 Safikur Rahman
Safikur Rahman Ayyagari Archana
Ayyagari Archana Arif Tasleem Jan
Arif Tasleem Jan Rinki Minakshi
Rinki Minakshi- 1Department of Medical Biotechnology, Yeungnam University, Gyeongsan, South Korea
- 2Department of Microbiology, Swami Shraddhanand College, University of Delhi, New Delhi, India
- 3School of Biosciences and Biotechnology, Baba Ghulam Shah Badshah University, Rajouri, India
- 4Institute of Home Economics, University of Delhi, New Delhi, India
Alzheimer’s disease (AD), a neurodegenerative disorder, is most common cause of dementia witnessed among aged people. The pathophysiology of AD develops as a consequence of neurofibrillary tangle formation which consists of hyperphosphorylated microtubule associated tau protein and senile plaques of amyloid-β (Aβ) peptide in specific brain regions that result in synaptic loss and neuronal death. The feeble buffering capacity of endoplasmic reticulum (ER) proteostasis in AD is evident through alteration in unfolded protein response (UPR), where UPR markers express invariably in AD patient’s brain samples. Aging weakens UPRER causing neuropathology and memory loss in AD. This review highlights molecular signatures of UPRER and its key molecular alliance that are affected in aging leading to the development of intriguing neuropathologies in AD. We present a summary of recent studies reporting usage of small molecules as inhibitors or activators of UPRER sensors/effectors in AD that showcase avenues for therapeutic interventions.
Introduction
Alzheimer’s disease (AD), the most common form of dementia faced by more than 40 million people worldwide, significantly affect morbidity and mortality in aged people (Alzheimer’s Association, 2016; Fiest et al., 2016; Scheltens et al., 2016; Cass, 2017). The most vulnerable group falling as target is above 65 years, which puts aging as the crucial risk factor associated with development of the disease (Alzheimer’s Association, 2016; Fiest et al., 2016; Scheltens et al., 2016; Cass, 2017). AD is a progressively neurodegenerative disorder, characterized by cognitive alterations and behavioral changes that owe to synaptic impairment and loss of neurons (Alzheimer’s Association, 2016; Scheltens et al., 2016). Mutations in genes encoding APP (amyloid precursor protein), presenilin 1 and 2 (PS1 and PS2 respectively), as well as ε4 allele of Apolipoprotein E are reported to be linked to rare familial and early development of AD (Selkoe, 2001a,b; Scheltens et al., 2016). AD leads to the formation of neurofibrillary tangles having hyperphosphorylated microtubule associated tau protein and senile plaques of amyloid-β (Aβ) peptide in specific brain regions, result in brain inflammation, astrogliosis and microglial proliferation (Citron, 2002; Selkoe, 2004a,b; Cleary et al., 2005; Haass and Selkoe, 2007; Atwood and Bowen, 2015; Minter et al., 2016; Sami et al., 2017). Gradual accumulation of Aβ peptide attributed to β- and γ-secretases action on the APP, results in synaptic loss and neuronal death (Chyung et al., 2005; Tatarnikova et al., 2015).
The expression pattern of neurodegenerative pathologies shows distinct molecular signatures, such as misfolded Aβ aggregation and tau protein hyperphosphorylation in the brain (Jiang et al., 2010; Atwood and Bowen, 2015; Sami et al., 2017). How this load of protein aggregates disrupt the neuronal function is still a mystery to medical science? In this review, we have tried to focus on the role of ER stress and the ensuing unfolded protein response (UPRER) imposed on the neuronal cell due to misfolded protein aggregates. Also, we have discussed various therapeutic interventions targeting the molecules involved in UPR pathways aiming at averting the neuropathologies of AD.
ER Stress and UPRER
Adversities in the endoplasmic reticulum (ER) microenvironment like nutrient deprivation, changes in redox potential, calcium homeostasis, hypoxia and accumulation of unfolded/misfolded protein triggers the UPRER (Schroder and Kaufman, 2005; Moneim, 2015). UPRER is a highly conserved signaling cascade in all eukaryotes involved in the cellular homeostasis (Ellgaard and Helenius, 2003; Mori, 2009; Walter and Ron, 2011) through transcriptional remodeling of ER proteostasis pathways (Lee et al., 2003; Yamamoto et al., 2007; Shoulders et al., 2013; Genereux et al., 2015). The ER lumen harbors various molecular chaperones like the Glucose Regulated Protein 78 kDa (GRP78) that are recruited to misfolded nascent peptides for aiding in their proper folding (Bertolotti et al., 2000; Shen et al., 2002). A plethora of studies have reported UPRER upregulation in the brain samples of Alzheimer’s patients (Hamos et al., 1991; Hoozemans et al., 2005, 2009).
The UPRER embodies a complex network comprised of three stress-responsive transmembrane proteins, Protein Kinase RNA like ER kinase (PERK), Inositol Requiring Element 1 (IRE1) and Activating Transcription Factor 6 (ATF6; Figure 1; Schroder and Kaufman, 2005; Walter and Ron, 2011; Minakshi et al., 2017; Rahman et al., 2017). PERK, a type 1 transmembrane kinase protein, gets trans-autophosphorylated and homodimerized after activation, thereby promoting phosphorylation of serine residues on cytoplasmic eIF2α (eukaryotic initiation factor 2 alpha; Harding et al., 1999; Bertolotti et al., 2000; Ma et al., 2002; Marciniak et al., 2006). Despite the general translational halt induced by the phosphorylated eIF2α (eIF2α-P), certain specific mRNAs bearing internal ribosome entry site (IRES), like the Activating Transcription Factor 4 (ATF4) mRNAs continues to be translated (Harding et al., 2000a; Baumeister et al., 2005). ATF4 regulates genes for various foldases, chaperones, regulatory proteins of the redox and autophagy, cholesterol metabolism etc. (Harding et al., 2003; Fusakio et al., 2016). CCAAT enhancer-binding (C/EBP) protein homologous protein (CHOP) is also a direct target of ATF4 and represents the pro-apoptotic component of the UPRER (Han et al., 2013). In a study, wild type mice subjected to tunicamycin injection showed higher degrees of apoptosis in their renal epithelium as compared to CHOP knockout mice (Marciniak et al., 2004; Onuki et al., 2004). PERK also induces the activation of another transcription factor nuclear factor (erythroid derived 2)-like 2 (Nrf2) independent of eIF2α, which regulates the antioxidant response (Cullinan et al., 2003; Cullinan and Diehl, 2004).
