 Zhiming Li
Zhiming Li Zaozao Zheng
Zaozao Zheng Jun Ruan
Jun Ruan Zhi Li
Zhi Li Chi-Meng Tzeng
Chi-Meng Tzeng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging Neurosci., 03 June 2016
Sec. Neuroinflammation and Neuropathy
Volume 8 - 2016 | https://doi.org/10.3389/fnagi.2016.00126
An increasing number of genetic studies suggest that the pathogenesis of Parkinson’s disease (PD) and cancer share common genes, pathways, and mechanisms. Despite a disruption in a wide range of similar biological processes, the end result is very different: uncontrolled proliferation and early neurodegeneration. Thus, the links between the molecular mechanisms that cause PD and cancer remain to be elucidated. We propose that chronic inflammation in neurons and tumors contributes to a microenvironment that favors the accumulation of DNA mutations and facilitates disease formation. This article appraises the key role of microglia, establishes the genetic role of COX2 and CARD15 in PD and cancer, and discusses prevention and treatment with this new perspective in mind. We examine the evidence that chronic inflammation is an important link between cancer and PD.
The three early, typical symptoms of Parkinson’s disease (PD) are static tremors, muscle rigidity, and bradykinesia, which result from the degeneration of dopaminergic neurons in the midbrain. In contrast, cancer is a disease caused by the clonal proliferation of selectively advantageous cells. Although the two may appear distinct, epidemiological studies have revealed some parallels. Doshay (1954) found a decreased frequency of cancer incidence in patients with PD, but the reason for this was unknown. Case-controlled and large prospective studies reported a lower rate of smoking-related and non-smoking-related cancers in PD patients. Melanoma, thyroid, and breast cancer were revealed to have an increased incidence in PD patients (D’Amelio et al., 2009; Liu et al., 2011). There are a number of potential explanations for the inverse association. One of theories between PD and skin cancer may be associated with the mode of therapy, such as Levodopa, rather than with the disease itself (Paisan-Ruiz and Houlden, 2010). However, some observations do not agree with the view that this may be a result of drug treatment (Fiala et al., 2003; Zanetti et al., 2006). Moreover, it was suggested that PD patients with a low incidence of cancer may be associated with the negative correlation between PD and smoking (Hernan et al., 2002). Although the reduced risk of smoking-related cancers can be explained in PD patients, it cannot decipher the risk of non-smoking-related cancers. Thus, more data are needed to confirm the epidemiological relationship between PD and cancer.
Recently, the unusual epidemiological information between PD and cancer is increasingly becoming of interest to many investigators. Genetic studies provide additional understanding, since many familial PD genes have been associated with cancer, such as parkin (PARK2), PINK1 (PARK6), DJ-1 (PARK7), and LRRK2 (PARK8; Table 1). However, the same gene’s variants may lead to distinct effects due to the different cell backgrounds associated with dopaminergic neurons and cancer cells. PD-related genes participate in a wide variety of cellular processes, including the misfolding and degradation of proteins, mitochondrial damage, response to oxidative stress, cell cycle control, and DNA repair (Figure 1). These processes all have an important influence on the occurrence and development of PD and cancer. The PI3K/AKT/mTOR pathway also plays important roles in both cell growth and death. Some common cellular pathways and genes have been explained in our work (Zhiming et al., 2014). Notably, neuroinflammation is increasingly considered a double-edged sword (Wyss-Coray and Mucke, 2002). Acute inflammation can repair damage and promote healing, but long-term chronic inflammation can severely damage the body. Chronic inflammation is considered a driving force behind many chronic diseases, certainly including cancer and neurodegenerative disease. In this article, we further discuss the relationship between PD and cancer from the perspective of chronic inflammation. This will provide an increased understanding and improved treatment approaches for both diseases.
