 Pradoldej Sompol
Pradoldej Sompol Christopher M. Norris
Christopher M. Norris- 1Sanders-Brown Center on Aging, University of Kentucky College of Medicine, Lexington, KY, United States
- 2Department of Pharmacology and Nutritional Sciences, University of Kentucky College of Medicine, Lexington, KY, United States
Mounting evidence supports a fundamental role for Ca2+ dysregulation in astrocyte activation. Though the activated astrocyte phenotype is complex, cell-type targeting approaches have revealed a number of detrimental roles of activated astrocytes involving neuroinflammation, release of synaptotoxic factors and loss of glutamate regulation. Work from our lab and others has suggested that the Ca2+/calmodulin dependent protein phosphatase, calcineurin (CN), provides a critical link between Ca2+ dysregulation and the activated astrocyte phenotype. A proteolyzed, hyperactivated form of CN appears at high levels in activated astrocytes in both human tissue and rodent tissue around regions of amyloid and vascular pathology. Similar upregulation of the CN-dependent transcription factor nuclear factor of activated T cells (NFAT4) also appears in activated astrocytes in mouse models of Alzheimer’s disease (ADs) and traumatic brain injury (TBI). Major consequences of hyperactivated CN/NFAT4 signaling in astrocytes are neuroinflammation, synapse dysfunction and glutamate dysregulation/excitotoxicity, which will be covered in this review article.
Introduction
The central role of Ca2+ dysregulation in age-related memory deficits and neurodegenerative disease, proposed more than 30 years ago (Gibson and Peterson, 1987; Khachaturian, 1987; Landfield, 1987; Abdul et al., 2009), has been supported time and again by molecular, electrophysiological, biochemical and behavioral studies and is the subject of many excellent reviews (Alzheimer’s Association Calcium Hypothesis Workgroup, 2017; Frazier et al., 2017; Gibson and Thakkar, 2017; Pchitskaya et al., 2018). Neurons are often the focus of studies on Ca2+ dysregulation, and for good reason. Ca2+ signaling is an absolutely essential mechanism for both intra- and interneuronal communication. Moreover, disruption of any of the many neuronal Ca2+ regulatory checkpoints can lead to the structural deterioration of neurons and neuronal death, which are defining features of most neurodegenerative diseases. Nonetheless, it is becoming increasingly clear that Ca2+ dysregulation underlies altered function and viability of other non-neuronal cells during aging and disease, especially astrocytes. Several recent articles have provided comprehensive reviews of Ca2+ signaling mechanisms and Ca2+ dyregulation in astrocytes as a function of disease (Vardjan et al., 2017; Verkhratsky et al., 2017; Zorec et al., 2018). The following review will instead focus on the protein phosphatase calcineurin (CN) as an emerging mechanism for linking astrocytic Ca2+ dysregulation to neuroinflammation, glutamate dysregulation, amyloid pathology and synaptotoxicity. Particular emphasis will be placed on CN interactions with the nuclear factor of activated T cells (NFATs), though other CN-sensitive transcription factors such as nuclear factor κB (NFκB) and forkhead O3 (FOXO3) will also be considered.
Ca2+ Dysregulation in Activated Astrocytes
Astrocytes are abundant and versatile cells that play critical roles in brain metabolism, vascular regulation, interneuronal signaling and defense. Fundamental to many of these duties are Ca2+ ions, which are handled by a sophisticated network of plasma membrane channels, Ca2+ pumps, Ca2+ binding proteins and intracellular stores (for recent comprehensive reviews see; Rusakov, 2015; Bazargani and Attwell, 2016; Shigetomi et al., 2016; Guerra-Gomes et al., 2017). Together, these mechanisms, and others, coordinate dynamic Ca2+ responses (e.g., Ca2+ waves and sparks) that can be propagated within the confines of individual astrocytes and also across large astrocyte syncytia via interconnecting gap junction channels (De Bock et al., 2014; Zheng et al., 2015; Fujii et al., 2017). The recent application of three-dimensional multiphoton imaging to astrocyte Ca2+ transients has highlighted the complexity and heterogeneity of Ca2+ signaling within different astrocyte compartments (e.g., soma, processes and endfeet) and perhaps points to an approaching renaissance in our understanding of the role of astrocytes in brain function and disease. Astrocytic Ca2+ dysregulation appears to be indelibly linked to morphologic transformations (i.e., astrocyte “activation” or “reactivity”) characterized by hypertrophic somata and processes and upregulation of the intermediate filament protein, GFAP (Pekny and Nilsson, 2005; Sofroniew, 2009; Rodríguez-Arellano et al., 2016; Bindocci et al., 2017). Astrocyte activation is triggered by a diverse range of injurious stimuli and is frequently localized to regions of frank pathology (e.g., damaged blood vessels, necrotic tissues and protein aggregates). Along with activated microglia, activated astrocytes provide one of the best neuroanatomical hallmarks of neuroinflammation.
Immunohistochemical studies have revealed the upregulation of numerous Ca2+ signaling mediators in activated astrocytes including: Ca2+ related proteases (Shields et al., 1998, 2000; Feng et al., 2011), L-type voltage-sensitive Ca2+ channels (Xu et al., 2007, 2010; Willis et al., 2010; Daschil et al., 2013; Wang et al., 2015), transient receptor potential vanilloid channels (Shirakawa et al., 2010; Butenko et al., 2012), endoplasmic reticulum Ca2+-release channels and Ca2+ pumps (Grolla et al., 2013), Ca2+-dependent K+ channels (Yi et al., 2016), and Ca2+ binding proteins (McAdory et al., 1998). Most extracellular factors that promote robust astrocyte activation in vivo (e.g., cytokines, reactive oxygen species, protein aggregates, excitotoxins, …etc) also trigger Ca2+ dysregulation (e.g., elevated Ca2+ levels, augmented Ca2+ transients) in primary culture and brain slices (Sama and Norris, 2013). Similar functional indices of Ca2+ dysregulation have been noted in animal models of AD (Takano et al., 2007; Kuchibhotla et al., 2009; Delekate et al., 2014), brain edema (Thrane et al., 2011), stroke (Ding et al., 2009; Rakers and Petzold, 2017) and epilepsy (Ding et al., 2007; Tian et al., 2010). The relationship between Ca2+ dysregulation and astrocyte activation is very likely to be bi-directional in nature. Indeed, Ca2+ modulates the activity of numerous transcription factor pathways (Mellstrom et al., 2008), several of which (e.g., NFκB, JAK/STAT, FOX proteins, peroxisome proliferator-activated receptors (PPARs) and activator protein-1 (AP-1), among others) have been implicated in shaping gene expression programs involved in astrocyte activation (Perez-Nievas and Serrano-Pozo, 2018). So, once astrocytic Ca2+ dysregulation is set in motion by injurious and/or neuroinflammatory factors, there are multiple routes through which Ca2+ could maintain astrocytes in an activated state. Perhaps the most direct link between Ca2+ and the gene regulatory machinery in astrocytes (and most other cell types) is provided by NFAT transcription factors, which are directly activated by the Ca2+-dependent protein phosphatase, CN. Mounting evidence, discussed below, shows that CN/NFATs exhibit clear signs of hyperactivation, and/or increased expression, in subsets of activated astrocytes, while cell-specific targeting approaches suggest that CN/NFAT signaling drives or exacerbates multiple forms of neuropathology.
CN Dysregulation and Neurodegenerative Disease
CN is a highly abundant protein found throughout the brain, appearing at high levels in neurons and low levels in glia in healthy adult animals (Goto et al., 1986a,b; Polli et al., 1991; Kuno et al., 1992). Hyperactive CN signaling is observed in human postmortem brain tissue at early stages of cognitive decline associated with AD, ramping up in later disease stages in parallel with worsening amyloid pathology, neurofibrillary pathology and/or cognitive decline (Liu et al., 2005; Abdul et al., 2009; Wu et al., 2010; Mohmmad Abdul et al., 2011; Qian et al., 2011; Watanabe et al., 2015; Pleiss et al., 2016b). Other human neurodegenerative conditions associated with increased CN signaling include Parkinson’s disease (Caraveo et al., 2014), dementia with Lewy bodies (Martin et al., 2012; Caraveo et al., 2014) and vascular pathology (Pleiss et al., 2016b). Similar changes are often recapitulated to a significant degree in corresponding animal models of aging and neurodegeneration (Foster et al., 2001; Huang et al., 2005; Norris et al., 2005; Shioda et al., 2006; Reese et al., 2008; Mukherjee et al., 2010; Wu et al., 2010; D’Amelio et al., 2011; Martin et al., 2012; Rosenkranz et al., 2012; Furman et al., 2016; Sompol et al., 2017). Moreover, inhibition of CN signaling with the commercial immunosuppressant drugs, tacrolimus and cyclosporine, commonly imparts neuroprotection in experimental models of injury and disease (Kuchibhotla et al., 2008; Wu et al., 2010; Rozkalne et al., 2011; O’Donnell et al., 2016; Xiong et al., 2018), reduces neuroinflammation (Yoshiyama et al., 2007; Rojanathammanee et al., 2015; Fields et al., 2016; Manocha et al., 2017a; Shah et al., 2017), improves synapse function (Chen et al., 2002; Dineley et al., 2010; Cavallucci et al., 2013; Kim et al., 2015), inhibits cognitive loss (Taglialatela et al., 2009; Dineley et al., 2010; Kumar and Singh, 2017; Liu et al., 2017), and may even extend lifespan (Yoshiyama et al., 2007). Consistent with the animal literature, an epidemiological investigation found that daily tacrolimus use reduced the risk of dementia in kidney transplant patients relative to age-matched healthy individuals in the general population (Taglialatela et al., 2015).
The CN holoenzyme consists of a catalytic subunit and a Ca2+-binding regulatory subunit (Norris, 2014). The catalytic subunit contains a critical autoinhibitory domain (AID) and a calmodulin binding site. Ca2+/calmodulin binding to CN is the primary stimulus for driving maximal CN phosphatase activity (Figure 1). When cellular Ca2+ levels are low, the AID masks the catalytic core and maintains CN in an inactive state. Cooperative binding of Ca2+ to the CN regulatory subunit and to calmodulin lead to the rapid displacement of the AID and robust activation of CN. When Ca2+ levels fall, Ca2+/calmodulin rapidly dissociates from CN, reinstating inhibition by the AID. CN is highly sensitive to Ca2+, with a Kd to Ca2+-saturated calmodulin in the picomolar range (Quintana et al., 2005). Thus, even small perturbations in cellular Ca2+ can lead to hyperactivation of CN. Under these conditions, CN activity can still be normalized if Ca2+ levels recover. However, large surges in Ca2+ can trigger the calpain-mediated proteolytic removal or disruption of the CN AID. Without the AID, CN becomes partially (but permanently) uncoupled from local Ca2+ changes and exhibits constitutively high levels of activity (i.e., in the presence or absence of Ca2+). Appearance of the CN proteolytic fragment (ΔCN) is one of the most clear-cut indicators of hyperactive CN signaling (Figure 1). Many commercial CN antibodies (directed to the CN carboxyl terminus) do not detect ΔCN in Western blot applications, which may explain why earlier studies failed to observe elevated CN in neurodegenerative conditions like AD (Gong et al., 1993; Ladner et al., 1996; Lian et al., 2001). In contrast, more recent work (using N terminus antibodies) has found that ΔCN is increased in human AD tissue (Liu et al., 2005; Wu et al., 2010; Mohmmad Abdul et al., 2011; Watanabe et al., 2015), in parallel with calpain activation (Liu et al., 2005; Mohmmad Abdul et al., 2011). The ΔCN fragment has been reported in numerous other experimental models of brain injury and disease including traumatic brain injury (TBI), ischemia and glaucoma (Norris, 2014).
