
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 13 July 2018
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 10 - 2018 | https://doi.org/10.3389/fnagi.2018.00194
 Alireza Majdi1
Alireza Majdi1 Saeed Sadigh-Eteghad1*
Saeed Sadigh-Eteghad1* Mahnaz Talebi1
Mahnaz Talebi1 Fereshteh Farajdokht1Marjan Erfani1
Fereshteh Farajdokht1Marjan Erfani1 Javad Mahmoudi1
Javad Mahmoudi1 Albert Gjedde1,2,3,4,5
Albert Gjedde1,2,3,4,5Here, we tested the claim that nicotine attenuates the signs of brain dysfunction in the model of brain aging induced by D-galactose (DGal) in mice. We administered nicotine at doses of 0.1, 0.5 and 1 mg/kg by the subcutaneous (s.c.) or at 0.1 mg/kg by the intranasal (i.n.) routes in mice that had received DGal at the dose of 500 mg/kg subcutaneous (s.c.) for 6 weeks. We assessed animal withdrawal signs as the number of presented somatic signs, thermal hyperalgesia, elevated plus maze (EPM) and open field tests. We evaluated spatial memory and recognition with Barnes maze and novel object recognition (NOR) tests. We tested brain tissue for reactive oxygen species (ROS), mitochondrial membrane potential, caspase-3, Bax, Bcl-2, cytochrome C, brain-derived neurotrophic factor and nerve growth factor levels. Nicotine administration in model groups (0.5 mg/kg s.c. and 0.1 mg/kg i.n. doses) significantly attenuated impairment of spatial and episodic memories in comparison to normal saline-received model group. These doses also reduced mito-oxidative damage as well as apoptosis and raised neurotrophic factors level in model groups in comparison to normal saline-received model group. The 1 mg/kg s.c. dose nicotine revealed withdrawal signs compared with the other nicotine-received groups. Nicotine at specific doses and routes has the potential to attenuate age-related cognitive impairment, mito-oxidative damage, and apoptosis. The doses raise neurotrophic factors without producing withdrawal signs.
In aging, the progressive loss of physiological integrity and the decline of functional capacity lead to a range of disabilities. The cardinal aspects of aging of the brain include cognitive impairment, anxiety and depression. Oxidative stress, mitochondrial damage, apoptosis, neurotrophic factor loss, and cholinergic system dysfunction are known to be associated with aging of the brain and age-related deficits of learning and memory (Paradies et al., 2011; Gleichmann et al., 2012; Guarente, 2014; Ali et al., 2015; Richter et al., 2017). Multiple strategies have been tested with the goal of attenuating dysfunction of the aging brain, including the use of neuroprotective agents (He et al., 2009), inhibition of neuroinflammatory processes, prevention of oxidative stress (Zhang et al., 2009; Lu et al., 2010b; Ali et al., 2015) and activation of cholinergic neurotransmission (Lu et al., 2010b).
Nicotine is a nAChR agonist and pharmacological chaperone that stimulates cholinergic activity in the brain (Jackson et al., 2010; Sadigh-Eteghad et al., 2015a). It has been shown that nicotine improves working memory, executive function, and cognitive performance, both in human and animals (Rushforth et al., 2011; Jansari et al., 2013; Vafaee et al., 2015; Majdi et al., 2017). Nicotine also reduces reactive oxygen species (ROS) generation by brain mitochondria and prevents oxidative stress in a dose-dependent manner (Cormier et al., 2003; Guan et al., 2003). Nicotine further protects neurons against mitochondrial apoptosis (Garrido et al., 2001). There is evidence that nicotine is neuroprotective and regulates neurotrophic factors in the brain and that it affects the development and maturation of neurons (Xiaoyu, 2015). Experimental and clinical studies both, have revealed that nicotine administartion renders pro-cognitive effects (Bontempi et al., 2003; Myers et al., 2008). This finding has implications in the treatment of disorders which primarily affect cognition including Alzheimer’s disease (AD; Levin et al., 2006).
Nonetheless, it is clear that nicotine may induce dependency and subsequent withdrawal symptoms in animals and humans that can be prevented by use of limited doses and selective routes of delivery to the brain (Matta et al., 2007). Intranasal (i.n.) drug delivery is one such alternative to conventional routes of administration to the brain. The delivery is non-invasive, bypasses blood-brain barrier (BBB), allowing the drug to target the olfactory region as the direct avenue from nose to the brain. In the case of nicotine, the intranasal route is an alternative choice for delivery of nicotine to the brain (Farzampour et al., 2016; Pourmemar et al., 2017).
Chronic administration of D-galactose (DGal) to animals has consequences that mimic the characteristics of the aging brain and the related learning and memory impairment in humans (Kumar et al., 2010; Lu et al., 2010a; Lan et al., 2012; Tsai and Yin, 2012; Salehpour et al., 2017), with extensive application of this animal model to the study of the aging brain (Wei et al., 2005; Hsieh et al., 2009; Lu et al., 2010b; Zhou et al., 2013; Ali et al., 2015; Sadigh-Eteghad et al., 2017). It has been reported that DGal administration could recapulate the changes that happen during accelerated aging (Song et al., 1999). Also, evidence suggests that prolonged DGal-injected animals resemble their 16- to 24-month-old senescent animals (Gong and Xu, 1991; Li et al., 1995; Zhang et al., 1996). These characteristics make DGal-induced aging model a valid tool to recapitulate human aging in rodent (Song et al., 1999).
Here, we tested the claim that chronic nicotine administration attenuates memory impairment, apoptosis, and oxidative damage by cholinergic receptor activation, and that it may raise neurotrophic factors, independently of its addictive potential in a mouse model of aging of the brain induced by DGal.
Seventy-two male BALB/c mice weighing 25–30 g were obtained from Tabriz University of Medical Sciences laboratory animal care center. Animals were socially housed in standard polypropylene cages (five in each cage) under the controlled condition of constant humidity and temperature on a 12 h light/12 h dark schedule before and through study with access to water and standard pellet food ad libitum.
All efforts were made to minimize animal suffering and the number of animals used. Also, all procedures were performed in accordance with the recommendations of the guide for the care and use of laboratory animals of the National Institutes of Health (NIH; Publication No. 85-23, revised 1985) and approved by the regional ethics committee of Tabriz University of Medical Sciences (IR.TBZMED.REC.1395.61).
The animals were randomly divided into six groups with 12 mice in each; in the control group, mice did not receive any injection or treatment. In the DGal-induced aging group, for modeling of brain aging, mice were injected with DGal (500 mg/kg s.c. for 6 weeks; Sigma-Aldrich, St. Louis, MO, USA). Separate groups of DGal-injected mice received either NS or nicotine (Santa Cruz Biotechnology, Santa Cruz, CA, USA) through either s.c. (0.1, 0.5 and 1 mg/kg) or i.n. (0.1 mg/kg) routes for 6 weeks. For i.n. administration of nicotine drops containing 5–6 μl were administered through nasal mucosa with alternation between left and right nares for 2 min to reach the total desired volume (Pourmemar et al., 2017). At the end of the treatment administration period, the behavioral tests and biochemical analyses were performed (Figure 1). All tests were performed by an experimenter that was unaware of the identity of experimentations. All solutions were freshly prepared on the day of experimentation by dissolving drugs in NS (0.9% NaCl). All injections had a volume of 8 ml/kg body weight.
Figure 1. The experimental design and timescale of the study.
Twenty-four hours after the administration of the last dose of treatments, the withdrawal signs of mice was assessed using the following tests (Damaj et al., 2003).
Mice were placed in the Plexiglas cages, and their behavior was recorded and controlled for somatic withdrawal signs including paw tremors, head shakes, writhing, retropulsion, scratching and Straub tail for 20 min.
To assess animals’ anxiety, elevated plus-maze (EPM) test was used. Briefly, EPM consisted of two open and two fenced arms that stretched from a central platform raised 60 cm from the ground. Mice were placed in the center of the maze, their behavior was recorded for 5 min and analyzed using EthoVision video tracking system, and the percentage of time spent in the open arms (%OAT) and entries to the open arms (%OAE) were extracted. After each animal removal, the apparatus was completely cleaned with a 70% ethanol to remove olfactory or intra-apparatus cues.
Mice locomotor activity and anxiety were evaluated using open field test. The animals were placed in a black Plexiglas open-field box measuring 33 cm × 33 cm × 20 cm and their behavior was recorded using EthoVision video tracking system for 20 min. The total distance moved considered as a locomotor activity, and arena periphery locomotion was measured as anxiety index.
The hotplate test was used to assess animals’ thermal hyperalgesia. The animal was placed in a cylindrical glass (width: 20 cm) on a hot plate apparatus, and the temperature maintained at 48°C. The reaction time for paw-licking or jumping was recorded as indices of thermal hyperalgesia.
Novel object recognition (NOR) test was used to assess mice recognition memory. It was performed according to the previously explained method (Pourmemar et al., 2017). Briefly, the test apparatus was a black Plexiglas open-field box measuring 33 cm × 33 cm × 20 cm. One day before the training session, mice were placed in the test room for 15 min for acclimation, and then each animal was transferred to the test box for a 10-min habituation session. On the training session, two identical objects (A and A’) were placed in the box and introduced to the mice. The objects used were common and different in shape and texture, however, were counterbalanced in complexity. Then, each animal was placed in the middle of the box and the total time spent to explore each object was recorded over 10 min. The mice then returned to their home cage. The next day after training session mice were reintroduced to the same task, but one of the familiar objects applied during the training session was replaced by a novel object (B) for a single retention session. The mice were considered to be exploring when the animal nose was toward the object (a distance of ≤2 cm), and there was rearing up against it. After each trial, the test box and objects were cleaned with a 70% ethanol to eliminate the presence of any olfactory cues. The cut-off time for exploration of two objects was 20 s over a period of 10 min. The recognition memory was measured by the time spent exploring of novel object. Data were acquired using a video camera that was fixed above the center of the task apparatus and analyzed using a video tracking program Etho Vision™ (Noldus, Netherlands).
