 Patricia M. Chege
Patricia M. Chege Gawain McColl
Gawain McColl- The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Parkville, VIC, Australia
Parkinson's disease (PD) is characterized by progressive motor impairment attributed to progressive loss of dopaminergic (DAergic) neurons in the substantia nigra pars compacta. Additional clinical manifestations include non-motor symptoms such as insomnia, depression, psychosis, and cognitive impairment. PD patients with mild cognitive impairment have an increased risk of developing dementia. The affected brain regions also show perturbed metal ion levels, primarily iron. These observations have led to speculation that metal ion dyshomeostasis plays a key role in the neuronal death of this disease. However, the mechanisms underlying this metal-associated neurodegeneration have yet to be completely elucidated. Mammalian models have traditionally been used to investigate PD pathogenesis. However, alternate animal models are also being adopted, bringing to bear their respective experimental advantage. The nematode, Caenorhabditis elegans, is one such system that has well-developed genetics, is amenable to transgenesis and has relatively low associated experimental costs. C. elegans has a well characterized neuronal network that includes a simple DAergic system. In this review we will discuss mechanisms thought to underlie PD and the use of C. elegans to investigate these processes.
Introduction
Parkinson's disease (PD) is the second most prevalent age-related neurodegenerative disorder of the central nervous system, after Alzheimer's disease (AD). Idiopathic or sporadic PD affects approximately 1% of people over 65 years old (Hirtz et al., 2007). PD is characterized by severe motor impairment, which is attributed to profound depletion of striatal dopamine (DA) due to progressive loss of dopaminergic (DAergic) neurons in the substantia nigra pars compacta, a region in the basal ganglia that is crucial in voluntary motor functions (Hornykiewicz and Kish, 1987; Wooten, 1997; Braak et al., 2003). PD is also characterized by proteinaceous neuronal inclusions known as Lewy bodies (Irizarry et al., 1998). Current PD therapies focus mainly on correcting this DA depletion. Although effective in alleviating symptoms, these treatments lose their efficacy over time and do not halt the underlying neurodegeneration (Smith et al., 2012). Determining the mechanisms contributing to PD neurodegeneration is critical to facilitate the design of effective therapies to halt further neuronal loss.
While some PD cases are monogenic, arising from single point mutation in a specific gene, more than 90% of the cases are idiopathic (Table 1). The mechanisms underlying idiopathic PD are not fully understood. However, increasing evidence suggests that oxidative stress may be a major contributing factor to neuronal loss. This is evidenced by increased levels of oxidized lipids, proteins and nucleic acids in PD brains (Dexter et al., 1989a, 1994; Jenner and Olanow, 1996; Yoritaka et al., 1996; Alam et al., 1997a,b). Oxidative stress is thought to arise from a variety of mechanisms including mitochondrial dysfunction, neuroinflammation, perturbed DA metabolism and environmental toxins (Thomas and Beal, 2007; Hwang, 2013). Metal ion dyshomeostasis has also been hypothesized to cause oxidative stress, following evidence that PD brains exhibit increased total iron concentration (Dexter et al., 1991; Gotz et al., 2004; Oakley et al., 2007). In addition, levels of zinc are increased and copper decreased in the substantia nigra (Dexter et al., 1991).
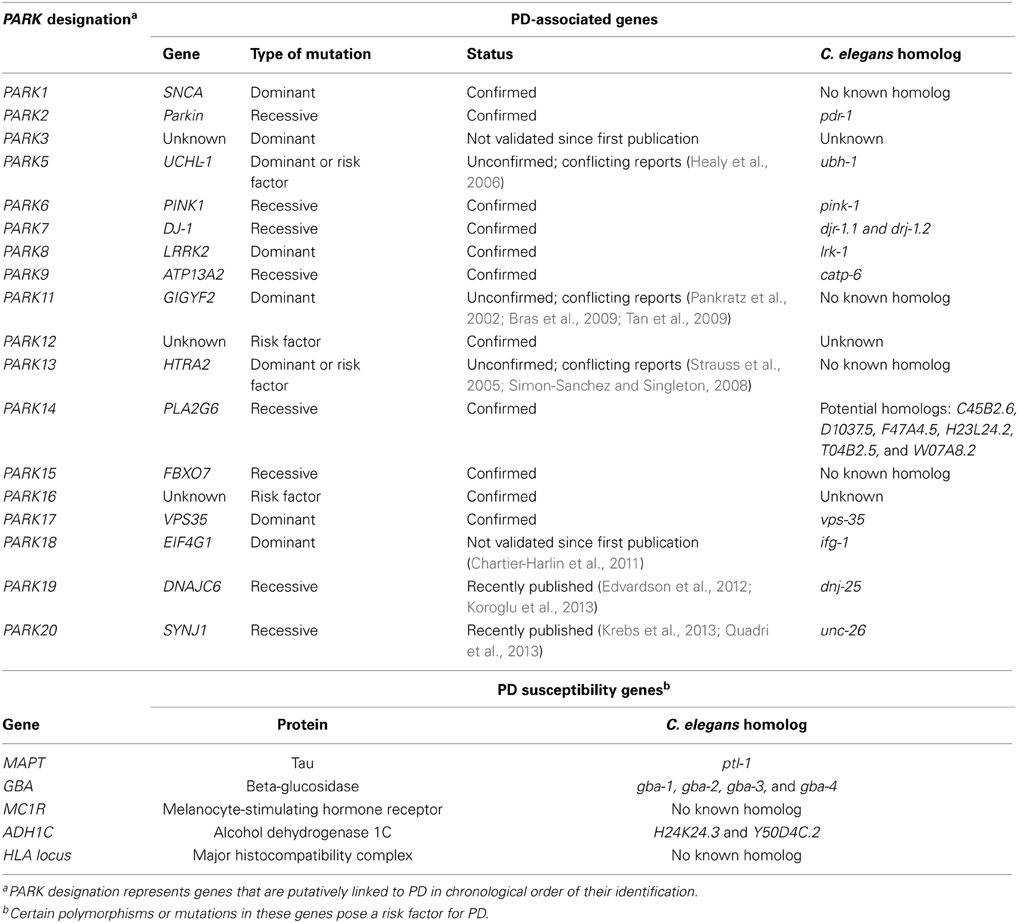
Table 1. PD associated and susceptibility genes and corresponding C. elegans homologs.
Investigating the molecular basis of neurodegeneration in vivo relies on animal models, with mammalian models typically being used. All animal models have inherent experimental limitations and none fully replicate all aspects of a disease such as PD. As greater understanding of PD is gained and new hypotheses proposed there is a parallel need for animal models to be updated and modified to further our understanding. Establishing new transgenic models can have a significant lead-time with some animal systems being less suited to genetic modification. These particular limitations can be alleviated by use of a less complex animal, such as Caenorhabditis elegans.
C. elegans as a Neurodegeneration Model
C. elegans is a free-living nematode, approximately 1 mm in length, which exists as either a self-fertilizing hermaphrodite or as a male (Figure 1). C. elegans can be cultured inexpensively on an E. coli lawn on agar media and has a short defined life cycle (Brenner, 1974). The rapid life cycle coupled with a high reproductive capacity makes C. elegans a suitable tool for mutagenesis and compound screening approaches. C. elegans also has an adult lifespan of approximately 3 weeks and is an established model of biological aging. Additionally, the C. elegans genome has been fully sequenced which has revealed that about 80% of C. elegans genes have human homologs and at least 42% of human disease-related genes have a C. elegans homolog (Consortium, 1998; Culetto and Sattelle, 2000; Lai et al., 2000). Functional studies of corresponding or related human genes can be done via mutation (where available) or RNA interference (RNAi) (Fire et al., 1998; Hamamichi et al., 2008; Ruan et al., 2010).
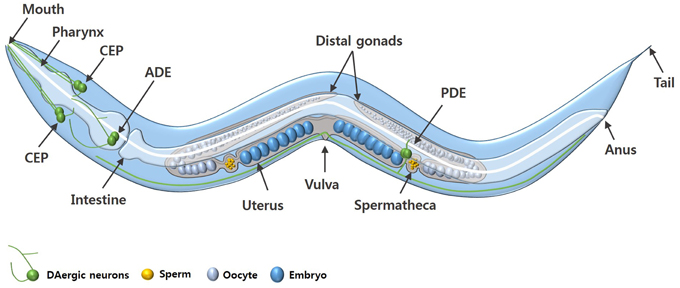
Figure 1. An adult C. elegans hermaphrodite. The diagram shows the key anatomical features and the DAergic neurons (green) of C. elegans. The DAergic neurons include four cephalic (CEP) neurons, two anterior deirid (ADE) neurons, and two posterior deirid (PDE) neurons. Males have six additional DAergic neurons located in the tail (not shown).
To complement these approaches or in the absence of endogenous homologs, C. elegans can be transgenically manipulated to express human disease associated genes in specific cell types, including neurons (Faber et al., 1999; Lakso et al., 2003; Brandt et al., 2009; McColl et al., 2009, 2012). Adult hermaphrodite C. elegans have 302 neurons, a neuronal network that is stereotypical between animals and which possesses most of the major neurotransmitter systems found in mammals, including DAergic neurons (White et al., 1986; Rand and Nonet, 1997; Bargmann, 1998). C. elegans are also optically transparent, which in conjunction with fluorescent protein reporters, allows for in vivo visualization of neurons, such as in Figure 2 (Chalfie et al., 1994; Nass et al., 2002; Chew et al., 2013).
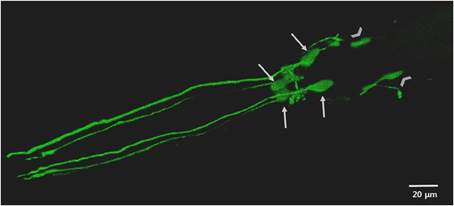
Figure 2. The anterior DAergic neurons of an adult C. elegans hermaphrodite. The neurons are visualized by the translational expression of GFP driven by the promoter of the DA transporter (Pdat-1::GFP). The key features highlighted include the cell bodies and dendritic processes of the four CEP neurons (arrows) and the cell bodies of the two ADE (chevrons).
As with other animal models, use of C. elegans to model disease is always tempered by an awareness of the limitations of cellular and anatomical differences. For example, C. elegans lack a vascular system and the somatic tissues of adult C. elegans are post-mitotic. Despite these obvious differences, key discoveries in C. elegans have been readily translated to vertebrate research. C. elegans was used to identify genes that are involved in regulating programmed cell death (Hedgecock et al., 1983; Ellis and Horvitz, 1986). dsRNA gene expression regulation was characterized in C. elegans and led to development of RNAi, a tool widely used in functional genomics (Fire et al., 1998). Additionally, the conserved effects of the insulin/insulin growth factor-1 signaling pathway on longevity were first noted in C. elegans mutants (Friedman and Johnson, 1988; Kenyon et al., 1993; Dorman et al., 1995; Murakami and Johnson, 1996; Kimura et al., 1997). C. elegans research has also linked iron metabolism to restless leg syndrome (Catoire et al., 2011). The findings in C. elegans were predictive of the role of ferritin in human tissue.
