 Simon D’Alton*
Simon D’Alton* Jada Lewis
Jada Lewis- Department of Neuroscience, Center for Translational Research in Neurodegenerative Disease, College of Medicine, University of Florida, Gainesville, FL, USA
In the search for therapeutic modifiers, frontotemporal dementia (FTD) has traditionally been overshadowed by other conditions such as Alzheimer’s disease (AD). A clinically and pathologically diverse condition, FTD has been galvanized by a number of recent discoveries such as novel genetic variants in familial and sporadic forms of disease and the identification of TAR DNA binding protein of 43 kDa (TDP-43) as the defining constituent of inclusions in more than half of cases. In combination with an ever-expanding knowledge of the function and dysfunction of tau—a protein which is pathologically aggregated in the majority of the remaining cases—there exists a greater understanding of FTD than ever before. These advances may indicate potential approaches for the development of hypothetical therapeutics, but FTD remains highly complex and the roles of tau and TDP-43 in neurodegeneration are still wholly unclear. Here the challenges facing potential therapeutic strategies are discussed, which include sufficiently accurate disease diagnosis and sophisticated technology to deliver effective therapies.
Introduction
Frontotemporal dementia (FTD) is the third most common dementia in modern society, rendering it a critical public health issue (Onyike and Diehl-Schmid, 2013). However there is currently a dearth of therapeutics in clinical trials that are intended for FTD. Although tau has long been recognized as the principle component of neurofibrillary tangles (NFTs) in FTD (Joachim et al., 1987) and mutations in the microtubule associated protein tau (MAPT) are responsible for a subset of FTD cases (Hutton et al., 1998; Spillantini et al., 1998; D’Souza et al., 1999), investment in tau therapies has traditionally lagged somewhat due to the focus on proteotoxicity of other aggregated proteins such as amyloid-beta (Aβ) in Alzheimer’s disease (AD; Schneider et al., 2014). Furthermore, the majority of advances in genetics—so often a guiding hand for neurodegenerative research in the identification of therapeutic targets—have occurred only relatively recently in FTD (Hutton et al., 1998; Watts et al., 2004; Baker et al., 2006; Cruts et al., 2006; Sreedharan et al., 2008; DeJesus-Hernandez et al., 2011; Renton et al., 2011), and have uncovered genes such as progranulin (GRN) and Tar DNA binding protein (TARDBP) with little preexisting neurological literature. For example, mutation of the gene TARDBP is pathogenic in amyotrophic lateral sclerosis (ALS; Sreedharan et al., 2008) and the encoded protein TAR DNA binding protein of 43 kDa (TDP-43) has been identified as a major component of the ubiquitinated inclusions that characterize approximately half of all FTD cases (Taniguchi et al., 2004; Shi et al., 2005; Neumann et al., 2006); however, only 12 publications on this protein existed prior to this discovery.
Findings such as these can drive promising therapeutic research. Our understanding of the clinical symptoms, neuropathological features and genetic contributions to FTD has vastly expanded. At this point, we should be asking if we have reached the critical mass of knowledge necessary to develop effective therapeutics for clinical trials and, if not, we must assess what the remaining hurdles are for therapeutic efforts to have the highest probability of success. The ability to do so will depend not only on developing bioactive compounds, but also on sufficiently accurate diagnosis to determine those persons in early, if not asymptomatic, stages of degenerative disease. While strides are being made in many of these areas, there are still numerous challenges that must be met prior to effective therapeutic intervention to treat this chronic, multifaceted condition. Fortunately, lessons learned from the failures of clinical trials in neurodegenerative diseases such as AD can inform clinical efforts for FTD.
Current Pharmaceutical Intervention in FTD
The current trialed therapy to date for FTD is based on management of symptoms and does not address cause, and the rationale for their use is based on efficacy in treating other neurodegenerative disorders or psychiatric conditions with similar behavioral phenotypes. All of the clinical trials have been small, and only a handful has been placebo-controlled, double blind trials (Rabinovici and Miller, 2010). Generally, there is some evidence to suggest that selective serotonin reuptake inhibitors or serotonin norepinephrine reuptake inhibitors may be beneficial for some patients, particularly those with behavioral disturbances (Boxer and Boeve, 2007; Vossel and Miller, 2008). Antipsychotics and drugs targeting the cholinergic system appear to be largely ineffective (Mendez et al., 2007). Furthermore, despite its off-label use to treat FTD, the largest clinical study to date found no benefit to patients of taking memantine (Boxer et al., 2013). Consequently, there is no definitive treatment for FTD, and there is a dire need for novel therapeutics that target the underlying causes.
Clinical FTD and Pathological FTLD
Clinical and Pathological Heterogeneity of FTD/FTLD
FTD is a clinical term for a cluster of syndromes resulting from degeneration of the frontal and temporal lobes. Although historically regarded as a disorder with presenile onset (prior to 65 years of age), perhaps 25% of pathologically confirmed cases present clinically later than this (Baborie et al., 2011; Onyike and Diehl-Schmid, 2013).
Several syndromes can present in FTD which are associated with specific regional atrophy (Kril et al., 2005; Pereira et al., 2009; Seelaar et al., 2011). Behavioral-variant frontotemporal dementia (bvFTD) is characterized by changes in personality and behavior, and patients are apathetic (loss of interest in responsibilities, social withdrawal) and disinhibited (demonstrating socially inappropriate behavior) (Neary et al., 1998; Rascovsky et al., 2011). Semantic dementia (SD) and progressive nonfluent aphasia (PNFA) comprise the two syndromes that are characterized by changes in language function. SD presents with impaired comprehension and anomia, in contrast to PNFA in which language comprehension is spared, but speech is effortful and grammatically erroneous (Gorno-Tempini et al., 2011). These three variants often overlap, particularly at later stages of disease as atrophy becomes more widespread (Mesulam et al., 2003; Marczinski et al., 2004; Banks and Weintraub, 2008). The FTD umbrella also includes corticobasal syndrome (CBS) and progressive supranuclear palsy syndrome (PSPS; Boeve et al., 2003; Josephs, 2008), which while originally identified as atypical movement disorders also frequently present with features of bvFTD or PNFA. Similarly, individuals initially showing typical bvFTD/PNFA can develop motor disturbances (Rabinovici and Miller, 2010). bvFTD is frequently accompanied by an element of motor dysfunction as in motor neuron disease (FTD-MND) (Geser et al., 2010). Thus, the clinical picture of FTD is a spectrum of overlapping syndromes.
Underlying these clinical syndromes is a cluster of diseases collectively termed frontotemporal lobar degeneration (FTLD). In keeping with the historical approach to neurodegenerative conditions, definitive diagnosis of underlying disease is reserved for postmortem analysis based on pathological findings (i.e., the nature of the protein aggregate in specific brain regions). Pathologically, FTLD can now be broadly split into two major categories; FTLD-TDP with neuronal and glial inclusions immunoreactive for TDP-43, and FTLD-tau containing fibrillar, hyperphosphorylated tau (Joachim et al., 1987; Cairns et al., 2007; Neumann et al., 2007a). A small number of tau-negative, TDP-43-negative cases comprise the remainder which are immunoreactive to components of the ubiquitin proteasome system (FTLD-UPS), the fused in sarcoma protein (FTLD-FUS), ubiquitin only (FTLD-U) or lack inclusions completely (FTLD-ni/FTLD-other) and which will not be discussed here (Holm et al., 2009; Urwin et al., 2010; Josephs et al., 2011).
Heterogeneity of FTLD-Tau
Further complexity of FTLD is evident in the nature of the TDP-43 or tau inclusions themselves, which appear as a variety of subtypes. In the case of tau lesions, FTLD-tau can be subdivided further to CBD (Dickson et al., 2002), progressive supranuclear palsy (PSP; Hauw et al., 1994), Pick’s disease (PiD; Dickson, 1998), argyrophilic grains disease (AGD; Braak and Braak, 1987, 1989), sporadic multisystem tauopathy (MST; Bigio et al., 2001) and diffuse NFT dementia with calcifications (DNTC; Kosaka, 1994). PSP, CBD and PiD comprise by far the majority of cases of FTLD-tau (Josephs et al., 2011). In addition to these sporadic diseases, FTD and Parkinsonism linked to chromosome 17 (FTDP-17t) is caused by mutations in MAPT—this genetic form representing the cornerstone piece of evidence for the involvement of tau in non-familial disease (Hutton et al., 1998; Spillantini et al., 1998; D’Souza et al., 1999). Each of these conditions is characterized by the appearance of hyperphosphorylated tau that self-aggregates into fibrillar paired helical filaments (PHFs) and consequently neurofibrillary tangles (NFTs), although the morphology of the inclusion differs amongst diseases. Some of this heterogeneity is derived from alternative splicing of exon 10 of MAPT, which encodes one of the four “repeat” regions that act as microtubule binding domains (Goedert et al., 1989; Kosik et al., 1989). The deposited tau in PiD is rounded in nature and cytoplasmic, and is unique in that it primarily contains only 3 of the repeats (exclusion of exon 10, “3R” tau) (Delacourte et al., 1998; de Silva et al., 2006). Inclusions of PSP and CBD are both primarily composed of tau containing exon 10 and thus all four repeat regions (“4R” tau) (Sergeant et al., 1999). Using numerous antibodies to phosphorylated epitopes of tau, it has been proposed that the antigenicity of inclusions in PSP and CBD are similar to one another yet distinct from other tauopathies such as AD (Berry et al., 2004). Subtle differences between the two may be discerned on the basis of the nature of the inclusion. Neuronal inclusions in PSP are large and globose, and are accompanied by astrocytic lesions termed “tufted astrocytes”; whereas, CBD is characterized by more rounded “cortico-basal bodies” in neurons and “astrocytic plaques” (Yamada et al., 1992, 1993; Dickson et al., 1996; Josephs et al., 2011). It is worth reiterating here that there is a distinction between the pathological terms such as FTLD, CBD and PSP and the clinical terms FTD, CBS and PSPS as they are often incorrectly used interchangeably and can lead to confusion if, for example, CBD or PSP is the underlying pathological disease in a clinical case of PNFA (Josephs et al., 2011 and see also “Current Clinical, Imaging and Biochemical Diagnostics” section below on correlation between clinical symptom and neuropathologically diagnosed disease).
Heterogeneity of FTLD-TDP
A major component of inclusions in cases of FTLD with ubiquitinated aggregates, which represents approximately 50% of FTLD cases, was identified in 2006 to be TDP-43, giving rise to the name FTLD-TDP (Neumann et al., 2006). There are four subtypes of FTLD-TDP, associated with distinct TDP-43 pathological morphology and distribution—Type A is characterized by many aggregated, juxtanuclear, neuronal cytoplasmic inclusions (NCI) and short dystrophic neurites (DN) predominantly in layer 2 of the cortex, Type B by fewer NCI and DN that are dispersed more widely throughout the cortex, Type C by long DN in cortical layer 2 and Type D by the appearance of neuronal intranuclear inclusions (Mackenzie et al., 2011). Type D is typically found in rare, familial cases involving mutations in the valosin containing protein (VCP) gene (Forman et al., 2006), thus the defining hallmark of sporadic FTLD-TDP is cytoplasmic aggregation as NCI or DN. This is accompanied by a loss of nuclear TDP-43 expression (Neumann et al., 2006). In FTLD-TDP, TDP-43 is also hyperphosphorylated and truncated to produce ~25 kDa C-terminal fragments, which appear to be enriched in inclusions relative to the N-terminus (Igaz et al., 2008). Therefore, as is the case for FTLD-tau, TDP-43 neuropathology appears as a number of distinct entities and pathological subtypes. The toxicity of TDP-43 and tau in disease is not evident from neuropathology alone, and critical evidence for the involvement of both in disease hinge on compelling genetic data.
FTLD Genetics as a Driver of Translational Research
Studies of the genetic component of FTLD have provided important breakthroughs in our understanding of disease pathogenesis. As noted above, familial mutations in the MAPT gene encoding the tau protein explain a subset of FTLD cases and provide compelling evidence for the involvement of tau in sporadic disease, which share some of the same pathological hallmarks (Hutton et al., 1998; Spillantini et al., 1998; D’Souza et al., 1999). Whilst inherited mutations in the TARDBP gene encoding TDP-43 are not found in heritable FTLD, familial cohorts do exist in ALS, a second TDP-43 proteinopathy (Sreedharan et al., 2008), implicating TDP-43 in neuronal dysfunction or death. This is used, in a manner analogous to that for tau and AD, as evidence that TDP-43 dysfunction is a driver of FTLD also. Since ALS and FTD are now considered at opposite ends of the same disease spectrum, the connection seems justified on a biological level also. Adding further weight to this argument are reports of TARDBP mutations in sporadic FTLD (Borroni et al., 2009a, 2010; Synofzik et al., 2014).
Interestingly, familial cohorts do exist for TDP-43 positive FTLD, although the mutations in this case are in the VCP, C9ORF72 and GRN genes (Watts et al., 2004; Baker et al., 2006; Cruts et al., 2006; Neumann et al., 2007b; DeJesus-Hernandez et al., 2011; Renton et al., 2011). From a therapeutic standpoint, by far the most intriguing of these is the secreted glycoprotein progranulin, primarily due to its neurotrophic activity (Van Damme et al., 2008). It also has roles in the periphery including in inflammation and wound healing (He et al., 2003; Ahmed et al., 2007). GRN mutations can be insertions, deletions, splice site, missense or nonsense and are scattered throughout the gene (Baker et al., 2006; Cruts et al., 2006; Gass et al., 2006; Mukherjee et al., 2006; Bronner et al., 2007; Bruni et al., 2007; Rademakers and Rovelet-Lecrux, 2009). However, all are predicted to result in reduction of functional progranulin (Baker et al., 2006; Cruts et al., 2006; Rademakers and Rovelet-Lecrux, 2009). Loss of progranulin function in these familial cases causes type A TDP-43 pathology (Mackenzie et al., 2006). Common genetic variants in progranulin are also potential risk factors for TDP-43 proteinopathies including sporadic FTLD (Brouwers et al., 2008; Sleegers et al., 2008; Galimberti et al., 2010). Currently, mechanisms by which reduction in progranulin results in neurodegeneration are unclear but principally center on a reduced ability to recover from age-related stressors or damage (Ryan et al., 2009; Laird et al., 2010; Kao et al., 2011; Jackman et al., 2013).
The discovery of mutations in GRN in 2006 left only one linkage region on chromosome 9 unaccounted for in hereditary cases of FTD. The delay in identifying the responsible C9ORF72 gene can be attributed to the nature of the mutation itself—an intronic, hexanucleotide (GGGGCC) repeat expansion that was undetectable by commonly used sequencing methods (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Both the antisense and sense strands are transcribed from this genomic locus and form nuclear RNA aggregates or “RNA foci” (DeJesus-Hernandez et al., 2011; Gendron et al., 2013; Zu et al., 2013), which may cause cell death via sequestration of functional RNA binding proteins (Lee et al., 2013b). The expanded RNA also undergoes repeat-associated non-ATG translation (RAN translation) to produce aggregation prone polypeptides that form inclusions in human FTD (Ash et al., 2013; Gendron et al., 2013; Mori et al., 2013; Zu et al., 2013). Reducing levels of expanded transcripts is an obvious therapeutic concept and one that has already proven successful in vitro (Donnelly et al., 2013). Another group has demonstrated antisense efficacy in motor neurons derived from C9ORF72 carriers (Sareen et al., 2013). However, future studies that clarify the mechanistic link between RNA foci, RAN proteins, and neurodegeneration will be essential in order to determine if such approaches are likely to fully address the neurotoxicity in C9ORF72 cases. It should be also noted that the neuropathology unique to C9ORF72 is not present in FTD lacking repeat expansion mutations. Although C9ORF72 mutations constitute a fascinating additional mechanism by which genetic aberration induces neuronal demise, therapeutic implications for FTD cannot currently be extrapolated to the population at large.