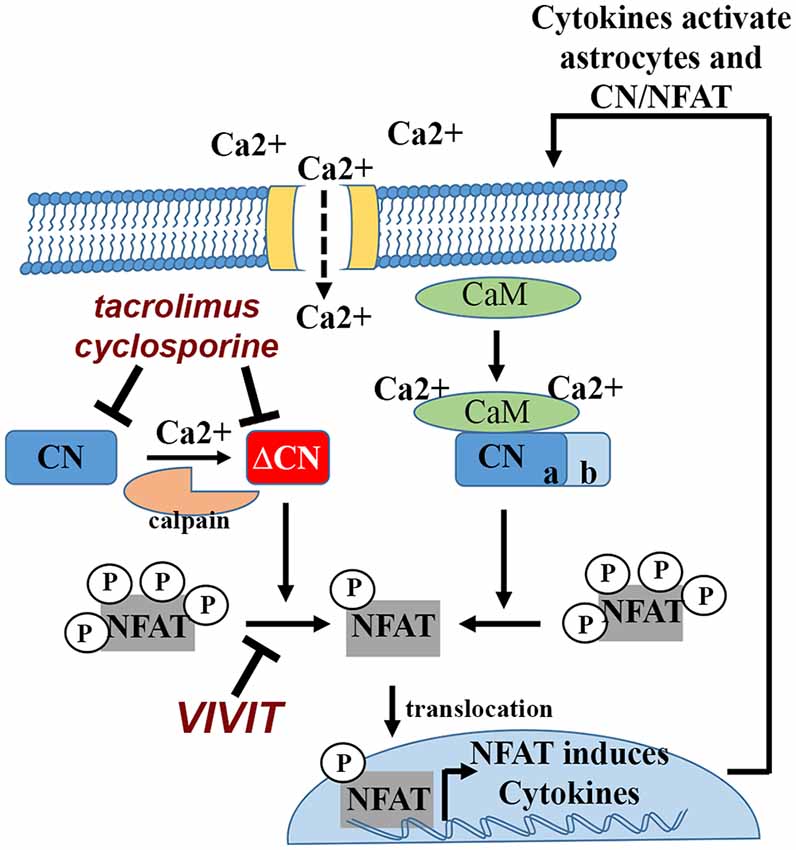
Figure 1. Calcineurin (CN)/nuclear factor of activated T cells (NFATs) signaling in astrocytes and bidirectional interactions with cytokines. Cytokines and other inflammatory factors lead to Ca2+ elevations in astrocytes. Ca2+ binds to calmodulin (CaM), which in turn, binds to and activates CN. CN dephosphorylates NFAT transcription factors, leading to nuclear translocation and induction of cytokine genes. CN activity can be inhibited using the commercially available immunosuppressants, tacrolimus and cyclosporine. Physical interactions between CN and NFATs can be blocked using peptide based reagents like VIVIT. Many cytokines that are induced by the CN/NFAT pathway can stimulate astrocytes in an autocrine or paracrine manner, triggering elevations in intracellular Ca2+, which can lead to further CN activation. Severe Ca2+ dysregulation can convert CN into a constitutively active proteolytic fragment (ΔCN) via calpain dependent proteolysis. Hyperactivation of CN/NFAT maintains chronic neuroinflammation (and astrocyte activation) through continued induction (i.e., a positive feedback loop) of pro-inflammatory cytokine genes.
CN Expression Is Increased in Activated Astrocytes in Humans and Animal Models
Cell-specific expression patterns of CN in both humans and animal models can exhibit striking changes characterized by intense upregulation in subsets of activated astrocytes (Hashimoto et al., 1998; Celsi et al., 2007; Abdul et al., 2009; Lim et al., 2013; Liu et al., 2015; Watanabe et al., 2015; Pleiss et al., 2016b; Sompol et al., 2017). Recent work using custom antibodies generated toward calpain-dependent proteolysis sites in the CN catalytic subunit, observed extensive labeling for a 45–48 kDa ΔCN proteolytic fragment in astrocytes and, to a seemingly lesser extent, neurons (Pleiss et al., 2016b). ΔCN was especially prominent in activated astrocytes bordering amyloid deposits and microinfarcts in human specimens (Pleiss et al., 2016b). Interestingly, ΔCN-positive and ΔCN-negative astrocytes were often found in the same regions (sometimes side-by-side) and appeared morphologically similar, highlighting the biochemical heterogeneity of activated astrocytes. In an aggressive mouse model of AD (i.e., 5xFAD mice), ΔCN was similarly observed in activated astrocytes in the hippocampus, increasing in direct proportion to elevated GFAP levels (Sompol et al., 2017). These observations are consistent with previous reports that found high levels of calpain activity in activated astrocytes (Shields et al., 1998, 2000; Feng et al., 2011) and suggest that Ca2+ dependent proteolysis of CN is a major outcome of astrocytic Ca2+ dysregulation.
NFATs
There are five primary NFAT family members: NFAT1 (or NFATp, NFATc2), NFAT2 (or NFATc, NFATc1), NFAT3 (or NFATc4), NFAT4 (or NFATc3) and NFAT5, all of which exhibit DNA-binding domains that are structurally similar to the Rel/NFκB family of transcription factors (Rao et al., 1997). Elevations in Ca2+ activate CN, which binds to and dephosphorylates NFATs 1–4 in the cytosol (Figure 1). NFAT5 is activated by osmotic stress and does not interact with CN. Dephosphorylation of NFATs exposes a nuclear localization signal, leading to transport into the nucleus and interaction with specific DNA binding elements. Similar to CN, NFAT activation is typically elevated under neurodegenerative conditions like AD (Abdul et al., 2009; Wu et al., 2010), Parkinson’s disease (Caraveo et al., 2014), and acute brain injury (Serrano-Pérez et al., 2011; Furman et al., 2016). As with other previously mentioned transcription factors (e.g., NFkB, JAK-STAT, AP-1,…etc), NFATs exert broad control over several transcriptional programs via the up- and downregulation of numerous genes, many of which involve cytokines and other classic inflammatory mediators (Im and Rao, 2004; Figure 1). NFATs are very strongly inhibited by the CN-inhibiting drugs tacrolimus and cyclosporine, but can be specifically targeted by a variety of peptide-based reagents. The VIVIT peptide, based on the endogenous CN docking sequence (PxIxIT) located in the N terminus of the regulatory region of NFATs 1–4, prevents CN from binding to and dephosphorylating NFATs (Aramburu et al., 1999). Thus, unlike commercial CN inhibitors, VIVIT impairs CN-mediated activation of NFATs without inhibiting CN activity per se, providing a powerful reagent for teasing apart NFAT-dependent signaling from the broader NFAT-independent actions of CN (Figure 1).
In peripheral tissues, NFATs play key roles in phenotype switching. Activation/anergy of T lymphocytes (Hogan, 2017), myotube formation and fiber-type commitment (Horsley and Pavlath, 2002; McCullagh et al., 2004; Rana et al., 2008), cardiomyocyte hypertrophy (Molkentin, 2004), vascular smooth muscle cell migration and proliferation (Liu et al., 2004; Karpurapu et al., 2010; Kundumani-Sridharan et al., 2013), and bone and joint remodeling (Sitara and Aliprantis, 2010) all depend critically on the NFAT pathway. Though not as extensively investigated in the CNS, several studies suggest that NFATs play a key role in the activation of astrocytes and microglia, as well (Nagamoto-Combs and Combs, 2010; Furman and Norris, 2014). All four CN-dependent NFATs have been identified in primary astrocytes at the mRNA and protein levels (Canellada et al., 2008). NFAT1 was found at higher levels in astrocyte nuclei in postmortem brain sections taken from human subjects with mild cognitive impairment (Abdul et al., 2009). NFAT1 has also been identified in microglia of AD mouse models (Manocha et al., 2017b). However, relative to all other NFAT isoforms, NFAT4 appears to show the greatest association with astrocytes in intact animals, with comparatively much less expression in neurons (Filosa et al., 2007; Serrano-Pérez et al., 2011; Neria et al., 2013; Caraveo et al., 2014; Yan et al., 2014; Furman et al., 2016; Sompol et al., 2017). While one study observed a reduction in NFAT4 protein levels in rats exposed to TBI (Yan et al., 2014), several other studies found that NFAT4 is strongly induced in activated astrocytes as a result of acute injury or progressive amyloid or synuclein pathology (Serrano-Pérez et al., 2011; Neria et al., 2013; Caraveo et al., 2014; Furman et al., 2016; Sompol et al., 2017).
Glial CN/NFAT Pathway and Neuroinflammatory Signaling
Similar to actions in lymphocytes, glial CN/NFAT activity appears to play a critical role in regulating immune/inflammatory responses. In primary astrocytes and microglia, the CN/NFAT pathway is robustly activated by many key inflammatory mediators, including cytokines, Aβ, glutamate and vascular injury-associated factors (Fernandez et al., 2007; Canellada et al., 2008; Pérez-Ortiz et al., 2008; Sama et al., 2008; Abdul et al., 2009; Furman et al., 2010; Nagamoto-Combs and Combs, 2010; Rojanathammanee et al., 2015). Little is known about the Ca2+ sources that are responsible for glial CN activation but L-type voltage sensitive Ca2+ channels have been specifically implicated in astrocytes (Canellada et al., 2008; Sama et al., 2008). Overexpression of the hyperactive ΔCN fragment in astrocytes leads to the upregulation of numerous immune/inflammatory related genes (Norris et al., 2005; Fernandez et al., 2007) and functional gene categories linked to the activated astrocyte phenotype (i.e., morphogenesis, cell adhesion and immune response; Norris et al., 2005). Interestingly, many of the genes identified in Norris et al. (2005) are part of the A1 “neurotoxic” astrocyte transcriptional signature described by the Barres lab (Zamanian et al., 2012; Liddelow et al., 2017). Of note, ΔCN triggered a two-to-three fold increase in the A1-associated complement component C3, found recently to drive microglia-mediated synapse loss in mouse models of AD (Hong et al., 2016; Shi et al., 2017). In addition to CN-activation studies, inhibitory approaches in primary cultures have revealed similar roles for CN/NFATs in neuroinflammation. Immune/inflammatory factors sensitive to CN/NFAT inhibition in glial cells include TNFα, GM-CSF, IL-6, CCL2 and Cox2, among others (Canellada et al., 2008; Sama et al., 2008; Nagamoto-Combs and Combs, 2010; Kim et al., 2011; Watanabe et al., 2015; Manocha et al., 2017b).
Bidirectional interactions between CN/NFAT and cytokine factors suggest that the CN/NFAT pathway is ideally suited to maintain positive feedback cycles underlying chronic neuroinflammation (Griffin et al., 1998; Figure 1). Consistent with this possibility, hyperactive CN/NFAT activity has been shown to propagate across local astrocyte networks through a paracrine signaling mechanism (Sama et al., 2008). A significant question remains about the mechanisms that keep these feedback cycles in check. One possibility is that CN/NFAT activity is limited by the expression of endogenous CN inhibitors. Regulator of CN 1 (RCAN1), for instance, is strongly induced by NFAT activity in multiple cell types including astrocytes (Canellada et al., 2008; Sobrado et al., 2012). RCANs are widely considered as CN inhibitors, though, it deserves noting that several studies have revealed permissive effects of RCAN on CN, depending on the presence of key accessory proteins (Liu et al., 2009). Whether RCANs provide a negative feedback mechanism for guarding against progressive Ca2+ dysregulation and neuroinflammation in astrocytes, in the context of neurodegeneration, will require further investigation.
