 Giuseppina Amadoro1,2*†
Giuseppina Amadoro1,2*† Veronica Corsetti1†
Veronica Corsetti1† Fulvio Florenzano2†
Fulvio Florenzano2† Anna Atlante3
Anna Atlante3 Antonella Bobba3
Antonella Bobba3 Vanessa Nicolin4
Vanessa Nicolin4 Stefania L. Nori5
Stefania L. Nori5 Pietro Calissano2
Pietro Calissano2- 1Institute of Translational Pharmacology – National Research Council, Rome, Italy
- 2European Brain Research Institute, Rome, Italy
- 3Institute of Biomembrane and Bioenergetics – National Research Council, Bari, Italy
- 4Clinical Department of Medical, Surgical and Health Science, University of Trieste, Trieste, Italy
- 5Department of Medicine and Surgery, University of Salerno, Baronissi, Italy
Evidence suggests a striking causal relationship between changes in quality control of neuronal mitochondria and numerous devastating human neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. Contrary to replicating mammalian cells with a metabolism essentially glycolytic, post-mitotic neurons are distinctive owing to (i) their exclusive energetic dependence from mitochondrial metabolism and (ii) their polarized shape, which entails compartmentalized and distinct energetic needs. Here, we review the recent findings on mitochondrial dynamics and mitophagy in differentiated neurons focusing on how the exceptional characteristics of neuronal populations in their morphology and bioenergetics needs make them quite different to other cells in controlling the intracellular turnover of these organelles.
Introduction
Mitochondria exhibit dynamic properties (fusion, fission, transport, biogenesis, and degradation) and their homeostasis in a healthy, functional network is a process which involves an intimate crosstalk between quality control and selective autophagy (Twig et al., 2008; Westermann, 2012). All these events are closely coordinated and reciprocally interact in an integrated system, constantly monitored in every cell to maintain a well-performing mitochondrial population which can sustain a proper bioenergetic status and, consequently, viability. The quality control of mitochondria relies on several different pathways, including: (i) degradation of misfolded proteins located in the matrix and intermembrane space by intrinsic mitochondrial proteases; (ii) clearance by ubiquitin–proteasome systems (UPS) of damaged proteins located in outer membrane or of nuclear-coded mitochondrial proteins which are not correctly imported; (iii) removal of oxidated proteins/lipids by mitochondria-derived vesicles which are directly targeted for lysosomal or peroxisomal degradation. Besides, recent studies have uncovered a novel autophagic process addressed selectively toward mitochondria and named mitophagy, which is – up to date – the only identified mechanism by which these defective and malfunctioning organelles are entirely recycled or degraded. Remarkably, mitophagy can also play a key physiological role by providing for the developmental maturation of reticulocytes as well for the exclusive maternal inheritance of mt DNA upon oocytes fertilization (Ding et al., 2012; Ashrafi and Schwarz, 2013).
The classical regulation of autophagy is governed by the mammalian target of rapamycin (mTOR) pathway, which negatively controls this process. The serine/threonine kinase mTOR belongs to the phosphatidylinositol kinase-related kinase (PIKK) family and is in charge of important intracellular processes such as translation, metabolism, and transcription in response to nutrients and/or growth factors (Sarkar, 2013). In mammalian cells, mTOR binds several proteins to form two distinct protein multi-complexes. mTORC1 (mTOR complex 1) contains the scaffolding protein Raptor and additional proteins that also function in association with TOR in a second complex, called mTORC2 (mTOR complex 2), that is not directly involved in autophagy. mTORC1 is a key regulator of translation and ribosome biogenesis as well is responsible for autophagy induction in response to starvation (Ghavami et al., 2014). On the other hand, although being originally reported as rapamycin-insensitive, mTORC2 is also likely targeted by this drug and is involved in the regulation of phosphorylation and activation of Akt/PKB, protein kinase C, serum- and glucocorticoid-induced protein kinase 1. In addition, as Akt positively regulates mTORC1, it would be possible that mTORC2 also acts as a negative regulator of autophagy (Jung et al., 2010) Accumulating data also highlight the crucial role of mTOR-dependent signaling pathways in neurodegenerative diseases, including Huntington’s disease (HD), Parkinson’s disease (PD), and Alzheimer’s disease (AD; Sarkar, 2013), as its pharmacological inhibition by rapamycin attenuates the accumulation of misfolded/aggregated proteins and protects against neuronal loss, in vivo and in vitro (Ghavami et al., 2014). Nevertheless, as compelling evidence demonstrate that canonical non-selective autophagy and mitophagy share the similar core autophagosome/mitophagosome initiation and formation machinery (Hayashi-Nishino et al., 2009; Yoshii et al., 2011), a two-step mitophagy model has been proposed to occur in mammalian cells involving the initial induction of autophagy-related genes (Atg)-dependent macroautophagy followed by the mitochondrial priming. In the first step, the reactive oxygen species (ROS) accumulation and the ATP depletion (indirectly via AMPK, AMP-activated protein kinase, activation) as result of damaged and dysfunctional mitochondria, inhibit the induction of mTOR which under physiological conditions blocks the autophagy by restraining the kinase activity of ubiquitin-like kinase (ULK; Laplante and Sabatini, 2009; Ghavami et al., 2014). Upon escaping from mTOR suppression, the ULK complex, including ULK-1 (mammalian Atg1 ortholog), Atg13, Atg101, and FIP200 promotes the de novo formation of the initiation complex by regulating the activity of the class III phosphoinositide-3 kinase (PI3K) including Beclin-1 (mammalian Atg6), Atg14, Ambra1 (activating molecule in Beclin-1-regulated autophagy), vacuolar protein sorting 34 (Vps34), and Vps15, to form PI3P, which further recruits several PI3P-binding proteins to drive the formation of the initiation membrane. The Atg12–Atg5–Atg16L1 multi complex and LC3 (microtubule-associated protein 1A/1B-light chain 3)–PE (phosphatidylethanolamine) conjugates are later involved in the elongation and closure of the initiation membrane (Itakura and Mizushima, 2010; Feng et al., 2013; Sarkar, 2013). In the second step, the priming of mitochondria is mediated by different mechanisms that could be Parkin-dependent, involving the Parkin–Pink1-mediated pathway (Youle and Narendra, 2011), or Parkin-independent, involving the ubiquitin E3 ligase SMURF1 (Orvedahl et al., 2011), the outer mitochondrial membrane (OMM) protein Nix (Schweers et al., 2007; Sandoval et al., 2008; Novak et al., 2010) and FUNDC1 (Liu et al., 2012a), the HSP90–Cdc37 chaperone complex stabilizing and activating ULK-1 (Joo et al., 2011) and the Atg9A/ULK-1 complex (Itakura et al., 2012). An Atg-independent mitophagy, involving the 15-lipoxygenase, has been also described (van Leyen et al., 1998) but the precise role of this enzyme in organelles degradation is still not completely clarified.
Moreover, as the mitophagy pathway has been mainly studied in non-neuronal cell lines, this process is still not fully clarified in terminally differentiated neurons. In polarized neurons mitochondria have a longer half-life than in other post-mitotic tissues (Menzies and Gold, 1971; Miwa et al., 2008; O’Toole et al., 2008) and, although the translation of a subset of mitochondrial proteins may occur in axons (Kaplan et al., 2009), the import of most of those newly synthesized that are stably localized on these organelles occurs in the cell body followed by their transport toward distal axons, dendrites and synaptic sites. Removal of damaged mitochondria is as well a bioenergetically demanding task for neuronal populations because these organelles need to be actively retro-transported to the cell body in order to fuse with locally resident lysosomes (Wang et al., 2006). Besides, although physiological aging has been associated with decreased mitochondrial functions and with mitophagic processes (Batlevi and La Spada, 2011; Green et al., 2011), functional as well as morphological impairment of these organelles – especially for those neuronal populations with poorly myelinated, long, thin axons located in selective brain areas (Verstreken et al., 2005) – have been causally connected to several human neurological disorders (Lin and Beal, 2006; Johri and Beal, 2012). An unbalanced turnover, recycling/elimination of the entire mitochondria through selective autophagy is indeed considered an early event involved in the pathogenesis of Charcot–Marie–Tooth (CMT) disease, PD, AD, HD (Lin and Beal, 2006; Wang et al., 2009; Batlevi and La Spada, 2011; Imai and Lu, 2011; Karbowski and Neutzner, 2012; Nunnari and Suomalainen, 2012; Sheng and Cai, 2012; Chaturvedi and Beal, 2013; Itoh et al., 2013), amyotrophic lateral sclerosis (ALS; Cozzolino and Carrì, 2012), cerebral ischemic models (Calo et al., 2013), schizophrenia, and depression (Deheshi et al., 2013). To this regard, it is worth mentioning that patogenetic and/or misfolded/aggregated proteins such as mutated superoxide dismutase in ALS (Israelson et al., 2010), mutant huntingtin in HD (Rockabrand et al., 2007), β-amyloid (Aβ), and tau in AD (Caspersen et al., 2005; Manczak et al., 2006; Hansson Petersen et al., 2008; Amadoro et al., 2010, 2012; Du et al., 2012; Schmitt et al., 2012), mutant α-synuclein in PD (Devi et al., 2008) in vivo accumulate on mitochondria causing their functional decline which, in turn, may affect their mitophagic elimination (Moreira et al., 2007; Chinta et al., 2010; Choubey et al., 2011; Amadoro et al., 2014; Figure 1). Furthermore, any direct change in the mitochondrial biogenesis pathway [peroxisome proliferator-activated receptor α coactivator/nuclear respiratory factor 1 (PGC-1α/NFR-1)] in neurons may also contribute itself to accumulation of the above-mentioned pathogenetic proteins at these organelles – affecting consequently their metabolic functions – as recently shown for the long insulin-degrading enzyme (IDE) isoform which is involved in mitochondrial Aβ catabolism (Leal et al., 2013).
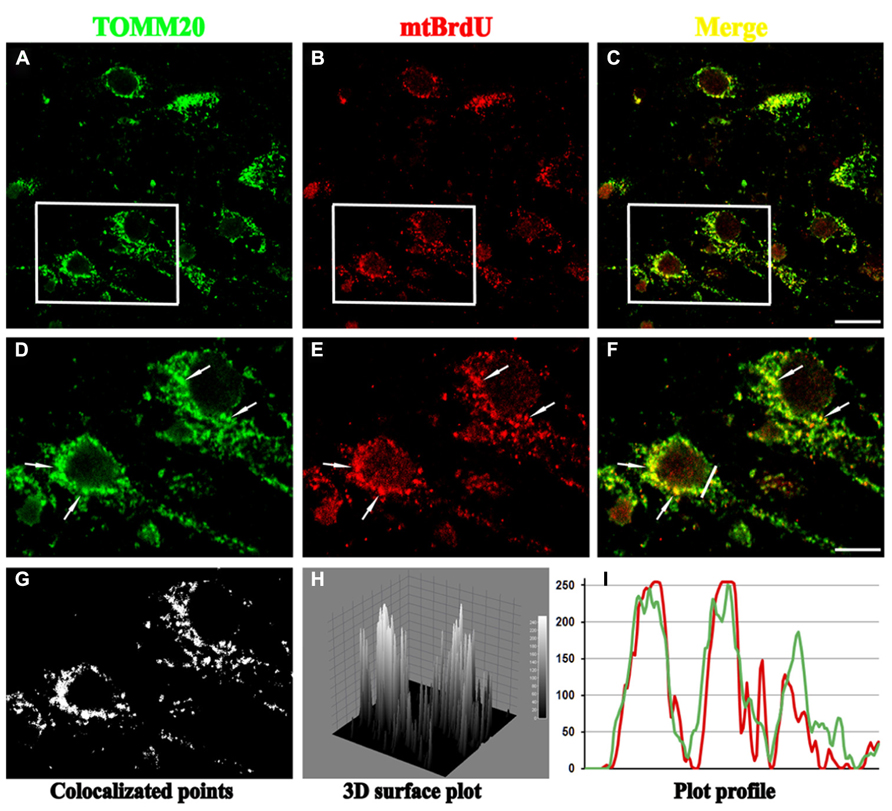
FIGURE 1. Confocal microscopy of mature hippocampal neuronal culture (15 DIV) double stained for TOMM20 (a mitochondrial marker; green channel) and BrdU (for visualization of newly synthesized mito-chondrial DNA; red channel). BrdU puncta colocalize with several TOMM20 positive mitochondria (arrows) in cell body, showing that biogenesis of these organelles mainly occurs in perinuclear compartment. (A–C) Lower magnification of TOMM20/BrdU immunofluorescence. (D–F) Inset higher magnification. (G–I) Colocalization analysis performed with ImageJ, including the spatial pattern of colocalized points (G), the luminance intensity height of the colocalized points (H), the spatial intensity profile for both fluorescence channels (I) of the white line positioned in (F). Scale bar 15 and 5 μm. Figure referring to data from Amadoro et al. (2014).
Interestingly, the relevance in assuring a proper quality control of neuronal mitochondria clearly comes into view considering that the mitochondrial dynamics which occur in a restricted hypothalamic populations can exert systemic effects and contribute to the physiological response of the whole-body energetic balance. To this regard, two recent papers provide striking evidence that alterations in mitochondrial number and morphology (Dietrich et al., 2013) or in mitochondria–endoplasmic reticulum (ER) connectivity (Schneeberger et al., 2013), both respectively occurring in orexigenic [agouti-related peptide (Agrp)- and neuropeptide Y (NPY)-producing] and antiorexigenic [pro-opiomelanocortin (POMC)-producing] hypothalamic neurons, allow the metabolic adaptation of feeding mice to different diet conditions.
In the present review, we focus on several specific and peculiar aspects of the mitochondrial clearance mediated by the Pink1–Parkin pathway in neurons and on the pathological implications that their inappropriate regulation may have in the onset and/or progression of human neurodegeneration. Furthermore, we will take advantage of the fact that a growing body of information on the mechanisms involved in mitophagy in non-neuronal cells has been extensively reported by other authors (Detmer and Chan, 2007; Palmer et al., 2011; Wang and Klionsky, 2011; Ashrafi and Schwarz, 2013).
