 Gabriela Martínez
Gabriela Martínez Claudia Duran-Aniotz
Claudia Duran-Aniotz Felipe Cabral-Miranda
Felipe Cabral-Miranda Claudio Hetz
Claudio Hetz- 1Center for Geroscience, Brain Health and Metabolism, Santiago, Chile
- 2Biomedical Neuroscience Institute, Faculty of Medicine, University of Chile, Santiago, Chile
- 3Program of Cellular and Molecular Biology, Institute of Biomedical Sciences, University of Chile, Santiago, Chile
- 4Center for Integrative Biology, Universidad Mayor, Santiago, Chile
- 5Buck Institute for Research on Aging, Novato, CA, USA
- 6Department of Immunology and Infectious diseases, Harvard School of Public Health, Boston, MA, USA
A commentary on
XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity
by Taylor, R. C., and Dillin A. (2013). Cell 153, 1435–1447. doi: 10.1016/j.cell.2013.05.042
The life expectancy in the world's population is increasing, highlighting the need of better understanding of the cellular and molecular pathways that drive the aging process. Because aging is the major risk factor to develop neurodegenerative conditions such as Alzheimer's and Parkinson's disease, the number of patients affected is constantly increasing, representing a major social and economic problem. Importantly, abnormal protein aggregation is a transversal pathological event of most aging-related brain diseases, suggesting that the ability of neurons to handle alterations in the proteome is specifically altered (Kaushik and Cuervo, 2015). Several hallmarks of aging have been identified at the cellular and molecular level (Lopez-Otin et al., 2013; Kennedy et al., 2014), highlighting alterations in protein homeostasis or proteostasis. In fact, studies in simple model organisms indicate that the buffering capacity of the proteostasis network (PN) is reduced during aging (Douglas and Dillin, 2010; Mardones et al., 2015). The PN can be decomposed in different interrelated sub-networks including mechanisms responsible for protein synthesis, translation, folding, trafficking, quality control, secretion, and degradation (Balch et al., 2008). Sustained dysfunction of one or more components of the PN may translate into cell dysfunction and even proteotoxicity (Figure 1).
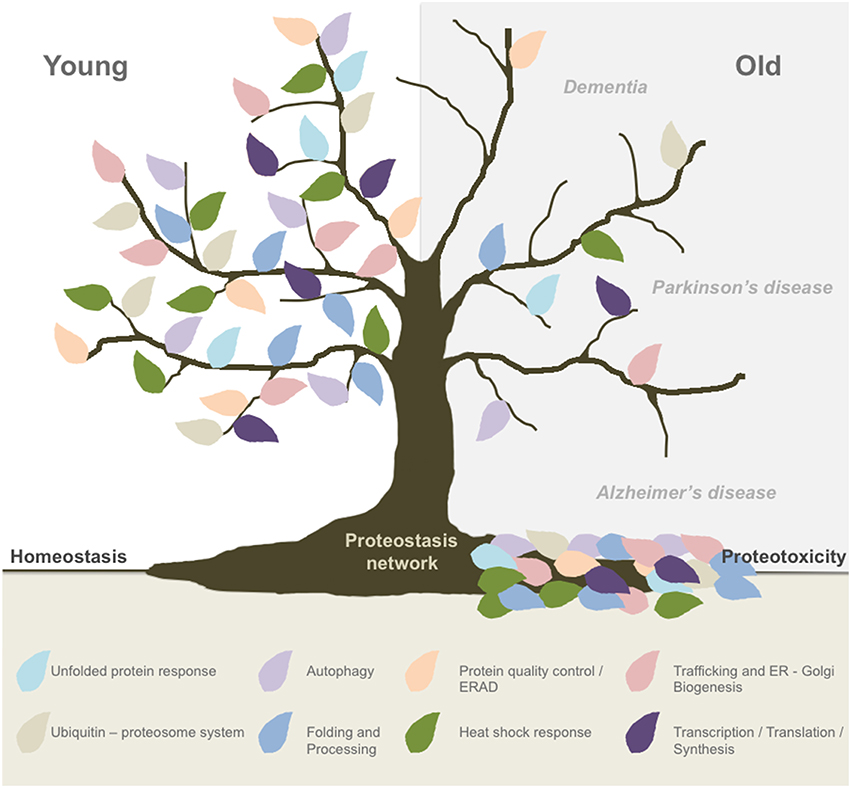
Figure 1. Global proteostasis network impairment during aging. Aging is the main risk factor to develop most neurodegenerative conditions and new evidence has pointed out to a progressive decline in the buffering capacity of the proteostasis network (PN) to handle cellular stress. The PN is formed by different interrelated sub-networks including mechanisms responsible for protein translation, folding, synthesis, protein quality control, trafficking, secretion, and degradation (ERAD, proteasome, autophagy). Proteostasis breakdown during aging may result in proteotoxicity and the development of neurodegenerative diseases such as Alzheimer's and Parkinson's disease.