The Barnes maze was used to assess spatial learning and memory in the animals (Sunyer et al., 2007). The maze consisted of a circular platform 100 cm in diameter raised 50 cm from the ground, with 20 holes (hole diameter: 5 cm) around the maze. An escape box (20 cm × 15 cm × 5 cm) was placed under the target hole. The test was carried out in a room with special spatial cues located on the walls and a buzzer (80 dB) as a negative stimulus.
The Barnes maze test included habituation, acquisition and probe sessions lasting 6 days. The first day of the test comprised habituation session. During this session, the animal was located in the center of the maze in a black cylindrical start chamber. After 10 s, the start chamber was lifted, the buzzer was switched on, and the animal was gently guided to the escape box, the buzzer was turned off, and the mice stayed there for 1 min.
The consequent days consisted of four acquisition trials per day, divided by a 3-min interval for 4 days. In the spatial acquisition session, the same condition repeated and animals were free to explore arena and find the escape box for 3 min. After entering the box, the buzzer was turned off, and the mice remained there for 1 min. The last day of the experiment had one probe trial session lasting 3 min. The Barnes maze arena without the escape box was used to assess mice reference memory.
After each session and trial, the entire maze and escape box were cleaned with 70% alcohol to remove olfactory cues. The time that took the mice to find the escape box (latency time) during the training sessions, time spent in the target quadrant, and correct to error time (time spent in the target hole/time spent in the error holes) during the probe session were evaluated using EthoVision™ software.
Twenty-four hours after the last behavioral test, mice were decapitated after deep anesthesia with ketamine (100 mg/kg) and xylazine (10 mg/kg). The animal brain except cerebellum was then extracted and transferred to the freezing beaker for further evaluations and biochemical assays. For mitochondria isolation, fresh brain samples placed in ice-cold isolation buffer containing 200-mM mannitol, 70-mM sucrose, 10-mM HEPES, and 2-mM EDTA, pH 7.5. Thereafter, the tissue was homogenized in ice-cold extraction buffer having 2 mg/mL albumin (10% w/v). The samples were centrifuged at 600 g in 4°C for 5 min. Then, the supernatant was transferred into another tube and centrifuged at 12,000 g in 4°C for 15 min. Finally, the pellet was resuspended in storage buffer containing 10-mM HEPES, pH 7.4, 250-mM sucrose, 1-mM ATP, 0.08-mM ADP, 5-mM sodium succinate, 2-mM K2HPO4 and 1-mM DTT. Protein level was determined by Bradford method (Bradford, 1976).
Mitochondrial membrane potentials (MMP) or Δψm changes were detected using JC-1 vital dye (Mitochondria Staining Kit; Sigma-Aldrich, St. Louis, MO, USA). Under normal condition, the concentration of dye in the mitochondrial matrix produces red fluorescent. Any situation during which the MMP is dissipated results in shifting from red to green fluorescence responsible for J-aggregates and JC-1 monomers respectively. Briefly according to the kit instructions, JC-1 stain (0.6 μM) was used. Fluorescence intensity in the stained samples was measured by the fluorimetric assay. The ratio of red (λex = 490 nm, λem = 590 nm) to green (λex = 488 nm, λem = 530 nm) fluorescence intensity was considered as the Δψm which was normalized to the sample proteins.
To determine the mitochondrial ROS level, the fluorescent vital dye dichlorohydro-fluorescein diacetate (DCFDA) was used (Novalija et al., 2003). This fluorescent probe (2-μM at 37°C for 20 min) is oxidized by mitochondrial ROS to Dichlorodihydrofluorescein (DCF), and the resulting fluorescence intensity was measured according to the DCF spectral characteristics (λex = 485 nm, λem = 530 nm). The resulting ROS level was expressed as fluorescence intensity which was normalized to the sample proteins.
Western blotting was performed using the previously explained method (Sadigh-Eteghad et al., 2015b). For total and cytosolic fractions preparations, 100 mg of fresh brain tissue was homogenized using ice-cold mitochondria isolation buffer [200 mM mannitol, 80 mM HEPES-KOH (pH 7.4), and the protease inhibitor cocktail]. Homogenates were centrifuged at 750× for 10 min at 4°C. After removing half of the supernatants that were used as total fractions, the rest of the supernatants were centrifuged at 12,000× g for 20 min at 4°C which was the mitochondria free fraction including cytosol.
Radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors plus total or cytosolic fractions were homogenized. The protein concentration of the sample was determined using the Bradford assay (Li et al., 2008). Electrophoresis was performed using 12.5% polyacrylamide gel, and the isolated proteins were transferred onto a polyvinylidene difluoride (PVDF; Roche, United Kingdom). These membranes were then incubated with the primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) including anti-caspase-3 (1:500, sc-7148), anti-Bcl-2 (1:500, sc-492), anti-Bax (1:500, sc-493), and anti-cytochrome C (1:500, sc-7159) antibodies. After three times of washing lasting 5 min each, the membranes were incubated with the horseradish peroxidase conjugated goat anti-rabbit IgG secondary antibody for 60 min (1:5000, sc-2004). The membranes were finally positioned in ECL prime western blotting detection reagent (Amersham, United Kingdom) and the resulting signals visualization was achieved using Kodak autoradiography film (Kodak, Rochester, NY, USA). Anti β-actin (1:500, sc-130656) antibody was applied for internal control of the procedure. The quantification of the signal intensity of each band was performed using ImageJ 1.62 software (National Institutes of Health, Bethesda, MD, USA) and was normalized to the matching internal control.
ELISA method was used to determine mouse BDNF (Elabscience Biotechnology, China) and brain NGF (Shanghai Crystal day Biotech, China) levels in the brain homogenate. All procedures and calculations were performed using a commercial kit according to the manufacturer’s instructions. Briefly, the tissues were minced to small pieces and rinsed in ice-cold PBS (0.01 M, pH = 7.4) to eliminate excess blood entirely. Tissue pieces were weighed and then homogenized in PBS with a glass homogenizer on ice. To further break the cells, the suspension was sonicated with an ultrasonic cell disrupter. The homogenates were then centrifuged for 5 min at 5000× g to get the supernatant.
Descriptive data were expressed as mean ± standard error of mean (SEM). Comparison of different groups was carried out by a one-way analysis of variance (ANOVA) followed by the post hoc Tukey test or two-way ANOVA when applicable. All analyses were performed using GraphPad Prism software (version 7 for Windows; GraphPad Software Inc., La Jolla, CA, USA). For all comparisons, p < 0.05 was considered to be significant.
One-way ANOVA showed a statistically significant difference between groups in the somatic signs presented by the animals (F(5,66) = 18.55, p < 0.001). Post hoc analysis revealed that spontaneous withdrawal of nicotine had not a significant impact on the number of somatic signs at the administered doses except for 1 mg/kg s.c. dose which significantly increased the number of somatic signs compared to other groups (p < 0.01; Figure 2A).
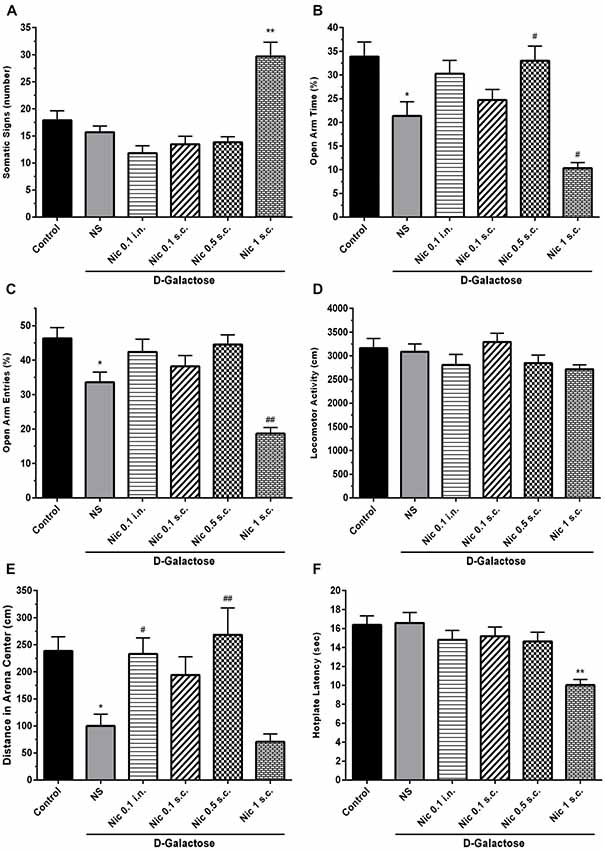
Figure 2. Withdrawal signs in study groups. (A) Number of somatic signs, (B) percentage of time spent in the open arms of EPM, (C) percentage of entries to the open arms of EPM, (D) locomotor activity, (E) distance moved in the center of the arena and (F) hotplate latency times. Each bar represents the mean ± standard error of mean (SEM), (n = 12). Significant differences tested by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test; *p < 0.05 and **p < 0.01 compared to the control group. #p < 0.05 and ##p < 0.01 compared to the DG+NS group; NS, normal saline; DG, D-galactose; Nic, nicotine; s.c., subcutaneous; i.n., intranasal; EPM, elevated plus maze.
