 Scott S. Shafiei
Scott S. Shafiei Marcos J. Guerrero-Muñoz
Marcos J. Guerrero-Muñoz Diana L. Castillo-Carranza
Diana L. Castillo-Carranza
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging Neurosci. , 04 April 2017
Sec. Alzheimer's Disease and Related Dementias
Volume 9 - 2017 | https://doi.org/10.3389/fnagi.2017.00083
Aging has long been considered as the main risk factor for several neurodegenerative disorders including a large group of diseases known as tauopathies. Even though neurofibrillary tangles (NFTs) have been examined as the main histopathological hallmark, they do not seem to play a role as the toxic entities leading to disease. Recent studies suggest that an intermediate form of tau, prior to NFT formation, the tau oligomer, is the true toxic species. However, the mechanisms by which tau oligomers trigger neurodegeneration remain unknown. This review summarizes recent findings regarding the role of tau oligomers in disease, including release from cells, propagation from affected to unaffected brain regions, uptake into cells, and toxicity via mitochondrial dysfunction. A greater understanding of tauopathies may lead to future advancements in regards to prevention and treatment.
Neurodegenerative diseases are a leading cause of death and disability affecting millions of elderly. As life expectancies rise, growing numbers of elderly become affected. This emerges as a major global issue as there are currently no adequate remedies for these diseases. A large group of these diseases, which are related to tau, a microtubule-associated protein, are known as tauopathies. Most notably, tauopathies include Alzheimer's disease (AD), progressive supranuclear palsy (PSP), Pick's disease, frontotemporal dementia (FTD), corticobasal degeneration, and variants of Parkinson's disease (PD) and Lewy body dementia (LBD; Goedert et al., 2000; Hutton, 2000; Spillantini et al., 2000). These share a common histopathological hallmark known as neurofibrillary tangles (NFTs) that consist of an accumulation of fibrillar tau deposits initially produced from tau protein aggregation (Ballatore et al., 2007).
While functional tau is an unfolded monomeric protein that stabilizes microtubules, regulates neurite growth, and monitors axonal transport of organelles (Medina and Avila, 2014), dysfunctional tau acquires a new toxic function.
NFTs, although considered a histopathological hallmark in tauopathies, do not appear to be the main toxic entities leading to disease (Gerson et al., 2014b). In AD, tau pathology and neuronal cell loss coincide in the same brain regions, and as brain dysfunction progresses, NFTs are found in greater anatomical distributions (Ihara, 2001). However, the role of NFTs in the progression of the disease is poorly understood. Compared to non-demented controls, AD brains exhibit up to 50% of neuronal loss in the cortex, exceeding the number of NFTs (Gómez-Isla et al., 1997). In addition, neurons containing NFTs are functionally intact in vivo (Kuchibhotla et al., 2014) and have been found in brains of cognitively normal individuals. Further, intra-neuronal NFTs do not affect post-synaptic function and signaling cascades responsible for long-term synaptic plasticity in tauopathy mice overexpressing P301L mutant tau (Rudinskiy et al., 2014), suggesting that synaptic deficits cannot be attributed to NFTs.
While evidence has linked FTD with parkinsonism in patients to tau mutations on chromosome 17 (FTDP-17), implying that tau dysfunction alone can cause neurodegeneration (Reed et al., 2001), studies in animal models have shown that overexpression of tau can lead to cell death (Lee et al., 2001; Tanemura et al., 2001, 2002; Tatebayashi et al., 2002) and exhibit behavioral abnormalities and synaptic dysfunction without the presence of NFTs (Wittmann et al., 2001; Andorfer et al., 2003; Santacruz et al., 2005; Spires et al., 2006; Berger et al., 2007; Yoshiyama et al., 2007; Cowan et al., 2010). Others have noted neuronal loss without NFT presence in a Drosophila model overexpressing tau (Wittmann et al., 2001). These studies provide evidence that progressive tau accumulation in neurodegeneration may not require NFT formation (Maeda et al., 2006). Indeed, reducing tau overexpression in mutant tau transgenic mice decreases neuronal cell loss even though NFTs continue to form (Santacruz et al., 2005). This indicates that NFT formation is not essential for neuronal loss.
While evidence indicates that these deposits are not toxic, many studies suggest that the tau oligomer, an intermediate entity, is likely responsible for disease onset. Hyper-phosphorylated tau assembles into small aggregates known as tau oligomers in route of NFT formation. As hyper-phosphorylated tau dislodges from microtubules, its affinity for other tau monomers leads individual tau to bind each other, forming oligomeric tau, a detergent-soluble aggregate. These tau oligomers potentiate neuronal damage, leading to neurodegeneration and traumatic brain injury (Hawkins et al., 2013; Gerson et al., 2014a, 2016; Sengupta et al., 2015). Moreover, they have been implicated in synaptic loss as shown in studies of wild-type human tau transgenic mice (Spires et al., 2006; Berger et al., 2007; Clavaguera et al., 2013). When the oligomer lengthens, it adapts a β-sheet structure and transforms into a detergent-insoluble aggregate with granular appearance under Atomic Force Microscopy (AFM). As these granular tau oligomers fuse together, they form tau fibrils, which ultimately form NFTs (Takashima, 2013). These steps hint that tau oligomers may be involved in neuronal dysfunction prior to NFT formation (Maeda et al., 2006).