Around 30% of the total proteome is synthetized at the endoplasmic reticulum (ER), an essential compartment involved in calcium handling, lipid synthesis among other functions. Different physiological and pathological stimuli can alter the function of this organelle, resulting in the accumulation of misfolded proteins. Importantly ER stress has been proposed as a central driver of several neurodegenerative conditions (Hetz and Mollereau, 2014). ER stress triggers the activation of the unfolded protein response (UPR), a central homeostatic pathway that orchestrates cells adaptation (Hetz et al., 2015). Studies in Caenorhabditis elegans and rats indicate that the activity of the UPR is drastically ablated during aging (Paz Gavilan et al., 2006; Naidoo et al., 2008; Ben-Zvi et al., 2009; Gavilan et al., 2009; Taylor and Dillin, 2013). The UPR is mediated by three main stress sensors located at the ER membrane including ATF6, PERK, and IRE1 (Ron and Walter, 2007). In brief, activation of IRE1 controls to the expression of the transcription factor XBP1s, leading to the upregulation of genes related with protein quality control, folding, ERAD, among other targets (Hetz et al., 2015). PERK phosphorylates eIF2α; inhibiting the translation of proteins into the ER, in addition to induce the expression of the transcription factor ATF4 regulating genes involved in the antioxidant response, amino acid metabolism and folding. Under irreversible ER stress ATF4 is essential to trigger apoptosis. ATF6 encodes a transcription factor in its cytosolic domain that upon processing is realized to control gene expression. Altogether, the activation of the UPR enforces adaptive mechanisms to sustain proteostasis or trigger cell demise when protein misfolding cannot be mitigated determining cell fate.
Several studies in model organisms have uncovered the significance of UPR signaling to the aging process. IRE1 is the only ER stress sensor expressed in yeast and contributes to lifespan extension (Labunskyy et al., 2014), consistent with the fact that UPR activation in this organism is a relevant feature involved in the health span control triggered by caloric restriction (Choi et al., 2013). Similarly, genetic modifications that enhance the activity of the UPR improve replicative lifespan in Saccharomyces cerevisiae (Cui et al., 2015). Studies in C. elegans demonstrated that ablating the expression of XBP1 reduces life expectancy, associated with altered FOXO and insulin/IGF-1 signaling, a canonical aging pathway (Henis-Korenblit et al., 2010). Importantly, another report indicated that the ectopic expression of XBP1s in neurons has a significant effect in increasing lifespan in C. elegans (around 30%), representing one of the strongest aging modulator described so far in this specie (Taylor and Dillin, 2013). In D. melanogaster, the occurrence of ER stress and chronic inflammation alters the stem cell pool in the gut, affecting intestinal homeostasis during aging (Wang et al., 2014). Unexpectedly, a recent study indicated that chronic PERK signaling limits lifespan by controlling intestinal homeostasis, having important consequences to organismal health (Wang et al., 2015). In mammals, it was reported that the capacity to response to ER stress and activate IRE1 is attenuated in macrophages during aging, increasing the susceptibility to apoptosis (Song et al., 2013). Accordantly, aged rats present more pro-apoptotic UPR components as opposed to adaptive mediators such as BIP, calnexin, and PDI after ER stress induction (Paz Gavilan et al., 2006; Naidoo et al., 2008). In contrast, during the aging process B cells, osteoclasts, adipocyte tissue, the retina, and muscle experience elevated levels of ER stress and UPR activation (Chalil et al., 2015; Ghosh et al., 2015; Lenox et al., 2015; Baehr et al., 2016; Kannan et al., 2016). These observations suggest that aging maybe associated with accumulative damage to the ER rather than an attenuation of UPR responses. However, the role of ER proteostasis impartment in mammalian aging needs to be functionally defined.
The UPR is emerging as a key player in the integration of systemic responses to handle proteostasis alterations at the whole organism, governed by the central nervous system (Sun et al., 2012; Taylor and Dillin, 2013). In addition to regulate the intrinsic capacity of the cell to respond to ER stress, activation of IRE1 in neurons engages an organismal reaction to promote stress resistance and longevity on a cell-nonautonomous manner (Taylor and Dillin, 2013). Interestingly, the activation of XBP1s in neurons per se was irrelevant to sustain organismal homeostasis, suggesting that the nervous system operates as a global adjustor of proteostasis, where the effectors in terms of enforcing aging resistance operate in the periphery, highlighting the intestine. Importantly, other studies have shown a similar mode of control for the heat shock response and the innate immunity in C. elegans (reviewed in Mardones et al., 2015). Similarly, in flies activation of PERK engages cell-nonautonomous responses in the gut during aging (Wang et al., 2015). The concept cell-nonautonomous UPR was recently validated in mammals, where the expression of XBP1s in the hypothalamus propagates signals to the periphery (i.e., the liver) to adjust energy metabolism (Williams et al., 2014). However, the specific mechanism of proteostasis control in mammals and the neuronal circuits mediating the propagation of UPR signals between cells remain to be determined. Importantly, in C. elegans the propagation of ER stress signals to the periphery depends on neurotransmitters, suggesting that signaling mechanisms may mediate the activation of UPR-like responses in the targeted tissue probably on a stress-independent manner (Taylor and Dillin, 2013). In this line, we recently reported that XBP1s has a novel function in controlling synaptic plasticity and behavior in mammals, where growth factors like BDNF can engage the pathway (Martinez et al., 2016).