Regulation of Mitochondrial Dynamics and Bioenergetic Status is of Particular Relevance to Post-Mitotic Neurons
The mitochondria quality control regulates in post-mitotic neurons several vital metabolic functions such as their proper distribution to synaptic terminals, maintenance of electron transport chain (ETC) activity and electrical connectivity (Stowers et al., 2002; Verstreken et al., 2005; Chen and Chan, 2006; Liu and Shio, 2008; Bereiter-Hahn and Jendrach, 2010; Ferree and Shirihai, 2012; Misko et al., 2012), protection of mtDNA integrity (Westermann, 2002; Parone et al., 2008), apoptosis (Suen et al., 2008), formation and function of synapses and dendritic spines (Li et al., 2004). As other cell types, neuronal populations also continually modulate size and number of these organelles, according to the variable energy demands and metabolic states throughout the entire lifetime and/or different sub-cellular compartments (Chen and Chan, 2009; Santos et al., 2010; Vives-Bauza and Przedborski, 2011; Van Laar and Berman, 2013). However, a tight control of the interplay between their mitochondrial dynamic and bioenergetic status is of particular relevance for post-mitotic neurons because they have a unique metabolic as well as morphological profile which entails specialized and compartimentalized energetic needs. Indeed, neuronal populations are typically characterized by: (i) high bioenergetic needs as the ATP production classically depends in these cells on OxPhos respiration rather than glycolysis (Rolfe and Brown, 1997; Attwell and Laughlin, 2001; Mironov, 2009; Bolanos et al., 2010); (ii) a very polarized morphology with extensive neuritic projections which are crucial for neuronal survival via a proper maintenance of their mitochondrial biomass. Remarkably, although the human brain consists of only 2% of the volume of the body, it is roughly responsible for 25% of net oxygen consumption in resting conditions (Magistretti and Pellerin, 1999) with neurons generating as much as 95% of their ATP exclusively from mitochondrial OXPhos (Erecińska et al., 1994). However, it is worth noticing that neurons and astrocytes – which are the two major types of brain cells – exhibit a different preference for glucose utilization since its metabolism in neuron is diverted mainly to the pentose phosphate pathway in order to regenerate antioxidants (reduced glutathione) and to promote survival (Bolanos et al., 2010). As they constantly require an active and efficient defense mechanism against oxidative stress (Almeida et al., 2005; Herrero-Mendez et al., 2009), neuronal populations are indeed unable to switch to anaerobic glycolytic metabolism (as an ATP-generating mechanism) during an acute mitochondrial stress, relying on lactate as an alternative substrate for their mitochondria-derived bioenergetic purposes (Pellerin et al., 2007). In addition, it is generally assumed that only 0.2% of the neuronal cellular volume is in the cytoplasmic soma while about 99.8% is constituted by the axonal and dendritic compartments (Devor, 1999; Fjell and Walhovd, 2010; Florenzano, 2012). For instance, the axonal length of projection neurons – such as those of rat dopaminergic neurons that are localized in the substantia nigra which is an area selectively affected in PD – can be as long as 470 μm or more and can provide several axon collaterals each of them making in turn contacts with approximately 400 synapses (Matsuda et al., 2009). Besides, in order to locally provide ATP supply and calcium buffering required for the neuronal activity of high energy-demanding terminal synapses (Schon and Przedborski, 2011), an high number of mitochondria resides far away from soma being localized in distal axon and dendritic processes (Hollenbeck, 2005) such as presynaptic terminals, including the active zones where synaptics vesicles (SVs) are released (Rowland et al., 2000; Perkins et al., 2010), post-synaptic densities, nodes of Ranvier and in growth cones (Fabricius et al., 1993; Morris and Hollenbeck, 1993). Collectively, such peculiar requirements imply that a high number of mitochondria spend the majority of their time in traveling up-and-down between the sites of their biogenesis, which are mainly localized into cell bodies (Davis and Clayton, 1996; Saxton and Hollenbeck, 2012; Figure 2), and those of their functional utilization which instead are close to terminal endings (Li et al., 2004). Interestingly there’s a positive correlation between the frequency and/or the intensity of synapse electrical activity and the number of metabolically active mitochondria at presynaptic compartment (Dubinsky, 2009). Furthermore, provided that the mitochondria size and mass are not the same for all neurons (Dubinsky, 2009; Lu, 2009) and that intrinsic mitophagic capacity has been found to be brain region-specific (Diedrich et al., 2011), any perturbation in controlling the dynamics properties of these organelles (Verstreken et al., 2005; Kann and Kovács, 2007; Nunnari and Suomalainen, 2012) can seriously and selectively compromise their survival (Xue et al., 2001). Different morphologies and ultrastructural profiles of mitochondria have been also correlated with distinct bioenergetic demands of the tissues they occupy and mitochondrial network in neurons is demonstrated to be distinct from those of other tissues in morphology, interconnectivity as well as cytoplasmatic pattern distribution (Dubinsky, 2009; Kuznetsov et al., 2009; Mironov, 2009). Neurons critically depend on autophagy for differentiation and survival (Komatsu et al., 2006) and are particularly prone to autophagic stress (Chu, 2006) so that basal autophagy appears to be more efficient than in other proliferating non-neuronal cells types (Boland et al., 2008) and is also likely to be regulated in alternative and quite different ways (Ashrafi and Schwarz, 2013; Van Laar and Berman, 2013). Being terminally differentiated and producing high levels of ROS against relatively fewer antioxidant molecules (Cui et al., 2004; Fatokun et al., 2008), neurons need to refurbish “old” mitochondrial pool to prevent the deleterious accumulation of oxidative damage, as suggested by the fact that antioxidant treatment supports the survival of cerebellar Purkinje cells (PCs) from knockout mice for dynamin-1-like protein (Drp1) fission protein (Kageyama et al., 2012). In contrast, proliferating cells constantly generate “new” mitochondria during continuous cell replication cycles so that oxidative stress may be diluted and maintained at relatively low levels, even without any mitochondrial division (Kageyama et al., 2012). A proper interplay between the bioenergetic – which is mainly regulated by energy requirement and substrate availability – and the mitochondrial quality control and autophagy – which control the overall health of mitochondrial population and their relative abundance – is specifically critical in neurons as it impinges on their intrinsic resistance to stress and, then, on their survival. Indeed, the relative sensitivity of different neuronal populations to mitochondrial inhibitors is tightly correlated with their “spare respiratory capacity,” which is an index of general mitochondrial health taking into account the ratio of the glucose utilization rate to the expression level of respiratory chain complexes (Fern, 2003). Interestingly, the vulnerability to a complex I inhibitor such as rotenone of cultured striatal neurons, which are selectively affected in HD pathology, is primarily determined by their “spare respiratory capacity” rather than oxidative stress (Yadava and Nicholls, 2007). Likewise, presynaptic mitochondria from hippocampus and cortex show a lower spare respiratory capacity to rotenone when compared to non-synaptic mitochondria from the same regions (Davey et al., 1997), in favor with the finding that the bioenergetic failure of peripheral mitochondria in vivo initiates the loss of synaptic terminals in neurodegenerative diseases.
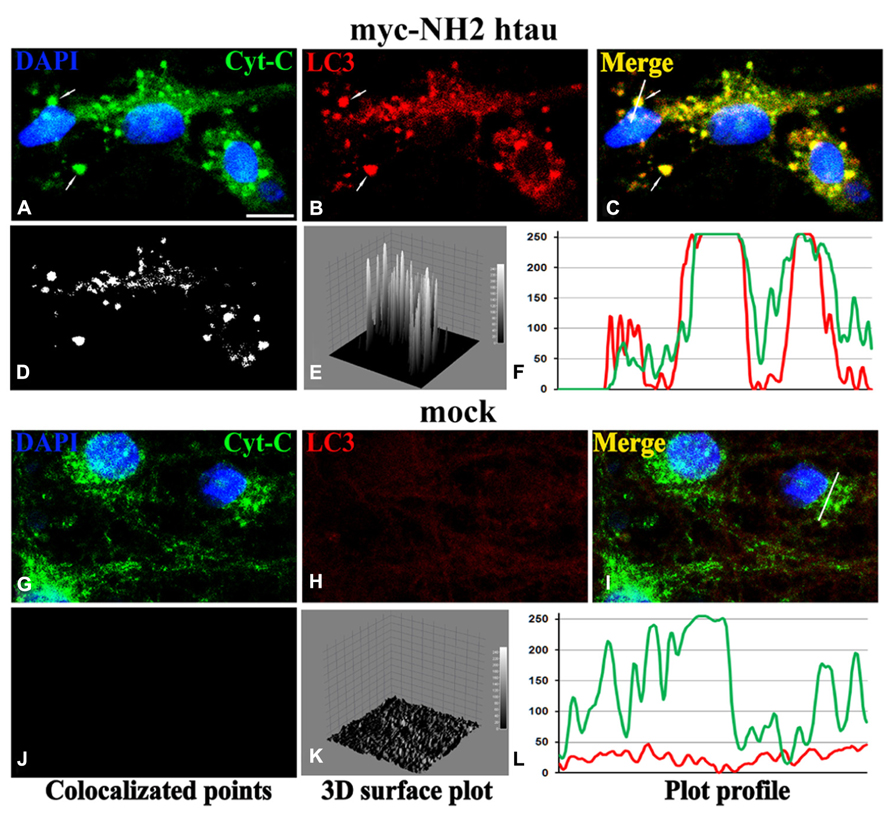
FIGURE 2. Confocal microscopy and image analysis of double immunofluorescence for cyt C (a mitochondrial marker; green channel) and LC3 (for visualization of autophagosomes; red channel), carried out on primary mature hippocampal cultures (15 DIV) at 12 h post-infection (MOI 50) with mock- and myc-NH2 26-230 human tau vectors. Nuclei were stained with DAPI (blue channel). (A–C) Numerous labeled LC3 stained vesicles intensely positive (colocalized) for cyt C were observed in myc-NH2htau neurons. Arrows point to two large mitophagosomal structures. (G–I) LC3 immunofluorescence is very faint in mock-treated cultures (D–F); (J–L) colocalization analysis performed with ImageJ, including the spatial pattern of colocalized points of (D,J), the intensity height of the luminance in the colocalized points (E,K), the spatial profile of the fluorescence intensity for both fluorescence channels for the white line positioned on several mitophagosomes in the second row (F,L). Scale bar 7 μm. Figure referring to data from Amadoro et al. (2014).
Nevertheless, despite the regulation of the physiological mitochondrial turnover is of particular importance for neuronal populations, it is not completely clear, as yet, whether an up-regulation or a down-regulation of mitophagic processes accompanied to an uncoordinated biogenesis can critically contribute to the onset and/or progression of human neurodegeneration (Batlevi and La Spada, 2011; Zhu et al., 2012, 2013).
The Pink/Parkin Pathway in Neurons: Energetic Metabolism, Nature, and Severity of Injury Dictate the Regulation of Mitochondrial Degradation in Mature Neurons
The Pink/Parkin pathway involves the interplay of two recessive Parkinson’s-linked genes PTEN-induced kinase 1 (PINK1) – a mitochondrially targeted serine/threonine kinase – and Parkin – an E3 ubiquitin ligase – which cooperate in maintaining the mitochondrial integrity by regulating several physiological processes of these organelles, including their membrane potential, calcium homeostasis, cristae structure, respiratory activity, and mtDNA integrity (Trempe and Fon, 2013). In addition, the Pink/Parkin pathway is crucial for autophagy-dependent clearance of dysfunctional mitochondria (Narendra et al., 2008; Matsuda et al., 2010; Figure 3) as, in the absence of PINK1 or Parkin, cells often develop fragmented mitochondria (Büeler, 2010). Specifically, in mammalian cells, cytosolic Parkin is selectively recruited to dysfunctional, depolarized mitochondria upon dissipation of ΔΨm by chemical uncoupler CCCP (carbonyl cyanide m-chlorophenyl hydrazone), then promoting their autophagic-lysosomal-mediated degradation. The Parkin translocation to depolarized mitochondria requires the activity of PINK which – in basal conditions – constantly undergoes a ΔΨm-dependent import into these organelles followed by specific maturation mediated by intrinsic protease(s), including matrix metalloproteinase (MMP), presenilin-associated-rhomboid-like (PARL), matrix-oriented AAA protease (m-AAA), and caseinolytic peptidase XP (ClpXP). Conversely, in response to low potential, full-length PINK is not imported/cleaved but rapidly stabilized, accumulating thus on outer membrane TOM (translocase of the outer membrane) complex (Batlevi and La Spada, 2011; Ashrafi and Schwarz, 2013; Grenier et al., 2013) where it engages Parkin which, in turn, triggers the mitochondria deliver toward autophagic-lysosomal pathway. Pink indeed recruits, directly or indirectly, Parkin from cytosol in close proximity on depolarized mitochondria and critically promotes its E3 ubiquitin ligase activity, likely by Ser65-phosphorylation, in the initial step of mitophagy (Kondapalli et al., 2012; Shiba-Fukushima et al., 2012; Iguchi et al., 2013). Parkin recruitment to mitochondria induces ubiquitination of several targets such as mitochondrial fusion proteins mitofusins (Mfns1/2) and voltage-dependent-activated channel (VDAC; Gegg et al., 2010; Geisler et al., 2010) whose proteasomal degradation (Chan et al., 2011; Yoshii et al., 2011) provokes the fragmentation of the organelle followed by its engulfment by autophagosomes (Deas et al., 2011). In addition p97, an AAA+ATPase, also accumulates on mitochondria in a Parkin-dependent manner to promote the degradation of OMM proteins and then mitophagy (Tanaka et al., 2010). Concerning the role of Parkin-mediated ubiquitination of Mfns1/2 in promoting mitophagy, two different but not mutually exclusive models have been proposed. To start with, the degradation of Mfns1/2 by UPS might disperse the clustered mitochondria and facilitate their engulfment by autophagosomes (Chan et al., 2011). Secondly, the removal of this pro-fusion mitochondrial protein shifts the balance toward the fragmentation – which is crucial in triggering mitophagy (Twig et al., 2008) – likely by physically interfering with the formation of Mfns1/2 trans-homodimers needed for tethering of these organelles (Ziviani and Whitworth, 2010). Alternatively, Parkin-mediated degradation might remove several negative regulators localized on the mitochondrial surface, thereby unmasking a molecular signal for recruitment of depolarized mitochondria by autophagosomes (Chan et al., 2011). Interestingly, the Parkin-induced elimination of Mfns1/2 is necessary for mitophagy to occur as it prevents the activation of an inhibitory default pathway in which depolarized and fragmented mitochondria undergo a drastic conformational change to become large spheroids that are not recognized by autophagosomes (Ding et al., 2012).