We found statistically significant differences between groups in OAT (F(5,66) = 11.70, p < 0.001) and OAE (F(5,66) = 14.05, p < 0.001) tasks in elevated plus maze (EPM) test. Accordingly, post hoc analysis showed that chronic DGal+NS administration reduced both OAT and OAE in comparison to the control group (p < 0.05). On the other hand, chronic nicotine injection increased OAT at 0.5 mg/kg s.c. dose in DGal-received animals. Also, chronic nicotine administration at 1 mg/kg s.c. dose significantly decreased OAT and OAE compared with the DGal+NS group (p < 0.05 and p < 0.01 respectively). Other used doses of nicotine did not significantly affect OAT and OAE (p > 0.05; Figures 2B,C).
There was no significant difference between groups in the total distance moved in open filed test (F(5,66) = 1.824, p = 0.12; Figure 2D). However, difference for the time spent in the center was significant between groups (F(5,66) = 8.206, p < 0.001). Chronic DGal+NS treatment significantly reduced distance moved in the center compared to the control group (p < 0.05). However, chronic nicotine administration at 0.5 mg/kg s.c. and 0.1 mg/kg i.n. doses remarkably increased distance moved in the center in comparison to the NS-received DGal group (p < 0.01 and p < 0.05 respectively; Figure 2E).
We found a significant difference between study groups in hotplate latency time (F(5,66) = 7.766, p < 0.001). Nicotine administration at 1 mg/kg s.c. dose significantly decreased latency time in hotplate test in comparison to other groups (p < 0.01). Other used doses of nicotine did not have an impact on the hotplate latency time (p > 0.05; Figure 2F).
In the Barnes maze training session, when the mean escape latency was analyzed across the 4 days of training and treated groups, a two-way ANOVA revealed significant effects of group (F(5,264) = 12.38, p < 0.001), day (F(3,264) = 191.7, p < 0.001) and group-day interaction (F(15,264) = 1.778, p = 0.037). Our results showed that chronic DGal+NS administration delayed the finding of escape box in the 3rd and 4th days of the training session in comparison to the control group (p < 0.05, and p < 0.01, respectively). Conversely, chronic administration of nicotine significantly decreased escape latency time on the 3rd and 4th day of the training session at 0.5 mg/kg s.c. dose (p < 0.05 and p < 0.01 respectively) and on the 4th day at 0.1 mg/kg i.n. dose compared to the NS-received DGal group (p < 0.01; Figure 3A).
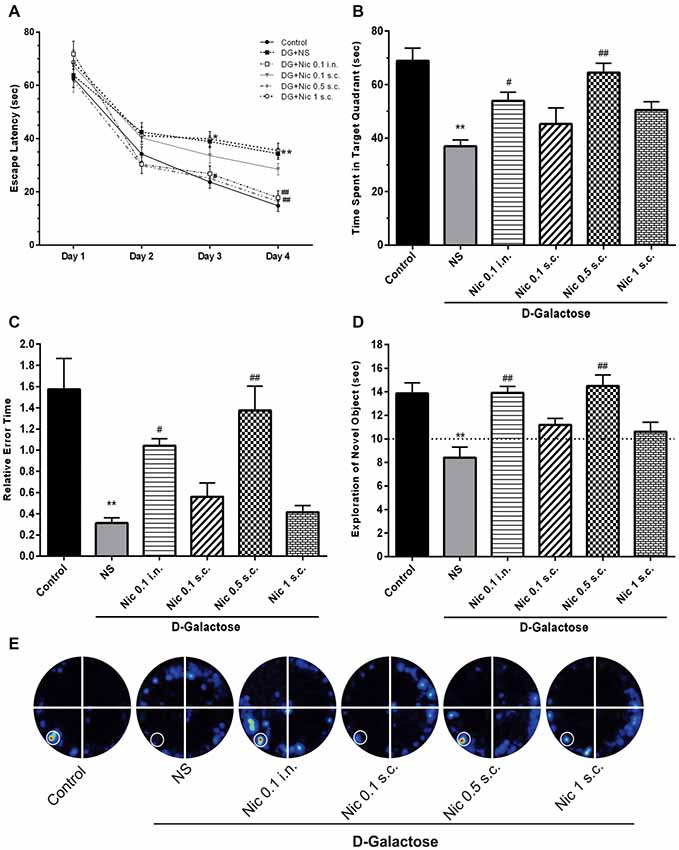
Figure 3. Learning and memory profile in study groups. (A) Mean escape latency time during 4 days of training session of MWM, (B) mean time spent in the target quadrant in the probe trial session of MWM, (C) mean relative error time in the probe trial session of MWM, (D) exploration time of the novel object during the retention session of NOR test, (E) corresponding heat maps show the combined traces of the mice from each group during the probe test of MWM (left lower part was considered as target quadrant). Values represent the mean ± SEM, (n = 12). Significant differences tested by two- (A) or one-way ANOVA followed by Tukey’s post hoc test; *p < 0.05 and **p < 0.01 compared to the control group. #p < 0.05 and ##p < 0.01 compared to the DG+NS group; NS, normal saline; DG, D-galactose; Nic, nicotine; s.c., subcutaneous; i.n., intranasal; MWM, Morris water maze; NOR, novel object recognition.
Also, one-way ANOVA revealed a significant difference in the time spent in the target quadrant between groups in the probe session (F(5,66) = 11.331, p < 0.001). Post hoc analysis showed that chronic injection of DGal+NS remarkably decreased the time spent in the target quadrant compared to the control group (p < 0.01). Also, it was shown that chronic administration of at 0.5 mg/kg s.c. and 0.1 mg/kg i.n. doses significantly increased the time spent in the target quadrant compared to the NS-received DGal group (p < 0.01 and p < 0.05 respectively; Figures 3B,E).
We found a significant difference between groups in mean relative error time (F(5,66) = 13.465, p < 0.001). We also found that mean relative error time was significantly lower in the DGal+NS group in comparison to the control group (p < 0.01). Chronic nicotine administration increased mean relative error time at 0.5 mg/kg s.c. and 0.1 mg/kg i.n. doses in comparison to the NS-received DGal group (p < 0.01 and p < 0.05 respectively; Figure 3C).
During the training phase, the exploratory preference for the objects was not influenced by the treatments (Data not shown; p > 0.05). In the retention phase of NOR test, analysis showed a significant difference between groups in the exploratory preference of the animals (F(5,66) = 9.263, p < 0.001). Chronic DGal+NS injection significantly decreased the exploratory preference of the animals for the novel object compared to the control group (p < 0.01). Additionally, chronic nicotine administration at 0.5 mg/kg s.c. and 0.1 mg/kg i.n. doses remarkably decreased DGal-induced recognition memory impairment compared to the DGal+NS -received group (p < 0.01).
All values were substantially different from the chance exploration (10 s) in the retention (p < 0.05), but not training phase (p > 0.05) as illustrated by the dashed line in the Figure 3D.
We found a significant difference between study groups in MMP (F(5,42) = 25.164, p < 0.001). A significant decrease in the MMP was revealed by post hoc analysis in the DGal+NS group in comparison to the control group (p < 0.01). Also, treatment with nicotine at 0.5 mg/kg s.c. and 0.1 mg/kg i.n. doses remarkably increased MMP in comparison to the NS-received DGal group (p < 0.01 and p < 0.05 respectively; Figure 4A).
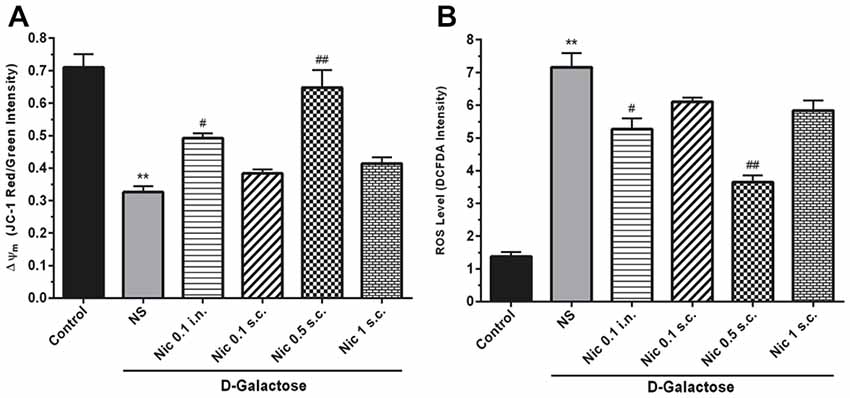
Figure 4. Mito-oxidative profile in study groups. (A) Mean of JC-1 red to green intensity as mitochondrial membrane potential (Δψm) index, (B) ROS level, Each bar represents the mean ± SEM, (n = 8). Significant differences tested by one-way ANOVA followed by Tukey’s post hoc test; **p < 0.01 compared to the control group. #p < 0.05 and ##p < 0.01 compared to the DG+NS group; ROS, reactive oxygen species; NS, normal saline; DG, DGal; Nic, nicotine; s.c., subcutaneous; i.n., intranasal.
Also, we found a significant difference between groups in the ROS level, (F(5,42) = 54.811, p < 0.001). Also, chronic administration of DGal+NS increased ROS level compared to the control group (p < 0.01). In addition, chronic nicotine treatment at 0.1 mg/kg i.n. and 0.5 mg/kg s.c. reduced ROS level in comparison to NS-received DGal group (p < 0.05 and p < 0.01 respectively; Figure 4B).
