

- Laboratory of Cerebrovascular Research, Neurology and Neurosurgery, Montreal Neurological Institute, McGill University, Montreal, QC, Canada
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear transcription factors that regulate peripheral lipid and glucose metabolism. Three subtypes make up the PPAR family (α, γ, β/δ), and synthetic ligands for PPARα (fibrates) and PPARγ (Thiazolidinediones, TZDs) are currently prescribed for the respective management of dyslipidemia and type 2 diabetes. In contrast to the well characterized action of PPARs in the periphery, little was known about the presence or function of these receptors in the brain and cerebral vasculature until fairly recently. Indeed, research in the last decade has uncovered these receptors in most brain cell types, and has shown that their activation, particularly that of PPARγ, is implicated in normal brain and cerebrovascular physiology, and confers protection under pathological conditions. Notably, accumulating evidence has highlighted the therapeutic potential of PPARγ ligands in the treatment of brain disorders such as Alzheimer’s disease (AD), leading to the testing of the TZDs pioglitazone and rosiglitazone in AD clinical trials. This review will focus on the benefits of PPARγ agonists for vascular, neuronal and glial networks, and assess the value of these compounds as future AD therapeutics in light of evidence from transgenic mouse models and recent clinical trials.
Introduction
In the 1960s and 1970s, a class of hepatocarcinogens that caused peroxisome proliferation in the liver of mice and rats, while exerting lipid-lowering effects, was discovered (Hess et al., 1965 ; Svoboda and Azarnoff, 1966 ; Reddy and Krishnakantha, 1975 ). A couple of decades later, the molecular target of these agents was identified as a member of the nuclear hormone receptor superfamily, and termed ‘peroxisome proliferator-activated receptor’ (PPAR) (Issemann and Green, 1990 ). Three PPAR isoforms (α, γ, and β/δ), each encoded by a different gene, were subsequently characterized in mammals (Chen et al., 1993 ; Zhu et al., 1993 ; Kliewer et al., 1994 ; Tontonoz et al., 1994 ) and in Xenopus laevis (Dreyer et al., 1992 ). PPARα was the receptor responsible for the hypolipidemic and peroxisome proliferative effects initially observed in rodents, and PPARδ corresponded to the Xenopus PPARβ, hence the mixed β/δ nomenclature. The search for the natural ligands of PPARs pointed to fatty acids and eicosanoids derived from the diet or metabolism (Issemann et al., 1993 ; Kliewer et al., 1997 ), which fit nicely with the proposed role of PPARs in regulating lipid storage and glucose homeostasis following dietary intake. Concurrently, PPARγ was identified as the receptor mediating the insulin sensitizing effects of a class of oral anti-diabetics, the thiazolidinediones (TZDs) that were being tested in rodent models of non-insulin dependent diabetes (Lehmann et al., 1995 ). TZDs were commercialized in the late nineties. However, the first of these, troglitazone (Rezulin), caused severe liver failure and was quickly withdrawn from the UK, US and Japanese markets. Soon thereafter, rosiglitazone (Avandia®, GlaxoSmithKline) and pioglitazone (Actos®, Takeda pharmaceuticals), which do not share the hepatotoxic profile of troglitazone (Scheen, 2001 ), were released and continue to be prescribed today in the management of type II diabetes. In more recent years, the focus on PPARγ has intensified, as new ligands and novel biological roles have emerged for the receptor, particularly for its therapeutic potential in neurodegenerative disorders, such as Alzheimer’s disease (AD). Previous excellent reviews have highlighted the benefits of PPARγ activation for the treatment of AD (Landreth et al., 2008 ; Jiang et al., 2008 ). The present review incorporates both supportive and contradictory evidence for the beneficial role of PPARγ ligands on afflicted neuronal, glial, and notably, cerebrovascular networks in AD. Gaps in our knowledge are addressed, and the value of PPARγ agonists as future AD therapeutics is discussed, in view of the latest clinical trials.
Mechanism of Action
Peroxisome proliferator-activated receptors are members of the nuclear receptor superfamily, an evolutionarily conserved family of receptors that also includes the thyroid, estrogen, and glucocorticoid receptors, each playing vital roles in development, endocrine signaling and metabolism. There are 48 nuclear receptors in humans, and PPARs belong to the class of ligand-dependent receptors (24) that convey a variety of hormonal and metabolic signals directly to the genome by activating or repressing transcription via their ligand-binding (LBD) and DNA-binding (DBD) domains (Nagy and Schwabe, 2004 ). The remaining receptors comprise the orphan receptors, for which ligands are unknown or may not exist (‘true orphans’) or are only recently being discovered (‘adopted orphans’) (Germain et al., 2006 ). Upon binding ligand, PPARs heterodimerize with the common retinoid X receptor (RXR, Issemann et al., 1993 ), and recruit coregulators and chromatin-modifying enzymes to specific DNA sequences, termed peroxisome proliferator response elements (PPREs), in the promoters of target genes (coregulator biology reviewed in McKenna and O’Malley, 2002 ). Normally, non-liganded receptor is constitutively associated with corepressors, for example, nuclear receptor corepressor (NCoR). These recruit histone deacetylase complexes that generate a condensed chromatin structure over the target promoter, effectively blocking the transcriptional machinery in a process called transcriptional repression. When ligand binds, the receptor undergoes a conformational change, causing corepressors to dissociate and coactivators to bind, resulting in transcriptional activation. Coactivators such as cAMP response element-binding protein (CBP) and p300 are thought to act as histone acetyltransferases that cause chromatin decondensation to prepare promoters for the recruitment of the RNA polymerase. The exchange of corepressors and coactivators defines a highly dynamic cycle of transcriptional modulation (Nagy and Schwabe, 2004 ). Finally, many nuclear receptors can regulate gene programs, not only by interaction with their specific targets, but through cross-talk with other signal transduction pathways, in a process called transrepression. Accumulating evidence suggests that PPARs can inhibit gene expression through this mechanism, by interfering with activator protein-1 (AP-1) and nuclear factor-kappaB (NF-κB) activities (Ricote et al., 1998 ; Pascual et al., 2005 ). Transrepression can be achieved through PPARγ sequestration of NF-κB or of the limited pool of coactivators available to NF-κB-dependent promoters. A more detailed account of transrepression mechanisms can be found in Jiang et al. (2008) and references therein. It is worth mentioning that PPAR ligands exert many of their biological actions without binding to their receptor, through direct interactions with mitochondrial proteins or smooth muscle ion channels, for example (Zhang et al., 1994 ; Chawla et al., 2001 ; Ryan et al., 2004 ). The pervasiveness of such receptor-independent effects is a topic of ongoing investigations, and has been reviewed elsewhere (Feinstein et al., 2005 ).
Relevance for Alzheimer’s disease
Alzheimer’s disease is characterized by a gradual failure of memory, language, orientation and judgment underlined by dysfunctional synapses and neuronal loss (Cummings, 2004 ). Its diagnosis is confirmed postmortem by the presence of amyloid-beta (Aβ) plaques and neurofibrillary tangles in the brain, respectively composed of aggregated Aβ derived from the amyloid precursor protein (APP) and of the hyperphosphorylated microtubule-associated protein tau (τ). At the site of plaques are activated microglial and astroglial cells secreting cytokines and chemokines that contribute to an inflammatory and pro-oxidant environment (Akiyama et al., 2000 ; Butterfield et al., 2001 ; Selkoe, 2001 ). AD patients also characteristically feature a decrease in basal cerebral glucose utilization (CGU) in parietotemporal and posterior cingulate cortex (Fukuyama et al., 1994 ; Mosconi et al., 2004 ; Langbaum et al., 2009 ), altered neurometabolic coupling (Melrose et al., 2009 ), and reduced levels and activity of the endothelial glucose transporter 1 (GLUT-1) that delivers glucose to the brain from the blood (Kalaria and Harik, 1989 ; Simpson et al., 1994 ). More recently, the hypothesis that AD represents a state of diabetes unique to the central nervous system (CNS) or type III diabetes was put forth (de la Monte et al., 2006 ), whereby insulin resistance and altered insulin receptor signaling in neurons would be expected to interfere with synaptic plasticity (Zhao et al., 2004 ). In support of this claim, intranasal insulin administration improved verbal recall in a subgroup of AD patients, suggesting a therapeutic potential of insulin sensitizers, like TZDs, in AD (Reger et al., 2008 ). Invariably, AD is marked by chronic resting hypoperfusion (Farkas and Luiten, 2001 ; Bell and Zlokovic, 2009 ), and a limited ability of the cerebral circulation to match the needs of activated neurons, that is, impaired neurovascular coupling or functional hyperemia (Hock et al., 1997 ; Rosengarten et al., 2006 ). As the brain has both high energy demands and no fuel reserves, these conditions threaten basic brain function and disrupt protein synthesis underlying learning (Hermann et al., 2001 ; Iadecola, 2004 ). Finally, AD brain vessels display structural alterations, ranging from Aβ deposition on small to medium arteries (cerebral amyloid angiopathy, CAA, Roher et al., 1993 ), and cholinergic deafferentation (Tong and Hamel, 1999 ), to accumulation of extracellular matrix (ECM) proteins, such as collagen, in vascular basement membranes (Christov et al., 2008 ), resulting in vascular fibrosis. The latter has been ascribed to increased levels of the ECM organizer, transforming growth factor-beta 1 (TGF-β1) in AD brain and vessels (Wyss-Coray et al., 1997 ; Grammas and Ovase, 2002 ), and of its effector molecules, endothelin-1 (ET-1) and connective tissue growth factor (CTGF) (Clozel and Salloukh, 2005 ). Thus, the pathological alterations are numerous in the AD brain, but as the following sections will show, PPARγ activation can counter many of these in both animal models and AD patients.