Finally, caution should be taken when interpreting immune/inflammatory actions of CN/NFATs in primary glia which are very sensitive to culturing conditions. When investigated in serum-containing media, primary astrocytes may exhibit a quasi-activated state characterized by elevated basal levels of CN/NFAT activity (Furman et al., 2010). Indeed, addition of standard (10% fetal calf) serum alone induces robust CN/NFAT activity in primary astrocytes previously maintained in serum-free media (Furman et al., 2010). Moreover, treatment with IL1-β, IF-γ, or TNFα, which strongly induce NFAT activity in the absence of sera, elicited significantly muted responses when delivered in the presence of sera. Similar caution is also warranted in studies on intact animals, where the effects of CN/NFAT inhibition may have very different effects on glial activity and neuroinflammation, depending on the nature of the insult. For instance, intracerebroventricular delivery of the VIVIT peptide, or astrocyte-specific expression of VIVIT using adeno-associated virus (AAV), reduced signs of astrocyte and microglial activation in mouse models of AD characterized by progressive amyloid pathology (Abdul et al., 2010; Furman et al., 2012; Rojanathammanee et al., 2015; Sompol et al., 2017), but not in a rat model of TBI characterized by acute trauma (Furman et al., 2016). The reason for these discrepancies is unclear, but could involve CN/NFAT interactions with multiple other transcription factors and signaling pathways (as discussed further below). In any case, the results highlight the importance of context in understanding astrocytic CN/NFAT signaling.
Astrocytic CN/NFAT Pathway in Glutamate Dysregulation
Mounting evidence suggest that activated astrocytes may lose protective glutamate buffering properties in some forms of injury and disease. Astrocytes control extracellular glutamate levels, in part, through the use of several excitatory amino acid transporters (EAATs) located in the astrocyte plasmalemma. The EAAT2/GLT-1 protein is responsible for the bulk of glutamate uptake in several brain regions, including hippocampus (Robinson and Jackson, 2016). Loss of EAAT2 has been observed in several human neurodegenerative conditions including AD (Masliah et al., 1996; Abdul et al., 2009; Simpson et al., 2010), Alexander disease (Tian et al., 2010), epilepsy with hippocampal sclerosis (Mathern et al., 1999; Proper et al., 2002), and TBI (van Landeghem et al., 2006). Similar changes have been reported in corresponding animal models (Masliah et al., 2000; Mookherjee et al., 2011; Schallier et al., 2011; Hefendehl et al., 2016; Sompol et al., 2017). Functional knockdown of EAAT2/GLT-1 very typically causes synaptic hyperexcitability, altered synaptic plasticity, excitotoxicity and a variety of functional deficits depending on the brain region affected (Rothstein et al., 1996; Rao et al., 2001; Selkirk et al., 2005; Petr et al., 2015; Moidunny et al., 2016). In contrast, increased expression/function of EAAT2/GLT-1 provides strong neuroprotection from exogenously delivered excitotoxins as well as from acute and chronic CNS injury and disease (Harvey et al., 2011; Rozkalne et al., 2011; Zumkehr et al., 2015; Karklin Fontana et al., 2016).
The human EAAT2 promoter has putative binding sites for numerous transcription factors linked to neuroinflammation, including NFATs (Kim et al., 2003; Su et al., 2003; Mallolas et al., 2006), and is activated (and in some cases, inhibited) by a number of cytokine factors. Several studies suggest that the CN/NFAT pathway provides a putative link between Ca2+ dysregulation, neuroinflammation and glutamate dysregulation in activated astrocytes through modulation of EAAT/GLT-1 expression. Recent work found that overexpression of the ΔCN fragment significantly reduced EAAT-mediated glutamate uptake in primary astrocytes (Sompol et al., 2017). In contrast, inhibition of CN/NFAT activity with the VIVIT peptide protected EAAT2-GLT-1 protein levels and reduced extracellular glutamate and/or neuronal hyperexcitability in primary cultures following treatment with either IL1-β or oligomeric Aβ (Sama et al., 2008; Abdul et al., 2009). Under the same treatment conditions, significantly greater neuronal survival was observed when astrocytic CN/NFAT activity was inhibited with VIVIT. Similar effects were found following VIVIT treatment in an intact mouse model of AD (Sompol et al., 2017). Specifically, VIVIT increased protein levels of the astrocytic glutamate transporter, GLT-1, especially around Aβ deposits, and reduced the frequency and duration of spontaneous glutamate transients in intact 5xFAD mice. VIVIT also quelled hyperactive synaptic transients in in situ brain slices from 5xFAD mice and reduced the augmented NMDA receptor-mediated component of basal synaptic transmission. The reduction in glutamate hyperexcitability in 5xFAD mice was accompanied by the normalization of dendrite morphology and integrity, suggesting that astrocyte activation and astrocytic CN/NFAT signaling can drive excitotoxic damage in some disease states, like AD.
Astrocytic CN/NFAT Pathway in Amyloid Pathology
Amyloid pathology has long been recognized as a potent stimulus for CN and/or NFAT activity in multiple neural cell types (Agostinho et al., 2008; Reese et al., 2008; Abdul et al., 2009; Li et al., 2010; Wu et al., 2010, 2012; Mohmmad Abdul et al., 2011; Fang et al., 2016). Mice with parenchymal amyloid pathology show clear Ca2+ dysregulation in astrocytes: i.e., higher basal Ca2+ levels and bigger and more frequent Ca2+ transients (Kuchibhotla et al., 2009), providing a permissive environment for CN/NFAT activity. In human postmortem tissue, elevations in CN/NFAT activity increase in direct proportion with soluble Aβ levels, within the same subjects (Abdul et al., 2009). In primary neuron/astrocyte cultures, Aβ stimulates CN/NFAT activity and generates ΔCN proteolytic fragments (Mohmmad Abdul et al., 2011). Moreover, CN/ΔCN is found at especially high levels in activated astrocytes surrounding amyloid deposits in both mouse and human tissue (Norris et al., 2005; Celsi et al., 2007; Abdul et al., 2009; Jin et al., 2012; Lim et al., 2013; Watanabe et al., 2015; Pleiss et al., 2016b).
In addition to responding to Aβ, several studies have suggested that astrocytic CN/NFAT activity stimulates the generation of Aβ peptides (Hong et al., 2010; Furman et al., 2012; Jin et al., 2012; Sompol et al., 2017). Peripheral administration of the CN inhibitor, tacrolimus, to 8-month-old APP/PS1 transgenic mice over a period of 2 months led to a large (>75%) significant reduction in amyloid plaque burden in both the hippocampus and cortex (Hong et al., 2010). A smaller (20%–30%), but statistically significant decrease in amyloid plaque load and soluble Aβ peptide levels was also observed when CN/NFAT activity was specifically inhibited in hippocampal astrocytes of 2x and 5xAPP/PS1 mice using AAV-mediated delivery of VIVIT (Furman et al., 2012; Sompol et al., 2017). Though reductions in Aβ could have simply stemmed from the increased viability of neurons in tacrolimus/VIVIT treated mice, an additional report by Sompol et al. (2017) demonstrated that Ca2+ overload can lead to elevated Aβ production—specifically within astrocytes—through a CN/NFAT4-dependent mechanism. In this study, NFAT4 was shown to bind to the promoter of BACE1 (the rate limiting enzyme for Aβ generation) and induce BACE1 transcription. These results suggest that astrocytic CN/NFATs may help to drive parenchymal Aβ plaque pathology in AD. Given the intimate association between astrocytes and the cerebrovasculature, it would be interesting to determine if astrocytic CN/NFATs play a particularly important role in cerebral amyloid angiopathy.
Astrocytic CN/NFAT Pathway in Synapse Dysfunction
As discussed, commercial CN inhibitors are commonly associated with neuroprotective, anti-inflammatory and nootropic properties across a wide-range of experimental models of neural injury and disease. Within our lab, synaptoprotection has emerged as the single most consistent functional outcome of inhibiting CN/NFAT activity in astrocytes. To inhibit CN/NFATs, we have relied heavily on AAV vectors expressing the NFAT inhibitor, VIVIT, under the control of the human GFAP promoter (Gfa2). Delivery of AAVGfa2-VIVIT to the hippocampus of adult rodents results in widespread, astrocyte-selective transgene expression, coincident with the inhibition of NFAT4 nuclear translocation (Furman et al., 2012, 2016; Sompol et al., 2017). AAV-Gfa2-VIVIT improves basal hippocampal synaptic strength in double transgenic APP/PS1 transgenic mice (Furman et al., 2012), 5xFAD mice (Sompol et al., 2017), rats with TBI (Furman et al., 2016), and mice with hyperhomocysteinemia (HHcy)-associated vascular pathology (Pleiss et al., 2016a). In regards to synaptic plasticity, AAV-Gfa2-VIVIT improves long-term potentiation (LTP) in double transgenic APP/PS1 mice (Furman et al., 2012) and suppresses the induction of long-term depression in TBI rats (Furman et al., 2016). Investigations on LTP in HHcy mice have shown very similar outcomes. In contrast, hyperactivation of CN in astrocytes of otherwise healthy adult rats, using AAV-Gfa delivery of the ΔCN fragment, induces local deficits in CA3-CA1 synaptic strength (Pleiss et al., 2016b). Though not investigated in every study, we have also found that delivery of AAV-Gfa2-VIVIT to hippocampal astrocytes of AD mouse models improves hippocampal-dependent cognition (Furman et al., 2012; Sompol et al., 2017).
It is presently unclear how or why astrocytic CN/NFAT signaling negatively affects synapses. Many of the CN-dependent cytokines released from astrocytes are known to disrupt synaptic viability under certain conditions. In fact, several cytokine-inhibiting drugs appear to have remarkably similar effects to astrocyte-VIVIT treatment in AD mouse models (Kotilinek et al., 2008; Bachstetter et al., 2012; MacPherson et al., 2017). In addition, CN-dependent TGF-β release from astrocytes was recently found to suppress PSD-95 levels in nearby neurons (Tapella et al., 2018). In addition to cytokines, gene microarray studies in primary cells and protein measurements in TBI rats suggest that CN/NFATs drive the induction of factors involved in synapse turnover and/or remodeling, including complement cascade components (e.g., C3) and matricellular factors (e.g., SPARC and hevin; Norris et al., 2005; Furman et al., 2016). As mentioned, C3 was recently identified as a key component of the “neurotoxic” A1 activated astrocyte phenotype (Liddelow et al., 2017). During development, C3 release from astrocytes tags synapses for microglia-mediated phagocytosis, leading to synapse removal/remodeling (Stevens et al., 2007). C3 levels drop during maturation, but then reappear under pathological conditions, like AD (Eikelenboom and Veerhuis, 1996; Zabel and Kirsch, 2013). Recent work found that C3 upregulation in activated astrocytes in an APP/PS1 mouse model of AD guides microglia-mediated synapse loss, similar to that observed during development (Lian et al., 2015; Hong et al., 2016; Shi et al., 2017). In the Lian et al.’s (2015) study, C3 induction in astrocytes was attributable to the activation of NFκB (which can be activated by CN, see below), though a role for NFAT was not investigated. The matricellular proteins SPARC and hevin are also developmentally regulated factors that become induced in activated astrocytes in mature brain following injury and/or disease (Jones and Bouvier, 2014; Blakely et al., 2015; Furman et al., 2016). These factors regulate adhesion and de-adhesion of astrocytes with the extracellular matrix where they influence interactions with the vasculature, with other astrocytes, and also with neurons, especially at synapses, leading to synaptogenesis and re-modeling (Jones et al., 2011; Kucukdereli et al., 2011; Jones and Bouvier, 2014; Blakely et al., 2015). Hevin, a pro-synaptogenic factor, is very strongly induced in TBI rats treated with AAV-Gfa2-VIVIT, suggesting that activated astrocytes and hyperactive CN/NFAT signaling inhibit the formation of new synapses by suppressing hevin levels, at least in the context of acute neural injury (Furman et al., 2016). Finally, it seems likely that glutamate dysregulation and Aβ pathology play a significant and non-specific role in synapse dysfunction. Indeed, synapses are very sensitive to excitotoxic insults and circulating oligomeric Aβ peptide levels. By contributing to glutamate dysregulation and amyloid toxicity, activated astrocytes and hyperactive CN/NFAT signaling may simply promote an inhospitable working environment for synapses. Of course, all of these mechanisms could be working in concert as part of a broader neurotoxic astrocyte phenotype, with Ca2+ dysregulation and hyperactive CN/NFAT4 activity as central driving features (Figure 2).
