 Yotam Raz
Yotam Raz Vered Raz
Vered Raz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging Neurosci. , 10 November 2014
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 6 - 2014 | https://doi.org/10.3389/fnagi.2014.00317
This article is part of the Research Topic Pathophysiological Mechanisms of Sarcopenia in Aging and in Muscular Dystrophy: a Translational Approach View all 28 articles
Symptoms in late-onset neuromuscular disorders initiate only from midlife onward and progress with age. These disorders are primarily determined by identified hereditable mutations, but their late-onset symptom manifestation is not fully understood. Here, we review recent research developments on the late-onset autosomal dominant oculopharyngeal muscular dystrophy (OPMD). OPMD is caused by an expansion mutation in the gene encoding for poly-adenylate RNA binding protein1 (PABPN1). The molecular pathogenesis for the disease is still poorly understood. Despite a ubiquitous expression of PABPN1, symptoms in OPMD are limited to skeletal muscles. We discuss recent studies showing that PABPN1 levels in skeletal muscles are lower compared with other tissues, and specifically in skeletal muscles, PABPN1 expression declines from midlife onward. In OPMD, aggregation of expanded PABPN1 causes an additional decline in the level of the functional protein, which is associated with severe muscle weakness in OPMD. Reduced PABNPN1 expression in muscle cell culture causes myogenic defects, suggesting that PABPN1 loss-of-function causes muscle weakness in OPMD and in the elderly. Molecular signatures of OPMD muscles are similar to those of normal muscle aging, although expression trends progress faster in OPMD. We discuss a working hypothesis that aging-associated factors trigger late-onset symptoms in OPMD, and contribute to accelerated muscle weakness in OPMD. We focus on the pharyngeal and eyelid muscles, which are often affected in OPMD patients. We suggest that muscle weakness in OPMD is a paradigm for muscle aging.
In aging populations, late-onset disorders are highly common, among which late-onset neuromuscular (NM) disorders are a subset. NM disorders affect muscle fibers and/or the central and peripheral nervous system and the NM junction that control the muscle fibers. At present, these disorders are often incurable. As life expectancy rises, the prevalence of late-onset disorders causing chronic muscle weakness increases. Muscle symptoms can manifest from midlife onward, leading to a drastic functional decline with social and economic burdens. This suggests that in addition to the fundamental genetic defect(s), possible similar aging-associated regulators trigger the late onset of symptoms and progression thereof. For example, protein catabolism, which discards defective or redundant proteins (mainly through the ubiquitin proteasome and autophagy systems), has been implicated as a predominant regulator in normal aging and late-onset diseases [reviewed in Low (2011) and Bonaldo and Sandri (2013)]. Reduced protein catabolism can also lead to an accumulation of aggregation-prone proteins and formation of insoluble protein aggregates. These aggregates are often the pathological hallmark in a number of late-onset neurological and/or muscular disorders (Ruegg and Glass, 2011; Bonaldo and Sandri, 2013). Although symptoms of these diseases widely vary, symptoms often initiate in a small subset of muscular or neuronal tissues (Ross and Poirier, 2005). While the primary genetic causes for these disorders are known, why symptoms initiate from midlife onward in specific tissues and how symptoms progress with age is still obscure.
Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant and rare myopathy. The estimated prevalence in Western countries is 1:100,000 [reviewed in Raz et al. (2013)]. Due to founder effects, clusters with a higher prevalence are found in French-Canadians and in the Bukhara community in Israel (1:1000 and 1:600, respectively) (Blumen et al., 2000; Laberge et al., 2005). It has been suggested, however, that outside these communities, the disease remains underdiagnosed (Ruegg et al., 2005). In OPMD, skeletal muscles are predominantly affected, whereby initial symptoms are manifested in only a subset of muscles. Most commonly, this leads to lowering (ptosis) of the eyelids and swallowing difficulties (dysphagia). With disease progression, additional skeletal muscles can be affected including the proximal muscles of the lower limb (including the quadriceps muscles) (Fischmann et al., 2012). OPMD is a monogenic disorder and its etiology is found in an alanine expansion mutation in the gene encoding for poly-adenylate (poly(A)) binding protein nuclear 1 (PABPN1) (Brais et al., 1998). Formation of insoluble inclusions in the cell nucleus is the pathological hallmark of OPMD muscles (Tome and Fardeau, 1980). Under physiological expression levels, expanded (exp)PABPN1 is more prone to aggregation compared with the wild-type PABPN1 (Raz et al., 2011a). High overexpression of expPABPN1 in muscles of various animal models causes muscle weakness, and it is suggested that accumulation of aggregates could be the cause for the muscle dysfunction (Davies et al., 2005). High overexpression of expPAPBN1 in animal models, as well as in cellular models leads to cell death, suggesting that expPABPN1 aggregates are toxic (Davies et al., 2008). Importantly, treatments that may reduce aggregation lead to less cell death and reduced muscle weakness in these animal models (Davies et al., 2006, 2010; Catoire et al., 2008; Chartier et al., 2009). Based on those models, it was suggested that muscle symptoms in OPMD are caused by a PABPN1 gain-of-function. However, it is not resolved whether animal models with high overexpression in muscles are relevant to OPMD. For example, high overexpression of expPABPN1 was not reported in OPMD heterozygous patients. Moreover, aggregates of wild-type PABPN1 were found in unaffected rat neural cells (Berciano et al., 2004). Altogether, it is striking that despite the well-known genetic cause for OPMD, the molecular mechanisms and physiological conditions that lead to muscle symptoms are poorly understood. Here, we discuss four questions for OPMD pathophysiology.