Although several studies are placing the ER PN as a relevant adjustor of organismal aging in several species, its actual impact to human aging remains to be established. Many important questions need to be solved in this emerging field: Why is the UPR buffering capacity attenuated during aging? How does the nervous system control organismal proteostasis? Is there a connection between ER stress and aging in protein misfolding disorders affecting the nervous system? Can we exploit the control of cell-nonautonomous UPR as a therapeutic strategy to delay aging? Importantly, recent studies suggest that oxidative damage could directly modify UPR stress sensors, ablating adaptive responses (Nakato et al., 2015). In addition, the redox status of the ER is altered during aging in C. elegans, suggesting that intrinsic physiological alterations to this subcellular compartment may underlay the reduced capacity of the pathway to handle proteostasis alterations when cells get old (Kirstein et al., 2015). Several novel drugs are available to fine-tune the UPR and reduce ER stress levels (Hetz et al., 2013), which promises new avenues to intervene brain aging which may reduce the risk to develop neurodegenerative diseases, improving health span.
Author Contributions
GM: conceptualization, editing, and writing of the manuscript. CD: editing of manuscript, FC: editing of manuscript. CH: conceptualization, editing, and writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is funded by Millennium Institute No. P09-015-F, and FONDAP 15150012, the Frick Foundation 20014-15, ALS Therapy Alliance 2014-F-059, Muscular Dystrophy Association 382453, CONICYT-USA 2013-0003, Michael J. Fox Foundation for Parkinson's Research – Target Validation grant No. 9277, COPEC-UC Foundation 2013.R.40, Ecos-Conicyt C13S02, FONDECYT no. 1140549, Office of Naval Research-Global (ONR-G) N62909-16-1-2003 and ALSRP Therapeutic Idea Award AL150111 (CH). FONDECYT No. 3150637 (GM) and FONDECYT No. 3140466 (CD).
References
Baehr, L. M., West, D. W., Marcotte, G., Marshall, A. G., De Sousa, L. G., Baar, K., et al. (2016). Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany. NY). 8, 127–146. doi: 10.18632/aging.100879
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Ben-Zvi, A., Miller, E. A., and Morimoto, R. I. (2009). Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. U.S.A. 106, 14914–14919. doi: 10.1073/pnas.0902882106
Chalil, S., Jaspers, R. T., Manders, R. J., Klein-Nulend, J., Bakker, A. D., and Deldicque, L. (2015). Increased endoplasmic reticulum stress in mouse osteocytes with aging alters Cox-2 response to mechanical stimuli. Calcif. Tissue Int. 96, 123–128. doi: 10.1007/s00223-014-9944-6
Choi, K. M., Kwon, Y. Y., and Lee, C. K. (2013). Characterization of global gene expression during assurance of lifespan extension by caloric restriction in budding yeast. Exp. Gerontol. 48, 1455–1468. doi: 10.1016/j.exger.2013.10.001
Cui, H. J., Liu, X. G., McCormick, M., Wasko, B. M., Zhao, W., He, X., et al. (2015). PMT1 deficiency enhances basal UPR activity and extends replicative lifespan of Saccharomyces cerevisiae. Age (Dordr). 37, 9788. doi: 10.1007/s11357-015-9788-7
Douglas, P. M., and Dillin, A. (2010). Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 190, 719–729. doi: 10.1083/jcb.201005144
Gavilan, M. P., Pintado, C., Gavilan, E., Jimenez, S., Rios, R. M., Vitorica, J., et al. (2009). Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell 8, 654–665. doi: 10.1111/j.1474-9726.2009.00519.x
Ghosh, A. K., Garg, S. K., Mau, T., O'Brien, M., Liu, J., and Yung, R. (2015). Elevated endoplasmic reticulum stress response contributes to adipose tissue inflammation in aging. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1320–1329. doi: 10.1093/gerona/glu186
Henis-Korenblit, S., Zhang, P., Hansen, M., McCormick, M., Lee, S. J., Cary, M., et al. (2010). Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc. Natl. Acad. Sci. U.S.A. 107, 9730–9735. doi: 10.1073/pnas.1002575107
Hetz, C., Chevet, E., and Harding, H. P. (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719. doi: 10.1038/nrd3976
