 Hui-Ming Yang
Hui-Ming Yang Su Yang
Su Yang Shan-Shan Huang
Shan-Shan Huang Bei-Sha Tang1,4,5,6
Bei-Sha Tang1,4,5,6 Ji-Feng Guo
Ji-Feng Guo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 19 June 2017
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 9 - 2017 | https://doi.org/10.3389/fnagi.2017.00193
This article is part of the Research Topic Microglial Polarization in the Pathogenesis and Therapeutics of Neurodegenerative Diseases View all 24 articles
Huntington’s disease (HD) is an autosomal dominantly inherited neurodegenerative disorder caused by expanded CAG trinucleotide repeats (>36) in exon 1 of HTT gene that encodes huntingtin protein. Although HD is characterized by a predominant loss of neurons in the striatum and cortex, previous studies point to a critical role of aberrant accumulation of mutant huntingtin in microglia that contributes to the progressive neurodegeneration in HD, through both cell-autonomous and non-cell-autonomous mechanisms. Microglia are resident immune cells in the central nervous system (CNS), which function to surveil the microenvironment at a quiescent state. In response to various pro-inflammatory stimuli, microglia become activated and undergo two separate phases (M1 and M2 phenotype), which release pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α), anti-inflammatory cytokines, and growth factors (TGF-β, CD206, and Arg1), respectively. Immunoregulation by microglial activation could be either neurotoxic or neuroprotective. In this review, we summarized current understanding about microglial activation in the pathogenesis and progression of HD, with a primary focus of M1 and M2 phenotype of activated microglia and their corresponding signaling pathways.
Huntington’s disease (HD) is a devastating neurodegenerative disease characterized by symptoms of cognitive disorder, motor impairment and mental disturbance. The prevalence of HD is 4–10 per 100, 000 in the western population, and the mean age of onset is 40 years old (Ross and Tabrizi, 2011). It is an autosomal dominant monogenic disease caused by CAG trinucleotide repeats expansion in the HTT gene, which translates into huntingtin (HTT) protein with an expanded polyglutamine (polyQ) tract that is prone to misfolding. Although the pathogenic mutant huntingtin (mHTT) is ubiquitously expressed in different types of neural cells in the brain (Jansen et al., 2017), it causes a preferential loss of medium spiny neurons (MSNs) in the striatum (Graveland et al., 1985), and the atrophy of caudate and putamen could be clinically imaged by magnetic resonance imaging (MRI) prior to motor symptom onset (Bates et al., 2015).
Neuroinflammation, characterized by remarkable gliosis and inflammatory reactions in the central nervous system (CNS), has been described as a prominent sign in various neurodegenerative diseases such as Alzheimer’s disease (AD) (Villegas-Llerena et al., 2016), Parkinson’s disease (PD) (Le et al., 2016), and Amyotrophic Lateral Sclerosis (ALS) (Philips and Robberecht, 2011). HD is no exception, as mounting evidence indicates that microglia activation could be detected in the brains from pre-symptomatic HD carriers to post-mortem HD patients. Specifically, elevated inflammatory cytokines could be detected in both the CNS and plasma from HD patients (Tai et al., 2007a,b; Bjorkqvist et al., 2008; Silvestroni et al., 2009; Politis et al., 2015).
Microglia, the resident macrophage of the CNS, monitor the microenvironment in the CNS and respond to neural damage or degeneration by switching to different activation states (Hanisch and Kettenmann, 2007). Moreover, CX3CL1-CX3CR1 and CD200-CD200R are two major signaling pathways that function to mediate neuron-microglia interaction and facilitate immunomodulatory and phagocytic activities of microglia in response to degenerated neurons in the CNS (Gomez-Nicola and Perry, 2015). Under normal conditions, resting microglia typically display small cell bodies with fine processes and work as the first line of defense in the CNS (Hanisch and Kettenmann, 2007). In the presence of adverse stimuli, microglia retract the processes and exhibit swollen shapes (Moller, 2010). Microglia exhibiting large amoeboid-like cell bodies with no processes or with short stout extensions were seen in both HD patient brain (Sapp et al., 2001) and YAC128 HD mouse model (Franciosi et al., 2012). In addition, the abnormal morphology of microglia is exacerbated in an age-dependent manner, throughout the course of HD progression. Here, we review the immune-regulatory role of microglia in HD pathology, and discuss potential cellular mechanisms of microglia activation underlying HD pathogenesis.
Expanded polyQ tract at the N-terminus of HTT is responsible for protein misfolding and the subsequent aberrant accumulation and aggregates formation. Aggregated mHTT could be detected as inclusion bodies either in the cytoplasm or nuclei (DiFiglia et al., 1997; Gutekunst et al., 1999). The relative contribution of soluble and aggregated forms of mHTT to the pathogenesis in HD is still unclear. Nevertheless, it is commonly believed that the expression of N-terminal mHTT fragments could lead to HD pathogenesis, and the sizes of the fragments correlate inversely with neurotoxicity (Mangiarini et al., 1996; Chang et al., 2015).
To facilitate investigation of HD, a variety of HD experimental models with the expression of either full-length or N-terminal fragments of mHTT were generated and studied, such as transgenic animal models (Chang et al., 2015), human embryonic stem cell derived models (Lu and Palacino, 2013) and HD patients derived induced pluripotent stem cells (Xu et al., 2017). Among them, mouse model is one of the most commonly used, due to its relative ease of genetic manipulation and physiological resemblance to human. In Table 1, we provide a summary of most commonly studied HD mouse models, in terms of their genetic background and mHTT expression pattern and level.
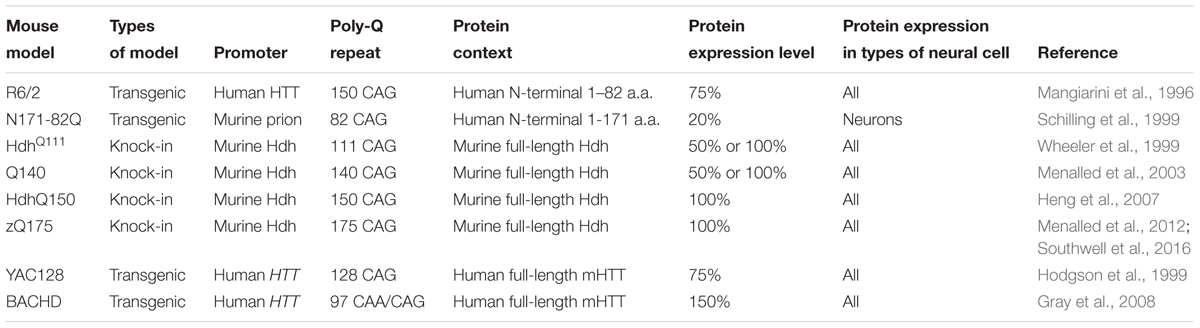
TABLE 1. Mouse models of Huntington’s disease.
