1
Arizona Research Laboratories Division of Neural Systems, Memory and Aging and Evelyn F. McKnight Brain Institute, University of Arizona, Tucson, AZ, USA
2
Department of Neurobiology and Evelyn F. McKnight Brain Institute, University of Alabama at Birmingham, Birmingham, AL, USA
3
Departments of Psychology and Neurology, University of Arizona, Tucson, AZ, USA
This brief review will focus on a new hypothesis for the role of epigenetic mechanisms in aging-related disruptions of synaptic plasticity and memory. Epigenetics refers to a set of potentially self-perpetuating, covalent modifications of DNA and post-translational modifications of nuclear proteins that produce lasting alterations in chromatin structure. These mechanisms, in turn, result in alterations in specific patterns of gene expression. Aging-related memory decline is manifest prominently in declarative/episodic memory and working memory, memory modalities anatomically based largely in the hippocampus and prefrontal cortex, respectively. The neurobiological underpinnings of age-related memory deficits include aberrant changes in gene transcription that ultimately affect the ability of the aged brain to be “plastic”. The molecular mechanisms underlying these changes in gene transcription are not currently known, but recent work points toward a potential novel mechanism, dysregulation of epigenetic mechanisms. This has led us to hypothesize that dysregulation of epigenetic control mechanisms and aberrant epigenetic “marks” drive aging-related cognitive dysfunction. Here we focus on this theme, reviewing current knowledge concerning epigenetic molecular mechanisms, as well as recent results suggesting disruption of plasticity and memory formation during aging. Finally, several open questions will be discussed that we believe will fuel experimental discovery.
Age-associated cognitive decline is an increasing biomedical concern in the US. According to the Administration on Aging, over 12% of the United States population is age 65 and older, and by 2030 this estimate is projected to reach 20%. In the general field of aging research, substantial effort has been focused on pathological aging associated with diseases such as Alzheimer’s disease. While this disease and many others are certainly devastating, it is also true that most people will not suffer from a dementing condition as they age; the prevalence of dementia among individuals aged 71 and older is 13.9% (Plassman et al., 2007
). Instead, most individuals will develop milder cognitive deficits known as age-associated memory impairment (AAMI; Crook et al., 1986
; Crook and Ferris, 1992
; Ferris and Kluger, 1996
). The memory deficits associated with AAMI are relatively subtle compared to those associated with a dementing condition, but they are nevertheless troublesome for the affected individual and their families, negatively affecting quality of life. Thus, with the average life span increasing, understanding the brain mechanisms that underlie age-associated memory decline is of increasing importance. The study of the mechanisms that contribute to age-related memory decline will not only lead to a better general understanding of memory function, but promises hope of more effective prevention strategies and improvement of the quality of life for our aging population.
Work in model systems ranging from rodents to humans indicates that aging-related memory decline is manifest in at least two memory modalities: (1) declarative/episodic memory, which is anatomically based largely in the hippocampus; and, (2) working memory/executive function, which is anatomically based in the prefrontal cortex (see Burke and Barnes, 2006
). A complete description of the neurobiological substrates of these age-related memory deficits have yet to be elucidated, but at least in the case of the hippocampus, certainly include aberrant changes in gene transcription that ultimately affect the ability of the aged brain to learn and remember (e.g. Blalock et al., 2003
; Small et al., 2004
; Rowe et al., 2007
). Indeed, it has been well-established that memory and synaptic plasticity processes in the cognitively healthy adult require transcription of immediate-early genes (IEGs), including Arc (activity-regulated cytoskeletal gene), zif268 (also known as nerve growth factor inducible-A), and bdnf (brain-derived neurotrophic factor) (Guzowski et al., 2000
; Hall et al., 2000
; French et al., 2001
; Steward and Worley, 2001
). Moreover, blocking the expression of these genes in adult animals prevents the consolidation of memory (Linnarsson et al., 1997
; Guzowski et al., 2000
; French et al., 2001
), and decreased IEG expression is prevalent in many models of memory disorders (Dickey et al., 2003
; Palop et al., 2005
; Rosi et al., 2005
) and a result of the normal aging process (e.g. Blalock et al., 2003
; Small et al., 2004
; Rowe et al., 2007
).