Analysis showed a significant difference between groups in total cytochrome C levels of the brain tissue (F(5,30) = 14.87, p < 0.001). Chronic administration of DGal+NS decreased total cytochrome C levels compared to the control group in the mice brain (p < 0.05). However, chronic administration of nicotine at 0.1 mg/kg i.n. and 0.5 mg/kg s.c. doses increased total cytochrome C level in comparison to the NS-received DGal group (p < 0.05 and p < 0.01 respectively; Figures 5A,E).
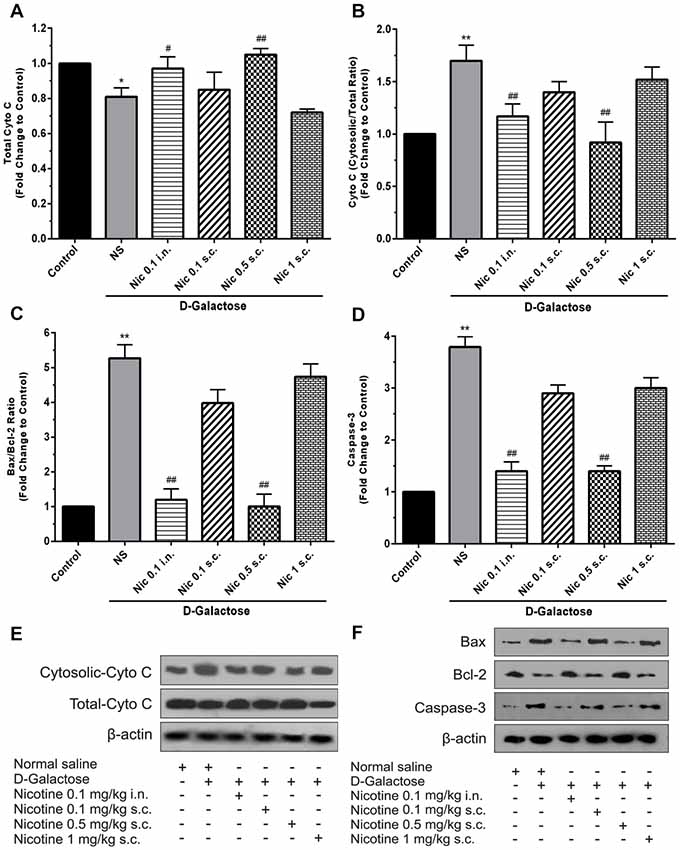
Figure 5. Brain cytochrome C and apoptosis markers levels in different study groups (A) mean total cytochrome C, (B) cytosolic to total cytochrome C level, (C) mean fold change of Bax to Bcl-2 ratio, (D) mean fold change of caspase-3 levels, (E) representative image of total and cytosolic cytochrome c, (F) representative image of Bax to Bcl-2 protein and caspase-3. Each bar represents the mean ± SEM, (n = 6). Significant differences tested by one-way ANOVA followed by Tukey’s post hoc test; *p < 0.05 and **p < 0.01 compared to the control group; #p < 0.05 and ##p < 0.01 compared to the DG+NS group; NS, normal saline; DG, D-galactose; Nic, nicotine; s.c., subcutaneous; i.n., intranasal.
A significant difference was revealed by one-way ANOVA in cytosolic to total cytochrome C ratio (F(5,30) = 24.34, p < 0.001). Accordingly, post hoc analysis showed that chronic DGal+NS administration increases cytosolic to total cytochrome C ratio in comparison to the control group (p < 0.01). On the other hand, chronic administration of nicotine at 0.1 mg/kg i.n. and 0.5 mg/kg s.c. doses decreased this item compared to the NS-received DGal group (p < 0.01; Figures 5B,E).
We found a significant difference between study groups in Bax/Bcl-2 ratio (F(5,30) = 149.7, p < 0.001) and caspase-3 levels (F(5,30) = 133.2, p < 0.001). Subsequently, analysis showed that chronic DGal+NS injection increased Bax/Bcl-2 ratio and caspase-3 levels compared to the control group (p < 0.01). In addition, chronic nicotine treatment at 0.1 mg/kg i.n. and 0.5 mg/kg s.c. doses decreased Bax/Bcl-2 ratio and caspase-3 levels compared to the NS-received DGal group (p < 0.01; Figures 5C,D,F).
We found a significant difference in the BDNF (F(5,42) = 13.24, p < 0.001) and NGF (F(5,42) = 17.39, p < 0.001) levels in the brain tissue in study groups. Chronic treatment with DGal+NS decreased BDNF and NGF levels in comparison to the control group (p < 0.01). However, chronic nicotine administration at 0.1 mg/kg i.n. and 0.5 mg/kg s.c. doses increased BDNF level compared to the DGal+NS group (p < 0.01 and p < 0.05; Figure 6A). Also, chronic nicotine administration at all doses increased NGF level compared to the NS-received DGal group (p < 0.01; Figure 6B).
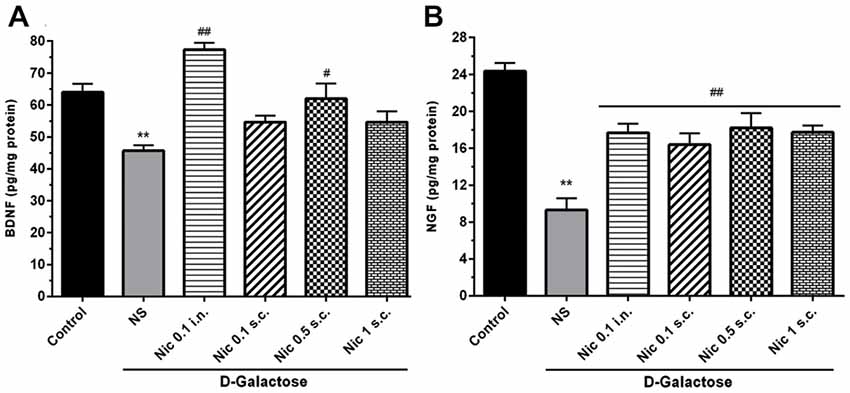
Figure 6. Neurotrophic factors level in study groups. (A) BDNF and (B) NGF level in different groups. Each bar represents the mean ± SEM, (n = 8). Significant differences tested by one-way ANOVA followed by Tukey’s post hoc test; **p < 0.01 compared to the control group, #p < 0.05 and ##p < 0.01 compared to the DG+NS group; BDNF, brain-derived neurotrophic factor; NGF, nerve growth factor; NS, normal saline; DG, D-galactose; Nic, nicotine; s.c., subcutaneous; i.n., intranasal.
Here, we showed that chronic administration of nicotine at doses of 0.1 mg/kg intranasally and 0.5 mg/kg subcutaneously decreased cognitive impairment induced by chronic administration of DGal to mice. We interpret the results as effects of nicotine against oxidative damage, apoptosis and neurodegenerative lesions in the mouse brain. Nicotine at these doses caused no detectable withdrawal signs in the mice. In contrast, the highest dose of nicotine (1 mg/kg) administered subcutaneously not only had no positive effects on cognition or neurochemical factors in brain tissue but also elicited signs of withdrawal in the treated mice. The lowest dose of nicotine (0.1 mg/kg) administered subcutaneously, in turn, had no effects on cognitive function or signs of withdrawal of the mice.
Age-related memory and learning impairment is thought to be the result of increased neurodegeneration and decline in the neuronal function (Haddadi et al., 2014). The resulting cognitive decline is a multifactorial process and involves oxidative stress, altered brain neurotransmitters (Haider et al., 2014), apoptosis (Kim et al., 2010), mitochondrial dysfunction, et cetera (Martínez et al., 2000).
The DGal model of brain aging is widely-used in rodent studies of features of human brain aging (Haider et al., 2015; Gong et al., 2016; Pourmemar et al., 2017) such as cognitive function impairment (Lu et al., 2010b; Han et al., 2014). The Barnes maze task tested the spatial learning and reference memory associated with hippocampal function in the animals. The Barnes task is less anxiogenic than other tests that assess spatial memory (Harrison et al., 2009) and is widely used to assess cognitive performance in aged animals. Also, the NOR task was used to assess recognition memory that is linked to connections between hippocampus and cortex and is impaired during aging (Gallagher et al., 1993; Sadigh-Eteghad et al., 2015b). These tests have been formerly used to assess rodent cognitive function in neurodegenerative disorders such as AD and their responses to various treatments (O’Leary and Brown, 2009; Sadigh-Eteghad et al., 2015b).
The results showed that chronic administration of DGal with NS raised the escape latency time and decreased the time spent in the target quadrant of the Barnes maze test. The results indicate impairment of working and reference memory in the DGal model of aging in mice, in agreement with results obtained by Ashour et al. (2016). Consistent with other studies (Wei et al., 2005; Pourmemar et al., 2017), we found that long-term administration of DGal with NS shortened explorations of novel object in comparison to the control group, indicating impairment of recognition memory in NS-treated DGal mice.