Although mHTT is ubiquitously expressed, HD elicits selective cortical and striatal neurodegeneration. To understand mechanisms underlying the preferential neuronal vulnerability, mouse models that express mHTT in selective cell populations were generated using the Cre-Loxp system. For instance, transgenic mice carrying Cre recombinase with different promoters, such as Emx1 or Rgs9, were crossed with BACHD mice, allowing restricted expression of mHTT in the cortex and striatum, respectively (Wang et al., 2014). Similarly, mice that express exon1 of mHTT in neurons or only in cortical pyramidal neurons were created by crossing RosaHD mice with Nestin-Cre mice or Emx1-Cre mice (Gu et al., 2005). In addition, there are transgenic mice that selectively express mHTT in astrocytes (Bradford et al., 2009) or oligodendrocytes (Huang et al., 2015), which demonstrate the mHTT expression in non-neuronal cells also contributes to the pathogenesis of HD. Mice that express mHTT specifically in microglia have been generated by crossing RosaHD with Cx3cr1-Cre miceD (Crotti et al., 2014).
Marked astrogliosis and microgliosis were observed in the post-mortem brains of HD patients but not in normal control brains (Singhrao et al., 1999). It was reported that microglia accumulated in all grades of HD patients’ brains, and the density correlated to the degree of neuronal loss (Sapp et al., 2001). Indeed, in vivo positron emission tomography (PET) studies revealed that microglial activation was significant in affected HD brain regions and this pathology was more pronounced in more severe cases of HD (Pavese et al., 2006; Politis et al., 2011). Furthermore, PET also detected microglial activation in HD patients prior to disease manifestations (Tai et al., 2007b). These studies provide strong evidence indicating that microglial activation is an integral part of HD pathogenesis. Nonetheless, whether the activated microglia plays a protective or detrimental role in HD pathogenesis remains to be illustrated.
Although mHTT is widely expressed, MSNs in the striatum are the most susceptible cell type to mHTT-mediated neurotoxicity. Furthermore, mHTT is more abundantly accumulated in neuronal dendrites and nerve terminals than in soma and other cell types, and in vitro studies showed that mHTT preferentially formed aggregates along neuronal processes and axonal terminals (Li et al., 2000; Zhao et al., 2016). Microglia, as an active sensor in the CNS, transform to activated states in response to pathological changes in the CNS (Hanisch and Kettenmann, 2007). In cortico-striatal brain slice and primary neuronal culture models, neuronal expression of mHTT initiates a local response of microglia, resulting in elevated numbers and activated morphological phenotypes. In particular, these proliferative microglia were prone to position along irregular neurites, but did not directly contributed to neuronal degeneration (Kraft et al., 2012). Over-activated microglia were demonstrated to be neurotoxic, due to their capacity to release toxins. Therefore, decreased number of reactive microglia coupled with downregulation of inflammatory cytokines was viewed as an indicator of pathologic alleviation in HD (Wang et al., 2014; Ochaba et al., 2016), and therapeutic methods aimed at lowering neuronal mHTT expression led to drastic amelioration of concomitant microglia activation (Wang et al., 2014). On the other hand, adding exogenous primary microglia to mHTT-expressing neurons increased neuronal survival, and this effect was proportional to the amount of microglia added (Kraft et al., 2012). Moreover, a recent in vivo study showed supplementing normal human glia to transgenic R6/2 HD mice exhibited neuronal protection as well as phenotypic improvement (Benraiss et al., 2016). The above evidence indicates that normal microglia can rescue mHTT-expressing neurons. Nonetheless, whether mHTT expression in microglia results in its phagocytic or immunoregulatory dysfunction, and how this contributes to HD neuropathology remain to be investigated.
Although mHTT is ubiquitously expressed in all types of cells, the frequency of inclusion bodies was observed at a much lower rate in microglia compared to others (Jansen et al., 2017), probably owing to the increased immunoproteasome subunits (Orre et al., 2013) and autophagosomes (Su et al., 2016), which renders microglia a higher capacity to degrade mHTT. In spite of less mHTT aggregates formed in microglia, a cell autonomous effect induced by intrinsic mutant protein may be responsible for microglia activation and contribute to the pathology in HD as well. It was demonstrated that mHTT expression altered the function of immune cells both centrally and peripherally, and isolated microglia from R6/2 mice were described to be much more hyperactive than wild type microglia in response to stimulation (Bjorkqvist et al., 2008). Intriguingly, using genome-wide approaches, expression of mHTT in microglia was found to promote cell-autonomous pro-inflammatory gene expression in the absence of sterile inflammation and is dependent on the expression and transcriptional activities of the myeloid lineage-determining factors PU.1 and C/EBPs. In addition, mHTT-expressing microglia-mediated cell-autonomous activation exhibits enhanced toxic effects on wild type neurons in comparison to wild type microglia ex vivo and after sterile inflammation in vivo (Crotti et al., 2014). However, whether restricting the expression of mHTT in microglia is sufficient to exert neuropathological and behavioral deficits in vivo still needs to be investigated.
Previous work suggests over-activation of N-methyl-D-aspartate receptor (NMDAR) by extracellular glutamate is involved in the degeneration of medium-sized spiny neurons in HD (Zeron et al., 2002). Astrocytes, the major type of glia in brain, express glutamate transporters that uptake extracellular glutamate while mHTT expression in astrocytes diminished this protection against glutamate neurotoxicity (Shin et al., 2005). Moreover, selective mHTT expression in astrocytes caused age-dependent neurological symptoms and exacerbated neuronal loss in vivo (Bradford et al., 2009, 2010). Markedly, reactive astrocytes are accumulated in proximity to degenerated neurons in HD brain, characterized by increased proliferation, cell hypertrophy and the induction of astroglial markers [e.g., glial fibrillary acidic protein (GFAP)]. Furthermore, activated astrocytes instead of microglia were shown to upregulate the pro-inflammation in N171-82 HD mice that express mHTT only in neurons, but not in glial cells (Hsiao et al., 2013). However, a recent study revealed that neurotoxic reactive astrocytes could be induced by activated microglia and consequently resulted in a similar expression profile of pro-inflammatory cytokines, which led to a more rapid death of neurons and oligodendrocytes (Liddelow et al., 2017). Herein, the relative contribution by activated astrocytes and activated microglia to neuroinflammation and the eventual HD pathogenesis remains to be elucidated.
