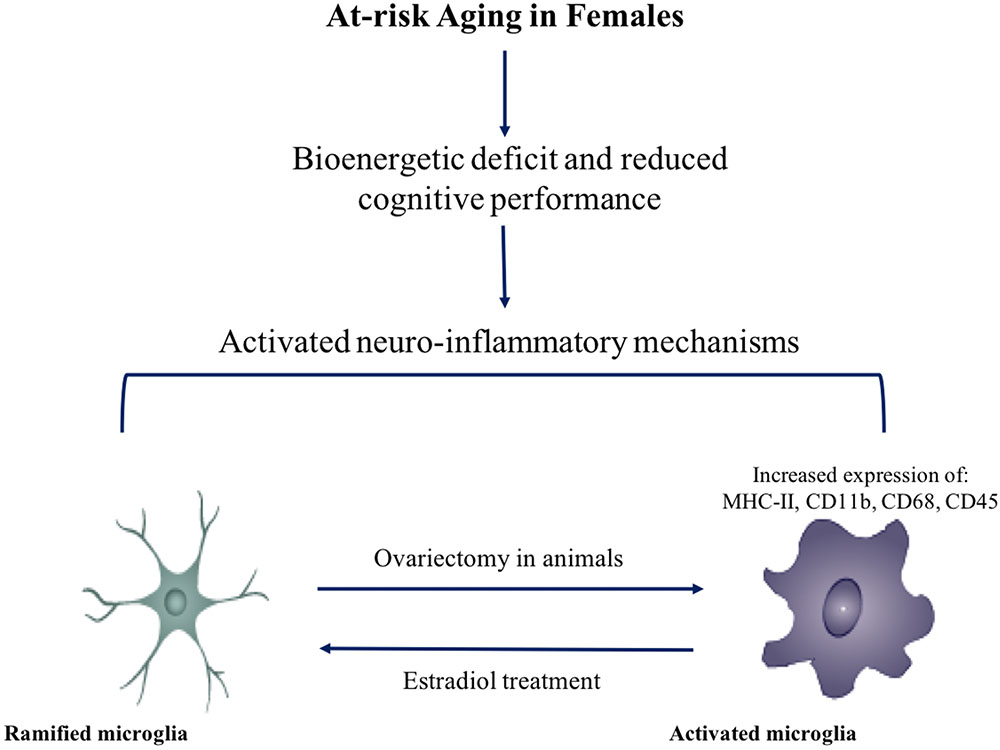
FIGURE 3. Effect of menopause on immunophenotype. Decline in ovarian hormones induces a pro-inflammatory state in microglia. Activation of microglia and neuro-inflammatory mechanisms may be involved in at-risk aging females.
Hippocampal inflammatory gene expression in middle-aged rats drastically changed upon ovariectomy. A similar upregulation of microglial markers (CD45, IBA1, CD68, CD11b, CD18, Fcgr1a, and Fcgr2b) to that witnessed in the cortex was observed in the hippocampus. The gene expression results imply a possible activation of microglia. This effect was mitigated by treatment with estradiol and selective estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) agonists (Sarvari et al., 2015). Human post-menopausal gene expression in the hippocampus corroborated the inflammatory gene expression pattern in ovariectomized rats, with an upregulation of microglial reactivity markers CD11b, CD18, IBA1, CD14, and complement C3 (Sarvari et al., 2015).
In parallel, aging is associated with a marked upregulation in genes encoding the major histocompatibility complex class I and class II (MHC-I and MHC-II) (VanGuilder et al., 2011), alterations in Toll-like receptors (TLRs) (Shaw et al., 2011), and the complement pathway (Reichwald et al., 2009). This effect is more pronounced in women and represents the sexual dimorphism of the immune system (Blalock et al., 2003; Berchtold et al., 2008). The dynamics involved between age and menopause-related increase in myelin degeneration and microglial priming can be a tipping point in the neuro-inflammatory system. The increased myelin antigen load and upregulation of antigen presentation by microglia can set forth a cascade that leads to dysregulated glial metabolism and hypertrophy, eventually causing altered extracellular matrix (Blalock et al., 2003). Each event is pivotal in the development of AD.
Hormone therapy (HT) promotes neuronal survival and has been shown to improve cognitive function and episodic memory in perimenopausal and postmenopausal women (Morrison et al., 2006; Brinton, 2008). Epidemiological studies have shown that HT delays the onset of AD as well as reduces the risk of developing AD (Tang et al., 1996; Persad et al., 2009; Dye et al., 2012). Women transitioning through their menopause benefit most from HT as compared to women who have already transitioned (Girard et al., 2017). Results from several clinical trials have emphasized on the timing of treatment with HT and the drawbacks of missing the window of treatment (Hogervorst et al., 2000; Zandi et al., 2002; Shumaker et al., 2003). This effect of estradiol in HT has been explained by two theories: the healthy cell bias of estrogen action (Brinton, 2008) and critical window hypothesis (Maki, 2013). Healthy cell bias highlights that neuronal viability and health at baseline are important for estradiol to exert its therapeutic efficacy, whereas the critical window hypothesis focuses on the perimenopausal transition, when cells are still healthy, being a key phase for using HT. The use of estradiol in HT provides a therapeutic opportunity to target inflammatory pathways that simultaneously modulate metabolic functions, thereby providing a supportive milieu for neuronal survival and growth (Vegeto et al., 2008; Zhao et al., 2014). HT restores the hormonal levels in post-menopausal women to those of premenopausal women. Post-menopausal women using HT have higher lymphocyte numbers and higher circulating monocytes in comparison to post-menopausal women who are not on HT (Kamada et al., 2000). Likewise, levels of B2 cells involved in antibody production are significantly higher in HT users in comparison to non-users (Kamada et al., 2001).
Estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) are abundantly expressed in astrocytes, microglia, and neurons, and both ERα and ERβ are involved in regulating the immunomodulatory responses exerted by astrocytes and microglia (Liu et al., 2003; Khan and Ansar Ahmed, 2015). In ovariectomized middle-aged rats, estradiol induces downregulation of the complement pathway and macrophage-associated genes in the frontal cortex. This effect was mediated through ERα and ERβ (Sárvári et al., 2011). Estradiol treatment in microglial cells induces a dose-dependent attenuation in superoxide release, phagocytic activity, and a concomitant increase in iNOS activity, without altering NF-κB expression (Bruce-Keller et al., 2000; Drew and Chavis, 2000). Some studies have shown that sex steroids reduce neuroinflammation via inhibiting the inflammasome complex, a possible downstream effect mediated by ERα and ERβ (Slowik and Beyer, 2015).
Astrocytes also participate in mediating the neuroprotective anti-inflammatory effect of estradiol via ERα (Spence et al., 2011). In contrast to microglial cells, estradiol inhibits NF-κB expression in astrocytes (Giraud et al., 2010; Acaz-Fonseca et al., 2014). Estradiol inhibits secretion of proinflammatory cytokines IL-6, TNF-α, IL-1β, expression of matrix metalloproteinases 9 (MMP-9), and interferon gamma-inducible protein 10 (IP-10) in astrocytes. Estradiol also reduces proinflammatory cytokines secreted by astrocytes when exposed to Aβ (Giraud et al., 2010; Acaz-Fonseca et al., 2014).
Much like estradiol, selective estrogen receptor modulators (SERMs) exert a neuroprotective effect by reducing neuroinflammation. Tamoxifen and raloxifene both reduce microgliosis, astrogliosis, and the production of proinflammatory cytokines IL-6 and IP-10 induced by LPS administration (Arevalo et al., 2012). They have also been demonstrated to protect neurons against neurotoxicity caused by neuroinflammation through an ER mediated pathway (Ishihara et al., 2015). SERMs reduce the proinflammatory response produced by astrocytes and are helpful in potentiating their neurotrophic function (Tapia-Gonzalez et al., 2008; Cerciat et al., 2010; Arevalo et al., 2012; Ishihara et al., 2015).
APOEε4 is the primary genetic risk factor for the late onset form of AD (Scacchi et al., 1995; Liu et al., 2014; Manning et al., 2014). The human form of the APOE gene possesses three polymorphic alleles: E2, E3, and E4. The E3 allele occurs more frequently (77%) than E2 (8%) and E4 (14%) (Eisenberg et al., 2010). The E4 allele occurs in 40% of AD patients (Farrer et al., 1997). However, 91% of homozygous E4 carriers and 47% heterozygous carriers go on to develop AD (Corder et al., 1993).
Female APOEε4 carriers are more susceptible to developing AD than males (Altmann et al., 2014; Ungar et al., 2014; Neu et al., 2017). Prospective cohort studies have also suggested that female APOEε4 carriers are at greater risk of converting from MCI to AD than males (Altmann et al., 2014). Female APOEε4 carriers also have a higher rate of cognitive decline than APOEε3 carriers (Holland et al., 2013). Leukocyte telomere length is also greatly reduced in APOEε4 carrier’s relative to age-matched controls, reflecting premature aging of female APOEε4 carriers (Jacobs et al., 2013).
Apolipoprotein E (ApoE) is a key regulator of lipid homeostasis and in the brain, it functions to shuttle lipid molecules from astrocytes and microglia to neurons, via lipoprotein complexes (Liu et al., 2013). In the periphery, ApoE is expressed in macrophages and liver. In the central nervous system, ApoE is mainly produced by astrocytes and microglia (Liu et al., 2013).