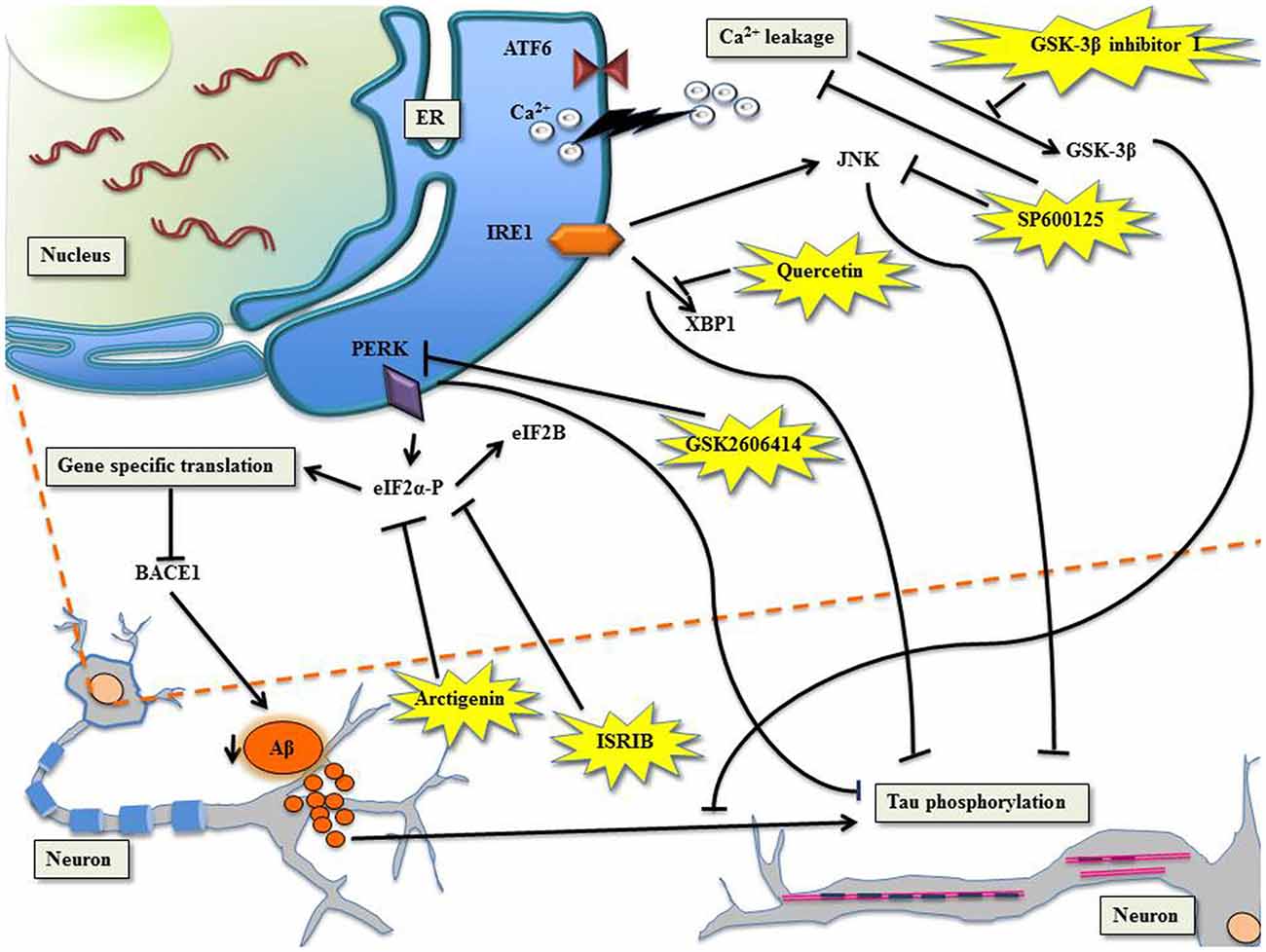
Figure 1. Targeting unfolded protein response (UPR) to manage Alzheimer’s disease (AD) with specific molecules. PERK/eIF2α: the phosphorylation of eIF2α shuts down global translation in the cell but for gene-specific translation upregulation of mRNA with internal ribosome entry site (IRES), for example β-site APP cleaving enzyme-1 (BACE1), the β-secretase enzyme (Ohno, 2014). Arctigenin, targets eIF2α-P thereby downregulating BACE1, consequently protects neurons from amyloid-β (Aβ) toxicity. ISRIB, affects eIF2B leading to inhibition of eIF2α-P, which comprehensively restores protein translation and hence enhances long term memory. PERK can be directly inhibited by GSK2606414, leading to halt in tau phosphorylation. Ca2+ leakage induced activation of glycogen synthase kinase-3β (GSK-3β) can be checked by GSK-3β inhibitor I, which prevents Aβ induced phosphorylation of tau. IRE1/XBP1: Quercetin, activates endoribonuclease activity of IRE1 inhibiting tau hyperphosphorylation. The c-Jun N-terminal kinase (JNK) inhibitor, SP600125, inhibits Ca2+ leakage and inhibits Aβ-induced c-Jun phosphorylation.
IRE1 is the most evolutionarily conserved ER stress transducer (Tirasophon et al., 1998), which upon activation, undergoes dimerization and trans-autophosphorylation, leading to the activation of its cytosolic endoribonuclease activity that splices a 26-nucleotide intron from the mRNA encoding transcription factor X box binding protein 1 (XBP1) forming XBP1(S) (Yoshida et al., 2001, 2003). The XBP1(S) upregulates genes involved in ER protein maturation and ER-associated degradation (ERAD; Lee et al., 2003; Acosta-Alvear et al., 2007). Cells lacking XBP1 are more sensitive to hypoxia-induced apoptosis (Romero-Ramirez et al., 2004). Upon activation, IRE1 also activates c-Jun N-terminal kinase (JNK) through tumor necrosis factor receptor-associated factor 2 (TRAF2); Zeng et al., 2015). IRE1-mediated JNK activation has been demonstrated to trigger autophagy under ER-stress (Urano et al., 2000).
ATF6 is a type II transmembrane protein, with a basic leucine zipper (bZIP) domain (Yoshida et al., 1998). During the imposed stress, luminal domain of ATF6 loses its association with GRP78, triggering the translocation of ATF6 into the Golgi apparatus where two intramembrane Golgi specific proteases, site 1 protease (S1P) and site 2 protease (S2P), process it. The N-terminal cleaved product p50ATF6 of full length ATF6 (p90ATF), then acts as a transcription factor, which upregulates several genes, including GRP78, Protein Disulfide Isomerase (PDI), XBP1 and CHOP (Haze et al., 1999; Walter and Ron, 2011).
UPRER in Alzheimer’s Disease
In neuronal pathophysiology, the activation of UPRER can have paradoxical affects. During stress condition, activation of UPRER could reactivate proteostasis; thereby rescuing the neurons by escalating the rate of protein folding through molecular chaperones, or may trigger neurodegeneration and neuronal collapse through the expression of apoptotic markers.
Evidences support the presence of abundant hyperphosphorylated tau protein and ER stress markers in the neurons of the cortex in postmortem brain samples of AD patients (Scheper and Hoozemans, 2015). It is presumed that ER stress is a cell death mechanism triggered by Aβ, and is linked to changes in ER calcium homeostasis (Cornejo and Hetz, 2013). Under the influence of Aβ imposed ER stress, Ca2+ leaching from ER is taken up by mitochondria leading to activation of apoptotic death of neurons (Fonseca et al., 2013). The presenilins are responsible for passive ER Ca2+ outflow. Documents support that aging neurons fail to maintain tight Ca2+ homeostasis across plasma membrane and ER (Supnet and Bezprozvanny, 2010). Such effects paved the way for “calcium hypothesis of brain aging and AD” (Khachaturian, 1989). Rise in prolonged imbalanced Ca2+ invites ROS accumulation and mitochondrial dysfunction resulting in neuronal death (Supnet and Bezprozvanny, 2010). ER stress may display binary role in AD, firstly modulating the production kinetics of amyloid plaques and secondly altering the cognitive functions in a distinct way (Halliday and Mallucci, 2015). Neurons of AD patients were also characterized by GRP78 induction in temporal cortex and hippocampus and phosphorylation of PERK (p-PERK; Hoozemans et al., 2005).
Active protein synthesis is a hallmark feature of synaptic plasticity and consolidation of memory (Costa-Mattioli et al., 2009). PERK signaling and protein translation control was linked to the cognitive impairment observed in AD models (Devi and Ohno, 2013, 2014). Impairment of cognitive functions due to the reduction in synaptic protein synthesis is displayed during increased phosphorylation of eIF2α (Costa-Mattioli et al., 2005, 2009; Jiang et al., 2010). Mitigating the expression of PERK improves cognitive function and synaptic plasticity in an AD model (Devi and Ohno, 2014). Moreover, targeting other eIF2α kinases like General Control Nonderepressible-2 (GCN2) and dsRNA-dependent protein kinase R (PKR) was also witnessed not only to improve learning and memory processes (Devi and Ohno, 2013), but also reduced inflammation (Lourenco et al., 2013). These results significantly indicate that genetic manipulation of PERK improved cognitive ability of cells to survive under stress conditions induced by Aβ deposition.