In response to neurodegeneration and the accumulation of misfolded proteins, microglia multiply and adopt a process referred to as priming. Priming makes the microglia susceptible to a secondary inflammatory stimulus, which then triggers an exaggerated inflammatory response (Perry and Holmes, 2014). As the main dynamic component of neuroinflammation in the CNS, activated microglia exist along a continuum of two functional states of polarization in which they are able to either expand the damage to neighboring cells, or clear the cell debris followed by tissue repair (Jha et al., 2016). In reference to peripheral macrophage polarization, microglia share comparative properties in reaction to acute or prolonged stimuli, and are classified into two extreme phenotypes: the classically activated M1 phenotype and the alternatively activated M2 phenotype. Differences between these two phenotypes range from morphological changes to alteration of representative cytokines, determined by protein or gene expression profiling (Durafourt et al., 2012; Hu et al., 2015). To modify this simplified nomenclature, there are other proposals of classifying macrophages into more informative populations (Mosser and Edwards, 2008) or refining M2 categorization further into alternative activation (M2a), type II alternative activation (M2b) and acquired deactivation (M2c) (Walker and Lue, 2015). Yet the existence of these refined cell populations has not been fully examined in HD brain.
When triggered by extracellular or intracellular stimuli, resident microglia conferred classically activated state named M1 phenotype to initiate and augment the innate immune function in the CNS. Classically activated M1 microglia release pro-inflammatory biomarkers such as redox molecules (NO, iNOS), cytokines (IL-1β, IL-6, IL-8, and TNF-α et al.), chemokines (CCL2, CCL20, and CXCL-1 et al.) and surface receptors (CD16, CD32, CD36, CD68, and CD86 et al.) which can be used to identify M1 phenotype of microglia (Franco and Fernandez-Suarez, 2015; Orihuela et al., 2016). In vitro and in vivo studies declared that M1 microglia could be induced by the treatment of LPS alone, or combining LPS/IFN-γ followed by increased levels of pro-inflammatory cytokines (Durafourt et al., 2012; Kroner et al., 2014; Cunha et al., 2016; Wang et al., 2016). M1 microglia relevant biomarkers were widely detected in HD brain, which indicates that M1 microglia may play a crucial role in the pathogenesis of HD. Primary glia cells including microglia, isolated from R6/2 transgenic mouse model, showed that iNOS, IL-1β, IL-6, and TNF-α were significantly elevated after LPS treatment (Hsiao et al., 2014), and IL-6 was increased in the microglia of R6/2 mouse brain (Bjorkqvist et al., 2008). In addition, higher levels of IL-1β and IL-8 were secreted by microglia in HD transgenic porcine model (Valekova et al., 2016). Furthermore, pro-inflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α were elevated both centrally (in the striatum and cerebrospinal fluid) and peripherally (in the plasma) in HD patients (Bjorkqvist et al., 2008; Chang et al., 2015; Rodrigues et al., 2016). M1 related markers involved in HD were summarized as Table 2.
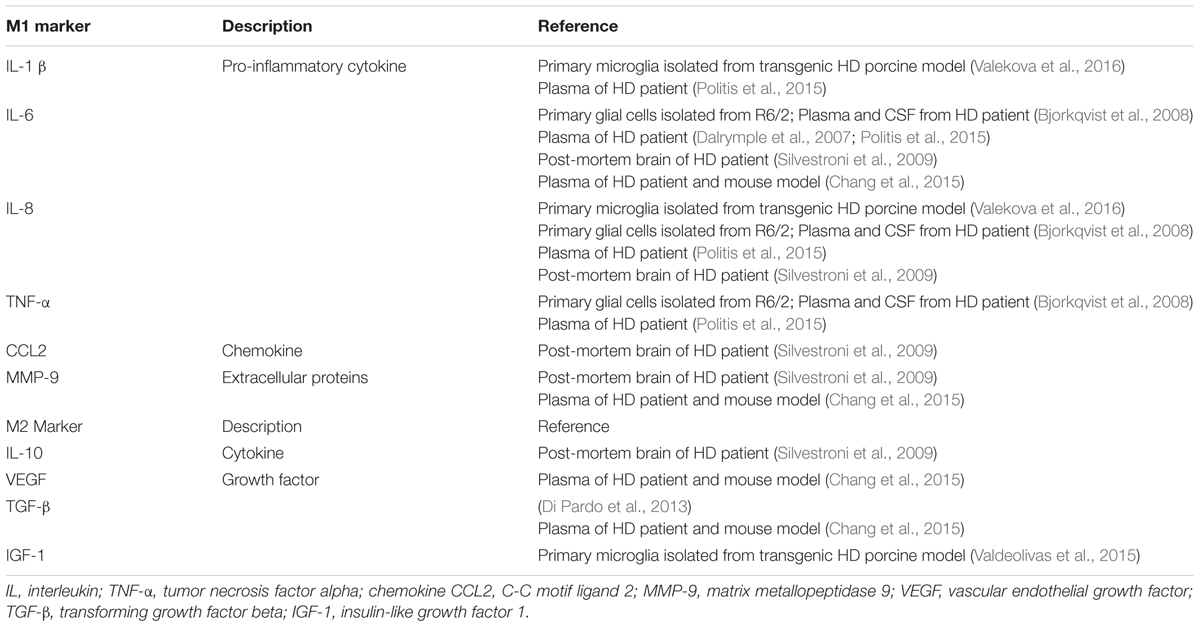
TABLE 2. Implications of M1 and M2 microglia relevant markers in HD.
In contrast to M1 microglia, alternative activated M2 microglia enable phagocytosis and initiate tissue repair and neural regeneration in the CNS (Miron et al., 2013). M2 microglia are characterized by producing anti-inflammatory cytokines and growth factors such as IL-10, TGF-β, CD206, Arg1, Ym1 (Chi3l3 in human) and Fizz1 (Franco and Fernandez-Suarez, 2015; Orihuela et al., 2016). Of these molecules, IL-10 played a key role in phagocytic microglia to engulf apoptotic cells (Chhor et al., 2013; Cianciulli et al., 2015). CD206, Arg1, and Ym1 were considered as typical markers for the identification of M2 microglia (Chhor et al., 2013; Miron et al., 2013; Zanier et al., 2014). To recapitulate the phenotype of M2 microglia, manipulation of IL-10 alone or combining IL-4/IL-13 was performed to establish the experimental models (Ponomarev et al., 2007; Chhor et al., 2013; Miron et al., 2013). Much less M2 markers were found in HD, as only VEGF and TGF-β were reported to coexist with M1 markers in the plasma and post-mortem brain tissues of HD patients (Di Pardo et al., 2013; Chang et al., 2015). M2 related markers in HD were summarized as Table 2.