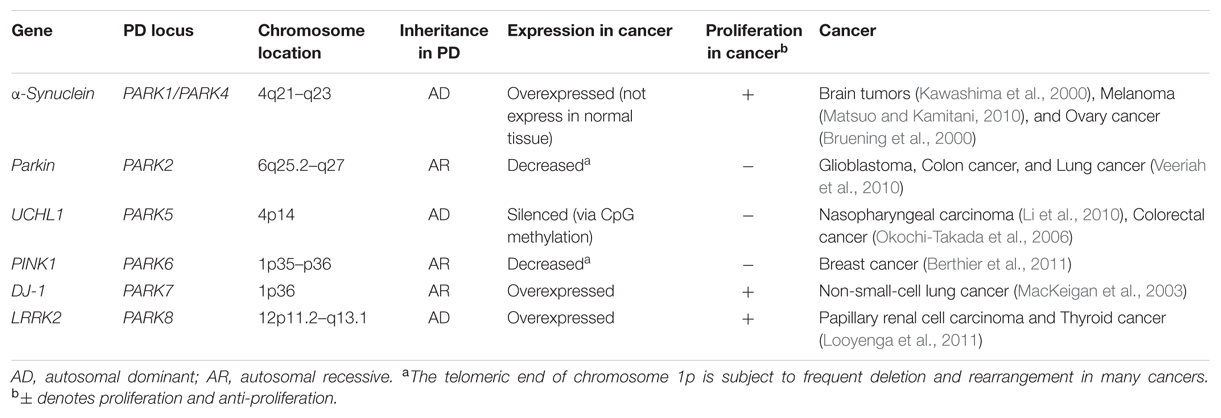
TABLE 1. Parkinson’s disease (PD) involved genes identified in cancer.
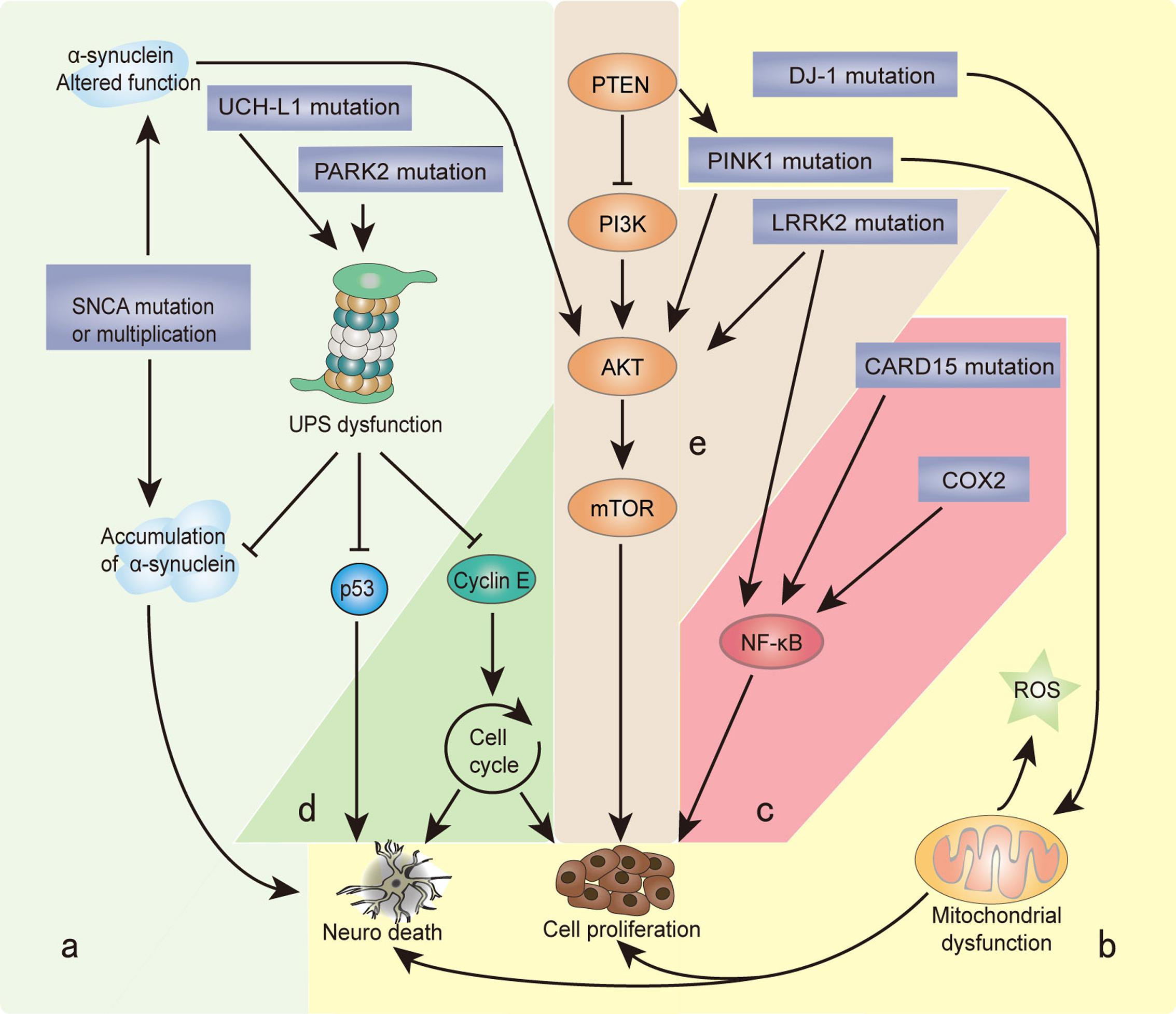
FIGURE 1. Genes and biological processes shared in Parkinson’s disease (PD) and cancer. Overlapping biological processes of PD and cancer mainly include the following: (a) misfolding and degradation of proteins, (b) mitochondrial damage and oxidative stress response, (c) chronic inflammation, (d) cell cycle control and DNA repair, and (e) PI3K/AKT/mTOR pathway regulation. The α-synuclein polymer attributed to SNCA multiplication is the main component of Lewy bodies (LBs). SNCA mutations alter the normal function of α-synuclein, which induces the PI3K/AKT/mTOR pathway and promotes cell proliferation. Under the cascade of phosphorylating AKT, mTOR is activated by PINK1 and LRRK2. PARK2 and UCH-L1 mutations disrupt the degradation function of the ubiquitin proteasome system (UPS), leading to the misfolding and aggregation of α-synuclein, cyclin E, and p53. PINK1 and DJ-1 mutations result in the overproduction of free radicals and oxidative stress in the mitochondria, which causes neuron defects and stimulates cell proliferation. COX2 and CARD15 mutations activate the NF-κB pathway and induce chronic inflammation, leading to a wide range of genetic mutations and abnormal cellular signaling. The different cellular backgrounds of cancer cells and neurons (mitotic vs. post-mitotic cells) bring completely distinct reactions to external stimuli and internal changes: some undergo cell proliferation, and others neuron death. The final result is two serious diseases: cancer and PD.
Inflammation and PD have an established link via microglia, which are the resident immune cells of the central nervous system (CNS). Under physiological conditions, microglia are inactive, have a small cell body, and exhibit highly ramified morphology. In response to injury or toxic factors, microglia transform into activated microglia, migrate to the lesion area, and phagocytose cellular debris or damaged neurons. It is now well documented that activated microglial surround the lost dopaminergic neurons in patients with PD and PD animal models, which provides for the speculation that microglia activation results in initiation and development of PD (Tansey and Goldberg, 2010). A recent investigation revealed that neurons could release α-synuclein oligomers, the main component of Lewy bodies (LBs) in the substantia nigra pars compacta (SNc) of the midbrain, which can bind to toll-like receptors (TLRs) to activate microglia, mediating the nuclear factor kappa B (NF-κB) pathway and causing the secretion