In addition to these familial, causative mutations, there are a number of genes that modulate the risk of FTD or phenotypic outcome. Notably, homozygous mutations in TREM2 are associated with atypical FTD presenting with epilepsy and parkinsonism (Giraldo et al., 2013; Guerreiro et al., 2013; Le Ber et al., 2014). Rare heterozygous mutations in this gene may increase risk of FTD (Lattante et al., 2013; Borroni et al., 2014; Cuyvers et al., 2014). Several genes identified in genome wide association studies may also modify risk (Ferrari et al., 2014).
Tau and Tau-Mediated Mechanisms of Toxicity
Tau Biology and Function
The MAPT gene generates six isoforms of the microtubule binding protein tau via alternative splicing of exons 2, 3 and 10 (Kosik et al., 1989). Exon 10 contains one of four microtubule binding domains in tau, such that exon inclusion generates the 4R isoform with higher affinity than 3R for microtubule binding, these two forms being expressed at approximately 1:1 ratio in human adult brain (Goedert et al., 1989; Goedert and Jakes, 1990; Butner and Kirschner, 1991). The binding of tau stabilizes microtubules and promotes assembly (Weingarten et al., 1975; Cleveland et al., 1977). Under physiological conditions in healthy neurons, this activity is further regulated by phosphorylation of tau at 47 confirmed sites (Martin et al., 2013) by a variety of major cellular kinases including glycogen synthase kinase 3 beta (GSK-3β; Wagner et al., 1996), microtubule affinity regulating kinase (MARK; Drewes et al., 1997) cyclin dependent kinase 5 (CDK5; Baumann et al., 1993), Protein Kinase A and C (PKA/PKC; Michel et al., 1998) and by phosphatases such as protein phosphatase 2a (PP2A; Sontag et al., 1996). Furthermore, our lab has recently demonstrated that the Parkinson’s disease associated kinase LRRK2 can phosphorylate tau (Bailey et al., 2013). Tau is regulated both in normal and disease states by phosphorylation, with phosphorylated tau having reduced affinity for microtubules, preventing their stabilization (Drechsel et al., 1992).
Although microtubule binding is the major function ascribed to tau, it has a number of other roles, for example in influencing cell signaling via kinases such as Fyn, Src (Sharma et al., 2007) and phospholipase Cγ (Hwang et al., 1996). A nuclear form of tau is also implicated in protection from stress (Sultan et al., 2011), indicating that our current knowledge of tau function is probably incomplete. Additionally, our knowledge of “what” tau does is often interpreted without the context of “when”, an important consideration when aiming to modify any activity but particularly so in an age-related condition. For example, there are numerous external inputs that may induce tau phosphorylation, including oxidative stress (Melov et al., 2007), hypoxia (Fang et al., 2010), hypothermia (Planel et al., 2007a), administration of drugs (Whittington et al., 2011), insulin dysfunction (Planel et al., 2007b) and viral infection (Patrick et al., 2011). Splice exclusion of tau exon 10 also occurs in response to hypoxia (Suh et al., 2010). These observations imply that tau is dynamically regulated in response to external cues, which may be important when evaluating scientific theories and therapeutic avenues.
Tau Aggregation and Toxicity
The identification of which form(s) of tau are actually toxic remains the largest question in the field, and is a key determinant of future therapeutic avenues. Originally, tangles themselves or smaller fibrillar antecedents were considered the species most likely to be neurotoxic, and theories regarding direct toxicity via physical blockade of cellular function such as trafficking, disruption to organelle distribution and inhibition of proteasome activity remain intuitive and often well-researched hypotheses (Figure 1; Keck et al., 2003; Lin et al., 2003; Ren et al., 2007).
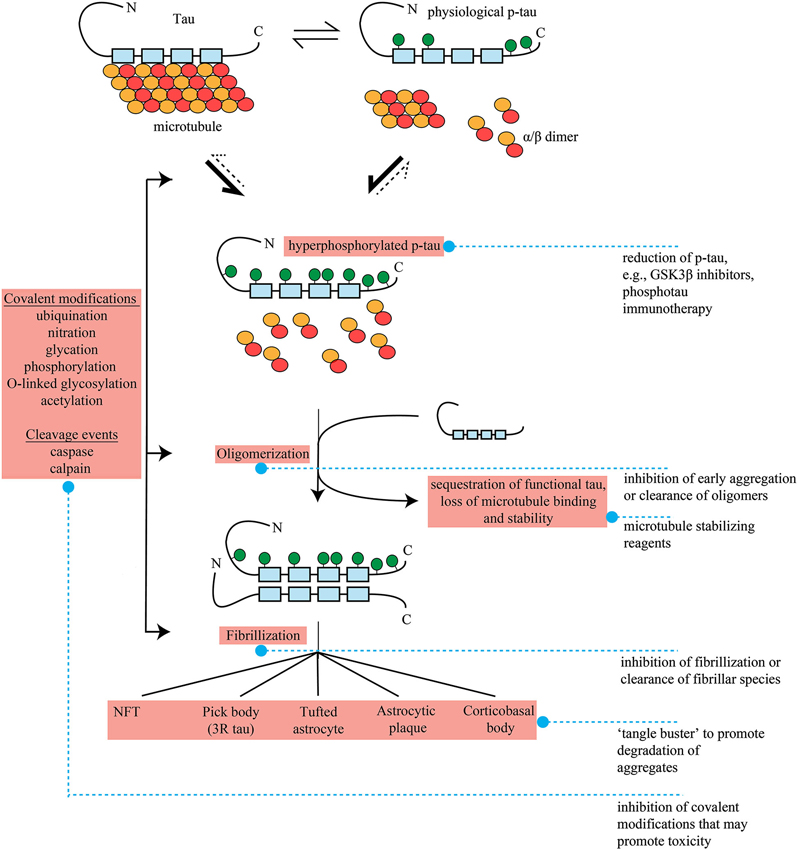
Figure 1. Formation of pathological tau and potential therapeutic interventions in FTLD. Tau bound to microtubules through repeat regions (blue boxes, 4 repeat tau shown) exists in dynamic equilibrium with physiological, phosphorylated tau (p-tau). P-tau formation is accompanied by microtubule disengagement and depolymerization to tubulin heterodimers. The point of equilibrium between tau and p-tau is influenced by genetics and external factors such as oxidative stress and hypothermia as well as stages of development. It is unclear if hyperphosphorylated, disease-associated tau results from tipping of this equilibrium to favor further p-tau formation, completely novel phosphorylation pathways, or a mixture of the two. In the hyperphosphorylated state, microtubule destabilzation beyond physiological levels results in neuronal dysfunction. This loss of function may be enhanced further by sequestration of functional tau by hyperphosphorylated tau into oligomers, and subsequently fibrils and aggregates. The repeat region of tau appears to form the core of these insoluble species. A number of other covalent modifications (ubiquination, nitration, glycation, phosphorylation, O-linked glycosylation) and cleavage events (caspase and calpain) may also occur at ill-defined junctures, and these may potentiate the dynamics of oligomerization or aggregate formation. As there are a range of pathological tau lesions which differ in appearance, these and other undefined pathways and potentially factors such as genetics, environmental inputs and cell type specific expression must differ in pathogenic tau formation. Pick bodies are entirely three repeat tau. Pathogenic species or potentially toxic mechanisms related to tau are highlighted in red. Blue dashed lines denote potential therapeutic interventions, see main text for more detail.
Despite this, several lines of evidence weigh against tangles as a form of tau toxicity, or at least suggest that they are not solely responsible. For example, neurons survive for perhaps decades with neurofibrillary pathology (Morsch et al., 1999) and silencing of the tau transgene in a mouse model of tauopathy following formation of NFT pathology rescues cognitive phenotype, although tangle formation continues unabated (Santacruz et al., 2005). Furthermore, several transgenic lines of mice and tau transgenic flies expressing various tau isoforms display a distinct dissociation between cell death and the appearance of tangle pathology (Wittmann et al., 2001; Andorfer et al., 2005; Spires et al., 2006). In fact, tangle-bearing neurons in mice expressing mutant P301L tau appear to be functionally identical to both wild type mice and to neighboring neurons that do not contain NFTs (Kuchibhotla et al., 2014). Additionally, study of human disease has demonstrated that despite greater tissue loss in FTDP-17t, the aggregated tau burden in these cases is approximately 10% of the level of that found in AD (Shiarli et al., 2006). While these studies do not exclude the possibility that tangles are ultimately toxic, they do suggest that tangles are not the initial neurotoxic species. Rather, such findings have shifted philosophy towards a less visible form of pre-fibrillar species, with aggregated material representing compensatory adaptation to sequester these smaller toxic forms, at least initially. Several groups have reported the existence of what might be referred to as tau oligomers (Berger et al., 2007; Maeda et al., 2007; Sahara et al., 2007). Although the nature and importance of these species are contentious, several studies have demonstrated an association between their levels and neurotoxicity. For example, heat-shock protein mediated stabilization of oligomers in the rTg4510 model of tauopathy is associated with enhanced neurotoxicity but reduced fibril formation (Blair et al., 2013). Conversely, immunotherapy in the JNPL3 transgenic mouse line of tauopathy using an antibody to oligomeric forms of tau improves motor and memory phenotypes without altering NFTs (Castillo-Carranza et al., 2014).
Post-Translational Modifications, Hyperphosphorylation and Tau Loss-of-Function
Tau modifications that occur prior to or concomitant with fibril formation or oligomerization may potentiate toxicity. These processing events include cleavage by caspases (Canu et al., 1998; Rohn et al., 2002) and calpain (Park and Ferreira, 2005; Ferreira and Bigio, 2011), O-linked glycosylation (Arnold et al., 1996; Liu et al., 2004), ubiquitination (Morishima-Kawashima et al., 1993), nitration (Horiguchi et al., 2003; Reyes et al., 2012), acetylation (Min et al., 2010) and glycation (Ledesma et al., 1994; Yan et al., 1994); however, the timing and significance of these modifications in pathogenesis is frequently debated (reviewed in Gendron and Petrucelli, 2009). The addition of such moieties and/or proteolytic cleavage have been generally linked in some way to either facilitation of tau into fibrillar species, the enhancement of subsequent events such as phosphorylation or to modification of function which might therefore facilitate the evolution of tau from functional molecule to pathological toxin.
Of these, tau hyperphosphorylation is by far the most cited candidate for a mediator of toxicity. Although phosphorylation has been implicated in facilitation of fibril formation and aggregation, which appears to differ between phosphorylation sites (Alonso et al., 1996; Necula and Kuret, 2004, 2005; Wang et al., 2007), pseudophosphorylated tau is also toxic in cell culture without the need for fibril formation, demonstrating a pathogenic effect of phospho-tau without fibrillization (Fath et al., 2002). This has driven an alternative hypothesis that abnormal phosphorylation of tau results in inappropriate disengagement from microtubules and consequent destabilization, which in turn results in defects such as reduced axonal transport—in essence, a tau loss of function. The absence of any robust neurodegenerative phenotype in tau knockout mice may contradict this hypothesis, although it seems plausible that constitutive loss of tau may be compensated for by in utero-programmed upregulation of other microtubule binding proteins such as MAP1a (Harada et al., 1994). Interestingly, the absence of phenotype in this mouse is also often cited as evidence that a potential therapy which reduces total tau, and therefore any toxic species, would not yield adverse consequences. The simultaneous assertion that loss of function is a possible mechanism of disease and that reduction in total tau should be considered for tau therapeutics is both highly contradictory and highlights the current lack of clarity regarding disease mechanism(s). In this specific instance, a conditional tau knockout mouse may provide greater insight.
Nevertheless, loss of microtubule stability remains a plausible neurodegenerative mechanism and phosphorylation of tau is a proven destabilizer. Hyperphosphorylated tau extracted from AD, another tauopathy, is a poor promoter of microtubule assembly and sequesters normal functional tau and other microtubule binding proteins in vitro (Alonso et al., 1996, 1997). Importantly, this effect is lost upon fibrillization or treatment with phosphatases (Wang et al., 1996; Alonso Adel et al., 2006). Activation of cdk5 in vivo using overexpression of p25 induces tau phosphorylation with accompanying features characteristic of loss of microtubule integrity (Ahlijanian et al., 2000; Bian et al., 2002). Genetic evidence also supports the tau loss of function and microtubule destabilization hypothesis. Mutations in MAPT that are the cause of FTDP-17t typically are either intronic and alter the splicing of exon 10 encoding one of the microtubule binding domains or cause changes in coding sequences situated close to the microtubule binding domain (Goedert, 2005). This suggests that correct splicing of tau and consequently tau-mediated microtubule stability is an important part of healthy aging. In many of these familial cases, the aggregated tau is almost entirely of the 4R form (Spillantini et al., 1997).
From the evidence above, it can be concluded that tau undergoes a host of conformational and biochemical changes indicating that in all likelihood there are a wide range of pathways acting upon it at a number of different junctures (Figure 1). During this evolution, tau may be toxic at one or several points via a number of different mechanisms that may occur simultaneously, and there is currently evidence supporting and undermining each.
Hypothetical Therapeutic Strategies for FTLD-Tau
The critical issues surrounding toxicity pose significant confounds for the development of therapeutic approaches for FTLD-tau. Hypothetical therapeutic strategies are at present based on a highly complex toxicity that is not fully understood. At this time, none of the aforementioned toxic mechanisms has emerged as the dominant candidate in the push for therapy development. Consequently, although there are a number of potential therapeutic possibilities (Figure 1) there is no clear cut candidate above all others for therapeutic intervention.
Under the gain of function aggregate toxicity model, determining toxic species will be highly influential in future direction. For example, the removal of NFTs using a pharmacological “tangle buster” of sorts might provide relief from the deleterious effects of large aggregates. Nevertheless, if fibrillar or aggregate forms of tau represent initial compensatory mechanisms to sequester more upstream oligomeric forms, then it may be that removal of this “toxic sink” would outweigh relief from tangle-associated toxicity and would be consequently more harmful than beneficial. Alternatively, small molecule inhibitors of oligomer formation or fibrillization may prevent accumulation of all toxic species at their origin. LMTX, a modified derivative of the aggregation inhibitor methylthionine chloride (methylene blue), is entering Phase III trials for bvFTD (clinical trial #NCT01626378, and see Wischik et al., 2014). Other therapeutic avenues targeting toxic species may be even more specific; immunotherapy is a potential method for the removal of pathological tau, and in several instances has been demonstrated to suppress tau pathology and improve phenotype in transgenic models of tauopathy (Asuni et al., 2007; Sigurdsson, 2008; Yanamandra et al., 2013; Castillo-Carranza et al., 2014). This strategy relies on identifying precisely the species, or epitope of the species, responsible for toxicity and successful application in human disease will rely on the resolution of this issue. If multiple toxic species exist, which is quite possible given the wide range of modifications that tau undergoes, then multiple epitopes may need to be targeted.