Anti-Inflammatory Effects
Aside from its function in lipid and glucose metabolism, one of the new and now well-documented roles attributed to PPARγ was the ability to silence inflammatory gene expression in peripheral immune cells producing natural PPARγ ligands, like 15-deoxy-δ;12,14 prostaglandin J2 (15d-PGJ2) (Jiang et al., 1998 ; Ricote et al., 1998 ). Around the same time, non-steroidal anti-inflammatory drugs (NSAIDs), such as indomethacin, ibuprofen, and fenoprofen, that had been associated with decreased AD risk (McGeer et al., 1990 ; In’t Veld et al., 2001 ), were demonstrated to be PPARγ ligands (Lehmann et al., 1997 ). Furthermore, expression of the receptor was documented, not only in peripheral organs, but also in the CNS (Moreno et al., 2004 ; Inestrosa et al., 2005 ) and human brain microvessels (Ramirez et al., 2008 ), with a report of increased protein levels in the postmortem AD cortex (Kitamura et al., 1999 ). Collectively, this led to the idea that PPARγ could be an important target for brain inflammation in AD. Indeed, various PPARγ ligands, both natural (15d-PGJ2, docosahexaenoic acid) and synthetic (NSAIDs and TZDs), were shown to inhibit the expression of interleukin-6 (IL-6), tumor necrosis factor α (TNFα) and cyclooxygenase-2 (COX-2) from monocytic and microglial cell cultures stimulated with Aβ (Combs et al., 2000 ). Similar results were obtained in transgenic mice carrying various mutations of human APP associated with early-onset familial AD. Studies in ∼10-month-old Tg2576 mice on 4- to 6-month ibuprofen treatment reported reduced interleukin-1β (IL-1β) and glial fibrillary acidic protein (GFAP) levels (Lim et al., 2000 ), and decreased expression of the microglial markers of activation CD45 and CD11b (Yan et al., 2003 ). In the latter study, pioglitazone, at a dose of 20 mg/kg/day, was ineffective. Yet in a subsequent study using twice the pioglitazone dose and an acute 7-day treatment, COX-2 and inducible nitric oxide synthase (iNOS) expression was attenuated in the cortex and hippocampus of 10-month-old APPV717I mice, and comparable results were seen with ibuprofen (Heneka et al., 2005 ) (Figure 1 A). A recent study by our group employing a different transgenic APP model, but the original dose of Yan et al. (2003) , corroborated the anti-inflammatory effects of pioglitazone: the percentage of cortex occupied by GFAP-immunoreactive astrocytes was significantly reduced in ∼15-month-old animals that had been treated for 6–8 weeks (Nicolakakis et al., 2008 ). Anti-inflammatory effects of PPARγ agonists were also more recently confirmed in various APP models (Escribano et al., 2010 ; Toledo and Inestrosa, 2010 ). In TGF-β1-overexpressing transgenic (TGF) mice that reproduce elements of the AD cerebrovascular pathology (Wyss-Coray et al., 2000 ; Tong et al., 2005 ), PPARγ agonists pioglitazone and ibuprofen effectively reduced microglial and astrocyte activation (Lacombe et al., 2004 ; Nicolakakis et al., 2009 ) (Figure 1 B).
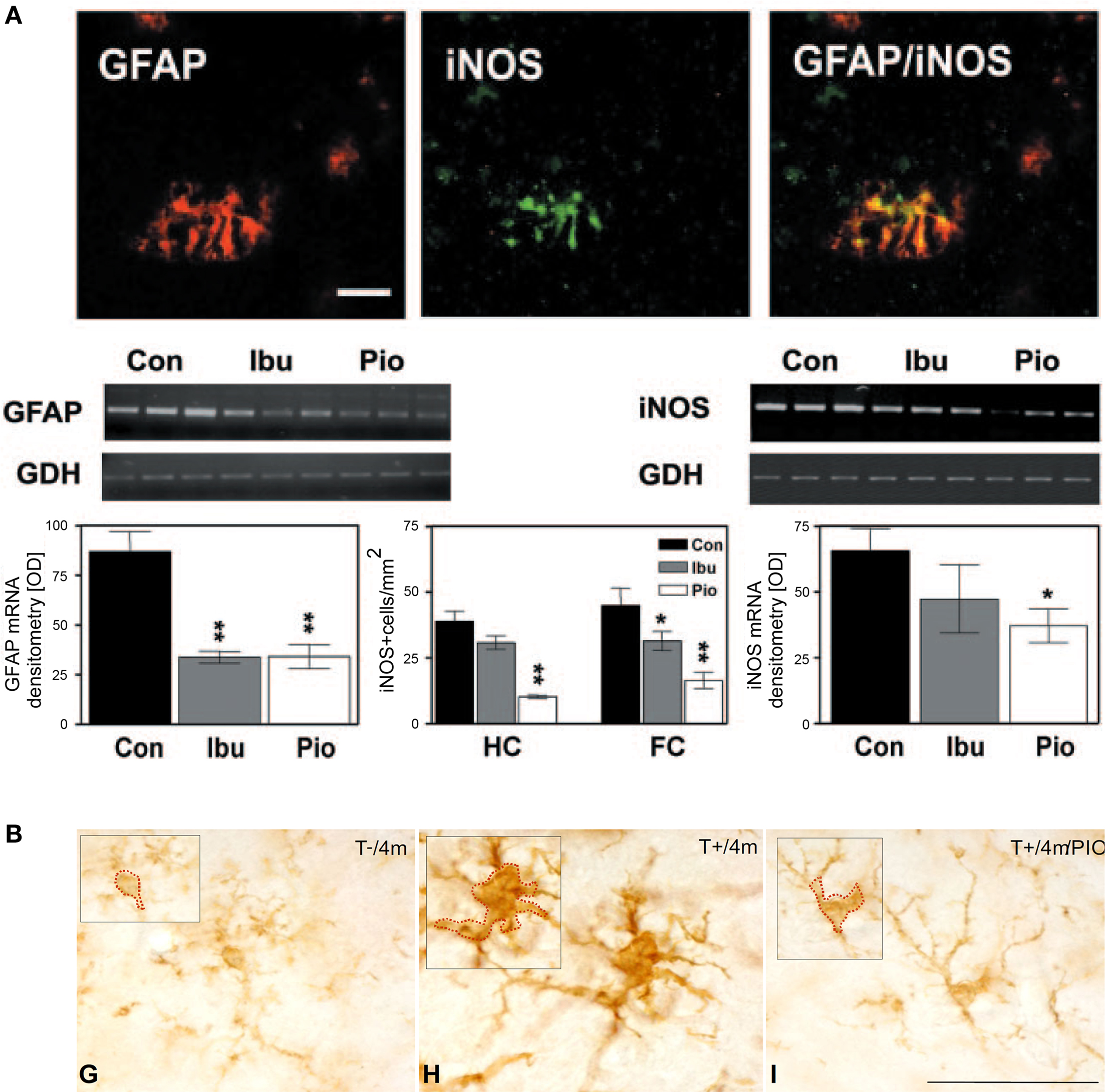
Figure 1. Anti-inflammatory effects of PPARγ agonists in transgenic mouse models of AD or of the cerebrovascular pathology of the disease. (A) Pioglitazone (Pio) and ibuprofen (Ibu) attenuated GFAP and iNOS expression in astrocytes of the hippocampus (HC) and frontal cortex (FC) of treated APPV717I mice. Bar, 50 μm. Reproduced with kind permission from Heneka et al. (2005) and Elsevier. (B) Pioglitazone countered microglial activation in TGF mice (T+) relative to age-matched controls (T−), as seen by the shrinking of the cell soma in animals treated for 2 months starting at 2 months of age. Bar, 50 μm. Reproduced with permission from Lacombe et al. (2004) and BioMed Central publisher.