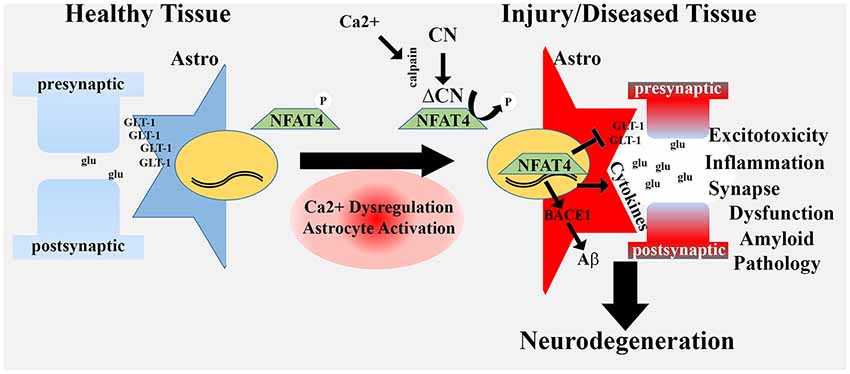
Figure 2. Hyperactivated CN/NFAT signaling in astrocytes may give rise to a neurotoxic astrocyte phenotype. In healthy tissue, astrocytes fine-tune synaptic communication and protect neuronal viability through numerous mechanisms, including uptake of excitotoxic glutamate (glu) at synapses, via GLT-1 transporters. During aging, injury and disease, many astrocytes exhibit an activated phenotype that includes Ca2+ dysregulation, proteolysis of CN to a high activity fragment (ΔCN) and induction of the NFAT4 isoform. Hyperactivation of NFAT4 leads to the downregulation of GLT-1, production and release of numerous pro-inflammatory cytokines, and induction of BACE1. These changes underlie a neurotoxic astrocyte phenotype associated with glutamate dysregulation/excitotoxicity, neuroinflammation, synapse dysfunction and amyloid pathology. Neurotoxic astrocytes contribute to or hasten neurodegenerative processes leading to dementia.
Non-NFAT Targets of CN in Astrocytes and Current Controversies
NFATs may be the most studied, but they are certainly not the only substrates for CN. In fact, CN has been shown to interact with most transcription factors involved in immune/inflammatory signaling. NFκB, for instance, is strongly regulated by CN activity, though in a fairly indirect manner. CN does not appear to physically bind to or dephosphorylate NFκB, but instead interacts with upstream targets that drive NFκB activation (Pons and Torres-Aleman, 2000; Frischbutter et al., 2011; Palkowitsch et al., 2011). CN-dependent activation of NFκB in astrocytes has been shown to modulate the expression of immune/inflammatory genes (Fernandez et al., 2007) and genes involved in Ca2+ signaling and homeostasis (e.g., mGluR5 type glutamate receptors and inositol triphosphate (IP3)-dependent Ca2+ release channels; Lim et al., 2013). IP3-receptors play an important role in regulating intracellular Ca2+ transients and waves in astrocytes (Filosa et al., 2004; Wu et al., 2017) and have been suggested to mediate neurotoxic actions of activated astrocytes in Alexander disease (Saito et al., 2018). CN/NFκB-dependent upregulation of mGluR5 and IP3 receptors occurs in direct response to pathogenic Aβ peptides and provides an intriguing CN-based mechanism for driving astrocytic Ca2+ dysregulation in AD mouse models (Kuchibhotla et al., 2009).
In addition to NFATs and NFκB, recent work suggests that CN can exert transcriptional control in astrocytes through novel interactions with the forkhead transcription factor, FOX03 (Fernandez et al., 2012, 2016). Proinflammatory cytokines, like TNFα, or Aβ peptides, stimulated the physical association between CN and FOX03, leading to dephosphorylation of FOXO3 and association with NFκB. The CN/FOX03/NFκB complex is thought to drive gene programs underlying deleterious neuro-immune/inflammatory signaling. Using an approach similar to the VIVIT strategy for inhibiting CN-NFAT interactions, Fernandez et al. (2016) developed a mimetic peptide that selectively disrupts CN-FOXO3 interactions. When delivered to primary astrocytes the CN-FOX03 interfering peptide reduced Aβ production and reduced the expression of pro-inflammatory cytokines. Interestingly, treatment of astrocytes with the neurotrophic factor, insulin like growth factor 1 (IGF-1), inhibited CN/FOX03/NFκB interactions and instead promoted the association of CN with NFκB and the peroxisome proliferator-activated receptor-γ (PPAR-γ). Formation of the CN/PPAR-γ/NFκB complex in astrocytes was associated with reduced amyloid pathology and improved cognitive function in an AD mouse model (Fernandez et al., 2012). These results suggest that CN activation in astrocytes can drive either deleterious or protective processes depending on which transcription factors are engaged. This work is consistent with other studies that find both beneficial and detrimental actions of activated astrocytes in disease models (Pekny and Pekna, 2014; Pekny et al., 2016). Moreover, there is good precedence for divergent actions of CN on gene expression programs in other non-neural cell types. For instance, CN activation in T lymphocytes can drive or inhibit expression of immune/inflammatory factors by interacting with different transcription factors in different T cell subtypes or in response to changing environmental conditions (Im and Rao, 2004; Wu et al., 2006).
Interestingly, overexpression of a ΔCN proteolytic fragment in astrocytes using a GFAP promoter was shown to have similar effects as IGF-1 stimulation, yielding beneficial effects in an AD mouse model, and in mice exposed to acute stab wound or LPS insult (Fernandez et al., 2007, 2012). The mechanisms of ΔCN’s beneficial effects are unclear. It is unknown whether ΔCN interacts with the PPARγ/NFκB complex, or if ΔCN opposes the interaction of NFκB with FOXO3, or if NFATs are involved in any of these pathways. The beneficial effects of ΔCN from the Fernandez et al. (2007, 2012) studies are especially unusual as this fragment is largely uncoupled from its normal mode of regulation (Ca2+/calmodulin) and is most commonly associated with cellular dysfunction and cell death in many different cell types, though there are some rare exceptions e.g., (Bousette et al., 2010). These results are also in apparent contrast to recent work showing that ΔCN expression in healthy rats drives (rather than prevents) local synapse dysfunction (Pleiss et al., 2016b). An alternative possibility for ΔCN-mediated neuroprotection in the Fernandez et al. (2007, 2012) studies may relate to an interaction between existing brain pathology and the over-expression system used (i.e., genetically modified ΔCN under the control of a GFAP promoter). Preexisting injury or amyloid pathology may be expected to strongly induce the GFAP promoter, leading to the intense upregulation of ΔCN in target cells, which could, possibly, lead to the death/deterioration of the most reactive and/or the most harmful astrocytes. Loss of harmful astrocytes may ultimately improve the viability of nearby neurons. Clearly, further research will be necessary to test this possibility.
Finally, CN is a versatile enzyme with numerous functions that are independent of transcriptional regulation. Nonetheless, very few non-transcription factor substrates of CN have been investigated in astrocytes. In most cases, CN’s interactions with other targets has been implied based on sensitivity to commercial CN inhibitors. For instance, tacrolimus and cyclosporine partially blocked the dephosphorylation of GFAP and vimentin in primary astrocytes and in brain slices from neonatal rat pups (Vinadé et al., 1997; Carvalho et al., 2016), suggesting that CN may regulate astrocyte morphology through a posttranscriptional mechanism. These results are reminiscent of studies in neurons, where CN has been long-known to regulate rapid cytoskeletal reorganization in dendritic spines and growth cones (Halpain et al., 1998; Wen et al., 2004). Given the dynamic nature of astrocyte processes and endfeet, it seems likely that CN would play a similar role in cytoskeletal reorganization in astrocytes. In addition to intermediate filaments, the astrocyte hemichannel protein, connexin 43, has also been revealed as a potential CN substrate (Li and Nagy, 2000; Tence et al., 2012). The cytoplasmic tail of connexin 43 is dephosphorylated in a tacrolimus/cyclosporine sensitive manner during hypoxic/ischemic insults. Interestingly, this dephosphorylation was associated with reduced gap junction coupling, which could have important implications for potassium and glutamate buffering during neural injury and disease. And, as with many other cell types, mitochondria function in astrocytes appears to be very sensitive to tacrolimus/cyclosporine (Kahraman et al., 2011; O’Donnell et al., 2016), though, it should be noted that cyclosporine can inhibit formation of the mitochondrial transition pore in a CN-independent manner (Halestrap et al., 1997). In one recent study, both tacrolimus and cyclosporine prevented the loss of mitochondria from astrocyte processes during hypoxic/ischemic insult (O’Donnell et al., 2016). However, it remains unclear how CN specifically contributed to this loss.
Summary
Mounting evidence suggests that the CN/NFAT pathway links astrocytic Ca2+ dysregulation to molecular and phenotypic changes involved with neuroinflammation, glutamate dysregulation, amyloid pathology and synapse dysfunction. We hypothesize that the increased expression and/or hyperactivation of CN/NFAT in activated astrocytes—found in human neurodegenerative disease and animal models of disease—plays a predominantly deleterious role in the brain, arising early in neurodegenerative diseases, like AD, and progressing as disease symptoms worsen (Figure 2). The numerous beneficial effects reported in disease models treated with CN and/or NFAT inhibitors is largely consistent with this hypothesis. These observations provide a very important extension and/or reconceptualization of the Ca2+ hypothesis of aging and disease to include glial Ca2+ dyshomeostasis and altered CN signaling as a critical component in the initiation and progression of neurodegeneration. Further work will be needed to tease apart the actions of CN on different transcriptional pathways and how these pathways interact to modulate neural function in healthy and diseased brain.
Author Contributions
PS and CN researched and wrote this manuscript.