The primary therapeutic alternative is based on the loss of function model, making inhibition of phosphorylation and stabilization of microtubules potential therapeutic options. Numerous studies have demonstrated reduced hyperphosphorylation and aggregation of tau with alleviation of motor phenotype or axonal degeneration in murine models of tauopathy by inhibition of tau kinases such as GSK3β, PKA and PKC (Nakashima et al., 2005; Noble et al., 2005; Le Corre et al., 2006). Temsirolimus, one of the newest potential therapies for tauopathy, appears to work on two levels, through reducing tau hyperphosphorylation and increasing autophagic clearance (Jiang et al., 2014). Although there is concern regarding chronic inhibition of major regulatory kinases with an approach such as this (McKnight et al., 2012), initial studies suggest that GSK3 inhibitors such as lithium and tideglusib are well tolerated in the short term (6–12 weeks) and perhaps up to a year in patients with AD (MacDonald et al., 2008; Hampel et al., 2009; Forlenza et al., 2011; del Ser et al., 2013). Notably, lithium is already FDA-approved and could therefore avoid many regulatory steps that typically retard drug availability. Although these studies have demonstrated a lack of efficacy in AD, duration of treatment has typically been short or using low participant numbers. Microtubule stabilizing agents are currently being actively pursued, and some appear to be brain penetrant and improve microtubule density, axonal stability and cognitive performance in mouse models of tauopathy (Brunden et al., 2010a,b). However, the microtubule binding agent davunetide (NCT01110720), which promotes assembly and affords neuroprotection in a number of animal models, has recently failed in Phase III trials for PSP (Shiryaev et al., 2009; Boxer et al., 2014).
These strategies represent early therapeutic endeavors in FTLD-tau. It is common in the literature to conclude research articles by advancing new forms of tau pathology or the pathways affecting or affected by tau as potential therapeutic targets. However, it is important to note the lack of evidence for a single, dominant toxic mechanism. The complexity of FTLD-tau may be similar to that recently posed for AD, in which a heterogeneous Aβ “soup” confers toxicity, rather than a single pathogenic species (Benilova et al., 2012). In future, it will be important to refine theories and identify mechanisms that contribute most significantly to neuronal demise and that consequently have a higher chance of success in clinical trials.
TDP-43 and TDP-43 Mediated Mechanisms of Toxicity
TDP-43 Biology and Function
Previously a relatively little-studied protein, the stature of TDP-43 has grown considerably due to description of mutations in the TARDBP gene in sporadic and familial cases of ALS and the discovery that TDP-43 is a major constituent of ubiquitin-positive inclusions in FTD and ALS (Neumann et al., 2006; Sreedharan et al., 2008). Subsequent demonstration that TDP-43 positive aggregates are found in numerous other neurodegenerative disorders including Parkinson’s disease and AD have consequently raised the possibility that TDP-43 may be an intriguing therapeutic target outside of FTLD-TDP (Amador-Ortiz et al., 2007; Nakashima-Yasuda et al., 2007). TDP-43 is also one of a number of disease-related proteins with a known role in RNA metabolism (Polymenidou et al., 2011), a field that over the last decade has grown considerably in impact due to an increasing appreciation for its complexity and influence in multiple biological systems.
The TARDBP gene encoding TDP-43 gives rise to a 414 amino acid nucleic acid binding protein containing a number of functional domains including two RNA Recognition Motifs (RRM), a glycine rich C-terminus required for binding to a host of cofactors such as members of the hnRNP family and nuclear localization and export sequences that contribute towards its ability to shuttle continuously between nucleus and cytoplasm (Buratti and Baralle, 2001; Ayala et al., 2008b; Winton et al., 2008; Ling et al., 2010). A highly mobile protein, it is redistributed from a predominantly nuclear localization to the cytoplasm in response to diverse stress stimuli (Ayala et al., 2011) such as oxidative stress (Liu-Yesucevitz et al., 2010), proteasome inhibition (van Eersel et al., 2011), endoplasmic reticulum stress (Leggett et al., 2012) and physical injury (Moisse et al., 2009) and relocalizes at synapses upon depolarization (Wang et al., 2008). Studies in humans and mice have demonstrated that TDP-43 binds a host of RNA substrates, equal to approximately 30% of transcripts in the mouse genome (Polymenidou et al., 2011). In addition to mRNA, substrates also include non-coding elements such as long noncoding, small nucleolar, small nuclear, ribosomal, micro and telomeric RNA, and in some cases binding was increased in cases of FTLD-TDP (Tollervey et al., 2011). In both mouse and human studies, reduction in TDP-43 resulted in alterations in splicing of bound transcripts (Polymenidou et al., 2011; Tollervey et al., 2011). Binding of TDP-43 to pre-mRNA with large introns results in stabilization in mice, and many of these transcripts are involved in synaptic activity (Polymenidou et al., 2011). Given that TDP-43 associates with a range of other proteins that influence microRNA processing, splicing, polyadenylation and RNA editing (Ling et al., 2010), it is likely that it is involved in a plethora of cellular functions by sheer virtue of regulating expression of transcripts in a number of ways that feed in to numerous biological pathways. This is supported by functional studies demonstrating that TDP-43 influences processes as diverse as neurite outgrowth (Iguchi et al., 2009), cell cycle dynamics (Ayala et al., 2008a), mitochondrial function (Duan et al., 2010), metabolism (Chiang et al., 2010) and autophagy (Bose et al., 2011). Evidently, TDP-43 is a critical regulator of cellular dynamics.
TDP-43 Aggregation and Toxicity
Given the relatively recent discovery of TDP-43, understanding the nature of its contribution to neurodegeneration is still in its infancy. Current hypotheses regarding TDP-43 pathogenesis stem from observation of post-mortem changes in protein immunohistochemistry and biochemistry, and thus center on direct toxicity from cytoplasmic aggregates or C-terminal fragments and loss of nuclear function. These are not necessarily mutually exclusive and may act in concert. C-terminal fragments of TDP-43 are aggregate-prone, undergo phosphorylation and ubiquitination and are cytotoxic in cell culture (Igaz et al., 2009; Nonaka et al., 2009; Zhang et al., 2009). Disease linked mutations found in ALS appear to increase the production of C-terminal fragments, and their aggregation and toxicity (Johnson et al., 2009; Nonaka et al., 2009; Barmada et al., 2010). The nature of these fragments and their generation is contentious. TDP-43 can be cleaved by caspases to generate ~25 kDa and ~35 kDa fragments, and immunopositive inclusions for the caspase-cleaved conformer are found in human cases of FTLD-TDP (Zhang et al., 2007; Dormann et al., 2009), with one group suggesting that this cleavage may be neuroprotective in some way, perhaps by limiting TDP-43 activity (Suzuki et al., 2011). However, sequencing of fragments from FTLD-TDP cases has yet to identify a single consensus N-terminus amino acid for these fragments (Nonaka et al., 2009). This may be consistent with observations using phospho-specific antibodies which demonstrate multiple fragments from 17–25 kDa, the intensity of which also may be distinct for each TDP-43 subtype (Hasegawa et al., 2008). Such heterogeneity may ultimately be important given that cleavage site determines extent of phosphorylation, solubility and aggregation in vitro (Furukawa et al., 2011a). Although cleavage and aggregation of TDP-43 is a pathological event, such properties may also be involved in normal cellular function. Two groups have reported on the prion-like nature of the C-terminus, which self-interacts to form insoluble yet functional nuclear aggregates that are repeatedly formed and destroyed (Wang et al., 2012; Nonaka et al., 2013). Inhibiting or stabilizing this interaction is proposed to cause degradation to 25 kDa or oligomerization respectively. This segment of protein also confers structural beta-sheet conformations that appear to promote the formation of amyloid fibrils, and this propensity is enhanced by the A315T mutation found in familial ALS (Guo et al., 2011). Thus, function and pathogenicity may be two sides of the same coin, and the prion nature of TDP-43 opens up the intriguing possibility of prion-like spreading of disease (Nonaka et al., 2013). One group has further suggested that prion-like aggregation of TDP-43 or C-terminal fragments may be modulated by a dynamic interaction with heat shock proteins (Udan-Johns et al., 2014).
TDP-43 Loss of Function and Toxicity
At least three studies using aggregate-prone C-terminal fragments or in vitro-generated fibrillar TDP-43 have demonstrated that aggregates are capable of sequestering the full length protein, and therefore depleting endogenous nuclear function (Nonaka et al., 2009; Che et al., 2011; Furukawa et al., 2011b), although this is not true for all studies (Zhang et al., 2009). Consequently, the reason for nuclear depletion is not fully understood, but is likely to be deleterious given that numerous studies in vitro and in vivo suggest TDP-43 levels necessitate tight regulation. Overexpression and knockdown in vitro can confer toxicity (Ayala et al., 2008a; Suzuki et al., 2011) and most persuasively knockout in vivo results in lethality. This occurs both in conditional knockout animals in which deletion is postponed until adulthood and in utero in constitutive Tardbp–/– mice (Chiang et al., 2010; Kraemer et al., 2010; Sephton et al., 2010; Wu et al., 2010). Loss of TDP-43 specifically in motor neurons results in cell death and an ALS-like phenotype in mice (Wu et al., 2012) and reduced TDP-43 expression in Drosophila and zebrafish results in motor deficits (Feiguin et al., 2009; Kabashi et al., 2010). These data suggest that TDP-43 may be a critical regulator of cellular function and pathological loss of function may have severe consequences for cellular integrity. Furthermore, loss of TDP-43 activity may result in altered response to cellular stress, as reduced TDP-43 levels results in reduction in the number and size of stress granules (SGs), transient cytoplasmic bodies involved in RNA triage that form in response to oxidative stressors (McDonald et al., 2011). Reduction of TDP-43 in primary neuronal cell culture also increases sensitivity to toxic proteasome inhibition (van Eersel et al., 2011). Both of these cellular insults may be involved in the initial formation of TDP-43 pathology. SGs, which contain TDP-43 itself, may form the initial “seeds” for subsequently greater aggregation whilst proteasomal inhibition also induces nuclear clearance and cytoplasmic aggregation of TDP-43 in vivo and in vitro (Liu-Yesucevitz et al., 2010; Meyerowitz et al., 2011; Parker et al., 2012; Tashiro et al., 2012).
Thus, as for tau, there are potentially a number of different mechanisms by which TDP-43 dysfunction contributes to neuronal demise, and which may not be mutually exclusive. Recently the tau field has reached a point of equilibrium with regards to the loss of function vs. gain of toxicity debate. It will be intriguing to see in the near future if there is a definitive answer for TDP-43 proteinopathies.
Therapeutic Strategies for FTLD-TDP
Understanding of TDP-43 proteinopathy or even TDP-43 endogenous function is currently limited and thus so are therapeutic expectations. However, plausible logical suggestions would be restoration of nuclear function and reduction in formation of or increased clearance of aggregates. As loss of TDP-43 has significant implications for cellular integrity, aiming to reduce expression and therefore aggregate formation, as proposed for tau, is a conceptual non-starter. One may hypothesize that preventing cytoplasmic accumulation and retaining nuclear function may prove beneficial. In this case, simply defining the mechanism by which loss of nuclear function occurs is critical. If it is underpinned by cytoplasmic aggregate sequestration, then targeting aggregated material would logically provide relief from both nuclear loss and any direct cytotoxicity. At least in vitro dimebon and methylene blue, two compounds that were successful in Phase II clinical trials for AD, are capable of reducing TDP-43 aggregation (Yamashita et al., 2009). Currently, our knowledge of TDP-43 biology is probably too limited to make TDP-targeted therapies a realistic short term goal. Increased knowledge of basic function and the development of more sophisticated study tools such as animal models recapitulating TDP-43 pathology and patient specific induced pluripotent stem cells (iPSCs) should provide a greater understanding of pathways amenable to therapy (Bilican et al., 2012; Egawa et al., 2012). Indeed, iPSCs have already been used to screen drug candidates, identifying anacardic acid as a potential modifier of insoluble TDP-43 species (Egawa et al., 2012).
Progranulin as an Unbiased Therapeutic Candidate
For both tau and TDP-43 directed therapies, there is a broader question raised by the presence of multiple disease subtypes (for example, CBD, PSP and PiD in FTLD-tau and type A-D for FTLD-TDP). As the pathology for each disease differs, the mechanisms leading to their formation may also be dissimilar. Would a different therapeutic strategy be required for each, or is there a keystone mechanism underpinning them all? This question is analogous to that posed in the cancer field where optimal breast cancer treatment paradigms depend on the type of cancer present (Kelly and Buzdar, 2013). A therapy that could be used more globally and that is not dependent on the type of pathology might therefore have greater application. The discovery of loss-of-function mutations in progranulin as a cause of TDP-43 proteinopathy may provide such a target. When compared with the challenge of identifying toxicity due to tau and TDP-43, raising levels of the potentially neuroprotective progranulin is a conceptually simple solution. Recent identification of TNF (Tang et al., 2011) [although this interaction is disputed (Chen et al., 2013)] and sortilin (Zheng et al., 2011) receptors that bind progranulin offer potentially modifiable and druggable targets. Indeed, small molecule inhibition of sortilin-mediated endocytosis restores extracellular progranulin levels in iPSCs derived from progranulin mutation carriers to wild type levels (Lee et al., 2013a). Additionally, chemicals that upregulate progranulin levels through enhanced transcription (suberoylanilide hydroxamic acid) or the stabilization of intra- and consequently extracellular progranulin by the alkalization of intracellular compartments have been identified (chloroquine, bepridil, and amiodarone), some of which are already FDA-approved (Capell et al., 2011; Cenik et al., 2011). Finally, protective genetic variants of the transmembrane protein TMEM106b were identified in a genome wide association study of GRN mutation carriers. These variants delay age of onset or reduce penetrance in GRN mutation carriers and correlate with plasma progranulin levels, implicating TMEM106b or interacting pathways as therapeutic targets (Van Deerlin et al., 2010; Cruchaga et al., 2011; Finch et al., 2011). The applicability of these or other future progranulin therapies to neurodegenerative diseases outside of FTLD-TDP could be tested in vivo in animal models, with transgenic models of tauopathy/FTLD-tau an obvious candidate.
A number of challenges present themselves. Primarily, little is known regarding how progranulin is processed in the brain, with the majority of knowledge coming from studies in the periphery. In this environment, the progranulin precursor—composed of seven and a half repeats of a common 12-cysteine module called granulin—can be processed proteolytically by elastase and proteinase 3 to produce the individual granulins (Kessenbrock et al., 2008), which may have distinct effects on biological processes. At present, it is unknown whether the same mechanisms occur in the brain or whether loss of full-length progranulin or individual granulin(s) is the cause of FTLD in familial forms. At least two studies have suggested that the neurotrophic properties of progranulin may be due to module granulin activity rather than the full-length form (Gass et al., 2012; De Muynck et al., 2013). If distinct biological effects exist for the full-length progranulin precursor and each individual granulin, the results of elevating the former in FTD patients may be a complex and multifaceted mix of therapeutic and unintended side effects. It is consequently imperative to either deconstruct the desirable mechanisms underlying progranulin function or to demonstrate in vivo the safety, tolerability and physiological benefits of increased progranulin levels. Encouragingly, mice overexpressing progranulin are significantly rescued from the behavioral deficits induced by middle cerebral artery occlusion stroke paradigms coincident with a shift to a more anti-inflammatory profile in transgenic animals (Tao et al., 2012; Egashira et al., 2013). This shift in inflammatory profile is consistent with a role for progranulin in modulating neuroinflammatory responses (Cenik et al., 2012; Martens et al., 2012; Tanaka et al., 2013). Progranulin knockout mice—a model of inherited FTLD due to GRN mutation—provide a useful tool for studying progranulin deficiency but lack overt TDP-43 aggregation and nuclear clearance as observed in humans (Ahmed et al., 2010). Pathology is restricted to hyperphosphorylated, insoluble TDP-43 that remains largely nuclear (Wils et al., 2012). Such tools may be necessary to fine tune elevation of progranulin levels, given its potential role in tumorigenesis (Tangkeangsirisin and Serrero, 2004; Matsumura et al., 2006; Göbel et al., 2013). In sum, progranulin therapy is an attractive target for FTLD-TDP, particularly so for families with heritable progranulin mutations, but gaps remain in our understanding of disease that may need to be resolved.