of inflammatory mediators, such as cytokines, eicosanoids, and chemokines, altering immunological functions (Beraud and Maguire-Zeiss, 2012). These immune factors not only directly act on dopaminergic neurons to cause neuronal death but also aggravate the inflammatory reaction and continue to activate microglia. Activated microglia also generate and release a wide range of reactive-free radicals and increasing evidence has shown that the oxidative stress response plays a crucial role in PD. The sustained production of free radicals and noxious compounds, like superoxide or peroxynitrite, can interact with biological macromolecules, leading to disruption to the redox balance of neurons. Additionally, levels of pro-inflammatory mediators, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and eicosanoids are enhanced in peripheral blood mononuclear cells (PBMCs) of PD patients (Bessler et al., 1999). Recent studies have shown that inhibiting the microglia cascade reactions could prevent the degradation of neurons (Burguillos et al., 2011). The immunohistochemical demonstration of reactive microglia suggests that chronic inflammation occurs in affected brain regions in PD.
The brain is a common metastatic site for various types of cancers, especially lung cancer, which suggests failure of the immune defense in the brain environment. Microglia are believed to be the most important immune cells in the CNS. Interestingly, significant microglial activation has been observed in the vicinity of glioma tumor cells (Roggendorf et al., 1996). In contrast to CNS inflammation, microglia associated with brain tumors do not seem to be active in inducing an effective antitumor response. The exact mechanism of this microglial inactivation remains unclear. There is evidence that some cytokines, such as IL-10 released from activated microglia, play important roles in local immunosuppression and progression of glioma, particularly promoting proliferation of tumor cells and their infiltration into surrounding normal brain tissue (Wagner et al., 1999). Microglia also play an important role in phagocytosing tumor cells. During the past decades, more attention has been paid to the secretory property and chemotaxis of microglia, but microglial phagocytosis is not well-studied. The activity of phagocytosis can be modulated by cancer cells or the cancer environment. However, whether microglial phagocytosis functions to contribute to cancer or cancer defense still needs further investigation. A better understanding of microglia function is important for the development of immune-based treatment strategies against malignant brain tumors.