The onset of clinical symptoms in AD and PSP brains correlate with elevated levels of tau oligomer (Maeda et al., 2006, 2007; Patterson et al., 2011; Lasagna-Reeves et al., 2012b; Gerson et al., 2014a). When tau oligomers, rather than tau monomers or fibrils, are injected into the brain of wild-type mice, cognitive, synaptic, and mitochondrial abnormalities follow (Lasagna-Reeves et al., 2011; Castillo-Carranza et al., 2014b). Additionally, studies have discovered that aggregated tau inhibits fast axonal transport in the anterograde direction at all physiological tau levels, whereas tau monomers have had no effect in either direction (LaPointe et al., 2009; Morfini et al., 2009). This suggests that monomers are not the toxic entity either. Most noteworthy, tau oligomers induce endogenous tau to misfold and propagate from affected to unaffected brain regions in mice, whereas fibrils do not (Lasagna-Reeves et al., 2012a,b; Wu et al., 2013). This indicates that tauopathies progress via a prion-like mechanism dependent upon tau oligomers (Gerson and Kayed, 2013; Castillo-Carranza et al., 2014b). With this concept, tau may be able to translocate between neurons and augment toxic tau components; in fact, evidence suggests probability of tau oligomer propagation between synaptically connected neurons (Gendreau and Hall, 2013; Pooler et al., 2013b). If true, then pathology begins in a small area and becomes symptomatic as it spreads to other areas of the brain (Medina and Avila, 2014). Studies show that tau pathology progresses from the entorhinal cortex to the hippocampus, eventually leading to the limbic and association cortex; this progression explains the individuals clinical cognitive status (Nelson et al., 2012). Further, mice injected with tau oligomers in the proximity of the hippocampus experienced immediate memory impairment (Lasagna-Reeves et al., 2011). These studies demonstrate that tau oligomers may be the toxic entities responsible for neurodegeneration in tauopathies (Ward et al., 2012).
Tau predominantly presents as an axonal cytoplasmic protein, however evidence has shown tau at the pre- and post-synapse in human brains (Tai et al., 2012) as well as at the post-synapse in mouse brains (Ittner et al., 2010). Further, tau directly interacts with synaptic proteins, including the NMDA receptor (Ittner et al., 2010; Mondragón-Rodríguez et al., 2012). This hints that tau plays a role in monitoring intracellular signaling pathways (Pooler and Hanger, 2010). Related evidence indicates that synaptic activity leads to tau monomer release (Pooler et al., 2013a; Yamada et al., 2014). Yet whether the movement of aggregates across the synapse occurs easily from nearby cells taking up released aggregated material at the axon terminal or whether the movement depends on activity is unknown.
Tau is also present outside the cell in brain fluids, including cerebrospinal fluid. In AD, the quantity of tau identified in the CSF increases with disease progression (Hampel et al., 2010). However, the mechanism of tau propagation from the brain to the CSF remains elusive. Recently, tau was discovered in the interstitial fluid of awake, wild-type mice, suggesting its release by neurons in the absence of neurodegeneration (Yamada et al., 2011). This evidence suggests that tau secretion is an active neuronal process separate from cell death (Saman et al., 2012; Pooler et al., 2013a).
In AD, misfolded and hyper-phosphorylated tau concentrates in various components of the neuron: dendrites, cell body, and axons (Avila et al., 2004). Hence, propagation of disease may depend on transport within neurons. Further, transgenic mouse lines expressing human tau aggregates in the entorhinal cortex have shown that tau is mislocalized from axons to cell bodies and dendrites as the mice age (Pooler et al., 2013b). Nevertheless, given that tau is detected in both axons and dendrites, it is possible that either region may be involved in its secretion (Pooler et al., 2013b). Extracellular tau is implicated as the primary agent during propagation of neurofibrillary lesions and spreading of tau toxicity (Medina and Avila, 2014). In trans-synaptic propagation, tau can be released and taken up by a synaptically-connected neuron (Clavaguera et al., 2009; Dujardin et al., 2014b; Dennissen et al., 2016). A recent study showed that neuronal networks facilitate cell-to-cell transfer of tau via synapses; using a microfluidic device they demonstrated that decreasing synaptic connections weakens tau transfer and the subsequent aggregation on the acceptor cell (Calafate et al., 2015).
Determining the mechanism behind the propagation of misfolded tau protein from one cell to another is currently of great significance in research. In AD, tau pathology has been found to spread from the transentorhinal cortex to the neocortex in a sequential pathway. This prion-like spreading of tau may occur throughout neuronal connections. However, the method of tau oligomer release via the cell and its spread is still unknown.
The prion concept suggests that a protein can be transformed into a disease-causing form when in contact with a pathogenic protein “seed.” The mechanism for the transformation is not well-understood; however, it includes templated conformational change (Telling et al., 1996) and is demonstrated to propagate through neural networks. Thus, the prion hypothesis serves as a useful model when testing ideas regarding propagation of protein pathology.
In trans-cellular propagation, tau aggregates escape from afflicted neurons into the extracellular space prior to entering adjacent or synaptically-connected cells. This suggests that extracellular tau may be susceptible to antibody-mediated therapies. According to this model, misfolded tau is released into the extracellular space and then gains entry into adjacent or synaptically-connected cells to trigger further aggregate formation via templated conformational change.
Recently, more evidence implies that the secretion of tau occurs through unconventional cellular pathways via vesicles known as exosomes (Saman et al., 2012) and ectosomes (Dujardin et al., 2014a).