Poly-adenylate RNA binding protein1 is multifunctional regulator of RNA metabolism. Initially, it was identified in vitro as a regulator of poly(A) tail length (Kerwitz et al., 2003), subsequently was validated in vivo (Benoit et al., 2005), and more recently, it was shown to have an impact on mRNA decay (Bresson and Conrad, 2013). PABPN1 knockdown in mouse muscle cells causes reduced poly(A) tail length that is associated with myogenesis defects (Apponi et al., 2010); however, the relevance for OPMD and muscle aging is unsettled. A change in poly(A) tail length was not found in muscles from OPMD patients (Calado et al., 2000). Recent studies revealed additional molecular functions for PABPN1. A genome-wide shift from distal to proximal alternative polyadenylation site (PAS) and accumulation of shortened transcripts were found in the mouse model for OPMD, A17.1, which was generated by expPABPN1 overexpression in muscles, and in cells with reduced PABPN1 expression (de Klerk et al., 2012; Jenal et al., 2012). Similar alternative PAS utilization was found in models with expPABPN1 overexpression and PABPN1 downregulation, suggesting that PABPN1 loss-of-function causes defects in RNA metabolism (de Klerk et al., 2012; Jenal et al., 2012). In OPMD muscles, PABPN1 downregulation was found to be comparable to age-matching controls (Anvar et al., 2013). Reduced PABPN1 levels in cellular models cause myogenic defects (Apponi et al., 2010; Anvar et al., 2013). In addition, PABPN1 was also found to regulate long non-coding RNA expression (Beaulieu et al., 2012). However, so far, alternative PAS or long non-coding RNA expression was not reported in OPMD patient muscles. To adequately understand how PABPN1 regulates changes in RNA metabolism in OPMD with an impact on muscle weakness, experiments should be conducted in models with physiological levels of PABPN1.
Aberrant RNA metabolism is not specific to OPMD, but is found in a wide spectrum of unrelated late-onset neurological and/or muscular disorders (O’Rourke and Swanson, 2009; Anthony and Gallo, 2010). Since these disorders share a late onset of symptoms and progression with age, age-associated regulators of RNA metabolism could be affected. It is still unclear whether similar regulators of RNA metabolism are affected in these disorders. In OPMD, affected muscles as well as muscles from pre-symptomatic family members can be accessible for research (Fischmann et al., 2012; Anvar et al., 2013). Therefore, OPMD could be used as a paradigm to study the functional contribution of RNA metabolism to symptoms in late-onset neurological and/or muscular disorders and to study a possible regulatory role in the age-associated progression of the symptoms.
Poly-adenylate RNA binding protein1 is essential for cell vitality (Bhattacharjee and Bag, 2012) and is expressed in all cells; however symptoms are predominantly manifested in skeletal muscles. In OPMD patients, PABPN1 expression is specifically reduced in affected Vastus lateralis muscles, while PABPN1 levels in whole blood are unchanged between OPMD patients and healthy controls (Anvar et al., 2013). In Dutch and Danish OPMD patients, weakness of the quadriceps muscles is reported as one of the initial symptoms (Sluijs et al., 2003; Witting et al., 2014). PAPBN1 level is lower in skeletal muscles of both human being and mouse, compared with a spectrum of tissues (Apponi et al., 2013). Since symptoms in OPMD are predominant in skeletal muscles, this suggests that below a certain crucial level, a functional impact is manifested. Although a threshold for functional PABPN1 is yet to be defined, due to low PABPN1 expression levels in skeletal muscles (Apponi et al., 2013) this crucial level could reach a functional impact. In other tissues, however, PABPN1 levels are sufficiently high (Apponi et al., 2013), and thus, if any aging-associated decline may occur (Anvar et al., 2013), they would be spared from a functional deficiency (Figure 2). Indeed, altered PABPN1 at 40, 60, or 80% expression level causes reciprocal decrease in the expression of sarcomeric genes (Anvar et al., 2013). This working model requires additional in vivo experiments.