As illustrated above, M1 and M2 microglia are defined by their contradictory functions and distinctive biomarkers. However, in traumatic brain injury (TBI), up-regulated expression of both M1 and M2 microglia associated genes can be found simultaneously. And co-localization of representative biomarkers of both activation states in the same microglia indicated concurrence of M1 and M2 phenotype following TBI (Morganti et al., 2016). Consistent phenomenon of concurrent M1/M2 response was found in ALS (Chiu et al., 2013) and spinal cord injury (SCI) (Shechter et al., 2013) as well. In addition, M1 microglia are able to transform to M2 microglia, as cyclic adenosine monophosphate (cAMP) has been reported to play a key role in converting LPS/IFN-γ induced M1 microglia to M2 microglia, which augments the anti-inflammatory effects in the presence of IL-4 with increased levels of Arg1 (Ghosh et al., 2016). In addition, switching M1 microglia to M2 microglia was demonstrated to be protective by initiating remyelination in multiple sclerosis (MS) (Miron et al., 2013). Concurrent inflammatory profiles of M1 and M2 microglia related biomarkers were also indicated in HD as shown as Table 2.
Microglia express toll like receptors (TLRs) to recognize extracellular stimuli. TLR-2 is the most expressed TLR in microglia, which is responsible for IL-6 and IL-10 secretion. TLR-3 and TLR-4 are two receptors, which most potently stimulate proinflammatory cytokines secretion, such as IL-12, TNF-α, IL-6, CXCL-10, and IL-10 (Jack et al., 2005). Studies showed that TLR-2, TLR-3, and TLR-4 activate microglia and astrocytes in a sequential manner, coupled with elevated IL-1β level (Facci et al., 2014). Experimental M1 microglia, obtained by the treatment of LPS or LPS/IFN-γ, were capable to increase TLR-2 and TLR-4 gene expression (Marinelli et al., 2015). Classical TLR signaling involves the intracellular adaptor protein myeloid differentiated 88 (MyD88) to trigger downstream NF-κB signaling cascade and thus upregulates the production of proinflammatory cytokines (Takeda and Akira, 2004). NF-κB is sequestered in the cytoplasm by I-κBs, a group of inhibitory proteins (Ghosh et al., 1998). When I-κBs are phosphorylated by I-κB kinase, NF-κB is dissociated from I-κBs and translocated to the nucleus (IKK including subunits of IKKα, IKKβ, and IKKγ) (Ghosh and Karin, 2002). Consequently, NF-κB regulates downstream inflammatory cytokines gene expression. Soluble mHTT was shown to activate IKK, which in turn upregulates the NF-κB signaling pathway (Khoshnan et al., 2004). Trager et al. used proximity ligation assays and offered evidence that mHTT interacted with IKK, which also mediated transcriptional changes of NF-κB signaling with increased levels of IL-1β, IL-6, IL-8, and TNFα. Furthermore, lowering mHTT level by siRNA was shown to ameliorate NF-κB transcriptional dysregulation, and downregulate the expression of pro-inflammatory cytokines in HD (Trager et al., 2014).
In mammalian cells, Kynurenine pathway (KP) mediates degradation of the majority of cellular tryptophan through different enzyme branches (Fujigaki et al., 2017). The 3-hydroxyanthranilate 3, 4-dioxygenase (KMO) enzyme, which is predominantly expressed in microglia, metabolizes tryptophan to neuroactive 3-hydroxykynurenine (3HK) and quinolinic acid (QUIN) (Heyes et al., 1992). QUIN could lead to neurotoxicity by acting as a selective agonist on NMDA receptors (Szalardy et al., 2012), and it has been associated with neuroinflammation in various neurological diseases (Maddison and Giorgini, 2015). Through a genome wide screening in yeast, BNA1, which encodes the enzyme Bna1 [3, 4-dioxygenase (KMO)] involved in the kynurenine pathway, was found to mediate mHTT toxicity by producing high levels of QUIN and 3HK (Giorgini et al., 2005), which activate NMDA receptors and generate toxic free radical, respectively (Schwarcz and Pellicciari, 2002). However, the first and rate-limiting enzyme of the kynurenine pathway, indoleamine 2,3-dioxygenase (IDO), preferentially localizes in microglia (Heyes et al., 1996), but not in astrocytes or neurons of human brain (Guillemin et al., 2005). A recent study using transgenic N171-82Q HD mouse model revealed that IDO activity was significantly elevated in HD mice compared to wild type control mice (Donley et al., 2016). Moreover, previous studies (Guidetti et al., 2004, 2006) revealed that IDO activity was elevated in the early stage of HD, which contributed to increased levels of HK and QUIN- and NMD-mediated neurotoxicity. Taking into account of the studies above, drugs that can inhibit KP enzymes may offer potential therapeutic approaches by preventing neurotoxicity caused by downstream toxic products (Schwarcz and Pellicciari, 2002). As KMO and QUIN production showed cell type specific expression in microglia, chronic administration of KMO inhibitor JM6 was shown to prevent synaptic degeneration and increase the survival of R6/2 HD mouse model, which is associated with amelioration of microglial activation (Zwilling et al., 2011). In addition, suppressing the expression of tryptophan-2, 3-dioxygenase (TDO), another rate limiting enzyme in KP, was demonstrated to be neuroprotective in MS (Lanz et al., 2017). Additionally, inhibiting KP is likely to be protective in several neurologic diseases (Fujigaki et al., 2017), probably by modulating microglial activation mediated neurotoxicity.
Cannabinoid receptors are a group of G-protein coupled receptors including cannabinoid type 1 (CB1) receptors and cannabinoid type 2 (CB2) receptors (Glass and Northup, 1999). In particular, CB2 receptors were involved in regulating cell proliferation, differentiation and survival through several signaling pathways, which include adenylyl cyclase and cyclic AMP-protein kinase A (PKA), extracellular signal-regulated kinase 1 (ERK1) and ERK2, p38 mitogen-activated protein kinase and JUN N-terminal kinases (JNKs) (Bisogno et al., 2016). In R6/2 HD mouse model, CB1 receptors were abundantly expressed in striatal MSNs (Chiarlone et al., 2014) and exerted a neuroprotective role through induction of brain-derived neurotrophic factor (BDNF) expression (Blazquez et al., 2015). CB2 receptors were reported to be mainly expressed in periphery immune cells, and were barely detected in brain tissue (Schatz et al., 1997). However, a significant increase of CB2 receptors expression was seen in traumatic mouse brain (Donat et al., 2014) and in activated microglia in AD mouse model (Benito et al., 2003). In addition, activation of CB2 receptors in microglia was shown to influence the acquisition of M2 polarization, and this effect was dampened by knocking out CB2 receptors (Mecha et al., 2015). As for HD, a study showed genetic ablation of CB2 receptors in R6/2 mouse model exacerbated the behavioral abnormalities and reduced life span, along with accelerated microglial activation (Palazuelos et al., 2009). The further impairment in motor activities caused by loss of CB2 receptors was recapitulated in BACHD mouse model as well (Bouchard et al., 2012). Therefore, therapeutic approaches targeting CB2 receptors in microglia might be promising in treating HD. Indeed, R6/2 mice have been treated with cannabigerol (CBG), a non-psychotropic phytocannabinoid that exerted neuroprotective effects through both CB1/CB2 receptors-dependent and -independent mechanisms. Administration of CBG significantly improved motor impairment and increased the expression of BDNF and insulin-like growth factor-1 (IGF-1) in treated R6/2 mouse model (Valdeolivas et al., 2015). In addition, VCE-003, a CBG quinone derivative, also attenuated striatal neuron loss, motor impairment and microglial activation in quinolinic acid (QA) and 3-NP induced HD mouse models (Diaz-Alonso et al., 2016). A CB2 receptor specific agonist, SR144528, has also been used for treating mice, which reduced the generation of proinflammatory molecules such as TNF-α (Sagredo et al., 2009).