Paradoxical Effects on Amyloid Pathology
A major effort in AD research has been to counter the amyloid pathology that hallmarks the disease, especially to reduce high levels of soluble Aβ oligomers shown detrimental to neuronal and synaptic function well before their assembly into parenchymal plaques (Hsia et al., 1999 ; Mucke et al., 2000 ; Lesné et al., 2008 ). To this end, several groups have investigated the effectiveness of PPARγ ligands against amyloidosis. Table 1 non-exhaustively summarizes the somewhat conflicting results, which likely reflect differences in model system (cell culture vs. in vivo), severity of deficits, dose and duration of treatment or method of quantification (e.g. ELISA, Western blot, immunohistochemistry). A more extensive review of NSAID effects in animal models and various cell lines, including those of neuronal origin, can be found in Gasparini et al. (2004) . The NSAIDs in Table 1 were given at doses believed to activate PPARγ. Of note, NSAIDs were shown to be effective independently of action on their classical targets, the COX enzymes, as evidenced in COX-1 and COX-2-deficient mouse embryonic fibroblasts (Weggen et al., 2001 ). They likely acted through PPARγ on β- and/or γ-secretase promoters to alter the proteolytic release of Aβ from its precursor protein, APP (Eriksen et al., 2003 ; Heneka et al., 2005 ; Sastre et al., 2003 , 2006 ). Also notable is the lack of studies investigating the effects of PPARγ ligands on vascular Aβ deposition, that is, CAA.
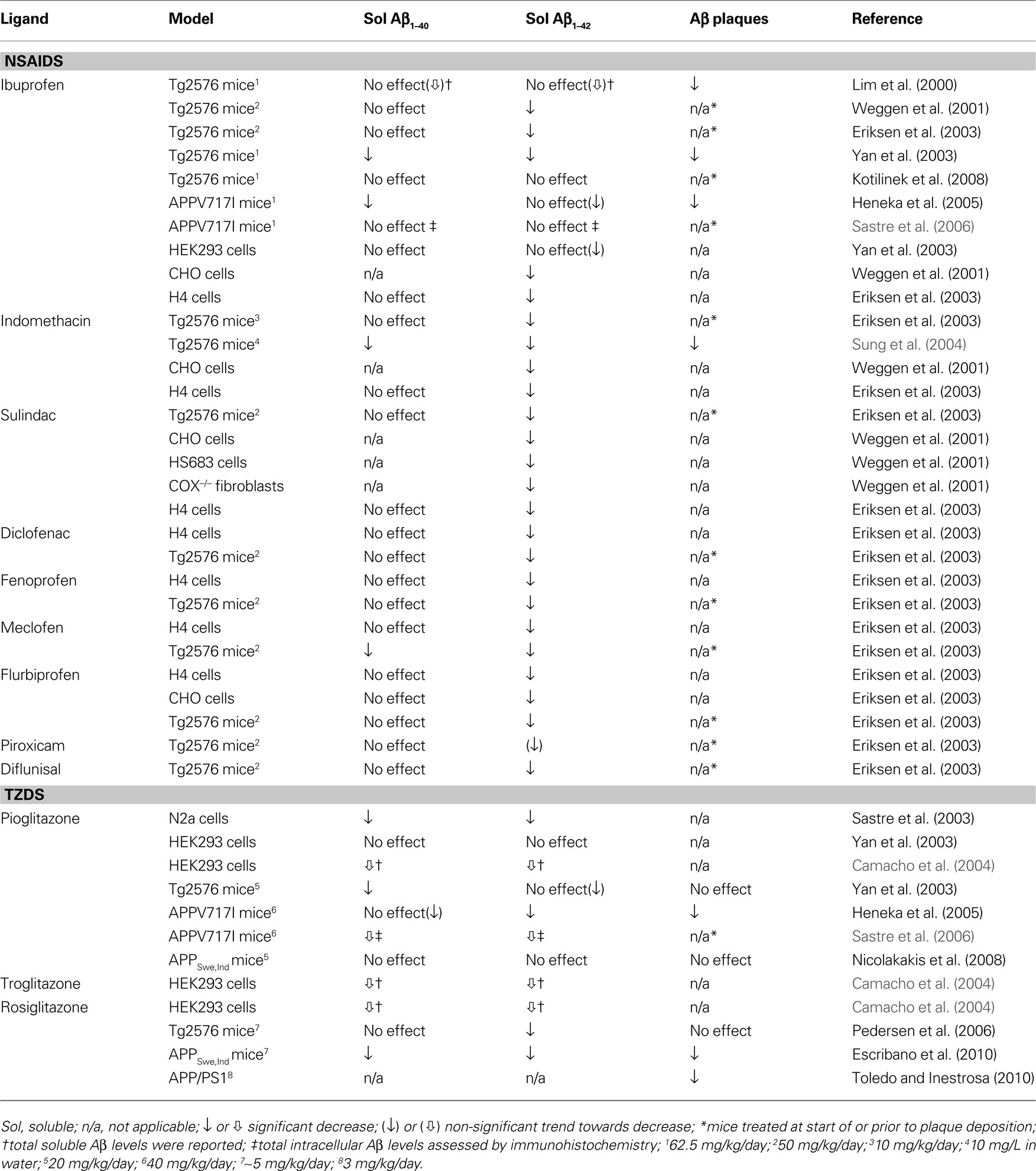
Table 1. Effects of PPARγ ligands on amyloidosis in transgenic mice or APP-expressing cells
Cerebrovascular Protection
Peroxisome proliferator-activated receptorγ plays an integral role in maintaining cerebrovascular health. Its genetic inactivation in mice results in enhanced free radical generation, endothelial dysfunction and hypertrophy of cerebral arterioles (Beyer et al., 2008 ; Ketsawatsomkron et al., 2010 ) (Figure 2 A). Free radicals such as superoxide (O2•−) are particularly deleterious for vascular function, as they sequester endothelium-derived vasodilators or disrupt smooth muscle ion channels, resulting in diminished dilatory ability. Accordingly, the use of TZDs to activate PPARγ can enhance antioxidant systems in human endothelial cells (Inoue et al., 2001 ), and suppress free radical and inflammatory pathways in the peripheral vasculature of humans and animal models of hypertension and diabetes, thereby restoring vascular function (Diep et al., 2002 ; Bagi et al., 2004 ; Martens et al., 2005 ; Hwang et al., 2007 ). PPARγ-induced changes in gene expression include upregulation of the O2•− scavenger, superoxide dismutase1 (SOD1) and downregulation of Nox-1, -2, and -4 genes encoding subunits of the O2•−-generating NADPH oxidase (Inoue et al., 2001 ; Hwang et al., 2005 , 2007 ), as measured with real-time PCR. PPARγ may also be involved in the induction of oxidative stress-response genes, such as heme oxygenase-1 (HO-1) through the nuclear factor E2-related factor/antioxidant response element (Nrf2/ARE) pathway, as shown in peripheral tissues (reviewed in Kang et al., 2005 ). The relevance of this pathway in the brain and cerebral vessels remains to be demonstrated. Moreover, PPARγ ligands can regulate the expression of ECM molecules, such as CTGF and collagen IV (Fu et al., 2001 ; Zafiriou et al., 2005 ), which are upregulated in AD brain vessels and implicated in the thickening of cerebrovascular basement membranes. Further, PPARγ overexpression or activation with ligands can reduce adhesion molecule and matrix metalloproteinase expression and activity in the vasculature, and prevent Rac1 and RhoA activation (Keen et al., 2004 ; Ramirez et al., 2008 ; Huang et al., 2009 ), thus protecting levels of tight junction proteins, and countering leukocyte–endothelial interactions and blood–brain barrier (BBB) compromise that occur in AD (Fiala et al., 2002 ; Zipser et al., 2007 ). Finally, ligand effects independent of PPARγ activation could also contribute to protecting the AD cerebral circulation; these include inhibition of the L-type Ca2+ current (Zhang et al., 1994 ) and opening of K+ channels (Nomura et al., 2008 ) in smooth muscle cells, promoting vasorelaxation.