Parkinson's Disease
The hallmark PD symptoms are motor deficits, which include resting tremor, rigidity, slowness in movement (bradykinesia) and posture instability. In the majority of PD cases, these clinical manifestations only appear when approximately 50–70% of nigral neurons are lost and approximately 80% of striatal DA is depleted (Hornykiewicz and Kish, 1987; Kish et al., 1988; Orth and Schapira, 2002). This DA deficiency leads to the observed motor impairments because DA is an essential motor control neurotransmitter. In addition to DAergic degeneration, extensive neurodegeneration and atrophy occurs in other nerve cell types and brain regions as PD advances. The regions affected include the hippocampus, thalamus, and neocortex. This additional neurodegeneration leads to non-motor symptoms that include insomnia, depression, psychosis and cognitive impairment (Braak et al., 2003; Weintraub et al., 2011; Smith et al., 2012). These non-motor symptoms worsen over time, for example, an estimated 80% of PD patients with mild cognitive impairment develop dementia (Janvin et al., 2006; Buter et al., 2008; Hely et al., 2008). The etiology of the neurodegeneration leading to cognitive impairment remains unclear.
PD is also characterized by neuronal inclusions, Lewy bodies and Lewy neurites, which mainly contain aggregated α-synuclein (Forno, 1996; Spillantini et al., 1997; Irizarry et al., 1998). Alpha-synuclein is a 140-amino acid peptide encoded by the SNCA gene and is predominantly located at presynaptic terminals. It is highly expressed in the substantia nigra, hippocampus, neocortex, thalamus and cerebellum (Ueda et al., 1993; Nakajo et al., 1994; Iwai et al., 1995; Recchia et al., 2004). These brain regions are highly impacted by neurodegeneration in PD pathology. Several heritable point mutations, A30P, A53T, E46K, H50Q, and G51D, and a triplication of the SNCA gene are implicated in autosomal dominant forms of familial PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Singleton et al., 2003; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Proukakis et al., 2013). These findings have initiated numerous studies into the involvement of α-synuclein in idiopathic PD pathology.
Although several possibilities have been proposed, the function of α-synuclein remains unknown. Alpha-synuclein KO mice have impaired spatial learning and working memory suggesting some involvement in cognitive function (Kokhan et al., 2012). Sequestration of the protein in Lewy bodies may contribute to cognitive impairment seen in advanced PD. Alpha-synuclein over-expression in transgenic mice inhibits DA synaptic release while α-synuclein deficiency causes decreased vesicle-bound striatal DA (Abeliovich et al., 2000; Nemani et al., 2010). This suggests that α-synuclein is involved in synaptic transmission by regulating DA release. Alpha-synuclein deficiency may lead to unregulated DA release, which when coupled with loss of DAergic neurons, leads to the striatal DA depletion observed in PD. Under normal physiological conditions, α-synuclein negatively modulates the dopamine transporter (DAT), which is required for re-uptake of synaptically released DA (Wersinger and Sidhu, 2003). This implies that α-synuclein deficiency caused by sequestration in Lewy bodies may lead to increased DA re-uptake, causing increased concentration of intracellular DA. High levels of unbound intracellular DA have been shown to be neurotoxic (Olanow and Arendash, 1994; Luo et al., 1998; Offen et al., 1999; Lee et al., 2001).
Other studies suggest that α-synuclein may be a microtubule-associated protein (MAP) as it interacts with tubulin (Alim et al., 2002, 2004), with α-synuclein deficiency postulated to lead to microtubule dysfunction. Microtubules provide structural scaffolding in neurons and so their dysfunction would compromise neuronal integrity leading to neuron death. Alpha-synuclein sequestration in Lewy bodies appears to have significant implications in PD pathology, potentially by inhibiting the normal functions of α-synuclein, which may include facilitating cognitive function, synaptic transmission and stabilizing neuronal morphology. However, the underlying mechanisms that trigger DAergic neuronal death and α-synuclein aggregation in idiopathic PD require further investigation.
C. elegans and Daergic Neurons
C. elegans hermaphrodites have a comparatively simple DAergic system comprising eight neurons in total: six anterior DAergic neurons, which include four cephalic (CEP) neurons and two anterior deirid (ADE) neurons, and two posterior deirid (PDE) neurons (Figures 1, 2). Males have six additional DAergic neurons located in the tail (Sulston et al., 1975). DA synthesis, storage and transport mechanisms are conserved in C. elegans and DAergic nerve endings and synaptic vesicles have DA levels similar to those in mammalian neurons (Fuxe and Jonsson, 1973; Bargmann, 1998).
The functions of DAergic neurons have been investigated using laser ablation, a technique which can target a specific neuron while leaving neighboring neurons intact. The loss of DAergic neurons revealed that they are important for food searching and the basal slowing response upon sensing food (Sawin et al., 2000; Hills et al., 2004). Exposure to exogenous DA resulted in decreased egg laying, slowed defecation and paralysis (Schafer and Kenyon, 1995; Weinshenker et al., 1995; Hills et al., 2004; McDonald et al., 2006). Studies of mutations in cat-2, the tyrosine hydroxylase which is the rate limiting enzyme in DA synthesis, showed loss of basal slowing response and decreased touch habituation suggesting that DA signaling is necessary for mechanosensation (Sawin et al., 2000; Sanyal et al., 2004). These findings suggest that DAergic neurons are important for locomotion, associative learning, food searching, food sensing, egg-laying and defecation.
Most models of DAergic neurodegeneration in C. elegans are induced through exposure to neurotoxins and some metals, which selectively ablate DAergic neurons. These toxins include 6-hydroxydopamine (6-OHDA), l-methyl-4-phenylpyridinium (MPP+), methylmercury (MeHg), and manganese (Table 2) (Nass et al., 2002; Braungart et al., 2004; Settivari et al., 2009; VanDuyn et al., 2010). When exposed to 6-OHDA, C. elegans show a progressive and selective DAergic neuron degeneration and loss as evidenced by formation of blebs in axonal and dendritic membranes. (Nass et al., 2002; VanDuyn et al., 2010).
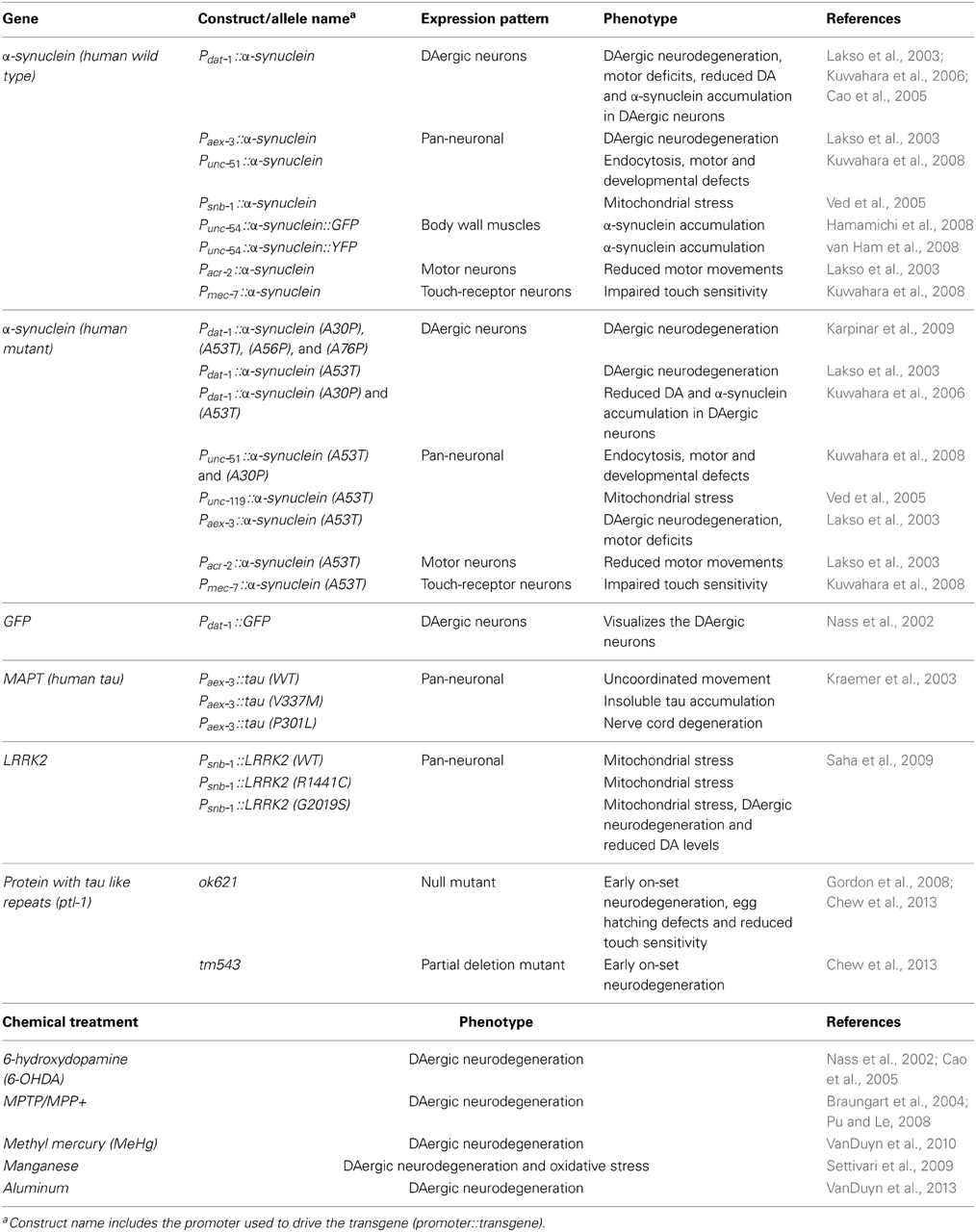
Table 2. C. elegans Parkinson's disease models.
C. elegans and α-Synuclein
Although C. elegans lacks a human α-synuclein homolog, α-synuclein expression has been investigated in transgenic C. elegans. The targeting of transgene expression in C. elegans body wall muscle cells has been used to explore the toxicity of several disease-associated proteins. Body wall muscles run longitudinally along the length of the nematode and are essential for locomotion. Functional disruption of these cells causes a clear and robust paralysis phenotype (McColl et al., 2009, 2012). In PD research, green or yellow fluorescent protein-tagged α-synuclein was expressed in the body-wall muscle of C. elegans to visualize α-synuclein aggregation in vivo (Hamamichi et al., 2008; van Ham et al., 2008). These lines have been used to screen RNAi libraries, revealing 20 neuroprotective genes whose knock down enhanced α-synuclein aggregation. One of these genes was the ortholog of human VSP41, a key lysosomal trafficking protein that protects against toxicity of DA-derived neurotoxins (Hamamichi et al., 2008; Ruan et al., 2010). Another genome-wide RNAi screen revealed 80 genes that when knocked down accelerated formation of α-synuclein inclusions. These genes, which appear to suppress inclusion formation, are predominantly involved in vesicular transport and lipid metabolism (van Ham et al., 2008). These findings suggest that defects in the endosomal-lysosomal and ER-Golgi vesicular trafficking system pathways may be implicated in α-synuclein toxicity.
Additionally, neurodegenerative processes can also be studied directly in C. elegans neurons. Over-expression of wild type and A53T mutant α-synuclein under the control of pan-neuronal promoter, aex-3 and under the DAergic neuron specific promoter, dat-1, caused loss of DAergic neurons (Lakso et al., 2003; Cao et al., 2005). Two neuroprotective endoplasmic reticulum (ER) associated proteins, TorsinA and Rab1 A, were found to ameliorate α-synuclein toxicity and prevent neuron loss (Cao et al., 2005; Cooper et al., 2006), suggesting that α-synuclein toxicity affects the ER-Golgi vesicular trafficking system. Another model over-expressing wild type or mutant α-synuclein under the control of the pan-neuronal promoter, unc-51, was used to screen an RNAi library for genetic modifiers that either suppress or exacerbate α-synuclein toxicity. Knock down of four genes that are involved in synaptic endocytosis enhanced α-synuclein toxicity (Kuwahara et al., 2008), suggesting that impaired endocytosis may contribute to α-synuclein dysfunction seen in PD pathology.