Inflammation is known as the seventh most important sign of cancer (Colotta et al., 2009). Sometimes, inflammatory mediators are referred to as genetic mutagens, which disturb DNA repair pathways and cell-cycle checkpoints, threatening the stability of the genome and accumulating chromosomal alterations. Inflammatory mediators in the microenvironment during chronic inflammation not only help cancer cells proliferate and escape from immune surveillance but also cause a large number of random mutations (Grivennikov et al., 2010). A major advantage of the cell cycle is careful checking and repair of the damaged DNA, but post-mitotic neurons are particularly vulnerable to the abnormal levels of mediators and accumulation of mutations. Some mutations in two genes unequivocally linked to chronic inflammation, namely COX2 and CARD15, were identified in tissue samples from patients with PD and cancer.
It is well known that prostanoids have critical functions in many cellular processes and pathophysiologic process, including inflammation, arthritis, and pain. COX-1 and -2 are two key enzymes in the synthesis of prostaglandins from of arachidonic acid. COX1 is constitutively expressed in the majority of tissues and is responsible for maintenance the normal physiological functions. Inversely, COX2 expression is low in most normal tissues, but it can be induced by various inflammatory stimuli (Smith et al., 2000). High levels of COX2 have been detected in many cancers, including gastric, breast, lung, esophageal, and hepatocellular carcinomas, particularly in colon cancer (Liu et al., 2003). Several lines of evidence suggest that non-steroidal anti-inflammatory drugs (NSAIDs) and selective COX2 inhibitors are promising as anti-cancer drugs. Genetic studies suggested that there is a strong causality between COX2 and tumorigenesis. Polymorphisms in the COX2 gene could alter enzyme expression, function, and/or the response to NSAIDs. The COX2 V511A polymorphism was very near to the active site of a molecular model (Fritsche et al., 2001). In African Americans, the COX2 V511A polymorphism was reported to confer a reduced susceptibility to colon cancer (Lin et al., 2002). COX2 has been shown to activate the NF-κB pathway and the p38 and JNK MAPK pathways (Hunot et al., 2004). Polymorphisms in the COX2 and other genes these pathways may account for inter-individual differences in cancer susceptibility or response to NSAIDs.
Cyclooxygenase 2 is inducible by growth factors, cytokines, and pro-inflammatory molecules. COX2 expression in brain has been associated with pro-inflammatory activities. COX2 has been observed to increase in brain dopaminergic neurons of both PD post-mortem specimens and in the PD mouse model induced by 1-methyl-4-phenyl-1,2,3,6-tertrahydropyridine (MPTP). The involvement of COX2 in PD pathogenesis was further evidenced by the observation that MPTP neurodegeneration was alleviated in COX2, but not in COX1, knock out mice (Minghetti, 2004). Recently, the COX2 1195G > A polymorphism was considered to be a protective factor in the onset of PD in the Chinese Han population (Dai et al., 2015). However, the role of the COX2 variant on the generation of PD has rarely been studied. Previous studies found that different polymorphisms of some known genes related to PD may result in different diseases: PD or cancer due to the differentiated background of neuronal and cancer cells (Veeriah et al., 2010; Vasseur et al., 2012). Lopez de Maturana et al. (2014) found that LRRK2 also affected the inflammatory response in PD patients through regulating the expression of the COX2 enzyme. By extension, researchers propose that the overlap between PD and cancer is chronic inflammation, and COX2 may be the key enzyme in the inflammatory response to combine them.
Caspase recruitment domain protein 15, also known as nucleotide-binding oligomerization domain protein 2 (NOD2), is a protein that is encoded by the CARD15 gene located on human chromosome 16q12 (Hampe et al., 2001; Hugot et al., 2001; Ogura et al., 2001). CARD15 is the receptor of muramyl peptides (MDP), a component of bacterial peptidoglycan. The binding of MDP and CARD15 activates NF-κB, resulting in inflammatory reactions. It was shown that CARD15 mutations are involved in the innate immune system and pathogen recognition in terms of other complex polygenic diseases. In 2001, three laboratories identified genetic variants related to Crohn’s disease (CD), an inflammatory bowel disease. About 40% of CD patients in Western countries have at least one of the three SNPs: R702W, G908R, and L1007fsinsC. The heterozygous mutation of any of these SNPs increases the risk of CD by two- to fourfold. In addition, multiple-locus heterozygous mutations or homozygous mutations may lead to a more than 20-fold increased CD risk. Nevertheless, it was reported that none of these SNPs are involved in CD in Chinese (Han; Gao et al., 2005), Korean (Croucher et al., 2003), and Japanese patients (Yamazaki et al., 2002). Bialecka et al. (2007) showed that three SNPs in CARD15 (R702W, G908R, and L1007fsinsC) were significantly related to PD patients in a Polish population. Our group identified P268S, an additional CARD15 SNP that might be a risk factor for PD in a Chinese population (Ma et al., 2013). Additionally, Crane et al. (2002) reported that P268S was related to susceptibility of ankylosing spondylitis. Proell et al. (2008) performed sequence comparisons and found that CARD15 shared a high degree of similarity with apoptotic protease activating factor 1 (Apaf-1). They simulated the homologous structure of CARD15 based on the structure of Apaf-1 and found that P268S was located at the connexon (ligand-binding position) before the first helix of the NOD. Replacing Pro with Ser changed the conformation of the connexon and affected its binding to the substrate. These experimental observations show a possible linkage between the dysfunction of CARD15 and neurodegenerative disease.