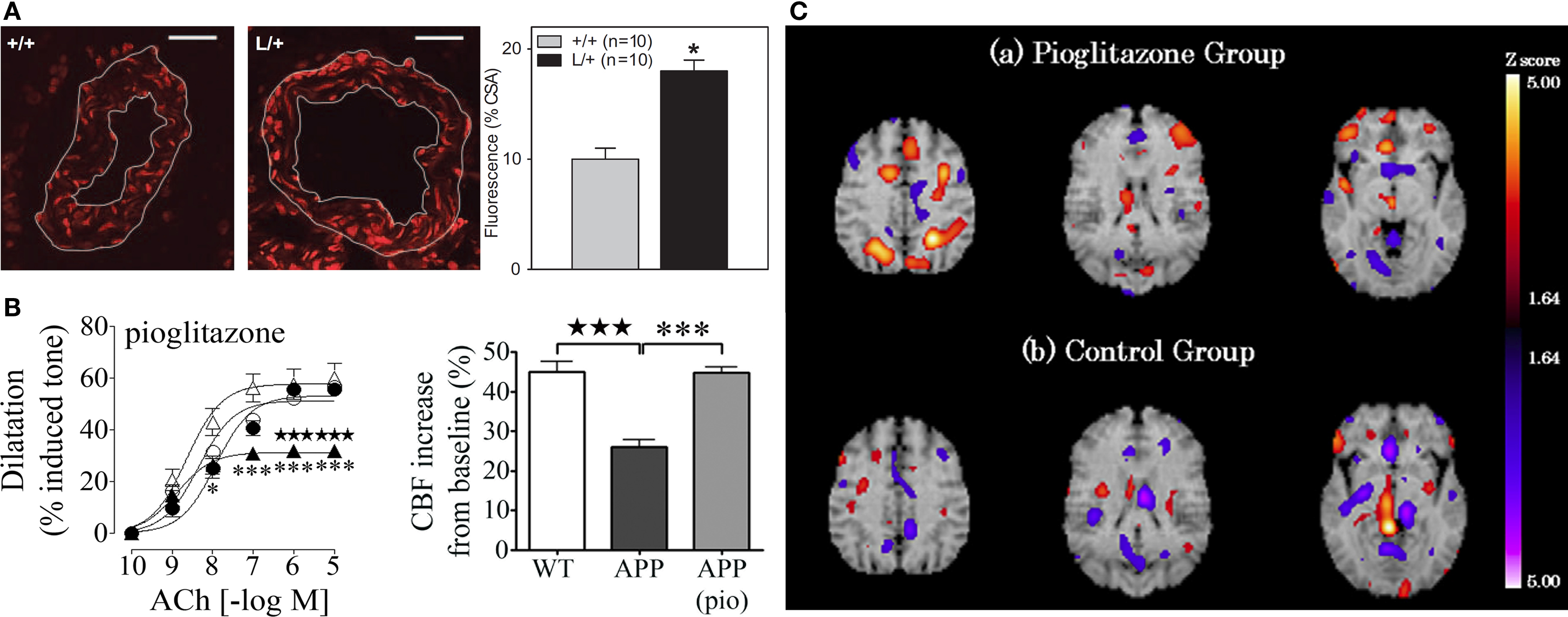
Figure 2. (A) Genetic inactivation of PPARγ in cerebral arterioles (L/+) leads to an increase in O2− content relative to control vessels (+/+), as detected with dihydroethidine fluorescence. Bar, 20 μm. Reproduced with permission from Beyer et al. (2008) and Wolters Kluwer. (B) Pioglitazone (pio) rescued cerebrovascular dilatations to acetylcholine (ACh) and the neurovascular coupling response to whisker stimulation in aged APP mice. •, wild-type (WT); ○, treated WT; ▲, APP; △, treated APP. Reproduced from Nicolakakis et al. (2008) . (C) Pioglitazone restored CBF in the parietal and frontal lobes of AD patients in single photon emission computed tomography (SPECT) studies. Reproduced with generous permission from Sato et al. (2009) and Elsevier.
Collectively, the evidence suggests that PPARγ ligands offer therapeutic potential against many aspects of the AD cerebrovascular pathology. In aged APP mice, in vivo pioglitazone treatment rescued evoked cerebrovascular dilatations, basal availability of the vasodilator nitric oxide, as well as the neurovascular coupling response to whisker stimulation (Figure 2 B). The improvements were accompanied by normalization of SOD2 levels in cerebral vessels, a sensitive marker of oxidative stress (Nicolakakis et al., 2008 ). Arterial responsiveness and neurovascular coupling were similarly rescued by pioglitazone in aged TGF mice, even though vascular structure remained altered and antioxidants were ineffective (Tong et al., 2005 ; Nicolakakis et al., 2009 ). The latter findings suggest that pioglitazone may be able to restore the function of an aged, fibrotic arterial tree that is insensitive to pure antioxidants. This may bear significance for the AD cerebral circulation that is characterized by aging, vascular wall thickening and TGF-β1 elevations (Grammas and Ovase, 2002 ). Recently, Sato et al. (2009) reported improved cerebral blood flow (CBF) in AD patients treated with pioglitazone (Figure 2 C), positioning the TZD as a valuable tool against chronic hypoperfusion in AD, along with the cholinesterase inhibitors, the currently prescribed AD drugs that have been re-evaluated and also shown to restore basal and evoked perfusion responses (Rosengarten et al., 2006 ; Yoshida et al., 2007 ).
Benefits on Neurons and Memory
Ultimately, it is hoped that the aforementioned benefits will promote a more favorable environment for neuronal cells, with lessened inflammatory and oxidative injury, and improved nutrient delivery by the cerebral circulation. PPARγ activation is also expected to confer direct protection to neurons from the AD process, since they express the receptor, and its activation was shown to modulate pathways leading to neurodegeneration (Inestrosa et al., 2005 ; Fuenzalida et al., 2007 ; Pancani et al., 2009 ). Specifically, rosiglitazone and troglitazone prevented Aβ-induced cell death, morphological neurodegenerative changes, and rise in cytosolic Ca2+ in rat hippocampal neurons, concomitantly with the restoration of cytoplasmic β-catenin levels and inhibition of glycogen synthase kinase-3β (GSK3β) activity implicated in τ hyperphosphorylation (Inestrosa et al., 2005 ). The benefits were not seen in the presence of the PPARγ inhibitor GW9662, confirming that neuroprotection was mediated by the receptor. In another study, rosiglitazone protected hippocampal and dorsal root ganglion neurons against Aβ-induced mitochondrial damage and nerve growth factor (NGF) deprivation-induced apoptosis, respectively. This was associated with upregulation of the anti-apoptotic protein Bcl-2. In the same study, PC12 cells constitutively overexpressing PPARγ were resistant to Aβ-induced injury and showed no increase in oxidative stress following hydrogen peroxide challenge. The cells showed a four to fivefold increase in Bcl-2 levels, but nearly no Bcl-2 or protection against injury when PPARγ or Bcl-2 was inactivated (Fuenzalida et al., 2007 ).
Peroxisome proliferator-activated receptorγ activation was similarly beneficial in the streptozotocin-injected rat model of type III diabetes. In this model, cerebral insulin is depleted while pancreatic insulin is spared, and animals share many features in common with AD, including brain insulin resistance, neurodegeneration, and impairments in learning and memory. Chronic treatment with PPARγ agonists reversed these pathological alterations by increasing insulin receptor binding, countering oxidative stress and τ phosphorylation, and augmenting choline acetyltransferase (ChAT) expression (de la Monte et al., 2006 ). It also restored glucose uptake in brain slices, and improved spatial memory in the Morris water maze (Pathan et al., 2006 ). However, in young TGF mice, pioglitazone failed to reverse the TGF-β1-induced decrease in basal CGU, and it unexpectedly reduced CGU in the most metabolically active brain areas of wild-type mice (Galea et al., 2006 ), a finding that could be related to PPARγ-independent effects on mitochondrial function (Feinstein et al., 2005 ). In this regard, it has been postulated that direct binding of PPARγ ligands to mitochondrial proteins in a receptor-independent fashion could indirectly cause protective changes in gene expression. This would occur through a decrease in pyruvate-driven respiration, leading to an increase in lactate production, free radicals and oxidative stress, eliciting, in turn, a stress response, characterized by increased expression of heat shock proteins, as well as anti-inflammatory and cytoprotective genes (Feinstein et al., 2005 ).