It is not fully understood why levels of PABPN1 are lower in skeletal muscles. PABPN1 mRNA is less stable in muscles compared with other tissues (Apponi et al., 2013). PABPN1 mRNA binds to PABPN1 protein (Raz et al., 2014), which potentially affects PABPN1 mRNA stability, nuclear export, and translation (Figure 1). As yet, regulators of mRNA stability in muscles and aging-associated changes are poorly understood. In addition, PABPN1 protein turnover is regulated by the ubiquitin proteasome system (UPS) (Raz et al., 2011b). Differences in poly-ubiquitination levels between wild type and expPABPN1 result in higher protein turnover of wild-type PAPBN1 as compared with expPABPN1 (Raz et al., 2011b). Since PABPN1 is prone to aggregation, higher protein accumulation leads to aggregate formation and reduced availability of the functional protein. PABPN1 protein accumulation is regulated by ARIH2 E3-ligase, whose level also declines from midlife onward in skeletal muscles (Raz et al., 2014). In OPMD muscles, ARIH2 level is lower compared to age-matching controls, which in part could result by the alternative PAS utilization in ARIH2 3′-UTR that is directly regulated by PABPN1 level. In addition, as ARIH2 protein is entrapped in expPABPN1 aggregates, functional protein levels would be depleted (Raz et al., 2014). Protein entrapment in PABPN1 aggregates was reported for other E3 ligases and the proteasome (Corbeil-Girard et al., 2005; Tavanez et al., 2005; Anvar et al., 2011), suggesting that the UPS machinery is dysregulated in OPMD. In turn, mRNA dysregulation of the UPS was found to be the most consistent dysregulated cellular system in OPMD (Anvar et al., 2011; de Klerk et al., 2012). Although a mechanistic model for regulation of PABPN1 levels in aging and muscle disease calls for additional research, the current literature suggests that changes in PABPN1 levels in muscles are regulated by both mRNA stability and protein turnover and that these machineries are specifically affected in OPMD (Figure 1). Regulators of PABPN1 mRNA stability and protein turnover should be identified in future studies.
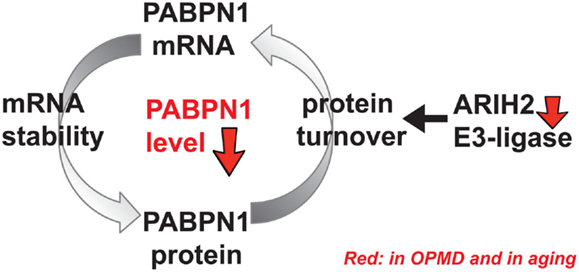
Figure 1. A schematic working model for regulation of PABPN1 levels. PABPN1 levels are established by a feed-forward loop combining at least regulation of mRNA stability and protein turnover. PABPN1 protein accumulation is regulated by the ubiquitin proteasome system (UPS), including ARIH2 E3-ligase. An aging-associated distortion of the UPS causes impairment of PABPN1 protein turnover affecting its mRNA stability and genes of the UPS, including ARIH2 E3-ligase. In turn, PABPN1 mRNA levels, which are self-regulated, also decrease with aging. However, the molecular factors regulating PABPN1 mRNA stability are unknown as yet. In OPMD and in aging, reduced levels of PABPN1 eventually cause genome-wide changes in gene expression.