The activation of UPRER in early stages of AD could be protective through activation of autophagy. However, sustained UPRER activation may be detrimental to the neurons (Hoozemans et al., 2005; Nijholt et al., 2011). The expression of XBP1 in Drosophila where the AD-associated Aβ peptide was expressed in neurons, led to reduced neurotoxicity, supporting the cytoprotective role of XBP1 (Casas-Tinto et al., 2011). In Caenorhabditis elegans (C. elegans) models expressing aggregation-prone mutant tau variants, XBP-1 was identified to be playing a similar protective role (Kraemer et al., 2006; Loewen and Feany, 2010). However, reports also suggest that IRE1 interacts with PS1 leading to activation of proapoptotic signaling by JNK (Shoji et al., 2000). The JNK3 (member of JNK family) localized in brain, is highly expressed in brain tissue and cerebrospinal fluid sample from AD patients (Gourmaud et al., 2015) and the activation of JNK3 exacerbates stress perpetuating AD pathology (Yoon and Jo, 2012).
Aging, UPRER and Alzheimer’s Disease
Aging is the single most important risk factor for AD. Decline in the UPRER with advancing age marked by the oxidative damage of ER chaperones, leads to disempowering of protein folding capacity (Rabek et al., 2003; Nuss et al., 2008). Studies report that the levels of GRP78 were low in murine cortex, in rat hippocampus, cortex, cerebellum, as well as in a multitude of organs (Paz Gavilán et al., 2006; Hussain and Ramaiah, 2007; Naidoo et al., 2008). Transcription of PERK mRNA were lowered in the aging rat hippocampus, while an increment was reported in the expression of growth arrest and DNA damage protein 34 (GADD34), because it escapes the effect of eIF2α-P translational inhibition (Paz Gavilán et al., 2006). Studies on C. elegans revealed that the activation of IRE1 branch of the UPRER diminishes during the fertile period of adulthood, manifesting in lowered immunity against ER stress (Taylor and Dillin, 2013). The implication of IRE1/XBP1 tier in aging was proven in C. elegans where IRE1 defect reduced life span (Chen et al., 2009).
Mitochondria, Oxidative Stress and Alzheimer’s Disease
Under the imposed stress, apart from UPRER coming to the rescue, the herald of mitochondrial UPR (UPRmt) ensuing after accumulation of unfolded peptide load is well documented. The pathway focuses on invigorating folding and degradation of misfolded peptides in mitochondrial matrix through the execution of retrograde transcriptional activation (Arnould et al., 2015). AD being a multifactorial malady, the accumulation of Aβ not only affects ER but also mitochondria. There are accumulating evidences, which support deposition of Aβ in mitochondrial matrix disrupting signaling of the organelle thereby leading to neurodegeneration (Kawamata and Manfredi, 2017). Impairment in the production and functionalities of metabolic enzymes preferentially of TCA cycle disturbs energy metabolism of the brain. Mitochondrial dysfunction causes depletion of cellular ATP pool and enhanced ROS production, which is well implicated in the pathogenesis of AD (Swerdlow et al., 2014; Hoekstra et al., 2016). Besides, impairment of mitochondrial turnover and function in brain, aging potentiates oxidative stress, leading to significant decrease in the cytochrome C oxidase activity that is associated with rise in oxygen radicals in different regions of postmortem AD brain (Figure 2; Hirai et al., 2001; Mosconi et al., 2007; Krishnan et al., 2012). A strong correlation of the cognitive decline with increase in oxidative stress is observed in AD patients (Revel et al., 2015). Incidence of aberrant Aβ processing ensues after the oxidation of mitochondrial DNA (mtDNA) under stressful circumstances (Volgyi et al., 2015).
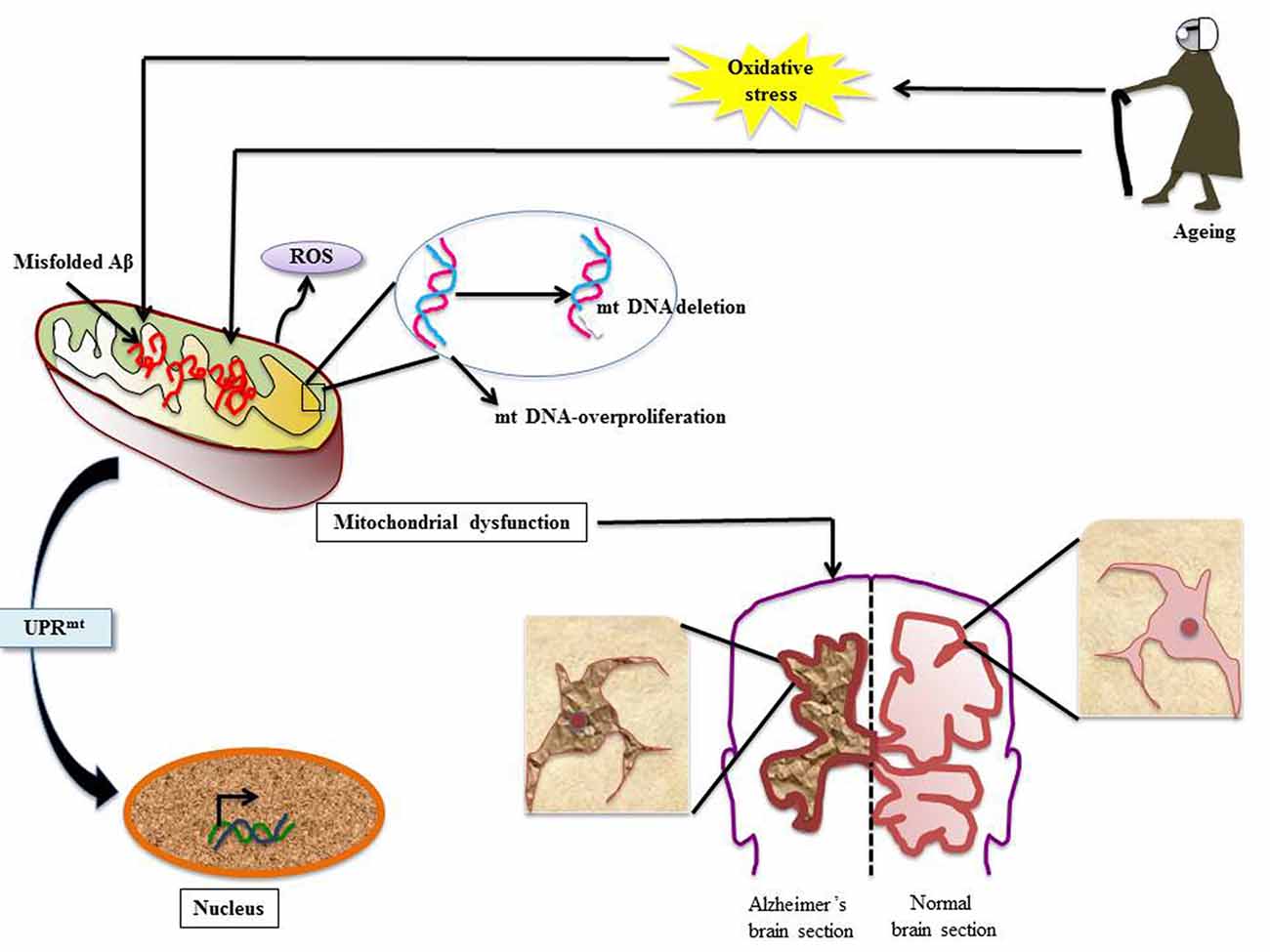
Figure 2. Mitochondrial dysfunction in AD: various stress insults like aging and oxidative stress disrupt client protein folding in mitochondria thereby invoking mitochondrial UPR (UPRmt). Numerous events line up; there is mitochondrial DNA (mtDNA) proliferation and deletion, misfolded Aβ overload and ROS generation. This leads to the condition of mitochondrial dysfunction and to rescue the ailing cell UPRmt is stimulated. The effect of mitochondrial dysfunction leads to development of neuropathologies associated with AD (Aliev et al., 2008, 2013; Onyango et al., 2016).