Microglial activation has been found for over a decade in HD brain and indicated as a strong component of neuroinflammation in HD pathogenesis (Crotti and Glass, 2015). Microglial activation in HD was reported to be triggered by neuronal mHTT-mediated excitotoxicity or mHTT expression in microglia, however, whether microglial activation makes a significant contribution to HD pathology has not been fully investigated. Polarized M1 phenotype and M2 phenotype of microglia, which have been illustrated in neurological diseases, especially traumatic brain injury (Morganti et al., 2016) or SCI (Kroner et al., 2014), exert coherent morphologic and functional features with alteration of central or peripheral inflammatory profiles. Although M1 and M2 microglia relevant biomarkers are found in HD, the definition and identification of these polarized phenotypes of microglia in HD remain to be elucidated. It is possible that microglial activation play dual roles in HD, either detrimental or beneficial, and thus targeting signaling pathways specific in protective properties of microglia may offer potential therapeutics.
H-MY conceived the study and wrote the manuscript. H-MY, SY, S-SH, B-ST, and J-FG discussed and revised the manuscript. H-MY prepared the tables. All authors read and approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer EP-N and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
This work was supported by grants 81371405, 81571248 from the National Natural Science Foundation of China (to J-FG), grant 2016CX025 from innovation-driven plan of Central South University (to J-FG), grants 81430023 from the National Natural Science Foundation of China (to B-ST), grant 2016YFC1306000 from the national key plan for scientific research and development of China.
Bates, G. P., Dorsey, R., Gusella, J. F., Hayden, M. R., Kay, C., Leavitt, B. R., et al. (2015). Huntington disease. Nat. Rev. Dis. Primers 1, 15005. doi: 10.1038/nrdp.2015.5
Benito, C., Nunez, E., Tolon, R. M., Carrier, E. J., Rabano, A., Hillard, C. J., et al. (2003). Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 23, 11136–11141.
Benraiss, A., Wang, S., Herrlinger, S., Li, X. J., Chandler-Militello, D., Mauceri, J., et al. (2016). Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 7:11758. doi: 10.1038/ncomms11758
Bisogno, T., Oddi, S., Piccoli, A., Fazio, D., and Maccarrone, M. (2016). Type-2 cannabinoid receptors in neurodegeneration. Pharmacol. Res. 111, 721–730. doi: 10.1016/j.phrs.2016.07.021
Bjorkqvist, M., Wild, E. J., Thiele, J., Silvestroni, A., Andre, R., Lahiri, N., et al. (2008). A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 205, 1869–1877. doi: 10.1084/jem.20080178
Blazquez, C., Chiarlone, A., Bellocchio, L., Resel, E., Pruunsild, P., Garcia-Rincon, D., et al. (2015). The CB(1) cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 22, 1618–1629. doi: 10.1038/cdd.2015.11
Bouchard, J., Truong, J., Bouchard, K., Dunkelberger, D., Desrayaud, S., Moussaoui, S., et al. (2012). Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of huntington’s disease. J. Neurosci. 32, 18259–18268. doi: 10.1523/JNEUROSCI.4008-12.2012
Bradford, J., Shin, J. Y., Roberts, M., Wang, C. E., Li, X. J., and Li, S. (2009). Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. U.S.A. 106, 22480–22485. doi: 10.1073/pnas.0911503106
Bradford, J., Shin, J. Y., Roberts, M., Wang, C. E., Sheng, G., Li, S., et al. (2010). Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J. Biol. Chem. 285, 10653–10661. doi: 10.1074/jbc.M109.083287
Chang, K. H., Wu, Y. R., Chen, Y. C., and Chen, C. M. (2015). Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav. Immun. 44, 121–127. doi: 10.1016/j.bbi.2014.09.011
Chang, R., Liu, X., Li, S., and Li, X. J. (2015). Transgenic animal models for study of the pathogenesis of Huntington’s disease and therapy. Drug Des. Devel. Ther. 9, 2179–2188. doi: 10.2147/DDDT.S58470
Chhor, V., Le Charpentier, T., Lebon, S., Ore, M. V., Celador, I. L., Josserand, J., et al. (2013). Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 32, 70–85. doi: 10.1016/j.bbi.2013.02.005
Chiarlone, A., Bellocchio, L., Blazquez, C., Resel, E., Soria-Gomez, E., Cannich, A., et al. (2014). A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc. Natl. Acad. Sci. U.S.A. 111, 8257–8262. doi: 10.1073/pnas.1400988111
Chiu, I. M., Morimoto, E. T. A., Goodarzi, H., Liao, J. T., O’Keeffe, S., Phatnani, H. P., et al. (2013). A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385–401. doi: 10.1016/j.celrep.2013.06.018
Cianciulli, A., Dragone, T., Calvello, R., Porro, C., Trotta, T., Lofrumento, D. D., et al. (2015). IL-10 plays a pivotal role in anti-inflammatory effects of resveratrol in activated microglia cells. Int. Immunopharmacol. 24, 369–376. doi: 10.1016/j.intimp.2014.12.035
Crotti, A., Benner, C., Kerman, B. E., Gosselin, D., Lagier-Tourenne, C., Zuccato, C., et al. (2014). Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 17, 513–521. doi: 10.1038/nn.3668
Crotti, A., and Glass, C. K. (2015). The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 36, 364–373. doi: 10.1016/j.it.2015.04.007
Cunha, C., Gomes, C., Vaz, A. R., and Brites, D. (2016). Exploring new inflammatory biomarkers and pathways during LPS-induced M1 polarization. Mediators Inflamm. 2016:6986175. doi: 10.1155/2016/6986175
Dalrymple, A., Wild, E. J., Joubert, R., Sathasivam, K., Bjorkqvist, M., Petersen, A., et al. (2007). Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J. Proteome Res. 6, 2833–2840. doi: 10.1021/pr0700753
Di Pardo, A., Alberti, S., Maglione, V., Amico, E., Cortes, E. P., Elifani, F., et al. (2013). Changes of peripheral TGF-beta1 depend on monocytes-derived macrophages in Huntington disease. Mol. Brain 6:55. doi: 10.1186/1756-6606-6-55
Diaz-Alonso, J., Paraiso-Luna, J., Navarrete, C., Del Rio, C., Cantarero, I., Palomares, B., et al. (2016). VCE-003.2, a novel cannabigerol derivative, enhances neuronal progenitor cell survival and alleviates symptomatology in murine models of Huntington’s disease. Sci. Rep. 6, 29789. doi: 10.1038/srep29789
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Donat, C. K., Fischer, F., Walter, B., Deuther-Conrad, W., Brodhun, M., Bauer, R., et al. (2014). Early increase of cannabinoid receptor density after experimental traumatic brain injury in the newborn piglet. Acta Neurobiol. Exp. (Wars) 74, 197–210.