Diagnostic Progress and the Hurdle of Heterogeneity
Current Clinical, Imaging and Biochemical Diagnostics
Of equal importance in therapeutic consideration is the ability to diagnose individuals with specific disease. In this regard, neuropathology remains the dominant driver of disease mechanism theory and consequently therapeutic hypotheses, but determination of underlying FTLD-tau or FTLD-TDP early in the disease process will be essential in any therapeutic endeavor. As alluded to above, hypothetical therapies may also need to be designed to individual subtypes of TDP and tau pathology, if no singular process can be commonly delineated or effectively targeted. There are a number of diagnostic approaches under consideration that can be categorized as clinical, biological and those using imaging technologies.
Clinical-pathological studies of FTD have advanced rapidly, resulting in several changes in neuropathologic diagnostic and nosologic criteria for FTLD to reflect these findings (Cairns et al., 2007). There are probabilistic correlations between the syndromes and diseases underlying them, which invites some optimism in disease and subtype diagnosis in symptomatic individuals (Figure 2; Josephs et al., 2011). SD and FTD-MND are almost exclusively FTLD-TDP whereas PSPS and CBS are almost always characterized by tau pathology. PNFA and bvFTD are more neuropathologically heterogeneous, which is unfortunate given the high prevalence of bvFTD (in the largest clinicopathological study to date, bvFTD represented around 45% of FTD). Furthermore, in some cases there are correlations—albeit weaker ones—between specific subtypes of tau or TDP-43 pathology and clinical phenotype. For example, FTD-MND is not only purely FTLD-TDP but is approximately 70% type B and TDP-43 positive SD almost 85% type C (Josephs et al., 2011). Again, bvFTD is a much more complex syndrome in terms of either TDP-43 or tau subtype. However, one intriguing suggestion is that FTLD-TDP and FTLD-tau may affect distinct, distant brain networks and that detailed neuropsychological analysis may be used to differentiate the network—regardless of the syndrome—and hence pathology (Grossman et al., 2008; Listerud et al., 2009; Seeley et al., 2009).
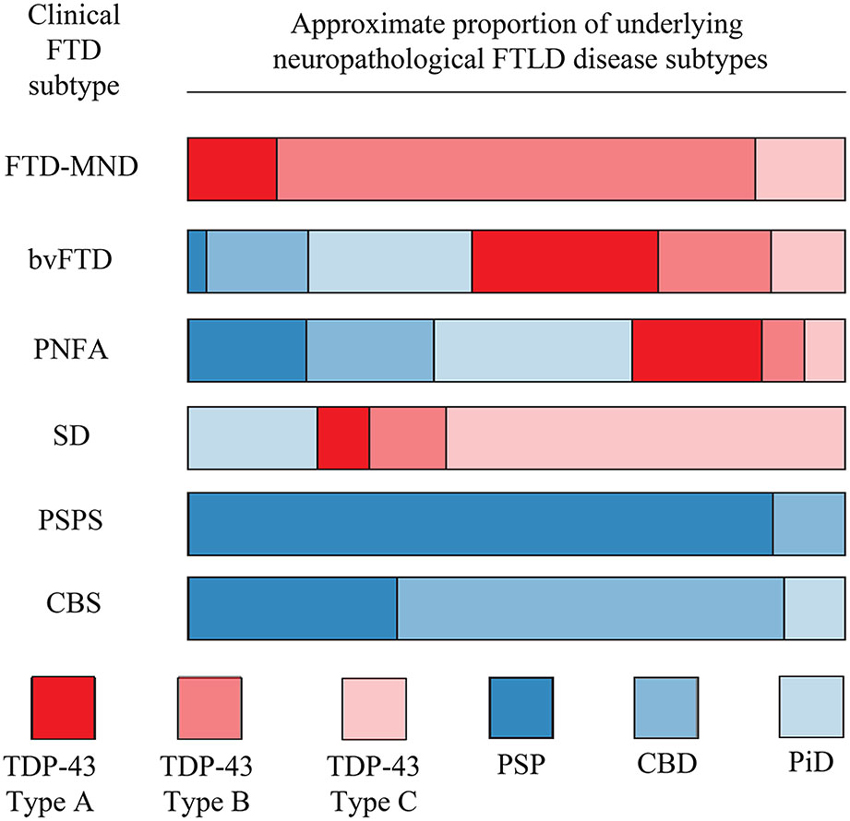
Figure 2. Relationship between clinical FTD phenotype and underlying FTLD disease neuropathology. For each clinical subtype on the left, the bar is divided to show the approximate proportion of cases which are underpinned by the indicated disease neuropathology. Profound neuropathological heterogeneity exists for many clinical subtypes, particularly bvFTD and PNFA. This figure is based on data from Josephs et al. (2011).
While such correlations are interesting on a population level, there is clear heterogeneity in disease/syndrome pairing and often overlap between syndromes, meaning there is currently minimal utility in diagnosing pathology in the clinic for individual patients based solely on phenotype. Furthermore, potentially the greatest issue for FTLD therapies—and neurodegenerative disease in general—is that of timing of intervention. Clinical trials in AD have generally reached the conclusion that the current pharmacological therapies in trial may require earlier administration if a clinical benefit is to be achieved. This may require studies in pre-symptomatic patients, as the appearance of pathology predates the manifestation of clinical symptoms by some time, potentially as much as 15–20 years (Golde et al., 2011; Bateman et al., 2012). Trials targeting earlier in AD now depend upon the use of diagnostic screening to identify patients at risk on the basis of preclinical disease biomarkers (Vellas et al., 2013). Although the length of presymptomatic phase may differ, this is distinctly probable in other chronic neurodegenerative diseases such as FTLD (Jacova et al., 2013), thus the early identification of people at risk of disease becomes crucial and will be underpinned by either biochemical and/or imaging techniques.
Antemortem neuroimaging analysis using the Aβ plaque binding radioligands florbetapir and Pittsburgh compound B have provided tremendous insight into pre-symptomatic disease progression in AD (Klunk et al., 2004; Bateman et al., 2012; Clark et al., 2012). Although the field currently lacks a substrate specific ligand for TDP-43, the recent development of a class of tau ligands will surely expedite studies in prodromal tauopathy patients (Maruyama et al., 2013). In the meantime, other imaging markers such as atrophy of specific brain regions are proving on the group level to correlate well with syndrome (Hu et al., 2011) and have been used, for example, to demonstrate that there is phenotypic heterogeneity linked to distinct atrophic patterns even within a subgroup such as bvFTD (Whitwell et al., 2009b). Specifically relating to disease prediction, results are currently inconclusive as to whether atrophy can predict FTLD-tau or FTLD-TDP pathotype (Grossman et al., 2007; Kim et al., 2007; Pereira et al., 2009; Whitwell et al., 2009a), although it appears that TDP-43 subtypes may be associated with distinct regional atrophy once FTLD-TDP is neuropathologically confirmed (Whitwell et al., 2010). These and other sophisticated techniques such as arterial spin labeling (Hu et al., 2010b) and 18F fluorodeoxyglucose positron emission tomography (Womack et al., 2011) may be useful in the future for diagnosing underlying disease or for following disease progression in therapeutic trials. However, as a diagnostic tool in screening a presymptomatic population, they are highly impractical due to expense and time consumption.
Ideally, the identification of biochemical biomarkers for the discrimination of FTLD-tau or FTLD-TDP would provide a more cost effective and less time consuming alternative to imaging and work has begun in probable cohorts based on syndromic presentation and pathologically confirmed cases of FTLD. Although TDP-43 appears to be raised in either plasma or CSF of clinically defined FTD or FTD-MND populations, there is significant overlap with control or AD subjects and it remains unclear if such an approach will prove clinically useful (Foulds et al., 2008; Steinacker et al., 2008). Similarly, results to date suggest that total tau or tau phosphorylated at threonine 181 may not be good candidates for discriminating FTLD-tau from control (Vanmechelen et al., 2000; Bian et al., 2008; Kapaki et al., 2008). Using unbiased proteomic approaches, it appears that FTLD-TDP and FTLD-tau might be distinguished from one another on the basis of several CSF analytes, although the specificity and reliability of these remain to be determined in larger studies (Hu et al., 2010a). It is also unknown whether the heterogeneity of TDP-43 pathology (subtypes A-C) and FTLD-tau (PiD, CBS, PSP) can be disaggregated at the biochemical level. For example, it appears PSP may have a unique truncated tau profile in CSF that separates it from other tau diseases (Borroni et al., 2009b) although this has not been replicated in additional, independent studies (Kuiperij and Verbeek, 2012).
Towards Multidimensional, Presymptomatic Diagnostics
While these studies represent early forays into biomarker evaluation, more powerful proteomic screening of CSF or plasma from clinically predicted FTLD-tau and FTLD-TDP may ultimately provide better biomarker possibilities and will require follow-up in larger, longitudinal cohorts to test their sensitivity and specificity. As such development occurs, it should do so with the heterogeneity of FTLD in mind and potentially involve other neurodegenerative diseases. Although one type of neuropathology may be dominant and one brain region afflicted more than others, cases of autopsy-confirmed FTLD may be further complicated by concomitant Aβ, vascular or alpha synuclein pathology (National Institute on Aging, 1997; McKeith et al., 2005; Toledo et al., 2013). This is already apparent in some biomarker studies in which perhaps as many as 30% of clinical FTLD patients are also AD biomarker positive (Schoonenboom et al., 2012) and in diagnostic imaging assays used for dementia with Lewy bodies (DLB) where there may be significant overlap with FTD (Morgan et al., 2012). In addition to AD and DLB, TDP-43 or phospho-TDP-43 pathology can be found co-occurring with tau pathology in cases of CBD, PSP, PiD and argyrophilic grain disease (Higashi et al., 2007; Uryu et al., 2008; Arai et al., 2009; Fujishiro et al., 2009; Rohn and Kokoulina, 2009; Yokota et al., 2010).
As a whole, these findings suggest that (1) clinical symptoms are not wholly reliable as an indicator of an underlying pathology or disease to be targeted for treatment; (2) an underlying disease pathology at autopsy is not a guaranteed predictor of a clinical phenotype; and (3) mixed pathologies and therefore diseases are often coexistent. Additionally, moving earlier in disease course towards “cognitively normal”—as will be the desired course for earlier administration of therapies—will increasingly blur the diagnostic line between “normal” and disease, creating greater chance of false positives or negatives. This “messy reality” of neurodegenerative disease means that a search for a single, definitive disease diagnosis in individuals will probably never achieve the same level of certitude as for diseases such as viral or bacterial agents. That being said, with the advancing sophistication of technology and data collection in medicine and the decreasing cost of genome sequencing, it is not unrealistic to envisage a combination of all or some of blood, CSF, and genetic “panels” that contain large numbers of analytes or alleles known to associate with different neurodegenerative pathology. Initial probable diagnoses could be refined by subsequent imaging studies. Such a multifaceted approach to disease diagnosis would certainly have far greater power than those relying on singular readouts, and may be the only way to discriminate early stage disease from “normal aging” as small deviations from the norm across multiple analytes synergize to create disease pattern readout (Figure 3). However, such an approach would still only return probabilistic diagnoses and has many considerations. Identifying individuals at risk who should undergo diagnostic examination is not a trivial issue. Determining who is screened, when, and how often, both in trials and using any eventual treatment identified is a priority. This may be based on a mixture of risk factors, including age, family history, common place biochemical screens, genetic risk alleles, or environmental risk factors that are currently unstudied (Onyike and Diehl-Schmid, 2013). Alternatively, suspending testing until clinical symptoms manifest may be too late in disease progression. Currently, we do not know at what stage intervention might be required and determining this by trial and error will be time consuming and financially costly. Furthermore, decades of unsuccessful clinical trials would also exact a human toll, and for this reason, it is important that social support resources be available to patients and families. Undoubtedly, clinical trials will necessitate great foresight and planning and such considerations will need to be debated prior to implementation of therapeutics.
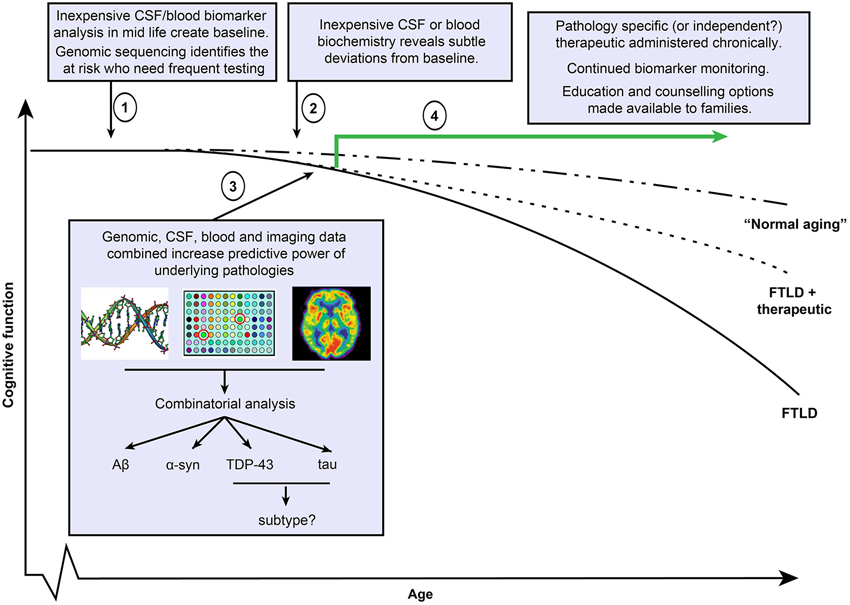
Figure 3. Potential FTLD diagnostic and therapeutic timeline. Clinical trials in Alzheimer’s disease (AD) suggest that therapeutics will be maximally effective when administered in early disease course. (1) Under this scenario, simple and economically viable blood or cerebrospinal fluid based screens at regular age intervals establish an individual’s biomarker status at cognitively healthy baseline and also with respect to general population. Screens include markers of all known disease pathologies including Aβ, α-syn, TDP-43 and tau. Inexpensive genomic sequencing may help to identify at risk individuals who require more regular evaluation. (2) As pathology (or multiple pathologies) progress, regular screening identifies deviations from the baseline that could potentially indicate early stage disease, triggering more frequent evaluations. (3) Continued biomarker positivity or progression justifies the use of more expensive imaging methods to determine regional atrophy or using pathology binding agents (such as florbetapir or as yet incompletely characterized tau binding agents). Such data, when combined with CSF, blood and potentially genetic alleles that associate with pathologies or sub types of pathology, may provide potentially more predictive power than a reliance on a single method. (4) Administration of single or multiple therapeutics depending on results, continued follow up evaluations to test response to treatment. As some individuals may be unresponsive to treatments, and future therapeutics may not entirely halt disease progression, social support structures including education and counselling for individuals and families should be integrated into care.