Aberrations in mtDNA have been well studied in AD. In an elegant study by Aliev et al. (2013) mtDNA-proliferation and deletion has been reported in AD tissues. Furthermore, the report also illustrates abnormal mitochondrial function in damaged hippocampal neurons in human AD as well as transgenic AD models. In another study using in situ hybridization, Aliev et al. (2008) detected a 5 kB deletion in mtDNA under oxidative stress in abnormal neurons. Such mitochondrial anomalies were also reported to help in AD pathogenesis in Aβ transgenic mice (Aliev et al., 2008).
In a study proving the existence of interlink between mitochondrial dysfunction and AD, the pharmacological/genetic targeting of mitochondrial translation process not only increased life span of GMC101 (model of Aβ proteotoxicity), but also showed reduction in beta-amyloid aggregation in worms and transgenic mouse models of AD (Sorrentino et al., 2017). Treatment of the mitochondrial division inhibitor-1 (mdiv-1) that inhibits mitochondrial fragmentation, thereby rescuing mitochondrial distribution, improves mitochondrial function in CRND8 (AD mouse model) neurons (Reddy et al., 2017; Wang et al., 2017). Treatment with mdivi-1 also causes a decrease in extracellular amyloid deposition and Aβ1–42/Aβ1–40 ratio (Wang et al., 2017). Additionally, SIRT-3, a sirtuin localized to inner mitochondrial membrane, has been found associated with enhancement in the levels of glutathione (Onyango et al., 2002; Someya et al., 2010). As downregulation of SIRT-3 was found to be having a retrograde effect on p53 mediated mitochondrial and neuronal damage in AD, its modulation by therapeutics was found to ameliorate mitochondrial pathology and neurodegeneration in AD (Lee et al., 2018).
Derangement of Glucose Metabolism in Alzheimer’s Disease: The Fallible UPRER
Among the many observed hallmarks of AD, positron emission tomography (PET) revealed a deranged glucose metabolism in brain regions. Aging registers diminished brain glucose utilization that surges in AD (Ivançević et al., 2000). Various reports suggest that UPRER is linked to abnormal glucose metabolism and insulin resistance (Hetz et al., 2015). Type 2 diabetes mellitus (T2DM) has been mechanistically linked to AD pathogenesis, where higher insulin resistance poses a greater risk of AD with reduced glucose uptake in the brain as well as memory loss (Willette et al., 2015; Wijesekara et al., 2017). In addition, there is decline in key neuronal glucose transporters, GLUT1 and GLUT3, as shown in AD mouse models (Ding et al., 2013). The exact molecular mechanism underlying the effect of glucose uptake in AD model is not completely understood, but evidences suggest a close link between AD and insulin signaling. Apart from controlling glucose metabolism, insulin also regulates neural development with respect to learning and memory (Ying et al., 2017).
The lowering in glucose concentration due to lack of active transporters (GLUT1 and GLUT3) instates mitigating effect on hexosamine pathway (HBP), due to which O-GlcNAcylation is compromised with hyperphosphorylation on tau protein (Liu et al., 2009). XBP1(S) is shown to directly target the rate limiting enzyme of HBP, glutamine fructose-6-phosphate aminotransferase (GFAT1; Wang et al., 2014), as XBP1(S) transgenics showed rise in O-GlcNAcylation (Wang et al., 2014). The situation of insulin resistance established in aging has also been shown to increase HBP flux (Einstein et al., 2008). A gain-of-function mutation in GFAT1 of C. elegans showed significant induction of ERAD and autophagy favoring longevity (Denzel et al., 2014).
Protein aggregation is a consequence of AD which is a result of abnormal proteostasis in the cell (Kaushik and Cuervo, 2015). An increase in the UPRER driven protein homeostasis was observed with the overexpression of GLUT1 as this promoted downregulation of expression of GRP78. GRP78, being the negative regulator of the UPRER, binds ATF6 and IRE1 thereby continuing them in an inactive state. One interesting study showed that flies (with increased glucose transport) when fed with the drug metformin showed mitigated levels of GRP78 with ensuing gain in lifespan, additionally the expression of GLUT1 and its association with the beginning of UPRER exerted neuroprotective effect (Niccoli et al., 2016).
Targeting UPRER to Manage AD
The involvement of ER stress and hence the UPRER in neuropathologies exposes the molecules of the pathway as attractive targets for therapeutic interventions. Here, we have compiled reports from studies that have targeted molecules of UPRER for managing the deterioration caused by AD (Figure 1).
eIF2α and PERK in AD
There are accumulating evidences that support increased phosphorylation of PERK and eIF2α in AD (Chang et al., 2002; Page et al., 2006; Kim et al., 2007). The processing of highly expressed single-pass transmembrane protein in brain, the amyloid precursor protein, leads to the generation of neurotoxic Aβ during neuropathogenesis. Reports suggest that the secretase β-site APP cleaving enzyme-1 (BACE1), increases APP cleavage as a result of eIF2α phosphorylation leading to the production of Aβ in neurons (O’Connor et al., 2008). The PERK tier of UPR when suppressed leads to the alleviation of synaptic plasticity and memory loss in AD (Ma et al., 2013). The administration of arctigenin, a bioactive product from Arctium lappa (L.), has been known to inhibit BACE1 translation through dephosphorylation of eIF2α-P (Zhu et al., 2013). The phosphorylation of eIF2α is central to integrated stress response (ISR) that modulates UPR (Harding et al., 2000b) and formation of memory proteins (Costa-Mattioli et al., 2005). ISR inhibitor (ISRIB) interferes with ISR by affecting eIF2B activity whose competitive inhibitor is eIF2α-P (Krishnamoorthy et al., 2001; Sekine et al., 2015; Bogorad et al., 2017). This comprehensively reverses the effect of eIF2α-P, which resulted in the restoration of translation and hence long term memory enhancement in rodents (Sidrauski et al., 2013, 2015). The genetic deletion of eIF2 kinases, PERK, GCN2 and dsRNA-dependent protein kinase (PKR) ameliorate synaptic plasticity and memory in AD models (Ma et al., 2013). The transient translational halt induced by PERK-P/eIF2α-P was challenged by GSK2606414, a PERK inhibitor, because of which tau phosphorylation could be checked, resulting in the amelioration of neurodegeneration (Axten et al., 2012; Radford et al., 2015). The development of AD manifested by Aβ accumulation forces tau hyper phosphorylation in sync with increased activity of glycogen synthase kinase-3β (GSK-3β) in the cortical neurons (Takashima et al., 1993, 1996; Tomidokoro et al., 2001; De Felice et al., 2008; Resende et al., 2008). Resende et al. (2008) showed that Aβ oligomers cause ER stress linked calcium leakage which in turn leads to GSK-3β activation, the later when inhibited by GSK-3β inhibitor I, led to the prevention of Aβ induced phosphorylation of tau.
IRE1/XBP1 in AD
The advantageous effects of XBP1 on memory was proven in neural-specific XBP1 knockout mice featuring impaired learning and synaptic plasticity deficit, where injections of adeno-associated viruses delivered XBP1(S) resulted in establishing long-term hippocampus memory (Martínez et al., 2016). In accordance with this finding, another study reinforced the neuro-protective role of XBP1 in AD mice (Casas-Tinto et al., 2011; Cisse et al., 2017). Nonetheless, a flavonol, called quercetin, activated endoribonuclease activity of IRE1 and inhibited tau hyperphosphorylation (de Boer et al., 2006; Suganthy et al., 2016). In cases of familial AD, deletions or mutations in presenilin genes accentuate ER Ca2+ leakage. The JNK inhibitor, SP600125, when challenged in PS1/PS2 double knockout mouse embryonic fibroblast, caused inhibition of Ca2+ leakage (Das et al., 2012). The neuroinflammation exhibited in AD through tau phosphorylation mediated by the kinase activity of JNK was inhibited by SP600125, consequently inhibiting Aβ-induced c-Jun phosphorylation (Vukic et al., 2009; Zhou et al., 2015).