In transgenic AD mouse models with prominent neuronal and behavioral deficits, 4-month rosiglitazone therapy attenuated cognitive impairments of 13-month-old Tg2576 animals in the radial arm maze (Pedersen et al., 2006 ). It was argued that this was due to normalization of increased glucocorticoid levels that are known to impair memory. The explanation offered a rationale for using rosiglitazone, which may not cross the BBB as readily as pioglitazone (Maeshiba et al., 1997 ), with the authors suggesting that rosiglitazone acted on molecules such as insulin and glucocorticoids that are capable of penetrating the brain. In APPSwe,Ind mice bearing the Indiana (Ind) in addition to the Swedish (Swe) APP mutation of Tg2576 mice, rosiglitazone restored performance in the object recognition and Morris water maze memory tests (Escribano et al., 2010 ). Rosiglitazone also improved performance of APP/presenilin1 (PS1) mice in a modified version of the Morris water maze (Toledo and Inestrosa, 2010 ). In the modified test, rosiglitazone reduced the number of trials to reach criterion (i.e. locate escape platform, <20 s, three successive trials), and harmonized swimming strategies of treated APP/PS1 mice to those of wild-type counterparts (Toledo and Inestrosa, 2010 ). In contrast to the rosiglitazone studies, we have found no effect of pioglitazone on memory. This was in spite of positive effects on glial cell activation and oxidative stress, and despite restored neurometabolic coupling during whisker stimulation, as measured with 18F-fluorodeoxyglucose positron emission tomography (FDG-PET), an indication of restored neuronal activity (Nicolakakis et al., 2008 ). The reason is unknown, but it could be related to different properties of the TZD agonists used, the advanced age of the animals, shorter treatment duration (6–8 weeks), insufficient cholinergic recovery and residual mitochondrial oxidative stress in neuronal perikarya, as detected by SOD2 immunostaining. Additional possibilities include the use of a stringent Morris water maze test that did not include visible platform pretraining, and finally, lack of a pioglitazone effect on toxic soluble Aβ oligomers. This is in contrast with the lowering of soluble Aβ species by rosiglitazone in the two studies reporting cognitive amelioration (Pedersen et al., 2006 ; Escribano et al., 2010 ; see Table 1 ). Whether a reduction in Aβ oligomers is required for mnemonic improvement is still debated, especially in light of the improved Morris water maze performance of elderly Tg2576 mice treated with COX-2 inhibitors, yet no reduction in levels of soluble and insoluble Aβ1–40/42 or Aβ*56 (Kotilinek et al., 2008 ), a 56-kDa Aβ1–42 oligomer linked to the memory deficits of these mice (Lesné et al., 2006 ).
The disparate results among mouse studies are made more difficult to interpret since most experiments lack a positive control against which to compare PPARγ therapeutic effects. For example, it would be desirable to compare the behavioral outcome of TZD therapy with that of cholinesterase inhibitors like donepezil (Aricept®), rivastigmine (Exelon®) or galantamine (Reminyl®), which are currently used to treat mild-to-moderate AD patients. Additionally, differences between the mouse models and human disease may explain benefits in the former, but not the latter, and vice versa. The efficacy of compounds in transgenic mouse models that lack neurofibrillary tangles and overt neurodegeneration (McGowan et al., 2006 ) may have limited predictive value for AD patients. A case in point is the positive outcome with TZDs in clinical trials, despite mixed results in the mouse models. To date, there have been two trials with rosiglitazone (Watson et al., 2005 ; Risner et al., 2006 ), and two with pioglitazone (Hanyu et al., 2009 ; Sato et al., 2009 ), and the reported benefits after 6 months were often comparable to those seen with cholinesterase inhibitors, in terms of point improvement in the Mini-Mental State Exam (MMSE) and/or Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog). For example, rosiglitazone improved the ADAS-Cog by 2.94 points in mild-to-moderate AD patients lacking the ApoE4 susceptibility factor (Risner et al., 2006 ), an improvement well in the range of that offered by cholinesterase inhibitors (0.84–3.99 points) (see references in Risner et al., 2006 and Yoshida et al., 2007 ). If larger studies corroborate these findings, TZDs may be considered as primary therapy for patients with diabetes, who are at greater risk of developing AD (Ott et al., 1999 ; Kroner, 2009 ) or for diabetic AD patients, who could forego cholinesterase inhibitors in favor of compounds that can additionally offer glycemic control.
Conclusion
The evidence reviewed herein suggests a beneficial role of PPARγ ligands on the cerebrovascular, glial and neuronal compartments affected by the AD process. Through the ability to regulate genes implicated in survival, brain glucose metabolism, oxidative stress, inflammation, vascular fibrosis, amyloid generation, and neurofibrillary tangle formation, PPARγ agonists offer therapeutic potential against most elements of the AD pathology (Figure 3 ). However, many of the aforementioned effects require further confirmation in appropriate AD models, and validation in patients. Positive cognitive outcomes reported by the first clinical trials with TZDs warrant larger studies to confirm whether PPARγ ligands should be considered as treatments for AD.
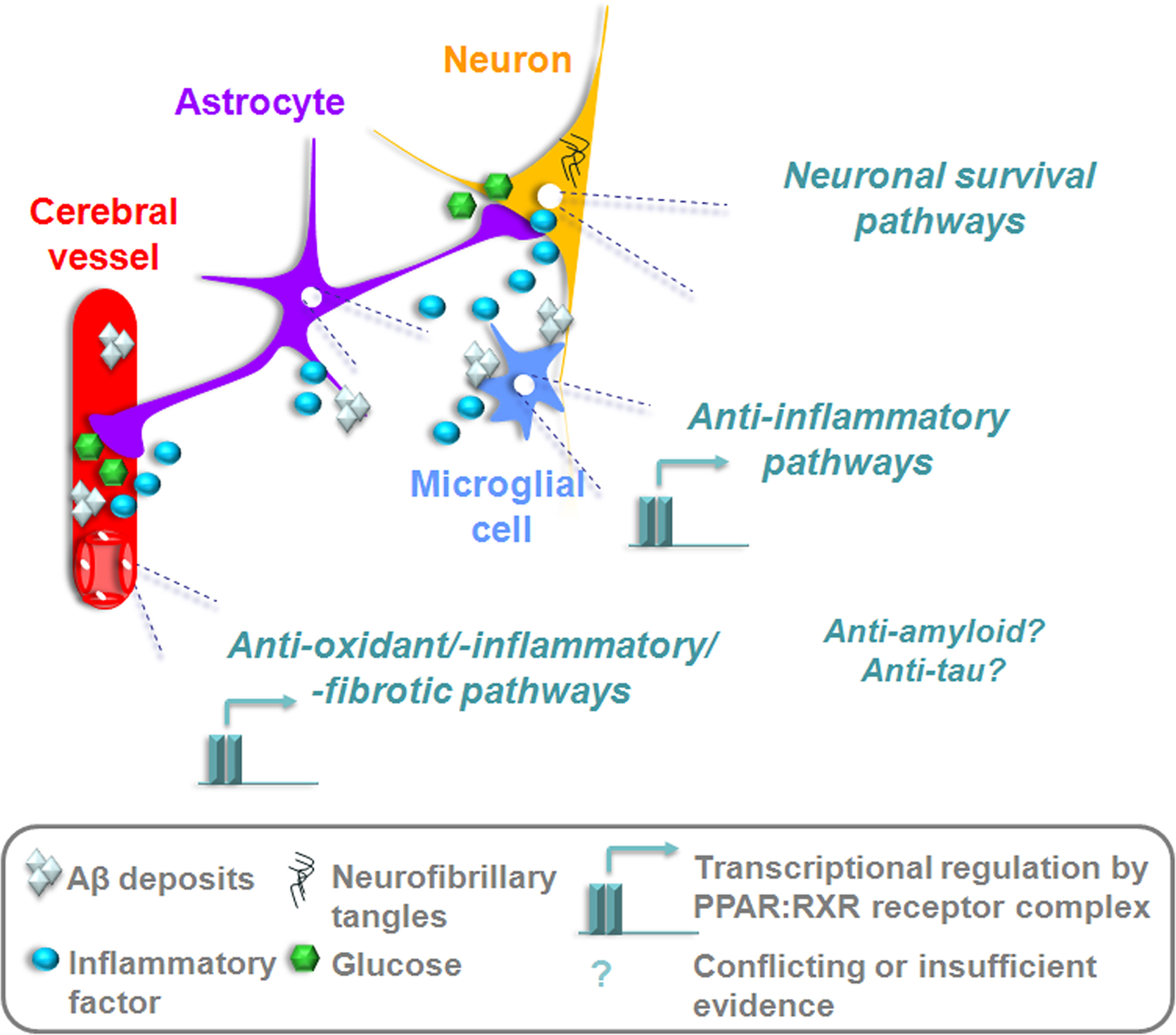
Figure 3. PPARγ activation could rescue afflicted vascular, glial, and neuronal compartments in the AD brain. Transcriptional suppression of inflammatory, oxidative, and fibrotic pathways would reduce tissue injury and reinstate adequate substrate delivery by the cerebral circulation. TZDs capable of passing the BBB could upregulate anti-apoptotic factors in neurons, re-sensitize cells to insulin and restore glucose utilization. The potential to counter amyloidogenesis and τ hyperphosphorylation could be disease-modifying, but requires further empirical support and confirmation in patients.
Conflict of Interest Statement
The authors received a research grant from Takeda Pharmaceuticals North America, Inc.