Exosomes are small membranous vesicles ranging from 30 to 100 nm, which are secreted from most cell types, including neurons. Exosomes have been identified in several body fluids (Witwer et al., 2013; Khalyfa and Gozal, 2014). They are made by the endocytosis of molecules and can assist in spreading pathology. Once taken up by a cell, the molecules inside the exosomes are either recycled to the plasma membrane or transported to multivesicular bodies (MVBs; Dujardin et al., 2014a). The fusion of MVBs with the plasma membrane results in exosomal release (Mathivanan et al., 2010). AD brain samples contain exosomal proteins within amyloid plaques hinting that exosomes play part in disease pathology (Rajendran et al., 2006). Tau, like other amyloidogenic proteins, may be secreted and spread via exosomal vesicles (Danzer et al., 2012; Asai et al., 2015). In support, tau associated with exosomes and phosphorylated at Thr-181 (AT270+ tau) has been identified in human CSF samples of AD patients. More recently, patients affected with FTD and AD, were found to have high levels of total tau and phosphorylated tau (p-T181 and p-S396; Saman et al., 2012). Further, peripheral exosomes extracted from AD cases, propagate tau pathology in the brain of normal mice (Winston et al., 2016). It was recently shown that microglial cells may facilitate tau spreading via exosomes. The authors speculated that microglia phagocytose tau-containing neurons or synapses and secrete tau protein via exosomes (Asai et al., 2015). Further investigation is needed to determine if predominant tau aggregates are released via this unconventional pathway.
Exosomes are not the only vesicles that may spread pathology. Ectosomes are also extracellular vesicles, but range from 50 to 1,000 nm. They directly shed from cells by budding from the plasma membrane (Piccin et al., 2007; Cocucci et al., 2009; Théry et al., 2009; Davizon et al., 2010). Ectosomes are contenders in secreting tau protein since they are released via cell membrane activation by fluctuating intracellular levels of calcium, inflammatory molecules, or oxidative stress (Piccin et al., 2007; Doeuvre et al., 2009). Significantly, evidence suggests that tau secretion is partly mediated by ectosomal vesicles and that pathological tau accumulation in cells leads to a deviation toward tau secretion by exosomal vesicles (Dujardin et al., 2014a). These studies provide further evidence that extracellular vesicles play an important role in disease pathogenesis (Figure 1).
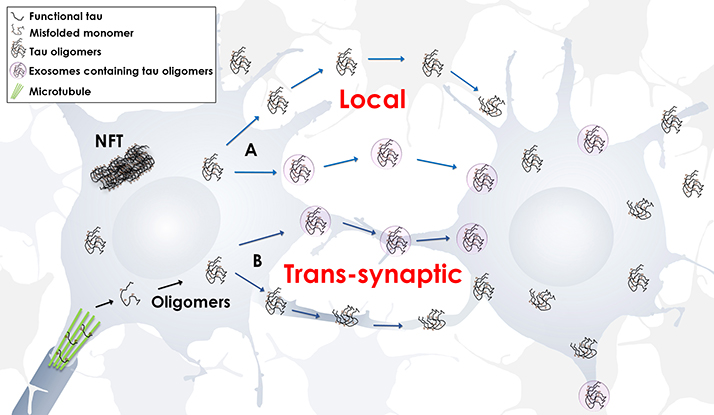
Figure 1. Propagation of tau oligomers. Schematic representation of free or exosomal tau oligomers release, local or trans-synaptic. In local transmission, tau oligomers are released from one neuron and taken up by another neuron in the vicinity. In trans-synaptic transmission, tau oligomers and/or exosomes containing tau oligomers are passed across the synapse of two neighboring neurons.
Quite a few mechanisms involving tau uptake have been proposed. These include (1) cell internalization of soluble, uncoated tau via receptor-mediated endocytosis (Gómez-Ramos et al., 2009), (2) dynamin-driven endocytosis of non-fibrillar, soluble tau aggregates (Wu et al., 2013), and (3) actin-dependent, proteoglycan-mediated macropinocytosis (Holmes et al., 2013; Figure 2).
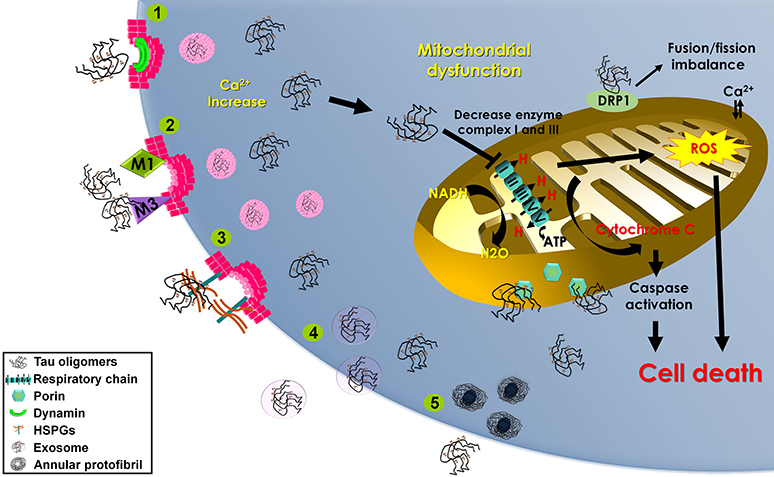
Figure 2. Mechanisms of tau oligomer internalization and mitochondrial damage. Schematic representation of the proposed mechanisms contributing to tau oligomer internalization that may lead to mitochondrial dysfunction and cell death. Methods of tau oligomer internalization include (1) dynamin-driven endocytosis; (2) muscarinic (M1 and M3) receptor-mediated endocytosis; (3) proteoglycan-mediated macropinocytosis (HSPGs); (4) exosomes; and (5) annular protofibrils. Internalized tau oligomers interfere with the mitochondrial respiratory chain, inducing cytochrome c release and stimulating reactive oxygen species (ROS) production. Tau oligomers induce mitochondrial fusion/fission imbalance by binding with dynamin-related protein 1 (DRP1). Tau oligomers interact with the outer membrane porin protein.