In OPMD, the ocular and pharyngeal muscles are initially the most commonly affected; however, with progression of the disease, more skeletal muscles are affected. These muscles are also often affected in otherwise healthy elderly. Most skeletal muscles are weakened during aging; however, it is unclear whether aging-associated muscle weakness is programed, concerning initially affected muscles and progression. Relevant to OPMD, dysphagia, resulting from weakness of pharyngeal muscles, and eyelid ptosis, resulting from weakness of the levator palpebrae muscle, is highly common in the elderly (Salvi et al., 2006; Fea et al., 2013; Iida et al., 2013). In contrast to the otherwise healthy population, in OPMD, symptoms start at an earlier age and seem more severe, possibly due to faster progression. Eyelid ptosis can also be caused by rare multiple mitochondrial DNA deletion syndromes, including progressive external ophthalmoplegia (PEO) (Van Goethem et al., 2003). Increased mitochondrial deletions were found in the extra-ocular muscles compared to other muscles and are aging associated (Yu-Wai-Man et al., 2010), indicating a prominent role for mitochondrial activity in ocular muscles during aging. A decrease in mitochondrial activity was reported in a mouse model for OPMD (Trollet et al., 2010) and in a muscle cell model with PABPN1 downregulation (Anvar et al., 2013). These studies suggest that in OPMD, mitochondrial dysfunction may contribute to the muscle weakness and future studies should explore a role for PAPBN1 in the regulation of mitochondrial genes and mitochondrial and metabolic cellular functionality.
Dysphagia in the elderly is determined by age-associated anatomical changes in the neck and throat, underpinning the swallowing function in otherwise healthy elderly (Ney et al., 2009). Furthermore, dysphagia is also common in different age-related diseases including NM disorders (Schindler and Kelly, 2002). Dysphagia in the otherwise healthy elderly manifests by a less efficient passage of food to the esophagus due to changes in swallowing pressures of the upper esophageal sphincter and delayed muscle movements (Yokoyama et al., 2000). Symptoms are suggested to be associated with an increase in muscle atrophy and a decrease in muscle strength of the pharyngeal muscles (Feng et al., 2013).
Although it is not clear why ocular and pharyngeal muscles are initially affected in OPMD, the ocular and pharyngeal muscles are constantly, and often unconsciously, routinely used in daily life. Interestingly, PABPN1 levels in mice pharyngeal muscle are lower compared with other skeletal muscles (Apponi et al., 2013). This suggests that this muscle could be more susceptible to a further decrease in PABPN1 levels. However, functional studies, especially in human beings, would lead to conclusive answers.
Muscle aging (sarcopenia) is marked, among others, by a decrease in muscle mass (atrophy), an increase in fat infiltration and inflammation leading to a decrease in muscle strength and physical performance (Cruz-Jentoft et al., 2010). In the A17.1 mouse model, muscle atrophy appears already from 12 weeks of age (24–month-old mice are considered as aged) and is complemented by expression dysregulation of known muscle atrophy genes such as Murf1 and Atrogin-1 (Trollet et al., 2010). Muscle atrophy in this mouse model is prominent in the fast glycolytic (type IIX/IIB) fibers, whereas the slow oxidative (type I) and fast oxidative (type IIA) fibers seem to be spared (Trollet et al., 2010). Although it is widely accepted that aged muscles of rodents are enriched in slow (type I) fibers compared to fast (type II) fibers, in aging human muscles, the literature concerning this fiber-type switch is not conclusive (Purves-Smith et al., 2014). Therefore, the implications of the muscle fiber-type switch for OPMD patients are unsettled. Studies in additional models are necessary in order to reveal whether reduced PABPN1 levels induce muscle atrophy and muscle fiber-type switch. Muscle aging can also be non-invasively quantified from magnetic resonance imaging (MRI) including muscle cross-sectional surface area, fatty infiltration (Fischmann et al., 2012; Willis et al., 2013), and inflammation (Eshed et al., 2007). These measures can be considered as approximations of muscular quality. With these measures, progressive muscular atrophy in OPMD was documented (Fischmann et al., 2012).
Although OPMD is often grouped within the muscular dystrophies, our studies of RNA expression profiles in OPMD muscles and OPMD models revealed higher similarities with muscles from the elderly rather than with muscular dystrophies or myopathies (Anvar et al., 2013), suggesting similar molecular signatures. Overlapping dysregulated genes were found for the mitochondria, the UPS, DNA repair, TGF-β signaling, and sarcomeric genes (Anvar et al., 2013). However, functional studies should investigate a role for additional aging-associated pathways. For example, the autophagy system has also been found to have an imperative regulatory role in many age-associated diseases, in healthy aging (Schneider and Cuervo, 2014), and in muscle atrophy (Bonaldo and Sandri, 2013). As PABPN1 can shuttle between the nucleus and the cytoplasm (Abu-Baker et al., 2005; Benoit et al., 2005), it should be investigated whether PABPN1 is also regulated by the autophagy system. Moreover, genes of the autophagy system could be regulated by PABPN1 (Anvar et al., 2013). More interestingly, age-associated changes in a cross-sectional data revealed a faster change in expression level for a subset of genes, among which many are known as aging genes, muscle-specific sarcomeric genes, and PABPN1 (Anvar et al., 2013). This suggests that muscle weakness in OPMD could represent accelerated muscular aging, and thus, OPMD muscles could be a paradigm for otherwise healthy muscle aging (Figure 2).