Funding
This work was supported by National Institutes of Health Grants AG027297, AG051945 and AG056998 and a gift from the Hazel Embry Research Trust.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdul, H. M., Furman, J. L., Sama, M. A., Mathis, D. M., and Norris, C. M. (2010). NFATs and Alzheimer’s disease. Mol. Cell. Pharmacol. 2, 7–14. doi: 10.4255/mcpharmacol.10.02
Abdul, H. M., Sama, M. A., Furman, J. L., Mathis, D. M., Beckett, T. L., Weidner, A. M., et al. (2009). Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 29, 12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009
Agostinho, P., Lopes, J. P., Velez, Z., and Oliveira, C. R. (2008). Overactivation of calcineurin induced by amyloid-β and prion proteins. Neurochem. Int. 52, 1226–1233. doi: 10.1016/j.neuint.2008.01.005
Alzheimer’s Association Calcium Hypothesis Workgroup. (2017). Calcium Hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 13, 178.e17–182.e17. doi: 10.1016/j.jalz.2016.12.006
Aramburu, J., Yaffe, M. B., López-Rodríguez, C., Cantley, L. C., Hogan, P. G., and Rao, A. (1999). Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285, 2129–2133. doi: 10.1126/science.285.5436.2129
Bachstetter, A. D., Norris, C. M., Sompol, P., Wilcock, D. M., Goulding, D., Neltner, J. H., et al. (2012). Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer’s disease-related pathology. J. Neurosci. 32, 10201–10210. doi: 10.1523/JNEUROSCI.1496-12.2012
Bazargani, N., and Attwell, D. (2016). Astrocyte calcium signaling: the third wave. Nat. Neurosci. 19, 182–189. doi: 10.1038/nn.4201
Bindocci, E., Savtchouk, I., Liaudet, N., Becker, D., Carriero, G., and Volterra, A. (2017). Three-dimensional Ca2+ imaging advances understanding of astrocyte biology. Science 356:eaai8185. doi: 10.1126/science.aai8185
Blakely, P. K., Hussain, S., Carlin, L. E., and Irani, D. N. (2015). Astrocyte matricellular proteins that control excitatory synaptogenesis are regulated by inflammatory cytokines and correlate with paralysis severity during experimental autoimmune encephalomyelitis. Front. Neurosci. 9:344. doi: 10.3389/fnins.2015.00344
Bousette, N., Chugh, S., Fong, V., Isserlin, R., Kim, K. H., Volchuk, A., et al. (2010). Constitutively active calcineurin induces cardiac endoplasmic reticulum stress and protects against apoptosis that is mediated by α-crystallin-B. Proc. Natl. Acad. Sci. U S A 107, 18481–18486. doi: 10.1073/pnas.1013555107
Butenko, O., Dzamba, D., Benesova, J., Honsa, P., Benfenati, V., Rusnakova, V., et al. (2012). The increased activity of TRPV4 channel in the astrocytes of the adult rat hippocampus after cerebral hypoxia/ischemia. PLoS One 7:e39959. doi: 10.1371/journal.pone.0039959
Canellada, A., Ramirez, B. G., Minami, T., Redondo, J. M., and Cano, E. (2008). Calcium/calcineurin signaling in primary cortical astrocyte cultures: Rcan1–4 and cyclooxygenase-2 as NFAT target genes. Glia 56, 709–722. doi: 10.1002/glia.20647
Caraveo, G., Auluck, P. K., Whitesell, L., Chung, C. Y., Baru, V., Mosharov, E. V., et al. (2014). Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl. Acad. Sci. U S A 111, E3544–E3552. doi: 10.1073/pnas.1413201111
Carvalho, R. V., da Silva Ferreira, F., Heimfarth, L., Pierozan, P., Fernandes, C., and Pessoa-Pureur, R. (2016). Acute hyperammonemia induces NMDA-mediated hypophosphorylation of intermediate filaments through PP1 and PP2B in cerebral cortex of young rats. Neurotox. Res. 30, 138–149. doi: 10.1007/s12640-016-9607-7
Cavallucci, V., Berretta, N., Nobili, A., Nisticò, R., Mercuri, N. B., and D’Amelio, M. (2013). Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer’s disease. Neuromolecular Med. 15, 541–548. doi: 10.1007/s12017-013-8241-2
Celsi, F., Svedberg, M., Unger, C., Cotman, C. W., Carrì, M. T., Ottersen, O. P., et al. (2007). β-amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol. Dis. 26, 342–352. doi: 10.1016/j.nbd.2006.12.022
Chen, Q. S., Wei, W. Z., Shimahara, T., and Xie, C. W. (2002). Alzheimer amyloid β-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol. Learn. Mem. 77, 354–371. doi: 10.1006/nlme.2001.4034
D’Amelio, M., Cavallucci, V., Middei, S., Marchetti, C., Pacioni, S., Ferri, A., et al. (2011). Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 14, 69–76. doi: 10.1038/nn.2709
Daschil, N., Obermair, G. J., Flucher, B. E., Stefanova, N., Hutter-Paier, B., Windisch, M., et al. (2013). CaV1.2 calcium channel expression in reactive astrocytes is associated with the formation of amyloid-β plaques in an Alzheimer’s disease mouse model. J. Alzheimers Dis. 37, 439–451. doi: 10.3233/JAD-130560
De Bock, M., Decrock, E., Wang, N., Bol, M., Vinken, M., Bultynck, G., et al. (2014). The dual face of connexin-based astroglial Ca2+ communication: a key player in brain physiology and a prime target in pathology. Biochim. Biophys. Acta 1843, 2211–2232. doi: 10.1016/j.bbamcr.2014.04.016
Delekate, A., Füchtemeier, M., Schumacher, T., Ulbrich, C., Foddis, M., and Petzold, G. C. (2014). Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 5:5422. doi: 10.1038/ncomms6422
Dineley, K. T., Kayed, R., Neugebauer, V., Fu, Y., Zhang, W., Reese, L. C., et al. (2010). Amyloid-β oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J. Neurosci. Res. 88, 2923–2932. doi: 10.1002/jnr.22445
Ding, S., Fellin, T., Zhu, Y., Lee, S. Y., Auberson, Y. P., Meaney, D. F., et al. (2007). Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. 27, 10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007
Ding, S., Wang, T., Cui, W., and Haydon, P. G. (2009). Photothrombosis ischemia stimulates a sustained astrocytic Ca2+ signaling in vivo. Glia 57, 767–776. doi: 10.1002/glia.20804
Eikelenboom, P., and Veerhuis, R. (1996). The role of complement and activated microglia in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 17, 673–680. doi: 10.1016/s0197-4580(96)00108-x
Fang, M., Zhang, P., Zhao, Y., Jin, A., and Liu, X. (2016). Aβ mediates Sigma receptor degradation via CaN/NFAT pathway. Am. J. Transl. Res. 8, 3471–3481.
Feng, Z. H., Hao, J., Ye, L., Dayao, C., Yan, N., Yan, Y., et al. (2011). Overexpression of mu-calpain in the anterior temporal neocortex of patients with intractable epilepsy correlates with clinicopathological characteristics. Seizure 20, 395–401. doi: 10.1016/j.seizure.2011.01.010
Fernandez, A. M., Fernandez, S., Carrero, P., Garcia-Garcia, M., and Torres-Aleman, I. (2007). Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J. Neurosci. 27, 8745–8756. doi: 10.1523/JNEUROSCI.1002-07.2007
Fernandez, A. M., Hervas, R., Dominguez-Fraile, M., Garrido, V. N., Gomez-Gutierrez, P., Vega, M., et al. (2016). Blockade of the interaction of calcineurin with FOXO in astrocytes protects against amyloid-β-induced neuronal death. J. Alzheimers Dis. 52, 1471–1478. doi: 10.3233/jad-160149
Fernandez, A. M., Jimenez, S., Mecha, M., Dávila, D., Guaza, C., Vitorica, J., et al. (2012). Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer’s pathology. Mol. Psychiatry 17, 705–718. doi: 10.1038/mp.2011.128
Fields, J. A., Overk, C., Adame, A., Florio, J., Mante, M., Pineda, A., et al. (2016). Neuroprotective effects of the immunomodulatory drug FK506 in a model of HIV1-gp120 neurotoxicity. J. Neuroinflammation 13:120. doi: 10.1186/s12974-016-0585-8
Filosa, J. A., Bonev, A. D., and Nelson, M. T. (2004). Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ. Res. 95, e73–e81. doi: 10.1161/01.res.0000148636.60732.2e
Filosa, J. A., Nelson, M. T., and Gonzalez Bosc, L. V. (2007). Activity-dependent NFATc3 nuclear accumulation in pericytes from cortical parenchymal microvessels. Am. J. Physiol. Cell Physiol. 293, C1797–C1805. doi: 10.1152/ajpcell.00554.2006
Foster, T. C., Sharrow, K. M., Masse, J. R., Norris, C. M., and Kumar, A. (2001). Calcineurin links Ca2+ dysregulation with brain aging. J. Neurosci. 21, 4066–4073. doi: 10.1523/JNEUROSCI.21-11-04066.2001
Frazier, H. N., Maimaiti, S., Anderson, K. L., Brewer, L. D., Gant, J. C., Porter, N. M., et al. (2017). Calcium’s role as nuanced modulator of cellular physiology in the brain. Biochem. Biophys. Res. Commun. 483, 981–987. doi: 10.1016/j.bbrc.2016.08.105
Frischbutter, S., Gabriel, C., Bendfeldt, H., Radbruch, A., and Baumgrass, R. (2011). Dephosphorylation of Bcl-10 by calcineurin is essential for canonical NF-kappaB activation in Th cells. Eur. J. Immunol. 41, 2349–2357. doi: 10.1002/eji.201041052
Fujii, Y., Maekawa, S., and Morita, M. (2017). Astrocyte calcium waves propagate proximally by gap junction and distally by extracellular diffusion of ATP released from volume-regulated anion channels. Sci. Rep. 7:13115. doi: 10.1038/s41598-017-13243-0
Furman, J. L., and Norris, C. M. (2014). Calcineurin and glial signaling: neuroinflammation and beyond. J. Neuroinflammation 11:158. doi: 10.1186/s12974-014-0158-7
Furman, J. L., Artiushin, I. A., and Norris, C. M. (2010). Disparate effects of serum on basal and evoked NFAT activity in primary astrocyte cultures. Neurosci. Lett. 469, 365–369. doi: 10.1016/j.neulet.2009.12.029
Furman, J. L., Sama, D. M., Gant, J. C., Beckett, T. L., Murphy, M. P., Bachstetter, A. D., et al. (2012). Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 16129–16140. doi: 10.1523/JNEUROSCI.2323-12.2012
Furman, J. L., Sompol, P., Kraner, S. D., Pleiss, M. M., Putman, E. J., Dunkerson, J., et al. (2016). Blockade of astrocytic calcineurin/NFAT signaling helps to normalize hippocampal synaptic function and plasticity in a rat model of traumatic brain injury. J. Neurosci. 36, 1502–1515. doi: 10.1523/JNEUROSCI.1930-15.2016
Gibson, G. E., and Peterson, C. (1987). Calcium and the aging nervous system. Neurobiol. Aging 8, 329–343. doi: 10.1016/0197-4580(87)90077-7
Gibson, G. E., and Thakkar, A. (2017). Interactions of mitochondria/metabolism and calcium regulation in Alzheimer’s disease: a calcinist point of view. Neurochem. Res. 42, 1636–1648. doi: 10.1007/s11064-017-2182-3
Gong, C. X., Singh, T. J., Grundke-Iqbal, I., and Iqbal, K. (1993). Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 61, 921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x
Goto, S., Matsukado, Y., Mihara, Y., Inoue, N., and Miyamoto, E. (1986a). Calcineurin in human brain and its relation to extrapyramidal system. Immunohistochemical study on postmortem human brains. Acta Neuropathol. 72, 150–156. doi: 10.1007/bf00685977
Goto, S., Matsukado, Y., Mihara, Y., Inoue, N., and Miyamoto, E. (1986b). The distribution of calcineurin in rat brain by light and electron microscopic immunohistochemistry and enzyme-immunoassay. Brain Res. 397, 161–172. doi: 10.1016/0006-8993(86)91381-8
Griffin, W. S., Sheng, J. G., Royston, M. C., Gentleman, S. M., Mckenzie, J. E., Graham, D. I., et al. (1998). Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 8, 65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x
Grolla, A. A., Fakhfouri, G., Balzaretti, G., Marcello, E., Gardoni, F., Canonico, P. L., et al. (2013). Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cells. Neurobiol. Aging 34, 511–522. doi: 10.1016/j.neurobiolaging.2012.05.005
Guerra-Gomes, S., Sousa, N., Pinto, L., and Oliveira, J. F. (2017). Functional roles of astrocyte calcium elevations: from synapses to behavior. Front. Cell. Neurosci. 11:427. doi: 10.3389/fncel.2017.00427
Halestrap, A. P., Connern, C. P., Griffiths, E. J., and Kerr, P. M. (1997). Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell. Biochem. 174, 167–172. doi: 10.1007/978-1-4615-6111-8_25
Halpain, S., Hipolito, A., and Saffer, L. (1998). Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J. Neurosci. 18, 9835–9844. doi: 10.1523/JNEUROSCI.18-23-09835.1998
Harvey, B. K., Airavaara, M., Hinzman, J., Wires, E. M., Chiocco, M. J., Howard, D. B., et al. (2011). Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS One 6:e22135. doi: 10.1371/journal.pone.0022135
Hashimoto, T., Kawamata, T., Saito, N., Sasaki, M., Nakai, M., Niu, S., et al. (1998). Isoform-specific redistribution of calcineurin Aα and Aβ in the hippocampal CA1 region of gerbils after transient ischemia. J. Neurochem. 70, 1289–1298. doi: 10.1046/j.1471-4159.1998.70031289.x
Hefendehl, J. K., LeDue, J., Ko, R. W., Mahler, J., Murphy, T. H., and Macvicar, B. A. (2016). Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Aβ plaques by iGluSnFR two-photon imaging. Nat. Commun. 7:13441. doi: 10.1038/ncomms13441
Hogan, P. G. (2017). Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 63, 66–69. doi: 10.1016/j.ceca.2017.01.014
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hong, H. S., Hwang, J. Y., Son, S. M., Kim, Y. H., Moon, M., and Inhee, M. J. (2010). FK506 reduces amyloid plaque burden and induces MMP-9 in AβPP/PS1 double transgenic mice. J. Alzheimers Dis. 22, 97–105. doi: 10.3233/jad-2010-100261
Horsley, V., and Pavlath, G. K. (2002). NFAT: ubiquitous regulator of cell differentiation and adaptation. J. Cell Biol. 156, 771–774. doi: 10.1083/jcb.200111073
Huang, W., Fileta, J. B., Dobberfuhl, A., Filippopolous, T., Guo, Y., Kwon, G., et al. (2005). Calcineurin cleavage is triggered by elevated intraocular pressure and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma. Proc. Natl. Acad. Sci. U S A 102, 12242–12247. doi: 10.1073/pnas.0505138102
Im, S. H., and Rao, A. (2004). Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol. Cells 18, 1–9.