Future Directions and Concluding Remarks
ER, being a central organelle in nerve cells, coordinates with the cellular homeostasis by managing translation/modification of proteins and Ca2+ equilibrium, thereby maintains the proper signaling in brain. The disruption in neuronal physiology is quite evident in age-related AD where ER dysfunctions are prominently expressed in the form of imbalance in proteostasis. Advancements in studies based on AD models have clearly shown how we can intervene the molecular pillars of UPRER and its associated signaling cascades to manage neurodegeneration in age-related AD. The present review is an attempt to revise functional relevance of the studies conducted in the field of management of age-related AD through therapeutic interventions on the UPRER pathway and its associate’s molecules. Studies reinforce that the strategies where intervening the molecules, which are involved in transposing effects of aging on neurodegeneration, will cause reduction in probability of AD pathology. The manifestation of ER proteostasis is a direct indication of healthy nervous system. Progression in AD witnesses glucose hypo-metabolism in brain, reduction in glucose transporters in neurons and endothelial cells of blood brain barrier in direct proportion with the amount of neurofibrillary tangles. Type 2 diabetics with higher insulin resistance are at a greater risk of AD. Recent reports elucidate that managing UPRER can exert neuroprotective effect in AD (Smith and Mallucci, 2016). Additionally, as evidenced in the study by Sorrentino et al. (2017), the recapitulation of mitochondrial function through activation of UPRmt can impede plaque formation. Aliev et al., also demonstrated link between cancer and AD where mtDNA over-proliferation and deletion induces cell cycle dysregulation prompting oncogenic pathway (Aliev et al., 2013). We have supporting literature that underpins the reversal of AD pathology by anticancer drugs (Cramer et al., 2012). Aiming at therapeutic intervention, the ailing mitochondria can be challenged with specific antioxidants like MitoQ, acetyl-L-carnitine and R-alpha lipoic acid to alleviate AD (Aliev et al., 2011; Volgyi et al., 2015). One remarkable study on astrocytes underpins the protective role of conditioned medium of human mesenchymal stem cells (CM-hMSCA) sourced from adipose tissue against neuropathologies (Baez-Jurado et al., 2017). The state of astrocyte mitochondrial dysfunction has been proven to be a start point for neuronal death (Baez et al., 2016). Pharmacological targeting of astrocytes has been proposed to be a potential way in therapeutics of AD (Baez et al., 2016). A transcriptomic analysis in astrocytes has put forward a conglomeration of various algorithms for strategic approaches in therapeutics of neuropathologies (Barreto et al., 2017).
We still need extensive and efficient model systems where the molecular intricacies of weakened UPRER in aging-induced neuropathology in AD can be ventured upon, so that pharmacological as well as genetic tools could underscore the significance of UPRER as well as UPRmt in aged brain.
Author Contributions
SR and RM conceived the idea. SR, ATJ, AA and RM contributed to writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors extend their thanks to colleagues for their criticism that helped to improve the quality of contents in the perspective of broader audience. No funding was availed to carry out the study.
References
Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M., Lents, N. H., Arias, C., et al. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66. doi: 10.1016/j.molcel.2007.06.011
Aliev, G., Gasimov, E., Obrenovich, M. E., Fischbach, K., Shenk, J. C., Smith, M. A., et al. (2008). Atherosclerotic lesions and mitochondria DNA deletions in brain microvessels: implication in the pathogenesis of Alzheimer’s disease. Vasc. Health Risk Manag. 4, 721–730. doi: 10.2147/vhrm.s2608
Aliev, G., Li, Y., Palacios, H. H., and Obrenovich, M. E. (2011). Oxidative stress induced mitochondrial DNA deletion as a hallmark for the drug development in the context of the cerebrovascular diseases. Recent Pat. Cardiovasc. Drug Discov. 6, 222–241. doi: 10.2174/157489011797376942
Aliev, G., Obrenovich, M. E., Tabrez, S., Jabir, N. R., Reddy, V. P., Li, Y., et al. (2013). Link between cancer and Alzheimer disease via oxidative stress induced by nitric oxide-dependent mitochondrial DNA overproliferation and deletion. Oxid. Med. Cell. Longev. 2013:962984. doi: 10.1155/2013/962984
Alzheimer’s Association. (2016). 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12, 459–509. doi: 10.1016/j.jalz.2016.03.001
Arnould, T., Michel, S., and Renard, P. (2015). Mitochondria retrograde signaling and the UPR mt: where are we in mammals? Int. J. Mol. Sci. 16, 18224–18251. doi: 10.3390/ijms160818224
Atwood, C. S., and Bowen, R. L. (2015). A unified hypothesis of early- and late-onset Alzheimer’s disease pathogenesis. J. Alzheimers Dis. 47, 33–47. doi: 10.3233/JAD-143210
Axten, J. M., Medina, J. R., Feng, Y., Shu, A., Romeril, S. P., Grant, S. W., et al. (2012). Discovery of 7-methyl-5-(1-[3-(trifluoromethyl)phenyl]acetyl-2,3-dihydro- 1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207. doi: 10.1021/jm300713s
Baez, E., Echeverria, V., Cabezas, R., Ávila-Rodriguez, M., Garcia-Segura, L. M., and Barreto, G. E. (2016). Protection by neuroglobin expression in brain pathologies. Front. Neurol. 7:146. doi: 10.3389/fneur.2016.00146
Baez-Jurado, E., Hidalgo-Lanussa, O., Guio-Vega, G., Ashraf, G. M., Echeverria, V., Aliev, G., et al. (2017). Conditioned medium of human adipose mesenchymal stem cells increases wound closure and protects human astrocytes following scratch assay in vitro. Mol. Neurobiol. doi: 10.1007/s12035-017-0771-4 [Epub ahead of print].
Barreto, G. E., Gomez, R. M., Bustos, R. H., Forero, D. A., Aliev, G., Tarasov, V. V., et al. (2017). Approaches of the transcriptomic analysis in astrocytes: potential pharmacological targets. Curr. Pharm. Des. 23, 4189–4197. doi: 10.2174/1381612823666170406113501
Baumeister, P., Luo, S., Skarnes, W. C., Sui, G., Seto, E., Shi, Y., et al. (2005). Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 25, 4529–4540. doi: 10.1128/mcb.25.11.4529-4540.2005
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Bogorad, A. M., Lin, K. Y., and Marintchev, A. (2017). Novel mechanisms of eIF2B action and regulation by eIF2α phosphorylation. Nucleic Acids Res. 45, 11962–11979. doi: 10.1093/nar/gkx845
Casas-Tinto, S., Zhang, Y., Sanchez-Garcia, J., Gomez-Velazquez, M., Rincon-Limas, D. E., and Fernandez-Funez, P. (2011). The ER stress factor XBP1s prevents amyloid-β neurotoxicity. Hum. Mol. Genet. 20, 2144–2160. doi: 10.1093/hmg/ddr100
Cass, S. P. (2017). Alzheimer’s disease and exercise: a literature review. Curr. Sports Med. Rep. 16, 19–22. doi: 10.1249/JSR.0000000000000332
Chang, R. C., Wong, A. K., Ng, H. K., and Hugon, J. (2002). Phosphorylation of eukaryotic initiation factor-2α (eIF2α) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13, 2429–2432. doi: 10.1097/00001756-200212200-00011
Chen, D., Thomas, E. L., and Kapahi, P. (2009). HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 5:e1000486. doi: 10.1371/journal.pgen.1000486
Chyung, J. H., Raper, D. M., and Selkoe, D. J. (2005). γ-secretase exists on the plasma membrane as an intact complex that accepts substrates and effects intramembrane cleavage. J. Biol. Chem. 280, 4383–4392. doi: 10.1074/jbc.M409272200