Whether CD-related CARD15 variants result in a loss or gain of effect of the CARD15 receptor is still unclear, and the mechanisms by which this alteration, in function, may enhance the susceptibility to CD need to be clarified. Patients with CD have an enhanced risk of developing colorectal cancer (Gasche and Carethers, 2004; D’Amelio et al., 2009). CARD15 mutations may also increase the susceptibility to colorectal cancer in non-CD Caucasians (Papaconstantinou et al., 2005; Roberts et al., 2006). These investigations demonstrated that immune system mechanisms are involved in the etiology of cell damage in CD and provided evidence for an ongoing active pathological process. Inflammation can be triggered by invading microbes and also internally within the organism by diseases that affect the nervous system. There are three common outcomes of inflammation. First, the offending agent or process is inactivated, and the injury is repaired. Second, the host loses the battle and dies or suffers irreparable tissue damage. Finally, neither the organism nor the injurious process prevails, resulting in a prolonged battle that provides a fertile ground for the development of chronic inflammatory conditions. This last outcome may be closely related to neurodegenerative diseases and cancer, two of the greatest public health problems of this century (Wyss-Coray and Mucke, 2002).
Lower rates of cancer mortality and incidence in patients with PD have given rise to speculation about risks or preventative factors common to both diseases. Thus, unraveling the link between PD and cancer may open a new therapeutic window for both diseases. Epidemiological investigations revealed that taking NSAIDs can help lower your risk of PD development, and patients with colorectal cancer treated with NSAIDs showed 40–50% decreased mortality compared with those not using these drugs (Subbaramaiah and Dannenberg, 2003). The important role of mitotic activation in neurodegeneration raises the possibility of the cell cycle as a therapeutic target. Drugs, such as retinoic acid, simvastatin, and IL-1, which lead to cell cycle arrest at the G0/G1 interface, might prevent neurons from committing themselves to mitosis and therefore apoptosis. Another intervention would be to interfere with cellular signaling cascades that drive cell cycle progression or apoptosis, like Mithramycin (Sleiman et al., 2011). A number of other compounds have anti-neoplastic and neuroprotective properties, including inhibitors of histone deacetylase (Sleiman et al., 2009), mTOR (mammalian target of rapamycin), and transglutaminase (Ravikumar et al., 2004). Along with the cell cycle, the ubiquitin proteasome system is a common link between cancer and neurodegeneration, and an exciting target for new therapies. Because defective proteolysis is common to all neurodegenerative diseases, the potential clinical effect of a proteasome activator is beneficial and should be a focus of drug development. Trials of proteasome activators in PD should be carefully designed to capture any result of a related increased risk for malignancy because upregulation of the proteasome is associated with many cancers.
In 2010, Datamonitor Inc., (USA) estimated that there were over 1.5 million PD patients in the United States, Japan, France, Germany, Italy, Spain, and UK combined, one third of which are in the United States. Combined with the aggravated aging problem, the incidence of PD is increasing annually (Huynh, 2011). Medication is usually the first treatment option for PD. Levodopa is currently the most effective medication, but long-term use can reduce its effectiveness and cause complications such as motor dysfunction. Most brain degenerative diseases are incurable, and it is difficult to obtain the brain tissues from the patients with significant impairment. However, large amounts of cancer-related studies have been carried out over the past 30 decades, and many small molecule agents have been demonstrated as cancer therapeutic drugs or are now being assessed in clinical trials. Therefore, the extensive therapeutic developments in cancer studies may help the identification of diagnostic and prognostic markers for neurodegeneration that could contribute to improve treatments for PD.