Acknowledgments
Work from our laboratory cited in the text was supported by research grants from the Canadian Institutes of Health Research (CIHR, MOP-84275), the Alzheimer Society of Canada, Takeda Pharmaceuticals North America, Inc., a CIHR studentship (N.N.), Jeanne Timmins Costello (T.A.), and Fonds de la Recherche en Santé du Québec (B.O.) fellowships. We thank Dr. L. Mucke (Gladstone Inst. of Neurological Disease and Dept. Neurology, UCSF, CA, USA) and the J. David Gladstone Institutes for the hAPPSw,Ind and TGF-β1 transgenic mouse breeders.
References
Akiyama, H., Barger, S. Barnum, S., Bradt, B., Bauer, J., Cole, G. M., Cooper, N. R., Eikelenboom, P., Emmerling, M., Fiebich, B. L., Finch, C. E., Frautschy, S., Griffin, W. S., Hampel, H., Hull, M., Landreth, G., Lue, L., Mrak, R., Mackenzie, I. R., McGeer, P. L., O’Banion, M. K., Pachter, J., Pasinetti, G., Plata-Salaman, C., Rogers, J., Rydel, R., Shen, Y., Streit, W., Strohmeyer, R., Tooyoma, I., Van Muiswinkel, F. L., Veerhuis, R., Walker, D., Webster, S., Wegrzyniak, B., Wenk, G., and Wyss-Coray, T. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421.
Bagi, Z., Koller, A., and Kaley, G. (2004). PPARγ activation, by reducing oxidative stress, increases NO bioavailability in coronary arterioles of mice with type 2 diabetes. Am. J. Physiol. Heart. Circ. Physiol. 286, H742–H748.
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 118, 103–113.
Beyer, A. M., Baumbach, G. L., Halabi, C. M., Modrick, M. L., Lynch, C. M., Gerhold, T. D., Ghoneim, S. M., de Lange, W. J., Keen, H. L., Tsai, Y. S., Maeda, N., Sigmund, C. D., and Faraci, F. M. (2008). Interference with PPARγ signaling causes cerebral vascular dysfunction, hypertrophy and remodeling. Hypertension 51, 867–871.
Butterfield, D. A., Drake, J., Pocernich, C., and Castegna, A. (2001). Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends. Mol. Med. 7, 548–554.
Camacho, I. E., Serneels, L., Spittaels, K., Merchiers, P., Dominguez, D., and De Strooper, B. (2004). Peroxisome proliferator-activated receptor γ induces a clearance mechanism for the amyloid-β peptide. J. Neurosci. 24, 10908–10917.
Chawla, A., Barak, Y., Nagy, L., Liao, D., Tontonoz, P., and Evans, R. M. (2001). PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 7, 48–52.
Chen, F., Law, S. W., and O’Malley, B. W. (1993). Identification of two mPPAR related receptors and evidence for the existence of five subfamily members. Biochem. Biophys. Res. Commun. 196, 671–677.
Christov, A., Ottman, J., Hamdheydari, L., and Grammas, P. (2008). Structural changes in Alzheimer’s disease brain microvessels. Curr. Alzheimer Res. 5, 392–395.
Clozel, M., and Salloukh, H. (2005). Role of endothelin in fibrosis and anti-fibrotic potential of bosentan. Ann. Med. 37, 2–12.
Combs, C. K., Johnson, D. E., Karlo, J. C., Cannady, S. B., and Landreth, G. E. (2000). Inflammatory mechanisms in Alzheimer’s disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J. Neurosci. 20, 558–567.
de la Monte, S. M., Tong, M., Lester-Coll, N., Plater, M. Jr., and Wands, J. R. (2006). Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J. Alzheimers Dis. 10, 89–109.
Diep, Q. N., El Mabrouk, M., Cohn, J. S., Endemann, D., Amiri, F., Virdis, A., Neves, M. F., and Schiffrin, E. L. (2002). Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-γ. Circulation 105, 2296–2302.
Dreyer, C., Krey, G., Keller, H., Givel, F., Helftenbein, G., and Wahli, W. (1992). Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell 68, 879–887.
Eriksen, J., Sagi, S. A., Smith, T. E., Weggen, S., Das, P., McLendon, D. C., Ozols, V. V., Jessing, K. W., Zavitz, K. H., Koo, E. H., and Golde, T. E. (2003). NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ42 in vivo. J. Clin. Invest. 112, 440–449.
Escribano, L., Simón, A. M., Gimeno, E., Cuadrado-Tejedor, M., de Maturana, R. L., García-Osta, A., Ricobaraza, A., Pérez-Mediavilla, A., Río, J. D., and Frechilla, D. (2010). Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology doi: 10.1038/npp.2010.32.
Farkas, E., and Luiten, P. G. (2001). Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 64, 575–611.
Feinstein, D. L., Spagnolo, A., Akar, C., Weinberg, G., Murphy, P., Gavrilyuk, V., and Dello Russo, C. (2005). Receptor-independent actions of PPAR thiazolidinedione agonists: Is mitochondrial function the key? Biochem. Pharmacol. 70, 177–188.
Fiala, M., Liu, Q. N., Sayre, J., Pop, V., Brahmandam, V., Graves, M. C., and Vinters, H. V. (2002). Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur. J. Clin. Inv. 32, 360–371.
Fu, M., Zhang, J., Zhu, X., Myles, D. E., Willson, T. M., Liu, X., and Chen, Y. E. (2001). Peroxisome proliferator-activated receptor γ inhibits transforming growth factor β-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3. J. Biol. Chem. 276, 45888–45894.
Fuenzalida, K., Quintanilla, R., Ramos, P., Piderit, D., Fuentealba, R. A., Martinez, G., Inestrosa, N. C., and Bronfman, M. (2007). Peroxisome proliferator-activated receptor γ up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J. Biol. Chem. 282, 37006–37015.
Fukuyama, H., Ogawa, M., Yamauchi, H., Yamaguchi, S., Kimura, J., Yonekura, Y., and Konishi, J. (1994). Altered cerebral energy metabolism in Alzheimer’s disease: a PET study. J. Nucl. Med. 35, 1–6.
Galea, E., Feinstein, D. L., and Lacombe, P. (2006). Pioglitazone does not increase cerebral glucose utilisation in a murine model of Alzheimer’s disease and decreases it in wild-type mice. Diabetologia 49, 2153–2161.
Gasparini, L., Ongini, E., and Wenk, G. (2004). Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of action. J. Neurochem. 91, 521–536.
Germain, P., Staels, B., Dacquet, C., Spedding, M., and Laudet, V. (2006). Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 58, 685–704.
Grammas, P., and Ovase, R. (2002). Cerebrovascular transforming growth factor-β contributes to inflammation in the Alzheimer’s disease brain. Am. J. Pathol. 160, 1583–1587.
Hanyu, H., Sato, T., Kiuchi, A., Sakurai, H., and Iwamoto, T. (2009). Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J. Am. Geriatr. Soc. 57, 177–179.
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., O’Banion, K., Klockgether, T., Van Leuven, F., and Landreth, G. E. (2005). Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1-42 levels in APPV717I transgenic mice. Brain 128, 1442–1453.
Hermann, D. M., Kilic, E., Hata, R., Hossmann, K. A., and Mies, G. (2001). Relationship between metabolic dysfunctions, gene responses and delayed cell death after mild focal cerebral ischemia in mice. Neuroscience 104, 947–955.
Hess, R., Stäubli, W., and Riess, W. (1965). Nature of the hepatomegalic effect produced by ethyl-chloro-phenoxy-isobutyrate in the rat. Nature 208, 856–858.
Hock, C., Villringer, K., Müller-Spahn, F., Wenzel, R., Heekeren, H., Schuh-Hofer, S., Hofmann, M., Minoshima, S., Schwaiger, M., Dirnagl, U., and Villringer, A. (1997). Decrease in parietal cerebral hemoglobin oxygenation during performance of a verbal fluency task in patients with Alzheimer’s disease monitored by means of near-infrared spectroscopy (NIRS)-correlation with simultaneous rCBF-PET measurements. Brain Res. 755, 293–303.
Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., Kholodenko, D., Malenka, R. C., Nicoll, R. A., and Mucke, L. (1999). Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 96, 3228–3233.
Huang, W., Eum, S. Y., András, I. E., Hennig, B., and Toborek, M. (2009). PPARα and PPARγ attenuate HIV-induced dysregulation of tight junction proteins by modulations of matrix metalloproteinase and proteasome activities. FASEB J. 23, 1596–1606.
Hwang, J., Kleinhenz, D. J., Lassègue, B., Griendling, K. K., Dikalov, S., and Hart, C. M. (2005). Peroxisome proliferator-activated receptor-{gamma} ligands regulate endothelial membrane superoxide production. Am. J. Physiol. Cell Physiol. 288, C899–C905.
Hwang, J., Kleinhenz, D. J., Rupnow, H. L., Campbell, A. G., Thulé, P. M., Sutliff, R. L., and Hart, C. M. (2007). The PPARγ ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vasc. Pharmacol. 46, 456–462.