Wild type and A53T mutant (human) α-synuclein have been transgenically over-expressed via the C. elegans DAergic neuron specific promoter, dat-1 (Lakso et al., 2003; Cao et al., 2005). A screen of 115,000 compounds in cells and then C. elegans identified four 1,2,3,4-tetrahydroquinolinones antagonists of α-synuclein toxicity (Su et al., 2010). Another larger screen revealed that several 8-hydroxyquinolines could ameliorate α-synuclein aggregation and toxicity in C. elegans (Tardiff et al., 2012). The underlying mechanism of protection is proposed to be via interplay between metal homeostasis and proteotoxicity of aggregation prone proteins. Interestingly another 8-hydroxyquinoline, PBT2, has been found to reduce (the Alzheimer's associated peptide) Aβ toxicity in transgenic C. elegans (McColl et al., 2012). This compound is currently under clinical trial as an AD therapeutic (Lannfelt et al., 2008; Crouch et al., 2011).
Oxidative Stress and Metal Homeostasis
Oxidative stress occurs from an imbalance between toxic oxidant production and antioxidant activity, which leads to cellular damage followed by apoptosis (Sies, 1991; Jenner, 2003). The main reactive oxidants are the reactive oxygen species (ROS) and the reactive nitrogen species (RNS). RNS have been comprehensively reviewed elsewhere (Jomova et al., 2010). ROS, such as superoxide (O•−2) and hydroxyl radical (•OH) are normal by-products of oxygen consumption during cellular metabolism, predominantly in the mitochondria (Kepp, 2012). ROS levels are tightly regulated by endogenous antioxidant enzymes, such as glutathione, superoxidase dismutase (SOD), and catalase (Bains and Shaw, 1997; Sohal and Orr, 2012). It is important to stress that ROS have essential functions in normal cell biology and are not always inherently detrimental. For example, ROS are a component of the innate immune system, particularly in phagocytes, which produce ROS to prevent colonization by microbes (Fang, 2004). ROS are also utilized in cellular signaling (Hekimi et al., 2011) to modulate the activity of kinases, phosphatases and transcription factors. However, ROS are detrimental when their production goes unchecked leading to damage of cellular lipids, proteins and nucleic acids, and ultimately cell death (Pattison et al., 2002; Niki, 2009).
A way to counter the detrimental effects of ROS overproduction could be to administer antioxidant supplements or drugs, such as, Vitamins A, C, and E and compounds that inhibit ROS production. However, antioxidant therapeutic interventions have not been successful in alleviating oxidative stress associated with neurodegenerative diseases. This is primarily due to the inability of these compounds to effectively cross the blood brain barrier (Halliwell, 2001). Additionally, these antioxidants when administered in high doses have negative side effects by affecting normal cellular processes that rely on ROS activity (Halliwell, 2001; Freeman and Keller, 2012). An understanding of the cause of oxidative stress is vital to design better therapies to prevent neurodegeneration.
Biological transition metals, such as iron, copper, zinc, magnesium, nickel, cobalt, and manganese, are essential co-factors for at least one-third to one-half of all proteins (Andreini et al., 2008; Waldron et al., 2009). Iron and copper are metabolically utilized due to their ability to redox cycle, with iron being the most abundant. However, in the event of metal ion misregulation, this redox ability has the potential to produce toxic radicals via Haber-Weiss and Fenton reactions leading to oxidative stress (Nunez et al., 2012). Levels of these metal ions are reported to be perturbed in brains affected by various neurodegenerative diseases. This has led to the metal ion dyshomeostasis hypothesis, which proposes that the metal ion imbalance triggers increased ROS production causing oxidative stress that eventually leads to neuronal death. It is plausible that the observed metal imbalance is just a symptom and not a cause of neurodegeneration. However, several heritable neurodegenerative diseases are directly caused by metal-ion misregulation. These progressive conditions include aceruloplasminaemia and neuroferritinopathy, which result from iron misregulation, and Menkes Disease and Wilson's Disease, which result from copper misregulation (Vulpe et al., 1993; Yoshida et al., 1995; Harris et al., 1998; Curtis et al., 2001). These diseases suggest that dyshomeostasis of brain metals is sufficient to initiate neurodegeneration.
Iron is an essential metal in organisms because of its redox ability (Cairo et al., 2002). For example, reactive iron is part of the cytochrome complex in the mitochondrial respiration chain, which is important for cellular energy production. It is a crucial co-factor for catalase, an antioxidant that regulates hydrogen peroxide levels and also for heme proteins, which are essential for vascular transport of oxygen and carbon-dioxide. In the substantia nigra, iron is essential for DA synthesis (Youdim et al., 1984). However, this reactivity also allows iron to catalyze production of toxic hydroxyl radicals via Fenton chemistry:
Therefore, the concentration of unbound intracellular iron must be kept low; a process regulated by iron storage and transport proteins, such as ferritin (iron storage), ferroportin (iron efflux), divalent metal transporter-1 (DMT-1, an iron transporter), and transferrin (iron shuttling/uptake) (Lee and Andersen, 2010; Gkouvatsos et al., 2012). Disruption in these homeostatic functions could result in iron accumulation leading to oxidative damage and loss of function of proteins that depend on iron as a co-factor. This could potentially disrupt cellular respiration, antioxidant activity, oxygen/carbon dioxide transport and DA synthesis.
Copper is an important co-factor in the activity of redox active proteins, such as ceruloplasmin (iron homeostasis), cytochrome c oxidase (mitochondrial cellular respiration), Cu/Zn-superoxide dismutase (antioxidant activity) and dopamine-b-hydroxylase and tyrosinase, which are key proteins in DA synthesis (Arredondo and Nunez, 2005; Kepp, 2012). Therefore, copper imbalance in neurons may affect the function of these proteins. Additionally, unbound copper concentration requires tight control due to its redox potential. Copper levels higher than 10−18 M can initiate oxidative damage by facilitating ROS production (Rae et al., 1999):
Copper levels are predominantly regulated by ion importers, copper efflux pumps (ATP7A and ATP7B), metallochaperones, metalloregulators and other copper regulating proteins, such as, ceruloplasmin (Cp), (Camakaris et al., 1999; Waldron et al., 2009; Pang et al., 2013). Defects in these systems may result in increased levels of unbound copper causing oxidative damage. In addition, copper misregulation may cause loss of function of the copper dependent proteins, with resultant negative implications on iron homeostasis, cellular energy metabolism, oxidative stress responses and DA synthesis.
Parkinson's Disease and Oxidative Stress
PD brains show increased levels of oxidized macromolecules, which can be used as an indirect measure of ROS levels. Malondialdehyde, lipid hydroperoxides and 4-hydroxynonenal, which are lipid peroxidation products, are increased in PD brains (Dexter et al., 1989a, 1994; Yoritaka et al., 1996). PD brains also show increased levels of 8-hydroxydeoxyguanosine (8-OHdG) and protein carbonyls, which are products of DNA and protein oxidation, respectively, (Alam et al., 1997a,b). Another marker of elevated ROS levels in PD brains is increased SOD activity in the substantia nigra (Marttila et al., 1988; Saggu et al., 1989). SOD catalyzes the dismutation of superoxide (O•−2) into oxygen and hydrogen peroxide, therefore its activity may increase as a neuroprotective measure to cope with increased ROS levels.
Increased ROS levels not only lead to cellular damage but also to production of oxidation by-products that are also potentially neurotoxic. For example, 4-hydroxynonenal irreversibly modifies α-synuclein aggregation in vitro, potentially leading to formation of protofibrils, which are neurotoxic to cultured DAergic neurons (Qin et al., 2007). Lipid hydroperoxides have been shown to lead to oxidation of DA to 6-OHDA, a known neurotoxin (Sauer and Oertel, 1994; Przedborski et al., 1995; Pezzella et al., 1997; Lotharius and O'Malley, 2000). Additionally, α-synuclein aggregation can be induced in vitro in the presence of hydrogen peroxide (Hashimoto et al., 1999). This suggests that increased ROS levels not only directly cause neuronal damage but also indirectly contribute to DA depletion and α-synuclein aggregation, which can further exacerbate PD progression.
Taken together, these findings suggest that PD brains are under oxidative stress, which leads to neurodegeneration. However, the mechanisms underlying the increase in ROS levels are not clearly understood. Mitochondrial dysfunction, neuroinflammation, DA autoxidation and environmental toxins have been implicated in the increase of ROS in PD brains (Thomas and Beal, 2007; Jomova et al., 2010; Hwang, 2013). Metal ion dyshomeostasis may also lead to increased ROS production in PD. Generally, the substantia nigra has the highest distribution of iron in the central nervous system. However, PD brains have more elevated levels of iron in this region (Dexter et al., 1989b; Riederer et al., 1989; Sofic et al., 1991; Good et al., 1992; Gerlach et al., 1994; Vymazal et al., 1999; Haacke et al., 2007). The infusion of iron into rat brains results in parkinsonism and behavioral changes (Ben-Shachar and Youdim, 1991; Sengstock et al., 1993). Additionally, in mice the 8-hydroxyquinoline metal ion chelator, clioquinol, and over-expression of ferritin, an iron storage protein, both prevent neurodegeneration in PD models (Kaur et al., 2003). These findings suggest that iron may play a significant role in PD neurodegeneration.
The elevated iron levels in the substantia nigra are proposed to directly and indirectly contribute to increased ROS production. Increased unbound iron levels can produce ROS, such as superoxide, via Fenton chemistry (Halliwell and Gutteridge, 1986). Additionally, ferric ions can precipitate oxidation of DA to 6-OHDA in the presence of hydrogen peroxide (Pezzella et al., 1997). Superoxide and 6-OHDA have the ability to release iron stored in ferritin and [4Fe-4S] cluster-containing enzymes (Liochev and Fridovich, 1994). This can potentially lead to a vicious cycle in which unbound iron increases levels of superoxide and 6-OHDA causing release of more unbound iron. This may contribute to the progressive neurodegeneration observed in PD.
Iron dyshomeostasis not only contributes to ROS production but also negatively impacts the function of proteins that use iron as a co-factor. For example, tyrosine hydroxylase, the rate-limiting enzyme in DA synthesis, depends on iron (Nagatsu, 1995; Ponting, 2001). Therefore, an increase in iron as seen in PD brains may increase DA synthesis, causing excess DA to be released into the cytoplasm, which may lead to increased ROS production. This iron-induced DA dysfunction not only inhibits the normal function of DA but may also lead to increased DA oxidation into the neurotoxin 6-OHDA (Pezzella et al., 1997; Jiang et al., 2013).
In addition to increased ROS production and iron dyshomeostasis, PD brains also exhibit a reduction in metal ion storage capacity and antioxidant activity. Ferritin is a key iron storage protein and disruption of its function perturbs iron homeostasis. PD brains have decreased ferritin levels (Dexter et al., 1991). This potentially leads to iron storage deficiency, which allows unbound reactive iron to accumulate in the substantia nigra, facilitating ROS production (White and Munro, 1988; Dexter et al., 1991; Connor et al., 1995). Neuroferritinopathy is a condition caused by a genetic mutation of the ferritin light chain which disrupts ferritin assembly, leading to iron accumulation and neurodegeneration in the basal ganglia, resulting in severe motor disorders (Curtis et al., 2001; Vidal et al., 2003). In addition to decreased ferritin levels, PD patients have decreased concentration and activity of Cp (Kristinsson et al., 2012). Cp is a multi-copper oxidase that oxidizes ferrous ions (Fe2+) to less reactive ferric ions (Fe3+). This oxidation is essential for cellular iron uptake and efflux by ferroportin and transferrin. Aceruloplasminaemia, a heritable condition resulting from Cp deficiency, leads to iron accumulation in the basal ganglia, neurodegeneration and motor problems including dystonia and tremors (Harris et al., 1998).