Both cancer and PD are considered to be the consequence of the interaction of genes and environmental factors. The key difference is that different reactions occur in the different cellular backgrounds of cell division and cell death. The inflammation hypothesis is one explanation for comparisons between PD and cancer. The immune factors and free radicals released from chronic inflammatory reactions not only promote disease occurrence but also allow cellular DNA to accumulate mutations more easily, forming proteins with aberrant functions. The abundant therapeutic achievements in cancer studies will allow researchers to identify diagnostic markers and treatments for neurodegenerative disease and vice versa.
ZZ and JR designed the figure. Zhi Li designed the table. ZL and C-MT drafted the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The project was supported by the Natural Science Foundation of Fujian Province (No. 2016J01171) of China and the National Natural Science Foundation of China (No. 81272445).
Beraud, D., and Maguire-Zeiss, K. A. (2012). Misfolded alpha-synuclein and Toll-like receptors: therapeutic targets for Parkinson’s disease. Parkinsonism Relat. Disord. 18(Suppl. 1), S17–S20. doi: 10.1016/S1353-8020(11)70008-6
Berthier, A., Navarro, S., Jimenez-Sainz, J., Rogla, I., Ripoll, F., Cervera, J., et al. (2011). PINK1 displays tissue-specific subcellular location and regulates apoptosis and cell growth in breast cancer cells. Hum. Pathol. 42, 75–87. doi: 10.1016/j.humpath.2010.05.016
Bessler, H., Djaldetti, R., Salman, H., Bergman, M., and Djaldetti, M. (1999). IL-1 beta, IL-2, IL-6 and TNF-alpha production by peripheral blood mononuclear cells from patients with Parkinson’s disease. Biomed. Pharmacother. 53, 141–145.
Bialecka, M., Kurzawski, M., Klodowska-Duda, G., Opala, G., Juzwiak, S., Kurzawski, G., et al. (2007). CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci. Res. 57, 473–476. doi: 10.1016/j.neures.2006.11.012
Bruening, W., Giasson, B. I., Klein-Szanto, A. J., Lee, V. M., Trojanowski, J. Q., and Godwin, A. K. (2000). Synucleins are expressed in the majority of breast and ovarian carcinomas and in preneoplastic lesions of the ovary. Cancer 88, 2154–2163. doi: 10.1002/(SICI)1097-0142(20000501)88:9<2154::AID-CNCR23>3.0.CO;2-9
Burguillos, M. A., Deierborg, T., Kavanagh, E., Persson, A., Hajji, N., Garcia-Quintanilla, A., et al. (2011). Caspase signalling controls microglia activation and neurotoxicity. Nature 472, 319–324. doi: 10.1038/nature09788
Colotta, F., Allavena, P., Sica, A., Garlanda, C., and Mantovani, A. (2009). Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081. doi: 10.1093/carcin/bgp127
Crane, A. M., Bradbury, L., van Heel, D. A., McGovern, D. P., Brophy, S., Rubin, L., et al. (2002). Role of NOD2 variants in spondylarthritis. Arthritis Rheum. 46, 1629–1633. doi: 10.1002/art.10329
Croucher, P. J., Mascheretti, S., Hampe, J., Huse, K., Frenzel, H., Stoll, M., et al. (2003). Haplotype structure and association to Crohn’s disease of CARD15 mutations in two ethnically divergent populations. Eur. J. Hum. Genet. 11, 6–16. doi: 10.1038/sj.ejhg.5200897
Dai, Y., Wu, Y., and Li, Y. (2015). Genetic association of cyclooxygenase-2 gene polymorphisms with Parkinson’s disease susceptibility in Chinese Han population. Int. J. Clin. Exp. Pathol. 8, 13495–13499.
D’Amelio, M., Ragonese, P., Sconzo, G., Aridon, P., and Savettieri, G. (2009). Parkinson’s disease and cancer: insights for pathogenesis from epidemiology. Ann. N. Y. Acad. Sci. 1155, 324–334. doi: 10.1111/j.1749-6632.2008.03681.x
Doshay, L. J. (1954). Problem situations in the treatment of paralysis agitans. J. Am. Med. Assoc. 156, 680–684. doi: 10.1001/jama.1954.02950070008003
Fiala, K. H., Whetteckey, J., and Manyam, B. V. (2003). Malignant melanoma and levodopa in Parkinson’s disease: causality or coincidence? Parkinsonism Relat. Disord. 9, 321–327. doi: 10.1016/S1353-8020(03)00040-3
Fritsche, E., Baek, S. J., King, L. M., Zeldin, D. C., Eling, T. E., and Bell, D. A. (2001). Functional characterization of cyclooxygenase-2 polymorphisms. J. Pharmacol. Exp. Ther. 299, 468–476.