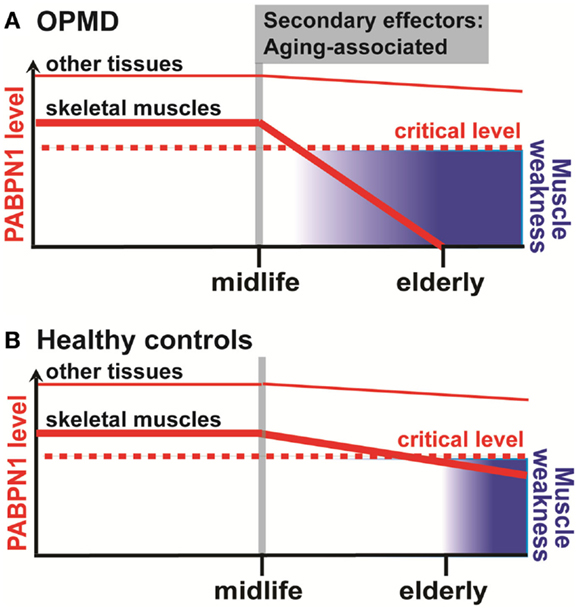
Figure 2. A schematic working model for the aging-associated decline of muscle strength and PABPN1 in OPMD (A) and healthy controls (B). In skeletal muscles, a decline in PABPN1 levels starts from midlife onward in both OPMD and healthy controls. The onset of decline from midlife onward is caused by secondary age-associated effectors, which mostly are unknown. Eventually, below a critical PABPN1 level, muscle weakness symptoms are manifested. This process is accelerated in OPMD due to aggregation of expanded PABPN1, which further depletes levels of functional PABPN1. In other tissues, however, levels of PABPN1 are higher and a possible age-related decline of PABPN1 does not reach the critical level for a functional impact.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the French Muscular Dystrophy Association, France, and from the Princess Beatrix Muscle Fund, The Netherlands for financial support on OPMD research.
Abu-Baker, A., Laganiere, S., Fan, X., Laganiere, J., Brais, B., and Rouleau, G. A. (2005). Cytoplasmic targeting of mutant poly(A)-binding protein nuclear 1 suppresses protein aggregation and toxicity in oculopharyngeal muscular dystrophy. Traffic 6, 766–779. doi: 10.1111/j.1600-0854.2005.00315.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anthony, K., and Gallo, J.-M. (2010). Aberrant RNA processing events in neurological disorders. Brain Res. 1338, 67–77. doi:10.1016/j.brainres.2010.03.008
Anvar, S. Y., Raz, Y., Verway, N., Van Der Sluijs, B., Venema, A., Goeman, J. J., et al. (2013). A decline in PABPN1 induces progressive muscle weakness in oculopharyngeal muscle dystrophy and in muscle aging. Aging (Albany NY). 5, 412–426.
Anvar, S. Y., ‘t Hoen, P. A., Venema, A., van der Sluijs, B., van Engelen, B., Snoeck, M., et al. (2011). Deregulation of the ubiquitin-proteasome system is the predominant molecular pathology in OPMD animal models and patients. Skelet. Muscle. 1, 15. doi:10.1186/2044-5040-1-15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Apponi, L. H., Corbett, A. H., and Pavlath, G. K. (2013). Control of mRNA stability contributes to low levels of nuclear poly(A) binding protein 1 (PABPN1) in skeletal muscle. Skelet. Muscle. 3, 23. doi:10.1186/2044-5040-3-23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Apponi, L. H., Leung, S. W., Williams, K. R., Valentini, S. R., Corbett, A. H., and Pavlath, G. K. (2010). Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum. Mol. Genet. 19, 1058–1065. doi:10.1093/hmg/ddp569
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beaulieu, Y. B., Kleinman, C. L., Landry-Voyer, A. M., Majewski, J., and Bachand, F. (2012). Polyadenylation-dependent control of long noncoding RNA expression by the poly(A)-binding protein nuclear 1. PLoS Genet. 8:e1003078. doi:10.1371/journal.pgen.1003078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benoit, B., Mitou, G., Chartier, A., Temme, C., Zaessinger, S., Wahle, E., et al. (2005). An essential cytoplasmic function for the nuclear poly(A) binding protein, PABP2, in poly(A) tail length control and early development in Drosophila. Dev. Cell 9, 511–522. doi:10.1016/j.devcel.2005.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berciano, M. T., Villagra, N. T., Ojeda, J. L., Navascues, J., Gomes, A., Lafarga, M., et al. (2004). Oculopharyngeal muscular dystrophy-like nuclear inclusions are present in normal magnocellular neurosecretory neurons of the hypothalamus. Hum. Mol. Genet. 13, 829–838. doi:10.1093/hmg/ddh101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhattacharjee, R. B., and Bag, J. (2012). Depletion of nuclear poly(A) binding protein PABPN1 produces a compensatory response by cytoplasmic PABP4 and PABP5 in cultured human cells. PLoS ONE. 7:e53036. doi:10.1371/journal.pone.0053036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blumen, S. C., Korczyn, A. D., Lavoie, H., Medynski, S., Chapman, J., Asherov, A., et al. (2000). Oculopharyngeal MD among Bukhara Jews is due to a founder (GCG)9 mutation in the PABP2 gene. Neurology. 55, 1267–1270. doi:10.1212/WNL.55.9.1267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonaldo, P., and Sandri, M. (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model Mech. 6, 25–39. doi:10.1242/dmm.010389