Conclusion
As outlined above, the ability to use biomarkers to identify specific subtypes of underlying FTLD-TDP or FTLD-tau in early stage disease (whether prodromal or asymptomatic phases) is a challenging issue, but one of paramount importance if the experiences of AD clinical trials in symptomatic patients also extrapolate to FTLD. Further complication arises regarding the complexity of disease. At the outset of studies focusing on tau, the prevailing theory was that removal of NFTs would provide a therapeutic breakthrough. Years on, that notion seems oversimplified and tauopathies are recognized as extremely complex, with an abundance of biochemical and structural tau alterations that may all, in some form or other, contribute to neuronal demise through mechanisms that are as yet not fully understood. Furthermore, such processes may not be a single linear mechanism, both in the formation of detrimental tau species and in their effect. It is inevitable that increased study will reveal an even greater complexity and this is highly probable for FTLD-TDP also. Although this means that we are increasingly knowledgeable about neurodegenerative mechanisms, it also suggests that these are multifactorial, multi-mechanistic diseases that current technology may find difficult to address. In this respect, it is possible that in addition to the “knowledge gaps” highlighted above, we may face somewhat of a “technology gap”. Compared to the myriad biochemical changes in tau, TDP-43 and other cellular processes, all of which occur in different cell types at various stages between healthy and degenerating, delivery of a single pharmaceutical compound aimed at a sole process or pathway to the whole brain may be oversimplified. The invention of novel technologies or use of multiple, simultaneous therapeutic strategies may be required. In the interim, care must be taken in the selection of therapeutics going forward as financing of trials for therapeutic interventions will not continue in aeternum if strategies based on unproven mechanisms fail repeatedly in clinical trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahlijanian, M. K., Barrezueta, N. X., Williams, R. D., Jakowski, A., Kowsz, K. P., McCarthy, S., et al. (2000). Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc. Natl. Acad. Sci. U S A 97, 2910–2915. doi: 10.1073/pnas.040577797
Ahmed, Z., Mackenzie, I. R., Hutton, M. L., and Dickson, D. W. (2007). Progranulin in frontotemporal lobar degeneration and neuroinflammation. J. Neuroinflammation 4:7. doi: 10.1186/1742-2094-4-7
Ahmed, Z., Sheng, H., Xu, Y. F., Lin, W. L., Innes, A. E., Gass, J., et al. (2010). Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 177, 311–324. doi: 10.2353/ajpath.2010.090915
Alonso, A. D., Grundke-Iqbal, I., Barra, H. S., and Iqbal, K. (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U S A 94, 298–303. doi: 10.1073/pnas.94.1.298
Alonso, A. C., Grundke-Iqbal, I., and Iqbal, K. (1996). Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 2, 783–787. doi: 10.1038/nm0796-783
Alonso Adel, C., Li, B., Grundke-Iqbal, I., and Iqbal, K. (2006). Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc. Natl. Acad. Sci. U S A 103, 8864–8869. doi: 10.1073/pnas.0603214103
Amador-Ortiz, C., Lin, W. L., Ahmed, Z., Personett, D., Davies, P., Duara, R., et al. (2007). TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 61, 435–445. doi: 10.1002/ana.21154
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454. doi: 10.1523/jneurosci.4637-04.2005
Arai, T., Mackenzie, I. R., Hasegawa, M., Nonoka, T., Niizato, K., Tsuchiya, K., et al. (2009). Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. 117, 125–136. doi: 10.1007/s00401-008-0480-1
Arnold, C. S., Johnson, G. V., Cole, R. N., Dong, D. L., Lee, M., and Hart, G. W. (1996). The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J. Biol. Chem. 271, 28741–28744. doi: 10.1074/jbc.271.46.28741
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Asuni, A. A., Boutajangout, A., Quartermain, D., and Sigurdsson, E. M. (2007). Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 27, 9115–9129. doi: 10.1523/jneurosci.2361-07.2007
Ayala, V., Granado-Serrano, A. B., Cacabelos, D., Naudi, A., Ilieva, E. V., Boada, J., et al. (2011). Cell stress induces TDP-43 pathological changes associated with ERK1/2 dysfunction: implications in ALS. Acta Neuropathol. 122, 259–270. doi: 10.1007/s00401-011-0850-y
Ayala, Y. M., Misteli, T., and Baralle, F. E. (2008a). TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc. Natl. Acad. Sci. U S A 105, 3785–3789. doi: 10.1073/pnas.0800546105
Ayala, Y. M., Zago, P., D’ambrogio, A., Xu, Y. F., Petrucelli, L., Buratti, E., et al. (2008b). Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785. doi: 10.1242/jcs.038950
Baborie, A., Griffiths, T. D., Jaros, E., Mckeith, I. G., Burn, D. J., Richardson, A., et al. (2011). Pathological correlates of frontotemporal lobar degeneration in the elderly. Acta Neuropathol. 121, 365–371. doi: 10.1007/s00401-010-0765-z
Bailey, R. M., Covy, J. P., Melrose, H. L., Rousseau, L., Watkinson, R., Knight, J., et al. (2013). LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol. 126, 809–827. doi: 10.1007/s00401-013-1188-4
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. doi: 10.1038/nature05016
Banks, S. J., and Weintraub, S. (2008). Neuropsychiatric symptoms in behavioral variant frontotemporal dementia and primary progressive aphasia. J. Geriatr. Psychiatry Neurol. 21, 133–141. doi: 10.1177/0891988708316856
Barmada, S. J., Skibinski, G., Korb, E., Rao, E. J., Wu, J. Y., and Finkbeiner, S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 30, 639–649. doi: 10.1523/JNEUROSCI.4988-09.2010
Bateman, R. J., Xiong, C., Benzinger, T. L., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Baumann, K., Mandelkow, E. M., Biernat, J., Piwnica-Worms, H., and Mandelkow, E. (1993). Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 336, 417–424. doi: 10.1016/0014-5793(93)80849-p
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Berger, Z., Roder, H., Hanna, A., Carlson, A., Rangachari, V., Yue, M., et al. (2007). Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 27, 3650–3662. doi: 10.1523/jneurosci.0587-07.2007
Berry, R. W., Sweet, A. P., Clark, F. A., Lagalwar, S., Lapin, B. R., Wang, T., et al. (2004). Tau epitope display in progressive supranuclear palsy and corticobasal degeneration. J. Neurocytol. 33, 287–295. doi: 10.1023/b:neur.0000044190.96426.b9
Bian, F., Nath, R., Sobocinski, G., Booher, R. N., Lipinski, W. J., Callahan, M. J., et al. (2002). Axonopathy, tau abnormalities and dyskinesia, but no neurofibrillary tangles in p25-transgenic mice. J. Comp. Neurol. 446, 257–266. doi: 10.1002/cne.10186
Bian, H., Van Swieten, J. C., Leight, S., Massimo, L., Wood, E., Forman, M., et al. (2008). CSF biomarkers in frontotemporal lobar degeneration with known pathology. Neurology 70, 1827–1835. doi: 10.1212/01.wnl.0000311445.21321.fc
Bigio, E. H., Lipton, A. M., Yen, S. H., Hutton, M. L., Baker, M., Nacharaju, P., et al. (2001). Frontal lobe dementia with novel tauopathy: sporadic multiple system tauopathy with dementia. J. Neuropathol. Exp. Neurol. 60, 328–341.
Bilican, B., Serio, A., Barmada, S. J., Nishimura, A. L., Sullivan, G. J., Carrasco, M., et al. (2012). Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. U S A 109, 5803–5808. doi: 10.1073/pnas.1202922109
Blair, L. J., Nordhues, B. A., Hill, S. E., Scaglione, K. M., O’leary, J. C. 3rd, Fontaine, S. N., et al. (2013). Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Invest. 123, 4158–4169. doi: 10.1172/JCI69003
Boeve, B. F., Lang, A. E., and Litvan, I. (2003). Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann. Neurol. 54(Suppl. 5), S15–S19. doi: 10.1002/ana.10570
Borroni, B., Archetti, S., Del Bo, R., Papetti, A., Buratti, E., Bonvicini, C., et al. (2010). TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features and disease course. Rejuvenation Res. 13, 509–517. doi: 10.1089/rej.2010.1017
Borroni, B., Bonvicini, C., Alberici, A., Buratti, E., Agosti, C., Archetti, S., et al. (2009a). Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 30, E974–E983. doi: 10.1002/humu.21100
Borroni, B., Ferrari, F., Galimberti, D., Nacmias, B., Barone, C., Bagnoli, S., et al. (2014). Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 35, 934.e7–934.e10. doi: 10.1016/j.neurobiolaging.2013.09.017
Borroni, B., Gardoni, F., Parnetti, L., Magno, L., Malinverno, M., Saggese, E., et al. (2009b). Pattern of Tau forms in CSF is altered in progressive supranuclear palsy. Neurobiol. Aging 30, 34–40. doi: 10.1016/j.neurobiolaging.2007.05.009
Bose, J. K., Huang, C. C., and Shen, C. K. (2011). Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 286, 44441–44448. doi: 10.1074/jbc.M111.237115
Boxer, A. L., and Boeve, B. F. (2007). Frontotemporal dementia treatment: current symptomatic therapies and implications of recent genetic, biochemical and neuroimaging studies. Alzheimer Dis. Assoc. Disord. 21, S79–S87. doi: 10.1097/wad.0b013e31815c345e
Boxer, A. L., Knopman, D. S., Kaufer, D. I., Grossman, M., Onyike, C., Graf-Radford, N., et al. (2013). Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 12, 149–156. doi: 10.1016/S1474-4422(12)70320-4
Boxer, A. L., Lang, A. E., Grossman, M., Knopman, D. S., Miller, B. L., Schneider, L. S., et al. (2014). Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 13, 676–685. doi: 10.1016/S1474-4422(14)70088-2
Braak, H., and Braak, E. (1987). Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci. Lett. 76, 124–127. doi: 10.1016/0304-3940(87)90204-7
Braak, H., and Braak, E. (1989). Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol. Appl. Neurobiol. 15, 13–26. doi: 10.1111/j.1365-2990.1989.tb01146.x
Bronner, I. F., Rizzu, P., Seelaar, H., van Mil, S. E., Anar, B., Azmani, A., et al. (2007). Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur. J. Hum. Genet. 15, 369–374. doi: 10.1038/sj.ejhg.5201772
Brouwers, N., Sleegers, K., Engelborghs, S., Maurer-Stroh, S., Gijselinck, I., van der Zee, J., et al. (2008). Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71, 656–664. doi: 10.1212/01.wnl.0000319688.89790.7a
Brunden, K. R., Yao, Y., Potuzak, J. S., Ferrer, N. I., Ballatore, C., James, M. J., et al. (2010a). The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol. Res. 63, 341–351. doi: 10.1016/j.phrs.2010.12.002
Brunden, K. R., Zhang, B., Carroll, J., Yao, Y., Potuzak, J. S., Hogan, A. M., et al. (2010b). Epothilone D improves microtubule density, axonal integrity and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 30, 13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010
Bruni, A. C., Momeni, P., Bernardi, L., Tomaino, C., Frangipane, F., Elder, J., et al. (2007). Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology 69, 140–147. doi: 10.1212/01.wnl.0000265220.64396.b4
Buratti, E., and Baralle, F. E. (2001). Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 276, 36337–36343. doi: 10.1074/jbc.m104236200
Butner, K. A., and Kirschner, M. W. (1991). Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 115, 717–730. doi: 10.1083/jcb.115.3.717
Cairns, N. J., Bigio, E. H., Mackenzie, I. R., Neumann, M., Lee, V. M., Hatanpaa, K. J., et al. (2007). Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol. 114, 5–22. doi: 10.1007/s00401-007-0237-2
Canu, N., Dus, L., Barbato, C., Ciotti, M. T., Brancolini, C., Rinaldi, A. M., et al. (1998). Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J. Neurosci. 18, 7061–7074.
Capell, A., Liebscher, S., Fellerer, K., Brouwers, N., Willem, M., Lammich, S., et al. (2011). Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J. Neurosci. 31, 1885–1894. doi: 10.1523/JNEUROSCI.5757-10.2011
Castillo-Carranza, D. L., Sengupta, U., Guerrero-Munoz, M. J., Lasagna-Reeves, C. A., Gerson, J. E., Singh, G., et al. (2014). Passive immunization with tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J. Neurosci. 34, 4260–4272. doi: 10.1523/JNEUROSCI.3192-13.2014
Cenik, B., Sephton, C. F., Dewey, C. M., Xian, X., Wei, S., Yu, K., et al. (2011). Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J. Biol. Chem. 286, 16101–16108. doi: 10.1074/jbc.M110.193433
Cenik, B., Sephton, C. F., Kutluk Cenik, B., Herz, J., and Yu, G. (2012). Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J. Biol. Chem. 287, 32298–32306. doi: 10.1074/jbc.r112.399170
Che, M. X., Jiang, Y. J., Xie, Y. Y., Jiang, L. L., and Hu, H. Y. (2011). Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J. 25, 2344–2353. doi: 10.1096/fj.10-174482
Chen, X., Chang, J., Deng, Q., Xu, J., Nguyen, T. A., Martens, L. H., et al. (2013). Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J. Neurosci. 33, 9202–9213. doi: 10.1523/JNEUROSCI.5336-12.2013
Chiang, P. M., Ling, J., Jeong, Y. H., Price, D. L., Aja, S. M., and Wong, P. C. (2010). Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity and alters body fat metabolism. Proc. Natl. Acad. Sci. U S A 107, 16320–16324. doi: 10.1073/pnas.1002176107
Clark, C. M., Pontecorvo, M. J., Beach, T. G., Bedell, B. J., Coleman, R. E., Doraiswamy, P. M., et al. (2012). Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 11, 669–678. doi: 10.1016/s1474-4422(12)70142-4
Cleveland, D. W., Hwo, S. Y., and Kirschner, M. W. (1977). Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 116, 207–225. doi: 10.1016/0022-2836(77)90213-3
Cruchaga, C., Graff, C., Chiang, H. H., Wang, J., Hinrichs, A. L., Spiegel, N., et al. (2011). Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch. Neurol. 68, 581–586. doi: 10.1001/archneurol.2010.350
Cruts, M., Gijselinck, I., van der Zee, J., Engelborghs, S., Wils, H., Pirici, D., et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. doi: 10.1038/nature05017
Cuyvers, E., Bettens, K., Philtjens, S., Van Langenhove, T., Gijselinck, I., van der Zee, J., et al. (2014). Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 35, 726.e711–726.e729. doi: 10.1016/j.neurobiolaging.2013.09.009
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Delacourte, A., Sergeant, N., Wattez, A., Gauvreau, D., and Robitaille, Y. (1998). Vulnerable neuronal subsets in Alzheimer’s and pick’s disease are distinguished by their tau isoform distribution and phosphorylation. Ann. Neurol. 43, 193–204. doi: 10.1002/ana.410430209
del Ser, T., Steinwachs, K. C., Gertz, H. J., Andres, M. V., Gomez-Carrillo, B., Medina, M., et al. (2013). Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J. Alzheimers Dis. 33, 205–215. doi: 10.3233/JAD-2012-120805
De Muynck, L., Herdewyn, S., Beel, S., Scheveneels, W., Van Den Bosch, L., Robberecht, W., et al. (2013). The neurotrophic properties of progranulin depend on the granulin E domain but do not require sortilin binding. Neurobiol. Aging 34, 2541–2547. doi: 10.1016/j.neurobiolaging.2013.04.022
de Silva, R., Lashley, T., Strand, C., Shiarli, A. M., Shi, J., Tian, J., et al. (2006). An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol. 111, 329–340. doi: 10.1007/s00401-006-0048-x
Dickson, D. W. (1998). Pick’s disease: a modern approach. Brain Pathol. 8, 339–354. doi: 10.1111/j.1750-3639.1998.tb00158.x
Dickson, D. W., Bergeron, C., Chin, S. S., Duyckaerts, C., Horoupian, D., Ikeda, K., et al. (2002). Office of rare diseases neuropathologic criteria for corticobasal degeneration. J. Neuropathol. Exp. Neurol. 61, 935–946.