Jin, S. M., Cho, H. J., Kim, Y. W., Hwang, J. Y., and Mook-Jung, I. (2012). Aβ-induced Ca2+ influx regulates astrocytic BACE1 expression via calcineurin/NFAT4 signals. Biochem. Biophys. Res. Commun. 425, 649–655. doi: 10.1016/j.bbrc.2012.07.123
Jones, E. V., Bernardinelli, Y., Tse, Y. C., Chierzi, S., Wong, T. P., and Murai, K. K. (2011). Astrocytes control glutamate receptor levels at developing synapses through SPARC-β-integrin interactions. J. Neurosci. 31, 4154–4165. doi: 10.1523/JNEUROSCI.4757-10.2011
Jones, E. V., and Bouvier, D. S. (2014). Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural Plast. 2014:321209. doi: 10.1155/2014/321209
Kahraman, S., Bambrick, L. L., and Fiskum, G. (2011). Effects of FK506 and cyclosporin a on calcium ionophore-induced mitochondrial depolarization and cytosolic calcium in astrocytes and neurons. J. Neurosci. Res. 89, 1973–1978. doi: 10.1002/jnr.22709
Karklin Fontana, A. C., Fox, D. P., Zoubroulis, A., Valente Mortensen, O., and Raghupathi, R. (2016). Neuroprotective effects of the glutamate transporter activator (R)-(−)-5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) following traumatic brain injury in the adult rat. J. Neurotrauma 33, 1073–1083. doi: 10.1089/neu.2015.4079
Karpurapu, M., Wang, D., Van Quyen, D., Kim, T. K., Kundumani-Sridharan, V., Pulusani, S., et al. (2010). Cyclin D1 is a bona fide target gene of NFATc1 and is sufficient in the mediation of injury-induced vascular wall remodeling. J. Biol. Chem. 285, 3510–3523. doi: 10.1074/jbc.M109.063727
Khachaturian, Z. S. (1987). Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol. Aging 8, 345–346. doi: 10.1016/0197-4580(87)90073-x
Kim, S. Y., Chao, W., Choi, S. Y., and Volsky, D. J. (2003). Cloning and characterization of the 3′-untranslated region of the human excitatory amino acid transporter 2 transcript. J. Neurochem. 86, 1458–1467. doi: 10.1046/j.1471-4159.2003.01958.x
Kim, B., Jeong, H. K., Kim, J. H., Lee, S. Y., Jou, I., and Joe, E. H. (2011). Uridine 5′-diphosphate induces chemokine expression in microglia and astrocytes through activation of the P2Y6 receptor. J. Immunol. 186, 3701–3709. doi: 10.4049/jimmunol.1000212
Kim, S., Violette, C. J., and Ziff, E. B. (2015). Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 M146V mutant. Neurobiol. Aging 36, 3239–3246. doi: 10.1016/j.neurobiolaging.2015.09.007
Kotilinek, L. A., Westerman, M. A., Wang, Q., Panizzon, K., Lim, G. P., Simonyi, A., et al. (2008). Cyclooxygenase-2 inhibition improves amyloid-β-mediated suppression of memory and synaptic plasticity. Brain 131, 651–664. doi: 10.1093/brain/awn008
Kuchibhotla, K. V., Goldman, S. T., Lattarulo, C. R., Wu, H. Y., Hyman, B. T., and Bacskai, B. J. (2008). Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59, 214–225. doi: 10.1016/j.neuron.2008.06.008
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T., and Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215. doi: 10.1126/science.1169096
Kucukdereli, H., Allen, N. J., Lee, A. T., Feng, A., Ozlu, M. I., Conatser, L. M., et al. (2011). Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. U S A 108, E440–E449. doi: 10.1073/pnas.1104977108
Kumar, A., and Singh, N. (2017). Calcineurin inhibitors improve memory loss and neuropathological changes in mouse model of dementia. Pharmacol. Biochem. Behav. 153, 147–159. doi: 10.1016/j.pbb.2016.12.018
Kundumani-Sridharan, V., Singh, N. K., Kumar, S., Gadepalli, R., and Rao, G. N. (2013). Nuclear factor of activated T cells c1 mediates p21-activated kinase 1 activation in the modulation of chemokine-induced human aortic smooth muscle cell F-actin stress fiber formation, migration, and proliferation and injury-induced vascular wall remodeling. J. Biol. Chem. 288, 22150–22162. doi: 10.1074/jbc.M113.454082
Kuno, T., Mukai, H., Ito, A., Chang, C. D., Kishima, K., Saito, N., et al. (1992). Distinct cellular expression of calcineurin Aα and Aβ in rat brain. J. Neurochem. 58, 1643–1651. doi: 10.1111/j.1471-4159.1992.tb10036.x
Ladner, C. J., Czech, J., Maurice, J., Lorens, S. A., and Lee, J. M. (1996). Reduction of calcineurin enzymatic activity in Alzheimer’s disease: correlation with neuropathologic changes. J. Neuropathol. Exp. Neurol. 55, 924–931. doi: 10.1097/00005072-199608000-00008
Landfield, P. W. (1987). ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol. Aging 8, 346–347. doi: 10.1016/0197-4580(87)90074-1
Li, Q., Fang, J., Yang, M., Wu, D., Zhang, L., and Zhang, Y. (2010). Galantamine inhibits calpain-calcineurin signaling activated by β-amyloid in human neuroblastoma SH-SY5Y cells. Neurosci. Lett. 480, 173–177. doi: 10.1016/j.neulet.2010.06.005
Li, W. E., and Nagy, J. I. (2000). Connexin43 phosphorylation state and intercellular communication in cultured astrocytes following hypoxia and protein phosphatase inhibition. Eur. J. Neurosci. 12, 2644–2650. doi: 10.1046/j.1460-9568.2000.00162.x
Lian, Q., Ladner, C. J., Magnuson, D., and Lee, J. M. (2001). Selective changes of calcineurin (protein phosphatase 2B) activity in Alzheimer’s disease cerebral cortex. Exp. Neurol. 167, 158–165. doi: 10.1006/exnr.2000.7534
Lian, H., Yang, L., Cole, A., Sun, L., Chiang, A. C., Fowler, S. W., et al. (2015). NFκB-activated astroglial release of compement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85, 101–115. doi: 10.1016/j.neuron.2014.11.018
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lim, D., Iyer, A., Ronco, V., Grolla, A. A., Canonico, P. L., Aronica, E., et al. (2013). Amyloid β deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-κB. Glia 61, 1134–1145. doi: 10.1002/glia.22502
Liu, Q., Busby, J. C., and Molkentin, J. D. (2009). Interaction between TAK1-TAB1-TAB2 and RCAN1-calcineurin defines a signalling nodal control point. Nat. Cell Biol. 11, 154–161. doi: 10.1038/ncb1823
Liu, Z., Dronadula, N., and Rao, G. N. (2004). A novel role for nuclear factor of activated T cells in receptor tyrosine kinase and G protein-coupled receptor agonist-induced vascular smooth muscle cell motility. J. Biol. Chem. 279, 41218–41226. doi: 10.1074/jbc.M406917200
Liu, F., Grundke-Iqbal, I., Iqbal, K., Oda, Y., Tomizawa, K., and Gong, C. X. (2005). Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J. Biol. Chem. 280, 37755–37762. doi: 10.1074/jbc.M507475200
Liu, J., Li, X., Chen, L., Xue, P., Yang, Q., and Wang, A. (2015). Increased calcineurin expression after pilocarpine-induced status epilepticus is associated with brain focal edema and astrogliosis. Int. J. Neurosci. doi: 10.3109/00207454.2015.1045975 [Epub ahead of print].