Donley, D. W., Olson, A. R., Raisbeck, M. F., Fox, J. H., and Gigley, J. P. (2016). Huntingtons disease mice infected with Toxoplasma gondii demonstrate early kynurenine pathway activation, altered CD8+ T-cell responses, and premature mortality. PLoS ONE 11:e0162404. doi: 10.1371/journal.pone.0162404
Durafourt, B. A., Moore, C. S., Zammit, D. A., Johnson, T. A., Zaguia, F., Guiot, M. C., et al. (2012). Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 60, 717–727. doi: 10.1002/glia.22298
Facci, L., Barbierato, M., Marinelli, C., Argentini, C., Skaper, S. D., and Giusti, P. (2014). Toll-like receptors 2, -3 and -4 prime microglia but not astrocytes across central nervous system regions for ATP-dependent interleukin-1beta release. Sci. Rep. 4, 6824. doi: 10.1038/srep06824
Franciosi, S., Ryu, J. K., Shim, Y., Hill, A., Connolly, C., Hayden, M. R., et al. (2012). Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 45, 438–449. doi: 10.1016/j.nbd.2011.09.003
Franco, R., and Fernandez-Suarez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Fujigaki, H., Yamamoto, Y., and Saito, K. (2017). L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: focus on cell type differences. Neuropharmacology 112, 264–274. doi: 10.1016/j.neuropharm.2016.01.011
Ghosh, M., Xu, Y., and Pearse, D. D. (2016). Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 13:9. doi: 10.1186/s12974-015-0463-9
Ghosh, S., and Karin, M. (2002). Missing pieces in the NF-kappaB puzzle. Cell 109, S81–S96. doi: 10.1016/S0092-8674(02)00703-1
Ghosh, S., May, M. J., and Kopp, E. B. (1998). NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16, 225–260. doi: 10.1146/annurev.immunol.16.1.225
Giorgini, F., Guidetti, P., Nguyen, Q., Bennett, S. C., and Muchowski, P. J. (2005). A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 37, 526–531. doi: 10.1038/ng1542
Glass, M., and Northup, J. K. (1999). Agonist selective regulation of G proteins by cannabinoid CB(1) and CB(2) receptors. Mol. Pharmacol. 56, 1362–1369.
Gomez-Nicola, D., and Perry, V. H. (2015). Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist 21, 169–184. doi: 10.1177/1073858414530512
Graveland, G. A., Williams, R. S., and DiFiglia, M. (1985). Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773. doi: 10.1126/science.3155875
Gray, M., Shirasaki, D. I., Cepeda, C., Andre, V. M., Wilburn, B., Lu, X. H., et al. (2008). Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 28, 6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008
Gu, X., Li, C., Wei, W., Lo, V., Gong, S., Li, S. H., et al. (2005). Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 46, 433–444. doi: 10.1016/j.neuron.2005.03.025
Guidetti, P., Bates, G. P., Graham, R. K., Hayden, M. R., Leavitt, B. R., MacDonald, M. E., et al. (2006). Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol. Dis. 23, 190–197. doi: 10.1016/j.nbd.2006.02.011
Guidetti, P., Luthi-Carter, R. E., Augood, S. J., and Schwarcz, R. (2004). Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol. Dis. 17, 455–461. doi: 10.1016/j.nbd.2004.07.006
Guillemin, G. J., Smythe, G., Takikawa, O., and Brew, B. J. (2005). Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 49, 15–23. doi: 10.1002/glia.20090
Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R., et al. (1999). Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 19, 2522–2534.
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Heng, M. Y., Tallaksen-Greene, S. J., Detloff, P. J., and Albin, R. L. (2007). Longitudinal evaluation of the Hdh((CAG)150) knock-in murine model of Huntington’s disease. J. Neurosci. 27, 8989–8998. doi: 10.1523/JNEUROSCI.1830-07.2007
Heyes, M. P., Achim, C. L., Wiley, C. A., Major, E. O., Saito, K., and Markey, S. P. (1996). Human microglia convert L-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 320, 595–597. doi: 10.1042/bj3200595
Heyes, M. P., Saito, K., Crowley, J. S., Davis, L. E., Demitrack, M. A., Der, M., et al. (1992). Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 115(Pt 5), 1249–1273. doi: 10.1093/brain/115.5.1249
Hodgson, J. G., Agopyan, N., Gutekunst, C. A., Leavitt, B. R., LePiane, F., Singaraja, R., et al. (1999). A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23, 181–192. doi: 10.1016/S0896-6273(00)80764-3
Hsiao, H. Y., Chen, Y. C., Chen, H. M., Tu, P. H., and Chern, Y. (2013). A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet. 22, 1826–1842. doi: 10.1093/hmg/ddt036
Hsiao, H. Y., Chiu, F. L., Chen, C. M., Wu, Y. R., Chen, H. M., Chen, Y. C., et al. (2014). Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 23, 4328–4344. doi: 10.1093/hmg/ddu151
Hu, X., Leak, R. K., Shi, Y., Suenaga, J., Gao, Y., Zheng, P., et al. (2015). Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 11, 56–64. doi: 10.1038/nrneurol.2014.207
Huang, B., Wei, W., Wang, G., Gaertig, M. A., Feng, Y., Wang, W., et al. (2015). Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron 85, 1212–1226. doi: 10.1016/j.neuron.2015.02.026
Jack, C. S., Arbour, N., Manusow, J., Montgrain, V., Blain, M., McCrea, E., et al. (2005). TLR signaling tailors innate immune responses in human microglia and astrocytes. J. Immunol. 175, 4320–4330. doi: 10.4049/jimmunol.175.7.4320
Jansen, A. H., van Hal, M., Op den Kelder, I. C., Meier, R. T., de Ruiter, A. A., Schut, M. H., et al. (2017). Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia 65, 50–61. doi: 10.1002/glia.23050
Jha, M. K., Lee, W. H., and Suk, K. (2016). Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem. Pharmacol. 103, 1–16. doi: 10.1016/j.bcp.2015.11.003
Khoshnan, A., Ko, J., Watkin, E. E., Paige, L. A., Reinhart, P. H., and Patterson, P. H. (2004). Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J. Neurosci. 24, 7999–8008. doi: 10.1523/JNEUROSCI.2675-04.2004