Dickson, D. W., Feany, M. B., Yen, S. H., Mattiace, L. A., and Davies, P. (1996). Cytoskeletal pathology in non-Alzheimer degenerative dementia: new lesions in diffuse Lewy body disease, pick’s disease and corticobasal degeneration. J. Neural Transm. Suppl. 47, 31–46. doi: 10.1007/978-3-7091-6892-9_2
Donnelly, C. J., Zhang, P. W., Pham, J. T., Heusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.055
Dormann, D., Capell, A., Carlson, A. M., Shankaran, S. S., Rodde, R., Neumann, M., et al. (2009). Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J. Neurochem. 110, 1082–1094. doi: 10.1111/j.1471-4159.2009.06211.x
Drechsel, D. N., Hyman, A. A., Cobb, M. H., and Kirschner, M. W. (1992). Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 3, 1141–1154. doi: 10.1091/mbc.3.10.1141
Drewes, G., Ebneth, A., Preuss, U., Mandelkow, E. M., and Mandelkow, E. (1997). MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 89, 297–308. doi: 10.1016/s0092-8674(00)80208-1
D’Souza, I., Poorkaj, P., Hong, M., Nochlin, D., Lee, V. M., Bird, T. D., et al. (1999). Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. U S A 96, 5598–5603. doi: 10.1073/pnas.96.10.5598
Duan, W., Li, X., Shi, J., Guo, Y., Li, Z., and Li, C. (2010). Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 169, 1621–1629. doi: 10.1016/j.neuroscience.2010.06.018
Egashira, Y., Suzuki, Y., Azuma, Y., Takagi, T., Mishiro, K., Sugitani, S., et al. (2013). The growth factor progranulin attenuates neuronal injury induced by cerebral ischemia-reperfusion through the suppression of neutrophil recruitment. J. Neuroinflammation 10:105. doi: 10.1186/1742-2094-10-105
Egawa, N., Kitaoka, S., Tsukita, K., Naitoh, M., Takahashi, K., Yamamoto, T., et al. (2012). Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4:145ra104. doi: 10.1126/scitranslmed.3004052
Fang, H., Zhang, L. F., Meng, F. T., Du, X., and Zhou, J. N. (2010). Acute hypoxia promote the phosphorylation of tau via ERK pathway. Neurosci. Lett. 474, 173–177. doi: 10.1016/j.neulet.2010.03.037
Fath, T., Eidenmuller, J., and Brandt, R. (2002). Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. J. Neurosci. 22, 9733–9741.
Feiguin, F., Godena, V. K., Romano, G., D’ambrogio, A., Klima, R., and Baralle, F. E. (2009). Depletion of TDP-43 affects drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592. doi: 10.1016/j.febslet.2009.04.019
Ferrari, R., Hernandez, D. G., Nalls, M. A., Rohrer, J. D., Ramasamy, A., Kwok, J. B., et al. (2014). Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 13, 686–699. doi: 10.1016/S1474-4422(14)70065-1
Ferreira, A., and Bigio, E. H. (2011). Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol. Med. 17, 676–685. doi: 10.2119/molmed.2010.00220
Finch, N., Carrasquillo, M. M., Baker, M., Rutherford, N. J., Coppola, G., Dejesus-Hernandez, M., et al. (2011). TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76, 467–474. doi: 10.1212/WNL.0b013e31820a0e3b
Forlenza, O. V., Diniz, B. S., Radanovic, M., Santos, F. S., Talib, L. L., and Gattaz, W. F. (2011). Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br. J. Psychiatry 198, 351–356. doi: 10.1192/bjp.bp.110.080044
Forman, M. S., MacKenzie, I. R., Cairns, N. J., Swanson, E., Boyer, P. J., Drachman, D. A., et al. (2006). Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J. Neuropathol. Exp. Neurol. 65, 571–581. doi: 10.1097/00005072-200606000-00005
Foulds, P., McAuley, E., Gibbons, L., Davidson, Y., Pickering-Brown, S. M., Neary, D., et al. (2008). TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol. 116, 141–146. doi: 10.1007/s00401-008-0389-8
Fujishiro, H., Uchikado, H., Arai, T., Hasegawa, M., Akiyama, H., Yokota, O., et al. (2009). Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol. 117, 151–158. doi: 10.1007/s00401-008-0463-2
Furukawa, Y., Kaneko, K., and Nukina, N. (2011a). Molecular properties of TAR DNA binding protein-43 fragments are dependent upon its cleavage site. Biochim. Biophys. Acta 1812, 1577–1583. doi: 10.1016/j.bbadis.2011.09.005
Furukawa, Y., Kaneko, K., Watanabe, S., Yamanaka, K., and Nukina, N. (2011b). A seeding reaction recapitulates intracellular formation of sarkosyl-insoluble TAR DNA binding protein-43 inclusions. J. Biol. Chem. 286, 18664–18672. doi: 10.1074/jbc.M111.231209
Galimberti, D., Fenoglio, C., Cortini, F., Serpente, M., Venturelli, E., Villa, C., et al. (2010). GRN variability contributes to sporadic frontotemporal lobar degeneration. J. Alzheimers Dis. 19, 171–177. doi: 10.3233/JAD-2010-1225
Gass, J., Cannon, A., Mackenzie, I. R., Boeve, B., Baker, M., Adamson, J., et al. (2006). Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 15, 2988–3001. doi: 10.1093/hmg/ddl241
Gass, J., Lee, W. C., Cook, C., Finch, N., Stetler, C., Jansen-West, K., et al. (2012). Progranulin regulates neuronal outgrowth independent of sortilin. Mol. Neurodegener. 7:33. doi: 10.1186/1750-1326-7-33
Gendron, T. F., Bieniek, K. F., Zhang, Y. J., Jansen-West, K., Ash, P. E., Caulfield, T., et al. (2013). Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844. doi: 10.1007/s00401-013-1192-8
Gendron, T. F., and Petrucelli, L. (2009). The role of tau in neurodegeneration. Mol. Neurodegener. 4:13. doi: 10.1186/1750-1326-4-13
Geser, F., Lee, V. M., and Trojanowski, J. Q. (2010). Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology 30, 103–112. doi: 10.1111/j.1440-1789.2009.01091.x
Giraldo, M., Lopera, F., Siniard, A. L., Corneveaux, J. J., Schrauwen, I., Carvajal, J., et al. (2013). Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer’s disease. Neurobiol. Aging 34, 2077.e2011–2077.e2078. doi: 10.1016/j.neurobiolaging.2013.02.016
Göbel, M., Eisele, L., Mollmann, M., Huttmann, A., Johansson, P., Scholtysik, R., et al. (2013). Progranulin is a novel independent predictor of disease progression and overall survival in chronic lymphocytic leukemia. PLoS One 8:e72107. doi: 10.1371/journal.pone.0072107
Goedert, M. (2005). Tau gene mutations and their effects. Mov. Disord. 20(Suppl. 12), S45–S52. doi: 10.1002/mds.20539
Goedert, M., and Jakes, R. (1990). Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230.
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., and Crowther, R. A. (1989). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399.
Golde, T. E., Schneider, L. S., and Koo, E. H. (2011). Anti-aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron 69, 203–213. doi: 10.1016/j.neuron.2011.01.002
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Grossman, M., Libon, D. J., Forman, M. S., Massimo, L., Wood, E., Moore, P., et al. (2007). Distinct antemortem profiles in patients with pathologically defined frontotemporal dementia. Arch. Neurol. 64, 1601–1609. doi: 10.1001/archneur.64.11.1601
Grossman, M., Xie, S. X., Libon, D. J., Wang, X., Massimo, L., Moore, P., et al. (2008). Longitudinal decline in autopsy-defined frontotemporal lobar degeneration. Neurology 70, 2036–2045. doi: 10.1212/01.wnl.0000303816.25065.bc
Guerreiro, R. J., Lohmann, E., Bras, J. M., Gibbs, J. R., Rohrer, J. D., Gurunlian, N., et al. (2013). Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 70, 78–84. doi: 10.1001/jamaneurol.2013.579
Guo, W., Chen, Y., Zhou, X., Kar, A., Ray, P., Chen, X., et al. (2011). An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 18, 822–830. doi: 10.1038/nsmb.2053
Hampel, H., Ewers, M., Burger, K., Annas, P., Mortberg, A., Bogstedt, A., et al. (2009). Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931. doi: 10.4088/jcp.08m04606
Harada, A., Oguchi, K., Okabe, S., Kuno, J., Terada, S., Ohshima, T., et al. (1994). Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369, 488–491. doi: 10.1038/369488a0
Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70. doi: 10.1002/ana.21425
Hauw, J. J., Daniel, S. E., Dickson, D., Horoupian, D. S., Jellinger, K., Lantos, P. L., et al. (1994). Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44, 2015–2019. doi: 10.1212/WNL.44.11.2015
He, Z., Ong, C. H., Halper, J., and Bateman, A. (2003). Progranulin is a mediator of the wound response. Nat. Med. 9, 225–229. doi: 10.1038/nm816
Higashi, S., Iseki, E., Yamamoto, R., Minegishi, M., Hino, H., Fujisawa, K., et al. (2007). Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 1184, 284–294. doi: 10.1016/j.brainres.2007.09.048
Holm, I. E., Isaacs, A. M., and Mackenzie, I. R. (2009). Absence of FUS-immunoreactive pathology in frontotemporal dementia linked to chromosome 3 (FTD-3) caused by mutation in the CHMP2B gene. Acta Neuropathol. 118, 719–720. doi: 10.1007/s00401-009-0593-1
Horiguchi, T., Uryu, K., Giasson, B. I., Ischiropoulos, H., Lightfoot, R., Bellmann, C., et al. (2003). Nitration of tau protein is linked to neurodegeneration in tauopathies. Am. J. Pathol. 163, 1021–1031. doi: 10.1016/s0002-9440(10)63462-1
Hu, W. T., Chen-Plotkin, A., Grossman, M., Arnold, S. E., Clark, C. M., Shaw, L. M., et al. (2010a). Novel CSF biomarkers for frontotemporal lobar degenerations. Neurology 75, 2079–2086. doi: 10.1212/WNL.0b013e318200d78d
Hu, W. T., Trojanowski, J. Q., and Shaw, L. M. (2011). Biomarkers in frontotemporal lobar degenerations–progress and challenges. Prog. Neurobiol. 95, 636–648. doi: 10.1016/j.pneurobio.2011.04.012
Hu, W. T., Wang, Z., Lee, V. M., Trojanowski, J. Q., Detre, J. A., and Grossman, M. (2010b). Distinct cerebral perfusion patterns in FTLD and AD. Neurology 75, 881–888. doi: 10.1212/WNL.0b013e3181f11e35
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Hwang, S. C., Jhon, D. Y., Bae, Y. S., Kim, J. H., and Rhee, S. G. (1996). Activation of phospholipase C-gamma by the concerted action of tau proteins and arachidonic acid. J. Biol. Chem. 271, 18342–18349. doi: 10.1074/jbc.271.31.18342
Igaz, L. M., Kwong, L. K., Chen-Plotkin, A., Winton, M. J., Unger, T. L., Xu, Y., et al. (2009). Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J. Biol. Chem. 284, 8516–8524. doi: 10.1074/jbc.m809462200
Igaz, L. M., Kwong, L. K., Xu, Y., Truax, A. C., Uryu, K., Neumann, M., et al. (2008). Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 173, 182–194. doi: 10.2353/ajpath.2008.080003
Iguchi, Y., Katsuno, M., Niwa, J., Yamada, S., Sone, J., Waza, M., et al. (2009). TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J. Biol. Chem. 284, 22059–22066. doi: 10.1074/jbc.M109.012195
Jackman, K., Kahles, T., Lane, D., Garcia-Bonilla, L., Abe, T., Capone, C., et al. (2013). Progranulin deficiency promotes post-ischemic blood-brain barrier disruption. J. Neurosci. 33, 19579–19589. doi: 10.1523/JNEUROSCI.4318-13.2013
Jacova, C., Hsiung, G. Y., Tawankanjanachot, I., Dinelle, K., McCormick, S., Gonzalez, M., et al. (2013). Anterior brain glucose hypometabolism predates dementia in progranulin mutation carriers. Neurology 81, 1322–1331. doi: 10.1212/WNL.0b013e3182a8237e
Jiang, T., Yu, J. T., Zhu, X. C., Zhang, Q. Q., Cao, L., Wang, H. F., et al. (2014). Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 85C, 121–130. doi: 10.1016/j.neuropharm.2014.05.032
Joachim, C. L., Morris, J. H., Kosik, K. S., and Selkoe, D. J. (1987). Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann. Neurol. 22, 514–520. doi: 10.1002/ana.410220411
Johnson, B. S., Snead, D., Lee, J. J., McCaffery, J. M., Shorter, J., and Gitler, A. D. (2009). TDP-43 is intrinsically aggregation-prone and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339. doi: 10.1074/jbc.m109.010264
Josephs, K. A. (2008). Frontotemporal dementia and related disorders: deciphering the enigma. Ann. Neurol. 64, 4–14. doi: 10.1002/ana.21426
Josephs, K. A., Hodges, J. R., Snowden, J. S., Mackenzie, I. R., Neumann, M., Mann, D. M., et al. (2011). Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 122, 137–153. doi: 10.1007/s00401-011-0839-6
Kabashi, E., Lin, L., Tradewell, M. L., Dion, P. A., Bercier, V., Bourgouin, P., et al. (2010). Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 19, 671–683. doi: 10.1093/hmg/ddp534
Kao, A. W., Eisenhut, R. J., Martens, L. H., Nakamura, A., Huang, A., Bagley, J. A., et al. (2011). A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. U S A 108, 4441–4446. doi: 10.1073/pnas.1100650108
Kapaki, E., Paraskevas, G. P., Papageorgiou, S. G., Bonakis, A., Kalfakis, N., Zalonis, I., et al. (2008). Diagnostic value of CSF biomarker profile in frontotemporal lobar degeneration. Alzheimer Dis. Assoc. Disord. 22, 47–53. doi: 10.1097/WAD.0b013e3181610fea
Keck, S., Nitsch, R., Grune, T., and Ullrich, O. (2003). Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 85, 115–122. doi: 10.1046/j.1471-4159.2003.01642.x
Kelly, C. M., and Buzdar, A. U. (2013). Using multiple targeted therapies in oncology: considerations for use and progress to date in breast cancer. Drugs 73, 505–515. doi: 10.1007/s40265-013-0044-0
Kessenbrock, K., Frohlich, L., Sixt, M., Lammermann, T., Pfister, H., Bateman, A., et al. (2008). Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J. Clin. Invest. 118, 2438–2447. doi: 10.1172/JCI34694
Kim, E. J., Rabinovici, G. D., Seeley, W. W., Halabi, C., Shu, H., Weiner, M. W., et al. (2007). Patterns of MRI atrophy in tau positive and ubiquitin positive frontotemporal lobar degeneration. J. Neurol. Neurosurg. Psychiatry 78, 1375–1378. doi: 10.1136/jnnp.2006.114231
Klunk, W. E., Engler, H., Nordberg, A., Wang, Y., Blomqvist, G., Holt, D. P., et al. (2004). Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann. Neurol. 55, 306–319. doi: 10.1002/ana.20009
Kosaka, K. (1994). Diffuse neurofibrillary tangles with calcification: a new presenile dementia. J. Neurol. Neurosurg. Psychiatry 57, 594–596. doi: 10.1136/jnnp.57.5.594
Kosik, K. S., Orecchio, L. D., Bakalis, S., and Neve, R. L. (1989). Developmentally regulated expression of specific tau sequences. Neuron 2, 1389–1397. doi: 10.1016/0896-6273(89)90077-9
Kraemer, B. C., Schuck, T., Wheeler, J. M., Robinson, L. C., Trojanowski, J. Q., Lee, V. M., et al. (2010). Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 119, 409–419. doi: 10.1007/s00401-010-0659-0
Kril, J. J., Macdonald, V., Patel, S., Png, F., and Halliday, G. M. (2005). Distribution of brain atrophy in behavioral variant frontotemporal dementia. J. Neurol. Sci. 232, 83–90. doi: 10.1016/j.jns.2005.02.003
Kuchibhotla, K. V., Wegmann, S., Kopeikina, K. J., Hawkes, J., Rudinskiy, N., Andermann, M. L., et al. (2014). Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. U S A 111, 510–514. doi: 10.1073/pnas.1318807111
Kuiperij, H. B., and Verbeek, M. M. (2012). Diagnosis of progressive supranuclear palsy: can measurement of tau forms help? Neurobiol. Aging 33, 204.e17–204.e18. doi: 10.1016/j.neurobiolaging.2010.08.011
Laird, A. S., Van Hoecke, A., De Muynck, L., Timmers, M., Van Den Bosch, L., Van Damme, P., et al. (2010). Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One 5:e13368. doi: 10.1371/journal.pone.0013368
Lattante, S., Le Ber, I., Camuzat, A., Dayan, S., Godard, C., Van Bortel, I., et al. (2013). TREM2 mutations are rare in a French cohort of patients with frontotemporal dementia. Neurobiol. Aging 34, 2443.e2441–2443.e2442. doi: 10.1016/j.neurobiolaging.2013.04.030
Le Ber, I., De Septenville, A., Guerreiro, R., Bras, J., Camuzat, A., Caroppo, P., et al. (2014). Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 35, 2419.e23–2419.e25. doi: 10.1016/j.neurobiolaging.2014.04.010
Le Corre, S., Klafki, H. W., Plesnila, N., Hubinger, G., Obermeier, A., Sahagun, H., et al. (2006). An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl. Acad. Sci. U S A 103, 9673–9678. doi: 10.1073/pnas.0602913103
Ledesma, M. D., Bonay, P., Colaco, C., and Avila, J. (1994). Analysis of microtubule-associated protein tau glycation in paired helical filaments. J. Biol. Chem. 269, 21614–21619.