Tau may be endocytosed, promoting an increase in intracellular calcium that results in neuronal death. In the former theory, endocytosed tau may interact with various cellular products, including tau itself, and can be secreted, uncoated or in a membrane vesicle. Such secreted vesicles may interact with other cells and be endocytosed in an unspecific way. In the latter theory, during the secretion, vesicles and the cell membrane can be fused and uncoated tau protein can be released to the extracellular space (Clavaguera et al., 2009; Iba et al., 2013).
Tau can interact with muscarinic receptors; more specifically, M1 and M3 receptors have approximately a 10-fold higher affinity for tau than acetylcholine. Overstimulation with tau (as opposed to acetylcholine) does not desensitize the muscarinic receptors present on neurons of the hippocampus; hence, a repeat stimulus via tau increases intracellular calcium every time, thus altering intracellular calcium homeostasis and the following hyper-phosphorylation and misfolding of tau. Coupled with the fact that tau persists in the extracellular environment for a longer time than acetylcholine, a neurotoxic effect may occur. In other words, it is sensible to theorize that tauopathies progress via interaction of extracellular tau with M1 and M3 receptors on neurons leading to cytotoxic effects (Gómez-Ramos et al., 2009). Thus, blocking M1 and M3 receptors via receptor antagonists can prevent cytotoxic effects (Gómez-Ramos et al., 2008).
Exogenous tau aggregates may be taken up via an active process attenuated by dynamin inhibition, supporting endocytosis-mediated internalization. Dynamin is a GTPase essential for multiple intracellular functions, including formation of vesicles from the cell membrane, endocytosis, and synaptic vesicle recycling among others (Kozlov, 1999). Evidence shows that tau aggregates colocalize with dextran and HeLa cells, hinting that internalized aggregates are transported in endosomal vesicles and passed through the endosomal pathway to lysosomes (Wu et al., 2013).
Previous studies suggest that uptake of aggregated tau from the extracellular space depends on interaction with heparan sulfate proteoglycans (HSPGs; Holmes and Diamond, 2014). HSPGs are cell-surface macromolecules of heparan sulfate glycosaminoglycan chains covalently attached to a core protein. HSPGs are ubiquitously expressed in many cell types including neurons, and have been previously associated with dense core plaques, cerebrovascular amyloid, and NFT formation (van Horssen et al., 2001). Consistently, HSPGs have been implicated in amyloid as well as tau fibril formation in vitro, presumably facilitated by anionic moieties. Whether deposition of amyloid-b or tau is preceded by HSPGs or vice versa, it is clear that HSPGs play a role in the stabilization and uptake of these aggregates.
The recruitment of exogenous tau starts with binding HSPGs on the cell surface, stimulating macropinocytosis and bringing pathogenic “seeds” into the cell to guide trans-cellular propagation (Holmes et al., 2013). This uptake is necessary for intracellular seeding and was previously described for the prion protein uptake (Hooper, 2011). Even though the mechanism by which HSPGs mediate tau uptake is unknown, it seems to be confined to a specific “size” aggregate. Studies agree that small misfolded tau oligomers are readily taken up by neuronal cells (Wu et al., 2013; Mirbaha et al., 2015). However, regardless of the multiple “sizes” of tau aggregates that interact with the cell surface via HSPGs, it is likely that an assembly of at least three tau molecules is required to initiate endocytosis via HSPGs (Mirbaha et al., 2015). Interestingly, trimers were identified as the toxic tau aggregate at low nanomolar concentrations in vitro (Tian et al., 2013). Thus, tau oligomers may act as “seeds” inducing endogenous tau misfolding, suggesting a unifying mechanism for the propagation of protein amyloids (Mirbaha et al., 2015).
In other words, the HSPGs serve as a receptor for the cellular uptake of tau, a critical step similar to prion-like propagation. Basically, pathogenic tau aggregates use HSPGs to bind the cell surface of a neuron. This actively stimulates macropinocytosis, leading to propagation of aggregates between cells in culture and aggregate uptake in vivo (Holmes et al., 2013). Further, another study implied that exosomes depend on HSPGs for internalization (Christianson et al., 2013). As delineated above, exosomes are a distinct mechanism for propagation of misfolded tau.
A handful of proteins implicated in neurodegenerative diseases have been found to produce pore-like amyloid structures known as annular protofibrils (APFs). APFs are similar to pore-forming protein toxins in that their properties lead to membrane disruption. A recent study showed the existence of tau APFs in human brain samples from patients with PSP and LBD as well as in mice brain samples which overexpressed mutated tau. The study discovered that APFs form after tau oligomer formation and bypass higher NFT aggregate formation. The findings showed that APF formation relies on mutations in tau, phosphorylation levels, and cell type (Lasagna-Reeves et al., 2014). Hence, tau APFs may play a significant role in tauopathies by linking pore formation to cell death.