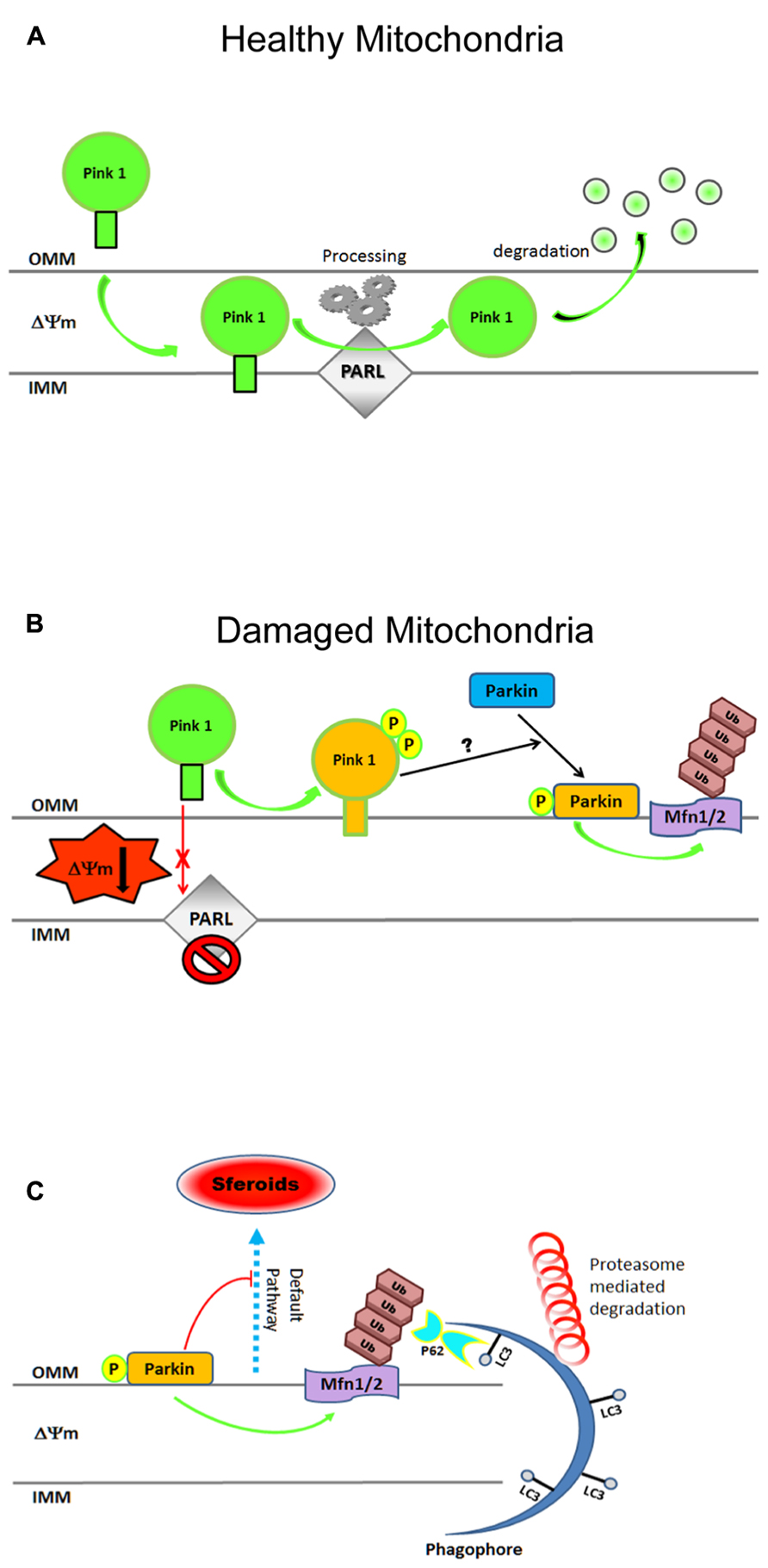
FIGURE 3. Cartoon illustrating steps in the mitochondrial clearance mediated by the Pink1–Parkin pathway. (A) In physiological conditions, Pink-1 is constitutively imported into healthy mitochondria via TIM/TOM complex to the inner membrane (IMM), cleaved by presenilin-associated rhomboid-like protease (PARL), and then proteolytically degraded. (B) Upon ΔΨ collapse, full-length Pink1 is not processed accumulating at the outer membrane (OMM) to recruit Parkin onto depolarized mitochondria. PINK1 autophosphorylation at Ser228 and Ser402 (P) is essential for efficient mitochondrial localization of Parkin. The PINK1-dependent Parkin phos-phorylation at Ser65, combined with unknown factor(s) (?), is required not only for its efficient translocation but also for the degradation of mito-chondrial proteins during mitophagy. (C) After being recruited on OMM, Parkin triggers mitophagy by ubiquitylating (Ub, K48, K63) several proteins including Mfns1/2, VDAC, TOM. Proteasome-mediated removal of Mfns1/2 not only inhibits mitochondrial fusion but also prevents its default rear-raggedright rangement into spheroids, allowing thus the damaged organelles to be recognized by the engulfing autophagosome. Cytosolic autophagy adaptor p62 (also known as sequestosome 1, SQSTM1) is also involved in mito-phagy as its K63-ubiquitin-binding domain (UBA) as well as an LC3- binding domain (LIR), recruits autophagosomes to ubiquitylated protein. For more information on Pink–Parkin-dependent mitophagy, please refer to recent excellent reviews (Twig and Shirihai, 2011; Vives-Bauza and Przedborski, 2011; Youle and Narendra, 2011; Ding and Yin, 2012; Jin and Youle, 2012). Freely adapted from Figure 6 of (Okatsu et al., 2012).
A large number of studies performed in non-neuronal cells lines have demonstrated that PINK1/Parkin pathway affects the autophagy clearance of damaged mitochondria in several ways: (i) by shifting the balance between fusion and fission of the mitochondrial network, allowing fragmented mitochondria to be taken up in autophagic vesicles; (ii) by modulating mitochondrial motility in order to switch the movement of mitochondria toward the autophagosome- and lysosome-rich perinuclear area, where the probability of being taken up by autophagosomes [AVs (autophagic vacuoles)] is higher and (iii) by directly recruiting the autophagic machinery to damaged mitochondria (de Vries and Przedborski, 2013). Neuronal mitochondria also undergo continuous reshaping by paired fission/fusion, transport, biogenesis, and selective degradation (Vives-Bauza et al., 2010; Fischer et al., 2012; Sheng and Cai, 2012; Youle and van der Bliek, 2012), as in immortalized cells lines (Youle and Narendra, 2011). Experimental data have indeed proved that PINK1/Parkin pathway regulating the mitochondrial quality control is active in primary neurons, as in replicating cell types (Seibler et al., 2011; Van Humbeeck et al., 2011; Yu et al., 2011; Cai et al., 2012; Joselin et al., 2012; Koyano et al., 2013; McCoy et al., 2014). However, other studies carried out in vitro (Van Laar et al., 2011; Rakovic et al., 2013) as well as in vivo (Sterky et al., 2011; Yu and Lu, 2011; Lee et al., 2012; Vincow et al., 2013) have yielded controversial results concerning the actual existence of mitophagy in neurons, especially those regarding the recruitment of Parkin to dysfunctional mitochondria. Indeed, in HeLa and MEF (mouse embryonic fibroblast) cell lines a general depolarization induced by pharmacological treatment with the uncoupler CCCP or other mito-toxins – such as antimycin A, valinomycin, and rotenone known to be ETC inhibitors – results in a rapid, robust, and faithfully reproducible re-localization of Parkin to mitochondria, followed by their prolonged and selective autophagic clearance (Narendra and Youle, 2011; Youle and Narendra, 2011; Ding and Yin, 2012; Youle and van der Bliek, 2012; Ashrafi and Schwarz, 2013). Likewise, immortalized cybrid cell lines carrying lethal mtDNA deletion – which causes a dysfunctional ETC assembly with decreased membrane potential – have also been shown to exhibit mitochondrial recruitment of Parkin at the beginning of mitophagy (Gilkerson et al., 2012). Conversely, in cultured primary neurons, a frank depolarization-evoked translocation of Parkin to mitochondria appears to be more variable. Discrepancies on (i) different type, handling and culture conditions (caspase(s) inhibitor, glial layer, antioxidant B27) used; (ii) expression of endogenous physiological level of proteins or transfection of exogenous transgenes; (iii) cell type-specific bioenergetic profile; (iv) type of injury and duration of treatment are indeed all important features that should be taken into account in evaluating studies on mitophagy mechanisms undergoing in post-mitotic neurons. As further discussed, the mitophagy pathways has turned out to differ – both spatially and kinetically – in cultured primary neurons from those found in immortalized lines (Table 1) and, perhaps, it might also diverge in its physiological function of removal of dysfunctional mitochondria.
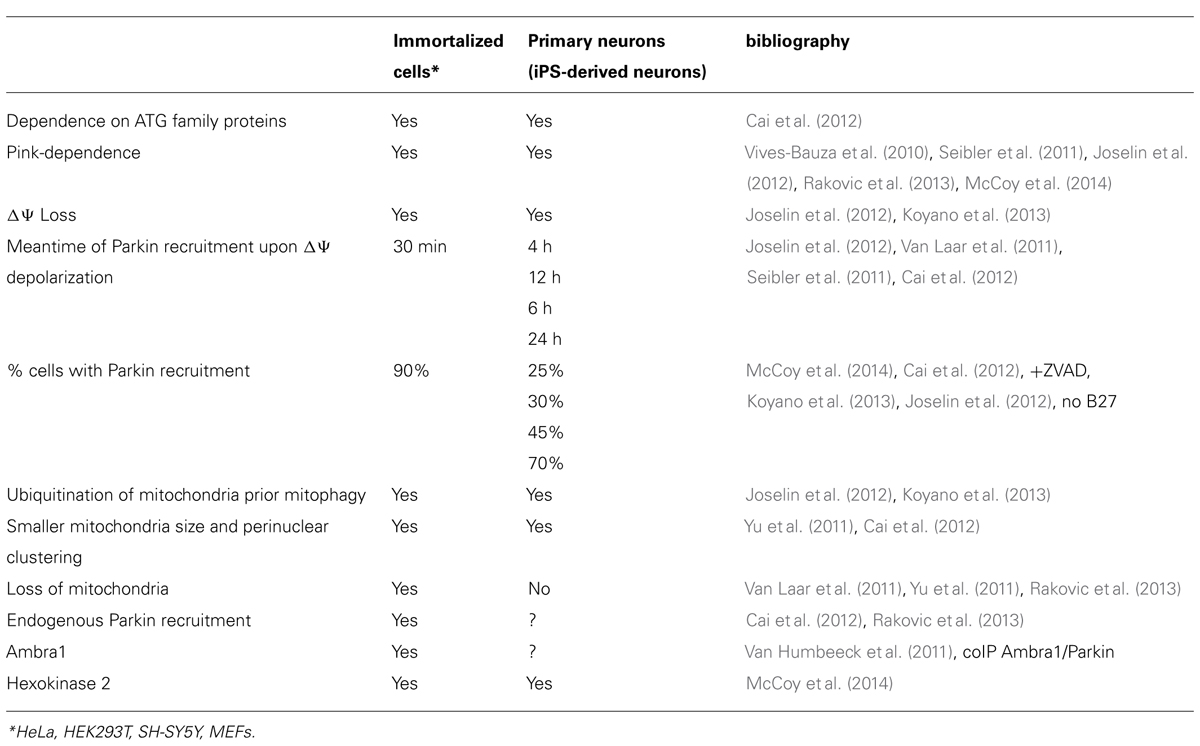
TABLE 1. Comparison of Pink–Parkin pathway between immortalized cells and post-mitotic neurons.