ApoE is known to exert an immunosuppressive effect by inhibiting lymphocyte proliferation, Ig synthesis, and neutrophil activation. ApoE also exerts this immunosuppressive property on microglial activation (Guo et al., 2004; Baitsch et al., 2011; Christensen et al., 2011). Relative to APOEε2 and APOEε3 carriers, APOEε4 carriers generate less ApoE protein in periphery and brain (Larson et al., 2000). Given the reduced amounts of ApoE protein in APOEε4 carriers, this population could be predisposed to a heightened inflammatory response when encountering a sterile inducer or infection (Ukkola et al., 2009; Gale et al., 2014; Tai et al., 2015).
Due to the differences in cysteine and arginine residues translated at positions 112 and 158, each of the three different isoforms of APOE exhibit different conformations. The conformational change in the protein affects its stability, folding characteristics, and the propensity to bind lipoprotein particles (Jofre-Monseny et al., 2008). APOEε4 has a globule-like structure and preferably binds to very low-density lipoprotein (vLDL) and low-density lipoprotein (LDL) particles. However, APOEε2 and APOEε3 tend to bind high-density lipoprotein (HDL) particles (Jofre-Monseny et al., 2008). This difference in protein structure affects the lipid shuttling ability of ApoE, which leads to hypercholesterolemia in APOEε4 carriers and increases the predisposition for generation of plaques (Jofre-Monseny et al., 2008). ApoE also exerts an inhibitory effect on the oxidation of LDL in an isoform-specific manner (E2>E3>E4) (Miyata and Smith, 1996). Among smokers, APOEε4 carriers have significantly higher amounts of oxidized LDL (ox-LDL) (Jofre-Monseny et al., 2008).
Cholesterol, ox-LDL, and Aβ are sterile inducers of inflammation called DAMPs (Miller et al., 2011; Clark and Vissel, 2015). DAMPs are recognized by PRRs expressed on macrophages, dendritic cells, monocytes, microglia, and neutrophils, which trigger the activation of an inflammatory process (Silverstein and Febbraio, 2009). One such PRR is CD36. In the context of recognizing DAMPs and plaque formation, it was recently demonstrated that CD36 (expressed on monocytes, macrophages, and microglia) recognizes soluble ligands such as oxidized LDL and soluble Aβ and converts them to crystals and fibrils, respectively. This leads to the assembly and activation of the NLRP3 inflammasome and the consequent release of the proinflammatory cytokine IL-1β (Sheedy et al., 2013; Oury, 2014).
Due to the increased probability of APOEε4 carriers to develop plaques, cholesterol crystals, and amyloid depositions, cellular immune function and reactivity are affected. In APOEε4 carriers there is a reduced clearance and efflux of cholesterol from macrophages (Cash et al., 2012). Moreover, increased nitric oxide production in APOEε4 causes increased platelet aggregation and secretion of adhesion molecules, further enabling the plaque formation in the periphery. In the brain, microglial and astrocytic clearance of debris is also diminished (Guo et al., 2006).
ApoE also affects Aβ uptake and oligomerization and thus can be a key factor in Aβ turnover. AD patients possessing the APOEε4 allele were found to have higher levels oligomeric Aβ in their brains as compared to APOEε3 carriers, implicating an association between ApoE with Aβ (Hashimoto et al., 2012). In vitro experiments correspond to clinical findings and have shown that APOEε4 has the greatest effect on promoting Aβ oligomerization in comparison to other isoforms (Hashimoto et al., 2012). Blocking Aβ and ApoE interaction by ApoE Aβ antagonist in hippocampal neuronal and astrocytic co-culture systems led to decreased accumulation and oligomerization of Aβ. Treatment with ApoE Aβ antagonists also inhibited the loss of synaptic proteins induced by Aβ accumulation (Kuszczyk et al., 2013; Liu S. et al., 2014).
Coupled with the Aβ oligomerization, ApoE also affects Aβ uptake. Astrocytes secrete ApoE as a lipoparticle into the interstitial fluid, where it binds with Aβ. Neurons endocytose and internalize these lipoparticles, thus promoting Aβ uptake. APOEε4 isoform has maximal binding affinity to Aβ and thus causes a greater uptake of Aβ by neurons in comparison to other isoforms (Mulder et al., 2014). APOEε4 prevents the uptake of oligomeric Aβ by astrocytes and the uptake of both oligomeric and fibrillar Aβ by microglia thereby inhibiting its clearance (Mulder et al., 2014).
ApoE also modulates Aβ clearance by microglia by regulating Aβ clearing enzymes such as neprilysin intracellularly and insulin degrading enzyme extracellularly. Effective degradation of Aβ depends on Liver X Receptor (LXR) activation, the isoform of APOE expressed, and the lipidation status of ApoE particles (Hashimoto et al., 2012; Kuszczyk et al., 2013; Liu S. et al., 2014). Activation of LXR/RXR (Retinoid X receptor) potentiates Aβ clearance as it compensates for the loss of APOEε4 function and induces the expression of ATP-binding cassette transporter subfamily A member 1 (ABCA1) and ApoE, thus inducing clearance by microglia and astrocytes (Lefterov et al., 2007; Terwel et al., 2011; Lee et al., 2012; Mandrekar-Colucci et al., 2012; Tai et al., 2014). Therefore, the presence of APOEε4 promotes the production of Aβ and uptake by neurons while preventing clearance and enabling the production of DAMPs and chronic low-grade inflammation.
Given the dysregulated lipid metabolism and impairment of cellular function with age, the APOEε4 allele is associated with increased systemic inflammation. It is expected that this would be reflected in cytokine levels, which are inflammatory markers such as C-Reactive Protein (CRP) measured in plasma/serum. Surprisingly, however, this is not the case, at least with CRP. Studies have consistently shown that CRP levels are lower in APOEε4 carriers (Ukkola et al., 2009; Lima et al., 2014; Metti et al., 2014; Yun et al., 2015). Marz et al. (2004) proposed that the metabolism of CRP might be associated with the mevalonate/cholesterol synthetic pathway, which might be downregulated in APOEε4 carriers. On the other hand, studies have shown that levels of IL-1β and vascular inflammatory marker: vascular cell adhesion molecule-1 (VCAM-1) are higher in E4 carriers (Olgiati et al., 2010; see Figure 4).
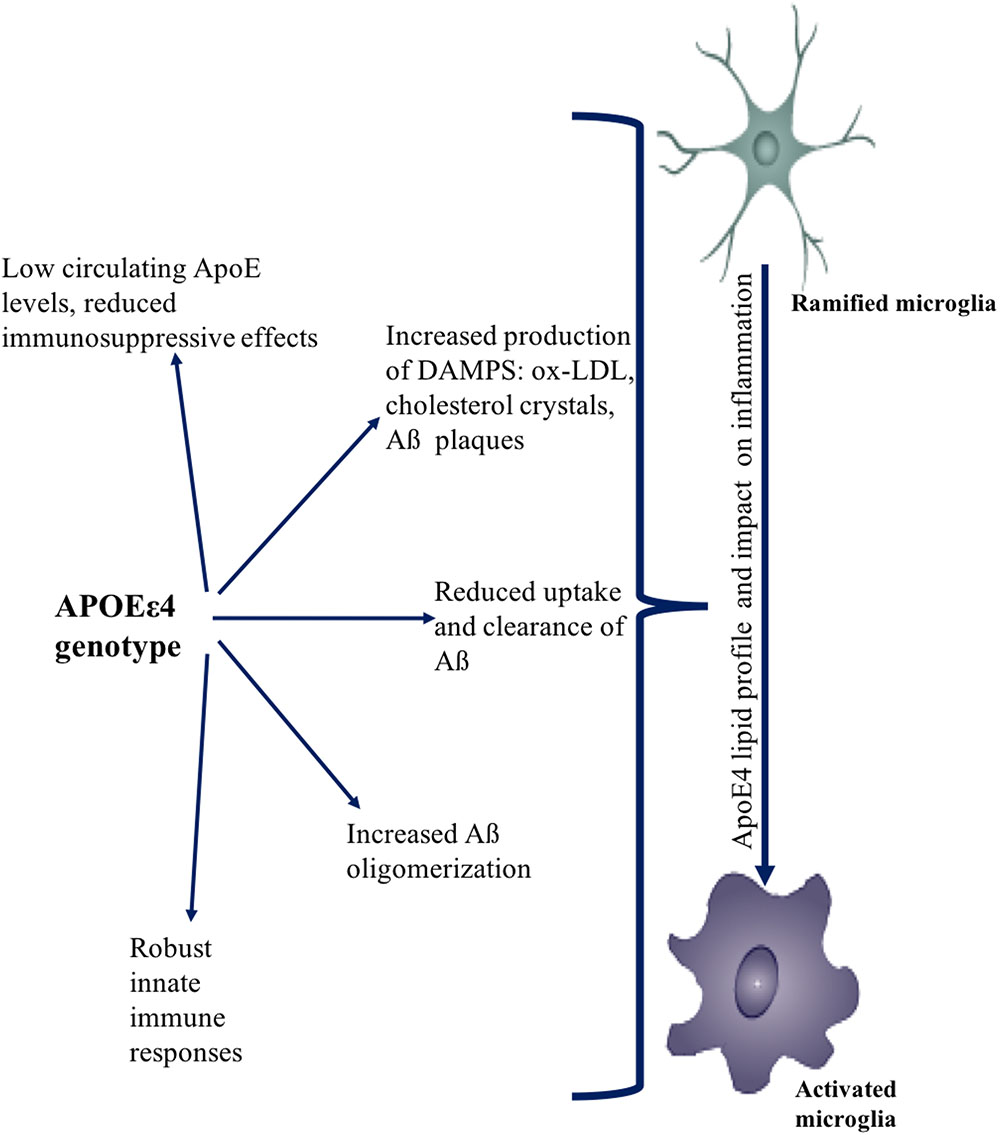
FIGURE 4. Effect of APOEε4 on immune function. APOEε4 genotype affects the uptake, clearance, and production of sterile triggers of inflammation. APOEε4 causes increased accumulation of DAMPs such as Aβ, ox-LDL, cholesterol. APOEε4 also causes more robust innate immune responses. Collectively, the effects of APOEε4 genotype promotes an overall neuro-inflammatory state.