Gao, M., Cao, Q., Luo, L. H., Wu, M. L., Hu, W. L., and Si, J. M. (2005). [NOD2/CARD15 gene polymorphisms and susceptibility to Crohn’s disease in Chinese Han population]. Zhonghua Nei Ke Za Zhi 44, 210–212.
Gasche, C., and Carethers, J. M. (2004). NOD2 and colorectal cancer: guilt by non-association. Cancer Res. 64, 5525; author reply 5525–5526.
Grivennikov, S. I., Greten, F. R., and Karin, M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. doi: 10.1016/j.cell.2010.01.025
Hampe, J., Cuthbert, A., Croucher, P. J., Mirza, M. M., Mascheretti, S., Fisher, S., et al. (2001). Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 357, 1925–1928. doi: 10.1016/S0140-6736(00)05063-7
Hernan, M. A., Takkouche, B., Caamano-Isorna, F., and Gestal-Otero, J. J. (2002). A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 52, 276–284. doi: 10.1002/ana.10277
Hugot, J. P., Chamaillard, M., Zouali, H., Lesage, S., Cezard, J. P., Belaiche, J., et al. (2001). Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603. doi: 10.1038/35079107
Hunot, S., Vila, M., Teismann, P., Davis, R. J., Hirsch, E. C., Przedborski, S., et al. (2004). JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 665–670. doi: 10.1073/pnas.0307453101
Huynh, T. (2011). The Parkinson’s disease market. Nat. Rev. Drug Discov. 10, 571–572. doi: 10.1038/nrd3515
Kawashima, M., Suzuki, S. O., Doh-ura, K., and Iwaki, T. (2000). alpha-Synuclein is expressed in a variety of brain tumors showing neuronal differentiation. Acta Neuropathol. 99, 154–160. doi: 10.1007/PL00007419
Li, L., Tao, Q., Jin, H., van Hasselt, A., Poon, F. F., Wang, X., et al. (2010). The tumor suppressor UCHL1 forms a complex with p53/MDM2/ARF to promote p53 signaling and is frequently silenced in nasopharyngeal carcinoma. Clin. Cancer Res. 16, 2949–2958. doi: 10.1158/1078-0432.CCR-09-3178
Lin, H. J., Lakkides, K. M., Keku, T. O., Reddy, S. T., Louie, A. D., Kau, I. H., et al. (2002). Prostaglandin H synthase 2 variant (Val511Ala) in African Americans may reduce the risk for colorectal neoplasia. Cancer Epidemiol. Biomarker. Prev. 11, 1305–1315.
Liu, R., Gao, X., Lu, Y., and Chen, H. (2011). Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology 76, 2002–2009. doi: 10.1212/WNL.0b013e31821e554e
Liu, W., Reinmuth, N., Stoeltzing, O., Parikh, A. A., Tellez, C., Williams, S., et al. (2003). Cyclooxygenase-2 is up-regulated by interleukin-1 beta in human colorectal cancer cells via multiple signaling pathways. Cancer Res. 63, 3632–3636.
Looyenga, B. D., Furge, K. A., Dykema, K. J., Koeman, J., Swiatek, P. J., Giordano, T. J., et al. (2011). Chromosomal amplification of leucine-rich repeat kinase-2 (LRRK2) is required for oncogenic MET signaling in papillary renal and thyroid carcinomas. Proc. Natl. Acad. Sci. U.S.A. 108, 1439–1444. doi: 10.1073/pnas.1012500108
Ma, Q., An, X., Li, Z., Zhang, H., Huang, W., Cai, L., et al. (2013). P268S in NOD2 associates with susceptibility to Parkinson’s disease in Chinese population. Behav. Brain Funct. 9, 19. doi: 10.1186/1744-9081-9-19
MacKeigan, J. P., Clements, C. M., Lich, J. D., Pope, R. M., Hod, Y., and Ting, J. P. (2003). Proteomic profiling drug-induced apoptosis in non-small cell lung carcinoma: identification of RS/DJ-1 and RhoGDIalpha. Cancer Res. 63, 6928–6934.