To this regard, the recruitment of endogenous Parkin to depolarized mitochondria has been first provided by Narendra et al. (2008), showing that an enhanced immunoreactivity level of Parkin is found by Western blotting on crude mitochondria-enriched fractions from rat cortical neurons treated for 1 h with CCCP, although its increase is not as much significant as in HEK293 (human embryonic kidney 293) cells exposed to similar experimental conditions. Afterward, another paper (Vives-Bauza et al., 2010) has proved that this event is strictly Pink-1 dependence since the CCCP-induced collapse of ΔΨm for 1 h is not longer associated with an endogenous relocalization of Parkin to mitochondria in primary neurons from PINK1 (-/-) mice, as well as in PINK1 – siRNA silenced HeLa cell line. More recently another group (Wang et al., 2011) reports that, as in non-neuronal HEK293T cells, treatment up to 1 h with a complex III inhibitor such as antimycin A also induces in cultured rat hippocampal neurons the translocation of exogenously expressed YFP-Parkin on axonal mitochondria, significantly decreasing their size and shape. In contrast, no evidence of a frank translocation of overexpressed as well as endogenous Parkin has been found to occur, neither in distal axons and dendrites nor in cell bodies mitochondria, in primary cortical neurons when they are acutely depolarized with CCCP up to 6 h, regardless of a clear evidence of active mitophagic pathway in these in vitro cultures (Van Laar et al., 2011). In agreement with the dispensable role of Parkin in degrading damaged mitochondria in vivo, overexpressed Parkin-independent accumulation and/or elimination of these organelles are allowed to occur in dopaminergic neurons of MitoPark mice carrying a severe respiration dysfunction due to their DNA (mtDNA) loss in mitochondrial transcription factor A (TFAM; Sterky et al., 2011). However, only recently, it has been clearly shown that the neuronal mitophagic response to an acute, excessive mitochondrial stressor is a process not only temporally slow but also spatially regulated (Cai et al., 2012). Compared with non-neuronal cells, CCCP-induced translocation of Parkin rarely occurs in mature neurons within 6 h and Parkin ring-like structures surrounding fragmented mitochondria are only occasionally detectable as early as 12 h of treatment, becoming increasingly frequent only at 18 h. In addition, the gradual and delayed Parkin recruitment to neuronal mitochondria (i) is compartmentally restricted to the somatodendritic regions and (ii) is coupled to reduced anterograde transport of these damaged organelles ending in an efficient lysosomal degradation into soma after 24 h exposure to CCCP (Cai et al., 2012). Interestingly, the fact that CCCP exposure does not initiate Parkin translocation and/or mitophagy in primary neurons up to 6 h might be in part explained because this process is regulated, to some extent, by bioenergetics dynamics since mammalian cells with a metabolism essentially glycolytic – if forced into OxPhos respiration – also recapitulate these effects, in a similar way of terminally differentiated neurons (Kanki and Klionsky, 2008; Van Laar et al., 2011). Indeed when glucose-fed HeLa cells were compelled to rely on mitochondrial oxidative phosphorylation in galactose/glutamine media – a culturing condition mimicking neuronal metabolism because it favors utilization of the citric acid cycle and oxidative phosphorylation over lactic acid-generating glycolysis for ATP production – they failed to recruit Parkin to their mitochondria upon CCCP-depolarization (Van Laar et al., 2011). The finding that Parkin translocates to depolarized mitochondria in glycolytic HeLa cells, which are grown in glucose-based media, but not in HeLa cells forced to rely on mitochondrial respiration, being grown in glucose-free media supplemented with galactose/glutamine, clearly shows that the induction of mitophagy is prevented by augmenting the cellular energy dependence on mitochondrial metabolism (Van Laar et al., 2011). Further, neither a robust association of overexpressed Parkin with mitochondria nor co-localization of these organelles with autophagic LC3 marker are clearly detected in cortical primary neurons at 1 h even after co-treatment with an ETC inhibitor such oligomycin which, blocking the reverse hydrolysis activity of mitochondrial ATP synthase, enable these in vitro cultures to temporarily hinder the massive energetic ATP drop caused by the CCCP-induced depolarization. Then again, the ATP loss induced by CCCP-depolarization is not the sole determinant in the inhibited Parkin translocation to mitochondria – and consequently in the induction of mitophagy – in oxidative phosphorylation-dependent neurons when compared to glycolytic HeLa cell lines (Van Laar et al., 2011). In view of these findings, it has been proposed that (i) the Parkin response is markedly suppressed in primary neurons in contrast to HeLa cells and that (ii) the rapid loss of ATP after a wide mitochondrial insult may prevent in neurons the full-scale Parkin-associated mitophagy, which may be quite important in conditions of slowly accumulating damages such as in aging-related human neurodegeneration. However, it is worth mentioning that even after 24 h, a selective damage to a small pool of mitochondria may still allow mitophagy processes since Parkin recruitment is seen in roughly 26.7% of neurons (Cai et al., 2012). Surprisingly, a restricted population of these mitochondria shows evidence of partial recovery of membrane potential – even after a prolonged depolarization induced by 24 h CCCP exposure – suggesting that a few, delayed bioenergetic compensatory changes occurring in primary neurons may yet permit these organelles to undergo elimination via autophagy (Cai et al., 2012). Finally a rapid (4 h) Parkin relocalization to mitochondria has been established to occur in mouse cortical neurons by exposing these cultures to a variety of mitochondrial damaging agents [i.e., CCCP, MPP+ (1-methyl-4-phenylpyridinium), and rotenone] in the absence of antioxidants, which are normally present in supplement B27 medium known to be routinely added to culturing medium of primary neuronal cultures. To this regard and consistently with a critical role of ROS in regulation of mitochondrial quality control, Joselin et al. (2012) report that mitochondrial translocation of Parkin is a ROS-dependent process in neurons because treatments with several ROS blockers can prevent at 6 h the recruitment of its overexpressed form in these cultures (as well as in MEFs). In addition, the same authors show that the loss of DJ-1 – a gene which is assumed to be critically linked to ROS management – can also trigger an accelerated stress-induced recruitment of Parkin to mitochondria facilitating thus their autophagic elimination via mitophagy. These findings not only undoubtedly confirm the active presence of Parkin/PINK1-mediated pathway in quality control of neuronal mitochondria but also (i) indicate that neuronal culturing conditions containing antioxidant supplements can prevent the pharmacologic effect of mitochondrial uncouplers and (ii) provide a possible explanation of the why several groups (Van Laar et al., 2011) are not able to detect significant Parkin translocation in neurons upon CCCP-depolarization. Interestingly TIGAR (TP53-induced glycolysis and apoptosis regulator), a biphosphatase which lowers the intracellular level of fructose-2,6-biphosphate (Fru-2,6-P2) with consequent inhibition of glycolysis and an overall decrease of intracellular ROS via increased production of NADPH through the pentose phosphate shunt, has also been recently identified as a novel negative regulator of neuronal mitophagy. In fact, antisense-mediated inactivation of TIGAR induces a complete normalization of impaired mitochondrial metabolism (morphology, complex I and III activity) in pink-/- zebrafish with ensuing rescue of dopaminergic neurons, suggesting that the manipulation of its physiological level can be a promising novel target for disease-modifying therapy in PINK1-related PD (Flinn et al., 2013). Finally, in favor with the proposal that the Pink1/Parkin is dependent on bioenergetic status (Van Laar et al., 2011), an elegant paper has recently reported that the apparent discrepancy in depolarization-induced relocalization of Parkin to mitochondria of primary neurons could be ascribed to their different amount of hexokinase activity (HK), an enzyme which catalyzes the conversion of glucose to glucose-6-phosphate. In agreement with their previous experiments in HeLa cells, McCoy et al. (2014) find that overexpression of wild-type but not kinase-inactive HK2 promotes the increase in mitochondrial translocation of overxpressed Parkin upon 6 h CCCP-depolarization, even in the presence of B27 in culturing medium. Data from these authors show in fact that (i) the activity of glycolytic hexokinase is upstream of Pink-1 and is crucial for recruitment of overexpressed Parkin to mitochondria in cortical primary neurons upon 6 h treatment with CCCP, acting thus as a modifier of Parkin relocalization to these organelles; (ii) the Parkin relocalization to mitochondria in these cultures in response to depolarizing conditions requires both ATP synthesis and active Pink1 signaling (McCoy et al., 2014).
An important point that needs also to be taken into account is that the type of insult may also impose the outcome of neuronal response to mitophagy. The mitochondrial complex III inhibitor, antimycin A, has been recently reported to trigger, up to 1 h, a rapid relocalization of Parkin to axonal mitochondria in primary rat hippocampal neurons (Wang et al., 2011). Conversely, in the same experimental conditions of overexpression and presence of B27, a retarded and sometimes insignificant Parkin-associated mitophagy has been described to occur at 12–24 h upon CCCP-depolarization throughout the whole neuronal body, including the soma and axonal compartments (Van Laar et al., 2011), or only in cell bodies and in dendrites but not in axons (Cai et al., 2012). Contradictory results in dopaminergic neurons derived from induced pluripotent stem cell (iPS) cells have also showed that, although recruitment of both endogenous and overexpressed Parkin to mitochondria is detectable in these in vitro cultures after 12 h exposure to another agent depolarizing such as valinomycin, this translocation is accompanied by no significant degradation of mitochondrial proteins located at the outer or inner membranes and at the matrix in contrast to a net reduction in mitochondrial DNA copy numbers (Seibler et al., 2011; Rakovic et al., 2013). To start with, it is worth noticing that a prolonged incubation with a chemical uncoupler in absence of caspase inhibitors – whose usage is conversely advised by Cai et al. (2012) – may promote apoptosis in these cultures. Secondly, the bioenergetics of cultured differentiated iPS cells requires still to be fully analyzed as it might be more different from that of primary neurons. Interestingly, in addition to a diminished or delayed translocation of Parkin to mitochondria in response to depolarizing CCCP, neurons show also a different temporal threshold for commitment of mitophagy in comparison to immortalized cell lines. In fact, it has very recently been demonstrated that redistribution of the cardiolipin from inner mitochondrial membrane (IMM) to OMM of damaged mitochondria can act as signal in cargo selection for their autophagic elimination and that injured neurons exhibit a larger percentage in membrane externalization of this phospholipid when compared to CCCP-treated HeLa cells (Chu et al., 2013).
However, it’s noteworthy that Parkin can regulate the mitochondrial physiology in neurons along different ways which are beyond their selective elimination via autophagy and do not necessarily require its stable recruitment to them such as (i) promoting their biogenesis (Shin et al., 2011) and optic atrophy 1 (OPA-1)-dependent integrity (Müller-Rischart et al., 2013) via activation of nuclear transcription factors; (ii) controlling their axonal trafficking (Wang et al., 2011; Liu et al., 2012b); (iii) suppressing apoptosis via Bax ubiquitination (Johnson et al., 2012); (iii) regulating the selective turnover of respiratory chain proteins (Vincow et al., 2013), likely by chaperone-mediated extraction followed by proteasomal degradation (Margineantu et al., 2007) and/or by direct transport of selected cargo to lysosomes through mitochondria-derived vesicles (Neuspiel et al., 2008; Soubannier et al., 2012).
The Pink/Parkin Pathway in Neurons: Morphology and Axonal Transport of Mitochondria, Nucleation of New Phagophores Through Direct Ambra1 Recruitment on Mitochondria
As far as the role of PINK1/Parkin pathway on mitochondrial morphology in neurons, evidence have demonstrated that PINK1 knock-down leads to elongation of mitochondria in rat hippocampal as well as in dopaminergic primary cultures while overexpression of either PINK1 or Parkin results in an increased number of mitochondria that are smaller in size (Yu et al., 2011). Quite the opposite, studies carried out in cell lines such as HeLa (Exner et al., 2007), SH-SY5Y (Lutz et al., 2009; Dagda et al., 2011), and N27 (Cui et al., 2010) have reported that mitochondria are found to be fragmented upon knock-down of PINK1 or Parkin and that this fragmentation is likely to be the result of an excessive fission because it is accompanied by an increase in mitochondrial fission proteins paralleled by an inverse decrease in their fusion proteins (Cui et al., 2010). However, no useful information are further provided by data from PD patient-derived fibroblasts carrying the PINK1Q126P or PINK1G309D pathogenic mutations which show no evident and significant changes in mitochondria morphology, although there is a slight tendency toward fragmentation in comparison with related control subjects (Exner et al., 2007). Nevertheless, a recent paper (Cronin-Furman et al., 2013) reports that the expression of three PD patients-derived mitochondrial DNAs in independent lines of cytoplasmic hybrid (cybrid) neural cells can provoke differential effects on mitochondrial morphology (from rod-like to swollen, fragmented, and globular shape), on movement of these organelles along axonal processes and on their oxygen consumption, assessed as basal measurements of oxygen consumption rate (OCR). Furthermore, neurons are able to activate pathways that affect mitochondrial mitophagy differently from those operating in replicating, non-neuronal cultures, even if in response to an identical insult. Indeed, when primary cortical neurons are exposed for 12 h to camptothecin in order to mimic the chronic genotoxic oxidative stress occurring in neurodegenerative diseases (Lin and Beal, 2006), a significant mitochondrial elongation with a suppressed expression of fission protein Drp1 and Parkin is clearly detectable in these in vitro cultures, in contrast to the opposite effect of other forms of stress (i.e., staurosporine and glutamate treatment) which are able to induce a marked fragmentation of these organelles. Interestingly, camptothecin-induced DNA damaging in non-neuronal cell lines – such as fibroblasts – does not evoke any significant mitochondrial elongation, providing the possible existence of intrinsic cell-specific differences in biological response of these organelles to exposure to similar stressor (Wang et al., 2013). Noteworthy, more and more compelling evidence outlines an interesting role of Parkin in managing the mitochondrial dynamics in response to insults of different severity. On one hand, Parkin can exert adaptive effects upon damage of neuronal mitochondria since, when these organelles are irreversibly injured in response to severe stress, can promote their mitophagic elimination mainly via K63-linked ubiquitination. On the other hand, under a moderate insult conditions causing only minor defects to these organelles, Parkin can support the maintenance of their integrity by activating specific nuclear signaling pathway, for instance the stimulation of their biogenesis (Shin et al., 2011) via NF-κB-dependent transcription of reshaping OPA-1 protein (Müller-Rischart et al., 2013). In the same way the long-term, neuron-specific overexpression of Parkin can positively modulate the mitochondrial dynamics/activity, likely by promoting their biogenesis via the transcriptional co-activator PGC-1α, extending thus the lifespan of the whole organism during the physiological aging (Rana et al., 2013).