PD brains have approximately 40% lower reduced glutathione (GSH), an antioxidant enzyme that catalyzes the reduction of ROS (Sofic et al., 1992). GSH also forms complexes with other enzymes, such as glutathione peroxidase and glutathione S-tranferases, to facilitate ROS reduction (Smeyne and Smeyne, 2013). Decreased antioxidant capacity likely contributes to the oxidative stress seen in PD brains. These findings suggest that defective metal ion transport and storage, decreased antioxidant activity and increased reactive metal ion accumulation contribute to oxidative stress leading to neurodegeneration.
C. elegans: Oxidative Stress and Metal Ion Hypothesis
Another link between PD and oxidative stress is gleaned from studies of mutations in DJ-1 and PINK1, which are associated with early onset PD (Bonifati et al., 2003; Valente et al., 2004). DJ-1 and PINK1 have been shown to protect against oxidative stress (Junn et al., 2005; Pridgeon et al., 2007). This was confirmed in C. elegans by studying the nematode homologs, djr1.1 and pink-1. The djr-1.1 knock-down and pink-1 mutant strains showed increased sensitivity to toxin-induced oxidative stress (Ved et al., 2005; Samann et al., 2009). These C. elegans models complement the familial PD studies and strengthen the hypothesis that oxidative stress contributes to PD pathology.
Transgenic C. elegans expressing α-synuclein in neurons exhibited mitochondrial fragmentation attributed to α-synuclein interaction with mitochondrial membranes, affecting membrane fusion (Kamp et al., 2010). Alpha-synuclein is localized in mitochondria, suggesting that α-synuclein dysfunction potentially contributes to mitochondrial dysfunction in PD (Li et al., 2007). In turn, mitochondrial dysfunction leads to ROS overproduction in the substantia nigra of PD brains, which leads to cellular damage and cell death.
A recent study using electron paramagnetic resonance demonstrated that unbound reactive iron levels increased during oxidative stress in C. elegans (Rangel et al., 2012). Increased iron levels in C. elegans resulted in increased protein oxidation, suggesting that iron triggers increased ROS production. Iron chelation using deferoxamine and over-expression of ferritin (ftn-1) reduced protein oxidation (Valentini et al., 2012). Knock down or deletion of C. elegans SMF-1/2/3 (orthologs of human iron transporter, DMT-1) partially inhibits DAergic neuronal death (Settivari et al., 2009; VanDuyn et al., 2013). PD brains have increased iron levels, decreased ferritin levels and increased DMT-1 levels, consistent with defective iron transport and storage systems in PD brains (Dexter et al., 1991; Salazar et al., 2008).
Knock down of SKN-1 (a C. elegans ortholog of Nrf2), a transcription factor that regulates expression of glutathione S-transferase, increased susceptibility to metal-induced neurodegeneration in DAergic neurons (VanDuyn et al., 2010; Settivari et al., 2013). This finding and the observation that PD brains show decreased levels of glutathione suggests that decreased antioxidant activity contributes to PD pathology (Sofic et al., 1992).
Metal dyshomeostasis and oxidative stress may represent an important component underlying idiopathic PD. C. elegans possesses homologs of some of the iron homeostasis proteins (Table 3) therefore the nematode can be used to further our understanding of metal homeostasis in relation to PD. Even more broadly, any findings can potentially be extended to familial autosomal PD because C. elegans also has homologs for the majority of genes implicated in familial PD (Table 1). These features may be exploited to investigate these genes and their interactions with metal homeostasis.

Table 3. C. elegans iron metal homeostasis proteins.
The Missing Link
Varied evidence supports the potential role of metal dyshomeostasis in PD neurodegeneration. However, the underlying mechanism that leads to metal imbalance still remains to be elucidated. The interplay between tau, α-synuclein and microtubules, may hold an answer to this question (Figure 3).
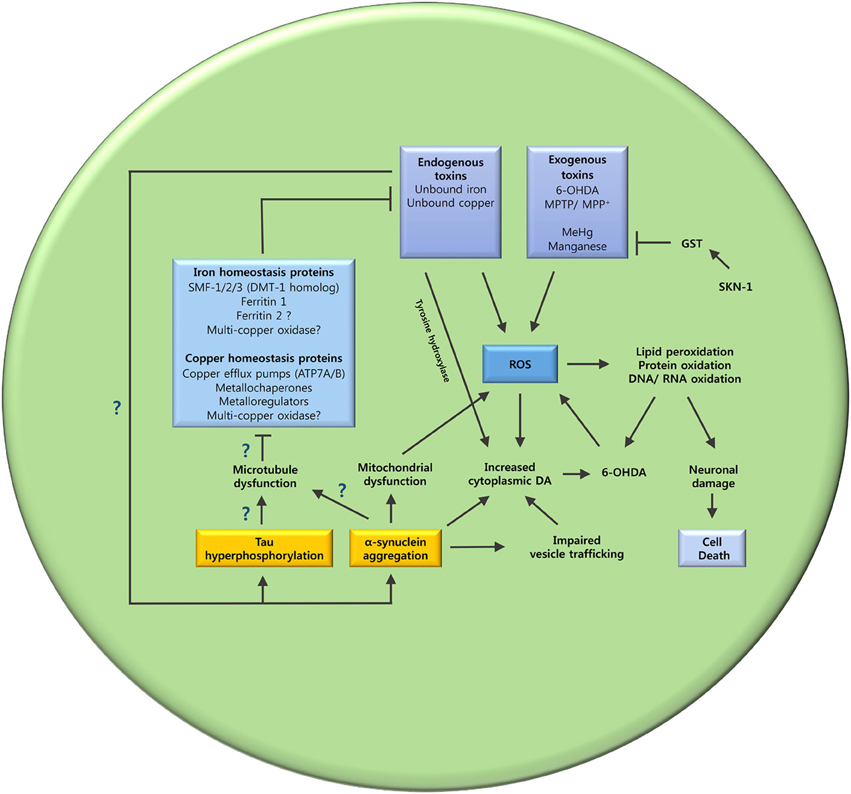
Figure 3. The oxidative stress and metal ion dyshomeostasis cascade. This schematic summarizes the hypothesized mechanisms that may lead to oxidative stress and metal dyshomeostasis in DAergic neurons of C. elegans PD models. The unanswered questions are also highlighted. Does tau hyperphosphorylation and α-synuclein aggregation cause microtubule dysfunction? Are key metal ion homeostasis proteins dependent on microtubules? Does metal ion dyshomeostasis contribute to tau hyperphosphorylation and α-synuclein aggregation? GST (glutathione S-transferase); SKN-1 (Nrf2 ortholog).
Tau, α-Synuclein, and Parkinson's Disease
Tau is a MAP predominantly expressed in axons and is thought to regulate the assembly of microtubules (Weingarten et al., 1975; Kosik and Finch, 1987). Neurofibrillary tangles (NFTs) comprised of hyperphosphorylated tau aggregates are a pathological hallmark of AD (Kidd, 1963; Wischik et al., 1988). Although not often emphasized, tau has also been implicated in PD pathology. Some PD patients have NFTs and in older people with parkinsonian symptoms, the severity of gait impairment appears to correlate with the degree of NFT accumulation (Joachim et al., 1987; Bancher et al., 1993; Schneider et al., 2006). Certain single-nucleotide polymorphisms in the tau gene pose an increased risk factor for PD (Zabetian et al., 2007; Edwards et al., 2010). Tau KO mice have recently been reported to exhibit neuronal iron accumulation, substantia nigra neuronal loss, parkinsonism and cognitive deficits (Lei et al., 2012). Anti-psychotic DA D2 receptor antagonists, such as azaperone, suppress insoluble tau aggregation in C. elegans (McCormick et al., 2013), suggesting an interplay between tau and DA.
Increasing evidence highlights the importance of tau and α-synuclein in PD pathology and indicates that the two proteins significantly interact. Tau is co-localized with α-synuclein in Lewy bodies (Arima et al., 1999). Tau and α-synuclein can seed and promote each other's polymerization to form insoluble aggregates (Giasson et al., 2003; Geddes, 2005). Alpha-synuclein has been shown to directly facilitate tau phosphorylation and also to mediate glycogen synthase kinase 3 (GSK-3β, a serine/threonine protein kinase) catalyzed tau phosphorylation, which is increased in PD brains (Jensen et al., 1999; Muntane et al., 2008; Duka et al., 2009). This indicates that α-synuclein may contribute to the increased GSK-3β activity, which leads to tau hyperphosphorylation.
Tau, α-Synuclein, and Microtubule Dysfunction
Based on the interaction between tau and α-synuclein, the dysfunction of the two proteins may disrupt two key functions of microtubules: axonal transport and maintaining neuronal morphology. Microtubule dysfunction precedes impaired axonal transport (Cartelli et al., 2013). This was deduced from altered mitochondria distribution and neurodegeneration in DAergic neurons of mice exposed to MPTP. MPTP is known to destabilize microtubules and impair axonal transport specifically in DAergic neurons (Cappelletti et al., 2005; Ren et al., 2005; Morfini et al., 2007). Administration of a microtubule stabilizer, Epothilone D, attenuated further nigrostriatal neurodegeneration (Cartelli et al., 2013), highlighting a potential link between axonal transport disruption, microtubule dysfunction and neurodegeneration.
As a MAP, tau not only stabilizes microtubules but also regulates transport by serving as a physical barrier and by interacting with transport motor proteins, dynein and kinesin, to regulate microtubule attachment and detachment (Jancsik et al., 1996; Trinczek et al., 1999; Stamer et al., 2002; Mandelkow et al., 2003; Magnani et al., 2007; Dixit et al., 2008). Tau over-expression disrupts the transport of mitochondria and vesicles leading to accumulation of mitochondria in distal parts of the neuron (Ebneth et al., 1998; Stamer et al., 2002; Mandelkow et al., 2003). Hyperphosphorylated tau filaments have been shown to phosphorylate the kinesin light chain thereby triggering the dissociation of kinesin from its cargo (Lapointe et al., 2009). Phosphorylation of tau at the amino terminus can also impact its inhibitory effect on axonal transport (Kanaan et al., 2012). Mutant tau has been shown to cause “traffic jams” which inhibit axonal transport (Shemesh et al., 2008). The tau dysfunction observed in PD may negatively impact axonal transport, contributing to neurodegeneration.
Alpha-synuclein is co-localized with tubulin in Lewy bodies and co-purifies with microtubules. Additionally, when incubated with tubulin, α-synuclein polymerizes tubulin into microtubules (Alim et al., 2002, 2004). Immunofluorescence staining of α-synuclein transfected COS-1 cells with α-synuclein and tubulin antibodies, showed that α-synuclein co-localized predominantly with microtubules (Alim et al., 2004). Alpha-synuclein binds synaptic vesicles via its amino terminus and is involved in vesicle trafficking (Jensen et al., 1999; Cooper et al., 2006). These findings suggest that α-synuclein, like tau, is a MAP and is involved in axonal transport of vesicles. Alpha-synuclein dysfunction likely leads to impaired axonal transport.