Oligomeric tau intermediates decrease cell viability (Flach et al., 2012). In aging, a protein involved in mitochondrial fission, dynamin-related protein 1 (DRP1), can bind tau abnormally, inducing neurodegeneration via mitochondrial dysfunction (Figure 2; DuBoff et al., 2012). Specifically, studies have shown reduced levels of mitochondrial proteins and activity in the brains of AD patients (Kim et al., 2001). One study showed diminished NADH-ubiquinone oxidoreductase (complex I) activity and injury to mitochondrial respiration and ATP synthesis (complex V) with age in P301L mice (David et al., 2005). Another study showed that expression of tau (truncated at Asp-421 to mimic caspase cleavage) caused mitochondrial dysfunction (Quintanilla et al., 2009).
Recently, data has shown that injected tau oligomers co-localize with the mitochondrial marker porin, suggesting a pathological relationship. In fact, tau oligomers might disrupt microtubule stability and trafficking, thus affecting organelle distribution. Mitochondria navigate long distances to provide for synaptic energy demand; therefore, inhibiting transport systems impairs energy production routes (Lasagna-Reeves et al., 2011).
Also, data shows low levels of complex I in brain hemispheres injected with tau oligomers when compared to brains injected with monomers or fibrils. This implies that alterations of complex I subunit mRNA, minimization in protein levels of complex I subunits (Kim et al., 2001), and other effects of mitochondrial damage (David et al., 2005) can be observed due to tau accumulation without the presence of NFT formation. Further, since tau oligomers hinder energy production through complex I, alterations in synaptically-localized mitochondria may result. However, recent data considering complex V levels suggested that tau oligomers do not implicate ATP synthesis initially. These results imply that tau oligomers initially affect complex I activity and may directly or indirectly disturb the later stage of complex V ATP synthesis (Lasagna-Reeves et al., 2011).
Mitochondrial damage can lead to activation of the apoptotic pathway. Hemispheres injected with tau oligomers were found to have increased levels of caspase-9 activation (Lasagna-Reeves et al., 2011). Suggestively, as tau oligomers concentrate at the mitochondrial membrane, cytochrome C is released, leading to caspase-9 activation via a complex with apoptotic-peptidase-activating-factor-1 (Apaf-1; Li et al., 1997). The relationship between cytochrome C and caspase-9 has been noted for other amyloidogenic proteins (Hashimoto et al., 1999; Simoneau et al., 2007). Moreover, caspase activation occurs before NFT formation, implying that a soluble tau entity may be the main toxic moiety (de Calignon et al., 2010).
Overall, evidence suggests that tau oligomers are the toxic entities in tau aggregation and that alteration of the mitochondrial membrane, minimization in complex I levels, and activation of the apoptotic-related caspase-9 can cause toxicity, impeding synaptic energy production (Lasagna-Reeves et al., 2011).
Discovering the pathological role of tau oligomers within the brain along with related mechanisms of cellular tau oligomer secretion, propagation, and uptake will allow for a better understanding of tauopathies (Castillo-Carranza et al., 2013, 2014a; Gerson et al., 2014b). Further, mitochondrial dysfunction caused by internalized tau oligomers may play an important role in pathogenesis. Admittedly, little is known regarding cellular tau oligomer release. Yet with greater knowledge regarding disease pathogenesis, better therapeutic approaches can be generated. We hypothesize that preventing tau oligomers from cellular release and uptake via exosomal or ectosomal pathways will relieve some toxic effects induced by tau oligomers in tauopathies.
SS, MG, and DC wrote the manuscript. DC and MG reviewed intellectual content. DC prepared the figures. SS, MG, DC, approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by funds from NIH National Institute for Minority Health & Health Disparities (NIMHD) award 4U54MD008621-04. We thank Ms. Urmi Sengupta and Julia Gerson for contributing to the editing process.
Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K. L., Barde, Y. A., et al. (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590. doi: 10.1046/j.1471-4159.2003.01879.x
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384. doi: 10.1152/physrev.00024.2003
Ballatore, C., Lee, V. M., and Trojanowski, J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672. doi: 10.1038/nrn2194
Berger, Z., Roder, H., Hanna, A., Carlson, A., Rangachari, V., Yue, M., et al. (2007). Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 27, 3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007
Calafate, S., Buist, A., Miskiewicz, K., Vijayan, V., Daneels, G., de Strooper, B., et al. (2015). Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. 11, 1176–1183. doi: 10.1016/j.celrep.2015.04.043
Castillo-Carranza, D. L., Gerson, J. E., Sengupta, U., Guerrero-Muñoz, M. J., Lasagna-Reeves, C. A., and Kayed, R. (2014b). Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J. Alzheimers Dis. 40(Suppl. 1), S97–S111. doi: 10.3233/JAD-132477