Another recently identified aspect of mitochondrial biology regulated by the PINK1–Parkin pathway in neurons is the axonal transport of these organelles (Wang et al., 2011; Liu et al., 2012b). That a proper control in the functional connections between mitochondrial fission–fusion dynamics and axonal transport is critical for sustaining the neuronal survival has been largely documented (Frederick and Shaw, 2007; Saxton and Hollenbeck, 2012), as proved by the fact that inhibition of mitochondrial fission protein Drp1 greatly reduces the number of these organelles in synaptic terminals (Verstreken et al., 2005; Kageyama et al., 2012). In fact, when Drp1 is deleted in post-mitotic neurons such as in cerebellar PCs, mitochondria appear elongated like swollen spheroids which accumulate oxidative damage, become respiration-incompetent and co-localize with mitophagic markers (i.e., LC3 and p62/sequestosome 1, p62/SQSTM1) and ubiquitin. Interestingly the activity of Parkin appears dispensable for mitochondrial ubiquitination in Drp1KO PCs, suggesting that other E3 ubiquitin ligases may also operate in the quality control of neuronal mitochondria (Kageyama et al., 2012). Likewise, the deficiency of Mfn2 in PCs also causes an anomalous development/maturation of the dendritic arborization which exhibits few spines generally lacking mitochondria which are, in contrast, mainly distributed in cell body (Chen et al., 2007). Furthermore, a large fraction of the mitochondrial mass resides in distal axons and dendritic processes – far away from the soma where lysosomes are localized (Holtzman and Novikoff, 1965) – suggesting thus that mitochondrial degradation in neurons is likely to require a long-distance retrograde transport of these organelles toward the cell body (Ashrafi and Schwarz, 2013). However, even if still now it is not completely clear when malfunctioning mitochondria are actually retrieved from axon, an engulfment of these depolarized organelles has been also described to occur locally in neurons within autophagosome before their transport to soma (Wang et al., 2011). Additional supports in favor of a causal link between mis-regulation of mitochondrial dynamics and neurodegeneration have been recently put forward by the finding that the coordinated ability of the Pink1/Parkin pathway in immobilizing unhealthy and faulty mitochondria prior their autophagic elimination has been evolutionary conserved (Wang et al., 2011). To this regard, in Drosophila motor neurons as well in primary rodent hippocampal neurons, the overexpression of PINK1 or Parkin causes a general decline of axonal motility of mitochondria in association with the loss of Miro, a mitochondrial GTPase interacting with the adapter protein Milton which, in turn, binds the kinesin motor protein KIF5 (Hirokawa et al., 2010). Indeed, upon PINK1 or Parkin overexpression or Parkin-induced recruitment to mitochondria, Miro is ubiquitinated by Parkin and targeted for proteasomal degradation so forcing the disassembly of the Miro/Milton/kinesin complex which, in turn, quarantine the movement of damaged mitochondria prior to their selective clearance. The ability of PINK1/Parkin pathway in preventing mitochondrial movement prior to their clearance is an important step in assuring a proper quality control of these organelles in neurons. In fact sequestration and/or eventual engulfment of dysfunctional mitochondria would prevent their harmful accumulation at high-energy demand regions of the neurites thus (i) preventing further neuronal stress due to local ROS production; (ii) enabling the intracellular pool of these organelles to be replenished by healthier ones (Wang et al., 2011; Liu et al., 2012b). Interestingly, the existence of an active Pink1–Parkin pathway in primary neurons may reflect the exceptional challenges for neuronal populations in supplying their distal processes with healthy mitochondria which locally provide sufficient energy and Ca+-buffering capacity needed for the synaptic transmission. At the same time, an active Pink1–Parkin pathway could also explain why mutations in their genes are critically involved in the etiology of PD affecting dopaminergic neurons of substantia nigra, which show high rates of calcium influx and a marked susceptibility of dopamine oxidation (Surmeier et al., 2010). Alternatively, it is possible that the PINK1/Parkin pathway may also regulate the mitochondrial transport through potential interaction of Parkin with microtubules (Yang et al., 2005) or that the lysosomal degradation could be completed outside the soma, since mitochondrial markers have also been detected occasionally in axonal autophagosomes (Maday et al., 2012). Remarkably, it is particularly striking that Mfn2, a disease gene involved in CMT neurodegeneration that is well known for having a key role in Parkin-dependent mitophagy (Ziviani et al., 2010 ), facilitates the axonal transport of mitochondria in both directions by forming a complex with Miro (Baloh et al., 2007; Misko et al., 2010).
Finally, it has recently reported that the direct interaction between endogenous Parkin and Ambra1, a positive regulator of the Beclin-1-dependent autophagy (Fimia et al., 2007), enhances the dynamics and efficiency of Parkin-dependent clearance of neuronal mitochondria (Van Humbeeck et al., 2011), by stimulating the peri-mitochondrial nucleation of new phagophores and their consequent engulfment by autophagosomes. In details, endogenous Parkin and Ambra1 coimmunoprecipitates from HEK293 cells, SH-SY5Y cells, and adult mouse brain and their interaction is strongly increased during prolonged mitochondrial depolarization; although there’s no clear evidence for ubiquitination of Ambra1 by Parkin. Ambra1 is not required for Parkin translocation to depolarized mitochondria but it critically contributes to Parkin-mediated mitophagy, by locally stimulating the activity of the class III PI3K complex (Beclin-1, Vps34, p150, and the autophagy-specific subunit Atg14) that is essential for the formation of new phagophores (Tooze and Yoshimori, 2010). Thus, Ambra1 recruitment to mito-aggresomes contributes to the spatially restricted, selective nature of mitophagy, allowing the controlled engulfment of dysfunctional mitochondria but sparing the rest of the cell from unwanted autophagic degradation (Van Humbeeck et al., 2011). Interestingly, under normal conditions, a pool of Ambra1 is docked by Bcl-2 at the mitochondria, inhibiting its autophagic function. After induction of the autophagy, mitochondrial Ambra1 is released from Bcl-2, enabling it to bind to Beclin-1 in order to initiate the phagophore formation (nucleation) at the mitochondrion (Strappazzon et al., 2011). However, whether Parkin and Ambra1 are degraded along with the mitochondria or dissociate from mitochondria after setting the mitophagic process, as well the detailed biochemical basis for their increased binding upon mitochondrial depolarization, are all important questions that remain to be still addressed.
Different Regulation of Mitochondrial Dynamics Within Soma and Neuritic Processes in Neurons
A functional specialization into different sub-cellular compartments occurs in post-mitotic neurons since axon and dendrites, and not only the soma, exhibit unique biological and bioenergetic needs such as localized synthesis and degradation of proteins (Steward and Schuman, 2003; Piper and Holt, 2004). Furthermore axon and dendrites have each one their own specific transport mechanisms (Hirokawa et al., 2010; Namba et al., 2011), calcium regulation as well ER functions and localized ATP requirement (Hollenbeck and Saxton, 2005; Mironov, 2009; Wang and Schwarz, 2009; MacAskill and Kittler, 2010). Consequently, the regulation of mitochondrial turnover is more complex in neurons than in other mammalian non-neuronal cells because it has to provide for different sub-cellular compartments and locally regulate changeable concentrations of calcium and ADP (MacAskill et al., 2009; Mironov, 2009; Hirokawa et al., 2010; Cai et al., 2011; Figure 4). Synaptic activity (Rintoul et al., 2003; Sung et al., 2008), the levels of nitric oxide (NO; Zanelli et al., 2006) and calcium homeostasis, likely via the EF domains of Miro which operates as calcium sensor (MacAskill et al., 2009; Wang and Schwarz, 2009), tightly facilitate the trafficking and recruitment of neuronal mitochondria to high energy-demanding sub-compartments, such as dendrites and terminal fields. In response to the variable energy requirement of dendrites and axons, neuronal mitochondria can be locally anchored by interacting with neurofilament and neuron-specific intermediate filaments (Toh et al., 1980; Hirokawa and Takemura, 2005; Winter et al., 2008) or with microtubules via syntaphilin anchor (Kang et al., 2008). Interestingly, mitochondrial dynamics in axon and in distal dendrites of healthy post-mitotic neurons are less frequent of that observed in other non-neuronal cell or in cell bodies (Jendrach et al., 2005; Berman et al., 2009). This conclusion is supported by the fact that live-imaging quantifications show unexpectedly that in primary cultured neurons the process of fusion occurs in only 16% of mitochondria per hour in contrast to COS7 and INS1 cells where a fission event frequently occurs within 100 s after a coupled fusion (Twig et al., 2008). Furthermore, in highly polarized neurons, the mitochondrial fission/fusion balance is singularly regulated in different sub-cellular compartments depending on local energetic requirements in order to produce mainly elongated organelles in the somatodendritic compartment and more fragmented ones in distal axons (Overly et al., 1996; Popov et al., 2005). For instance synaptic mitochondria, which are localized at docking sites, significantly differ from other non-synaptic mitochondria as they are long-lived, undergo an increased oxidation during aging (Vos et al., 2010; Du et al., 2012) and contain higher levels of the matrix protein cyclophilin D which makes them more susceptible to calcium insult (Brown et al., 2006; Naga et al., 2007). These studies could also explain why the selective vulnerability of synapses (“dying-back” degeneration) is an early prominent characteristic of many human neurodegenerative disorders in which damage may begin at neuron terminals in the absence of any change in the cell body (Wishart et al., 2006; Bettini et al., 2007). Importantly, synaptic mitochondria are largely presynaptic (Gray and Whittaker, 1962; Nicholls, 1993) and residing mitochondria locally support the ATP synthesis and Ca2+ buffering in addition to the neurotransmitter synthesis and catabolism. Considering that (i) numerous neurodegenerative diseases are characterized by iron accumulation (Oshiro et al., 2011) which plays a crucial role in NMDA/NO-mediated neurotoxicity (Cheah et al., 2006; Chen et al., 2013); (ii) iron chelation is a strong Pink/parkin-independent activator of mitophagy (Allen et al., 2013), it has been proposed that an umbalance in homeostasis of this ion might affect in neurons the mitochondrial quality control processes (Allen et al., 2013). Interestingly, using primary cortical neurons which are chronically exposed to non-lethal low-concentration of rotenone, it has been proved that mitochondrial dynamics can be differentially affected in neurons over time and in specific sub-cellular regions. In fact, evidence outlines that the rates of fission and fusion of these organelles can modify during the in vitro cultures maturation (from 7 to 24 days of age) with changes which precede any sign of cell death, providing thus the existence of an important temporal window for new therapeutic opportunities prior to the onset of irreversible neurodegenerative phenomena. Besides, compartments-specific compensatory changes (i.e., homeostatic biogenesis) can be also possible in neurons in a attempt to balance the accumulating mitochondrial toxicity as demonstrated by the finding that the density of these organelles increases in more vulnerable and early affected distal neurites, prior to significant changes in cell bodies (Arnold et al., 2011). Furthermore, the morphology and function of ER, which plays a key role in mitochondrial fission (Friedman et al., 2011), may differ in somatodendritic compartment compared to distal axons (Ramírez and Couve, 2011) and the specialized sub-cellular contacts between ER and mitochondria [i.e., MAM (mitochondria-associated ER membrane)] involved in lipid synthesis and Ca2+ handling are also localized in hippocampal neurons at synapses to sustain locally their integrative activities and integrity (Hedskog et al., 2013). Finally, mitochondrial biogenesis occurs in neurons mainly in the soma (Davis and Clayton, 1996; Saxton and Hollenbeck, 2012) and the activation of its regulatory nuclear transcription factors – such as peroxisome proliferator-activated receptor gamma coactivator 1α (PPARGC1A/PGC-1α) and NFR-1/2 is controlled by neuronal activity (Yang et al., 2006; Scarpulla, 2008).
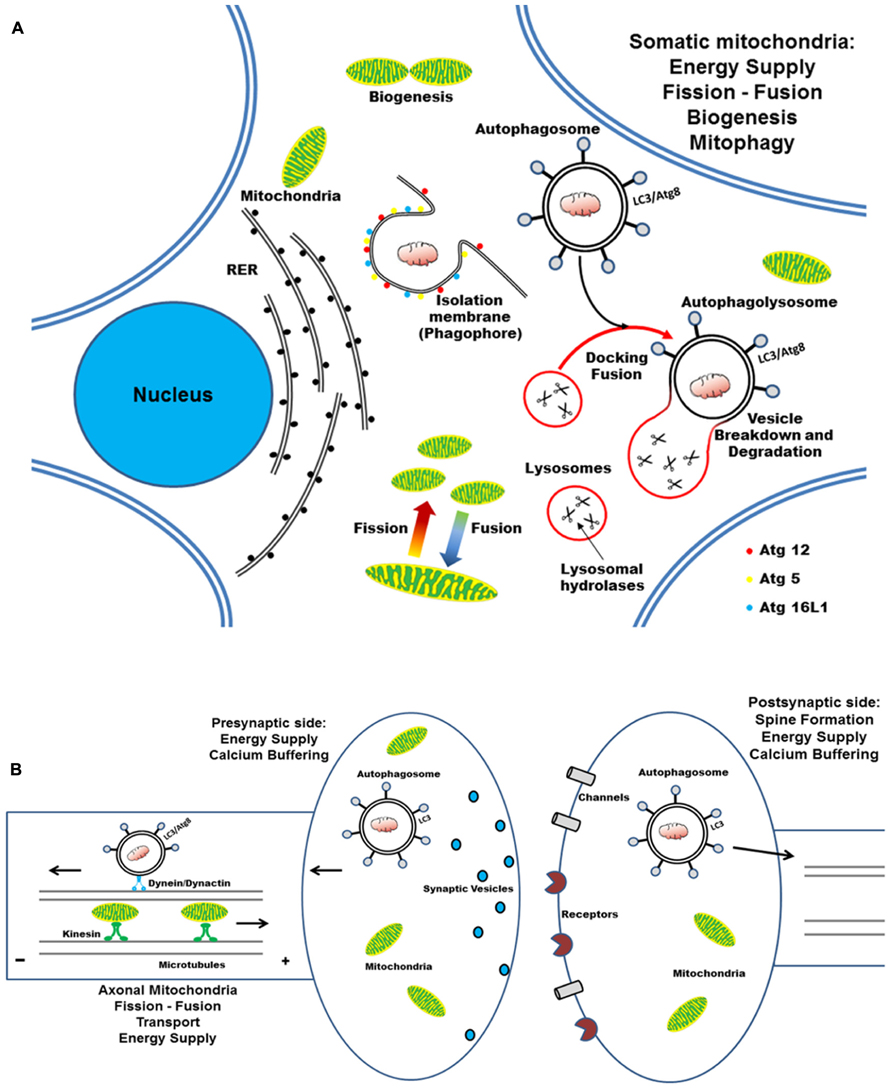
FIGURE 4. Cartoon illustrating the mitochondrial turnover which copes with the compartmentalized and distinct energetic requirements in the cell body, axon, and synaptic compartments of post-mitotic neurons. (A) In neurons, mitochondria travel long distances from the cell body out to distal dendritic and axonal terminals, where they subserve the ATP production and calcium homeostasis. The dynamic processes of biogenesis, fusion–fission regulate the mitochondrial function and quality control, by allowing them to adapt to spatial–temporal changes in cellular energy requirements. Selective autophagy begins with the nucleation of an isolation membrane (phagophore) which surrounds the damaged mitochondria to be degraded. RER could serve as membrane donors for autophagosome formation and the elongation of nascent double-membraned autophagic vesicle requires the coordinated assembly of Atg12–Atg5–Atg16L complex and LC3–PE conju-gates. (B) Newly formed autophagosomes move along microtubules in two directions – as a result of the opposing activities of the minus-end-directed motor protein dynein/dynactin and a plus-end-directed motor kinesin – and, finally, concentrate in perinuclear region (close to centrosome) where fuse with the lysosomes. Degradation of autophagosomal mitochondrial cargoes is then achieved by the acid hydrolases and the cathepsin proteases that are present in the lysosomal lumen.