Protein with tau-like repeats (PTL-1) is the only known tau/MAP2 ortholog in C. elegans (Goedert et al., 1996). PTL-1 is important for maintaining C. elegans neuronal morphology (Chew et al., 2013). Null mutants for ptl-1 show accelerated neurite branching and microtubule bundle disorganization in mechanosensory and GABAergic neurons (Chew et al., 2013). Microtubule changes in these neurons suggest a link between tau deficiency and compromised neuronal integrity. In addition, transfection of ptl-1 into non-neuronal cells promotes microtubule assembly and bundling (Goedert et al., 1996).
Loss of function of tau and α-synuclein may result in significant microtubule disruption that leads to neurodegeneration seen in PD. Although the downstream effect of microtubule dysfunction in neurons remains to be elucidated, we can speculate that cellular functions which rely upon microtubules will be disrupted. The function of metal ion regulating proteins, such as ferroportin and copper transporter (ATP7A), are dependent on axonal transport (Cobbold et al., 2004; Moos and Rosengren Nielsen, 2006). Microtubule disruption would likely lead to disrupted trafficking of these metal ion homeostasis proteins. This in turn disrupts metal ion homeostasis leading to accumulation of unbound reactive metal ions and metal ion deficiency, resulting in oxidative stress followed by neuronal loss.
Conclusion
Despite evidence pointing to the involvement of metal ion imbalance and microtubule dysfunction in neurodegeneration, few studies have attempted to link these two elements. We propose that disrupted axonal transport and neuronal integrity greatly impacts metal ion balance by hindering the trafficking of metal ion homeostasis proteins and neuronal anti-oxidants (Figure 3). Disrupting metal ion homeostasis is likely to result in oxidative stress leading to neuronal loss. In addition, microtubule disruption may result in loss of synaptic connections due to altered neuronal morphology causing synaptic transmission impairment. The interplay between tau, α-synuclein and metal dyshomeostasis offers a new avenue of investigation. C. elegans has homologs for many of the genes involved in iron regulation (Table 3) and can be genetically manipulated to express transgenes in the absence of homologs (Table 2); this may represent an ideal system in which to investigate these questions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Australian Research Council (ARC), the National Health and Medical Research Council (NHMRC) of Australia, and the Victorian Government's Operational Infrastructure Support Program supported this work.
References
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/S0896-6273(00)80886-7
Alam, Z. I., Daniel, S. E., Lees, A. J., Marsden, D. C., Jenner, P., and Halliwell, B. (1997a). A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J. Neurochem. 69, 1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x
Alam, Z. I., Jenner, A., Daniel, S. E., Lees, A. J., Cairns, N., Marsden, C. D., et al. (1997b). Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 69, 1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x
Alim, M. A., Hossain, M. S., Arima, K., Takeda, K., Izumiyama, Y., Nakamura, M., et al. (2002). Tubulin seeds alpha-synuclein fibril formation. J. Biol. Chem. 277, 2112–2117. doi: 10.1074/jbc.M102981200
Alim, M. A., Ma, Q.-L., Takeda, K., Aizawa, T., Matsubara, M., Nakamura, M., et al. (2004). Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimers Dis. 6, 435–442. discussion: 443–439.
Andreini, C., Bertini, I., Cavallaro, G., Holliday, G. L., and Thornton, J. M. (2008). Metal ions in biological catalysis: from enzyme databases to general principles. J. Biol. Inorg. Chem. 13, 1205–1218. doi: 10.1007/s00775-008-0404-5
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Arima, K., Hirai, S., Sunohara, N., Aoto, K., Izumiyama, Y., Ueda, K., et al. (1999). Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in lewy bodies in sporadic Parkinson's disease and in dementia with Lewy bodies. Brain Res. 843, 53–61. doi: 10.1016/S0006-8993(99)01848-X
Arredondo, M., and Nunez, M. T. (2005). Iron and copper metabolism. Mol. Aspects Med. 26, 313–327. doi: 10.1016/j.mam.2005.07.010
Bains, J. S., and Shaw, C. A. (1997). Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev. 25, 335–358. doi: 10.1016/S0165-0173(97)00045-3
Bancher, C., Braak, H., Fischer, P., and Jellinger, K. A. (1993). Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer's and Parkinson's disease patients. Neurosci. Lett. 162, 179–182. doi: 10.1016/0304-3940(93)90590-H
Bargmann, C. I. (1998). Neurobiology of the Caenorhabditis elegans genome. Science 282, 2028–2033. doi: 10.1126/science.282.5396.2028
Ben-Shachar, D., and Youdim, M. B. (1991). Intranigral iron injection induces behavioral and biochemical “parkinsonism” in rats. J. Neurochem. 57, 2133–2135. doi: 10.1111/j.1471-4159.1991.tb06432.x
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Brandt, R., Gergou, A., Wacker, I., Fath, T., and Hutter, H. (2009). A Caenorhabditis elegans model of tau hyperphosphorylation: induction of developmental defects by transgenic overexpression of Alzheimer's disease-like modified tau. Neurobiol. Aging 30, 22–33. doi: 10.1016/j.neurobiolaging.2007.05.011
Bras, J., Simon-Sanchez, J., Federoff, M., Morgadinho, A., Januario, C., Ribeiro, M., et al. (2009). Lack of replication of association between GIGYF2 variants and Parkinson disease. Hum. Mol. Genet. 18, 341–346. doi: 10.1093/hmg/ddn340
Braungart, E., Gerlach, M., Riederer, P., Baumeister, R., and Hoener, M. C. (2004). Caenorhabditis elegans MPP+ model of Parkinson's disease for high-throughput drug screenings. Neurodegener. Dis. 1, 175–183. doi: 10.1159/000080983
Buter, T. C., van den Hout, A., Matthews, F. E., Larsen, J. P., Brayne, C., and Aarsland, D. (2008). Dementia and survival in Parkinson disease: a 12-year population study. Neurology 70, 1017–1022. doi: 10.1212/01.wnl.0000306632.43729.24
Cairo, G., Recalcati, S., Pietrangelo, A., and Minotti, G. (2002). The iron regulatory proteins: targets and modulators of free radical reactions and oxidative damage. Free Radic. Biol. Med. 32, 1237–1243. doi: 10.1016/S0891-5849(02)00825-0
Camakaris, J., Voskoboinik, I., and Mercer, J. F. (1999). Molecular mechanisms of copper homeostasis. Biochem. Biophys. Res. Commun. 261, 225–232. doi: 10.1006/bbrc.1999.1073
Cao, S., Gelwix, C. C., Caldwell, K. A., and Caldwell, G. A. (2005). Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J. Neurosci. 25, 3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005
Cappelletti, G., Surrey, T., and Maci, R. (2005). The parkinsonism producing neurotoxin MPP+ affects microtubule dynamics by acting as a destabilising factor. FEBS Lett. 579, 4781–4786. doi: 10.1016/j.febslet.2005.07.058
Cartelli, D., Casagrande, F., Busceti, C. L., Bucci, D., Molinaro, G., Traficante, A., et al. (2013). Microtubule alterations occur early in experimental parkinsonism and the microtubule stabilizer epothilone D is neuroprotective. Sci. Rep. 3:1837. doi: 10.1038/srep01837
Catoire, H., Dion, P. A., Xiong, L., Amari, M., Gaudet, R., Girard, S. L., et al. (2011). Restless legs syndrome-associated MEIS1 risk variant influences iron homeostasis. Ann. Neurol. 70, 170–175. doi: 10.1002/ana.22435
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263, 802–805. doi: 10.1126/science.8303295
Chartier-Harlin, M. C., Dachsel, J. C., Vilarino-Guell, C., Lincoln, S. J., Lepretre, F., Hulihan, M. M., et al. (2011). Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 89, 398–406. doi: 10.1016/j.ajhg.2011.08.009
Chew, Y. L., Fan, X., Gotz, J., and Nicholas, H. R. (2013). PTL-1 regulates neuronal integrity and lifespan in C. elegans. J. Cell Sci. 126, 2079–2091. doi: 10.1242/jcs.jcs124404
Cobbold, C., Coventry, J., Ponnambalam, S., and Monaco, A. P. (2004). Actin and microtubule regulation of trans-Golgi network architecture, and copper-dependent protein transport to the cell surface. Mol. Membr. Biol. 21, 59–66. doi: 10.1080/096870310001607350
Connor, J. R., Snyder, B. S., Arosio, P., Loeffler, D. A., and Lewitt, P. (1995). A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer's diseased brains. J. Neurochem. 65, 717–724. doi: 10.1046/j.1471-4159.1995.65020717.x
Consortium, C. E. S. (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018. doi: 10.1126/science.282.5396.2012
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328. doi: 10.1126/science.1129462
Crouch, P. J., Savva, M. S., Hung, L. W., Donnelly, P. S., Mot, A. I., Parker, S. J., et al. (2011). The Alzheimer's therapeutic PBT2 promotes amyloid-beta degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 119, 220–230. doi: 10.1111/j.1471-4159.2011.07402.x
Culetto, E., and Sattelle, D. B. (2000). A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum. Mol. Genet. 9, 869–877. doi: 10.1093/hmg/9.6.869
Curtis, A. R., Fey, C., Morris, C. M., Bindoff, L. A., Ince, P. G., Chinnery, P. F., et al. (2001). Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet. 28, 350–354. doi: 10.1038/ng571
Dexter, D. T., Carayon, A., Javoy-Agid, F., Agid, Y., Wells, F. R., Daniel, S. E., et al. (1991). Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114(pt 4), 1953–1975. doi: 10.1093/brain/114.4.1953
Dexter, D. T., Carter, C. J., Wells, F. R., Javoy-Agid, F., Agid, Y., Lees, A., et al. (1989a). Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J. Neurochem. 52, 381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x
Dexter, D. T., Holley, A. E., Flitter, W. D., Slater, T. F., Wells, F. R., Daniel, S. E., et al. (1994). Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov. Disord. 9, 92–97. doi: 10.1002/mds.870090115
Dexter, D. T., Wells, F. R., Lees, A. J., Agid, F., Agid, Y., Jenner, P., et al. (1989b). Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J. Neurochem. 52, 1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x
Dixit, R., Ross, J. L., Goldman, Y. E., and Holzbaur, E. L. F. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. doi: 10.1126/science.1152993
Dorman, J. B., Albinder, B., Shroyer, T., and Kenyon, C. (1995). The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics 141, 1399–1406.
Duka, T., Duka, V., Joyce, J. N., and Sidhu, A. (2009). Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson's disease models. FASEB J. 23, 2820–2830. doi: 10.1096/fj.08-120410
Ebneth, A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B., and Mandelkow, E. (1998). Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J. Cell Biol. 143, 777–794. doi: 10.1083/jcb.143.3.777
Edvardson, S., Cinnamon, Y., Ta-Shma, A., Shaag, A., Yim, Y. I., Zenvirt, S., et al. (2012). A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 7:e36458. doi: 10.1371/journal.pone.0036458
Edwards, T. L., Scott, W. K., Almonte, C., Burt, A., Powell, E. H., Beecham, G. W., et al. (2010). Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 74, 97–109. doi: 10.1111/j.1469-1809.2009.00560.x
Ellis, H. M., and Horvitz, H. R. (1986). Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829. doi: 10.1016/0092-8674(86)90004-8
Faber, P. W., Alter, J. R., Macdonald, M. E., and Hart, A. C. (1999). Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc. Natl. Acad. Sci. U.S.A. 96, 179–184. doi: 10.1073/pnas.96.1.179
Fang, F. C. (2004). Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832. doi: 10.1038/nrmicro1004
Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., and Mello, C. C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. doi: 10.1038/35888
Forno, L. S. (1996). Neuropathology of Parkinson's disease. J. Neuropathol. Exp. Neurol. 55, 259–272. doi: 10.1097/00005072-199603000-00001
Freeman, L. R., and Keller, J. N. (2012). Oxidative stress and cerebral endothelial cells: regulation of the blood-brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta 1822, 822–829. doi: 10.1016/j.bbadis.2011.12.009
Friedman, D. B., and Johnson, T. E. (1988). A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 118, 75–86.