Liu, J., Si, Z., Li, S., Huang, Z., He, Y., Zhang, T., et al. (2017). The calcineurin inhibitor fk506 prevents cognitive impairment by inhibiting reactive astrogliosis in pilocarpine-induced status epilepticus rats. Front. Cell. Neurosci. 11:428. doi: 10.3389/fncel.2017.00428
MacPherson, K. P., Sompol, P., Kannarkat, G. T., Chang, J., Sniffen, L., Wildner, M. E., et al. (2017). Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases β-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 102, 81–95. doi: 10.1016/j.nbd.2017.02.010
Mallolas, J., Hurtado, O., Castellanos, M., Blanco, M., Sobrino, T., Serena, J., et al. (2006). A polymorphism in the EAAT2 promoter is associated with higher glutamate concentrations and higher frequency of progressing stroke. J. Exp. Med. 203, 711–717. doi: 10.1084/jem.20051979
Manocha, G. D., Floden, A. M., Puig, K. L., Nagamoto-Combs, K., Scherzer, C. R., and Combs, C. K. (2017a). Defining the contribution of neuroinflammation to Parkinson’s disease in humanized immune system mice. Mol. Neurodegener. 12:17. doi: 10.1186/s13024-017-0158-z
Manocha, G. D., Ghatak, A., Puig, K. L., Kraner, S. D., Norris, C. M., and Combs, C. K. (2017b). NFATc2 modulates microglial activation in the AβPP/PS1 Mouse model of Alzheimer’s disease. J. Alzheimers Dis. 58, 775–787. doi: 10.3233/JAD-151203
Martin, Z. S., Neugebauer, V., Dineley, K. T., Kayed, R., Zhang, W., Reese, L. C., et al. (2012). α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases. J. Neurochem. 120, 440–452. doi: 10.1111/j.1471-4159.2011.07576.x
Masliah, E., Alford, M., DeTeresa, R., Mallory, M., and Hansen, L. (1996). Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 40, 759–766. doi: 10.1002/ana.410400512
Masliah, E., Alford, M., Mallory, M., Rockenstein, E., Moechars, D., and Van Leuven, F. (2000). Abnormal glutamate transport function in mutant amyloid precursor protein transgenic mice. Exp. Neurol. 163, 381–387. doi: 10.1006/exnr.2000.7386
Mathern, G. W., Mendoza, D., Lozada, A., Pretorius, J. K., Dehnes, Y., Danbolt, N. C., et al. (1999). Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 52, 453–472. doi: 10.1212/wnl.52.3.453
McAdory, B. S., Van Eldik, L. J., and Norden, J. J. (1998). S100B, a neurotropic protein that modulates neuronal protein phosphorylation, is upregulated during lesion-induced collateral sprouting and reactive synaptogenesis. Brain Res. 813, 211–217. doi: 10.1016/s0006-8993(98)01014-2
McCullagh, K. J., Calabria, E., Pallafacchina, G., Ciciliot, S., Serrano, A. L., Argentini, C., et al. (2004). NFAT is a nerve activity sensor in skeletal muscle and controls activity-dependent myosin switching. Proc. Natl. Acad. Sci. U S A 101, 10590–10595. doi: 10.1073/pnas.0308035101
Mellstrom, B., Savignac, M., Gomez-Villafuertes, R., and Naranjo, J. R. (2008). Ca2+-operated transcriptional networks: molecular mechanisms and in vivo models. Physiol. Rev. 88, 421–449. doi: 10.1152/physrev.00041.2005
Mohmmad Abdul, H., Baig, I., Levine, H. III., Guttmann, R. P., and Norris, C. M. (2011). Proteolysis of calcineurin is increased in human hippocampus during mild cognitive impairment and is stimulated by oligomeric Aβ in primary cell culture. Aging Cell 10, 103–113. doi: 10.1111/j.1474-9726.2010.00645.x
Moidunny, S., Matos, M., Wesseling, E., Banerjee, S., Volsky, D. J., Cunha, R. A., et al. (2016). Oncostatin M promotes excitotoxicity by inhibiting glutamate uptake in astrocytes: implications in HIV-associated neurotoxicity. J. Neuroinflammation 13:144. doi: 10.1186/s12974-016-0613-8
Molkentin, J. D. (2004). Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 63, 467–475. doi: 10.1016/j.cardiores.2004.01.021
Mookherjee, P., Green, P. S., Watson, G. S., Marques, M. A., Tanaka, K., Meeker, K. D., et al. (2011). GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J. Alzheimers Dis. 26, 447–455. doi: 10.3233/JAD-2011-110503
Mukherjee, A., Morales-Scheihing, D., Gonzalez-Romero, D., Green, K., Taglialatela, G., and Soto, C. (2010). Calcineurin inhibition at the clinical phase of prion disease reduces neurodegeneration, improves behavioral alterations and increases animal survival. PLoS Pathog. 6:e1001138. doi: 10.1371/journal.ppat.1001138
Nagamoto-Combs, K., and Combs, C. K. (2010). Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells). J. Neurosci. 30, 9641–9646. doi: 10.1523/JNEUROSCI.0828-10.2010
Neria, F., del Carmen Serrano-Perez, M., Velasco, P., Urso, K., Tranque, P., and Cano, E. (2013). NFATc3 promotes Ca2+-dependent MMP3 expression in astroglial cells. Glia 61, 1052–1066. doi: 10.1002/glia.22494
Norris, C. M. (2014). “Calpain interactions with the protein phosphatase calcineurin in neurodegeneration,” in Advances in Biochemistry in Health and Disease, (Vol. 8) eds N. Dhalla and S. Chakraborti (New York, NY: Springer), 17–45.
Norris, C. M., Kadish, I., Blalock, E. M., Chen, K. C., Thibault, V., Porter, N. M., et al. (2005). Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci. 25, 4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005
O’Donnell, J. C., Jackson, J. G., and Robinson, M. B. (2016). Transient oxygen/glucose deprivation causes a delayed loss of mitochondria and increases spontaneous calcium signaling in astrocytic processes. J. Neurosci. 36, 7109–7127. doi: 10.1523/JNEUROSCI.4518-15.2016
Palkowitsch, L., Marienfeld, U., Brunner, C., Eitelhuber, A., Krappmann, D., and Marienfeld, R. B. (2011). The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-κB activation. J. Biol. Chem. 286, 7522–7534. doi: 10.1074/jbc.M110.155895
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. doi: 10.1016/j.ceca.2017.06.008
Pekny, M., and Nilsson, M. (2005). Astrocyte activation and reactive gliosis. Glia 50, 427–434. doi: 10.1002/glia.20207
Pekny, M., and Pekna, M. (2014). Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol. Rev. 94, 1077–1098. doi: 10.1152/physrev.00041.2013
Pekny, M., Pekna, M., Messing, A., Steinhauser, C., Lee, J. M., Parpura, V., et al. (2016). Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345. doi: 10.1007/s00401-015-1513-1
Perez-Nievas, B. G., and Serrano-Pozo, A. (2018). Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 10:114. doi: 10.3389/fnagi.2018.00114
Pérez-Ortiz, J. M., Serrano-Pérez, M. C., Pastor, M. D., Martin, E. D., Calvo, S., Rincon, M., et al. (2008). Mechanical lesion activates newly identified NFATc1 in primary astrocytes: implication of ATP and purinergic receptors. Eur. J. Neurosci. 27, 2453–2465. doi: 10.1111/j.1460-9568.2008.06197.x
Petr, G. T., Sun, Y., Frederick, N. M., Zhou, Y., Dhamne, S. C., Hameed, M. Q., et al. (2015). Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J. Neurosci. 35, 5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015
Pleiss, M. M., Sompol, P., Artiushin, I. A., Kraner, S. D., Powell, D. K., Bakshi, V., et al. (2016a). Inhibition of astrocytic calcineurin/NFAT signaling in a mouse model of vascular cognitive impairment and dementia. Soc. Neurosci. Abs. 46:482.09.
Pleiss, M. M., Sompol, P., Kraner, S. D., Abdul, H. M., Furman, J. L., Guttmann, R. P., et al. (2016b). Calcineurin proteolysis in astrocytes: implications for impaired synaptic function. Biochim. Biophys. Acta 1862, 1521–1532. doi: 10.1016/j.bbadis.2016.05.007
Polli, J. W., Billingsley, M. L., and Kincaid, R. L. (1991). Expression of the calmodulin-dependent protein phosphatase, calcineurin, in rat brain: developmental patterns and the role of nigrostriatal innervation. Dev. Brain Res. 63, 105–119. doi: 10.1016/0165-3806(91)90071-p
Pons, S., and Torres-Aleman, I. (2000). Insulin-like growth factor-I stimulates dephosphorylation of IκB through the serine phosphatase calcineurin (protein phosphatase 2B). J. Biol. Chem. 275, 38620–38625. doi: 10.1074/jbc.M004531200
Proper, E. A., Hoogland, G., Kappen, S. M., Jansen, G. H., Rensen, M. G., Schrama, L. H., et al. (2002). Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125, 32–43. doi: 10.1093/brain/awf001
Qian, W., Yin, X., Hu, W., Shi, J., Gu, J., Grundke-Iqbal, I., et al. (2011). Activation of protein phosphatase 2B and hyperphosphorylation of Tau in Alzheimer’s disease. J. Alzheimers Dis. 23, 617–627. doi: 10.3233/JAD-2010-100987
Quintana, A. R., Wang, D., Forbes, J. E., and Waxham, M. N. (2005). Kinetics of calmodulin binding to calcineurin. Biochem. Biophys. Res. Commun. 334, 674–680. doi: 10.1016/j.bbrc.2005.06.152
Rakers, C., and Petzold, G. C. (2017). Astrocytic calcium release mediates peri-infarct depolarizations in a rodent stroke model. J. Clin. Invest. 127, 511–516. doi: 10.1172/JCI89354
Rana, Z. A., Gundersen, K., and Buonanno, A. (2008). Activity-dependent repression of muscle genes by NFAT. Proc. Natl. Acad. Sci. U S A 105, 5921–5926. doi: 10.1073/pnas.0801330105
Rao, V. L., Dogan, A., Bowen, K. K., Todd, K. G., and Dempsey, R. J. (2001). Antisense knockdown of the glial glutamate transporter GLT-1 exacerbates hippocampal neuronal damage following traumatic injury to rat brain. Eur. J. Neurosci. 13, 119–128. doi: 10.1111/j.1460-9568.2001.01367.x
Rao, A., Luo, C., and Hogan, P. G. (1997). Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15, 707–747. doi: 10.1146/annurev.immunol.15.1.707
Reese, L. C., Zhang, W., Dineley, K. T., Kayed, R., and Taglialatela, G. (2008). Selective induction of calcineurin activity and signaling by oligomeric amyloid β. Aging Cell 7, 824–835. doi: 10.1111/j.1474-9726.2008.00434.x
Robinson, M. B., and Jackson, J. G. (2016). Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem. Int. 98, 56–71. doi: 10.1016/j.neuint.2016.03.014
Rodríguez-Arellano, J. J., Parpura, V., Zorec, R., and Verkhratsky, A. (2016). Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 323, 170–182. doi: 10.1016/j.neuroscience.2015.01.007
Rojanathammanee, L., Floden, A. M., Manocha, G. D., and Combs, C. K. (2015). Attenuation of microglial activation in a mouse model of Alzheimer’s disease via NFAT inhibition. J. Neuroinflammation 12:42. doi: 10.1186/s12974-015-0255-2
Rosenkranz, K., May, C., Meier, C., and Marcus, K. (2012). Proteomic analysis of alterations induced by perinatal hypoxic-ischemic brain injury. J. Proteome Res. 11, 5794–5803. doi: 10.1021/pr3005869
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., et al. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686. doi: 10.1016/s0896-6273(00)80086-0
Rozkalne, A., Hyman, B. T., and Spires-Jones, T. L. (2011). Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol. Dis. 41, 650–654. doi: 10.1016/j.nbd.2010.11.014
Rusakov, D. A. (2015). Disentangling calcium-driven astrocyte physiology. Nat. Rev. Neurosci. 16, 226–233. doi: 10.1038/nrn3878
Saito, K., Shigetomi, E., Yasuda, R., Sato, R., Nakano, M., Tashiro, K., et al. (2018). Aberrant astrocyte Ca2+ signals “AxCa signals” exacerbate pathological alterations in an Alexander disease model. Glia 66, 1053–1067. doi: 10.1002/glia.23300
Sama, M. A., Mathis, D. M., Furman, J. L., Abdul, H. M., Artiushin, I. A., Kraner, S. D., et al. (2008). Interleukin-1β-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J. Biol. Chem. 283, 21953–21964. doi: 10.1074/jbc.M800148200
Sama, D. M., and Norris, C. M. (2013). Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 12, 982–995. doi: 10.1016/j.arr.2013.05.008