Chronic nicotine treatment at 0.1 mg/kg i.n. and 0.5 mg/kg s.c. doses reversed the DGal-induced cognitive impairment both in Barnes maze and NOR tests. However, nicotine administration at 0.1 and 1 mg/kg s.c. doses did not have a statistically significant effect on the cognitive performance of the mice in Barnes and NOR tests. Similarly, in a study by French et al. (2006), it was revealed that chronic administration of nicotine to aged rats at 0.1 mg/kg and 0.3 mg/kg doses improved working and reference memories in Morris water maze (MWM) task. This study also showed that cognition-improving effect of nicotine was “dose-dependent or inverted U shape” and certain higher doses had superior effects on the cognitive function (French et al., 2006). Another study showed that effects of nicotine on cognitive function are dependent upon “duration/magnitude of nicotine exposure” and its procognitive impacts are limited under specific conditions (Ortega et al., 2013) which may explain why high (1 mg/kg s.c.) and low (0.1 mg/kg s.c.) doses used in this study did not have cognition-improving effects on mice. Similar studies using the same dose of nicotine, but shorter periods of administration have yielded the same results in the aged rodent both in MWM and NOR tests (Socci et al., 1995; Riekkinen and Riekkinen, 1997; Puma et al., 1999). In a study by Levin and Torry (1996), it was found that acute nicotine administration reduces cognitive deficits in aged rats. However, chronic nicotine injection did not have the same procognitive effects. On the other hand, Arendash et al. (1995) found that chronic nicotine treatment decreases learning/memory deficits in aged rats, and the effects were reproduced in several cognitive tasks. Similarly, Buccafusco and Jackson (1991) showed that nicotine injection in aged monkeys improves their performance in delayed matching-to-sample task.
We could not find any study regarding effects of i.n. nicotine on the cognitive performance of aged animals. In this study, we showed that none of the treatments had effect on the locomotor activity of the mice. These findings were in line with the findings of previous works (Kalejaiye et al., 2013; Pourmemar et al., 2017).
The results also revealed that chronic DGal administration remarkably decreased the number of OAEs in EPM and distance moved in the center in the open filed test suggesting the anxiogenic effects of DGal-induced aging on mice. Of direct relevance to this line of argument is a study by Bessa et al. (2005), demonstrating that aging is accompanied by increased signs of anxiety (Torres et al., 2013). Of the administered doses of nicotine in this study, nicotine at 1 mg/kg s.c. dose significantly increased withdrawal signs including somatic signs and thermal hyperalgesia. It also decreased OAE as well as OAT in EPM and reduced time spent in the arena center in the open field test all of which indicate nicotine potential to increase withdrawal signs and induce dependency at this dose. In line with that, a study by Malin et al. (1992) showed that administration of nicotine at 3 mg/kg dose induced withdrawal signs and thus might cause dependency in mice. In another study, they stated that 1–3 mg/kg daily dose of nicotine could result in dependency in rodents (Malin and Goyarzu, 2009). Other nicotine doses used in this study did not increase withdrawal signs in mice. However, in a study by Grabus et al. (2005), it was shown that chronic oral administration of nicotine (~0.1 mg/kg) increased withdrawal signs presented by the animal on days 1, 2, 3, but not 5. It is believed that “rapid drug delivery” and the concentration of the delivered drug are two main determining factors in the nicotine-induced dependency (Schneider et al., 1996). So, it appears that the administered doses of nicotine in this study (except 1 mg/kg s.c.) due to the specific rate of delivery and concentration of nicotine in the brain did not produce withdrawal signs in mice.
Our data demonstrated that chronically administered DGal increases ROS levels in the mice brain. In line with that many studies have found that chronic DGal administration increases ROS levels and causes oxidative stress in the brain (He et al., 2009; Kumar et al., 2010; Hao et al., 2014) which then predisposes the brain to neurodegeneration and aging (Floyd and Hensley, 2002; Majdi et al., 2016).
We also showed that chronic nicotine administration at 0.1 mg/kg i.n. and 0.5 mg/kg s.c. doses significantly reduced brain ROS levels in mice. In a study by Linert et al. (1999), the authors concluded that nicotine at 0.8 mg/kg i.p. dose does not change ROS levels in the brain. They also stated that the anti-oxidant properties of nicotine might be mediated through its blocking effects on Fenton’s reaction. Also, it has been shown that nicotine may have dual effects on oxidative stress and ROS formation in the brain depending upon the administered dose. It is believed that nicotine has “inverted U-shaped dose-response curves” meaning high doses increase oxidative stress whereas medium doses show antioxidant properties (Guan et al., 2003).
MMP perturbations may have a role in the progression of aging (Reddy and Beal, 2008). Similar to the Shen et al. (2014) results, the findings of the present study showed that chronic DGal administration decreases MMP and thus increases mito-oxidative damage. It is believed that decrease in the MMP increases mitochondrial permeability which subsequently increases ROS and cytochrome C release from mitochondria and results in apoptosis as well as neuronal death (Pollack and Leeuwenburgh, 2001; Zhang et al., 2010). Evidence suggests that ROS formation does not occur until MMP changes which proves MMP importance in the DGal-induced mito-oxidative stress (Nohl et al., 2005). Conversely, some studies have found that increase in the ROS levels causes mitochondrial membrane anisotropy and apoptosis. In fact, increased oxidative stress and ROS levels causes MMP collapse, and transient ROS release to the cytosol which triggers ROS reproduction and is called “ROS-induced ROS-release” (Zorov et al., 2006). We found that chronic nicotine administration at 0.1 mg/kg i.n. and 0.5 mg/kg s.c. doses reduce mito-oxidative damage through decreasing mitochondrial membrane anisotropy. In line with that Cormier et al. (2003), reported that chronically administered nicotine at 0.6 mg/kg dose had protective effects on mitochondria and prevented mitochondrial membrane anisotropy and mito-oxidative damage which happens in neurodegeneration and aging (Cormier et al., 2003; Reddy and Beal, 2008).
In this study, we also showed that chronic injection of DGal increases cytoplasmic to total cytochrome C ratio and decreases total cytochrome C levels in the brain. It also increased Bax/Bcl-2 ratio and caspase-3 levels in DGal-received mice brain. As we discussed earlier DGal treatment increases ROS levels in the brain which leads to MMP perturbations. This subsequently increases cytochrome C release to the cytoplasm. Cytoplasmic cytochrome C then links to other apoptotic factors and creates an apoptosome which then activates caspase-3 (Pollack and Leeuwenburgh, 2001; Pollack et al., 2002). In addition, DGal-induced increase in the Bax/Bcl-2 ratio further increase cytochrome C release to the cytoplasm and exacerbates the above-mentioned pathologic pathway. These mechanisms finally result in neuronal apoptosis as an important component of brain aging and neurodegeneration (Beal, 2005). DGal administration also decreased total cytochrome C levels which may indicate an age-related decrease in the number of active mitochondria (Navarro et al., 2002; O’Toole et al., 2010).
In this study, administration of nicotine at 0.5 mg/kg s.c. and 0.1 mg/kg i.n. doses was found to reverse the mentioned pathologic events. Nicotine decreased Bax/Bcl-2 ratio as well as cytochrome C release to the cytoplasm. This along with decreased caspase-3 levels is thought to prevent the subsequent apoptosis in the DGal-received mice brain. In line with that, nicotine has been shown to be the so-called “survival agonist” and inhibit pro-apoptotic pathways (Mai et al., 2003; Tizabi et al., 2005). In a direct relevance to this line of evidence, Marrero and Bencherif (2009) showed that nicotine increases the production of Bcl-2 and blocks the release of cytochrome C the cytosol and prevents apoptosis. Nicotine at the mentioned doses also increased total cytochrome C levels suggesting nicotine-induced improvement of mitochondrial function.
We confirmed that long-term DGal injection reduces NGF and BDNF levels as neuroprotective factors in the brain. Woo et al. (2014) found that chronic DGal administration impairs neurotrophic factors production in the brain resulting in cognitive impairment. Similarly, Erraji-Benchekroun et al. (2005) showed that the production of neurotrophic factors declines through time in the aging brain. The results of this study also showed that nicotine treatment chronically at 0.5 mg/kg s.c. and 0.1 mg/kg i.n. doses can increase BDNF levels in the DGal-received mice brain. Also, all the administered doses of nicotine increased NGF level in this aging model.
Nicotine-mediated enhancement of nuclear translocation and transcriptional activity of NF-κB amplifies the expression of NGF and protects the brain from aging-induced neuronal damage (Wongtrakool et al., 2014). Additionally, nicotine regulates NGF level possibly via glutamatergic neurones modulation (Rattray, 2001). The increased NGF levels in the brain could then improve learning, memory and cognitive performance (Fischer et al., 1991; De Rosa et al., 2005) which is impaired in aging. Also, evidence suggests that chronic nicotine administration at 0.5 mg/kg i.p. dose (Kenny et al., 2000) increases expression of BDNF through α7 nAChRs-mediated pathways. BDNF then involves in memory formation in the hippocampus and long-term potentiation (Tyler et al., 2002; Czubak et al., 2009).
Intranasal drug delivery is a novel and safe way to administer medications through the nasal mucosa (Farzampour et al., 2016). This route of drug administration shunts the BBB, is non-invasive and encompasses two separate pathways including trans-neuronal or immediate and para-neuronal or delayed (Mustafa et al., 2016). It has been shown that nicotine nasal spray improves continuous attention, working memory, and executing processing (Smith et al., 2002; Myers et al., 2008). Our results suggested the anti-aging effect of intranasal nicotine on mice through its anti-apoptosis, neuroprotective and anti mito-oxidative actions without increasing withdrawal signs presentation and causing dependency. This might be an alternative approach in the treatment of age-associated cognitive impairment.