Matsuo, Y., and Kamitani, T. (2010). Parkinson’s disease-related protein, alpha-synuclein, in malignant melanoma. PLoS ONE 5:e10481. doi: 10.1371/journal.pone.0010481
Minghetti, L. (2004). Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 63, 901–910. doi: 10.1093/jnen/63.9.901
Ogura, Y., Bonen, D. K., Inohara, N., Nicolae, D. L., Chen, F. F., Ramos, R., et al. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606. doi: 10.1038/35079114
Okochi-Takada, E., Nakazawa, K., Wakabayashi, M., Mori, A., Ichimura, S., Yasugi, T., et al. (2006). Silencing of the UCHL1 gene in human colorectal and ovarian cancers. Int. J. Cancer 119, 1338–1344. doi: 10.1002/ijc.22025
Paisan-Ruiz, C., and Houlden, H. (2010). Common pathogenic pathways in melanoma and Parkinson disease. Neurology 75, 1653–1655. doi: 10.1212/WNL.0b013e3181fb4466
Papaconstantinou, I., Theodoropoulos, G., Gazouli, M., Panoussopoulos, D., Mantzaris, G. J., Felekouras, E., et al. (2005). Association between mutations in the CARD15/NOD2 gene and colorectal cancer in a Greek population. Int. J. Cancer 114, 433–435. doi: 10.1002/ijc.20747
Proell, M., Riedl, S. J., Fritz, J. H., Rojas, A. M., and Schwarzenbacher, R. (2008). The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS ONE 3:e2119. doi: 10.1371/journal.pone.0002119
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Roberts, R. L., Gearry, R. B., Allington, M. D., Morrin, H. R., Robinson, B. A., and Frizelle, F. A. (2006). Caspase recruitment domain-containing protein 15 mutations in patients with colorectal cancer. Cancer Res. 66, 2532–2535. doi: 10.1158/0008-5472.CAN-05-4165
Roggendorf, W., Strupp, S., and Paulus, W. (1996). Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol. 92, 288–293. doi: 10.1007/s004010050520
Sleiman, S. F., Basso, M., Mahishi, L., Kozikowski, A. P., Donohoe, M. E., Langley, B., et al. (2009). Putting the ‘HAT’ back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Exp. Opin. Investig. Drugs 18, 573–584. doi: 10.1517/13543780902810345
Sleiman, S. F., Langley, B. C., Basso, M., Berlin, J., Xia, L., Payappilly, J. B., et al. (2011). Mithramycin is a gene-selective Sp1 inhibitor that identifies a biological intersection between cancer and neurodegeneration. J. Neurosci. 31, 6858–6870. doi: 10.1523/JNEUROSCI.0710-11.2011
Smith, W. L., DeWitt, D. L., and Garavito, R. M. (2000). Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182. doi: 10.1146/annurev.biochem.69.1.145
Subbaramaiah, K., and Dannenberg, A. J. (2003). Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol. Sci. 24, 96–102. doi: 10.1016/S0165-6147(02)00043-3
Tansey, M. G., and Goldberg, M. S. (2010). Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 37, 510–518. doi: 10.1016/j.nbd.2009.11.004
Vasseur, S., Afzal, S., Tomasini, R., Guillaumond, F., Tardivel-Lacombe, J., Mak, T. W., et al. (2012). Consequences of DJ-1 upregulation following p53 loss and cell transformation. Oncogene 31, 664–670. doi: 10.1038/onc.2011.268
Veeriah, S., Taylor, B. S., Meng, S., Fang, F., Yilmaz, E., Vivanco, I., et al. (2010). Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 42, 77–82. doi: 10.1038/ng.491
Wagner, S., Czub, S., Greif, M., Vince, G. H., Suss, N., Kerkau, S., et al. (1999). Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int. J. Cancer 82, 12–16. doi: 10.1002/(SICI)1097-0215(19990702)82:1<12::AID-IJC3>3.0.CO;2-O
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35, 419–432. doi: 10.1016/S0896-6273(02)00794-8
Yamazaki, K., Takazoe, M., Tanaka, T., Kazumori, T., and Nakamura, Y. (2002). Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn’s disease. J. Hum. Genet. 47, 469–472. doi: 10.1007/s100380200067
Zanetti, R., Loria, D., and Rosso, S. (2006). Melanoma, Parkinson’s disease and levodopa: causal or spurious link? A review of the literature. Melanoma Res. 16, 201–206.
Keywords: PD, cancer, chronic inflammation, microglia, COX2, CARD15
Citation: Li Z, Zheng Z, Ruan J, Li Z and Tzeng C-M (2016) Chronic Inflammation Links Cancer and Parkinson’s Disease. Front. Aging Neurosci. 8:126. doi: 10.3389/fnagi.2016.00126
Received: 15 February 2016; Accepted: 13 May 2016;
Published: 03 June 2016.
Edited by:
Ying Xu, The State University of New York at Buffalo, USAReviewed by:
Valentina Echeverria Moran, Bay Pines VA Healthcare System, USACopyright © 2016 Li, Zheng, Ruan, Li and Tzeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chi-Meng Tzeng, Y210emVuZ0B4bXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.