The molecular mechanisms driving these changes in gene transcription are not currently known, but recent work points to a potential novel mechanism, dysregulation of epigenetic mechanisms. Waddington (1957)
was the first to use the term epigenetics to describe a mechanism or mechanisms that are ‘on top of’ or ‘above’ the level of the genes encoded by the DNA sequence. These mechanisms, which can be influenced by environmental factors such as diet (e.g., Waterland and Jirtle, 2003
) and experiential factors such as maternal care (e.g. Weaver et al., 2004
), can ultimately control which genes will be expressed, and allow cells and even whole organisms with the same genome (e.g. identical twins, Fraga et al., 2005
) to become phenotypically distinct. The traditional view has been that once epigenetic marks (discussed below) have been laid down during development, they remain unchanged for the life of the organism. Recent work in the adult organism, however, has challenged this view, and thus the term ‘epigenetics’ may need to be expanded to include additional possibilities (see Bird, 2007
). For example, recent work (discussed in greater detail below) indicates that epigenetic marks can be rapidly (within minutes) and transiently (less than 24 h) changed to dynamically regulate gene transcription in the adult brain (Miller and Sweatt, 2007
). Our hypothesis is that this type of dynamic change is dysregulated in the aged brain, and contributes to cognitive impairments.
The relevant epigenetic mechanisms include DNA methylation and histone post-translational modifications, mechanisms that have recently been discovered to control hippocampal synaptic plasticity and long-term memory formation (Barrett and Wood, 2008
; Jiang et al., 2008
). These epigenetic changes involve the covalent chemical modification of DNA by DNA methyltransferases (DNMTs), and histones by histone acetyltransferases (HATs) and histone deacetylases (HDACs). These epigenetic mechanisms are powerful controllers of memory-associated gene transcription, and in general result in transcriptional silencing and loss of gene function through DNA methylation or transcriptional activation by histone acetylation, although numerous other modifications and effects are possible (Chahrour et al., 2008
; Suzuki and Bird, 2008
). Overall, it is now appreciated that DNA methylation plays a key role in dynamically regulating gene transcription in the adult CNS (Levenson et al., 2006
; Miller and Sweatt, 2007
; Lubin et al., 2008
), in concert with histone acetylation (Levenson et al., 2004
; Lubin et al., 2008
; Miller et al., 2008
), and that these processes are integral in long-term memory formation. What is not currently known is whether alterations in these mechanisms play a key role in the age-related changes in gene transcription and memory decline.
This has led us to hypothesize that dysregulation of epigenetic control mechanisms and aberrant epigenetic “marks” (i.e. the chemical additions to DNA and histone proteins) contribute to aging-related cognitive dysfunction. Specifically, given that the transcription of key memory-promoting genes are known to decline during aging (Blalock et al., 2003
; Small et al., 2004
; Rowe et al., 2007
), we propose that these changes are regulated by aberrant epigenetic marks and control mechanisms within brain regions particularly vulnerable to the aging process, which together result in age-related cognitive deficits. This is the central hypothesis examined in this review.