Kraft, A. D., Kaltenbach, L. S., Lo, D. C., and Harry, G. J. (2012). Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol. Aging 33, 621.e17–33. doi: 10.1016/j.neurobiolaging.2011.02.015
Kroner, A., Greenhalgh, A. D., Zarruk, J. G., Passos Dos Santos, R., Gaestel, M., and David, S. (2014). TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 83, 1098–1116. doi: 10.1016/j.neuron.2014.07.027
Lanz, T. V., Williams, S. K., Stojic, A., Iwantscheff, S., Sonner, J. K., Grabitz, C., et al. (2017). Tryptophan-2,3-Dioxygenase (TDO) deficiency is associated with subclinical neuroprotection in a mouse model of multiple sclerosis. Sci. Rep. 7:41271. doi: 10.1038/srep41271
Le, W. D., Wu, J. J., and Tang, Y. (2016). Protective microglia and their regulation in Parkinson’s Disease. Front. Mol. Neurosci. 9:89. doi: 10.3389/fnmol.2016.00089
Li, H., Li, S. H., Johnston, H., Shelbourne, P. F., and Li, X. J. (2000). Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat. Genet. 25, 385–389. doi: 10.1038/78054
Li, S. H., Schilling, G., Young, W. S., Li, X. J., Margolis, R. L., Stine, O. C., et al. (1993). Huntingtons-Disease gene (It-15) is widely expressed in human and rat-tissues. Neuron 11, 985–993. doi: 10.1016/0896-6273(93)90127-D
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lu, B., and Palacino, J. (2013). A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 27, 1820–1829. doi: 10.1096/fj.12-219220
Maddison, D. C., and Giorgini, F. (2015). The kynurenine pathway and neurodegenerative disease. Semin. Cell Dev. Biol. 40, 134–141. doi: 10.1016/j.semcdb.2015.03.002
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. doi: 10.1016/S0092-8674(00)81369-0
Marinelli, C., Di Liddo, R., Facci, L., Bertalot, T., Conconi, M. T., Zusso, M., et al. (2015). Ligand engagement of Toll-like receptors regulates their expression in cortical microglia and astrocytes. J. Neuroinflamm. 12, 244. doi: 10.1186/s12974-015-0458-6
Mecha, M., Feliu, A., Carrillo-Salinas, F. J., Rueda-Zubiaurre, A., Ortega-Gutierrez, S., de Sola, R. G., et al. (2015). Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 49, 233–245. doi: 10.1016/j.bbi.2015.06.002
Menalled, L. B., Kudwa, A. E., Miller, S., Fitzpatrick, J., Watson-Johnson, J., Keating, N., et al. (2012). Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS ONE 7:e49838. doi: 10.1371/journal.pone.0049838
Menalled, L. B., Sison, J. D., Dragatsis, I., Zeitlin, S., and Chesselet, M. F. (2003). Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 465, 11–26. doi: 10.1002/cne.10776
Miron, V. E., Boyd, A., Zhao, J. W., Yuen, T. J., Ruckh, J. M., Shadrach, J. L., et al. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211–1218. doi: 10.1038/nn.3469
Moller, T. (2010). Neuroinflammation in Huntington’s disease. J. Neural Trans. 117, 1001–1008. doi: 10.1007/s00702-010-0430-7
Morganti, J. M., Riparip, L. K., and Rosi, S. (2016). Call Off the Dog(ma): M1/M2 polarization is concurrent following traumatic brain injury. PLoS ONE 11:e0148001. doi: 10.1371/journal.pone.0148001
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Ochaba, J., Monteys, A. M., O’Rourke, J. G., Reidling, J. C., Steffan, J. S., Davidson, B. L., et al. (2016). PIAS1 regulates mutant huntingtin accumulation and Huntington’s Disease-associated phenotypes In Vivo. Neuron 90, 507–520. doi: 10.1016/j.neuron.2016.03.016
Orihuela, R., McPherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
Orre, M., Kamphuis, W., Dooves, S., Kooijman, L., Chan, E. T., Kirk, C. J., et al. (2013). Reactive glia show increased immunoproteasome activity in Alzheimer’s disease. Brain 136, 1415–1431. doi: 10.1093/brain/awt083
Palazuelos, J., Aguado, T., Pazos, M. R., Julien, B., Carrasco, C., Resel, E., et al. (2009). Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain 132, 3152–3164. doi: 10.1093/brain/awp239
Pavese, N., Gerhard, A., Tai, Y. F., Ho, A. K., Turkheimer, F., Barker, R. A., et al. (2006). Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology 66, 1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17
Perry, V. H., and Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 10, 217–224. doi: 10.1038/nrneurol.2014.38
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/S1474-4422(11)70015-1
Politis, M., Lahiri, N., Niccolini, F., Su, P., Wu, K., Giannetti, P., et al. (2015). Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 83, 115–121. doi: 10.1016/j.nbd.2015.08.011
Politis, M., Pavese, N., Tai, Y. F., Kiferle, L., Mason, S. L., Brooks, D. J., et al. (2011). Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum. Brain. Mapp. 32, 258–270. doi: 10.1002/hbm.21008
Ponomarev, E. D., Maresz, K., Tan, Y., and Dittel, B. N. (2007). CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 27, 10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007
Raper, J., Bosinger, S., Johnson, Z., Tharp, G., Moran, S. P., and Chan, A. W. (2016). Increased irritability, anxiety, and immune reactivity in transgenic Huntington’s disease monkeys. Brain Behav. Immun. 58, 181–190. doi: 10.1016/j.bbi.2016.07.004
Rodrigues, F. B., Byrne, L. M., McColgan, P., Robertson, N., Tabrizi, S. J., Zetterberg, H., et al. (2016). Cerebrospinal fluid inflammatory biomarkers reflect clinical severity in Huntington’s Disease. PLoS ONE 11:e0163479. doi: 10.1371/journal.pone.0163479
Ross, C. A., and Tabrizi, S. J. (2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. doi: 10.1016/S1474-4422(10)70245-3
Sagredo, O., Gonzalez, S., Aroyo, I., Pazos, M. R., Benito, C., Lastres-Becker, I., et al. (2009). Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: relevance for Huntington’s Disease. Glia 57, 1154–1167. doi: 10.1002/glia.20838
Sapp, E., Kegel, K. B., Aronin, N., Hashikawa, T., Uchiyama, Y., Tohyama, K., et al. (2001). Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 60, 161–172. doi: 10.1093/jnen/60.2.161