The APOEε4 allele also accelerates aging, which is reflected in the shorter telomeres in APOEε4 women in comparison to APOEε3 women (Jacobs et al., 2013). The accelerated aging phenotype is also evident in the reduction of T cell numbers. Age-related reduction in T cells for women is more dramatic during menopause, which is even more pronounced if the woman is an APOEε4 carrier (Begum et al., 2014). In comparison to APOEε3 derived microglia, estradiol has a reduced anti-inflammatory effect on microglia derived from APOEε4 (Brown et al., 2008). The APOEε4 allele is also a risk factor for metabolic syndrome and has been associated with RA, thus increasing the risk of comorbidities that can, in turn, affect systemic inflammation (Sima et al., 2007; Gungor et al., 2012; Toms et al., 2012; Johnson et al., 2013). APOEε4 also affects immune function by causing blood barrier dysfunction (Nelson et al., 2016).
Inflammatory responses triggered by innate immune agonists are highest in APOEε4 carriers (Vitek et al., 2009; Gale et al., 2014). This finding holds true in cells isolated from both humans and rodents and in both the periphery and the brain (Gale et al., 2014; Li X. et al., 2015). Therefore, inflammatory triggers such as TBI, infection, and DAMPs produced from metabolic syndrome significantly increase the inflammatory response in APOEε4 carriers. This may potentially lead to the incomplete resolution of inflammation to initiate chronic inflammatory processes that result in neurotoxicity. This trend is evident in HIV-associated dementia, for which E4 carriers have increased risk (Chang et al., 2011).
A heightened inflammatory response occurred in microglia derived from humanized APOEε4 knock-in mice upon on treatment with the TLR3 and TLR4 activator LPS compared to APOEε3 mice (Vitek et al., 2009; Heneka et al., 2015). The inflammatory response was characterized by altered cell morphology, increased nitric oxide production, COX-2 expression, prostaglandin E2 (PGE2) expression, and cytokine production (IL-6, TNF-α, and IL-12p40). In contrast, TREM2 expression was decreased (Vitek et al., 2009; Heneka et al., 2015). A comparable inflammatory response was observed in peripheral macrophages isolated from APOEε4 mice. The E4 allele increases the reactivity of glial and peripheral immune cells, thus aggravating the neurotoxic proinflammatory response (Vitek et al., 2009; Heneka et al., 2015).
The etiology of the prodromal phase of AD presents as a complex interplay between several risk factors, which is relevant to therapeutic interventions and preventive strategies (see Figures 5, 6). This implies that therapeutic strategies should employ stratification of patient populations regarding parameters of age, sex, and APOE genotype. It also calls for the use of combination therapies that modulate inflammation, lipid-based metabolism in APOEε4 carriers, and loss of estrogenic control in menopausal women. For example, preventive strategies to reduce age-related inflammation could include a combination of NSAIDs and statins in middle-aged adults (45–55 years). In women, this therapy could be modified to include HT during their perimenopausal transition. Patient’s medical histories and electronic health records are a source of indicators for chronic inflammation. These strategies should be tailored to the patient’s metabolic profile, genetic history, and endocrine-related transition states (see Figures 5, 6).
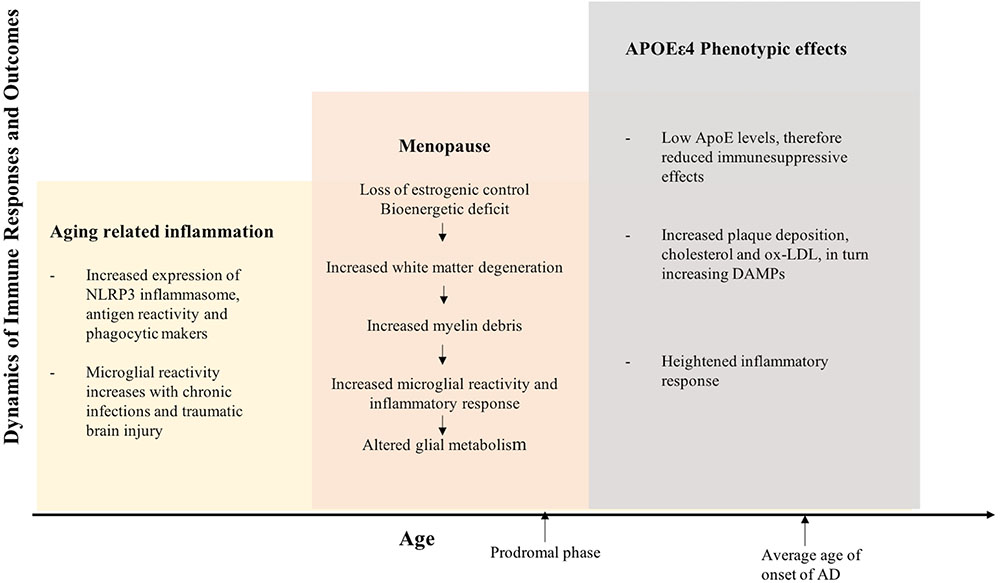
FIGURE 5. Inflammation integrates Alzheimer’s disease risk factors of female sex, chronological age, endocrine aging, and APOEε4 genotype. The three hit model of Alzheimer’s risk: aging, menopause, and APOEε4 genotype collectively induce a compromised bioenergetic system in brain that is impacted by the chronic low grade innate inflammation of aging coupled with APOEε4 dysregulated cholesterol homeostasis lead to activation of the adaptive immune response. The inflammatory immune response is the factor that bridges across each of the risk factors for AD. Immune system regulators that are specific to stage of disease and inflammatory phenotype would provide a therapeutic strategy to disconnect the bridge that drives disease.
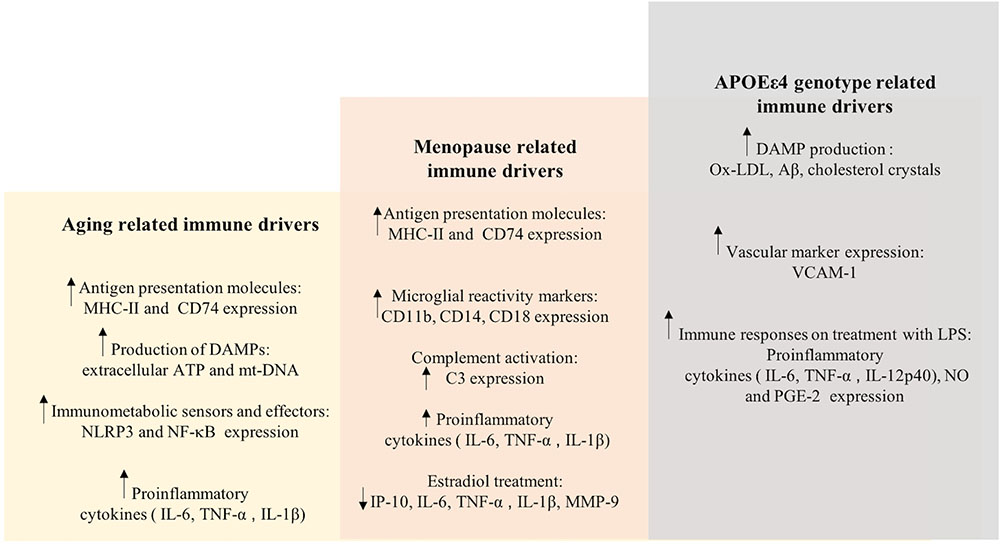
FIGURE 6. Immune drivers involved in aging, menopause, and APOEε4 genotype related inflammation. Key immune drivers contributing to inflammation due to aging, menopause and APOEε4 genotype related inflammation are detailed to give a global picture of immune dynamics (upward and downward arrows indicate increased and decreased expression, respectively).
This understanding of the disease progression also calls for change in design of clinical trials that target the amyloidogenic pathway and treat later stages of the disease pathogenesis. Trial design should incorporate the identification of persons with an increased risk of developing AD, and utilize a risk-factor-based responder analysis. Inflammation-mediated therapeutic and preventive strategies will largely depend upon this stratification of patient populations.
The inflammatory response is influenced by age, chromosomal sex, endocrine transition – menopause and APOE genotype. Inflammation is characteristic of each of these modifying factors and can be a driving force for development of AD. Thus, inflammation has been a therapeutic target in multiple clinical trials for AD. However, each of these trials have failed to meet primary endpoints. Reviewed herein is a consideration of the multiple factors that contribute to and modify the inflammatory phenotype. Going forward, in both discovery and clinical science, it will be important to delineate the etiology of the inflammatory response, the stage of the inflammatory cascade, and the activated network of inflammatory signaling. Inflammation is a moving target and thus requires a precision approach to identifying etiology, stage and appropriate therapeutic target.
In summary, neuro-inflammatory processes are evident early in the latent prodromal phase and worsen during the course of the Alzheimer’s. Disparate results from epidemiological and clinical trials targeting inflammation, highlight the complexity of the inflammatory process. The inflammatory processes that occur during aging, midlife endocrine transitions, and in the APOEε4 carrier contribute to risk and progression of AD. The chronic inflammatory processes that are activated during midlife chronological and endocrine aging, ultimately limit the clearance capacity of microglia and lead to immune senescence. The loss of estrogenic control of bioenergetic function in the brain coupled with dysregulated lipid metabolism in the APOEε4 genotype adversely impact microglial function and clearance mechanisms. The dynamic and context specific activation pattern of the inflammatory processes provide plausible mechanisms underlying failed clinical trials of anti-inflammatory agents in Alzheimer’s patients. Collectively, these considerations highlight the rationale for stratifying AD clinical trial cohorts based on their inflammatory phenotype. Combination therapies that include targeted use of anti-inflammatory agent’s specific to the immune phenotype could have a higher probability of successfully modifying risk and progression of Alzheimer’s disease.