Schallier, A., Smolders, I., Van Dam, D., Loyens, E., De Deyn, P. P., Michotte, A., et al. (2011). Region- and age-specific changes in glutamate transport in the AβPP23 mouse model for Alzheimer’s disease. J. Alzheimers Dis. 24, 287–300. doi: 10.3233/JAD-2011-101005
Selkirk, J. V., Nottebaum, L. M., Vana, A. M., Verge, G. M., Mackay, K. B., Stiefel, T. H., et al. (2005). Role of the GLT-1 subtype of glutamate transporter in glutamate homeostasis: the GLT-1-preferring inhibitor WAY-855 produces marginal neurotoxicity in the rat hippocampus. Eur. J. Neurosci. 21, 3217–3228. doi: 10.1111/j.1460-9568.2005.04162.x
Serrano-Pérez, M. C., Martín, E. D., Vaquero, C. F., Azcoitia, I., Calvo, S., Cano, E., et al. (2011). Response of transcription factor NFATc3 to excitotoxic and traumatic brain insults: identification of a subpopulation of reactive astrocytes. Glia 59, 94–107. doi: 10.1002/glia.21079
Shah, S. Z. A., Zhao, D., Taglialatela, G., Khan, S. H., Hussain, T., Dong, H., et al. (2017). Early minocycline and late FK506 treatment improves survival and alleviates neuroinflammation, neurodegeneration, and behavioral deficits in prion-infected hamsters. Neurotherapeutics 14, 463–483. doi: 10.1007/s13311-016-0500-0
Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., et al. (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9:eaaf6295. doi: 10.1126/scitranslmed.aaf6295
Shields, D. C., Schaecher, K. E., Hogan, E. L., and Banik, N. L. (2000). Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J. Neurosci. Res. 61, 146–150. doi: 10.1002/1097-4547(20000715)61:2<146::aid-jnr5>3.3.co;2-3
Shields, D. C., Tyor, W. R., Deibler, G. E., Hogan, E. L., and Banik, N. L. (1998). Increased calpain expression in activated glial and inflammatory cells in experimental allergic encephalomyelitis. Proc. Natl. Acad. Sci. U S A 95, 5768–5772. doi: 10.1073/pnas.95.10.5768
Shigetomi, E., Patel, S., and Khakh, B. S. (2016). Probing the complexities of astrocyte calcium signaling. Trends Cell Biol. 26, 300–312. doi: 10.1016/j.tcb.2016.01.003
Shioda, N., Moriguchi, S., Shirasaki, Y., and Fukunaga, K. (2006). Generation of constitutively active calcineurin by calpain contributes to delayed neuronal death following mouse brain ischemia. J. Neurochem. 98, 310–320. doi: 10.1111/j.1471-4159.2006.03874.x
Shirakawa, H., Sakimoto, S., Nakao, K., Sugishita, A., Konno, M., Iida, S., et al. (2010). Transient receptor potential canonical 3 (TRPC3) mediates thrombin-induced astrocyte activation and upregulates its own expression in cortical astrocytes. J. Neurosci. 30, 13116–13129. doi: 10.1523/JNEUROSCI.1890-10.2010
Simpson, J. E., Ince, P. G., Lace, G., Forster, G., Shaw, P. J., Matthews, F., et al. (2010). Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 31, 578–590. doi: 10.1016/j.neurobiolaging.2008.05.015
Sitara, D., and Aliprantis, A. O. (2010). Transcriptional regulation of bone and joint remodeling by NFAT. Immunol. Rev. 233, 286–300. doi: 10.1111/j.0105-2896.2009.00849.x
Sobrado, M., Ramirez, B. G., Neria, F., Lizasoain, I., Arbones, M. L., Minami, T., et al. (2012). Regulator of calcineurin 1 (Rcan1) has a protective role in brain ischemia/reperfusion injury. J. Neuroinflammation 9:48. doi: 10.1186/1742-2094-9-48
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi: 10.1016/j.tins.2009.08.002
Sompol, P., Furman, J. L., Pleiss, M. M., Kraner, S. D., Artiushin, I. A., Batten, S. R., et al. (2017). Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in aβ-bearing mice. J. Neurosci. 37, 6132–6148. doi: 10.1523/JNEUROSCI.0877-17.2017
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Su, Z. Z., Leszczyniecka, M., Kang, D. C., Sarkar, D., Chao, W., Volsky, D. J., et al. (2003). Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc. Natl. Acad. Sci. U S A 100, 1955–1960. doi: 10.1073/pnas.0136555100
Taglialatela, G., Hogan, D., Zhang, W. R., and Dineley, K. T. (2009). Intermediate- and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav. Brain Res. 200, 95–99. doi: 10.1016/j.bbr.2008.12.034
Taglialatela, G., Rastellini, C., and Cicalese, L. (2015). Reduced incidence of dementia in solid organ transplant patients treated with calcineurin inhibitors. J. Alzheimers Dis. 47, 329–333. doi: 10.3233/JAD-150065
Takano, T., Han, X., Deane, R., Zlokovic, B., and Nedergaard, M. (2007). Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann. N Y Acad. Sci. 1097, 40–50. doi: 10.1196/annals.1379.004
Tapella, L., Cerruti, M., Biocotino, I., Stevano, A., Rocchio, F., Canonico, P. L., et al. (2018). TGF-β2 and TGF-β3 from cultured β-amyloid-treated or 3xTg-AD-derived astrocytes may mediate astrocyte-neuron communication. Eur. J. Neurosci. 47, 211–221. doi: 10.1111/ejn.13819
Tence, M., Ezan, P., Amigou, E., and Giaume, C. (2012). Increased interaction of connexin43 with zonula occludens-1 during inhibition of gap junctions by G protein-coupled receptor agonists. Cell Signal 24, 86–98. doi: 10.1016/j.cellsig.2011.08.006
Thrane, A. S., Rappold, P. M., Fujita, T., Torres, A., Bekar, L. K., Takano, T., et al. (2011). Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc. Natl. Acad. Sci. U S A 108, 846–851. doi: 10.1073/pnas.1015217108
Tian, R., Wu, X., Hagemann, T. L., Sosunov, A. A., Messing, A., McKhann, G. M., et al. (2010). Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J. Neuropathol. Exp. Neurol. 69, 335–345. doi: 10.1097/NEN.0b013e3181d3cb52
van Landeghem, F. K., Weiss, T., Oehmichen, M., and von Deimling, A. (2006). Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J. Neurotrauma 23, 1518–1528. doi: 10.1089/neu.2006.23.1518
Vardjan, N., Verkhratsky, A., and Zorec, R. (2017). Astrocytic pathological calcium homeostasis and impaired vesicle trafficking in neurodegeneration. Int. J. Mol. Sci. 18:E358. doi: 10.3390/ijms18020358
Verkhratsky, A., Rodríguez-Arellano, J. J., Parpura, V., and Zorec, R. (2017). Astroglial calcium signalling in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 483, 1005–1012. doi: 10.1016/j.bbrc.2016.08.088
Vinadé, L., Gonçalves, C. A., Wofchuk, S., Gottfried, C., and Rodnight, R. (1997). Evidence for a role for calcium ions in the dephosphorylation of glial fibrillary acidic protein (GFAP) in immature hippocampal slices and in astrocyte cultures from the rat. Dev. Brain Res. 104, 11–17. doi: 10.1016/s0165-3806(97)00114-4
Wang, F., Du, T., Liang, C., Verkhratsky, A., and Peng, L. (2015). Ammonium increases Ca2+ signalling and upregulates expression of Cav1.2 gene in astrocytes in primary cultures and in the in vivo brain. Acta Physiol. 214, 261–274. doi: 10.1111/apha.12500
Watanabe, K., Uemura, K., Asada, M., Maesako, M., Akiyama, H., Shimohama, S., et al. (2015). The participation of insulin-like growth factor-binding protein 3 released by astrocytes in the pathology of Alzheimer’s disease. Mol. Brain 8:82. doi: 10.1186/s13041-015-0174-2
Wen, Z., Guirland, C., Ming, G. L., and Zheng, J. Q. (2004). A CaMKII/calcineurin switch controls the direction of Ca2+-dependent growth cone guidance. Neuron 43, 835–846. doi: 10.1016/j.neuron.2004.08.037
Willis, M., Kaufmann, W. A., Wietzorrek, G., Hutter-Paier, B., Moosmang, S., Humpel, C., et al. (2010). L-type calcium channel CaV 1.2 in transgenic mice overexpressing human AβPP751 with the London (V717I) and Swedish (K670M/N671L) mutations. J. Alzheimers Dis. 20, 1167–1180. doi: 10.3233/JAD-2010-091117
Wu, Y., Borde, M., Heissmeyer, V., Feuerer, M., Lapan, A. D., Stroud, J. C., et al. (2006). FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126, 375–387. doi: 10.1016/j.cell.2006.05.042
Wu, D. C., Chen, R. Y., Cheng, T. C., Chiang, Y. C., Shen, M. L., Hsu, L. L., et al. (2017). Spreading depression promotes astrocytic calcium oscillations and enhances gliotransmission to hippocampal neurons. Cereb. Cortex doi: 10.1093/cercor/bhx192 [Epub ahead of print].
Wu, H. Y., Hudry, E., Hashimoto, T., Kuchibhotla, K., Rozkalne, A., Fan, Z., et al. (2010). Amyloid β induces the morphological neurodegenerative triad of spine loss, dendritic simplification and neuritic dystrophies through calcineurin activation. J. Neurosci. 30, 2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010
Wu, H. Y., Hudry, E., Hashimoto, T., Uemura, K., Fan, Z. Y., Berezovska, O., et al. (2012). Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-β revealed by a novel fluorescence resonance energy transfer assay. J. Neurosci. 32, 5298–5309. doi: 10.1523/JNEUROSCI.0227-12.2012
Xiong, T. Q., Chen, L. M., Tan, B. H., Guo, C. Y., Li, Y. N., Zhang, Y. F., et al. (2018). The effects of calcineurin inhibitor FK506 on actin cytoskeleton, neuronal survival and glial reactions after pilocarpine-induced status epilepticus in mice. Epilepsy Res. 140, 138–147. doi: 10.1016/j.eplepsyres.2018.01.007
Xu, J. H., Long, L., Tang, Y. C., Hu, H. T., and Tang, F. R. (2007). Cav1.2, Cav1.3, and Cav2.1 in the mouse hippocampus during and after pilocarpine-induced status epilepticus. Hippocampus 17, 235–251. doi: 10.1002/hipo.20263
Xu, J. H., Long, L., Wang, J., Tang, Y. C., Hu, H. T., Soong, T. W., et al. (2010). Nuclear localization of Cav2.2 and its distribution in the mouse central nervous system, and changes in the hippocampus during and after pilocarpine-induced status epilepticus. Neuropathol. Appl. Neurobiol. 36, 71–85. doi: 10.1111/j.1365-2990.2009.01044.x
Yan, H. Q., Shin, S. S., Ma, X., Li, Y., and Dixon, C. E. (2014). Differential effect of traumatic brain injury on the nuclear factor of activated T Cells C3 and C4 isoforms in the rat hippocampus. Brain Res. 1548, 63–72. doi: 10.1016/j.brainres.2013.12.028
Yi, M., Yu, P., Lu, Q., Geller, H. M., Yu, Z., and Chen, H. (2016). KCa3.1 constitutes a pharmacological target for astrogliosis associated with Alzheimer’s disease. Mol. Cell. Neurosci. 76, 21–32. doi: 10.1016/j.mcn.2016.08.008
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Zabel, M. K., and Kirsch, W. M. (2013). From development to dysfunction: microglia and the complement cascade in CNS homeostasis. Ageing Res. Rev. 12, 749–756. doi: 10.1016/j.arr.2013.02.001
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zheng, K., Bard, L., Reynolds, J. P., King, C., Jensen, T. P., Gourine, A. V., et al. (2015). Time-resolved imaging reveals heterogeneous landscapes of nanomolar Ca2+ in neurons and astroglia. Neuron 88, 277–288. doi: 10.1016/j.neuron.2015.09.043
Zorec, R., Parpura, V., and Verkhratsky, A. (2018). Astroglial vesicular network: evolutionary trends, physiology and pathophysiology. Acta Physiol. 222:e12915. doi: 10.1111/apha.12915
Zumkehr, J., Rodriguez-Ortiz, C. J., Cheng, D., Kieu, Z., Wai, T., Hawkins, C., et al. (2015). Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer’s disease. Neurobiol. Aging 36, 2260–2271. doi: 10.1016/j.neurobiolaging.2015.04.005
Keywords: Alzheimer’s disease, Ca2+, glia, dementia, astrocytes, neuroinflammation, synapse
Citation: Sompol P and Norris CM (2018) Ca2+, Astrocyte Activation and Calcineurin/NFAT Signaling in Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 10:199. doi: 10.3389/fnagi.2018.00199
Received: 03 April 2018; Accepted: 12 June 2018;
Published: 09 July 2018.
Edited by:
Ashok Kumar, University of Florida, United StatesReviewed by:
Giulio Taglialatela, University of Texas Medical Branch, United StatesCarole Escartin, UMR9199 Laboratoire de Maladies Neurodégénératives Mécanismes, Thérapies, Imagerie, France
Copyright © 2018 Sompol and Norris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher M. Norris, Y25vcnIyQHVreS5lZHU=