Schatz, A. R., Lee, M., Condie, R. B., Pulaski, J. T., and Kaminski, N. E. (1997). Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicol. Appl. Pharmacol. 142, 278–287. doi: 10.1006/taap.1996.8034
Schilling, G., Becher, M. W., Sharp, A. H., Jinnah, H. A., Duan, K., Kotzuk, J. A., et al. (1999). Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 8, 397–407. doi: 10.1093/hmg/8.3.397
Schwarcz, R., and Pellicciari, R. (2002). Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J. Pharmacol. Exp. Ther. 303, 1–10. doi: 10.1124/jpet.102.034439
Shechter, R., Miller, O., Yovel, G., Rosenzweig, N., London, A., Ruckh, J., et al. (2013). Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38, 555–569. doi: 10.1016/j.immuni.2013.02.012
Shin, J. Y., Fang, Z. H., Yu, Z. X., Wang, C. E., Li, S. H., and Li, X. J. (2005). Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 171, 1001–1012. doi: 10.1083/jcb.200508072
Silvestroni, A., Faull, R. L., Strand, A. D., and Moller, T. (2009). Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport 20, 1098–1103. doi: 10.1097/WNR.0b013e32832e34ee
Singhrao, S. K., Neal, J. W., Morgan, B. P., and Gasque, P. (1999). Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp. Neurol. 159, 362–376. doi: 10.1006/exnr.1999.7170
Southwell, A. L., Smith-Dijak, A., Kay, C., Sepers, M., Villanueva, E. B., Parsons, M. P., et al. (2016). An enhanced Q175 knock-in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum. Mol. Genet. 25, 3654–3675. doi: 10.1093/hmg/ddw212
Su, P., Zhang, J., Wang, D., Zhao, F., Cao, Z., Aschner, M., et al. (2016). The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 319, 155–167. doi: 10.1016/j.neuroscience.2016.01.035
Szalardy, L., Klivenyi, P., Zadori, D., Fulop, F., Toldi, J., and Vecsei, L. (2012). Mitochondrial disturbances, tryptophan metabolites and neurodegeneration: medicinal chemistry aspects. Curr. Med. Chem. 19, 1899–1920. doi: 10.2174/092986712800167365
Tai, Y. F., Pavese, N., Gerhard, A., Tabrizi, S. J., Barker, R. A., Brooks, D. J., et al. (2007a). Imaging microglial activation in Huntington’s disease. Brain Res. Bull. 72, 148–151. doi: 10.1016/j.brainresbull.2006.10.029
Tai, Y. F., Pavese, N., Gerhard, A., Tabrizi, S. J., Barker, R. A., Brooks, D. J., et al. (2007b). Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 130, 1759–1766. doi: 10.1093/brain/awm044
Takeda, K., and Akira, S. (2004). TLR signaling pathways. Semin. Immunol. 16, 3–9. doi: 10.1016/j.smim.2003.10.003
The Huntington’s Disease Collaborative Research Group’ (1993). A novel gene containing a trinucleotide repeat that is expanded, and unstable on Huntington’s disease chromosomes. Cell 72, 971–983.
Trager, U., Andre, R., Lahiri, N., Magnusson-Lind, A., Weiss, A., Grueninger, S., et al. (2014). HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain 137, 819–833. doi: 10.1093/brain/awt355
Valdeolivas, S., Navarrete, C., Cantarero, I., Bellido, M. L., Munoz, E., and Sagredo, O. (2015). Neuroprotective properties of cannabigerol in Huntington’s Disease: studies in R6/2 Mice and 3-nitropropionate-lesioned mice. Neurotherapeutics 12, 185–199. doi: 10.1007/s13311-014-0304-z
Valekova, I., Jarkovska, K., Kotrcova, E., Bucci, J., Ellederova, Z., Juhas, S., et al. (2016). Revelation of the IFNalpha, IL-10, IL-8 and IL-1beta as promising biomarkers reflecting immuno-pathological mechanisms in porcine Huntington’s disease model. J. Neuroimmunol. 293, 71–81. doi: 10.1016/j.jneuroim.2016.02.012
Villegas-Llerena, C., Phillips, A., Garcia-Reitboeck, P., Hardy, J., and Pocock, J. M. (2016). Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 36, 74–81. doi: 10.1016/j.conb.2015.10.004
Walker, D. G., and Lue, L. F. (2015). Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res. Ther. 7, 56. doi: 10.1186/s13195-015-0139-9
Wang, H., Liu, C., Han, M., Cheng, C., and Zhang, D. (2016). TRAM1 promotes microglia M1 polarization. J. Mol. Neurosci. 58, 287–296. doi: 10.1007/s12031-015-0678-3
Wang, N., Gray, M., Lu, X. H., Cantle, J. P., Holley, S. M., Greiner, E., et al. (2014). Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat. Med. 20, 536–541. doi: 10.1038/nm.3514
Wheeler, V. C., Auerbach, W., White, J. K., Srinidhi, J., Auerbach, A., Ryan, A., et al. (1999). Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum. Mol. Genet. 8, 115–122. doi: 10.1093/hmg/8.1.115
Xu, X., Tay, Y., Sim, B., Yoon, S. I., Huang, Y., Ooi, J., et al. (2017). Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep. 8, 619–633. doi: 10.1016/j.stemcr.2017.01.022
Zanier, E. R., Pischiutta, F., Riganti, L., Marchesi, F., Turola, E., Fumagalli, S., et al. (2014). Bone marrow mesenchymal stromal cells drive protective M2 microglia polarization after brain trauma. Neurotherapeutics 11, 679–695. doi: 10.1007/s13311-014-0277-y
Zeron, M. M., Hansson, O., Chen, N., Wellington, C. L., Leavitt, B. R., Brundin, P., et al. (2002). Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 33, 849–860. doi: 10.1016/S0896-6273(02)00615-3
Zhao, T., Hong, Y., Li, S. H., and Li, X. J. (2016). Compartment-dependent degradation of mutant huntingtin accounts for its preferential accumulation in neuronal processes. J. Neurosci. 36, 8317–8328. doi: 10.1523/JNEUROSCI.0806-16.2016
Keywords: Huntington’s disease, microglia, microglial activation, M1 phenotype, M2 phenotype, pathogenesis
Citation: Yang H-M, Yang S, Huang S-S, Tang B-S and Guo J-F (2017) Microglial Activation in the Pathogenesis of Huntington’s Disease. Front. Aging Neurosci. 9:193. doi: 10.3389/fnagi.2017.00193
Received: 05 March 2017; Accepted: 30 May 2017;
Published: 19 June 2017.
Edited by:
Isidre Ferrer, University of Barcelona, SpainReviewed by:
Esther Pérez-Navarro, University of Barcelona, SpainCopyright © 2017 Yang, Yang, Huang, Tang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Feng Guo, Z3VvamlmZW5nMjAwM0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.