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. doi: 10.1038/ncb3184
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 1–19. doi: 10.1038/nrn3689
Kannan, S., Dawany, N., Kurupati, R., Showe, L. C., and Ertl, H. C. (2016). Age-related changes in the transcriptome of antibody-secreting cells. Oncotarget 7, 13340–13353. doi: 10.18632/oncotarget.7958
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. doi: 10.1038/nm.4001
Kennedy, B. K., Berger, S. L., Brunet, A., Campisi, J., Cuervo, A. M., Epel, E. S., et al. (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. doi: 10.1016/j.cell.2014.10.039
Kirstein, J., Morito, D., Kakihana, T., Sugihara, M., Minnen, A., Hipp, M. S., et al. (2015). Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J. 34, 2334–2349. doi: 10.15252/embj.201591711
Labunskyy, V. M., Gerashchenko, M. V., Delaney, J. R., Kaya, A., Kennedy, B. K., Kaeberlein, M., et al. (2014). Lifespan extension conferred by endoplasmic reticulum secretory pathway deficiency requires induction of the unfolded protein response. PLoS Genet. 10:e1004019. doi: 10.1371/journal.pgen.1004019
Lenox, A. R., Bhootada, Y., Gorbatyuk, O., Fullard, R., and Gorbatyuk, M. (2015). Unfolded protein response is activated in aged retinas. Neurosci. Lett. 609, 30–35. doi: 10.1016/j.neulet.2015.10.019
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Mardones, P., Martinez, G., and Hetz, C. (2015). Control of systemic proteostasis by the nervous system. Trends Cell Biol. 25, 1–10. doi: 10.1016/j.tcb.2014.08.001
Martinez, G., Vidal, R. L., Mardones, P., Serrano, F. G., Ardiles, A. O., Wirth, C., et al. (2016). Regulation of memory formation by the transcription factor XBP1. Cell Rep. 14, 1382–1394. doi: 10.1016/j.celrep.2016.01.028
Naidoo, N., Ferber, M., Master, M., Zhu, Y., and Pack, A. I. (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. doi: 10.1523/JNEUROSCI.5685-07.2008
Nakato, R., Ohkubo, Y., Konishi, A., Shibata, M., Kaneko, Y., Iwawaki, T., et al. (2015). Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci. Rep. 5:14812. doi: 10.1038/srep14812
Paz Gavilan, M., Vela, J., Castano, A., Ramos, B., del Rio, J. C., Vitorica, J., et al. (2006). Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 27, 973–982. doi: 10.1016/j.neurobiolaging.2005.05.010
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Song, Y., Shen, H., Du, W., and Goldstein, D. R. (2013). Inhibition of x-box binding protein 1 reduces tunicamycin-induced apoptosis in aged murine macrophages. Aging Cell 12, 794–801. doi: 10.1111/acel.12105
Sun, J., Liu, Y., and Aballay, A. (2012). Organismal regulation of XBP-1-mediated unfolded protein response during development and immune activation. EMBO Rep. 13, 855–860. doi: 10.1038/embor.2012.100
Taylor, R. C., and Dillin, A. (2013). XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. doi: 10.1016/j.cell.2013.05.042
Wang, L., Ryoo, H. D., Qi, Y., and Jasper, H. (2015). PERK limits Drosophila lifespan by promoting intestinal stem cell proliferation in response to ER Stress. PLoS Genet. 11:e1005220. doi: 10.1371/journal.pgen.1005220
Wang, L., Zeng, X., Ryoo, H. D., and Jasper, H. (2014). Integration of UPRER and oxidative stress signaling in the control of intestinal stem cell proliferation. PLoS Genet. 10:e1004568. doi: 10.1371/journal.pgen.1004568
Keywords: aging, proteostasis cell stress and aging, unfolded protein response (UPR), cell-nonautonomous, proteostasis deficiencies, protein misfolding and disease, protein misfolding disease
Citation: Martínez G, Duran-Aniotz C, Cabral-Miranda F and Hetz C (2016) Commentary: XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Front. Aging Neurosci. 8:182. doi: 10.3389/fnagi.2016.00182
Received: 23 May 2016; Accepted: 14 July 2016;
Published: 03 August 2016.
Edited by:
Agustin Ibanez, Instituto de Neurología Cognitiva, ArgentinaReviewed by:
Diego Ruano, University of Seville, SpainEmmanuel Moyse, François Rabelais University, France
Copyright © 2016 Martínez, Duran-Aniotz, Cabral-Miranda and Hetz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Hetz, Y2xhaGV0ekBtZWQudWNoaWxlLmNs