Brais, B., Bouchard, J. P., Xie, Y. G., Rochefort, D. L., Chretien, N., Tome, F. M., et al. (1998). Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 18, 164–167. doi:10.1038/ng0298-164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bresson, S. M., and Conrad, N. K. (2013). The human nuclear poly(a)-binding protein promotes RNA hyperadenylation and decay. PLoS Genet. 9:e1003893. doi:10.1371/journal.pgen.1003893
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calado, A., Tome, F. M., Brais, B., Rouleau, G. A., Kuhn, U., Wahle, E., et al. (2000). Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum. Mol. Genet. 9, 2321–2328. doi:10.1093/oxfordjournals.hmg.a018924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catoire, H., Pasco, M. Y., Abu-Baker, A., Holbert, S., Tourette, C., Brais, B., et al. (2008). Sirtuin inhibition protects from the polyalanine muscular dystrophy protein PABPN1. Hum. Mol. Genet. 17, 2108–2117. doi:10.1093/hmg/ddn109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chartier, A., Raz, V., Sterrenburg, E., Verrips, C. T., Van Der Maarel, S. M., and Simonelig, M. (2009). Prevention of oculopharyngeal muscular dystrophy by muscular expression of Llama single-chain intrabodies in vivo. Hum. Mol. Genet. 18, 1849–1859. doi:10.1093/hmg/ddp101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Corbeil-Girard, L. P., Klein, A. F., Sasseville, A. M., Lavoie, H., Dicaire, M. J., Saint-Denis, A., et al. (2005). PABPN1 overexpression leads to upregulation of genes encoding nuclear proteins that are sequestered in oculopharyngeal muscular dystrophy nuclear inclusions. Neurobiol. Dis. 18, 551–567. doi:10.1016/j.nbd.2004.10.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cruz-Jentoft, A. J., Baeyens, J. P., Bauer, J. M., Boirie, Y., Cederholm, T., Landi, F., et al. (2010). Sarcopenia: European consensus on definition and diagnosis: report of the European working group on sarcopenia in older people. Age Ageing. 39, 412–423. doi:10.1093/ageing/afq034
Davies, J. E., Rose, C., Sarkar, S., and Rubinsztein, D. C. (2010). Cystamine suppresses polyalanine toxicity in a mouse model of oculopharyngeal muscular dystrophy. Sci. Transl. Med. 2, 34ra40. doi:10.1126/scitranslmed.3000723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davies, J. E., Sarkar, S., and Rubinsztein, D. C. (2006). Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 15, 23–31. doi:10.1093/hmg/ddi422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davies, J. E., Sarkar, S., and Rubinsztein, D. C. (2008). Wild-type PABPN1 is anti-apoptotic and reduces toxicity of the oculopharyngeal muscular dystrophy mutation. Hum. Mol. Genet. 17, 1097–1108. doi:10.1093/hmg/ddm382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davies, J. E., Wang, L., Garcia-Oroz, L., Cook, L. J., Vacher, C., O’Donovan, D. G., et al. (2005). Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med. 11, 672–677. doi:10.1038/nm1242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Klerk, E., Venema, A., Anvar, S. Y., Goeman, J. J., Hu, O., Trollet, C., et al. (2012). Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res. 40, 9089–9101. doi:10.1093/nar/gks655
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eshed, I., Bollow, M., Mcgonagle, D. G., Tan, A. L., Althoff, C. E., Asbach, P., et al. (2007). MRI of enthesitis of the appendicular skeleton in spondyloarthritis. Ann. Rheum. Dis. 66, 1553–1559. doi:10.1136/ard.2007.070243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fea, A., Damato, D., Actis, A. G., De Sanctis, U., Actis, G., and Grignolo, F. M. (2013). Blepharoplastic: essential review. Minerva Chir. 68, 49–56.