In conclusion, this set of data showed that nicotine at certain controlled doses has a potential to attenuate age-induced cognitive impairment without producing withdrawal signs and dependency. These doses could also ameliorate age-induced mito-oxidative damage, apoptosis and neurotrophic factors level reduction. Further, our results indicated that i.n. nicotine delivery could be an alternative choice for the treatment of age-related cognitive decline.
AM, FF, ME and JM performed the experiments, interpreted the results and wrote the manuscript. AG, SS-E and MT designed the experiments. AG critically interpreted data and critically revised and approved the manuscript.
This research was in part supported by a grant from Neurosciences Research Center (NSRC), Tabriz University of Medical Sciences (TUOMS; grant number: 5/88/1523) to SS-E and grants from Department of Nuclear Medicine, Odense University Hospital, and from Alzheimer Foundation of Denmark to AG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ali, T., Badshah, H., Kim, T. H., and Kim, M. O. (2015). Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-κB/JNK signaling pathway in aging mouse model. J. Pineal Res. 58, 71–85. doi: 10.1111/jpi.12194
Arendash, G. W., Sanberg, P. R., and Sengstock, G. J. (1995). Nicotine enhances the learning and memory of aged rats. Pharmacol. Biochem. Behav. 52, 517–523. doi: 10.1016/0091-3057(95)00119-h
Ashour, F. A., Abdel-Razek, H., Youssef, G. S., Ewida, S. F., and Adel, M. M. (2016). Effect of exercise and/or melatonin on spatial learning and memory of D-galactose-treated rats. Menoufia Med. J. 29, 944–953. doi: 10.4103/1110-2098.202490
Beal, M. F. (2005). Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 58, 495–505. doi: 10.1002/ana.20624
Bessa, J., Oliveira, M., Cerqueira, J., Almeida, O., and Sousa, N. (2005). Age-related qualitative shift in emotional behaviour: paradoxical findings after re-exposure of rats in the elevated-plus maze. Behav. Brain Res. 162, 135–142. doi: 10.1016/j.bbr.2005.03.005
Bontempi, B., Whelan, K. T., Risbrough, V. B., Lloyd, G. K., and Menzaghi, F. (2003). Cognitive enhancing properties and tolerability of cholinergic agents in mice: a comparative study of nicotine, donepezil, and SIB-1553A, a subtype-selective ligand for nicotinic acetylcholine receptors. Neuropsychopharmacology 28, 1235–1246. doi: 10.1038/sj.npp.1300150
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1006/abio.1976.9999
Buccafusco, J. J., and Jackson, W. J. (1991). Beneficial effects of nicotine administered prior to a delayed matching-to-sample task in young and aged monkeys. Neurobiol. Aging 12, 233–238. doi: 10.1016/0197-4580(91)90102-p
Cormier, A., Morin, C., Zini, R., Tillement, J.-P., and Lagrue, G. (2003). Nicotine protects rat brain mitochondria against experimental injuries. Neuropharmacology 44, 642–652. doi: 10.1016/s0028-3908(03)00041-8
Czubak, A., Nowakowska, E., Kus, K., Burda, K., Metelska, J., Baer-Dubowska, W., et al. (2009). Influences of chronic venlafaxine, olanzapine and nicotine on the hippocampal and cortical concentrations of brain-derived neurotrophic factor (BDNF). Pharmacol. Rep. 61, 1017–1023. doi: 10.1016/s1734-1140(09)70163-x
Damaj, M., Kao, W., and Martin, B. R. (2003). Characterization of spontaneous and precipitated nicotine withdrawal in the mouse. J. Pharmacol. Exp. Ther. 307, 526–534. doi: 10.1124/jpet.103.054908
De Rosa, R., Garcia, A. A., Braschi, C., Capsoni, S., Maffei, L., Berardi, N., et al. (2005). Intranasal administration of nerve growth factor (NGF) rescues recognition memory deficits in AD11 anti-NGF transgenic mice. Proc. Natl. Acad. Sci. U S A 102, 3811–3816. doi: 10.1073/pnas.0500195102
Erraji-Benchekroun, L., Underwood, M. D., Arango, V., Galfalvy, H., Pavlidis, P., Smyrniotopoulos, P., et al. (2005). Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol. Psychiatry 57, 549–558. doi: 10.1016/j.biopsych.2004.10.034
Farzampour, S., Majdi, A., and Sadigh-Eteghad, S. (2016). Intranasal insulin treatment improves memory and learning in a rat amyloid-β model of Alzheimer’s disease. Physiol. Int. 103, 344–353. doi: 10.1556/2060.103.2016.3.7
Fischer, W., Björklund, A., Chen, K., and Gage, F. (1991). NGF improves spatial memory in aged rodents as a function of age. J. Neurosci. 11, 1889–1906. doi: 10.1523/JNEUROSCI.11-07-01889.1991
Floyd, R. A., and Hensley, K. (2002). Oxidative stress in brain aging: implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 23, 795–807. doi: 10.1016/s0197-4580(02)00019-2
French, K. L., Granholm, A.-C. E., Moore, A. B., Nelson, M. E., and Bimonte-Nelson, H. A. (2006). Chronic nicotine improves working and reference memory performance and reduces hippocampal NGF in aged female rats. Behav. Brain Res. 169, 256–262. doi: 10.1016/j.bbr.2006.01.008
Gallagher, M., Burwell, R., and Burchinal, M. R. (1993). Severity of spatial learning impairment in aging: development of a learning index for performance in the Morris water maze. Behav. Neurosci. 107, 618–626. doi: 10.1037//0735-7044.107.4.618
Garrido, R., Mattson, M. P., Hennig, B., and Toborek, M. (2001). Nicotine protects against arachidonic-acid-induced caspase activation, cytochrome C release and apoptosis of cultured spinal cord neurons. J. Neurochem. 76, 1395–1403. doi: 10.1046/j.1471-4159.2001.00135.x
Gleichmann, M., Zhang, Y., Wood, W. H., Becker, K. G., Mughal, M. R., Pazin, M. J., et al. (2012). Molecular changes in brain aging and Alzheimer’s disease are mirrored in experimentally silenced cortical neuron networks. Neurobiol. Aging 33, 205.e1–205.e18. doi: 10.1016/j.neurobiolaging.2010.08.012
Gong, Y.-S., Guo, J., Hu, K., Gao, Y.-Q., Xie, B.-J., Sun, Z.-D., et al. (2016). Ameliorative effect of lotus seedpod proanthocyanidins on cognitive impairment and brain aging induced by D-galactose. Exp. Gerontol. 74, 21–28. doi: 10.1016/j.exger.2015.11.020
Grabus, S. D., Martin, B. R., Batman, A. M., Tyndale, R. F., Sellers, E., and Damaj, M. I. (2005). Nicotine physical dependence and tolerance in the mouse following chronic oral administration. Psychopharmacology 178, 183–192. doi: 10.1007/s00213-004-2007-3
Guan, Z.-Z., Yu, W.-F., and Nordberg, A. (2003). Dual effects of nicotine on oxidative stress and neuroprotection in PC12 cells. Neurochem. Int. 43, 243–249. doi: 10.1016/s0197-0186(03)00009-3
Guarente, L. (2014). Aging research—where do we stand where are we going? Cell 159, 15–19. doi: 10.1016/j.cell.2014.08.041
Haddadi, M., Jahromi, S. R., Sagar, B. K., Patil, R. K., Shivanandappa, T., and Ramesh, S. R. (2014). Brain aging, memory impairment and oxidative stress: a study in Drosophila melanogaster. Behav. Brain Res. 259, 60–69. doi: 10.1016/j.bbr.2013.10.036
Haider, S., Liaquat, L., Shahzad, S., Sadir, S., Madiha, S., Batool, Z., et al. (2015). A high dose of short term exogenous D-galactose administration in young male rats produces symptoms simulating the natural aging process. Life Sci. 124, 110–119. doi: 10.1016/j.lfs.2015.01.016
Haider, S., Saleem, S., Perveen, T., Tabassum, S., Batool, Z., Sadir, S., et al. (2014). Age-related learning and memory deficits in rats: role of altered brain neurotransmitters, acetylcholinesterase activity and changes in antioxidant defense system. Age 36:9653. doi: 10.1007/s11357-014-9653-0
Han, C.-H., Lin, Y.-S., Lee, T.-L., Liang, H.-J., and Hou, W.-C. (2014). Asn-Trp dipeptides improve the oxidative stress and learning dysfunctions in D-galactose-induced BALB/c mice. Food Funct. 5, 2228–2236. doi: 10.1039/c4fo00205a
Hao, L., Huang, H., Gao, J., Marshall, C., Chen, Y., and Xiao, M. (2014). The influence of gender, age and treatment time on brain oxidative stress and memory impairment induced by D-galactose in mice. Neurosci. Lett. 571, 45–49. doi: 10.1016/j.neulet.2014.04.038
Harrison, F., Hosseini, A., and McDonald, M. (2009). Endogenous anxiety and stress responses in water maze and Barnes maze spatial memory tasks. Behav. Brain Res. 198, 247–251. doi: 10.1016/j.bbr.2008.10.015