Castillo-Carranza, D. L., Lasagna-Reeves, C. A., and Kayed, R. (2013). Tau aggregates as immunotherapeutic targets. Front. Biosci. 5, 426–438. doi: 10.2741/S381
Castillo-Carranza, D. L., Sengupta, U., Guerrero-Muñoz, M. J., Lasagna-Reeves, C. A., Gerson, J. E., Singh, G., et al. (2014a). Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J. Neurosci. 34, 4260–4272. doi: 10.1523/JNEUROSCI.3192-13.2014
Christianson, H. C., Svensson, K. J., van Kuppevelt, T. H., Li, J. P., and Belting, M. (2013). Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. U.S.A. 110, 17380–17385. doi: 10.1073/pnas.1304266110
Clavaguera, F., Akatsu, H., Fraser, G., Crowther, R. A., Frank, S., Hench, J., et al. (2013). Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U.S.A. 110, 9535–9540. doi: 10.1073/pnas.1301175110
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. doi: 10.1038/ncb1901
Cocucci, E., Racchetti, G., and Meldolesi, J. (2009). Shedding microvesicles: artefacts no more. Trends Cell Biol. 19, 43–51. doi: 10.1016/j.tcb.2008.11.003
Cowan, C. M., Bossing, T., Page, A., Shepherd, D., and Mudher, A. (2010). Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 120, 593–604. doi: 10.1007/s00401-010-0716-8
Danzer, K. M., Kranich, L. R., Ruf, W. P., Cagsal-Getkin, O., Winslow, A. R., Zhu, L., et al. (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 7:42. doi: 10.1186/1750-1326-7-42
David, D. C., Hauptmann, S., Scherping, I., Schuessel, K., Keil, U., Rizzu, P., et al. (2005). Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 280, 23802–23814. doi: 10.1074/jbc.M500356200
Davizon, P., Munday, A. D., and López, J. A. (2010). Tissue factor, lipid rafts, and microparticles. Semin. Thromb. Hemost. 36, 857–864. doi: 10.1055/s-0030-1267039
de Calignon, A., Fox, L. M., Pitstick, R., Carlson, G. A., Bacskai, B. J., Spires-Jones, T. L., et al. (2010). Caspase activation precedes and leads to tangles. Nature 464, 1201–1204. doi: 10.1038/nature08890
Dennissen, F. J., Anglada-Huguet, M., Sydow, A., Mandelkow, E., and Mandelkow, E. M. (2016). Adenosine A1 receptor antagonist rolofylline alleviates axonopathy caused by human Tau DeltaK280. Proc. Natl. Acad. Sci. U.S.A. 113, 11597–11602. doi: 10.1073/pnas.1603119113
Doeuvre, L., Plawinski, L., Toti, F., and Anglés-Cano, E. (2009). Cell-derived microparticles: a new challenge in neuroscience. J. Neurochem. 110, 457–468. doi: 10.1111/j.1471-4159.2009.06163.x
DuBoff, B., Götz, J., and Feany, M. B. (2012). Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75, 618–632. doi: 10.1016/j.neuron.2012.06.026
Dujardin, S., Bégard, S., Caillierez, R., Lachaud, C., Delattre, L., Carrier, S., et al. (2014a). Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS ONE 9:e100760. doi: 10.1371/journal.pone.0100760
Dujardin, S., Lécolle, K., Caillierez, R., Bégard, S., Zommer, N., Lachaud, C., et al. (2014b). Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2:14. doi: 10.1186/2051-5960-2-14
Flach, K., Hilbrich, I., Schiffmann, A., Gärtner, U., Krüger, M., Leonhardt, M., et al. (2012). Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 287, 43223–43233. doi: 10.1074/jbc.M112.396176
Gendreau, K. L., and Hall, G. F. (2013). Tangles, toxicity, and tau secretion in AD - new approaches to a vexing problem. Front. Neurol. 4:160. doi: 10.3389/fneur.2013.00160
Gerson, J. E., and Kayed, R. (2013). Formation and propagation of tau oligomeric seeds. Front. Neurol. 4:93. doi: 10.3389/fneur.2013.00093
Gerson, J. E., Castillo-Carranza, D. L., and Kayed, R. (2014b). Advances in therapeutics for neurodegenerative tauopathies: moving toward the specific targeting of the most toxic tau species. ACS Chem. Neurosci. 5, 752–769. doi: 10.1021/cn500143n
Gerson, J. E., Sengupta, U., Lasagna-Reeves, C. A., Guerrero-Muñoz, M. J., Troncoso, J., and Kayed, R. (2014a). Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol. Commun. 2:73. doi: 10.1186/2051-5960-2-73
Gerson, J., Castillo-Carranza, D. L., Sengupta, U., Bodani, R., Prough, D. S., DeWitt, D. S., et al. (2016). Tau oligomers derived from traumatic brain injury cause cognitive impairment and accelerate onset of pathology in htau mice. J. Neurotrauma 33, 2034–2043. doi: 10.1089/neu.2015.4262
Goedert, M., Ghetti, B., and Spillantini, M. G. (2000). Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Their relevance for understanding the neurogenerative process. Ann. N. Y. Acad. Sci. 920, 74–83. doi: 10.1111/j.1749-6632.2000.tb06907.x
Gómez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Gómez-Ramos, A., Díaz-Hernández, M., Rubio, A., Díaz-Hernández, J. I., Miras-Portugal, M. T., and Avila, J. (2009). Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 19, 708–717. doi: 10.1016/j.euroneuro.2009.04.006
Gómez-Ramos, A., Díaz-Hernández, M., Rubio, A., Miras-Portugal, M. T., and Avila, J. (2008). Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 37, 673–681. doi: 10.1016/j.mcn.2007.12.010
Hampel, H., Blennow, K., Shaw, L. M., Hoessler, Y. C., Zetterberg, H., and Trojanowski, J. Q. (2010). Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Exp. Gerontol. 45, 30–40. doi: 10.1016/j.exger.2009.10.010
Hashimoto, M., Takeda, A., Hsu, L. J., Takenouchi, T., and Masliah, E. (1999). Role of cytochrome c as a stimulator of alpha-synuclein aggregation in Lewy body disease. J. Biol. Chem. 274, 28849–28852. doi: 10.1074/jbc.274.41.28849