Cisse, M., Duplan, E., Lorivel, T., Dunys, J., Bauer, C., Meckler, X., et al. (2017). The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 22, 1562–1575. doi: 10.1038/mp.2016.152
Citron, M. (2002). Alzheimer’s disease: treatments in discovery and development. Nat. Neurosci. 5, 1055–1057. doi: 10.1038/nn940
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., et al. (2005). Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84. doi: 10.1038/nn1372
Cornejo, V. H., and Hetz, C. (2013). The unfolded protein response in Alzheimer’s disease. Semin. Immunopathol. 35, 277–292. doi: 10.1007/s00281-013-0373-9
Costa-Mattioli, M., Gobert, D., Harding, H., Herdy, B., Azzi, M., Bruno, M., et al. (2005). Translational control of hippocampal synaptic plasticity and memory by the eIF2α kinase GCN2. Nature 436, 1166–1173. doi: 10.1038/nature03897
Costa-Mattioli, M., Sossin, W. S., Klann, E., and Sonenberg, N. (2009). Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26. doi: 10.1016/j.neuron.2008.10.055
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi: 10.1126/science.1217697
Cullinan, S. B., and Diehl, J. A. (2004). PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 279, 20108–20117. doi: 10.1074/jbc.M314219200
Cullinan, S. B., Zhang, D., Hannink, M., Arvisais, E., Kaufman, R. J., and Diehl, J. A. (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23, 7198–7209. doi: 10.1128/mcb.23.20.7198-7209.2003
Das, H. K., Tchedre, K., and Mueller, B. (2012). Repression of transcription of presenilin-1 inhibits γ-secretase independent ER Ca2+ leak that is impaired by FAD mutations. J. Neurochem. 122, 487–500. doi: 10.1111/j.1471-4159.2012.07794.x
de Boer, V. C., de Goffau, M. C., Arts, I. C., Hollman, P. C., and Keijer, J. (2006). SIRT1 stimulation by polyphenols is affected by their stability and metabolism. Mech. Aging Dev. 127, 618–627. doi: 10.1016/j.mad.2006.02.007
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., et al. (2008). Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol. Aging 29, 1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029
Denzel, M. S., Storm, N. J., Gutschmidt, A., Baddi, R., Hinze, Y., Jarosch, E., et al. (2014). Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell 156, 1167–1178. doi: 10.1016/j.cell.2014.01.061
Devi, L., and Ohno, M. (2013). Deletion of the eIF2α Kinase GCN2 fails to rescue the memory decline associated with Alzheimer’s disease. PLoS One 8:e77335. doi: 10.1371/journal.pone.0077335
Devi, L., and Ohno, M. (2014). PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 35, 2272–2281. doi: 10.1016/j.neurobiolaging.2014.04.031
Ding, F., Yao, J., Rettberg, J. R., Chen, S., and Brinton, R. D. (2013). Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One 8:e79977. doi: 10.1371/journal.pone.0079977
Einstein, F. H., Fishman, S., Bauman, J., Thompson, R. F., Huffman, D. M., Atzmon, G., et al. (2008). Enhanced activation of a “nutrient-sensing” pathway with age contributes to insulin resistance. FASEB J. 22, 3450–3457. doi: 10.1096/fj.08-109041
Ellgaard, L., and Helenius, A. (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191. doi: 10.1038/nrm1052
Fiest, K. M., Roberts, J. I., Maxwell, C. J., Hogan, D. B., Smith, E. E., Frolkis, A., et al. (2016). The prevalence and incidence of dementia due to Alzheimer’s disease: a systematic review and meta-analysis. Can. J. Neurol. Sci. 43, S51–S82. doi: 10.1017/cjn.2016.36
Fonseca, A. C., Ferreiro, E., Oliveira, C. R., Cardoso, S. M., and Pereira, C. F. (2013). Activation of the endoplasmic reticulum stress response by the amyloid-β 1–40 peptide in brain endothelial cells. Biochim. Biophys. Acta 1832, 2191–2203. doi: 10.1016/j.bbadis.2013.08.007
Fusakio, M. E., Willy, J. A., Wang, Y., Mirek, E. T., Al Baghdadi, R. J., Adams, C. M., et al. (2016). Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 27, 1536–1551. doi: 10.1091/mbc.E16-01-0039
Genereux, J. C., Qu, S., Zhou, M., Ryno, L. M., Wang, S., Shoulders, M. D., et al. (2015). Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J. 34, 4–19. doi: 10.15252/embj.201488896
Gourmaud, S., Paquet, C., Dumurgier, J., Pace, C., Bouras, C., Gray, F., et al. (2015). Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: links to cognitive decline. J. Psychiatry Neurosci. 40, 151–161. doi: 10.1503/jpn.140062
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Halliday, M., and Mallucci, G. R. (2015). Modulating the unfolded protein response to prevent neurodegeneration and enhance memory. Neuropathol. Appl. Neurobiol. 41, 414–427. doi: 10.1111/nan.12211
Hamos, J. E., Oblas, B., Pulaski-Salo, D., Welch, W. J., Bole, D. G., and Drachman, D. A. (1991). Expression of heat shock proteins in Alzheimer’s disease. Neurology 41, 345–350. doi: 10.1212/WNL.41.3.345
Han, J., Back, S. H., Hur, J., Lin, Y.-H., Gildersleeve, R., Shan, J., et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490. doi: 10.1038/ncb2738
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., et al. (2000a). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. doi: 10.1016/s1097-2765(00)00108-8
Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H., and Ron, D. (2000b). Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897–904. doi: 10.1016/s1097-2765(00)80330-5
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. doi: 10.1038/16729
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. doi: 10.1016/s1097-2765(03)00105-9
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi: 10.1091/mbc.10.11.3787
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. doi: 10.1038/ncb3184
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023.
Hoekstra, J. G., Hipp, M. J., Montine, T. J., and Kennedy, S. R. (2016). Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann. Neurol. 80, 301–306. doi: 10.1002/ana.24709
Hoozemans, J. J., van Haastert, E. S., Nijholt, D. A., Rozemuller, A. J., Eikelenboom, P., and Scheper, W. (2009). The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 174, 1241–1251. doi: 10.2353/ajpath.2009.080814
Hoozemans, J. J., Veerhuis, R., Van Haastert, E. S., Rozemuller, J. M., Baas, F., Eikelenboom, P., et al. (2005). The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 110, 165–172. doi: 10.1007/s00401-005-1038-0
Hussain, S. G., and Ramaiah, K. V. (2007). Reduced eIF2α phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 355, 365–370. doi: 10.1016/j.bbrc.2007.01.156
Ivançević, V., Alavi, A., Souder, E., Mozley, P. D., Gur, R. E., Bénard, F., et al. (2000). Regional cerebral glucose metabolism in healthy volunteers determined by fluordeoxyglucose positron emission tomography: appearance and variance in the transaxial, coronal, and sagittal planes. Clin. Nucl. Med. 25, 596–602. doi: 10.1097/00003072-200008000-00005
Jiang, X. Y., Litkowski, P. E., Taylor, A. A., Lin, Y., Snider, B. J., and Moulder, K. L. (2010). A role for the ubiquitin-proteasome system in activity-dependent presynaptic silencing. J. Neurosci. 30, 1798–1809. doi: 10.1523/JNEUROSCI.4965-09.2010
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. doi: 10.1038/nm.4001
Kawamata, H., and Manfredi, G. (2017). Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J. Cell Biol. 216, 3917–3929. doi: 10.1083/jcb.201709172
Khachaturian, Z. S. (1989). Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann. N Y Acad. Sci. 568, 1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x
Kim, H. S., Choi, Y., Shin, K. Y., Joo, Y., Lee, Y. K., Jung, S. Y., et al. (2007). Swedish amyloid precursor protein mutation increases phosphorylation of eIF2α in vitro and in vivo. J. Neurosci. Res. 85, 1528–1537. doi: 10.1002/jnr.21267