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Neurosci. Rev. 5, 347–360.
In’t Veld, B. A., Ruitenberg, A., Hofman, A., Launer, L. J., van Duijn, C. M., Stijnen, T., Breteler, M. M. B., and Stricker, B. H. C. (2001). Nonsteroidal anti-inflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 345, 1515–1521.
Inestrosa, N. C., Godoy, J. A., Quintanilla, R. A., Koenig, C. S., and Bronfman, M. (2005). Peroxisome proliferator-activated receptor γ is expressed in hippocampal neurons and its activation prevents β-amyloid neurodegeneration: role of Wnt signaling. Exp. Cell. Res. 304, 91–104.
Inoue, I., Goto, S., Matsunaga, T., Nakajima, T., Awata, T., Hokari, S., Komoda, T., and Katayama, S. (2001). The ligands/activators for peroxisome proliferator-activated receptor α(PPARα) and PPARγ increase Cu2+, Zn2+-superoxide dismutase and decrease p22phox message expression in primary endothelial cells. Metab. Clin. Exp. 50, 3–11.
Issemann, I., and Green, S. (1990). Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347, 645–650.
Issemann, I., Prince, R. A., Tugwood, J. D., and Green, S. (1993). The peroxisome proliferator-activated receptor:retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J. Mol. Endocrinol. 11, 37–47.
Jiang, C., Ting, A. T., and Seed, B. (1998). PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 391, 82–86.
Jiang, Q., Heneka, M., and Landreth, G. E. (2008). The role of peroxisome proliferator-activated receptor-γ (PPARγ) in Alzheimer’s disease. CNS Drugs 22, 1–14.
Kalaria, R. N., and Harik, S. I. (1989). Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J. Neurochem. 53, 1083–1088.
Kang, K. W., Lee, S. J., and Kim, S. G. (2005). Molecular mechanism of Nrf2 activation by oxidative stess. Antioxid. Redox Signal. 7, 1664–1673.
Keen, H. L., Ryan, M. J., Beyer, A., Mathur, S., Scheetz, T. E., Gackle, B. D., Faraci, F. M., Casavant, T. L., and Sigmund, C. D. (2004). Gene expression profiling of potential PPARγ target genes in mouse aorta. Physiol. Genomics 18, 33–42.
Ketsawatsomkron, P., Pelham, C. J., Groh, S., Keen, H. L., Faraci, F. M., and Sigmund, C. D. (2010). Does peroxisome proliferator-activated receptor-γ (PPARγ) protect from hypertension directly through effects in the vasculature? J. Biol. Chem. 285, 9311–9316.
Kitamura, Y., Shimohama, S., Koike, H., Kakimura, J., Matsuoka, Y., Nomura, Y., Gebicke-Haerter, P. J., and Taniguchi, T. (1999). Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-γ in Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 254, 582–586.
Kliewer, S. A., Forman, B. M., Blumberg, B., Ong, E. S., Borgmeyer, U., Mangelsdorf, D. J., Umesono, K., and Evans, R. M. (1994). Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 91, 7355–7359.
Kliewer, S. A., Sundseth, S. S., Jones, S. A., Brown, P. J., Wisely, G. B., Koble, C. S., Devchand, P., Wahli, W., Willson, T. M., Lenhard, J. M., and Lehmann, J. M. (1997). Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. USA 94, 4318–4323.
Kotilinek, L. A., Westerman, M. A., Wang, Q., Panizzon, K., Lim, G. P., Simonyi, A., Lesné, S., Falinska, A., Younkin, L. H., Younkin, S. G., Rowan, M., Cleary, J., Wallis, R. A., Sun, G. Y., Cole, G., Frautschy, S., Anwyl, R., and Ashe, K. H. (2008). Cyclooxygenase-2 inhibition improves amyloid-β-mediated suppression of memory and synaptic plasticity. Brain 131, 651–664.
Kroner, Z. (2009). The relationship between Alzheimer’s disease and diabetes: type 3 diabetes? Altern. Med. Rev. 14, 373–379.
Lacombe, P., Mathews, P. M., Schmidt, S. D., Breidert, T., Heneka, M. T., Landreth, G. E., Feinstein, D., and Galea, E. (2004). Effect of anti-inflammatory agents on transforming growth factor beta over-expressing mouse brains: a model revised. J. Neuroinflammation 1, 11–27.
Landreth, G., Jiang, Q., Mandrekar, S., and Heneka, M. (2008). PPARγ agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 5, 481–489.
Langbaum, J. B., Chen, K., Lee, W., Reschke, C., Bandy, D., Fleisher, A. S., Alexander, G. E., Foster, N. L., Weiner, M. W., Koeppe, R. A., Jagust, W. J., and Reiman, E. M.; Alzheimer’s Disease Neuroimaging Initiative. (2009). Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography images from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Neuroimage 45, 1107–1116.
Lehmann, J. M., Lenhard, J. M., Oliver, B. B., Ringold, G. M., and Kliewer, S. A. (1997). Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 272, 3406–3410.
Lehmann, J. M., Moore, L. B., Smith-Oliver, T. A., Wilkison, W. O., Willson, T. M., and Kliewer, S. A. (1995). An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J. Biol. Chem. 270, 12953–12956.
Lesné, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., Gallagher, M., and Ashe, K. H. (2006). A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357.
Lesné, S., Kotilinek, L., and Ashe, K. H. (2008). Plaque-bearing mice with reduced levels of oligomeric amyloid-β assemblies have intact memory function. Neuroscience 151, 745–749.
Lim, G. P., Yang, F., Chu, T., Chen, P., Beech, W., Teter, B., Tran, T., Ubeda, O., Ashe, K. H., Frautschy, S. A., and Cole, G. M. (2000). Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 20, 5709–5714.
Maeshiba, Y., Kiyota, Y., Yamashita, K., Yoshimura, Y., Motohashi, M., and Tanayama, S. (1997). Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. Arzneimittelforschung 47, 29–35.
Martens, F. M., Visseren, F. L., de Koning, E. J., and Rabelink, T. J. (2005). Short-term pioglitazone treatment improves vascular function irrespective of metabolic changes in patients with type 2 diabetes. J. Cardiovasc. Pharmacol. 46, 773–778.
McGeer, P. L., McGeer, E., Rogers, J., and Sibley, J. (1990). Anti-inflammatory drugs and Alzheimer disease. Lancet 335, 1037.
McGowan, E., Eriksen, J., and Hutton, M. (2006). A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet. 22, 281–289.
McKenna, N. J., and O’Malley, B. W. (2002). Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108, 465–474.
Melrose, R. J., Campa, O. M., Harwood, D. G., Osato, S., Mandelkern, M. A., and Sultzer, D. L. (2009). The neural correlates of naming and fluency deficits in Alzheimer’s disease: an FDG-PET study. Int. J. Geriatr. Psychiatry 24, 885–893.
Moreno, S., Farioli-Vecchioli, S., and Cerù, M. P. (2004). Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 123, 131–145.
Mosconi, L., Perani, D., Sorbi, S., Herholz, K., Nacmias, B., Holthoff, V., Salmon, E., Baron, J. C., De Cristofaro, M. T., Padovani, A., Borroni, B., Franceschi, M., Bracco, L., and Pupi, A. (2004). MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology 63, 2332–2340.
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., Hu, K., Kholodenko, D., Johnson-Wood, K., and McConlogue, L. (2000). High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058.
Nagy, L., and Schwabe, J. W. R. (2004). Mechanism of the nuclear receptor molecular switch. Trends Biochem. Sci. 29, 317–324.
Nicolakakis, N., Aboulkassim, T., Ongali, B., Lecrux, C., Fernandes, P., Rosa-Neto, P., Tong, X. K., and Hamel, E. (2008). Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor γ agonist. J. Neurosci. 28, 9287–9296.
Nicolakakis, N., Aboulkassim, T., Ongali, B., Lecrux, C., Fernandes, P., Tong, X. K., Sandoe, C., and Hamel, E. (2009) Reversal of cerebrovascular dysfunction by pioglitazone in APP and TGF mice: relevance to Alzheimer’s disease cerebrovascular pathology. 38th Annual Meeting of the Society for Neurosciences. . Abstract no. 102909.
Nomura, H., Yamawaki, H., Mukohda, M., Okada, M., and Hara, Y. (2008). Mechanisms underlying pioglitazone-mediated relaxation in isolated blood vessel. J. Pharmacol. Sci. 108, 258–265.
Ott, A., Stolk, R. P., van Harskamp, F., Pols, H. A., Hofman, A., and Breteler, M. M. (1999). Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53, 1937–1942.
Pancani, T., Phelps, J. T., Searcy, J. L., Kilgore, M. W., Chen, K. C., Porter, N. M., and Thibault, O. (2009). Distinct modulation of voltage-gated and ligand-gated Ca2+ currents by PPAR-γ agonists in cultured hippocampal neurons. J. Neurochem. 109, 1800–1811.