Hawkins, B. E., Krishnamurthy, S., Castillo-Carranza, D. L., Sengupta, U., Prough, D. S., Jackson, G. R., et al. (2013). Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. J. Biol. Chem. 288, 17042–17050. doi: 10.1074/jbc.M113.472746
Holmes, B. B., and Diamond, M. I. (2014). Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J. Biol. Chem. 289, 19855–19861. doi: 10.1074/jbc.R114.549295
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Hooper, N. M. (2011). Glypican-1 facilitates prion conversion in lipid rafts. J. Neurochem. 116, 721–725. doi: 10.1111/j.1471-4159.2010.06936.x
Hutton, M. (2000). Molecular genetics of chromosome 17 tauopathies. Ann. N. Y. Acad. Sci. 920, 63–73. doi: 10.1111/j.1749-6632.2000.tb06906.x
Iba, M., Guo, J. L., McBride, J. D., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J. Neurosci. 33, 1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013
Ihara, Y. (2001). PHF and PHF-like fibrils–cause or consequence? Neurobiol. Aging 22, 123–126. doi: 10.1016/S0197-4580(00)00200-1
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Khalyfa, A., and Gozal, D. (2014). Exosomal miRNAs as potential biomarkers of cardiovascular risk in children. J. Transl. Med. 12:162. doi: 10.1186/1479-5876-12-162
Kim, S. H., Vlkolinsky, R., Cairns, N., Fountoulakis, M., and Lubec, G. (2001). The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer's disease. Life Sci. 68, 2741–2750. doi: 10.1016/S0024-3205(01)01074-8
Kozlov, M. M. (1999). Dynamin: possible mechanism of “Pinchase” action. Biophys. J. 77, 604–616. doi: 10.1016/S0006-3495(99)76917-1
Kuchibhotla, K. V., Wegmann, S., Kopeikina, K. J., Hawkes, J., Rudinskiy, N., Andermann, M. L., et al. (2014). Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, 510–514. doi: 10.1073/pnas.1318807111
LaPointe, N. E., Morfini, G., Pigino, G., Gaisina, I. N., Kozikowski, A. P., Binder, L. I., et al. (2009). The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res. 87, 440–451. doi: 10.1002/jnr.21850
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Clos, A. L., Jackson, G. R., and Kayed, R. (2011). Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 6:39. doi: 10.1186/1750-1326-6-39
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Guerrero-Muñoz, M. J., Kiritoshi, T., Neugebauer, V., et al. (2012a). Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2:700. doi: 10.1038/srep00700
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Sarmiento, J., Troncoso, J., Jackson, G. R., et al. (2012b). Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 26, 1946–1959. doi: 10.1096/fj.11-199851
Lasagna-Reeves, C. A., Sengupta, U., Castillo-Carranza, D., Gerson, J. E., Guerrero-Muñoz, M., Troncoso, J. C., et al. (2014). The formation of tau pore-like structures is prevalent and cell specific: possible implications for the disease phenotypes. Acta Neuropathol. Commun. 2:56. doi: 10.1186/2051-5960-2-56
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi: 10.1146/annurev.neuro.24.1.1121
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S., et al. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. doi: 10.1016/S0092-8674(00)80434-1
Maeda, S., Sahara, N., Saito, Y., Murayama, M., Yoshiike, Y., Kim, H., et al. (2007). Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861. doi: 10.1021/bi061359o
Maeda, S., Sahara, N., Saito, Y., Murayama, S., Ikai, A., and Takashima, A. (2006). Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci. Res. 54, 197–201. doi: 10.1016/j.neures.2005.11.009
Mathivanan, S., Ji, H., and Simpson, R. J. (2010). Exosomes: extracellular organelles important in intercellular communication. J. Proteomics 73, 1907–1920. doi: 10.1016/j.jprot.2010.06.006
Medina, M., and Avila, J. (2014). The role of extracellular Tau in the spreading of neurofibrillary pathology. Front. Cell. Neurosci. 8:113. doi: 10.3389/fncel.2014.00113
Mirbaha, H., Holmes, B. B., Sanders, D. W., Bieschke, J., and Diamond, M. I. (2015). Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J. Biol. Chem. 290, 14893–14903. doi: 10.1074/jbc.M115.652693
Mondragón-Rodríguez, S., Trillaud-Doppia, E., Dudilot, A., Bourgeois, C., Lauzon, M., Leclerc, N., et al. (2012). Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 287, 32040–32053. doi: 10.1074/jbc.M112.401240
Morfini, G. A., Burns, M., Binder, L. I., Kanaan, N. M., LaPointe, N., Bosco, D. A., et al. (2009). Axonal transport defects in neurodegenerative diseases. J. Neurosci. 29, 12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Patterson, K. R., Remmers, C., Fu, Y., Brooker, S., Kanaan, N. M., Vana, L., et al. (2011). Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 286, 23063–23076. doi: 10.1074/jbc.M111.237974
Piccin, A., Murphy, W. G., and Smith, O. P. (2007). Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 21, 157–171. doi: 10.1016/j.blre.2006.09.001
Pooler, A. M., and Hanger, D. P. (2010). Functional implications of the association of tau with the plasma membrane. Biochem. Soc. Trans. 38, 1012–1015. doi: 10.1042/BST0381012
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013a). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Pooler, A. M., Polydoro, M., Wegmann, S., Nicholls, S. B., Spires-Jones, T. L., and Hyman, B. T. (2013b). Propagation of tau pathology in Alzheimer's disease: identification of novel therapeutic targets. Alzheimers Res. Ther. 5:49. doi: 10.1186/alzrt214