Fuxe, K., and Jonsson, G. (1973). The histochemical fluorescence method for the demonstration of catecholamines. Theory, practice and application. J. Histochem. Cytochem. 21, 293–311. doi: 10.1177/21.4.293
Geddes, J. W. (2005). alpha-Synuclein: a potent inducer of tau pathology. Exp. Neurol. 192, 244–250. doi: 10.1016/j.expneurol.2004.12.002
Gerlach, M., Ben-Shachar, D., Riederer, P., and Youdim, M. B. (1994). Altered brain metabolism of iron as a cause of neurodegenerative diseases? J. Neurochem. 63, 793–807. doi: 10.1046/j.1471-4159.1994.63030793.x
Giasson, B. I., Forman, M. S., Higuchi, M., Golbe, L. I., Graves, C. L., Kotzbauer, P. T., et al. (2003). Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300, 636–640. doi: 10.1126/science.1082324
Gkouvatsos, K., Papanikolaou, G., and Pantopoulos, K. (2012). Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 1820, 188–202. doi: 10.1016/j.bbagen.2011.10.013
Goedert, C., Baur, C. P., Ahringer, J., Jakes, R., Hasegawa, M., Spillantini, M. G., et al. (1996). PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J. Cell Sci. 109, 2661–2672.
Good, P. F., Olanow, C. W., and Perl, D. P. (1992). Neuromelanin-containing neurons of the substantia nigra accumulate iron and aluminum in Parkinson's disease: a LAMMA study. Brain Res. 593, 343–346. doi: 10.1016/0006-8993(92)91334-B
Gordon, P., Hingula, L., Krasny, M. L., Swienckowski, J. L., Pokrywka, N. J., and Raley-Susman, K. M. (2008). The invertebrate microtubule-associated protein PTL-1 functions in mechanosensation and development in Caenorhabditis elegans. Dev. Genes Evol. 218, 541–551. doi: 10.1007/s00427-008-0250-z
Gotz, M. E., Double, K., Gerlach, M., Youdim, M. B., and Riederer, P. (2004). The relevance of iron in the pathogenesis of Parkinson's disease. Ann. N.Y. Acad. Sci. 1012, 193–208. doi: 10.1196/annals.1306.017
Haacke, E. M., Ayaz, M., Khan, A., Manova, E. S., Krishnamurthy, B., Gollapalli, L., et al. (2007). Establishing a baseline phase behavior in magnetic resonance imaging to determine normal vs. abnormal iron content in the brain. J. Magn. Reson. Imaging 26, 256–264. doi: 10.1002/jmri.22987
Halliwell, B. (2001). Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 18, 685–716. doi: 10.2165/00002512-200118090-00004
Halliwell, B., and Gutteridge, J. M. (1986). Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch. Biochem. Biophys. 246, 501–514. doi: 10.1016/0003-9861(86)90305-X
Hamamichi, S., Rivas, R. N., Knight, A. L., Cao, S., Caldwell, K. A., and Caldwell, G. A. (2008). Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson's disease model. Proc. Natl. Acad. Sci. U.S.A. 105, 728–733. doi: 10.1073/pnas.0711018105
Harris, Z. L., Klomp, L. W., and Gitlin, J. D. (1998). Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Am. J. Clin. Nutr. 67, 972S–977S.
Hashimoto, M., Hsu, L. J., Xia, Y., Takeda, A., Sisk, A., Sundsmo, M., et al. (1999). Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 10, 717–721. doi: 10.1097/00001756-199903170-00011
Healy, D. G., Abou-Sleiman, P. M., Casas, J. P., Ahmadi, K. R., Lynch, T., Gandhi, S., et al. (2006). UCHL-1 is not a Parkinson's disease susceptibility gene. Ann. Neurol. 59, 627–633. doi: 10.1002/ana.20757
Hedgecock, E. M., Sulston, J. E., and Thomson, J. N. (1983). Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 220, 1277–1279. doi: 10.1126/science.6857247
Hekimi, S., Lapointe, J., and Wen, Y. (2011). Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 21, 569–576. doi: 10.1016/j.tcb.2011.06.008
Hely, M. A., Reid, W. G., Adena, M. A., Halliday, G. M., and Morris, J. G. (2008). The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov. Disord. 23, 837–844. doi: 10.1002/mds.21956
Hills, T., Brockie, P. J., and Maricq, A. V. (2004). Dopamine and glutamate control area-restricted search behavior in Caenorhabditis elegans. J. Neurosci. 24, 1217–1225. doi: 10.1523/JNEUROSCI.1569-03.2004
Hirtz, D., Thurman, D. J., Gwinn-Hardy, K., Mohamed, M., Chaudhuri, A. R., and Zalutsky, R. (2007). How common are the “common” neurologic disorders? Neurology 68, 326–337. doi: 10.1212/01.wnl.0000252807.38124.a3
Hornykiewicz, O., and Kish, S. J. (1987). Biochemical pathophysiology of Parkinson's disease. Adv. Neurol. 45, 19–34.
Hwang, O. (2013). Role of oxidative stress in Parkinson's disease. Exp. Neurobiol. 22, 11–17. doi: 10.5607/en.2013.22.1.11
Irizarry, M. C., Growdon, W., Gomez-Isla, T., Newell, K., George, J. M., Clayton, D. F., et al. (1998). Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J. Neuropathol. Exp. Neurol. 57, 334–337. doi: 10.1097/00005072-199804000-00005
Iwai, A., Masliah, E., Yoshimoto, M., Ge, N., Flanagan, L., De Silva, H. A., et al. (1995). The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475. doi: 10.1016/0896-6273(95)90302-X
Jancsik, V., Filliol, D., and Rendon, A. (1996). Tau proteins bind to kinesin and modulate its activation by microtubules. Neurobiology (Bp) 4, 417–429.
Janvin, C. C., Larsen, J. P., Aarsland, D., and Hugdahl, K. (2006). Subtypes of mild cognitive impairment in Parkinson's disease: progression to dementia. Mov. Disord. 21, 1343–1349. doi: 10.1002/mds.20974
Jenner, P. (2003). Oxidative stress in Parkinson's disease. Ann. Neurol. 53(Suppl. 3), S26–S36. discussion: S36–S38. doi: 10.1002/ana.10483
Jenner, P., and Olanow, C. W. (1996). Oxidative stress and the pathogenesis of Parkinson's disease. Neurology 47, 161S–170S. doi: 10.1212/WNL.47.6_Suppl_3.161S
Jensen, P. H., Hager, H., Nielsen, M. S., Hojrup, P., Gliemann, J., and Jakes, R. (1999). alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 274, 25481–25489. doi: 10.1074/jbc.274.36.25481
Jiang, D., Shi, S., Zhang, L., Liu, L., Ding, B., Zhao, B., et al. (2013). Inhibition of the Fe(III)-catalyzed dopamine oxidation by ATP and its relevance to oxidative stress in Parkinson's disease. ACS Chem. Neurosci. 4, 1305–1313. doi: 10.1021/cn400105d
Joachim, C. L., Morris, J. H., Kosik, K. S., and Selkoe, D. J. (1987). Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann. Neurol. 22, 514–520. doi: 10.1002/ana.410220411
Jomova, K., Vondrakova, D., Lawson, M., and Valko, M. (2010). Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 345, 91–104. doi: 10.1007/s11010-010-0563-x
Junn, E., Taniguchi, H., Jeong, B. S., Zhao, X., Ichijo, H., and Mouradian, M. M. (2005). Interaction of DJ-1 with Daxx inhibits apoptosis signal-regulating kinase 1 activity and cell death. Proc. Natl. Acad. Sci. U.S.A. 102, 9691–9696. doi: 10.1073/pnas.0409635102
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Kanaan, N. M., Morfini, G., Pigino, G., Lapointe, N. E., Andreadis, A., Song, Y., et al. (2012). Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol. Aging 33, 826.e15–826.e30. doi: 10.1016/j.neurobiolaging.2011.06.006
Karpinar, D. P., Balija, M. B., Kugler, S., Opazo, F., Rezaei-Ghaleh, N., Wender, N., et al. (2009). Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 28, 3256–3268. doi: 10.1038/emboj.2009.257
Kaur, D., Yantiri, F., Rajagopalan, S., Kumar, J., Mo, J. Q., Boonplueang, R., et al. (2003). Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron 37, 899–909. doi: 10.1016/S0896-6273(03)00126-0
Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464. doi: 10.1038/366461a0
Kepp, K. P. (2012). Bioinorganic chemistry of Alzheimer's disease. Chem. Rev. 112, 5193–5239. doi: 10.1021/cr300009x
Kidd, M. (1963). Paired helical filaments in electron microscopy of Alzheimer's disease. Nature 197, 192–193. doi: 10.1038/197192b0
Kimura, K. D., Tissenbaum, H. A., Liu, Y., and Ruvkun, G. (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–946. doi: 10.1126/science.277.5328.942
Kish, S. J., Shannak, K., and Hornykiewicz, O. (1988). Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. N. Engl. J. Med. 318, 876–880. doi: 10.1056/NEJM198804073181402
Kokhan, V. S., Afanasyeva, M. A., and Van'kin, G. I. (2012). alpha-Synuclein knockout mice have cognitive impairments. Behav. Brain Res. 231, 226–230. doi: 10.1016/j.bbr.2012.03.026
Koroglu, C., Baysal, L., Cetinkaya, M., Karasoy, H., and Tolun, A. (2013). DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat. Disord. 19, 320–324. doi: 10.1016/j.parkreldis.2012.11.006
Kosik, K. S., and Finch, E. A. (1987). MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: an immunocytochemical study of cultured rat cerebrum. J. Neurosci. 7, 3142–3153.