Conclusion
Neurons – as well other non-neuronal cellular types – appear to be endowed by a common, basic molecular machinery involved in selective elimination of damaged mitochondria via an autophagic-lysosomal pathway named mitophagy. However, due to their specific morphological and bioenergetic necessities, differentiated neuronal populations display peculiar differences in mitophagic processes compared to other replicating cells and, sometimes, among the different brain regions. Increasing number of human neurodegenerative disorders are causally linked to an impaired mitochondrial turnover and two genes associated with autosomal recessive forms of PD, PINK1 and Parkin, have been recently found out to control the mitophagy also in primary post-mitotic neurons. Thus, the understanding how these unique neuronal features impinge on mitophagy and coordinated mitochondrial biogenesis will help to develop new disease-modifying approaches in future therapeutic interventions of neurodegenerative diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by grant from FIRB B81J07000070001 and Fondazione Roma to Pietro Calissano and by grant from PRIN 2010-2011 (prot. 2010M2JARJ-003) to Giuseppina Amadoro The funders had no role in study design, data collection and analysis, decision to publish or preparation of manuscript.
Abbreviations
AD, Alzheimer’s disease; Aβ, amyloid-β; ALS, amyotrophic lateral sclerosis; Ambra1, activating molecule in Beclin-1-regulated autophagy 1; AMPK, 5′-AMP-activated protein kinase; Atg, autophagy-related gene; BAX, Bcl-2-associated X protein; CCCP, carbonyl cyanide m-chlorophenyl hydrazone; ClpXP, caseinolytic peptidase XP; DJ-1/PARK7, Parkinson protein 7; Drp1, dynamin-1-like protein; E3, ubiquitin ligase; ETC, electron transport chain; FIP200, focal adhesion kinase (FAK) family interacting protein of 200 kDa; HD, Huntington’ disease; HDAC6, histone deacetylase; HK, hexokinase activity; HSP90–Cdc37, heat shock protein 90–cell division cycle 37; IDE, insulin-degrading enzyme; IMM, inner mitochondrial membrane; iPS, induced pluripotent stem cell; KIF5, kinesin family motor protein 5; LC3, microtubule-associated protein 1A/1B-light chain 3; LIR, LC3 interacting region; m-AAA, matrix-oriented AAA protease; Mfns1/2, mitofusins 1/2; Miro, mitochondrial Rho GTPase 1; MMP, matrix metalloproteinase; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; NFR-1, nuclear respiratory factor 1; Nix, NIP3-like protein X; OCR, oxygen consumption rate; OMM, outer mitochondrial membrane; OPA-1, optic atrophy 1; OxPhos, oxidative phosphorylation; p97, AAA-ATPase (also called VCP for valosin-containing protein); PARL, presenilin-associated-rhomboid-like; PCs, Purkinje cells; PD, Parkinson’s disease; PE, phosphatidylethanolamine; PGC-1α, peroxisome proliferator-activated receptor α coactivator; PI3K, phosphoinositide-3 kinase; PI3P, phosphatidylinositol 3-phosphate; PIKK, phosphatidylinositol kinase-related kinase; PINK-1, PTEN-induced putative kinase 1; Raptor, regulatory associated protein of mTOR; RER, rough endoplasmic reticulum; ROS reactive oxygen species; SMURF1, E3 ubiquitin-protein ligase; SQSTM1, sequestosome 1; TFAM, mitochondrial transcription factor A; TIGAR, TP53-induced glycolysis and apoptosis regulator; TOM, translocase of the outer membrane; Ub, ubiquitin; UBA, ubiquitin binding domain; ULK-1, UNC-51-like kinase 1; UPS, ubiquitin–proteasome system; VCP, valosin containing protein; VDAC, voltage-dependent-activated channel; Vps, vacuolar protein sorting.
References
Allen, G. F., Toth, R., James, J., and Ganley, I. G. (2013). Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 14, 1127–1135. doi: 10.1038/embor.2013.168
Almeida, A., Bolanos, J., and Moreno, S. (2005). Cdh1/Hct1-APC is essential for the survival of postmitotic neurons. J. Neurosci. 25, 8115–8121. doi: 10.1523/JNEUROSCI.1143-05.2005
Amadoro, G., Corsetti, V., Atlante, A., Florenzano, F., Capsoni, S., Bussani, R., et al. (2012). Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 33, 833.e1–833.e25. doi: 10.1016/j.neurobiolaging.2011.08.001
Amadoro, G., Corsetti, V., Florenzano, F., Atlante, A., Ciotti, M. T., Mongiardi, M. P., et al. (2014). AD-linked, toxic NH2 human tau increases the mitochondrial autophagy in neurons. Neurobiol. Dis. 62, 489–507. doi: 10.1016/j.nbd.2013.10.018
Amadoro, G., Corsetti, V., Stringaro, A., Colone, M., D’Aguanno, S., Meli, G., et al. (2010). A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J. Alzheimers Dis. 21, 445–470. doi: 10.3233/JAD-2010-100120
Arnold, B., Cassady, S. J., VanLaar, V. S., and Berman, S. B. (2011). Integrating multiple aspects of mitochondrial dynamics in neurons: age-related differences and dynamic changes in a chronic rotenone model. Neurobiol. Dis. 41, 189–200. doi: 10.1016/j.nbd.2010.09.006
Ashrafi, G., and Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42. doi: 10.1038/cdd.2012.81
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Baloh, R. H., Schmidt, R. E., Pestronk, A., and Milbrandt, J. (2007). Altered axonal mitochondrial transport in the pathogenesis of Charcot–Marie–Tooth disease from mitofusin 2 mutations. J. Neurosci. 27, 422–430. doi: 10.1523/JNEUROSCI.4798-06.2007
Batlevi, Y., and La Spada, A. R. (2011). Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol. Dis. 43, 46–51. doi: 10.1016/j.nbd.2010.09.009
Bereiter-Hahn, J., and Jendrach, M. (2010). Mitochondrial dynamics. Int. Rev. Cell Mol. Biol. 284, 1–65. doi: 10.1016/S1937-6448(10)84001-8
Berman, S. B., Chen, Y. B., Qi, B., McCaffery, J. M., Rucker, E. B. III, Goebbels, S., et al. (2009). Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J. Cell Biol. 184, 707–719. doi: 10.1083/jcb.200809060
Bettini, N. L., Moores, T. S., Baxter, B., Deuchars, J., and Parson, S. H. (2007). Dynamic remodelling of synapses can occur in the absence of the parent cell body. BMC Neurosci. 8:79. doi: 10.1186/1471-2202-8-79
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008
Bolanos, J. P., Almeida, A., and Moncada, S. (2010). Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci. 35, 145–149. doi: 10.1016/j.tibs.2009.10.006
Brown, M. R., Sullivan, P. G., and Geddes, J. W. (2006). Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J. Biol. Chem. 281, 11658–11668. doi: 10.1074/jbc.M510303200
Büeler, H. (2010). Mitochondrial dynamics, cell death and the pathogenesis of Parkinson’s disease. Apoptosis 15, 1336–1353. doi: 10.1007/s10495-010-0465-0
Cai, Q., Davis, M. L., and Sheng, Z. H. (2011). Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci. Res. 70, 9–15. doi: 10.1016/j.neures.2011.02.005
Cai, Q., Zakaria, H. M., Simone, A., and Sheng, Z. H. (2012). Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 22, 545–552. doi: 10.1016/j.cub.2012.02.005
Calo, L., Dong, Y., Kumar, R., Przyklenk, K., and Sanderson, T. H. (2013). Mitochondrial dynamics: an emerging paradigm in ischemia-reperfusion injury. Curr. Pharm. Des. 19, 6848–6857. doi: 10.2174/138161281939131127110701
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041.
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L., et al. (2011). Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Chaturvedi, R. K., and Beal, M. F. (2013). Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol. Cell. Neurosci. 55, 101–114. doi: 10.1016/j.mcn.2012.11.011
Cheah, J. H., Kim, S. F., Hester, L. D., Clancy, K. W., and Patterson, S. E. III. (2006). NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 51, 431–440. doi: 10.1016/j.neuron.2006.07.011
Chen, H., and Chan, D. C. (2006). Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell Biol. 18, 453–459. doi: 10.1016/j.ceb.2006.06.004
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics – fusion, fission, movement, and mitophagy – in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi: 10.1093/hmg/ddp326
Chen, H., McCaffery, J. M., and Chan, D. C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562. doi: 10.1016/j.cell.2007.06.026
Chen, Y., Khan, R. S., Cwanger, A., Song, Y., Steenstra, C., Bang, S., et al. (2013). Dexras1, a small GTPase, is required for glutamate-NMDA neurotoxicity. J. Neurosci. 33, 3582–3587. doi: 10.1523/JNEUROSCI.1497-12.2013
Chinta, S. J., Mallajosyula, J. K., Rane, A., and Andersen, J. K. (2010). Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 486, 235–239. doi: 10.1016/j.neulet.2010.09.061
Choubey, V., Safiulina, D., Vaarmann, A., Cagalinec, M., Wareski, P., Kuum, M., et al. (2011). Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 286, 10814–10824. doi: 10.1074/jbc.M110.132514
Chu, C. T. (2006). Autophagic stress in neuronal injury and disease. J. Neuropathol. Exp. Neurol. 65, 423–432. doi: 10.1097/01.jnen.0000229233.75253.be
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Cozzolino, M., and Carrì, M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66. doi: 10.1016/j.pneurobio.2011.06.003
Cronin-Furman, E. N., Borland, M. K., Bergquist, K. E., Bennett, J. P. Jr., and Trimmer, P. A. (2013). Mitochondrial quality, dynamics and functional capacity in Parkinson’s disease cybrid cell lines selected for Lewy body expression. Mol. Neurodegener. 8, 6. doi: 10.1186/1750-1326-8-6
Cui, K., Luo, X., Xu, K., and Ven Murthy, M. R. (2004). Role of oxidative stress in neurodegeneration: recent developments in assay methods for oxidative stress and nutraceutical antioxidants. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 771–799. doi: 10.1016/j.pnpbp.2004.05.023
Cui, M., Tang, X., Christian, W. V., Yoon, Y., and Tieu, K. (2010). Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 285, 11740–11752. doi: 10.1074/jbc.M109.066662
Dagda, R. K., Gusdon, A. M., Pien, I., Strack, S., Green, S., Li, C., et al. (2011). Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 18, 1914–1923. doi: 10.1038/cdd.2011.74
Davey, G. P., Canevari, L., and Clark, J. B. (1997). Threshold effects in synaptosomal and nonsynaptic mitochondria from hippocampal CA1 and paramedian neocortex brain regions. J. Neurochem. 69, 2564–2570. doi: 10.1046/j.1471-4159.1997.69062564.x
Davis, A. F., and Clayton, D. A. (1996). In situ localization of mitochondrial DNA replication in intact mammalian cells. J. Cell Biol. 135, 883–893. doi: 10.1083/jcb.135.4.883
Deas, E., Wood, N. W., and Plun-Favreau, H. (2011). Mitophagy and Parkinson’s disease: the PINK1–parkin link. Biochim. Biophys. Acta 1813, 623–633. doi: 10.1016/j.bbamcr.2010.08.007
Deheshi, S., Pasqualotto, B. A., and Rintoul, G. L. (2013). Mitochondrial trafficking in neuropsychiatric diseases. Neurobiol. Dis. 51, 66–71. doi: 10.1016/j.nbd.2012.06.015
Detmer, S. A., and Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879. doi: 10.1038/nrm2275
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Devor, M. (1999). Unexplained peculiarities of the dorsal root ganglion. Pain 6(Suppl.), S27–S35. doi: 10.1016/S0304-3959(99)00135-9
de Vries, R. L., and Przedborski, S. (2013). Mitophagy and Parkinson’s disease: be eaten to stay healthy. Mol. Cell. Neurosci. 55, 37–43. doi: 10.1016/j.mcn.2012.07.008
Diedrich, M., Kitada, T., Nebrich, G., Koppelstaetter, A., Shen, J., Zabel, C., et al. (2011). Brain region specific mitophagy capacity could contribute to selective neuronal vulnerability in Parkinson’s disease. Proteome Sci. 9, 59. doi: 10.1186/1477-5956-9-59
Dietrich, M. O., Liu, Z. W., and Horvath, T. L. (2013). Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 155, 188–199. doi: 10.1016/j.cell.2013.09.004
Ding, W. X., Guo, F., Ni, H. M., Bockus, A., Manley, S., Stolz, D. B., et al. (2012). Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J. Biol. Chem. 287, 42379–432388. doi: 10.1074/jbc.M112.413682
Ding, W. X., and Yin, X. M. (2012). Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564. doi: 10.1515/hsz-2012-0119
Du, H., Guo, L., and Yan, S. S. (2012). Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid. Redox Signal. 16, 1467–1475. doi: 10.1089/ars.2011.4277
Dubinsky, J. M. (2009). Heterogeneity of nervous system mitochondria: location, location, location! Exp. Neurol. 218, 293–307. doi: 10.1016/j.expneurol.2009.05.020
Erecińska, M., Nelson, D., Yudkoff, M., and Silver, I. A. (1994). Energetics of the nerve terminal in relation to central nervous system function. Biochem. Soc. Trans. 22, 959–965.