Lee, W. C., Almeida, S., Prudencio, M., Caulfield, T. R., Zhang, Y. J., Tay, W. M., et al. (2013a). Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Hum. Mol. Genet. 23, 1467–1478. doi: 10.1093/hmg/ddt534
Lee, Y. B., Chen, H. J., Peres, J. N., Gomez-Deza, J., Attig, J., Stalekar, M., et al. (2013b). Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins and are neurotoxic. Cell Rep. 5, 1178–1186. doi: 10.1016/j.celrep.2013.10.049
Leggett, C., McGehee, D. S., Mastrianni, J., Yang, W., Bai, T., and Brorson, J. R. (2012). Tunicamycin produces TDP-43 cytoplasmic inclusions in cultured brain organotypic slices. J. Neurol. Sci. 317, 66–73. doi: 10.1016/j.jns.2012.02.027
Lin, W. L., Lewis, J., Yen, S. H., Hutton, M., and Dickson, D. W. (2003). Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J. Neurocytol. 32, 1091–1105. doi: 10.1023/b:neur.0000021904.61387.95
Ling, S. C., Albuquerque, C. P., Han, J. S., Lagier-Tourenne, C., Tokunaga, S., Zhou, H., et al. (2010). ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. U S A 107, 13318–13323. doi: 10.1073/pnas.1008227107
Listerud, J., Powers, C., Moore, P., Libon, D. J., and Grossman, M. (2009). Neuropsychological patterns in magnetic resonance imaging-defined subgroups of patients with degenerative dementia. J. Int. Neuropsychol. Soc. 15, 459–470. doi: 10.1017/s1355617709090742
Liu, F., Iqbal, K., Grundke-Iqbal, I., Hart, G. W., and Gong, C. X. (2004). O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 101, 10804–10809. doi: 10.1073/pnas.0400348101
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderwyde, T., Citro, A., Mehta, T., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. doi: 10.3410/f.12796966.14073066
MacDonald, A., Briggs, K., Poppe, M., Higgins, A., Velayudhan, L., and Lovestone, S. (2008). A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 23, 704–711. doi: 10.1002/gps.1964
Mackenzie, I. R., Baker, M., Pickering-Brown, S., Hsiung, G. Y., Lindholm, C., Dwosh, E., et al. (2006). The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129, 3081–3090. doi: 10.1093/brain/awl271
Mackenzie, I. R., Neumann, M., Baborie, A., Sampathu, D. M., Du Plessis, D., Jaros, E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113. doi: 10.1007/s00401-011-0845-8
Maeda, S., Sahara, N., Saito, Y., Murayama, M., Yoshiike, Y., Kim, H., et al. (2007). Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861. doi: 10.1021/bi061359o
Marczinski, C. A., Davidson, W., and Kertesz, A. (2004). A longitudinal study of behavior in frontotemporal dementia and primary progressive aphasia. Cogn. Behav. Neurol. 17, 185–190.
Martens, L. H., Zhang, J., Barmada, S. J., Zhou, P., Kamiya, S., Sun, B., et al. (2012). Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest. 122, 3955–3959. doi: 10.1172/JCI63113
Martin, L., Latypova, X., Wilson, C. M., Magnaudeix, A., Perrin, M. L., Yardin, C., et al. (2013). Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res. Rev. 12, 289–309. doi: 10.1016/j.arr.2012.06.003
Maruyama, M., Shimada, H., Suhara, T., Shinotoh, H., Ji, B., Maeda, J., et al. (2013). Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 79, 1094–1108. doi: 10.1016/j.neuron.2013.07.037
Matsumura, N., Mandai, M., Miyanishi, M., Fukuhara, K., Baba, T., Higuchi, T., et al. (2006). Oncogenic property of acrogranin in human uterine leiomyosarcoma: direct evidence of genetic contribution in in vivo tumorigenesis. Clin. Cancer Res. 12, 1402–1411. doi: 10.1158/1078-0432.ccr-05-2003
McDonald, K. K., Aulas, A., Destroismaisons, L., Pickles, S., Beleac, E., Camu, W., et al. (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410. doi: 10.1093/hmg/ddr021
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’brien, J. T., Feldman, H., et al. (2005). Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65, 1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1
McKnight, R. F., Adida, M., Budge, K., Stockton, S., Goodwin, G. M., and Geddes, J. R. (2012). Lithium toxicity profile: a systematic review and meta-analysis. Lancet 379, 721–728. doi: 10.1016/S0140-6736(11)61516-X
Melov, S., Adlard, P. A., Morten, K., Johnson, F., Golden, T. R., Hinerfeld, D., et al. (2007). Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2:e536. doi: 10.1371/journal.pone.0000536
Mendez, M. F., Shapira, J. S., McMurtray, A., and Licht, E. (2007). Preliminary findings: behavioral worsening on donepezil in patients with frontotemporal dementia. Am. J. Geriatr. Psychiatry 15, 84–87. doi: 10.1097/01.JGP.0000231744.69631.33
Mesulam, M. M., Grossman, M., Hillis, A., Kertesz, A., and Weintraub, S. (2003). The core and halo of primary progressive aphasia and semantic dementia. Ann. Neurol. 54(Suppl. 5), S11–S14. doi: 10.1002/ana.10569
Meyerowitz, J., Parker, S. J., Vella, L. J., Ng, D., Price, K. A., Liddell, J. R., et al. (2011). C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol. Neurodegener. 6:57. doi: 10.1186/1750-1326-6-57
Michel, G., Mercken, M., Murayama, M., Noguchi, K., Ishiguro, K., Imahori, K., et al. (1998). Characterization of tau phosphorylation in glycogen synthase kinase-3beta and cyclin dependent kinase-5 activator (p23) transfected cells. Biochim. Biophys. Acta 1380, 177–182. doi: 10.1016/s0304-4165(97)00139-6
Min, S. W., Cho, S. H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Moisse, K., Volkening, K., Leystra-Lantz, C., Welch, I., Hill, T., and Strong, M. J. (2009). Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 1249, 202–211. doi: 10.1016/j.brainres.2008.10.021
Morgan, S., Kemp, P., Booij, J., Costa, D. C., Padayachee, S., Lee, L., et al. (2012). Differentiation of frontotemporal dementia from dementia with Lewy bodies using FP-CIT SPECT. J. Neurol. Neurosurg. Psychiatry 83, 1063–1070. doi: 10.1136/jnnp-2012-302577
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morishima-Kawashima, M., Hasegawa, M., Takio, K., Suzuki, M., Titani, K., and Ihara, Y. (1993). Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 10, 1151–1160. doi: 10.1016/0896-6273(93)90063-w
Morsch, R., Simon, W., and Coleman, P. D. (1999). Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 58, 188–197. doi: 10.1097/00005072-199902000-00008
Mukherjee, O., Pastor, P., Cairns, N. J., Chakraverty, S., Kauwe, J. S., Shears, S., et al. (2006). HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann. Neurol. 60, 314–322. doi: 10.1002/ana.20963
Nakashima, H., Ishihara, T., Suguimoto, P., Yokota, O., Oshima, E., Kugo, A., et al. (2005). Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 110, 547–556. doi: 10.1007/s00401-005-1087-4
Nakashima-Yasuda, H., Uryu, K., Robinson, J., Xie, S. X., Hurtig, H., Duda, J. E., et al. (2007). Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 114, 221–229. doi: 10.1007/s00401-007-0261-2
National Institute on Aging. (1997). Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The national institute on aging and reagan institute working group on diagnostic criteria for the neuropathological assessment of Alzheimer’s disease. Neurobiol. Aging 18, S1–S2.
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., et al. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. doi: 10.1212/wnl.51.6.1546
Necula, M., and Kuret, J. (2004). Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J. Biol. Chem. 279, 49694–49703. doi: 10.1074/jbc.m405527200
Necula, M., and Kuret, J. (2005). Site-specific pseudophosphorylation modulates the rate of tau filament dissociation. FEBS Lett. 579, 1453–1457. doi: 10.1016/j.febslet.2005.01.047
Neumann, M., Kwong, L. K., Sampathu, D. M., Trojanowski, J. Q., and Lee, V. M. (2007a). TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch. Neurol. 64, 1388–1394. doi: 10.1001/archneur.64.10.1388
Neumann, M., Mackenzie, I. R., Cairns, N. J., Boyer, P. J., Markesbery, W. R., Smith, C. D., et al. (2007b). TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol. 66, 152–157. doi: 10.1097/nen.0b013e31803020b9
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., et al. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U S A 102, 6990–6995. doi: 10.1073/pnas.0500466102
Nonaka, T., Kametani, F., Arai, T., Akiyama, H., and Hasegawa, M. (2009). Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 18, 3353–3364. doi: 10.1093/hmg/ddp275
Nonaka, T., Masuda-Suzukake, M., Arai, T., Hasegawa, Y., Akatsu, H., Obi, T., et al. (2013). Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134. doi: 10.1016/j.celrep.2013.06.007
Onyike, C. U., and Diehl-Schmid, J. (2013). The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry 25, 130–137. doi: 10.3109/09540261.2013.776523
Park, S. Y., and Ferreira, A. (2005). The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J. Neurosci. 25, 5365–5375. doi: 10.1523/jneurosci.1125-05.2005
Parker, S. J., Meyerowitz, J., James, J. L., Liddell, J. R., Crouch, P. J., Kanninen, K. M., et al. (2012). Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem. Int. 60, 415–424. doi: 10.1016/j.neuint.2012.01.019
Patrick, C., Crews, L., Desplats, P., Dumaop, W., Rockenstein, E., Achim, C. L., et al. (2011). Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am. J. Pathol. 178, 1646–1661. doi: 10.1016/j.ajpath.2010.12.033
Pereira, J. M., Williams, G. B., Acosta-Cabronero, J., Pengas, G., Spillantini, M. G., Xuereb, J. H., et al. (2009). Atrophy patterns in histologic vs clinical groupings of frontotemporal lobar degeneration. Neurology 72, 1653–1660. doi: 10.1212/WNL.0b013e3181a55fa2
Planel, E., Richter, K. E., Nolan, C. E., Finley, J. E., Liu, L., Wen, Y., et al. (2007a). Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J. Neurosci. 27, 3090–3097. doi: 10.1523/jneurosci.4854-06.2007
Planel, E., Tatebayashi, Y., Miyasaka, T., Liu, L., Wang, L., Herman, M., et al. (2007b). Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J. Neurosci. 27, 13635–13648. doi: 10.1523/jneurosci.3949-07.2007
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Rabinovici, G. D., and Miller, B. L. (2010). Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 24, 375–398. doi: 10.2165/11533100-000000000-00000
Rademakers, R., and Rovelet-Lecrux, A. (2009). Recent insights into the molecular genetics of dementia. Trends Neurosci. 32, 451–461. doi: 10.1016/j.tins.2009.05.005
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477. doi: 10.1093/brain/awr179
Ren, Q. G., Liao, X. M., Chen, X. Q., Liu, G. P., and Wang, J. Z. (2007). Effects of tau phosphorylation on proteasome activity. FEBS Lett. 581, 1521–1528. doi: 10.1016/j.febslet.2007.02.065
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Reyes, J. F., Geula, C., Vana, L., and Binder, L. I. (2012). Selective tau tyrosine nitration in non-AD tauopathies. Acta Neuropathol. 123, 119–132. doi: 10.1007/s00401-011-0898-8
Rohn, T. T., and Kokoulina, P. (2009). Caspase-cleaved TAR DNA-binding protein-43 in pick’s disease. Int. J. Physiol. Pathophysiol. Pharmacol. 1, 25–32.