Kraemer, B. C., Zhang, B., Leverenz, J. B., Thomas, J. H., Trojanowski, J. Q., and Schellenberg, G. D. (2003). Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. U.S.A. 100, 9980–9985. doi: 10.1073/pnas.1533448100
Krebs, C. E., Karkheiran, S., Powell, J. C., Cao, M., Makarov, V., Darvish, H., et al. (2013). The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat. 34, 1200–1207. doi: 10.1002/humu.22372
Kristinsson, J., Snaedal, J., Torsdottir, G., and Johannesson, T. (2012). Ceruloplasmin and iron in Alzheimer's disease and Parkinson's disease: a synopsis of recent studies. Neuropsychiatr. Dis. Treat. 8, 515–521. doi: 10.2147/NDT.S34729
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Kuwahara, T., Koyama, A., Gengyo-Ando, K., Masuda, M., Kowa, H., Tsunoda, M., et al. (2006). Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J. Biol. Chem. 281, 334–340. doi: 10.1074/jbc.M504860200
Kuwahara, T., Koyama, A., Koyama, S., Yoshina, S., Ren, C. H., Kato, T., et al. (2008). A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum. Mol. Genet. 17, 2997–3009. doi: 10.1093/hmg/ddn198
Lai, C. H., Chou, C. Y., Ch'ang, L. Y., Liu, C. S., and Lin, W. (2000). Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 10, 703–713. doi: 10.1101/gr.10.5.703
Lakso, M., Vartiainen, S., Moilanen, A. M., Sirvio, J., Thomas, J. H., Nass, R., et al. (2003). Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 86, 165–172. doi: 10.1046/j.1471-4159.2003.01809.x
Lannfelt, L., Blennow, K., Zetterberg, H., Batsman, S., Ames, D., Harrison, J., et al. (2008). Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7, 779–786. doi: 10.1016/S1474-4422(08)70167-4
Lapointe, N. E., Morfini, G., Pigino, G., Gaisina, I. N., Kozikowski, A. P., Binder, L. I., et al. (2009). The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res. 87, 440–451. doi: 10.1002/jnr.21850
Lee, D. W., and Andersen, J. K. (2010). Iron elevations in the aging Parkinsonian brain: a consequence of impaired iron homeostasis? J. Neurochem. 112, 332–339. doi: 10.1111/j.1471-4159.2009.06470.x
Lee, F. J., Liu, F., Pristupa, Z. B., and Niznik, H. B. (2001). Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 15, 916–926. doi: 10.1096/fj.00-0334com
Lei, P., Ayton, S., Finkelstein, D. I., Spoerri, L., Ciccotosto, G. D., Wright, D. K., et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295. doi: 10.1038/nm.2613
Li, W. W., Yang, R., Guo, J. C., Ren, H. M., Zha, X. L., Cheng, J. S., et al. (2007). Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 18, 1543–1546. doi: 10.1097/WNR.0b013e3282f03db4
Liochev, S. I., and Fridovich, I. (1994). The role of O2.- in the production of HO.: in vitro and in vivo. Free Radic. Biol. Med. 16, 29–33. doi: 10.1016/0891-5849(94)90239-9
Lotharius, J., and O'Malley, K. L. (2000). The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J. Biol. Chem. 275, 38581–38588. doi: 10.1074/jbc.M005385200
Luo, Y., Umegaki, H., Wang, X., Abe, R., and Roth, G. S. (1998). Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J. Biol. Chem. 273, 3756–3764. doi: 10.1074/jbc.273.6.3756
Magnani, E., Fan, J., Gasparini, L., Golding, M., Williams, M., Schiavo, G., et al. (2007). Interaction of tau protein with the dynactin complex. EMBO J. 26, 4546–4554. doi: 10.1038/sj.emboj.7601878
Mandelkow, E. M., Stamer, K., Vogel, R., Thies, E., and Mandelkow, E. (2003). Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 24, 1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007
Marttila, R. J., Lorentz, H., and Rinne, U. K. (1988). Oxygen toxicity protecting enzymes in Parkinson's disease. Increase of superoxide dismutase-like activity in the substantia nigra and basal nucleus. J. Neurol. Sci. 86, 321–331. doi: 10.1016/0022-510X(88)90108-6
McColl, G., Roberts, B. R., Gunn, A. P., Perez, K. A., Tew, D. J., Masters, C. L., et al. (2009). The Caenorhabditis elegans A beta 1-42 model of Alzheimer disease predominantly expresses A beta 3-42. J. Biol. Chem. 284, 22697–22702. doi: 10.1074/jbc.C109.028514
McColl, G., Roberts, B. R., Pukala, T. L., Kenche, V. B., Roberts, C. M., Link, C. D., et al. (2012). Utility of an improved model of amyloid-beta (Abeta(1)(-)(4)(2)) toxicity in Caenorhabditis elegans for drug screening for Alzheimer's disease. Mol. Neurodegener. 7, 57. doi: 10.1186/1750-1326-7-57
McCormick, A. V., Wheeler, J. M., Guthrie, C. R., Liachko, N. F., and Kraemer, B. C. (2013). Dopamine D2 receptor antagonism suppresses tau aggregation and neurotoxicity. Biol. Psychiatry 73, 464–471. doi: 10.1016/j.biopsych.2012.08.027
McDonald, P. W., Jessen, T., Field, J. R., and Blakely, R. D. (2006). Dopamine signaling architecture in Caenorhabditis elegans. Cell. Mol. Neurobiol. 26, 593–618. doi: 10.1007/s10571-006-9003-6
Moos, T., and Rosengren Nielsen, T. (2006). Ferroportin in the postnatal rat brain: implications for axonal transport and neuronal export of iron. Semin. Pediatr. Neurol. 13, 149–157. doi: 10.1016/j.spen.2006.08.003
Morfini, G., Pigino, G., Opalach, K., Serulle, Y., Moreira, J. E., Sugimori, M., et al. (2007). 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 104, 2442–2447. doi: 10.1073/pnas.0611231104
Muntane, G., Dalfo, E., Martinez, A., and Ferrer, I. (2008). Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer's disease, and in Parkinson's disease and related alpha-synucleinopathies. Neuroscience 152, 913–923. doi: 10.1016/j.neuroscience.2008.01.030
Murakami, S., and Johnson, T. E. (1996). A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditis elegans. Genetics 143, 1207–1218.
Nagatsu, T. (1995). Tyrosine hydroxylase: human isoforms, structure and regulation in physiology and pathology. Essays Biochem. 30, 15–35.
Nakajo, S., Shioda, S., Nakai, Y., and Nakaya, K. (1994). Localization of phosphoneuroprotein 14 (PNP 14) and its mRNA expression in rat brain determined by immunocytochemistry and in situ hybridization. Brain Res. Mol. Brain Res. 27, 81–86. doi: 10.1016/0169-328X(94)90187-2
Nass, R., Hall, D. H., Miller, D. M. 3rd., and Blakely, R. D. (2002). Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 99, 3264–3269. doi: 10.1073/pnas.042497999
Nemani, V. M., Lu, W., Berge, V., Nakamura, K., Onoa, B., Lee, M. K., et al. (2010). Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. doi: 10.1016/j.neuron.2009.12.023
Niki, E. (2009). Lipid peroxidation: physiological levels and dual biological effects. Free Radic. Biol. Med. 47, 469–484. doi: 10.1016/j.freeradbiomed.2009.05.032
Nunez, M. T., Urrutia, P., Mena, N., Aguirre, P., Tapia, V., and Salazar, J. (2012). Iron toxicity in neurodegeneration. Biometals 25, 761–776. doi: 10.1007/s10534-012-9523-0
Oakley, A. E., Collingwood, J. F., Dobson, J., Love, G., Perrott, H. R., Edwardson, J. A., et al. (2007). Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 68, 1820–1825. doi: 10.1212/01.wnl.0000262033.01945.9a
Offen, D., Hochman, A., Gorodin, S., Ziv, I., Shirvan, A., Barzilai, A., et al. (1999). Oxidative stress and neuroprotection in Parkinson's disease: implications from studies on dopamine-induced apoptosis. Adv. Neurol. 80, 265–269.
Olanow, C. W., and Arendash, G. W. (1994). Metals and free radicals in neurodegeneration. Curr. Opin. Neurol. 7, 548–558. doi: 10.1097/00019052-199412000-00013
Orth, M., and Schapira, A. H. (2002). Mitochondrial involvement in Parkinson's disease. Neurochem. Int. 40, 533–541. doi: 10.1016/S0197-0186(01)00124-3
Pang, W. L., Kaur, A., Ratushny, A. V., Cvetkovic, A., Kumar, S., Pan, M., et al. (2013). Metallochaperones regulate intracellular copper levels. PLoS Comput. Biol. 9:e1002880. doi: 10.1371/journal.pcbi.1002880
Pankratz, N., Nichols, W. C., Uniacke, S. K., Halter, C., Rudolph, A., Shults, C., et al. (2002). Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am. J. Hum. Genet. 71, 124–135. doi: 10.1086/341282
Pattison, D. I., Dean, R. T., and Davies, M. J. (2002). Oxidation of DNA, proteins and lipids by DOPA, protein-bound DOPA, and related catechol(amine)s. Toxicology 177, 23–37. doi: 10.1016/S0300-483X(02)00193-2
Pezzella, A., D'Ischia, M., Napolitano, A., Misuraca, G., and Prota, G. (1997). Iron-mediated generation of the neurotoxin 6-hydroxydopamine quinone by reaction of fatty acid hydroperoxides with dopamine: a possible contributory mechanism for neuronal degeneration in Parkinson's disease. J. Med. Chem. 40, 2211–2216. doi: 10.1021/jm970099t
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Ponting, C. P. (2001). Domain homologues of dopamine beta-hydroxylase and ferric reductase: roles for iron metabolism in neurodegenerative disorders? Hum. Mol. Genet. 10, 1853–1858. doi: 10.1093/hmg/10.17.1853
Pridgeon, J. W., Olzmann, J. A., Chin, L. S., and Li, L. (2007). PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 5:e172. doi: 10.1371/journal.pbio.0050172
Proukakis, C., Dudzik, C. G., Brier, T., Mackay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/WNL.0b013e31828727ba
Przedborski, S., Levivier, M., Jiang, H., Ferreira, M., Jackson-Lewis, V., Donaldson, D., et al. (1995). Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience 67, 631–647. doi: 10.1016/0306-4522(95)00066-R
Pu, P., and Le, W. (2008). Dopamine neuron degeneration induced by MPP+ is independent of CED-4 pathway in Caenorhabditis elegans. Cell Res. 18, 978–981. doi: 10.1038/cr.2008.279
Qin, Z., Hu, D., Han, S., Reaney, S. H., Di Monte, D. A., and Fink, A. L. (2007). Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. J. Biol. Chem. 282, 5862–5870. doi: 10.1074/jbc.M608126200
Quadri, M., Fang, M., Picillo, M., Olgiati, S., Breedveld, G. J., Graafland, J., et al. (2013). Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum. Mutat. 34, 1208–1215. doi: 10.1002/humu.22373
Rae, T. D., Schmidt, P. J., Pufahl, R. A., Culotta, V. C., and O'Halloran, T. V. (1999). Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284, 805–808. doi: 10.1126/science.284.5415.805
Rand, J. B., and Nonet, M. L. (1997). “Synaptic transmission,” in C. elegans II, 2nd Edn., eds D. L. Riddle, T. Blumenthal, B. J. Meyer, and J. R. Priess. (New York, NY: Cold Spring Harbor), 611–643.