Exner, N., Treske, B., Paquet, D., Holmström, K., Schiesling, C., Gispert, S., et al. (2007). Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 27, 12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007
Fabricius, C., Berthold, C. H., and Rydmark, M. (1993). Axoplasmic organelles at nodes of Ranvier. II. Occurrence and distribution in large myelinated spinal cord axons of the adult cat. J. Neurocytol. 22, 941–954. doi: 10.1007/BF01218352
Fatokun, A. A., Stone, T. W., and Smith, R. A. (2008). Oxidative stress in neurodegeneration and available means of protection. Front. Biosci. 13, 3288–3311. doi: 10.2741/2926
Feng, D., Liu, L., Zhu, Y., and Chen, Q. (2013). Molecular signaling toward mitophagy and its physiological significance. Exp. Cell Res. 319, 1697–1705. doi: 10.1016/j.yexcr.2013.03.034
Fern, R. (2003). Variations in spare electron transport chain capacity: the answer to an old riddle? J. Neurosci. Res. 71, 759–762. doi: 10.1002/jnr.10553
Ferree, A., and Shirihai, O. (2012). Mitochondrial dynamics: the intersection of form and function. Adv. Exp. Med. Biol. 748, 13–40. doi: 10.1007/978-1-4614-3573-0_2
Fimia, G. M., Stoykova, A., Romagnoli, A., Giunta, L., Di Bartolomeo, S., Nardacci, R., et al. (2007). Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125. doi: 10.1038/nature05925
Fischer, F., Hamann, A., and Osiewacz, H. D. (2012). Mitochondrial quality control: an integrated network of pathways. Trends Biochem. Sci. 37, 284–292. doi: 10.1016/j.tibs.2012.02.004
Fjell, A. M., and Walhovd, K. B. (2010). Structural brain changes in aging: courses, causes and cognitive consequences. Rev. Neurosci. 21, 187–221. doi: 10.1515/REVNEURO.2010.21.3.187
Flinn, L. J., Keatinge, M., Bretaud, S., Mortiboys, H., Matsui, H., De Felice, E., et al. (2013). TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann. Neurol. 74, 837–847. doi: 10.1002/ana.23999
Florenzano, F. (2012). Localization of axonal motor molecules machinery in neurodegenerative disorders. Int. J. Mol. Sci. 13, 5195–5206. doi: 10.3390/ijms13045195
Frederick, R. L., and Shaw, J. M. (2007). Moving mitochondria: establishing distribution of an essential organelle. Traffic 8, 1668–1675. doi: 10.1111/j.1600-0854.2007.00644.x
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Ghavami, S., Shojaei, S., Yeganeh, B., Ande, S. R., Jangamreddy, J. R., Mehrpour, M., et al. (2014). Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49. doi: 10.1016/j.pneurobio.2013.10.004
Gilkerson, R. W., De Vries, R. L., Lebot, P., Wikstrom, J. D., Torgyekes, E., Shirihai, O. S., et al. (2012). Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 21, 978–990. doi: 10.1093/hmg/ddr529
Gray, E. G., and Whittaker, V. P. (1962). The isolation of nerve endings from brain: an electron-microscopic study of cell fragments derived by homogenization and centrifugation. J. Anat. 96, 79–88.
Green, D. R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333, 1109–1112. doi: 10.1126/science.1201940
Grenier, K., McLelland, G. L., and Fon, E. A. (2013). Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front. Neurol. 4:100. doi: 10.3389/fneur.2013.00100
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 105, 13145–13150. doi: 10.1073/pnas.0806192105
Hayashi-Nishino, M., Fujita, N., Noda, T., Yamaguchi, A., Yoshimori, T., and Yamamoto, A. (2009). A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 11, 1433–1437. doi: 10.1038/ncb1991
Hedskog, L., Pinho, C. M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U.S.A. 110, 7916–7921. doi: 10.1073/pnas.1300677110
Herrero-Mendez, A., Almeida, A., Fernández, E., Maestre, C., Moncada, S., and Bolaños, J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol. 11, 747–752. doi: 10.1038/ncb1881
Hirokawa, N., Niwa, S., and Tanaka, Y. (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. doi: 10.1016/j.neuron.2010.09.039
Hirokawa, N., and Takemura, R. (2005). Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 6, 201–214. doi: 10.1038/nrn1624
Hollenbeck, P. J. (2005). Mitochondria and neurotransmission: evacuating the synapse. Neuron 47, 331–333. doi: 10.1016/j.neuron.2005.07.017
Hollenbeck, P. J., and Saxton, W. M. (2005). The axonal transport of mitochondria. J. Cell Sci. 118(Pt 23), 5411–5419. doi: 10.1242/jcs.02745
Holtzman, E., and Novikoff, A. B. (1965). Lysosomes in the rat sciatic nerve following crush. J. Cell Biol. 27, 651–669. doi: 10.1083/jcb.27.3.651
Iguchi, M., Kujuro, Y., Okatsu, K., Koyano, F., Kosako, H., Kimura, M., et al. (2013). Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 288, 22019–22032. doi: 10.1074/jbc.M113.467530
Imai, Y., and Lu, B. (2011). Mitochondrial dynamics and mitophagy in Parkinson’s disease: disordered cellular power plant becomes a big deal in a major movement disorder. Curr. Opin. Neurobiol. 21, 935–941. doi: 10.1016/j.conb.2011.10.016
Israelson, A., Arbel, N., Da Cruz, S., Ilieva, H., Yamanaka, K., Shoshan-Barmatz, V., et al. (2010). Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67, 575–587. doi: 10.1016/j.neuron.2010.07.019
Itakura, E., Kishi-Itakura, C., Koyama-Honda, I., and Mizushima, N. (2012). Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci. 125(Pt 6), 1488–1499. doi: 10.1242/jcs.094110
Itakura, E., and Mizushima, N. (2010). Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6, 764–776. doi: 10.4161/auto.6.6.12709
Itoh, K., Nakamura, K., Iijima, M., and Sesaki, H. (2013). Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 23, 64–71. doi: 10.1016/j.tcb.2012.10.006
Jendrach, M., Pohl, S., Vöth, M., Kowald, A., Hammerstein, P., and Bereiter-Hahn, J. (2005). Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech. Ageing Dev. 126, 813–821. doi: 10.1016/j.mad.2005.03.002
Jin, S. M., and Youle, R. J. (2012). PINK1 – and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795–799. doi: 10.1242/jcs.093849
Johnson, B. N., Berger, A. K., Cortese, G. P., and Lavoie, M. J. (2012). The ubiquitin E3 ligase parkin regulates the proapoptotic function of bax. Proc. Natl. Acad. Sci. U.S.A. 109, 6283–6288. doi: 10.1073/pnas.1113248109
Johri, A., and Beal, M. F. (2012). Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 342, 619–630. doi: 10.1124/jpet.112.192138
Joo, J. H., Dorsey, F. C., Joshi, A., Hennessy-Walters, K. M., Rose, K. L., McCastlain, K., et al. (2011). Hsp90–Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol. Cell 43, 572–585. doi: 10.1016/j.molcel.2011.06.018
Joselin, A. P., Hewitt, S. J., Callaghan, S. M., Kim, R. H., Chung, Y. H., Mak, T. W., et al. (2012). ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 21, 4888–4903. doi: 10.1093/hmg/dds325
Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., and Kim, D. H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. doi: 10.1016/j.febslet.2010.01.017
Kageyama, Y., Zhang, Z., Roda, R., Fukaya, M., Wakabayashi, J., Wakabayashi, N., et al. (2012). Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 197, 535–551. doi: 10.1083/jcb.201110034
Kang, J. S., Tian, J. H., Pan, P. Y., Zald, P., Li, C., Deng, C., et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. doi: 10.1016/j.cell.2007.11.024
Kanki, T., and Klionsky, D. J. (2008). Mitophagy in yeast occurs through a selective mechanism. J. Biol. Chem. 283, 32386–32393. doi: 10.1074/jbc.M802403200
Kann, O., and Kovács, R. (2007). Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 292, C641–C657. doi: 10.1152/ajpcell.00222.2006
Kaplan, B. B., Gioio, A. E., Hillefors, M., and Aschrafi, A. (2009). Axonal protein synthesis and the regulation of local mitochondrial function. Results Probl. Cell Differ. 48, 225–242. doi: 10.1007/400_2009_1
Karbowski, M., and Neutzner, A. (2012). Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 123, 157–171. doi: 10.1007/s00401-011-0921-0
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol. 2, 120080. doi: 10.1098/rsob.120080
Koyano, F., Okatsu, K., Ishigaki, S., Fujioka, Y., Kimura, M., Sobue, G., et al. (2013). The principal PINK1 and Parkin cellular events triggered in response to dissipation of mitochondrial membrane potential occur in primary neurons. Genes Cells 18, 672–681. doi: 10.1111/gtc.12066
Kuznetsov, A. V., Hermann, M., Saks, V., Hengster, P., and Margreiter, R. (2009). The cell-type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 41, 1928–1939. doi: 10.1016/j.biocel.2009.03.007
Laplante, M., and Sabatini, D. M. (2009). mTOR signaling at a glance. J. Cell Sci. 122(Pt 20), 3589–3594. doi: 10.1242/jcs.051011
Leal, M. C., Magnani, N., Villordo, S., Buslje, C. M., Evelson, P., Castaño, E. M., et al. (2013). Transcriptional regulation of insulin-degrading enzyme modulates mitochondrial amyloid β (Aβ) peptide catabolism and functionality. J. Biol. Chem. 288, 12920–12931. doi: 10.1074/jbc.M112.424820
Lee, S., Sterky, F. H., Mourier, A., Terzioglu, M., Cullheim, S., Olson, L., et al. (2012). Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 21, 4827–4835. doi: 10.1093/hmg/dds352
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012a). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, S., Sawada, T., Lee, S., Yu, W., Silverio, G., Alapatt, P., et al. (2012b). Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 8:e1002537. doi: 10.1371/journal.pgen.1002537
Liu, Q. A., and Shio, H. (2008). Mitochondrial morphogenesis, dendrite development, and synapse formation in cerebellum require both Bcl-w and the glutamate receptor delta2. PLoS Genet. 4:e1000097. doi: 10.1371/journal.pgen.1000097
Lu, B. (2009). Mitochondrial dynamics and neurodegeneration. Curr. Neurol. Neurosci. Rep. 9, 212–219. doi: 10.1007/s11910-009-0032-7
Lutz, A. K., Exner, N., Fett, M. E., Schlehe, J. S., Kloos, K., Lämmermann, K., et al. (2009). Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951. doi: 10.1074/jbc.M109.035774
MacAskill, A. F., and Kittler, J. T. (2010). Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 20, 102–212. doi: 10.1016/j.tcb.2009.11.002
MacAskill, A. F., Rinholm, J. E., Twelvetrees, A. E., Arancibia-Carcamo, I. L., Muir, J., Fransson, A., et al. (2009). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. doi: 10.1016/j.neuron.2009.01.030
Maday, S., Wallace, K. E., and Holzbaur, E. L. (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407–417. doi: 10.1083/jcb.201106120
Magistretti, P. J., and Pellerin, L. (1999). Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1155–1163. doi: 10.1098/rstb.1999.0471
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Margineantu, D. H., Emerson, C. B., Diaz, D., and Hockenbery, D. M. (2007). Hsp90 inhibition decreases mitochondrial protein turnover. PLoS ONE 2:e1066. doi: 10.1371/journal.pone.0001066
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Matsuda, W., Furuta, T., Nakamura, K. C., Hioki, H., Fujiyama, F., Arai, R., et al. (2009). Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 29, 444–453. doi: 10.1523/JNEUROSCI.4029-08.2009
McCoy, M. K., Kaganovich, A., Rudenko, I. N., Ding, J., and Cookson, M. R. (2014). Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum. Mol. Genet. 23, 145–156. doi: 10.1093/hmg/ddt407
Menzies, R. A., and Gold, P. H. (1971). The turnover of mitochondria in a variety of tissues of young adult and aged rats. J. Biol. Chem. 246, 2425–2429.
Mironov, S. L. (2009). Complexity of mitochondrial dynamics in neurons and its control by ADP produced during synaptic activity. Int. J. Biochem. Cell Biol. 41, 2005–2014. doi: 10.1016/j.biocel.2009.04.009
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J., and Baloh, R. H. (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 30, 4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010
Misko, A. L., Sasaki, Y., Tuck, E., Milbrandt, J., and Baloh, R. H. (2012). Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 32, 4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012
Miwa, S., Lawless, C., and von Zglinicki, T. (2008). Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model. Aging Cell 7, 920–923. doi: 10.1111/j.1474-9726.2008.00426.x
Moreira, P. I., Siedlak, S. L., Wang, X., Santos, M. S., Oliveira, C. R., Tabaton, M., et al. (2007). Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 3, 614–615.
Morris, R. L., and Hollenbeck, P. J. (1993). The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 104(Pt 3), 917–927.
Müller-Rischart, A. K., Pilsl, A., Beaudette, P., Patra, M., Hadian, K., Funke, M., et al. (2013). The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol. Cell 49, 908–921. doi: 10.1016/j.molcel.2013.01.036
Naga, K. K., Sullivan, P. G., and Geddes, J. W. (2007). High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J. Neurosci. 27, 7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007
Namba, T., Nakamuta, S., Funahashi, Y., and Kaibuchi, K. (2011). The role of selective transport in neuronal polarization. Dev. Neurobiol. 71, 445–457. doi: 10.1002/dneu.20876
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Narendra, D. P., and Youle, R. J. (2011). Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 14, 1929–1938. doi: 10.1089/ars.2010.3799
Neuspiel, M., Schauss, A. C., Braschi, E., Zunino, R., Rippstein, P., Rachubinski, R. A., et al. (2008). Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 18, 102–108. doi: 10.1016/j.cub.2007.12.038
Nicholls, D. G. (1993). The glutamatergic nerve terminal. Eur. J. Biochem. 212, 613–631. doi: 10.1111/j.1432-1033.1993.tb17700.x
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Okatsu, K., Oka, T., Iguchi, M., Imamura, K., Kosako, H., Tani, N., et al. (2012). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3:1016. doi: 10.1038/ncomms2016
Orvedahl, A., Sumpter, R. Jr., Xiao, G., Ng, A., Zou, Z., Tang, Y., et al. (2011). Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117. doi: 10.1038/nature10546
Oshiro, S., Morioka, M. S., and Kikuchi, M. (2011). Dysregulation of iron metabolism in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Adv. Pharmacol. Sci. 2011, 378278. doi: 10.1155/2011/378278
O’Toole, M., Latham, R., Baqri, R. M., and Miller, K. E. (2008). Modeling mitochondrial dynamics during in vivo axonal elongation. J. Theor. Biol. 255, 369–377. doi: 10.1016/j.jtbi.2008.09.009
Overly, C. C., Rieff, H. I., and Hollenbeck, P. J. (1996). Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J. Cell Sci. 109(Pt 5), 971–980.