AM and RB wrote and reviewed the manuscript.
This work was supported by National Institute on Aging (NIA) grants P01AG026572 and R37AG053589 to RB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AG and handling Editor declared their shared affiliation at the time of the review.
Acaz-Fonseca, E., Sanchez-Gonzalez, R., Azcoitia, I., Arevalo, M. A., and Garcia-Segura, L. M. (2014). Role of astrocytes in the neuroprotective actions of 17β-estradiol and selective estrogen receptor modulators. Mol. Cell. Endocrinol. 389, 48–57. doi: 10.1016/j.mce.2014.01.009
Ağaçayak, E., Yaman Görük, N., Küsen, H., Yaman Tunç, S., Başaranoğlu, S., Içe, M. S., et al. (2016). Role of inflammation and oxidative stress in the etiology of primary ovarian insufficiency. J. Turk. Soc. Obstet. Gynecol. 13, 109–115. doi: 10.4274/tjod.00334
Alisky, J. M. (2007). The coming problem of HIV-associated Alzheimer’s disease. Med. Hypotheses 69, 1140–1143. doi: 10.1016/j.mehy.2007.02.030
Altmann, A., Tian, L., Henderson, V. W., Greicius, M. D., and Alzheimer’s Disease Neuroimaging Initiative, I. (2014). Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75, 563–573. doi: 10.1002/ana.24135
Alzheimer’s Disease Anti-inflammatory Prevention Trial Research Group (2013). Results of a follow-up study to the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT). Alzheimers Dement. 9, 714–723. doi: 10.1016/j.jalz.2012.11.012
Amieva, H., Goff, M. L., Millet, X., Orgogozo, J. M., Pérès, K., Barberger-Gateau, P., et al. (2008). Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann. Neurol. 64, 492–498. doi: 10.1002/ana.21509
Arevalo, M. A., Diz-Chaves, Y., Santos-Galindo, M., Bellini, M. J., and Garcia-Segura, L. M. (2012). Selective oestrogen receptor modulators decrease the inflammatory response of glial cells. J. Neuroendocrinol. 24, 183–190. doi: 10.1111/j.1365-2826.2011.02156.x
Baeza, I., De Castro, N. M., Arranz, L., Fdez-Tresguerres, J., and De La Fuente, M. (2011). Ovariectomy causes immunosenescence and oxi-inflamm-ageing in peritoneal leukocytes of aged female mice similar to that in aged males. Biogerontology 12, 227–238. doi: 10.1007/s10522-010-9317-0
Baitsch, D., Bock, H. H., Engel, T., Telgmann, R., Muller-Tidow, C., Varga, G., et al. (2011). Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler. Thromb. Vasc. Biol. 31, 1160–1168. doi: 10.1161/ATVBAHA.111.222745
Begum, A. N., Cunha, C., Sidhu, H., Alkam, T., Scolnick, J., Rosario, E. R., et al. (2014). Women with the Alzheimer’s risk marker ApoE4 lose Abeta-specific CD4+ T cells 10-20 years before men. Transl. Psychiatry 4:e414. doi: 10.1038/tp.2014.51
Benedusi, V., Meda, C., Della Torre, S., Monteleone, G., Vegeto, E., and Maggi, A. (2012). A lack of ovarian function increases neuroinflammation in aged mice. Endocrinology 153, 2777–2788. doi: 10.1210/en.2011-1925
Berchtold, N. C., Cribbs, D. H., Coleman, P. D., Rogers, J., Head, E., Kim, R., et al. (2008). Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. U.S.A. 105, 15605–15610. doi: 10.1073/pnas.0806883105
Blalock, E. M., Chen, K.-C., Sharrow, K., Herman, J. P., Porter, N. M., Foster, T. C., et al. (2003). Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J. Neurosci. 23, 3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003
Bove, R. (2013). Autoimmune diseases and reproductive aging. Clin. Immunol. 149, 251–264. doi: 10.1016/j.clim.2013.02.010
Bove, R., Secor, E., Chibnik, L. B., Barnes, L. L., Schneider, J. A., Bennett, D. A., et al. (2014). Age at surgical menopause influences cognitive decline and Alzheimer pathology in older women. Neurology 82, 222–229. doi: 10.1212/WNL.0000000000000033
Boyd, T. D., Bennett, S. P., Mori, T., Governatori, N., Runfeldt, M., Norden, M., et al. (2010). GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J. Alzheimers Dis. 21, 507–518. doi: 10.3233/JAD-2010-091471
Breitner, J. C., Baker, L. D., Montine, T. J., Meinert, C. L., Lyketsos, C. G., Ashe, K. H., et al. (2011). Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement. 7, 402–411. doi: 10.1016/j.jalz.2010.12.014
Breunig, J. J., Guillot-Sestier, M. V., and Town, T. (2013). Brain injury, neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 5:26. doi: 10.3389/fnagi.2013.00026
Brinkmalm, A., Brinkmalm, G., Honer, W. G., Frölich, L., Hausner, L., Minthon, L., et al. (2014). SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s disease. Mol. Neurodegener. 9:53. doi: 10.1186/1750-1326-9-53
Brinton, R. D. (2008). The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 31, 529–537. doi: 10.1016/j.tins.2008.07.003
Brinton, R. D., Yao, J., Yin, F., Mack, W. J., and Cadenas, E. (2015). Perimenopause as a neurological transition state. Nat. Rev. Endocrinol. 11, 393–405. doi: 10.1038/nrendo.2015.82
Brooks-Asplund, E. M., Tupper, C. E., Daun, J. M., Kenney, W. L., and Cannon, J. G. (2002). Hormonal modulation of interleukin-6, tumor necrosis factor and associated receptor secretion in postmenopausal women. Cytokine 19, 193–200. doi: 10.1006/cyto.2002.1963
Brown, C. M., Choi, E., Xu, Q., Vitek, M. P., and Colton, C. A. (2008). The APOE4 genotype alters the response of microglia and macrophages to 17beta-estradiol. Neurobiol. Aging 29, 1783–1794. doi: 10.1016/j.neurobiolaging.2007.04.018
Bruce-Keller, A. J., Keeling, J. L., Keller, J. N., Huang, F. F., Camondola, S., and Mattson, M. P. (2000). Antiinflammatory effects of estrogen on microglial activation. Endocrinology 141, 3646–3656. doi: 10.1210/endo.141.10.7693
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn.3599
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Cash, J. G., Kuhel, D. G., Basford, J. E., Jaeschke, A., Chatterjee, T. K., Weintraub, N. L., et al. (2012). Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J. Biol. Chem. 287, 27876–27884. doi: 10.1074/jbc.M112.377549
Cerciat, M., Unkila, M., Garcia-Segura, L. M., and Arevalo, M. A. (2010). Selective estrogen receptor modulators decrease the production of interleukin-6 and interferon-gamma-inducible protein-10 by astrocytes exposed to inflammatory challenge in vitro. Glia 58, 93–102. doi: 10.1002/glia.20904
Chakradhar, S. (2018). A tale of two diseases: aging HIV patients inspire a closer look at Alzheimer’s disease. Nat. Med. 24, 376–377. doi: 10.1038/nm0418-376
Chang, L., Andres, M., Sadino, J., Jiang, C. S., Nakama, H., Miller, E., et al. (2011). Impact of apolipoprotein E epsilon4 and HIV on cognition and brain atrophy: antagonistic pleiotropy and premature brain aging. Neuroimage 58, 1017–1027. doi: 10.1016/j.neuroimage.2011.07.010
Chou, R. C., Kane, M., Ghimire, S., Gautam, S., and Gui, J. (2016). Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: a nested case-control analysis. CNS Drugs 30, 1111–1120. doi: 10.1007/s40263-016-0374-z
Christensen, D. J., Ohkubo, N., Oddo, J., Van Kanegan, M. J., Neil, J., Li, F., et al. (2011). Apolipoprotein E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J. Immunol. 186, 2535–2542. doi: 10.4049/jimmunol.1002847
Clark, I. A., and Vissel, B. (2015). Amyloid beta: one of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer’s disease. Br. J. Pharmacol. 172, 3714–3727. doi: 10.1111/bph.13181
Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Munoz-Planillo, R., Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255. doi: 10.1038/nm.3806
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Crain, J. M., Nikodemova, M., and Watters, J. J. (2013). Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J. Neurosci. Res. 91, 1143–1151. doi: 10.1002/jnr.23242
Cui, H., Kong, Y., and Zhang, H. (2012). Oxidative stress, mitochondrial dysfunction, and aging. J. Signal. Transduct. 2012:646354. doi: 10.1155/2012/646354
Damani, M. R., Zhao, L., Fontainhas, A. M., Amaral, J., Fariss, R. N., and Wong, W. T. (2011). Age-related alterations in the dynamic behavior of microglia. Aging Cell 10, 263–276. doi: 10.1111/j.1474-9726.2010.00660.x
Dickson, D. W., Lee, S. C., Mattiace, L. A., Yen, S. -H. C., and Brosnan, C. (1993). Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 7, 75–83. doi: 10.1002/glia.440070113
Ding, F., Yao, J., Rettberg, J. R., Chen, S., and Brinton, R. D. (2013). Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One 8:e79977. doi: 10.1371/journal.pone.0079977
Dixit, V. D. (2013). Nlrp3 inflammasome activation in type 2 diabetes: is it clinically relevant? Diabetes 62, 22–24. doi: 10.2337/db12-1115
Doran, M. F., Pond, G. R., Crowson, C. S., O’fallon, W. M., and Gabriel, S. E. (2002). Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum. 46, 625–631. doi: 10.1002/art.509
Drew, P. D., and Chavis, J. A. (2000). Female sex steroids: effects upon microglial cell activation. J. Neuroimmunol. 111, 77–85. doi: 10.1016/S0165-5728(00)00386-6
Dye, R. V., Miller, K. J., Singer, E. J., and Levine, A. J. (2012). Hormone replacement therapy and risk for neurodegenerative diseases. Int. J. Alzheimers Dis. 2012:258454. doi: 10.1155/2012/258454
Eikelenboom, P., Van Exel, E., Veerhuis, R., Rozemuller, A. J., Van Gool, W. A., and Hoozemans, J. J. (2012). Innate immunity and the etiology of late-onset Alzheimer’s disease. Neurodegener. Dis. 10, 271–273. doi: 10.1159/000334287
Eisenberg, D. T., Kuzawa, C. W., and Hayes, M. G. (2010). Worldwide allele frequencies of the human apolipoprotein E gene: climate, local adaptations, and evolutionary history. Am. J. Phys. Anthropol. 143, 100–111. doi: 10.1002/ajpa.21298
Etminan, M., Gill, S., and Samii, A. (2003). Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ 327:128. doi: 10.1136/bmj.327.7407.128
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356. doi: 10.1001/jama.1997.03550160069041
Ferretti, M. T., and Cuello, A. C. (2011). Does a pro-inflammatory process precede Alzheimer’s disease and mild cognitive impairment? Curr. Alzheimer Res. 8, 164–174. doi: 10.2174/156720511795255982
Floden, A. M., and Combs, C. K. (2011). Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J. Alzheimers Dis. 25, 279–293. doi: 10.3233/JAD-2011-101014
Fougère, B., Boulanger, E., Nourhashémi, F., Guyonnet, S., and Cesari, M. (2017). Chronic inflammation: accelerator of biological aging. J. Gerontol. Ser. A 72, 1218–1225.