He, M., Zhao, L., Wei, M.-J., Yao, W.-F., Zhao, H.-S., and Chen, F.-J. (2009). Neuroprotective effects of (−)-epigallocatechin-3-gallate on aging mice induced by D-galactose. Biol. Pharm. Bull. 32, 55–60. doi: 10.1248/bpb.32.55
Hsieh, H.-M., Wu, W.-M., and Hu, M.-L. (2009). Soy isoflavones attenuate oxidative stress and improve parameters related to aging and Alzheimer’s disease in C57BL/6J mice treated with D-galactose. Food Chem. Toxicol. 47, 625–632. doi: 10.1016/j.fct.2008.12.026
Jackson, K. J., Marks, M. J., Vann, R. E., Chen, X., Gamage, T. F., Warner, J. A., et al. (2010). Role of α5 nicotinic acetylcholine receptors in pharmacological and behavioral effects of nicotine in mice. J. Pharmacol. Exp. Ther. 334, 137–146. doi: 10.1124/jpet.110.165738
Jansari, A. S., Froggatt, D., Edginton, T., and Dawkins, L. (2013). Investigating the impact of nicotine on executive functions using a novel virtual reality assessment. Addiction 108, 977–984. doi: 10.1111/add.12082
Kalejaiye, O., Bhatti, B. H., Taylor, R. E., and Tizabi, Y. (2013). Nicotine blocks the depressogenic effects of alcohol: implications for drinking-smoking co-morbidity. J. Drug Alcohol Res. 2:235709. doi: 10.4303/jdar/235709
Kenny, P. J., File, S. E., and Rattray, M. (2000). Acute nicotine decreases, and chronic nicotine increases the expression of brain-derived neurotrophic factor mRNA in rat hippocampus. Mol. Brain Res. 85, 234–238. doi: 10.1016/s0169-328x(00)00246-1
Kim, S. E., Ko, I. G., Kim, B. K., Shin, M. S., Cho, S., Kim, C. J., et al. (2010). Treadmill exercise prevents aging-induced failure of memory through an increase in neurogenesis and suppression of apoptosis in rat hippocampus. Exp. Gerontol. 45, 357–365. doi: 10.1016/j.exger.2010.02.005
Kumar, A., Prakash, A., and Dogra, S. (2010). Naringin alleviates cognitive impairment, mitochondrial dysfunction and oxidative stress induced by D-galactose in mice. Food Chem. Toxicol. 48, 626–632. doi: 10.1016/j.fct.2009.11.043
Lan, Z., Liu, J., Chen, L., Fu, Q., Luo, J., Qu, R., et al. (2012). Danggui-Shaoyao-San ameliorates cognition deficits and attenuates oxidative stress-related neuronal apoptosis in D-galactose-induced senescent mice. J. Ethnopharmacol. 141, 386–395. doi: 10.1016/j.jep.2012.02.050
Levin, E. D., McClernon, F. J., and Rezvani, A. H. (2006). Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification and anatomic localization. Psychopharmacology 184, 523–539. doi: 10.1007/s00213-005-0164-7
Levin, E. D., and Torry, D. (1996). Acute and chronic nicotine effects on working memory in aged rats. Psychopharmacology 123, 88–97. doi: 10.1007/bf02246285
Li, L., Peng, L., and Zuo, Z. (2008). Isoflurane preconditioning increases B-cell lymphoma-2 expression and reduces cytochrome C release from the mitochondria in the ischemic penumbra of rat brain. Eur. J. Pharmacol. 586, 106–113. doi: 10.1016/j.ejphar.2008.02.073
Li, W., Wei, F., Fan, M., Zhang, J., Zhang, B., Ma, X., et al. (1995). Mimetic brain aging effect induced by D-galactose in mice. Chin. J. Pharmacol. Toxicol. 9, 93–95.
Linert, W., Bridge, M., Huber, M., Bjugstad, K., Grossman, S., and Arendash, G. (1999). In vitro and in vivo studies investigating possible antioxidant actions of nicotine: relevance to Parkinson’s and Alzheimer’s diseases. Biochim. Biophys. Acta 1454, 143–152. doi: 10.1016/s0925-4439(99)00029-0
Lu, J., Wu, D. M., Zheng, Y. L., Hu, B., and Zhang, Z. F. (2010a). Purple sweet potato color alleviates D-galactose-induced brain aging in old mice by promoting survival of neurons via PI3K pathway and inhibiting cytochrome C-mediated apoptosis. Brain Pathol. 20, 598–612. doi: 10.1111/j.1750-3639.2009.00339.x
Lu, J., Wu, D.-M., Zheng, Y.-L., Hu, B., Zhang, Z.-F., Ye, Q., et al. (2010b). Ursolic acid attenuates D-galactose-induced inflammatory response in mouse prefrontal cortex through inhibiting AGEs/RAGE/NF-κB pathway activation. Cereb. Cortex 20, 2540–2548. doi: 10.1093/cercor/bhq002
Mai, H., May, W. S., Gao, F., Jin, Z., and Deng, X. (2003). A functional role for nicotine in Bcl2 phosphorylation and suppression of apoptosis. J. Biol. Chem. 278, 1886–1891. doi: 10.1074/jbc.M209044200
Majdi, A., Kamari, F., Vafaee, M. S., and Sadigh-Eteghad, S. (2017). Revisiting nicotine’s role in the ageing brain and cognitive impairment. Rev. Neurosci. 28, 767–781. doi: 10.1515/revneuro-2017-0008
Majdi, A., Mahmoudi, J., Sadigh-Eteghad, S., Golzari, S. E., Sabermarouf, B., and Reyhani-Rad, S. (2016). Permissive role of cytosolic pH acidification in neurodegeneration: a closer look at its causes and consequences. J. Neurosci. Res. 94, 879–887. doi: 10.1002/jnr.23757
Malin, D. H., and Goyarzu, P. (2009). “Rodent models of nicotine withdrawal syndrome,” in Nicotine Psychopharmacology, eds J. E. Henningfield, E. Calvento and S. Pogun (Berlin: Springer), 401–434.
Malin, D. H., Lake, J. R., Nelin-Maultsby, P., Roberts, L. K., Lanier, J. G., Carter, V. A., et al. (1992). Rodent model of nicotine abstinence syndrome. Pharmacol. Biochem. Behav. 43, 779–784. doi: 10.1016/0091-3057(92)90408-8
Marrero, M. B., and Bencherif, M. (2009). Convergence of α7 nicotinic acetylcholine receptor-activated pathways for anti-apoptosis and anti-inflammation: central role for JAK2 activation of STAT3 and NF-κB. Brain Res. 1256, 1–7. doi: 10.1016/j.brainres.2008.11.053
Martínez, M., Hernández, A. I., and Martínez, N. (2000). N-Acetylcysteine delays age-associated memory impairment in mice: role in synaptic mitochondria. Brain Res. 855, 100–106. doi: 10.1016/s0006-8993(99)02349-5
Matta, S. G., Balfour, D. J., Benowitz, N. L., Boyd, R. T., Buccafusco, J. J., Caggiula, A. R., et al. (2007). Guidelines on nicotine dose selection for in vivo research. Psychopharmacology 190, 269–319. doi: 10.1007/s00213-006-0441-0
Mustafa, G., Alrohaimi, A. H., Bhatnagar, A., Baboota, S., Ali, J., and Ahuja, A. (2016). Brain targeting by intranasal drug delivery (INDD): a combined effect of trans-neural and para-neuronal pathway. Drug Deliv. 23, 923–929. doi: 10.3109/10717544.2014.923064
Myers, C. S., Taylor, R. C., Moolchan, E. T., and Heishman, S. J. (2008). Dose-related enhancement of mood and cognition in smokers administered nicotine nasal spray. Neuropsychopharmacology 33, 588–598. doi: 10.1038/sj.npp.1301425
Navarro, A., Sánchez Del Pino, M. J., Gómez, C., Peralta, J. L., and Boveris, A. (2002). Behavioral dysfunction, brain oxidative stress and impaired mitochondrial electron transfer in aging mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R985–R992. doi: 10.1152/ajpregu.00537.2001
Nohl, H., Gille, L., and Staniek, K. (2005). Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 69, 719–723. doi: 10.1016/j.bcp.2004.12.002
Novalija, E., Kevin, L. G., Eells, J. T., Henry, M. M., and Stowe, D. F. (2003). Anesthetic preconditioning improves adenosine triphosphate synthesis and reduces reactive oxygen species formation in mitochondria after ischemia by a redox dependent mechanism. Anesthesiology 98, 1155–1163. doi: 10.1097/00000542-200305000-00018
O’Leary, T. P., and Brown, R. E. (2009). Visuo-spatial learning and memory deficits on the Barnes maze in the 16-month-old APPswe/PS1dE9 mouse model of Alzheimer’s disease. Behav. Brain Res. 201, 120–127. doi: 10.1016/j.bbr.2009.01.039
Ortega, L. A., Tracy, B. A., Gould, T. J., and Parikh, V. (2013). Effects of chronic low-and high-dose nicotine on cognitive flexibility in C57BL/6J mice. Behav. Brain Res. 238, 134–145. doi: 10.1016/j.bbr.2012.10.032
O’Toole, J. F., Patel, H. V., Naples, C. J., Fujioka, H., and Hoppel, C. L. (2010). Decreased cytochrome C mediates an age-related decline of oxidative phosphorylation in rat kidney mitochondria. Biochem. J. 427, 105–112. doi: 10.1042/BJ20091373
Paradies, G., Petrosillo, G., Paradies, V., and Ruggiero, F. M. (2011). Mitochondrial dysfunction in brain aging: role of oxidative stress and cardiolipin. Neurochem. Int. 58, 447–457. doi: 10.1016/j.neuint.2010.12.016