Quintanilla, R. A., Matthews-Roberson, T. A., Dolan, P. J., and Johnson, G. V. (2009). Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J. Biol. Chem. 284, 18754–18766. doi: 10.1074/jbc.M808908200
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177. doi: 10.1073/pnas.0603838103
Reed, L. A., Wszolek, Z. K., and Hutton, M. (2001). Phenotypic correlations in FTDP-17. Neurobiol. Aging 22, 89–107. doi: 10.1016/S0197-4580(00)00202-5
Rudinskiy, N., Hawkes, J. M., Wegmann, S., Kuchibhotla, K. V., Muzikansky, A., Betensky, R. A., et al. (2014). Tau pathology does not affect experience-driven single-neuron and network-wide Arc/Arg3.1 responses. Acta Neuropathol. Commun. 2:63. doi: 10.1186/2051-5960-2-63
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Saman, S., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. doi: 10.1126/science.1113694
Sengupta, U., Guerrero-Muñoz, M. J., Castillo-Carranza, D. L., Lasagna-Reeves, C. A., Gerson, J. E., Paulucci-Holthauzen, A. A., et al. (2015). Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol. Psychiatry 78, 672–683. doi: 10.1016/j.biopsych.2014.12.019
Simoneau, S., Rezaei, H., Salès, N., Kaiser-Schulz, G., Lefebvre-Roque, M., Vidal, C., et al. (2007). In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 3:e125. doi: 10.1371/journal.ppat.0030125
Spillantini, M. G., Van Swieten, J. C., and Goedert, M. (2000). Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Neurogenetics 2, 193–205. doi: 10.007/s100489900084
Spires, T. L., Orne, J. D., SantaCruz, K., Pitstick, R., Carlson, G. A., Ashe, K. H., et al. (2006). Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607. doi: 10.2353/ajpath.2006.050840
Tai, H. C., Serrano-Pozo, A., Hashimoto, T., Frosch, M. P., Spires-Jones, T. L., and Hyman, B. T. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181, 1426–1435. doi: 10.1016/j.ajpath.2012.06.033
Takashima, A. (2013). Tauopathies and tau oligomers. J. Alzheimers Dis. 37, 565–568. doi: 10.3233/JAD-130653
Tanemura, K., Akagi, T., Murayama, M., Kikuchi, N., Murayama, O., Hashikawa, T., et al. (2001). Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol. Dis. 8, 1036–1045. doi: 10.1006/nbdi.2001.0439
Tanemura, K., Murayama, M., Akagi, T., Hashikawa, T., Tominaga, T., Ichikawa, M., et al. (2002). Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J. Neurosci. 22, 133–141.
Tatebayashi, Y., Miyasaka, T., Chui, D. H., Akagi, T., Mishima, K., Iwasaki, K., et al. (2002). Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. U.S.A. 99, 13896–13901. doi: 10.1073/pnas.202205599
Telling, G. C., Parchi, P., DeArmond, S. J., Cortelli, P., Montagna, P., Gabizon, R., et al. (1996). Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274, 2079–2082. doi: 10.1126/science.274.5295.2079
Théry, C., Ostrowski, M., and Segura, E. (2009). Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 9, 581–593. doi: 10.1038/nri2567
Tian, H., Davidowitz, E., Lopez, P., Emadi, S., Moe, J., and Sierks, M. (2013). Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell Biol. 2013:260787. doi: 10.1155/2013/260787
van Horssen, J., Otte-Höller, I., David, G., Maat-Schieman, M. L., van den Heuvel, L. P., Wesseling, P., et al. (2001). Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer's disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol. 102, 604–614. doi: 10.1007/s004010100414
Ward, S. M., Himmelstein, D. S., Lancia, J. K., and Binder, L. I. (2012). Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 40, 667–671. doi: 10.1042/BST20120134
Winston, C. N., Goetzl, E. J., Akers, J. C., Carter, B. S., Rockenstein, E. M., Galasko, D., et al. (2016). Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. 3, 63–72. doi: 10.1016/j.dadm.2016.04.001
Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M., et al. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714. doi: 10.1126/science.1062382
Witwer, K. W., Buzás, E. I., Bemis, L. T., Bora, A., Lässer, C., Lötvall, J., et al. (2013). Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2:20360. doi: 10.3402/jev.v2i0.20360
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Yamada, K., Cirrito, J. R., Stewart, F. R., Jiang, H., Finn, M. B., Holmes, B. B., et al. (2011). In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J. Neurosci. 31, 13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011
Yamada, K., Holth, J. K., Liao, F., Stewart, F. R., Mahan, T. E., Jiang, H., et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. doi: 10.1084/jem.20131685
Keywords: tauopathy, tau oligomers, tau secretion, propagation, mitochondrial dysfunction
Citation: Shafiei SS, Guerrero-Muñoz MJ and Castillo-Carranza DL (2017) Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 9:83. doi: 10.3389/fnagi.2017.00083
Received: 15 September 2016; Accepted: 16 March 2017;
Published: 04 April 2017.
Edited by:
Rodrigo Orlando Kuljiš, University of Miami School of Medicine, USAReviewed by:
Ralf J. Braun, University of Bayreuth, GermanyCopyright © 2017 Shafiei, Guerrero-Muñoz and Castillo-Carranza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana L. Castillo-Carranza, ZGlhbmEuY2FzdGlsbG9jYXJyYW56YUBoYW1wdG9udS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.