Franceschi, C., Bonafè, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging: an evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Gale, S. C., Gao, L., Mikacenic, C., Coyle, S. M., Rafaels, N., Murray Dudenkov, T., et al. (2014). APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 134, 127–134. doi: 10.1016/j.jaci.2014.01.032
Gameiro, C. M., Romao, F., and Castelo-Branco, C. (2010). Menopause and aging: changes in the immune system–a review. Maturitas 67, 316–320. doi: 10.1016/j.maturitas.2010.08.003
Girard, R., Metereau, E., Thomas, J., Pugeat, M., Qu, C., and Dreher, J. C. (2017). Hormone therapy at early post-menopause increases cognitive control-related prefrontal activity. Sci. Rep. 7:44917. doi: 10.1038/srep44917
Giraud, S. N., Caron, C. M., Pham-Dinh, D., Kitabgi, P., and Nicot, A. B. (2010). Estradiol inhibits ongoing autoimmune neuroinflammation and NFkappaB-dependent CCL2 expression in reactive astrocytes. Proc. Natl. Acad. Sci. U.S.A. 107, 8416–8421. doi: 10.1073/pnas.0910627107
Giuliani, N., Sansoni, P., Girasole, G., Vescovini, R., Passeri, G., Passeri, M., et al. (2001). Serum interleukin-6, soluble interleukin-6 receptor and soluble gp130 exhibit different patterns of age- and menopause-related changes. Exp. Gerontol. 36, 547–557. doi: 10.1016/S0531-5565(00)00220-5
Goetzl, E. J., Huang, M. -C., Kon, J., Patel, K., Schwartz, J. B., Fast, K., et al. (2010). Gender specificity of altered human immune cytokine profiles in aging. FASEB J. 24, 3580–3589. doi: 10.1096/fj.10-160911
Goronzy, J. J., and Weyand, C. M. (2012). Immune aging and autoimmunity. Cell. Mol. Life Sci. 69, 1615–1623. doi: 10.1007/s00018-012-0970-0
Group, A. R. (2007). Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68, 1800–1808. doi: 10.1212/01.wnl.0000260269.93245.d2
Group, A. R. (2008). Cognitive function over time in the alzheimer's disease anti-inflammatory prevention trial (adapt): results of a randomized, controlled trial of naproxen and celecoxib. Arch. Neurol. 65, 896–905. doi: 10.1001/archneur.2008.65.7.nct70006
Gulke, E., Gelderblom, M., and Magnus, T. (2018). Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 11:1756286418774254. doi: 10.1177/1756286418774254
Gungor, Z., Anuurad, E., Enkhmaa, B., Zhang, W., Kim, K., and Berglund, L. (2012). Apo E4 and lipoprotein-associated phospholipase A2 synergistically increase cardiovascular risk. Atherosclerosis 223, 230–234. doi: 10.1016/j.atherosclerosis.2012.04.021
Guo, L., Ladu, M. J., and Van Eldik, L. J. (2004). A dual role for apolipoprotein E in neuroinflammation. J. Mol. Neurosci. 23, 205–212. doi: 10.1385/JMN:23:3:205
Guo, S., Wang, S., Kim, W. J., Lee, S. R., Frosch, M. P., Bacskai, B. J., et al. (2006). Effects of apoE isoforms on beta-amyloid-induced matrix metalloproteinase-9 in rat astrocytes. Brain Res. 1111, 222–226. doi: 10.1016/j.brainres.2006.06.041
Habeck, C., Risacher, S., Lee, G. J., Glymour, M. M., Mormino, E., Mukherjee, S., et al. (2012). Relationship between baseline brain metabolism measured using [(1)(8)F]FDG PET and memory and executive function in prodromal and early Alzheimer’s disease. Brain Imaging Behav. 6, 568–583. doi: 10.1007/s11682-012-9208-x
Haberthur, K., Engelman, F., Barron, A., and Messaoudi, I. (2010). Immune senescence in aged nonhuman primates. Exp. Gerontol. 45, 655–661. doi: 10.1016/j.exger.2010.06.001
Habib, P., Slowik, A., Zendedel, A., Johann, S., Dang, J., and Beyer, C. (2014). Regulation of hypoxia-induced inflammatory responses and M1-M2 phenotype switch of primary rat microglia by sex steroids. J. Mol. Neurosci. 52, 277–285. doi: 10.1007/s12031-013-0137-y
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Hashimoto, T., Serrano-Pozo, A., Hori, Y., Adams, K. W., Takeda, S., Banerji, A. O., et al. (2012). Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J. Neurosci. 32, 15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012
Helenius, M., Hänninen, M., Lehtinen, S. K., and Salminen, A. (1996). Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor-kappa B. Biochem. J. 318, 603–608. doi: 10.1042/bj3180603
Heneka, M. T., Carson, M. J., Khoury, J. E., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hickman, S. E., Kingery, N. D., Ohsumi, T. K., Borowsky, M. L., Wang, L. C., Means, T. K., et al. (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. doi: 10.1038/nn.3554
Hogervorst, E., Williams, J., Budge, M., Riedel, W., and Jolles, J. (2000). The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience 101, 485–512. doi: 10.1016/S0306-4522(00)00410-3
Holland, D., Desikan, R. S., Dale, A. M., Mcevoy, L. K., and Alzheimer’s Disease Neuroimaging, I. (2013). Higher rates of decline for women and apolipoprotein E epsilon4 carriers. AJNR Am. J. Neuroradiol. 34, 2287–2293. doi: 10.3174/ajnr.A3601
Holtman, I. R., Raj, D. D., Miller, J. A., Schaafsma, W., Yin, Z., Brouwer, N., et al. (2015). Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol. Commun. 3:31. doi: 10.1186/s40478-015-0203-5
Ishihara, Y., Itoh, K., Ishida, A., and Yamazaki, T. (2015). Selective estrogen-receptor modulators suppress microglial activation and neuronal cell death via an estrogen receptor-dependent pathway. J. Steroid Biochem. Mol. Biol. 145, 85–93. doi: 10.1016/j.jsbmb.2014.10.002
Itagaki, S., Mcgeer, P. L., Akiyama, H., Zhu, S., and Selkoe, D. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 24, 173–182. doi: 10.1016/0165-5728(89)90115-X
Jack, C. R., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., et al. (2013). Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/S1474-4422(12)70291-0
Jacobs, E. G., Kroenke, C., Lin, J., Epel, E. S., Kenna, H. A., Blackburn, E. H., et al. (2013). Accelerated cell aging in female APOE-epsilon4 carriers: implications for hormone therapy use. PLoS One 8:e54713. doi: 10.1371/journal.pone.0054713
Jofre-Monseny, L., Minihane, A. M., and Rimbach, G. (2008). Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol. Nutr. Food Res. 52, 131–145. doi: 10.1002/mnfr.200700322
Johnson, L. A., Kim, H. S., Knudson, M. J., Nipp, C. T., Yi, X., and Maeda, N. (2013). Diabetic atherosclerosis in APOE∗4 mice: synergy between lipoprotein metabolism and vascular inflammation. J. Lipid Res. 54, 386–396. doi: 10.1194/jlr.M031435
Kamada, M., Irahara, M., Maegawa, M., Yasui, T., Takeji, T., Yamada, M., et al. (2000). Effect of hormone replacement therapy on post-menopausal changes of lymphocytes and T cell subsets. J. Endocrinol. Invest. 23, 376–382. doi: 10.1007/BF03343741
Kamada, M., Irahara, M., Maegawa, M., Yasui, T., Yamano, S., Yamada, M., et al. (2001). B cell subsets in postmenopausal women and the effect of hormone replacement therapy. Maturitas 37, 173–179. doi: 10.1016/S0378-5122(00)00180-8