Rangel, N. A., Lin, L., Rakariyatham, K., Bach, A., Trinh, K., Clement, M. H., et al. (2012). Unincorporated iron pool is linked to oxidative stress and iron levels in Caenorhabditis elegans. Biometals 25, 971–985. doi: 10.1007/s10534-012-9563-5
Recchia, A., Debetto, P., Negro, A., Guidolin, D., Skaper, S. D., and Giusti, P. (2004). Alpha-synuclein and Parkinson's disease. FASEB J. 18, 617–626. doi: 10.1096/fj.03-0338rev
Ren, Y., Liu, W., Jiang, H., Jiang, Q., and Feng, J. (2005). Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J. Biol. Chem. 280, 34105–34112. doi: 10.1074/jbc.M503483200
Riederer, P., Sofic, E., Rausch, W. D., Schmidt, B., Reynolds, G. P., Jellinger, K., et al. (1989). Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 52, 515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x
Ruan, Q., Harrington, A. J., Caldwell, K. A., Caldwell, G. A., and Standaert, D. G. (2010). VPS41, a protein involved in lysosomal trafficking, is protective in Caenorhabditis elegans and mammalian cellular models of Parkinson's disease. Neurobiol. Dis. 37, 330–338. doi: 10.1016/j.nbd.2009.10.011
Saggu, H., Cooksey, J., Dexter, D., Wells, F. R., Lees, A., Jenner, P., et al. (1989). A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J. Neurochem. 53, 692–697. doi: 10.1111/j.1471-4159.1989.tb11759.x
Saha, S., Guillily, M. D., Ferree, A., Lanceta, J., Chan, D., Ghosh, J., et al. (2009). LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J. Neurosci. 29, 9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009
Salazar, J., Mena, N., Hunot, S., Prigent, A., Alvarez-Fischer, D., Arredondo, M., et al. (2008). Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 105, 18578–18583. doi: 10.1073/pnas.0804373105
Samann, J., Hegermann, J., von Gromoff, E., Eimer, S., Baumeister, R., and Schmidt, E. (2009). Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 284, 16482–16491. doi: 10.1074/jbc.M808255200
Sanyal, S., Wintle, R. F., Kindt, K. S., Nuttley, W. M., Arvan, R., Fitzmaurice, P., et al. (2004). Dopamine modulates the plasticity of mechanosensory responses in Caenorhabditis elegans. EMBO J. 23, 473–482. doi: 10.1038/sj.emboj.7600057
Sauer, H., and Oertel, W. H. (1994). Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience 59, 401–415. doi: 10.1016/0306-4522(94)90605-X
Sawin, E. R., Ranganathan, R., and Horvitz, H. R. (2000). C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26, 619–631. doi: 10.1016/S0896-6273(00)81199-X
Schafer, W. R., and Kenyon, C. J. (1995). A calcium-channel homologue required for adaptation to dopamine and serotonin in Caenorhabditis elegans. Nature 375, 73–78. doi: 10.1038/375073a0
Schneider, J. A., Li, J. L., Li, Y., Wilson, R. S., Kordower, J. H., and Bennett, D. A. (2006). Substantia nigra tangles are related to gait impairment in older persons. Ann. Neurol. 59, 166–173. doi: 10.1002/ana.20723
Sengstock, G. J., Olanow, C. W., Menzies, R. A., Dunn, A. J., and Arendash, G. W. (1993). Infusion of iron into the rat substantia nigra: nigral pathology and dose-dependent loss of striatal dopaminergic markers. J. Neurosci. Res. 35, 67–82. doi: 10.1002/jnr.490350109
Settivari, R., Levora, J., and Nass, R. (2009). The divalent metal transporter homologues SMF-1/2 mediate dopamine neuron sensitivity in caenorhabditis elegans models of manganism and parkinson disease. J. Biol. Chem. 284, 35758–35768. doi: 10.1074/jbc.M109.051409
Settivari, R., VanDuyn, N., Levora, J., and Nass, R. (2013). The Nrf2/SKN-1-dependent glutathione S-transferase pi homologue GST-1 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of manganism. Neurotoxicology 38, 51–60. doi: 10.1016/j.neuro.2013.05.014
Shemesh, O. A., Erez, H., Ginzburg, I., and Spira, M. E. (2008). Tau-induced traffic jams reflect organelles accumulation at points of microtubule polar mismatching. Traffic 9, 458–471. doi: 10.1111/j.1600-0854.2007.00695.x
Sies, H. (1991). Oxidative stress: from basic research to clinical application. Am. J. Med. 91, 31S–38S. doi: 10.1016/0002-9343(91)90281-2
Simon-Sanchez, J., and Singleton, A. B. (2008). Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum. Mol. Genet. 17, 1988–1993. doi: 10.1093/hmg/ddn096
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson's disease. Science 302:841. doi: 10.1126/science.1090278
Smeyne, M., and Smeyne, R. J. (2013). Glutathione metabolism and Parkinson's disease. Free Radic. Biol. Med. 62, 13–25. doi: 10.1016/j.freeradbiomed.2013.05.001
Smith, Y., Wichmann, T., Factor, S. A., and Delong, M. R. (2012). Parkinson's disease therapeutics: new developments and challenges since the introduction of levodopa. Neuropsychopharmacology 37, 213–246. doi: 10.1038/npp.2011.212
Sofic, E., Lange, K. W., Jellinger, K., and Riederer, P. (1992). Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson's disease. Neurosci. Lett. 142, 128–130. doi: 10.1016/0304-3940(92)90355-B
Sofic, E., Paulus, W., Jellinger, K., Riederer, P., and Youdim, M. B. (1991). Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J. Neurochem. 56, 978–982. doi: 10.1111/j.1471-4159.1991.tb02017.x
Sohal, R. S., and Orr, W. C. (2012). The redox stress hypothesis of aging. Free Radic. Biol. Med. 52, 539–555. doi: 10.1016/j.freeradbiomed.2011.10.445
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063. doi: 10.1083/jcb.200108057
Strauss, K. M., Martins, L. M., Plun-Favreau, H., Marx, F. P., Kautzmann, S., Berg, D., et al. (2005). Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum. Mol. Genet. 14, 2099–2111. doi: 10.1093/hmg/ddi215
Su, L. J., Auluck, P. K., Outeiro, T. F., Yeger-Lotem, E., Kritzer, J. A., Tardiff, D. F., et al. (2010). Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson's disease models. Dis. Model. Mech. 3, 194–208. doi: 10.1242/dmm.004267
Sulston, J., Dew, M., and Brenner, S. (1975). Dopaminergic neurons in the nematode Caenorhabditis elegans. J. Comp. Neurol. 163, 215–226. doi: 10.1002/cne.901630207
Tan, E. K., Lin, C. H., Tai, C. H., Tan, L. C., Chen, M. L., Li, R., et al. (2009). Non-synonymous GIGYF2 variants in Parkinson's disease from two Asian populations. Hum. Genet. 126, 425–430. doi: 10.1007/s00439-009-0678-x
Tardiff, D. F., Tucci, M. L., Caldwell, K. A., Caldwell, G. A., and Lindquist, S. (2012). Different 8-hydroxyquinolines protect models of TDP-43 protein, alpha-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J. Biol. Chem. 287, 4107–4120. doi: 10.1074/jbc.M111.308668
Thomas, B., and Beal, M. F. (2007). Parkinson's disease. Hum. Mol. Genet. 16, R183–R194. doi: 10.1093/hmg/ddm159
Trinczek, B., Ebneth, A., Mandelkow, E. M., and Mandelkow, E. (1999). Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 112(pt 14), 2355–2367.
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., et al. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 11282–11286. doi: 10.1073/pnas.90.23.11282
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Valentini, S., Cabreiro, F., Ackerman, D., Alam, M. M., Kunze, M. B., Kay, C. W., et al. (2012). Manipulation of in vivo iron levels can alter resistance to oxidative stress without affecting ageing in the nematode C. elegans. Mech. Ageing Dev. 133, 282–290. doi: 10.1016/j.mad.2012.03.003
VanDuyn, N., Settivari, R., Levora, J., Zhou, S., Unrine, J., and Nass, R. (2013). The metal transporter SMF-3/DMT-1 mediates aluminum-induced dopamine neuron degeneration. J. Neurochem. 124, 147–157. doi: 10.1111/jnc.12072
VanDuyn, N., Settivari, R., Wong, G., and Nass, R. (2010). SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol. Sci. 118, 613–624. doi: 10.1093/toxsci/kfq285
van Ham, T. J., Thijssen, K. L., Breitling, R., Hofstra, R. M., Plasterk, R. H., and Nollen, E. A. (2008). C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 4:e1000027. doi: 10.1371/journal.pgen.1000027
Ved, R., Saha, S., Westlund, B., Perier, C., Burnam, L., Sluder, A., et al. (2005). Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J. Biol. Chem. 280, 42655–42668. doi: 10.1074/jbc.M505910200
Vidal, R., Delisle, M. B., Rascol, O., and Ghetti, B. (2003). Hereditary ferritinopathy. J. Neurol. Sci. 207, 110–111. doi: 10.1016/S0022-510X(02)00435-5
Vulpe, C., Levinson, B., Whitney, S., Packman, S., and Gitschier, J. (1993). Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 3, 6–12. doi: 10.1038/ng0193-7
Vymazal, J., Righini, A., Brooks, R. A., Canesi, M., Mariani, C., Leonardi, M., et al. (1999). T1 and T2 in the brain of healthy subjects, patients with Parkinson disease, and patients with multiple system atrophy: relation to iron content. Radiology 211, 489–495. doi: 10.1148/radiology.211.2.r99ma53489
Waldron, K. J., Rutherford, J. C., Ford, D., and Robinson, N. J. (2009). Metalloproteins and metal sensing. Nature 460, 823–830. doi: 10.1038/nature08300
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Weinshenker, D., Garriga, G., and Thomas, J. H. (1995). Genetic and pharmacological analysis of neurotransmitters controlling egg laying in C. elegans. J. Neurosci. 15, 6975–6985.
Weintraub, D., Doshi, J., Koka, D., Davatzikos, C., Siderowf, A. D., Duda, J. E., et al. (2011). Neurodegeneration across stages of cognitive decline in Parkinson disease. Arch. Neurol. 68, 1562–1568. doi: 10.1001/archneurol.2011.725
Wersinger, C., and Sidhu, A. (2003). Attenuation of dopamine transporter activity by α-synuclein. Neurosci. Lett. 340, 189–192. doi: 10.1016/S0304-3940(03)00097-1
White, J. G., Southgate, E., Thomson, J. N., and Brenner, S. (1986). The Structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1–340. doi: 10.1098/rstb.1986.0056
White, K., and Munro, H. N. (1988). Induction of ferritin subunit synthesis by iron is regulated at both the transcriptional and translational levels. J. Biol. Chem. 263, 8938–8942.
Wischik, C. M., Novak, M., Thogersen, H. C., Edwards, P. C., Runswick, M. J., Jakes, R., et al. (1988). Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 85, 4506–4510. doi: 10.1073/pnas.85.12.4506
Wooten, G. F. (1997). Functional anatomical and behavioral consequences of dopamine receptor stimulation. Ann. N.Y. Acad. Sci. 835, 153–156. doi: 10.1111/j.1749-6632.1997.tb48626.x
Yoritaka, A., Hattori, N., Uchida, K., Tanaka, M., Stadtman, E. R., and Mizuno, Y. (1996). Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 93, 2696–2701. doi: 10.1073/pnas.93.7.2696
Yoshida, K., Furihata, K., Takeda, S., Nakamura, A., Yamamoto, K., Morita, H., et al. (1995). A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat. Genet. 9, 267–272. doi: 10.1038/ng0395-267
Youdim, M. B., Ashkenazi, R., Ben-Shachar, D., and Yehuda, S. (1984). Modulation of dopamine receptor in the striatum by iron: behavioral and biochemical correlates. Adv. Neurol. 40, 159–170.
Zabetian, C. P., Hutter, C. M., Factor, S. A., Nutt, J. G., Higgins, D. S., Griffith, A., et al. (2007). Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson's disease. Ann. Neurol. 62, 137–144. doi: 10.1002/ana.21157
Keywords: C. elegans, oxidative stress, metals, Parkinson's disease, α-synuclein, tau, microtubules, axonal transport
Citation: Chege PM and McColl G (2014) Caenorhabditis elegans: a model to investigate oxidative stress and metal dyshomeostasis in Parkinson's disease. Front. Aging Neurosci. 6:89. doi: 10.3389/fnagi.2014.00089
Received: 03 March 2014; Accepted: 27 April 2014;
Published online: 19 May 2014.
Edited by:
Paul Adlard, The Mental Health Research Institute, AustraliaCopyright © 2014 Chege and McColl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gawain McColl, The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Kenneth Myer Building, 30 Royal Parade, Parkville, VIC 3052, Australia e-mail:Z21jY29sbEBmbG9yZXkuZWR1LmF1