Feng, X., Todd, T., Lintzenich, C. R., Ding, J., Carr, J. J., Ge, Y., et al. (2013). Aging-related geniohyoid muscle atrophy is related to aspiration status in healthy older adults. J. Gerontol. A Biol. Sci. Med. Sci. 68, 853–860. doi:10.1093/gerona/gls225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fischmann, A., Hafner, P., Fasler, S., Gloor, M., Bieri, O., Studler, U., et al. (2012). Quantitative MRI can detect subclinical disease progression in muscular dystrophy. J. Neurol. 259, 1648–1654. doi:10.1007/s00415-011-6393-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iida, T., Tohara, H., Wada, S., Nakane, A., Sanpei, R., and Ueda, K. (2013). Aging decreases the strength of suprahyoid muscles involved in swallowing movements. Tohoku J. Exp. Med. 231, 223–228. doi:10.1620/tjem.231.223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jenal, M., Elkon, R., Loayza-Puch, F., Van Haaften, G., Kuhn, U., Menzies, F. M., et al. (2012). The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 149, 538–553. doi:10.1016/j.cell.2012.03.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kerwitz, Y., Kuhn, U., Lilie, H., Knoth, A., Scheuermann, T., Friedrich, H., et al. (2003). Stimulation of poly(A) polymerase through a direct interaction with the nuclear poly(A) binding protein allosterically regulated by RNA. EMBO J. 22, 3705–3714. doi:10.1093/emboj/cdg347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laberge, A. M., Michaud, J., Richter, A., Lemyre, E., Lambert, M., Brais, B., et al. (2005). Population history and its impact on medical genetics in Quebec. Clin. Genet. 68, 287–301. doi:10.1111/j.1399-0004.2005.00497.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Low, P. (2011). The role of ubiquitin-proteasome system in ageing. Gen. Comp. Endocrinol. 172, 39–43. doi:10.1016/j.ygcen.2011.02.005
Ney, D. M., Weiss, J. M., Kind, A. J., and Robbins, J. (2009). Senescent swallowing: impact, strategies, and interventions. Nutr. Clin. Pract. 24, 395–413. doi:10.1177/0884533609332005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Rourke, J. R., and Swanson, M. S. (2009). Mechanisms of RNA-mediated disease. J. Biol. Chem. 284, 7419–7423. doi:10.1074/jbc.R800025200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purves-Smith, F. M., Sgarioto, N., and Hepple, R. T. (2014). Fiber typing in aging muscle. Exerc. Sport Sci. Rev. 42, 45–52. doi:10.1249/JES.0000000000000012
Raz, V., Abraham, T., Van Zwet, E. W., Dirks, R. W., Tanke, H. J., and Van Der Maarel, S. M. (2011a). Reversible aggregation of PABPN1 pre-inclusion structures. Nucleus. 2, 208–218. doi:10.4161/nucl.2.3.15736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raz, V., Routledge, S., Venema, A., Buijze, H., Van Der Wal, E., Anvar, S., et al. (2011b). Modeling oculopharyngeal muscular dystrophy in myotube cultures reveals reduced accumulation of soluble mutant PABPN1 protein. Am. J. Pathol. 179, 1988–2000. doi:10.1016/j.ajpath.2011.06.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raz, V., Buijze, H., Raz, Y., Verwey, N., Anvar, S. Y., Aartsma-Rus, A., et al. (2014). A novel feed-forward loop between ARIH2 E3-ligase and PABPN1 regulates aging-associated muscle degeneration. Am. J. Pathol. 30, 00016–00019. doi:10.1016/j.ajpath.2013.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raz, V., Butler-Browne, G., Van Engelen, B., and Brais, B. (2013). 191st ENMC international workshop: recent advances in oculopharyngeal muscular dystrophy research: from bench to bedside 8-10 June 2012, Naarden, The Netherlands. Neuromuscul. Disord. 23, 516–523. doi:10.1016/j.nmd.2013.03.001
Ross, C. A., and Poirier, M. A. (2005). What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 6, 891–898. doi:10.1038/nrm1742
Ruegg, M. A., and Glass, D. J. (2011). Molecular mechanisms and treatment options for muscle wasting diseases. Annu. Rev. Pharmacol. Toxicol. 51, 373–395. doi:10.1146/annurev-pharmtox-010510-100537
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ruegg, S., Lehky Hagen, M., Hohl, U., Kappos, L., Fuhr, P., Plasilov, M., et al. (2005). Oculopharyngeal muscular dystrophy - an under-diagnosed disorder? Swiss Med. Wkly. 135, 574–586.