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
Khan, A. M., Babcock, A. A., Saeed, H., Myhre, C. L., Kassem, M., and Finsen, B. (2015). Telomere dysfunction reduces microglial numbers without fully inducing an aging phenotype. Neurobiol. Aging 36, 2164–2175. doi: 10.1016/j.neurobiolaging.2015.03.008
Khan, D., and Ansar Ahmed, S. (2015). The immune system is a natural target for estrogen action: opposing effects of estrogen in two prototypical autoimmune diseases. Front. Immunol. 6:635. doi: 10.3389/fimmu.2015.00635
Kim, Y. S., and Joh, T. H. (2006). Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 38, 333–347. doi: 10.1038/emm.2006.40
Kireev, R. A., Vara, E., Vina, J., and Tresguerres, J. A. (2014). Melatonin and oestrogen treatments were able to improve neuroinflammation and apoptotic processes in dentate gyrus of old ovariectomized female rats. Age 36:9707. doi: 10.1007/s11357-014-9707-3
Klosinski, L. P., Yao, J., Yin, F., Fonteh, A. N., Harrington, M. G., Christensen, T. A., et al. (2015). White matter lipids as a ketogenic fuel supply in aging female brain: implications for Alzheimer’s disease. EBioMedicine 2, 1888–1904. doi: 10.1016/j.ebiom.2015.11.002
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9. doi: 10.1016/j.immuni.2017.08.008
Krstic, D., Madhusudan, A., Doehner, J., Vogel, P., Notter, T., Imhof, C., et al. (2012). Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflammation 9:151. doi: 10.1186/1742-2094-9-151
Kuh, D., Cooper, R., Moore, A., Richards, M., and Hardy, R. (2018). Age at menopause and lifetime cognition: findings from a British birth cohort study. Neurology 90, e1673–e1681. doi: 10.1212/WNL.0000000000005486
Kuszczyk, M. A., Sanchez, S., Pankiewicz, J., Kim, J., Duszczyk, M., Guridi, M., et al. (2013). Blocking the interaction between apolipoprotein E and Abeta reduces intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am. J. Pathol. 182, 1750–1768. doi: 10.1016/j.ajpath.2013.01.034
Lai, A. Y., Dibal, C. D., Armitage, G. A., Winship, I. R., and Todd, K. G. (2013). Distinct activation profiles in microglia of different ages: a systematic study in isolated embryonic to aged microglial cultures. Neuroscience 254, 185–195. doi: 10.1016/j.neuroscience.2013.09.010
Larbi, A., Franceschi, C., Mazzatti, D., Solana, R., Wikby, A., and Pawelec, G. (2008). Aging of the immune system as a prognostic factor for human longevity. Physiology 23, 64–74. doi: 10.1152/physiol.00040.2007
Larson, I. A., Ordovas, J. M., Deluca, C., Barnard, J. R., Feussner, G., and Schaefer, E. J. (2000). Association of apolipoprotein (Apo)E genotype with plasma apo E levels. Atherosclerosis 148, 327–335. doi: 10.1016/S0021-9150(99)00280-4
Latta, C. H., Brothers, H. M., and Wilcock, D. M. (2014). Neuroinflammation in Alzheimer’s disease; a source of heterogeneity and target for personalized therapy. Neuroscience 302, 103–111. doi: 10.1016/j.neuroscience.2014.09.061
Latta, C. H., Sudduth, T. L., Weekman, E. M., Brothers, H. M., Abner, E. L., Popa, G. J., et al. (2015). Determining the role of IL-4 induced neuroinflammation in microglial activity and amyloid-beta using BV2 microglial cells and APP/PS1 transgenic mice. J. Neuroinflammation 12:41. doi: 10.1186/s12974-015-0243-6
Lee, C. Y., Tse, W., Smith, J. D., and Landreth, G. E. (2012). Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 287, 2032–2044. doi: 10.1074/jbc.M111.295451
Lefterov, I., Bookout, A., Wang, Z., Staufenbiel, M., Mangelsdorf, D., and Koldamova, R. (2007). Expression profiling in APP23 mouse brain: inhibition of Abeta amyloidosis and inflammation in response to LXR agonist treatment. Mol. Neurodegener. 2:20. doi: 10.1186/1750-1326-2-20
Li, L., Ruau, D., Chen, R., Weber, S., and Butte, A. J. (2012). “Systematic identification of risk factors for alzheimer’s disease through shared genetic architecture and electronic medical records,” in Proceedings of the Pacific Symposium on Biocomputing 2013. (Singapore: World Scientific), 224–235. doi: 10.1142/9789814447973_0022
Li, M. D., Burns, T. C., Kumar, S., Morgan, A. A., Sloan, S. A., and Palmer, T. D. (2015). Aging-like changes in the transcriptome of irradiated microglia. Glia 63, 754–767. doi: 10.1002/glia.22782
Li, X., Montine, K. S., Keene, C. D., and Montine, T. J. (2015). Different mechanisms of apolipoprotein E isoform-dependent modulation of prostaglandin E(2) production and triggering receptor expressed on myeloid cells 2 (TREM2) expression after innate immune activation of microglia. FASEB J. 29, 1754–1762. doi: 10.1096/fj.14-262683
Lima, T. A., Adler, A. L., Minett, T., Matthews, F. E., Brayne, C., Marioni, R. E., et al. (2014). C-reactive protein, APOE genotype and longitudinal cognitive change in an older population. Age Ageing 43, 289–292. doi: 10.1093/ageing/aft193
Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, H.-B., Loo, K. K., Palaszynski, K., Ashouri, J., Lubahn, D. B., and Voskuhl, R. R. (2003). Estrogen receptor α mediates estrogen’s immune protection in autoimmune disease. J. Immunol. 171, 6936–6940. doi: 10.4049/jimmunol.171.12.6936
Liu, M., Bian, C., Zhang, J., and Wen, F. (2014). Apolipoprotein E gene polymorphism and Alzheimer’s disease in Chinese population: a meta-analysis. Sci. Rep. 4:4383. doi: 10.1038/srep04383
Liu, S., Breitbart, A., Sun, Y., Mehta, P. D., Boutajangout, A., Scholtzova, H., et al. (2014). Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid beta and tau pathology. J. Neurochem. 128, 577–591. doi: 10.1111/jnc.12484
Maki, P. M. (2013). Critical window hypothesis of hormone therapy and cognition: a scientific update on clinical studies. Menopause 20, 695–709. doi: 10.1097/GME.0b013e3182960cf8
Mandrekar-Colucci, S., Karlo, J. C., and Landreth, G. E. (2012). Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 32, 10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012
Manly, J. J., Merchant, C. A., Jacobs, D. M., Small, S. A., Bell, K., Ferin, M., et al. (2000). Endogenous estrogen levels and Alzheimer’s disease among postmenopausal women. Neurology 54, 833–837. doi: 10.1212/WNL.54.4.833
Manning, E. N., Barnes, J., Cash, D. M., Bartlett, J. W., Leung, K. K., Ourselin, S., et al. (2014). APOE ε4 is associated with disproportionate progressive hippocampal atrophy in AD. PLoS One 9:e97608. doi: 10.1371/journal.pone.0097608
Marz, W., Scharnagl, H., Hoffmann, M., Boehm, B., and Winkelmann, B. (2004). The apolipoprotein E polymorphism is associated with circulating C-reactive protein (the Ludwigshafen risk and cardiovascular health study). Eur. Heart J. 25, 2109–2119. doi: 10.1016/j.ehj.2004.08.024
Mattiace, L. A., Davies, P., and Dickson, D. W. (1990). Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. Am. J. Pathol. 136, 1101–1114.