Palmer, C. S., Osellame, L. D., Stojanovski, D., and Ryan, M. T. (2011). The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal. 23, 1534–1545. doi: 10.1016/j.cellsig.2011.05.021
Parone, P. A., Da Cruz, S., Tondera, D., Mattenberger, Y., James, D. I., Maechler, P., et al. (2008). Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 3:e3257. doi: 10.1371/journal.pone.0003257
Pellerin, L., Bouzier-Sore, A. K., Aubert, A., Serres, S., Merle, M., Costalat, R., et al. (2007). Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262. doi: 10.1002/glia.20528
Perkins, G. A., Tjong, J., Brown, J. M., Poquiz, P. H., Scott, R. T., Kolson, D. R., et al. (2010). The micro-architecture of mitochondria at active zones: electron tomography reveals novel anchoring scaffolds and cristae structured for high-rate metabolism. J. Neurosci. 30, 1015–1026. doi: 10.1523/JNEUROSCI.1517-09.2010
Piper, M., and Holt, C. (2004). RNA translation in axons. Annu. Rev. Cell Dev. Biol. 20, 505–523. doi: 10.1146/annurev.cellbio.20.010403.111746
Popov, V., Medvedev, N. I., Davies, H. A., and Stewart, M. G. (2005). Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: a three-dimensional ultrastructural study. J. Comp. Neurol. 492, 50–65. doi: 10.1002/cne.20682
Rakovic, A., Shurkewitsch, K., Seibler, P., Grünewald, A., Zanon, A., Hagenah, J., et al. (2013). Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 288, 2223–2237. doi: 10.1074/jbc.M112.391680
Ramírez, O. A., and Couve, A. (2011). The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol. 21, 219–227. doi: 10.1016/j.tcb.2010.12.003
Rana, A., Rera, M., and Walker, D. W. (2013). Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. U.S.A. doi: 10.1073/pnas.1216197110 [Epub ahead of print].
Rintoul, G. L., Filiano, A. J., Brocard, J. B., Kress, G. J., and Reynolds, I. J. (2003). Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J. Neurosci. 23, 7881–7888.
Rockabrand, E., Slepko, N., Pantalone, A., Nukala, V. N., Kazantsev, A., Marsh, J. L., et al. (2007). The first 17 amino acids of huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 16, 61–77. doi: 10.1093/hmg/ddl440
Rolfe, D. F., and Brown, G. C. (1997). Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 77, 731–758.
Rowland, K. C., Irby, N. K., and Spirou, G. A. (2000). Specialized synapse-associated structures within the calyx of Held. J. Neurosci. 20, 9135–9144.
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Santos, R. X., Correia, S. C., Wang, X., Perry, G., Smith, M. A., Moreira, P. I., et al. (2010). A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimers. Dis. 20(Suppl. 2), S401–S412. doi: 10.3233/JAD-2010-100666
Sarkar, S. (2013). Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 41, 1103–1130. doi: 10.1042/BST20130134
Saxton, W. M., and Hollenbeck, P. J. (2012). The axonal transport of mitochondria. J. Cell Sci. 125(Pt 9), 2095–2104. doi: 10.1242/jcs.053850
Scarpulla, R. C. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638. doi: 10.1152/physrev.00025.2007
Schmitt, K., Grimm, A., Kazmierczak, A., Strosznajder, J. B., Götz, J., and Eckert, A. (2012). Insights into mitochondrial dysfunction: aging, amyloid-β, and tau-A deleterious trio. Antioxid. Redox Signal. 16, 1456–1466. doi: 10.1089/ars.2011.4400
Schneeberger, M., Dietrich, M. O., Sebastián, D., Imbernón, M., Castaño, C., Garcia, A., et al. (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155, 172–187. doi: 10.1016/j.cell.2013.09.003
Schon, E. A., and Przedborski, S. (2011). Mitochondria: the next (neurode)generation. Neuron 70, 1033–1053. doi: 10.1016/j.neuron.2011.06.003
Schweers, R. L., Zhang, J., Randall, M. S., Loyd, M. R., Li, W., Dorsey, F. C., et al. (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U.S.A. 104, 19500–19505. doi: 10.1073/pnas.0708818104
Seibler, P., Graziotto, J., Jeong, H., Simunovic, F., Klein, C., and Krainc, D. (2011). Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 31, 5970–5976. doi: 10.1523/JNEUROSCI.4441-10.2011
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2, 1002. doi: 10.1038/srep01002
Shin, J. H., Ko, H. S., Kang, H., Lee, Y., Lee, Y. I., Pletinkova, O., et al. (2011). PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 144, 689–702. doi: 10.1016/j.cell.2011.02.010
Soubannier, V., McLelland, G. L., Zunino, R., Braschi, E., Rippstein, P., Fon, E. A., et al. (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141. doi: 10.1016/j.cub.2011.11.057
Sterky, F. H., Lee, S., Wibom, R., Olson, L., and Larsson, N. G. (2011). Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 12937–12942. doi: 10.1073/pnas.1103295108
Steward, O., and Schuman, E. M. (2003). Compartmentalized synthesis and degradation of proteins in neurons. Neuron 40, 347–359. doi: 10.1016/S0896-6273(03)00635-4
Stowers, R. S., Megeath, L. J., Górska-Andrzejak, J., Meinertzhagen, I. A., and Schwarz, T. L. (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. doi: 10.1016/S0896-6273(02)01094-2
Strappazzon, F., Vietri-Rudan, M., Campello, S., Nazio, F., Florenzano, F., Fimia, G. M., et al. (2011). Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 30, 1195–1208. doi: 10.1038/emboj.2011.49
Suen, D. F., Norris, K. L., and Youle, R. J. (2008). Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590. doi: 10.1101/gad.1658508
Sung, J. Y., Engmann, O., Teylan, M. A., Nairn, A. C., Greengard, P., and Kim, Y. (2008). WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 105, 3112–3116. doi: 10.1073/pnas.0712180105
Surmeier, D. J., Guzman, J. N., Sanchez-Padilla, J., and Goldberg, J. A. (2010). What causes the death of dopaminergic neurons in Parkinson’s disease? Prog. Brain Res. 183, 59–77. doi: 10.1016/S0079-6123(10)83004-3
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Toh, B. H., Lolait, S. J., Mathy, J. P., and Baum, R. (1980). Association of mitochondria with intermediate filaments and of polyribosomes with cytoplasmic actin. Cell Tissue Res. 211, 163–169. doi: 10.1007/BF00233731
Tooze, S. A., and Yoshimori, T. (2010). The origin of the autophagosomal membrane. Nat. Cell Biol. 12, 831–835. doi: 10.1038/ncb0910-831
Trempe, J. F., and Fon, E. A. (2013). Structure and function of parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front. Neurol. 4:38. doi: 10.3389/fneur.2013.00038
Twig, G., Hyde, B., and Shirihai, O. S. (2008). Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim. Biophys. Acta 1777, 1092–1097. doi: 10.1016/j.bbabio.2008.05.001
Twig, G., and Shirihai, O. S. (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 14, 1939–1951. doi: 10.1089/ars.2010.3779
Van Humbeeck, C., Cornelissen, T., Hofkens, H., Mandemakers, W., Gevaert, K., De Strooper, B., et al. (2011). Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 31, 10249–10261. doi: 10.1523/JNEUROSCI.1917-11.2011
Van Laar, V. S., Arnold, B., Cassady, S. J., Chu, C. T., Burton, E. A., and Berman, S. B. (2011). Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet. 20, 927–940. doi: 10.1093/hmg/ddq531
Van Laar, V. S., and Berman, S. B. (2013). The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson’s disease. Neurobiol. Dis. 51, 43–55. doi: 10.1016/j.nbd.2012.05.015
van Leyen, K., Duvoisin, R. M., Engelhardt, H., and Wiedmann, M. (1998). A function for lipoxygenase in programmed organelle degradation. Nature 395, 392–395. doi: 10.1038/26500
Verstreken, P., Ly, C. V., Venken, K. J., Koh, T. W., Zhou, Y., and Bellen, H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. doi: 10.1016/j.neuron.2005.06.018
Vincow, E. S., Merrihew, G., Thomas, R. E., Shulman, N. J., Beyer, R. P., Maccoss, M. J., et al. (2013). The PINK1–Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 6400–6405. doi: 10.1073/pnas.1221132110
Vives-Bauza, C., and Przedborski, S. (2011). Mitophagy: the latest problem for Parkinson’s disease. Trends Mol. Med. 17, 158–165. doi: 10.1016/j.molmed.2010.11.002
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383. doi: 10.1073/pnas.0911187107
Vos, M., Lauwers, E., and Verstreken, P. (2010). Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2:139. doi: 10.3389/fnsyn.2010.00139
Wang, D. B., Garden, G. A., Kinoshita, C., Wyles, C., Babazadeh, N., Sopher, B., et al. (2013). Declines in Drp1 and parkin expression underlie DNA damage-induced changes in mitochondrial length and neuronal death. J. Neurosci. 33, 1357–1365. doi: 10.1523/JNEUROSCI.3365-12.2013
Wang, K., and Klionsky, D. J. (2011). Mitochondria removal by autophagy. Autophagy 7, 297–300. doi: 10.4161/auto.7.3.14502
Wang, Q. J., Ding, Y., Kohtz, D. S., Mizushima, N., Cristea, I. M., Rout, M. P., et al. (2006). Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 26, 8057–8068. Erratum in: J. Neurosci. 26, 1 p preceding 8409. Kohtz, Stave [corrected to Kohtz, D Stave]. doi: 10.1523/JNEUROSCI.2261-06.2006
Wang, X., and Schwarz, T. L. (2009). The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. doi: 10.1016/j.cell.2008.11.046
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Westermann, B. (2002). Merging mitochondria matters: cellular role and molecular machinery of mitochondrial fusion. EMBO Rep. 3, 527–531. doi: 10.1093/embo-reports/kvf113
Westermann, B. (2012). Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 1817, 1833–1838. doi: 10.1016/j.bbabio.2012.02.033
Winter, L., Abrahamsberg, C., and Wiche, G. (2008). Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J. Cell Biol. 181, 903–911. doi: 10.1083/jcb.200710151
Wishart, T. M., Parson, S. H., and Gillingwater, T. H. (2006). Synaptic vulnerability in neurodegenerative disease. J. Neuropathol. Exp. Neurol. 65, 733–739. doi: 10.1097/01.jnen.0000228202.35163.c4
Xue, L., Fletcher, G. C., and Tolkovsky, A. M. (2001). Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr. Biol. 11, 361–365. doi: 10.1016/S0960-9822(01)00100-2
Yadava, N., and Nicholls, D. G. (2007). Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 27, 7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007
Yang, F., Jiang, Q., Zhao, J., Ren, Y., Sutton, M. D., and Feng, J. (2005). Parkin stabilizes microtubules through strong binding mediated by three independent domains. J. Biol. Chem. 280, 17154–17162. doi: 10.1074/jbc.M500843200
Yang, S. J., Liang, H. L., and Wong-Riley, M. T. (2006). Activity-dependent transcriptional regulation of nuclear respiratory factor-1 in cultured rat visual cortical neurons. Neuroscience 141, 1181–1192. doi: 10.1016/j.neuroscience.2006.04.063
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640. doi: 10.1074/jbc.M110.209338
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Yu, W., Sun, Y., Guo, S., and Lu, B. (2011). The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 20, 3227–3240. doi: 10.1093/hmg/ddr235
Zanelli, S. A., Trimmer, P. A., and Solenski, N. J. (2006). Nitric oxide impairs mitochondrial movement in cortical neurons during hypoxia. J. Neurochem. 97, 724–736. doi: 10.1111/j.1471-4159.2006.03767.x
Zhu, J. H., Gusdon, A. M., Cimen, H., Van Houten, B., Koc, E., and Chu, C. T. (2012). Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis. 3, e312. doi: 10.1038/cddis.2012.46
Zhu, J. H., Kent, Z. Q., and Chu, C. T. (2013). After the banquet: mitochondrial biogenesis, mitophagy and cell survival. Autophagy 9, 1–14. doi: 10.4161/auto.24135
Ziviani, E., Tao, R. N., and Whitworth, A. J. (2010). Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. U.S.A. 107, 5018–5023. doi: 10.1073/pnas.0913485107
Keywords: mitochondria dynamics, quality control, mitophagy, primary neurons, neurodegenerative diseases
Citation: Amadoro G, Corsetti V, Florenzano F, Atlante A, Bobba A, Nicolin V, Nori SL and Calissano P (2014) Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: the pink–parkin pathway. Front. Aging Neurosci. 6:18. doi: 10.3389/fnagi.2014.00018
Received: 23 October 2013; Accepted: 27 January 2014;
Published online: 18 February 2014.
Edited by:
Antonio Camins, University of Barcelona, SpainReviewed by:
Daniel Ortuño-Sahagun, Centro Universitario de Ciencias Biológicas y Agropecuarias, MexicoNibaldo C. Inestrosa, Pontifical Catholic University of Chile, Chile
Alberto Lazarowski, University of Buenos Aires, Argentina
Copyright © 2014 Amadoro, Corsetti, Florenzano, Atlante, Bobba, Nicolin, Nori and Calissano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppina Amadoro, Institute of Translational Pharmacology – National Research Council, Via Fosso del Cavaliere 100, 00133 Rome, Italy e-mail:Zy5hbWFkb3JvQGlubW0uY25yLml0
† Giuseppina Amadoro, Veronica Corsetti, and Fulvio Florenzano have contributed equally to this work.