Pascual, G., Fong, A. L., Ogawa, S., Gamliel, A., Li, A. C., Perissi, V., Rose, D. W., Willson, T. M., Rosenfeld, M. G., and Glass, C. K. (2005). A sumoylation-dependent pathway mediating transrepression of inflammatory response genes by PPARγ. Nature 437, 759–763.
Pathan, A. R., Viswanad, B., Sonkusare, S. K., and Ramarao, P. (2006). Chronic administration of pioglitazone attenuates intracerebroventricular streptozotocin induced-memory impairment in rats. Life Sci. 79, 2209–2216.
Pedersen, W. A., McMillan, P. J., Kulstad, J. J., Leverenz, J. B., Craft, S., and Haynatzki, G. R. (2006). Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp. Neurol. 199, 265–273.
Ramirez, S. H., Heilman, D., Morsey, B., Potula, R., Haorah, J., and Persidsky, Y. (2008). Activation of peroxisome proliferator-activated receptor γ (PPARγ) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes. J. Immunol. 180, 1854–1865.
Reddy, J. K., and Krishnakantha, T. P. (1975). Hepatic peroxisome proliferation: induction by two novel compounds structurally unrelated to clofibrate. Science 190, 787–789.
Reger, M. A., Watson, G. S., Green, P. S., Baker, L. D., Cholerton, B., Fishel, M. A., Plymate, S. R., Cherrier, M. M., Schellenberg, G. D., Frey, W.H. 2nd., and Craft, S. (2008). Intranasal insulin administration dose-dependently modulates verbal memory and plasma β-amyloid in memory-impaired older adults. J. Alzheimers Dis. 13, 323–331.
Risner, M. E., Saunders, A. M., Altman, J. F. B., Ormandy, G. C., Craft, S., Foley, I. M., Zvartau-Hind, M. E., Hosford, D. A., and Roses, A. D. (2006). Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 6, 246–254.
Ricote, M., Li, A. C., Willson, T. M., Kelly, C. J., and Glass, C. K. (1998). The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 391, 79–82.
Roher, A. E., Lowenson, J. D., Clarke, S., Woods, A. S., Cotter, R. J., Gowing, E., and Ball, M. J. (1993). β-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 90, 10836–10840.
Rosengarten, B., Paulsen, S., Molnar, S., Kaschel, R., Gallhofer, B., and Kaps, M. (2006). Acetylcholine esterase inhibitor donepezil improves dynamic cerebrovascular regulation in Alzheimer patients. J. Neurol. 253, 58–64.
Ryan, M. J., Didion, S. P., Mathur, S., Faraci, F. M., and Sigmund, C. D. (2004). PPARγ agonist rosiglitazone improves vascular function and lowers blood pressure in hypertensive transgenic mice. Hypertension 43, 661–666.
Sastre, M., Dewachter, I., Landreth, G. E., Willson, T. M., Klockgether, T., van Leuven, F., and Heneka, M. T. (2003). Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-γ agonists modulate immunostimulated processing of amyloid precursor protein through regulation of β-secretase. J. Neurosci. 23, 9796–9804.
Sastre, M., Dewachter, I., Rossner, S., Bogdanovic, N., Rosen, E., Borghgraef, P., Evert, B. O., Dumitrescu-Ozimek, L., Thal, D. R., Landreth, G., Walter, J., Klockgether, T., van Leuven, F., and Heneka, M. T. (2006). Nonsteroidal anti-inflammtory drugs repress β-secretase gene promoter activity by the activation of PPARγ. Proc. Natl. Acad. Sci. USA 103, 443–448.
Sato, T, Hanyu, H, Hirao, K, Kanetaka, H, Sakurai, H, Iwamoto, T. (2009). Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging in press. doi:10.1016/j.neurobiolaging.2009.10.009.
Simpson, I. A., Chundu, K. R., Davies-Hill, T., Honer, W. G., and Davies, P. (1994). Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 35, 546–551.
Sung, S., Yang, H., Uryu, K., Lee, E. B., Zhao, L., Shineman, D., Trojanowski, J. Q., Lee, V. M., and Praticò, D. (2004). Modulation of nuclear factor-κB activity by indomethacin influences Aβ levels but not Aβ precursor protein metabolism in a model of Alzheimer’s disease. Am. J. Pathol. 165, 2197–2206.
Svoboda, D. J., and Azarnoff, D. L. (1966). Response of hepatic microbodies to a hypolipidemic agent, ethyl chlorophenoxyisobutyrate (CPIB). J. Cell Biol. 30, 442–450.
Toledo, E. M., and Inestrosa, N. C. (2010). Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1δE9 mouse model of Alzheimer’s disease. Mol. Psychiatry 15, 272–285.
Tong, X. K., and Hamel, E. (1999). Regional cholinergic denervation of cortical microvessels and nitric oxide synthase-containing neurons in Alzheimer’s disease. Neuroscience 92, 163–175.
Tong, X. K., Nicolakakis, N., Kocharyan, A., and Hamel, E. (2005). Vascular remodeling versus amyloid β-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J. Neurosci. 25, 11165–11174.
Tontonoz, P., Hu, E., Graves, R. A., Budavari, A. I., and Spiegelman, B. M. (1994). mPPARγ2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 8, 1224–1234.
Watson, G., Cholerton, B., Reger, M., Baker, L., Plymate, S., Asthana, S., Fishel, M., Kulstad, J., Green, P., Cook, D., Kahn, S., Keeling, M., and Craft, S. (2005). Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am. J. Geriatr. Psychiatry 13, 950–958.
Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., Findlay, K. A., Smith, T. E., Murphy, M. P., Bulter, T., Kang, D. E., Marquez-Sterling, N., Golde, T. E., and Koo, E. H. (2001). A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216.
Wyss-Coray, T., Lin, C., Sanan, D. A., Mucke, L., and Masliah, E. (2000). Chronic overproduction of transforming growth factor-β1 by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am. J. Pathol. 156, 139–150.
Wyss-Coray, T., Masliah, E., Mallory, M., McConlogue, L., Johnson-Wood, K., Lin, C., and Mucke, L. (1997). Amyloidogenic role of cytokine TGF-β1 in transgenic mice and in Alzheimer’s disease. Nature 389, 603–606.
Yan, Q., Zhang, J., Liu, H., Babu-Khan, S., Vassar, R., Biere, A. L., Citron, M., and Landreth, G. (2003). Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer’s disease. J. Neurosci. 23, 7504–7509.
Yoshida, T., Ha-Kawa, S., Yoshimura, M., Nobuhara, K., Kinoshita, T., and Sawada, S. (2007). Effectiveness of treatment with donepezil hydrochloride and changes in regional cerebral blood flow in patients with Alzheimer’s disease. Ann. Nucl. Med. 21, 257–265.
Zafiriou, S., Stanners, S. R., Saad, S., Polhill, T. S., Poronnik, P., and Pollock, C. A. (2005). Pioglitazone inhibits cell growth and reduces matrix production in human kidney fibroblasts. J. Am. Soc. Nephrol. 16, 638–645.
Zhang, F., Sowers, J. R., Ram, J. L., Standley, P. R., and Peuler, J. D. (1994). Effects of pioglitazone on calcium channels in vascular smooth muscle. Hypertension 24, 170–175.
Zhao, W. Q., Chen, H., Quon, M. J., and Alkon, D. L. (2004). Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 490, 71–81.
Zhu, Y., Alvares, K., Huang, Q., Rao, M. S., and Reddy, J. K. (1993). Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J. Biol. Chem. 268, 26817–26820.
Keywords: pioglitazone, arterial reactivity, cerebral blood flow, vascular fibrosis, brain metabolism, oxidative stress, inflammation, spatial memory
Citation: Nicolakakis N and Hamel E (2010) The nuclear receptor PPARγ as a therapeutic target for cerebrovascular and brain dysfunction in Alzheimer’s disease. Front. Ag. Neurosci. 2:21. doi: 10.3389/fnagi.2010.00021
Received: 01 February 2010;
Paper pending published: 21 February 2010;
Accepted: 29 April 2010;
Published online: 21 May 2010
Edited by:
Elena Galea, Universitat Autònoma de Barcelona, SpainReviewed by:
Marilyn J. Cipolla, University of Vermont, USAMagdalena Sastre, Imperial College London, UK
Copyright: © 2010 Nicolakakis and Hamel. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Edith Hamel, Laboratory of Cerebrovascular Research, Department of Neurology and Neurosurgery, Montreal Neurological Institute, McGill University, 3801 University Street, Montreal, QC, H3A 2B4, Canada. e-mail:ZWRpdGguaGFtZWxAbWNnaWxsLmNh