McGeer, P. L., Itagaki, S., Boyes, B. E., and Mcgeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi: 10.1212/WNL.38.8.1285
McGeer, P. L., Itagaki, S., Tago, H., and Mcgeer, E. G. (1987). Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 79, 195–200. doi: 10.1016/0304-3940(87)90696-3
McGeer, P. L., Schulzer, M., and Mcgeer, E. G. (1996). Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 47, 425–432. doi: 10.1212/WNL.47.2.425
Metti, A. L., Yaffe, K., Boudreau, R. M., Simonsick, E. M., Carnahan, R. M., Satterfield, S., et al. (2014). Trajectories of inflammatory markers and cognitive decline over 10 years. Neurobiol. Aging 35, 2785–2790. doi: 10.1016/j.neurobiolaging.2014.05.030
Miller, Y. I., Choi, S. H., Wiesner, P., Fang, L., Harkewicz, R., Hartvigsen, K., et al. (2011). Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 108, 235–248. doi: 10.1161/CIRCRESAHA.110.223875
Miyata, M., and Smith, J. D. (1996). Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and [beta]-amyloid peptides. Nat. Genet. 14, 55–61. doi: 10.1038/ng0996-55
Morrison, J. H., Brinton, R. D., Schmidt, P. J., and Gore, A. C. (2006). Estrogen, menopause, and the aging brain: how basic neuroscience can inform hormone therapy in women. J. Neurosci. 26, 10332–10348. doi: 10.1523/JNEUROSCI.3369-06.2006
Mosconi, L., Berti, V., Guyara-Quinn, C., Mchugh, P., Petrongolo, G., Osorio, R. S., et al. (2017a). Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery. PLoS One 12:e0185926. doi: 10.1371/journal.pone.0185926
Mosconi, L., Berti, V., Quinn, C., Mchugh, P., Petrongolo, G., Varsavsky, I., et al. (2017b). Sex differences in Alzheimer risk: brain imaging of endocrine vs chronologic aging. Neurology 89, 1382–1390. doi: 10.1212/WNL.0000000000004425
Mulder, S. D., Nielsen, H. M., Blankenstein, M. A., Eikelenboom, P., and Veerhuis, R. (2014). Apolipoproteins E and J interfere with amyloid-beta uptake by primary human astrocytes and microglia in vitro. Glia 62, 493–503. doi: 10.1002/glia.22619
Muller, G. C., Gottlieb, M. G. V., Luz Correa, B., Filho, I. G., Moresco, R. N., and Bauer, M. E. (2015). The inverted CD4:CD8 ratio is associated with gender-related changes in oxidative stress during aging. Cell. Immunol. 296, 149–154. doi: 10.1016/j.cellimm.2015.05.006
Nelson, A. R., Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2016). Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 1862, 887–900. doi: 10.1016/j.bbadis.2015.12.016
Neu, S. C., Pa, J., Kukull, W., Beekly, D., Kuzma, A., Gangadharan, P., et al. (2017). Apolipoprotein E genotype and sex risk factors for alzheimer disease: a meta-analysis. JAMA Neurol. 74, 1178–1189. doi: 10.1001/jamaneurol.2017.2188
Ojo, J. O., Rezaie, P., Gabbott, P. L., and Stewart, M. G. (2015). Impact of age-related neuroglial cell responses on hippocampal deterioration. Front. Aging Neurosci. 7:57. doi: 10.3389/fnagi.2015.00057
Olgiati, P., Politis, A., Malitas, P., Albani, D., Dusi, S., Polito, L., et al. (2010). APOE epsilon-4 allele and cytokine production in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 25, 338–344. doi: 10.1002/gps.2344
Olsson, B., Hertze, J., Lautner, R., Zetterberg, H., Nagga, K., Hoglund, K., et al. (2013). Microglial markers are elevated in the prodromal phase of Alzheimer’s disease and vascular dementia. J. Alzheimers Dis. 33, 45–53. doi: 10.3233/JAD-2012-120787
Orre, M., Kamphuis, W., Osborn, L. M., Melief, J., Kooijman, L., Huitinga, I., et al. (2014). Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 35, 1–14. doi: 10.1016/j.neurobiolaging.2013.07.008
Oury, C. (2014). CD36: linking lipids to the NLRP3 inflammasome, atherogenesis and atherothrombosis. Cell. Mol. Immunol. 11, 8–10. doi: 10.1038/cmi.2013.48
Perry, V. H., and Teeling, J. (2013). Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 35, 601–612. doi: 10.1007/s00281-013-0382-8
Persad, C. C., Zubieta, J. K., Love, T., Wang, H., Tkaczyk, A., and Smith, Y. R. (2009). Enhanced neuroactivation during verbal memory processing in postmenopausal women receiving short-term hormone therapy. Fertil. Steril. 92, 197–204. doi: 10.1016/j.fertnstert.2008.04.040
Policicchio, S., Ahmad, A. N., Powell, J. F., and Proitsi, P. (2017). Rheumatoid arthritis and risk for Alzheimer’s disease: a systematic review and meta-analysis and a Mendelian Randomization study. Sci. Rep. 7:12861. doi: 10.1038/s41598-017-13168-8
Rajan, K. B., Wilson, R. S., Weuve, J., Barnes, L. L., and Evans, D. A. (2015). Cognitive impairment 18 years before clinical diagnosis of Alzheimer disease dementia. Neurology 85, 898–904. doi: 10.1212/WNL.0000000000001774
Reichwald, J., Danner, S., Wiederhold, K. H., and Staufenbiel, M. (2009). Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J. Neuroinflammation 6:35. doi: 10.1186/1742-2094-6-35
Rettberg, J. R., Yao, J., and Brinton, R. D. (2014). Estrogen: a master regulator of bioenergetic systems in the brain and body. Front. Neuroendocrinol. 35, 8–30. doi: 10.1016/j.yfrne.2013.08.001
Ritzel, R. M., Patel, A. R., Pan, S., Crapser, J., Hammond, M., Jellison, E., et al. (2015). Age- and location-related changes in microglial function. Neurobiol. Aging 36, 2153–2163. doi: 10.1016/j.neurobiolaging.2015.02.016
Rocca, W. A., Grossardt, B. R., and Maraganore, D. M. (2008). The long-term effects of oophorectomy on cognitive and motor aging are age dependent. Neurodegener. Dis. 5, 257–260. doi: 10.1159/000113718
Safaiyan, S., Kannaiyan, N., Snaidero, N., Brioschi, S., Biber, K., Yona, S., et al. (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998. doi: 10.1038/nn.4325
Salminen, A., Kaarniranta, K., and Kauppinen, A. (2012). Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging 4, 166–175. doi: 10.18632/aging.100444
Sárvári, M., Hrabovszky, E., Kalló, I., Solymosi, N., Likó, I., Berchtold, N., et al. (2012). Menopause leads to elevated expression of macrophage-associated genes in the aging frontal cortex: rat and human studies identify strikingly similar changes. J. Neuroinflammation 9:264. doi: 10.1186/1742-2094-9-264
Sárvári, M., Hrabovszky, E., Kalló, I., Solymosi, N., Tóth, K., Likó, I., et al. (2011). Estrogens regulate neuroinflammatory genes via estrogen receptors α and β in the frontal cortex of middle-aged female rats. J. Neuroinflammation 8:82. doi: 10.1186/1742-2094-8-82
Sarvari, M., Kallo, I., Hrabovszky, E., Solymosi, N., Rodolosse, A., Vastagh, C., et al. (2015). Hippocampal gene expression is highly responsive to estradiol replacement in middle-aged female rats. Endocrinology 156, 2632–2645. doi: 10.1210/en.2015-1109
Scacchi, R., De Bernardini, L., Mantuano, E., Donini, L. M., Vilardo, T., and Corbo, R. M. (1995). Apolipoprotein E (APOE) allele frequencies in late-onset sporadic Alzheimer’s disease (AD), mixed dementia and vascular dementia: lack of association of ε4 allele with AD in Italian octogenarian patients. Neurosci. Lett. 201, 231–234. doi: 10.1016/0304-3940(95)12190-0
Sharma, N., Sharma, R. K., Tewari, S., Chauhan, M., and Bhatia, A. (2018). Association of periodontal inflammation, systemic inflammation, and duration of menopausal years in postmenopausal women. Quintessence Int. 49, 123–131.
Shaw, A. C., Panda, A., Joshi, S. R., Qian, F., Allore, H. G., and Montgomery, R. R. (2011). Dysregulation of human Toll-like receptor function in aging. Ageing Res. Rev. 10, 346–353. doi: 10.1016/j.arr.2010.10.007
Sheedy, F. J., Grebe, A., Rayner, K. J., Kalantari, P., Ramkhelawon, B., Carpenter, S. B., et al. (2013). CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820. doi: 10.1038/ni.2639
Shumaker, S. A., Legault, C., Rapp, S. R., Thal, L., Wallace, R. B., Ockene, J. K., et al. (2003). Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 289, 2651–2662. doi: 10.1001/jama.289.20.2651
Silverstein, R. L., and Febbraio, M. (2009). CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2:re3. doi: 10.1126/scisignal.272re3
Sima, A., Iordan, A., and Stancu, C. (2007). Apolipoprotein E polymorphism–a risk factor for metabolic syndrome. Clin. Chem. Lab. Med. 45, 1149–1153. doi: 10.1515/CCLM.2007.258
Slowik, A., and Beyer, C. (2015). Inflammasomes are neuroprotective targets for sex steroids. J. Steroid Biochem. Mol. Biol. 153, 135–143. doi: 10.1016/j.jsbmb.2015.02.013
Smith, A. M., and Dragunow, M. (2014). The human side of microglia. Trends Neurosci. 37, 125–135. doi: 10.1016/j.tins.2013.12.001
Smith, R. G., Betancourt, L., and Sun, Y. (2005). Molecular endocrinology and physiology of the aging central nervous system. Endocr. Rev. 26, 203–250. doi: 10.1210/er.2002-0017
Spence, R. D., Hamby, M. E., Umeda, E., Itoh, N., Du, S., Wisdom, A. J., et al. (2011). Neuroprotection mediated through estrogen receptor-α in astrocytes. Proc. Natl. Acad. Sci. U.S.A. 108, 8867–8872. doi: 10.1073/pnas.1103833108
Stanley, L. C., Mrak, R. E., Woody, R. C., Perrot, L. J., Zhang, S., Marshak, D. R., et al. (1994). Glial cytokines as neuropathogenic factors in HIV infection: pathogenic similarities to Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 53, 231–238. doi: 10.1097/00005072-199405000-00003