Salvi, S. M., Akhtar, S., and Currie, Z. (2006). Ageing changes in the eye. Postgrad. Med. J. 82, 581–587. doi:10.1136/pgmj.2005.040857
Schindler, J. S., and Kelly, J. H. (2002). Swallowing disorders in the elderly. Laryngoscope. 112, 589–602. doi:10.1097/00005537-200204000-00001
Schneider, J. L., and Cuervo, A. M. (2014). Autophagy and human disease: emerging themes. Curr. Opin. Genet. Dev. 26c, 16–23. doi:10.1016/j.gde.2014.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sluijs, B., Hoefsloot, L., Padberg, G., Maarel, S., and Engelen, B. M. (2003). Oculopharyngeal muscular dystrophy with limb girdle weakness as major complaint. J. Neurol. 250, 1307–1312. doi:10.1007/s00415-003-0201-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tavanez, J. P., Calado, P., Braga, J., Lafarga, M., and Carmo-Fonseca, M. (2005). In vivo aggregation properties of the nuclear poly(A)-binding protein PABPN1. RNA. 11, 752–762. doi:10.1261/rna.7217105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tome, F. M., and Fardeau, M. (1980). Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol. 49, 85–87. doi:10.1007/BF00692226
Trollet, C., Anvar, S. Y., Venema, A., Hargreaves, I. P., Foster, K., Vignaud, A., et al. (2010). Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum. Mol. Genet. 19, 2191–2207. doi:10.1093/hmg/ddq098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Goethem, G., Martin, J. J., and Van Broeckhoven, C. (2003). Progressive external ophthalmoplegia characterized by multiple deletions of mitochondrial DNA: unraveling the pathogenesis of human mitochondrial DNA instability and the initiation of a genetic classification. Neuromolecular Med. 3, 129–146. doi:10.1385/NMM:3:3:129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Willis, T. A., Hollingsworth, K. G., Coombs, A., Sveen, M.-L., Andersen, S., Stojkovic, T., et al. (2013). Quantitative muscle MRI as an assessment tool for monitoring disease progression in LGMD2I: a multicentre longitudinal study. PLoS ONE 8:e70993. doi:10.1371/journal.pone.0070993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Witting, N., Mensah, A., Kober, L., Bundgaard, H., Petri, H., Duno, M., et al. (2014). Ocular, bulbar, limb, and cardiopulmonary involvement in oculopharyngeal muscular dystrophy. Acta Neurol. Scand. 130, 125–130. doi:10.1111/ane.12244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yokoyama, M., Mitomi, N., Tetsuka, K., Tayama, N., and Niimi, S. (2000). Role of laryngeal movement and effect of aging on swallowing pressure in the pharynx and upper esophageal sphincter. Laryngoscope. 110, 434–439. doi:10.1097/00005537-200003000-00021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu-Wai-Man, P., Lai-Cheong, J., Borthwick, G. M., He, L., Taylor, G. A., Greaves, L. C., et al. (2010). Somatic mitochondrial DNA deletions accumulate to high levels in aging human extraocular muscles. Invest. Ophthalmol. Vis. Sci. 51, 3347–3353. doi:10.1167/iovs.09-4660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: adult myopathy, muscle degeneration, OPMD, PABPN1, RNA metabolism
Citation: Raz Y and Raz V (2014) Oculopharyngeal muscular dystrophy as a paradigm for muscle aging. Front. Aging Neurosci. 6:317. doi: 10.3389/fnagi.2014.00317
Received: 18 June 2014; Accepted: 28 October 2014;
Published online: 10 November 2014.
Edited by:
Luciano Merlini, Istituto Ortopedico Rizzoli, Italy; Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), ItalyReviewed by:
Nir Barzilai, Albert Einstein College of Medicine, USACopyright: © 2014 Raz and Raz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vered Raz, Department of Human Genetics, Leiden University Medical Center, Einthovenweg 20, Building 2, Room R3-17, Postzone S4-P; P.O. Box 9600, Leiden 2300RC, Netherlands e-mail:di5yYXpAbHVtYy5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.