Kraemer, B. C., Burgess, J. K., Chen, J. H., Thomas, J. H., and Schellenberg, G. D. (2006). Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 15, 1483–1496. doi: 10.1093/hmg/ddl067
Krishnamoorthy, T., Pavitt, G. D., Zhang, F., Dever, T. E., and Hinnebusch, A. G. (2001). Tight binding of the phosphorylated α subunit of initiation factor 2 (eIF2α) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 21, 5018–5030. doi: 10.1128/mcb.21.15.5018-5030.2001
Krishnan, K. J., Ratnaike, T. E., De Gruyter, H. L., Jaros, E., and Turnbull, D. M. (2012). Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol. Aging 33, 2210–2214. doi: 10.1016/j.neurobiolaging.2011.08.009
Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/mcb.23.21.7448-7459.2003
Lee, J., Kim, Y., Liu, T., Hwang, Y. J., Hyeon, S. J., Im, H., et al. (2018). SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 17:e12679. doi: 10.1111/acel.12679
Liu, F., Shi, J., Tanimukai, H., Gu, J., Grundke-Iqbal, I., Iqbal, K., et al. (2009). Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 132, 1820–1832. doi: 10.1093/brain/awp099
Loewen, C. A., and Feany, M. B. (2010). The unfolded protein response protects from tau neurotoxicity in vivo. PLoS One 5:e13084. doi: 10.1371/journal.pone.0013084
Lourenco, M. V., Clarke, J. R., Frozza, R. L., Bomfim, T. R., Forny-Germano, L., Batista, A. F., et al. (2013). TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843. doi: 10.1016/j.cmet.2013.11.002
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi: 10.1038/nn.3486
Ma, K., Vattem, K. M., and Wek, R. C. (2002). Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J. Biol. Chem. 277, 18728–18735. doi: 10.1074/jbc.M200903200
Marciniak, S. J., Garcia-Bonilla, L., Hu, J., Harding, H. P., and Ron, D. (2006). Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 172, 201–209. doi: 10.1083/jcb.200508099
Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., et al. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077. doi: 10.1101/gad.1250704
Martínez, G., Vidal, R. L., Mardones, P., Serrano, F. G., Ardiles, A. O., Wirth, C., et al. (2016). Regulation of memory formation by the transcription factor XBP1. Cell Rep. 14, 1382–1394. doi: 10.1016/j.celrep.2016.01.028
Minakshi, R., Rahman, S., Jan, A. T., Archana, A., and Kim, J. (2017). Implications of aging and the endoplasmic reticulum unfolded protein response on the molecular modality of breast cancer. Exp. Mol. Med. 49:e389. doi: 10.1038/emm.2017.215
Minter, M. R., Taylor, J. M., and Crack, P. J. (2016). The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 136, 457–474. doi: 10.1111/jnc.13411
Moneim, A. E. (2015). Oxidant/Antioxidant imbalance and the risk of Alzheimer’s disease. Curr. Alzheimer Res. 12, 335–349. doi: 10.2174/1567205012666150325182702
Mori, K. (2009). Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 146, 743–750. doi: 10.1093/jb/mvp166
Mosconi, L., Brys, M., Switalski, R., Mistur, R., Glodzik, L., Pirraglia, E., et al. (2007). Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc. Natl. Acad. Sci. U S A 104, 19067–19072. doi: 10.1073/pnas.0705036104
Naidoo, N., Ferber, M., Master, M., Zhu, Y., and Pack, A. I. (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. doi: 10.1523/JNEUROSCI.5685-07.2008
Niccoli, T., Cabecinha, M., Tillmann, A., Kerr, F., Wong, C. T., Cardenes, D., et al. (2016). Increased glucose transport into neurons rescues Aβ toxicity in Drosophila. Curr. Biol. 26, 2291–2300. doi: 10.1016/j.cub.2016.07.017
Nijholt, D. A., de Graaf, T. R., van Haastert, E. S., Oliveira, A. O., Berkers, C. R., Zwart, R., et al. (2011). Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer’s disease. Cell Death Differ. 18, 1071–1081. doi: 10.1038/cdd.2010.176
Nuss, J. E., Choksi, K. B., Deford, J. H., and Papaconstantinou, J. (2008). Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 365, 355–361. doi: 10.1016/j.bbrc.2007.10.194
O’Connor, T., Sadleir, K. R., Maus, E., Velliquette, R. A., Zhao, J., Cole, S. L., et al. (2008). Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 60, 988–1009. doi: 10.1016/j.neuron.2008.10.047
Ohno, M. (2014). Roles of eIF2α kinases in the pathogenesis of Alzheimer’s disease. Front. Mol. Neurosci. 7:22. doi: 10.3389/fnmol.2014.00022
Onuki, R., Bando, Y., Suyama, E., Katayama, T., Kawasaki, H., Baba, T., et al. (2004). An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer’s disease. EMBO J. 23, 959–968. doi: 10.1038/sj.emboj.7600049
Onyango, P., Celic, I., McCaffery, J. M., Boeke, J. D., and Feinberg, A. P. (2002). SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. U S A 99, 13653–13658. doi: 10.1073/pnas.222538099
Onyango, I. G., Dennis, J., and Khan, S. M. (2016). Mitochondrial dysfunction in Alzheimer’s disease and the rationale for bioenergetics based therapies. Aging Dis. 7, 201–214. doi: 10.14336/AD.2015.1007
Page, G., Rioux Bilan, A., Ingrand, S., Lafay-Chebassier, C., Pain, S., Perault Pochat, M. C., et al. (2006). Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience 139, 1343–1354. doi: 10.1016/j.neuroscience.2006.01.047
Paz Gavilán, M., Vela, J., Castaño, A., Ramos, B., del Río, J. C., Vitorica, J., et al. (2006). Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 27, 973–982. doi: 10.1016/j.neurobiolaging.2005.05.010
Rabek, J. P., Boylston, W. H. III., and Papaconstantinou, J. (2003). Carbonylation of ER chaperone proteins in aged mouse liver. Biochem. Biophys. Res. Commun. 305, 566–572. doi: 10.1016/s0006-291x(03)00826-x
Radford, H., Moreno, J. A., Verity, N., Halliday, M., and Mallucci, G. R. (2015). PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 130, 633–642. doi: 10.1007/s00401-015-1487-z
Rahman, S., Jan, A. T., Ayyagari, A., Kim, J., Kim, J., and Minakshi, R. (2017). Entanglement of UPRER in aging driven neurodegenerative diseases. Front. Aging Neurosci. 9:341. doi: 10.3389/fnagi.2017.00341
Reddy, P. H., Manczak, M., and Yin, X. (2017). Mitochondria-division inhibitor 1 protects against amyloid-β induced mitochondrial fragmentation and synaptic damage in Alzheimer’s disease. J. Alzheimers Dis. 58, 147–162. doi: 10.3233/JAD-170051
Resende, R., Ferreiro, E., Pereira, C., and Oliveira, C. R. (2008). ER stress is involved in Aβ-induced GSK-3β activation and tau phosphorylation. J. Neurosci. Res. 86, 2091–2099. doi: 10.1002/jnr.21648
Revel, F., Gilbert, T., Roche, S., Drai, J., Blond, E., Ecochard, R., et al. (2015). Influence of oxidative stress biomarkers on cognitive decline. J. Alzheimers Dis. 45, 553–560. doi: 10.3233/JAD-141797
Romero-Ramirez, L., Cao, H., Nelson, D., Hammond, E., Lee, A. H., Yoshida, H., et al. (2004). XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 64, 5943–5947. doi: 10.1158/0008-5472.can-04-1606
Sami, N., Rahman, S., Kumar, V., Zaidi, S., Islam, A., Ali, S., et al. (2017). Protein aggregation, misfolding and consequential human neurodegenerative diseases. Int. J. Neurosci. 127, 1047–1057. doi: 10.1080/00207454.2017.1286339
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Scheper, W., and Hoozemans, J. J. (2015). The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331. doi: 10.1007/s00401-015-1462-8
Schroder, M., and Kaufman, R. J. (2005). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789. doi: 10.1146/annurev.biochem.73.011303.074134
Sekine, Y., Zyryanova, A., Crespillo-Casado, A., Fischer, P. M., Harding, H. P., and Ron, D. (2015). Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348, 1027–1030. doi: 10.1126/science.aaa6986