Streit, W. J., Mrak, R. E., and Griffin, W. S. (2004). Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 1:14. doi: 10.1186/1742-2094-1-14
Streit, W. J., and Xue, Q. S. (2014). Human CNS immune senescence and neurodegeneration. Curr. Opin. Immunol. 29, 93–96. doi: 10.1016/j.coi.2014.05.005
Suenaga, J., Hu, X., Pu, H., Shi, Y., Hassan, S. H., Xu, M., et al. (2015). White matter injury and microglia/macrophage polarization are strongly linked with age-related long-term deficits in neurological function after stroke. Exp. Neurol. 272, 109–119. doi: 10.1016/j.expneurol.2015.03.021
Tai, L. M., Ghura, S., Koster, K. P., Liakaite, V., Maienschein-Cline, M., Kanabar, P., et al. (2015). APOE-modulated Abeta-induced neuroinflammation in Alzheimer’s disease: current landscape, novel data, and future perspective. J. Neurochem. 133, 465–488. doi: 10.1111/jnc.13072
Tai, L. M., Koster, K. P., Luo, J., Lee, S. H., Wang, Y. T., Collins, N. C., et al. (2014). Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 289, 30538–30555. doi: 10.1074/jbc.M114.600833
Tang, M. -X., Jacobs, D., Stern, Y., Marder, K., Schofield, P., Gurland, B., et al. (1996). Effect of oestrogen during menopause on risk and age at onset of Alzheimer’s disease. Lancet 348, 429–432. doi: 10.1016/S0140-6736(96)03356-9
Tang, Y., Purkayastha, S., and Cai, D. (2015). Hypothalamic microinflammation: a common basis of metabolic syndrome and aging. Trends Neurosci. 38, 36–44. doi: 10.1016/j.tins.2014.10.002
Tapia-Gonzalez, S., Carrero, P., Pernia, O., Garcia-Segura, L. M., and Diz-Chaves, Y. (2008). Selective oestrogen receptor (ER) modulators reduce microglia reactivity in vivo after peripheral inflammation: potential role of microglial ERs. J. Endocrinol. 198, 219–230. doi: 10.1677/JOE-07-0294
Terwel, D., Steffensen, K. R., Verghese, P. B., Kummer, M. P., Gustafsson, J. A., Holtzman, D. M., et al. (2011). Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J. Neurosci. 31, 7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011
Thal, L. J., Ferris, S. H., Kirby, L., Block, G. A., Lines, C. R., Yuen, E., et al. (2005). A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 30, 1204–1215. doi: 10.1038/sj.npp.1300690
Toms, T. E., Smith, J. P., Panoulas, V. F., Blackmore, H., Douglas, K. M., and Kitas, G. D. (2012). Apolipoprotein E gene polymorphisms are strong predictors of inflammation and dyslipidemia in rheumatoid arthritis. J. Rheumatol. 39, 218–225. doi: 10.3899/jrheum.110683
Tutuncu, M., Tang, J., Zeid, N. A., Kale, N., Crusan, D. J., Atkinson, E. J., et al. (2013). Onset of progressive phase is an age dependent clinical milestone in multiple sclerosis. Mult. Scler. 19, 188–198. doi: 10.1177/1352458512451510
Ukkola, O., Kunnari, A., Jokela, M., Paivansalo, M., and Kesaniemi, Y. A. (2009). ApoE phenotype is associated with inflammatory markers in middle-aged subjects. Inflamm. Res. 58, 54–59. doi: 10.1007/s00011-008-8215-2
Ungar, L., Altmann, A., and Greicius, M. D. (2014). Apolipoprotein E, gender, and Alzheimer’s disease: an overlooked, but potent and promising interaction. Brain Imaging Behav. 8, 262–273. doi: 10.1007/s11682-013-9272-x
Uri-Belapolsky, S., Shaish, A., Eliyahu, E., Grossman, H., Levi, M., Chuderland, D., et al. (2014). Interleukin-1 deficiency prolongs ovarian lifespan in mice. Proc. Natl. Acad. Sci. U.S.A. 111, 12492–12497. doi: 10.1073/pnas.1323955111
Vadasz, Z., Haj, T., Kessel, A., and Toubi, E. (2013). Age-related autoimmunity. BMC Med. 11:94. doi: 10.1186/1741-7015-11-94
Van Den Heuvel, C., Thornton, E., and Vink, R. (2007). Traumatic brain injury and Alzheimer’s disease: a review. Prog. Brain Res. 161, 303–316. doi: 10.1016/S0079-6123(06)61021-2
Vandanmagsar, B., Youm, Y.-H., Ravussin, A., Galgani, J. E., Stadler, K., Mynatt, R. L., et al. (2011). The NALP3/NLRP3 inflammasome instigates obesity-induced autoinflammation and insulin resistance. Nat. Med. 17, 179–188. doi: 10.1038/nm.2279
VanGuilder, H. D., Bixler, G. V., Brucklacher, R. M., Farley, J. A., Yan, H., Warrington, J. P., et al. (2011). Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J. Neuroinflammation 8:138. doi: 10.1186/1742-2094-8-138
Vegeto, E., Benedusi, V., and Maggi, A. (2008). Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front. Neuroendocrinol. 29, 507–519. doi: 10.1016/j.yfrne.2008.04.001
Vitek, M. P., Brown, C. M., and Colton, C. A. (2009). APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360. doi: 10.1016/j.neurobiolaging.2007.11.014
Vlad, S. C., Miller, D. R., Kowall, N. W., and Felson, D. T. (2008). Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70, 1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63
von Bernhardi, R., Eugenin-Von Bernhardi, L., and Eugenin, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
Walsh, J. G., Muruve, D. A., and Power, C. (2014). Inflammasomes in the CNS. Nat. Rev. Neurosci. 15, 84–97. doi: 10.1038/nrn3638
Wilson, R. S., Leurgans, S. E., Boyle, P. A., and Bennett, D. A. (2011). Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch. Neurol. 68, 351–356. doi: 10.1001/archneurol.2011.31
Wirz, K. T., Keitel, S., Swaab, D. F., Verhaagen, J., and Bossers, K. (2014). Early molecular changes in Alzheimer disease: can we catch the disease in its presymptomatic phase? J. Alzheimers Dis. 38, 719–740. doi: 10.3233/JAD-130920
Wotton, C. J., and Goldacre, M. J. (2017). Associations between specific autoimmune diseases and subsequent dementia: retrospective record-linkage cohort study, UK. J. Epidemiol. Commun. Health 71, 576–583. doi: 10.1136/jech-2016-207809
Xiang, Z., Haroutunian, V., Ho, L., Purohit, D., and Pasinetti, G. M. (2006). Microglia activation in the brain as inflammatory biomarker of Alzheimer’s disease neuropathology and clinical dementia. Dis. Markers 22, 95–102. doi: 10.1155/2006/276239
Xie, F., Zhang, J. C., Fu, H., and Chen, J. (2013). Age-related decline of myelin proteins is highly correlated with activation of astrocytes and microglia in the rat CNS. Int. J. Mol. Med. 32, 1021–1028. doi: 10.3892/ijmm.2013.1486
Xu, J., and Ikezu, T. (2009). The comorbidity of HIV-associated neurocognitive disorders and Alzheimer’s disease: a foreseeable medical challenge in post-HAART Era. J. Neuroimmune Pharmacol. 4, 200–212. doi: 10.1007/s11481-008-9136-0
Yao, J., Hamilton, R. T., Cadenas, E., and Brinton, R. D. (2010). Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim. Biophys. Acta 1800, 1121–1126. doi: 10.1016/j.bbagen.2010.06.002
Yasui, T., Maegawa, M., Tomita, J., Miyatani, Y., Yamada, M., Uemura, H., et al. (2007). Changes in serum cytokine concentrations during the menopausal transition. Maturitas 56, 396–403. doi: 10.1016/j.maturitas.2006.11.002
Yin, F., Yao, J., Sancheti, H., Feng, T., Melcangi, R. C., Morgan, T. E., et al. (2015). The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol. Aging 36, 2282–2295. doi: 10.1016/j.neurobiolaging.2015.03.013
Youm, Y. H., Grant, R. W., Mccabe, L. R., Albarado, D. C., Nguyen, K. Y., Ravussin, A., et al. (2013). Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 18, 519–532. doi: 10.1016/j.cmet.2013.09.010
Youm, Y. H., Kanneganti, T. D., Vandanmagsar, B., Zhu, X., Ravussin, A., Adijiang, A., et al. (2012). The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 1, 56–68. doi: 10.1016/j.celrep.2011.11.005
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269. doi: 10.1038/nm.3804
Yun, Y. W., Kweon, S. S., Choi, J. S., Rhee, J. A., Lee, Y. H., Nam, H. S., et al. (2015). APOE polymorphism is associated with c-reactive protein levels but not with white blood cell count: dong-gu study and namwon study. J. Korean Med. Sci. 30, 860–865. doi: 10.3346/jkms.2015.30.7.860
Zandi, P. P., Carlson, M. C., Plassman, B. L., Welsh-Bohmer, K. A., Mayer, L. S., Steffens, D. C., et al. (2002). Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County Study. JAMA 288, 2123–2129. doi: 10.1001/jama.288.17.2123
Zhang, G., Li, J., Purkayastha, S., Tang, Y., Zhang, H., Yin, Y., et al. (2013). Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 497, 211–216. doi: 10.1038/nature12143
Zhao, J., Jiang, C. Q., Lam, T. H., Liu, B., Cheng, K. K., Kavikondala, S., et al. (2014). Genetically predicted 17beta-estradiol and systemic inflammation in women: a separate-sample Mendelian randomisation analysis in the Guangzhou Biobank Cohort Study. J. Epidemiol. Commun. Health 68, 780–785. doi: 10.1136/jech-2013-203451
Keywords: inflammation, Alzheimer’s disease, menopause, APOEε4, aging
Citation: Mishra A and Brinton RD (2018) Inflammation: Bridging Age, Menopause and APOEε4 Genotype to Alzheimer’s Disease. Front. Aging Neurosci. 10:312. doi: 10.3389/fnagi.2018.00312
Received: 08 August 2018; Accepted: 18 September 2018;
Published: 09 October 2018.
Edited by:
Anne Eckert, Universitäre Psychiatrische Kliniken Basel, SwitzerlandReviewed by:
Walter A. Rocca, Mayo Clinic, United StatesCopyright © 2018 Mishra and Brinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberta D. Brinton, cmJyaW50b25AZW1haWwuYXJpem9uYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.