Rohn, T. T., Rissman, R. A., Davis, M. C., Kim, Y. E., Cotman, C. W., and Head, E. (2002). Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol. Dis. 11, 341–354. doi: 10.1006/nbdi.2002.0549
Ryan, C. L., Baranowski, D. C., Chitramuthu, B. P., Malik, S., Li, Z., Cao, M., et al. (2009). Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci. 10:130. doi: 10.1186/1471-2202-10-130
Sahara, N., Maeda, S., Yoshiike, Y., Mizoroki, T., Yamashita, S., Murayama, M., et al. (2007). Molecular chaperone-mediated tau protein metabolism counteracts the formation of granular tau oligomers in human brain. J. Neurosci. Res. 85, 3098–3108. doi: 10.1002/jnr.21417
Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. doi: 10.1126/science.1113694
Sareen, D., O’rourke, J. G., Meera, P., Muhammad, A. K., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:208ra149. doi: 10.1126/scitranslmed.3007529
Schneider, L. S., Mangialasche, F., Andreasen, N., Feldman, H., Giacobini, E., Jones, R., et al. (2014). Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J. Intern. Med. 275, 251–283. doi: 10.1111/joim.12191
Schoonenboom, N. S., Reesink, F. E., Verwey, N. A., Kester, M. I., Teunissen, C. E., Van De Ven, P. M., et al. (2012). Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology 78, 47–54. doi: 10.1212/wnl.0b013e31823ed0f0
Seelaar, H., Rohrer, J. D., Pijnenburg, Y. A., Fox, N. C., and Van Swieten, J. C. (2011). Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J. Neurol. Neurosurg. Psychiatry 82, 476–486. doi: 10.1136/jnnp.2010.212225
Seeley, W. W., Crawford, R. K., Zhou, J., Miller, B. L., and Greicius, M. D. (2009). Neurodegenerative diseases target large-scale human brain networks. Neuron 62, 42–52. doi: 10.1016/j.neuron.2009.03.024
Sephton, C. F., Good, S. K., Atkin, S., Dewey, C. M., Mayer, P. 3rd, Herz, J., et al. (2010). TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 285, 6826–6834. doi: 10.1074/jbc.M109.061846
Sergeant, N., Wattez, A., and Delacourte, A. (1999). Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J. Neurochem. 72, 1243–1249. doi: 10.1046/j.1471-4159.1999.0721243.x
Sharma, V. M., Litersky, J. M., Bhaskar, K., and Lee, G. (2007). Tau impacts on growth-factor-stimulated actin remodeling. J. Cell Sci. 120, 748–757. doi: 10.1242/jcs.03378
Shi, J., Shaw, C. L., Du Plessis, D., Richardson, A. M., Bailey, K. L., Julien, C., et al. (2005). Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 110, 501–512. doi: 10.1007/s00401-005-1079-4
Shiarli, A. M., Jennings, R., Shi, J., Bailey, K., Davidson, Y., Tian, J., et al. (2006). Comparison of extent of tau pathology in patients with frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), frontotemporal lobar degeneration with pick bodies and early onset Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 32, 374–387. doi: 10.1111/j.1365-2990.2006.00736.x
Shiryaev, N., Jouroukhin, Y., Giladi, E., Polyzoidou, E., Grigoriadis, N. C., Rosenmann, H., et al. (2009). NAP protects memory, increases soluble tau and reduces tau hyperphosphorylation in a tauopathy model. Neurobiol. Dis. 34, 381–388. doi: 10.1016/j.nbd.2009.02.011
Sigurdsson, E. M. (2008). Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J. Alzheimers Dis. 15, 157–168.
Sleegers, K., Brouwers, N., Maurer-Stroh, S., Van Es, M. A., Van Damme, P., Van Vught, P. W., et al. (2008). Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71, 253–259. doi: 10.1212/01.wnl.0000289191.54852.75
Sontag, E., Nunbhakdi-Craig, V., Lee, G., Bloom, G. S., and Mumby, M. C. (1996). Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron 17, 1201–1207. doi: 10.1016/s0896-6273(00)80250-0
Spillantini, M. G., Goedert, M., Crowther, R. A., Murrell, J. R., Farlow, M. R., and Ghetti, B. (1997). Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. U S A 94, 4113–4118. doi: 10.1073/pnas.94.8.4113
Spillantini, M. G., Murrell, J. R., Goedert, M., Farlow, M. R., Klug, A., and Ghetti, B. (1998). Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U S A 95, 7737–7741. doi: 10.1073/pnas.95.13.7737
Spires, T. L., Orne, J. D., Santacruz, K., Pitstick, R., Carlson, G. A., Ashe, K. H., et al. (2006). Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607. doi: 10.2353/ajpath.2006.050840
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Steinacker, P., Hendrich, C., Sperfeld, A. D., Jesse, S., Von Arnim, C. A., Lehnert, S., et al. (2008). TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol. 65, 1481–1487. doi: 10.1001/archneur.65.11.1481
Suh, J., Im, D. S., Moon, G. J., Ryu, K. S., De Silva, R., Choi, I. S., et al. (2010). Hypoxic ischemia and proteasome dysfunction alter tau isoform ratio by inhibiting exon 10 splicing. J. Neurochem. 114, 160–170. doi: 10.1111/j.1471-4159.2010.06732.x
Sultan, A., Nesslany, F., Violet, M., Begard, S., Loyens, A., Talahari, S., et al. (2011). Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 286, 4566–4575. doi: 10.1074/jbc.m110.199976
Suzuki, H., Lee, K., and Matsuoka, M. (2011). TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by Caspase-mediated TDP-43 cleavage. J. Biol. Chem. 286, 13171–13183. doi: 10.1074/jbc.m110.197483
Synofzik, M., Born, C., Rominger, A., Lummel, N., Schols, L., Biskup, S., et al. (2014). Targeted high-throughput sequencing identifies a TARDBP mutation as a cause of early-onset FTD without motor neuron disease. Neurobiol. Aging 35, 1212.e1–1212.e5. doi: 10.1016/j.neurobiolaging.2013.10.092
Tanaka, Y., Matsuwaki, T., Yamanouchi, K., and Nishihara, M. (2013). Exacerbated inflammatory responses related to activated microglia after traumatic brain injury in progranulin-deficient mice. Neuroscience 231, 49–60. doi: 10.1016/j.neuroscience.2012.11.032
Tang, W., Lu, Y., Tian, Q. Y., Zhang, Y., Guo, F. J., Liu, G. Y., et al. (2011). The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484. doi: 10.1126/science.1199214
Tangkeangsirisin, W., and Serrero, G. (2004). PC cell-derived growth factor (PCDGF/GP88, progranulin) stimulates migration, invasiveness and VEGF expression in breast cancer cells. Carcinogenesis 25, 1587–1592. doi: 10.1093/carcin/bgh171
Taniguchi, S., Mcdonagh, A. M., Pickering-Brown, S. M., Umeda, Y., Iwatsubo, T., Hasegawa, M., et al. (2004). The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol. Appl. Neurobiol. 30, 1–18. doi: 10.1046/j.0305-1846.2003.00481.x
Tao, J., Ji, F., Wang, F., Liu, B., and Zhu, Y. (2012). Neuroprotective effects of progranulin in ischemic mice. Brain Res. 1436, 130–136. doi: 10.1016/j.brainres.2011.11.063
Tashiro, Y., Urushitani, M., Inoue, H., Koike, M., Uchiyama, Y., Komatsu, M., et al. (2012). Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem. 287, 42984–42994. doi: 10.1074/jbc.m112.417600
Toledo, J. B., Arnold, S. E., Raible, K., Brettschneider, J., Xie, S. X., Grossman, M., et al. (2013). Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the national Alzheimer’s coordinating centre. Brain 136, 2697–2706. doi: 10.1093/brain/awt188
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458. doi: 10.1038/nn.2778
Udan-Johns, M., Bengoechea, R., Bell, S., Shao, J., Diamond, M. I., True, H. L., et al. (2014). Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 23, 157–170. doi: 10.1093/hmg/ddt408
Urwin, H., Josephs, K. A., Rohrer, J. D., Mackenzie, I. R., Neumann, M., Authier, A., et al. (2010). FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 120, 33–41. doi: 10.1007/s00401-010-0698-6
Uryu, K., Nakashima-Yasuda, H., Forman, M. S., Kwong, L. K., Clark, C. M., Grossman, M., et al. (2008). Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 67, 555–564. doi: 10.1097/nen.0b013e31817713b5
Van Damme, P., Van Hoecke, A., Lambrechts, D., Vanacker, P., Bogaert, E., van Swieten, J., et al. (2008). Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 181, 37–41. doi: 10.1083/jcb.200712039
Van Deerlin, V. M., Sleiman, P. M., Martinez-Lage, M., Chen-Plotkin, A., Wang, L. S., Graff-Radford, N. R., et al. (2010). Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239. doi: 10.1038/ng.536
van Eersel, J., Ke, Y. D., Gladbach, A., Bi, M., Gotz, J., Kril, J. J., et al. (2011). Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS One 6:e22850. doi: 10.1371/journal.pone.0022850
Vanmechelen, E., Vanderstichele, H., Davidsson, P., Van Kerschaver, E., Van Der Perre, B., Sjogren, M., et al. (2000). Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci. Lett. 285, 49–52. doi: 10.1016/s0304-3940(00)01036-3
Vellas, B., Carrillo, M. C., Sampaio, C., Brashear, H. R., Siemers, E., Hampel, H., et al. (2013). Designing drug trials for Alzheimer’s disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD task force. Alzheimers Dement. 9, 438–444. doi: 10.1016/j.jalz.2013.03.007
Vossel, K. A., and Miller, B. L. (2008). New approaches to the treatment of frontotemporal lobar degeneration. Curr. Opin. Neurol. 21, 708–716. doi: 10.1097/wco.0b013e328318444d
Wagner, U., Utton, M., Gallo, J. M., and Miller, C. C. (1996). Cellular phosphorylation of tau by GSK-3 beta influences tau binding to microtubules and microtubule organisation. J. Cell Sci. 109(Pt. 6), 1537–1543.
Wang, I. F., Chang, H. Y., Hou, S. C., Liou, G. G., Way, T. D., and James Shen, C. K. (2012). The self-interaction of native TDP-43 C terminus inhibits its degradation and contributes to early proteinopathies. Nat. Commun. 3:766. doi: 10.1038/ncomms1766
Wang, J. Z., Grundke-Iqbal, I., and Iqbal, K. (1996). Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res. Mol. Brain Res. 38, 200–208. doi: 10.1016/0169-328x(95)00316-k
Wang, J. Z., Grundke-Iqbal, I., and Iqbal, K. (2007). Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 25, 59–68. doi: 10.1111/j.1460-9568.2006.05226.x
Wang, I. F., Wu, L. S., Chang, H. Y., and Shen, C. K. (2008). TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 105, 797–806. doi: 10.1111/j.1471-4159.2007.05190.x
Watts, G. D., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi: 10.1038/ng1332
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U S A 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Whittington, R. A., Virág, L., Marcouiller, F., Papon, M. A., El Khoury, N. B., Julien, C., et al. (2011). Propofol directly increases tau phosphorylation. PLoS One 6:e16648. doi: 10.1371/journal.pone.0016648
Whitwell, J. L., Jack, C. R. Jr., Parisi, J. E., Senjem, M. L., Knopman, D. S., Boeve, B. F., et al. (2010). Does TDP-43 type confer a distinct pattern of atrophy in frontotemporal lobar degeneration? Neurology 75, 2212–2220. doi: 10.1212/wnl.0b013e31820203c2
Whitwell, J. L., Jack, C. R. Jr., Senjem, M. L., Parisi, J. E., Boeve, B. F., Knopman, D. S., et al. (2009a). MRI correlates of protein deposition and disease severity in postmortem frontotemporal lobar degeneration. Neurodegener. Dis. 6, 106–117. doi: 10.1159/000209507
Whitwell, J. L., Przybelski, S. A., Weigand, S. D., Ivnik, R. J., Vemuri, P., Gunter, J. L., et al. (2009b). Distinct anatomical subtypes of the behavioural variant of frontotemporal dementia: a cluster analysis study. Brain 132, 2932–2946. doi: 10.1093/brain/awp232
Wils, H., Kleinberger, G., Pereson, S., Janssens, J., Capell, A., Van Dam, D., et al. (2012). Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J. Pathol. 228, 67–76. doi: 10.1002/path.4043
Winton, M. J., Igaz, L. M., Wong, M. M., Kwong, L. K., Trojanowski, J. Q., and Lee, V. M. (2008). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration and aggregate formation. J. Biol. Chem. 283, 13302–13309. doi: 10.1074/jbc.m800342200
Wischik, C. M., Harrington, C. R., and Storey, J. M. (2014). Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 88, 529–539. doi: 10.1016/j.bcp.2013.12.008
Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M., et al. (2001). Tauopathy in drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714. doi: 10.1126/science.1062382
Womack, K. B., Diaz-Arrastia, R., Aizenstein, H. J., Arnold, S. E., Barbas, N. R., Boeve, B. F., et al. (2011). Temporoparietal hypometabolism in frontotemporal lobar degeneration and associated imaging diagnostic errors. Arch. Neurol. 68, 329–337. doi: 10.1001/archneurol.2010.295
Wu, L. S., Cheng, W. C., Hou, S. C., Yan, Y. T., Jiang, S. T., and Shen, C. K. (2010). TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 48, 56–62. doi: 10.1002/dvg.20584
Wu, L. S., Cheng, W. C., and Shen, C. K. (2012). Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J. Biol. Chem. 287, 27335–27344. doi: 10.1074/jbc.m112.359000
Yamada, T., Calne, D. B., Akiyama, H., McGeer, E. G., and McGeer, P. L. (1993). Further observations on Tau-positive glia in the brains with progressive supranuclear palsy. Acta Neuropathol. 85, 308–315. doi: 10.1007/bf00227727
Yamada, T., McGeer, P. L., and McGeer, E. G. (1992). Appearance of paired nucleated, Tau-positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci. Lett. 135, 99–102. doi: 10.1016/0304-3940(92)90145-w
Yamashita, M., Nonaka, T., Arai, T., Kametani, F., Buchman, V. L., Ninkina, N., et al. (2009). Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Lett. 583, 2419–2424. doi: 10.1016/j.febslet.2009.06.042
Yan, S. D., Chen, X., Schmidt, A. M., Brett, J., Godman, G., Zou, Y. S., et al. (1994). Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc. Natl. Acad. Sci. U S A 91, 7787–7791. doi: 10.1073/pnas.91.16.7787
Yanamandra, K., Kfoury, N., Jiang, H., Mahan, T. E., Ma, S., Maloney, S. E., et al. (2013). Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80, 402–414. doi: 10.1016/j.neuron.2013.07.046
Yokota, O., Davidson, Y., Bigio, E. H., Ishizu, H., Terada, S., Arai, T., et al. (2010). Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol. 120, 55–66. doi: 10.1007/s00401-010-0702-1
Zhang, Y. J., Xu, Y. F., Cook, C., Gendron, T. F., Roettges, P., Link, C. D., et al. (2009). Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U S A 106, 7607–7612. doi: 10.1073/pnas.0900688106
Zhang, Y. J., Xu, Y. F., Dickey, C. A., Buratti, E., Baralle, F., Bailey, R., et al. (2007). Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci. 27, 10530–10534. doi: 10.1523/jneurosci.3421-07.2007
Zheng, Y., Brady, O. A., Meng, P. S., Mao, Y., and Hu, F. (2011). C-terminus of progranulin interacts with the beta-propeller region of sortilin to regulate progranulin trafficking. PLoS One 6:e21023. doi: 10.1371/journal.pone.0021023
Keywords: frontotemporal dementia, frontotemporal lobar degeneration, TDP-43, tau, therapeutics
Citation: D’Alton S and Lewis J (2014) Therapeutic and diagnostic challenges for frontotemporal dementia. Front. Aging Neurosci. 6:204. doi: 10.3389/fnagi.2014.00204
Received: 30 May 2014; Paper pending published: 20 June 2014;
Accepted: 25 July 2014; Published online: 19 August 2014.
Edited by:
Cristian Lasagna Reeves, Baylor College of Medicine, USAReviewed by:
Emmanuel Planel, Centre Hospitalier de l’université Laval, CanadaArianna Bellucci, University of Brescia, Italy
Copyright © 2014 D’Alton and Lewis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon D’Alton, Department of Neuroscience, Center for Translational Research in Neurodegenerative Disease, College of Medicine, University of Florida, 1275 Center Drive, Gainesville, FL 32610, USA e-mail:c2RhbHRvbjgyQHVmbC5lZHU=