Pollack, M., and Leeuwenburgh, C. (2001). Apoptosis and aging role of the mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 56, B475–B482. doi: 10.1093/gerona/56.11.B475
Pollack, M., Phaneuf, S., Dirks, A., and Leeuwenburgh, C. (2002). The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann. N Y Acad. Sci. 959, 93–107. doi: 10.1111/j.1749-6632.2002.tb02086.x
Pourmemar, E., Majdi, A., Haramshahi, M., Talebi, M., Karimi, P., and Sadigh-Eteghad, S. (2017). Intranasal cerebrolysin attenuates learning and memory impairments in D-galactose-induced senescence in mice. Exp. Gerontol. 87, 16–22. doi: 10.1016/j.exger.2016.11.011
Puma, C., Deschaux, O., Molimard, R., and Bizot, J.-C. (1999). Nicotine improves memory in an object recognition task in rats. Eur. Neuropsychopharmacol. 9, 323–327. doi: 10.1016/s0924-977x(99)00002-4
Rattray, M. (2001). Is there nicotinic modulation of nerve growth factor? Implications for cholinergic therapies in Alzheimer’s disease. Biol. Psychiatry 49, 185–193. doi: 10.1016/s0006-3223(00)01047-7
Reddy, P. H., and Beal, M. F. (2008). Amyloid β, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53. doi: 10.1016/j.molmed.2007.12.002
Richter, N., Michel, A., Onur, O. A., Kracht, L., Dietlein, M., Tittgemeyer, M., et al. (2017). White matter lesions and the cholinergic deficit in aging and mild cognitive impairment. Neurobiol. Aging 53, 27–35. doi: 10.1016/j.neurobiolaging.2017.01.012
Riekkinen, M., and Riekkinen, P. Jr. (1997). Nicotine and D-cycloserine enhance acquisition of water maze spatial navigation in aged rats. Neuroreport 8, 699–703. doi: 10.1097/00001756-199702100-00024
Rushforth, S. L., Steckler, T., and Shoaib, M. (2011). Nicotine improves working memory span capacity in rats following sub-chronic ketamine exposure. Neuropsychopharmacology 36, 2774–2781. doi: 10.1038/npp.2011.224
Sadigh-Eteghad, S., Majdi, A., McCann, S. K., Mahmoudi, J., Vafaee, M. S., and Macleod, M. R. (2017). D-galactose-induced brain ageing model: a systematic review and meta-analysis on cognitive outcomes and oxidative stress indices. PLoS One 12:e0184122. doi: 10.1371/journal.pone.0184122
Sadigh-Eteghad, S., Sabermarouf, B., Majdi, A., Talebi, M., Farhoudi, M., and Mahmoudi, J. (2015a). Amyloid-β: a crucial factor in Alzheimer’s disease. Med. Princ. Pract. 24, 1–10. doi: 10.1159/000369101
Sadigh-Eteghad, S., Talebi, M., Mahmoudi, J., Babri, S., and Shanehbandi, D. (2015b). Selective activation of α7 nicotinic acetylcholine receptor by PHA-543613 improves Aβ25–35-mediated cognitive deficits in mice. Neuroscience 298, 81–93. doi: 10.1016/j.neuroscience.2015.04.017
Salehpour, F., Ahmadian, N., Rasta, S. H., Farhoudi, M., Karimi, P., and Sadigh-Eteghad, S. (2017). Transcranial low-level laser therapy improves brain mitochondrial function and cognitive impairment in D-galactose-induced aging mice. Neurobiol. Aging 58, 140–150. doi: 10.1016/j.neurobiolaging.2017.06.025
Schneider, N. G., Lunell, E., Olmstead, R. E., and Fagerström, K.-O. (1996). Clinical pharmacokinetics of nasal nicotine delivery. Clin. Pharmacokinet. 31, 65–80. doi: 10.2165/00003088-199631010-00005
Shen, Y., Gao, H., Shi, X., Wang, N., Ai, D., Li, J., et al. (2014). Glutamine synthetase plays a role in D-galactose-induced astrocyte aging in vitro and in vivo. Exp. Gerontol. 58, 166–173. doi: 10.1016/j.exger.2014.08.006
Smith, R. C., Singh, A., Infante, M., Khandat, A., and Kloos, A. (2002). Effects of cigarette smoking and nicotine nasal spray on psychiatric symptoms and cognition in schizophrenia. Neuropsychopharmacology 27, 479–497. doi: 10.1016/s0893-133x(02)00324-x
Socci, D. J., Sanberg, P. R., and Arendash, G. W. (1995). Nicotine enhances Morris water maze performance of young and aged rats. Neurobiol. Aging 16, 857–860. doi: 10.1016/0197-4580(95)00091-r
Song, X., Bao, M., Li, D., and Li, Y. M. (1999). Advanced glycation in D-galactose induced mouse aging model. Mech. Ageing Dev. 108, 239–251. doi: 10.1016/s0047-6374(99)00022-6
Sunyer, B., Patil, S., Höger, H., and Lubec, G. (2007). Barnes maze, a useful task to assess spatial reference memory in the mice. Nat. Protoc. 390, 10–38. doi: 10.1038/nprot.2007.390
Tizabi, Y., Manaye, K. F., and Taylor, R. E. (2005). Nicotine blocks ethanol-induced apoptosis in primary cultures of rat cerebral cortical and cerebellar granule cells. Neurotox. Res. 7, 319–322. doi: 10.1007/bf03033888
Torres, O. V., Gentil, L. G., Natividad, L. A., Carcoba, L. M., and O’Dell, L. E. (2013). Behavioral, biochemical, and molecular indices of stress are enhanced in female versus male rats experiencing nicotine withdrawal. Front. Psychiatry 4:38. doi: 10.3389/fpsyt.2013.00038
Tsai, S.-J., and Yin, M.-C. (2012). Anti-oxidative, anti-glycative and anti-apoptotic effects of oleanolic acid in brain of mice treated by D-galactose. Eur. J. Pharmacol. 689, 81–88. doi: 10.1016/j.ejphar.2012.05.018
Tyler, W. J., Alonso, M., Bramham, C. R., and Pozzo-Miller, L. D. (2002). From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn. Mem. 9, 224–237. doi: 10.1101/lm.51202
Vafaee, M. S., Gjedde, A., Imamirad, N., Vang, K., Chakravarty, M. M., Lerch, J. P., et al. (2015). Smoking normalizes cerebral blood flow and oxygen consumption after 12-hour abstention. J. Cereb. Blood Flow Metab. 35, 699–705. doi: 10.1038/jcbfm.2014.246
Wei, H., Li, L., Song, Q., Ai, H., Chu, J., and Li, W. (2005). Behavioural study of the D-galactose induced aging model in C57BL/6J mice. Behav. Brain Res. 157, 245–251. doi: 10.1016/j.bbr.2004.07.003
Wongtrakool, C., Grooms, K., Bijli, K. M., Crothers, K., Fitzpatrick, A. M., and Hart, C. M. (2014). Nicotine stimulates nerve growth factor in lung fibroblasts through an NFκB-dependent mechanism. PLoS One 9:e109602. doi: 10.1371/journal.pone.0109602
Woo, J.-Y., Gu, W., Kim, K.-A., Jang, S.-E., Han, M. J., and Kim, D.-H. (2014). Lactobacillus pentosus var. plantarum C29 ameliorates memory impairment and inflammaging in a D-galactose-induced accelerated aging mouse model. Anaerobe 27, 22–26. doi: 10.1016/j.anaerobe.2014.03.003
Xiaoyu, W. (2015). The exposure to nicotine affects expression of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in neonate rats. Neurol. Sci. 36, 289–295. doi: 10.1007/s10072-014-1934-y
Zhang, X., Liu, W., Niu, X., and An, L. (2010). Systemic administration of catalpol prevents D-galactose induced mitochondrial dysfunction in mice. Neurosci. Lett. 473, 224–228. doi: 10.1016/j.neulet.2010.02.054
Zhang, X., Zhang, B., Yang, X., Zhang, W., and Li, L. (1996). Behavior and memory changes in D-galactose-induced aging rat model. Chin. J. Gerontol. 16, 230–232.
Zhang, Z.-F., Fan, S.-H., Zheng, Y.-L., Lu, J., Wu, D.-M., Shan, Q., et al. (2009). Purple sweet potato color attenuates oxidative stress and inflammatory response induced by D-galactose in mouse liver. Food Chem. Toxicol. 47, 496–501. doi: 10.1016/j.fct.2008.12.005
Zhou, Y., Dong, Y., Xu, Q., He, Y., Tian, S., Zhu, S., et al. (2013). Mussel oligopeptides ameliorate cognition deficit and attenuate brain senescence in D-galactose-induced aging mice. Food Chem. Toxicol. 59, 412–420. doi: 10.1016/j.fct.2013.06.009
Keywords: aging, nicotine, learning and memory, oxidative stress, mitochondrial dysfunction, neurotrophic factors
Citation: Majdi A, Sadigh-Eteghad S, Talebi M, Farajdokht F, Erfani M, Mahmoudi J and Gjedde A (2018) Nicotine Modulates Cognitive Function in D-Galactose-Induced Senescence in Mice. Front. Aging Neurosci. 10:194. doi: 10.3389/fnagi.2018.00194
Received: 20 December 2017; Accepted: 08 June 2018;
Published: 13 July 2018.
Edited by:
Ashok Kumar, University of Florida, United StatesReviewed by:
Karolina Pytka, Jagiellonian University, PolandCopyright © 2018 Majdi, Sadigh-Eteghad, Talebi, Farajdokht, Erfani, Mahmoudi and Gjedde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saeed Sadigh-Eteghad, c2FlZWQuc2FkaWdldGVnYWRAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.