Selkoe, D. J. (2001a). Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid β-protein. J. Alzheimers Dis. 3, 75–80. doi: 10.3233/jad-2001-3111
Selkoe, D. J. (2001b). Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766. doi: 10.1152/physrev.2001.81.2.741
Selkoe, D. J. (2004a). Alzheimer disease: mechanistic understanding predicts novel therapies. Ann. Intern. Med. 140, 627–638. doi: 10.7326/0003-4819-140-8-200404200-00047
Selkoe, D. J. (2004b). Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 6, 1054–1061. doi: 10.1038/ncb1104-1054
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111. doi: 10.1016/s1534-5807(02)00203-4
Shoji, M., Iwakami, N., Takeuchi, S., Waragai, M., Suzuki, M., Kanazawa, I., et al. (2000). JNK activation is associated with intracellular β-amyloid accumulation. Brain Res. Mol. Brain Res. 85, 221–233. doi: 10.1016/s0169-328x(00)00245-x
Shoulders, M. D., Ryno, L. M., Genereux, J. C., Moresco, J. J., Tu, P. G., Wu, C., et al. (2013). Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 3, 1279–1292. doi: 10.1016/j.celrep.2013.03.024
Sidrauski, C., Acosta-Alvear, D., Khoutorsky, A., Vedantham, P., Hearn, B. R., Li, H., et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2:e00498. doi: 10.7554/eLife.00498
Sidrauski, C., McGeachy, A. M., Ingolia, N. T., and Walter, P. (2015). The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 4:e05033. doi: 10.7554/eLife.05033
Smith, H. L., and Mallucci, G. R. (2016). The unfolded protein response: mechanisms and therapy of neurodegeneration. Brain 139, 2113–2121. doi: 10.1093/brain/aww101
Someya, S., Yu, W., Hallows, W. C., Xu, J., Vann, J. M., Leeuwenburgh, C., et al. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812. doi: 10.1016/j.cell.2010.10.002
Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J. S., Zhang, H., D’Amico, D., et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193. doi: 10.1038/nature25143
Suganthy, N., Devi, K. P., Nabavi, S. F., Braidy, N., and Nabavi, S. M. (2016). Bioactive effects of quercetin in the central nervous system: focusing on the mechanisms of actions. Biomed. Pharmacother. 84, 892–908. doi: 10.1016/j.biopha.2016.10.011
Supnet, C., and Bezprozvanny, I. (2010). The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 47, 183–189. doi: 10.1016/j.ceca.2009.12.014
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Takashima, A., Noguchi, K., Michel, G., Mercken, M., Hoshi, M., Ishiguro, K., et al. (1996). Exposure of rat hippocampal neurons to amyloid β peptide (25–35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 β. Neurosci. Lett. 203, 33–36. doi: 10.1016/s0006-3495(96)79570-x
Takashima, A., Noguchi, K., Sato, K., Hoshino, T., and Imahori, K. (1993). Tau protein kinase I is essential for amyloid β-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. U S A 90, 7789–7793. doi: 10.1073/pnas.90.16.7789
Tatarnikova, O. G., Orlov, M. A., and Bobkova, N. V. (2015). β-amyloid and tau-protein: structure, interaction, and prion-like properties. Biochemistry (Mosc) 80, 1800–1819. doi: 10.1134/S000629791513012X
Taylor, R. C., and Dillin, A. (2013). XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. doi: 10.1016/j.cell.2013.05.042
Tirasophon, W., Welihinda, A. A., and Kaufman, R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. doi: 10.1101/gad.12.12.1812
Tomidokoro, Y., Ishiguro, K., Harigaya, Y., Matsubara, E., Ikeda, M., Park, J. M., et al. (2001). Aβ amyloidosis induces the initial stage of tau accumulation in APP(Sw) mice. Neurosci. Lett. 299, 169–172. doi: 10.1016/s0304-3940(00)01767-5
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666. doi: 10.1126/science.287.5453.664
Volgyi, K., Juhász, G., Kovacs, Z., and Penke, B. (2015). Dysfunction of endoplasmic reticulum (ER) and mitochondria (MT) in Alzheimer’s disease: the role of the ER-MT cross-talk. Curr. Alzheimer Res. 12, 655–672. doi: 10.2174/1567205012666150710095035
Vukic, V., Callaghan, D., Walker, D., Lue, L. F., Liu, Q. Y., Couraud, P. O., et al. (2009). Expression of inflammatory genes induced by β-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 34, 95–106. doi: 10.1016/j.nbd.2008.12.007
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, Z. V., Deng, Y., Gao, N., Pedrozo, Z., Li, D. L., Morales, C. R., et al. (2014). Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156, 1179–1192. doi: 10.1016/j.cell.2014.01.014
Wang, W., Yin, J., Ma, X., Zhao, F., Siedlak, S. L., Wang, Z., et al. (2017). Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 26, 4118–4131. doi: 10.1093/hmg/ddx299
Wijesekara, N., Ahrens, R., Sabale, M., Wu, L., Ha, K., Verdile, G., et al. (2017). Amyloid-β and islet amyloid pathologies link Alzheimer disease and type 2 diabetes in a transgenic model. FASEB J. 31, 5409–5418. doi: 10.1096/fj.201700431R
Willette, A. A., Bendlin, B. B., Starks, E. J., Birdsill, A. C., Johnson, S. C., Christian, B. T., et al. (2015). Association of insulin resistance with cerebral glucose uptake in late middle-aged adults at risk for Alzheimer disease. JAMA Neurol. 72, 1013–1020. doi: 10.1001/jamaneurol.2015.0613
Yamamoto, K., Sato, T., Matsui, T., Sato, M., Okada, T., Yoshida, H., et al. (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 13, 365–376. doi: 10.1016/j.devcel.2007.07.018
Ying, M., Sui, X., Zhang, Y., Sun, Q., Qu, Z., Luo, X., et al. (2017). Identification of novel key molecules involved in spatial memory impairment in triple transgenic mice of Alzheimer’s disease. Mol. Neurobiol. 54, 3843–3858. doi: 10.1007/s12035-016-9959-2
Yoon, S. S., and Jo, S. A. (2012). Mechanisms of amyloid-β peptide clearance: potential therapeutic targets for Alzheimer’s disease. Biomol. Ther. (Seoul) 20, 245–255. doi: 10.4062/biomolther.2012.20.3.245
Yoshida, H., Haze, K., Yanagi, H., Yura, T., and Mori, K. (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749. doi: 10.1074/jbc.273.50.33741
Yoshida, H., Matsui, T., Hosokawa, N., Kaufman, R. J., Nagata, K., and Mori, K. (2003). A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4, 265–271. doi: 10.1016/s1534-5807(03)00022-4
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/s0092-8674(01)00611-0
Zeng, T., Peng, L., Chao, H., Xi, H., Fu, B., Wang, Y., et al. (2015). IRE1α-TRAF2-ASK1 complex-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to CXC195-induced apoptosis in human bladder carcinoma T24 cells. Biochem. Biophys. Res. Commun. 460, 530–536. doi: 10.1016/j.bbrc.2015.03.064
Zhou, Q., Wang, M., Du, Y., Zhang, W., Bai, M., Zhang, Z., et al. (2015). Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 77, 637–654. doi: 10.1002/ana.24361
Keywords: Alzheimer disease, neurodegenerative diseases, endoplasmic reticulum stress (ER), aging, UPR (unfolded protein response)
Citation: Rahman S, Archana A, Jan AT and Minakshi R (2018) Dissecting Endoplasmic Reticulum Unfolded Protein Response (UPRER) in Managing Clandestine Modus Operandi of Alzheimer’s Disease. Front. Aging Neurosci. 10:30. doi: 10.3389/fnagi.2018.00030
Received: 31 October 2017; Accepted: 24 January 2018;
Published: 06 February 2018.
Edited by:
Ghulam Md. Ashraf, King Abdulaziz University, Saudi ArabiaReviewed by:
Gulam M. Rather, Rutgers Cancer Institute of New Jersey, United StatesGjumrakch Aliev, GALLY International Biomedical Research, United States
Showkat Bhawani, UNIMAS Sarawak, Malaysia
Copyright © 2018 Rahman, Archana, Jan and Minakshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rinki Minakshi, cmlua2kubWluYWtzaGlAaG90bWFpbC5jb20=; bWluYWtzaGk0MDUwQGdtYWlsLmNvbQ==