Memory and LTP in the Aged Brain
Spatial cognition involves the ability of an organism to acquire and retain information critical for successful navigation through space. An abundance of research indicates that this behavior is dependent on the hippocampus, since damage to this structure, and other closely related structures, impairs performance on tasks that measure spatial learning and memory including the Morris swim task (Morris et al., 1982
), the 14-unit T-maze (Bresnahan et al., 1988
), the Y-maze (Aggleton et al., 1986
), the radial arm maze (Olton et al., 1978
), and the circular platform task (McNaughton et al., 1989
). Because the hippocampus is a brain region vulnerable to the normal aging process, it perhaps is not surprising that performance on tasks that require information processing within this structure is impaired with age. In fact, age-related decline in navigational skills are well documented in humans (e.g. Allen et al., 2004
; Driscoll et al., 2005
), and spatial memory deficits appear in aged monkeys (Lai et al., 1995
; Rapp et al., 1997
), dogs (Head et al., 1995
), rats (Gallagher and Rapp, 1997
), and mice (Bach et al., 1999
). In rats, age-related spatial learning and memory deficits have been consistently observed using the Barnes maze (Barnes, 1979
), the Morris swim task (e.g. Gage et al., 1984
; Rapp et al., 1987
; Markowska et al., 1989
; Barnes et al., 1997
; Shen et al., 1997
), the T-maze (e.g. Barnes et al., 1980
), and the 8-arm radial maze (e.g. Barnes et al., 1980
; de Toledo-Morrell et al., 1984
). These deficits often appear to mimic those observed in animals with direct lesions of the hippocampus, although the impairments observed in aged rats are less severe.
While it is certainly true that hippocampal-dependent memory is impaired in aged organisms, these impairments are not the result of massive cell death. In fact, with only a few exceptions, the hippocampus remains relatively structurally intact in the aged mouse, rat, monkey and human undergoing normal aging (for review see Rosenzweig and Barnes, 2003
; Burke and Barnes, 2006
). In addition, most electrical properties remain constant over the lifespan (Barnes, 1994
), including resting membrane potential, membrane time constant, input resistance, and threshold to reach an action potential (Barnes, 1979
; Barnes and McNaughton, 1980
; Segal, 1982
; Landfield and Pitler, 1984
; Niesen et al., 1988
; Kerr et al., 1989
; Reynolds and Carlen, 1989
; Pitler and Landfield, 1990
; Turner and Deupree, 1991
; Barnes et al., 1992
; Potier et al., 1992
, 1993
; Luebke and Rosene, 2003
), although Ca2+ dysregulation is evident, including an increase in Ca2+ conductance, and increased density of L-type Ca2+ channels (Landfield, 1988
; Thibault and Landfield, 1996
; Foster and Norris, 1997
; Toescu et al., 2004
). Despite the relative sparing of the electrical properties of the hippocampus, synaptic plasticity mechanisms do change in the aged brain, including long-term potentiation (LTP), which is widely believed to represent an experimental analog of memory.
Most current theories regarding the cellular basis of learning and memory propose that information is stored via activity-dependent changes at the synapse. Bliss and Lomo (1973)
were the first to describe a long-lasting increase in the strength of synaptic transmission within the hippocampus, a phenomenon that eventually became known as long-term potentiation (LTP; Douglas and Goddard, 1975
). The delivery of brief, high frequency stimulation to the perforant path inputs into the dentate gyrus, resulted in a subsequent change in the amplitude of population excitatory post synaptic potentials (EPSPs) that lasted for several hours. Since this initial observation, there are literally thousands of papers reporting on various aspects of the LTP phenomenon. Thus, much of what we know about synaptic plasticity within the hippocampus has come from studying the mechanisms of LTP induction (sometimes referred to as early-phase LTP) and maintenance (sometimes referred to late-phase LTP).
In the aged brain, when high-intensity stimulation protocols that are well above threshold for LTP induction are used, there are no age-related deficits in LTP induction at the CA3-CA1 Schaffer collateral synapse (e.g., Landfield and Lynch, 1977
; Landfield et al., 1978
) or the perforant-path-granule cell synapse (e.g., Barnes, 1979
). When, however, stimulus parameters are very close to the LTP induction threshold, aged rats consistently show LTP induction deficits at the Schaffer collateral CA1 synapse and the perforant path-granule cell synapse (e.g., Deupree et al., 1993
; Moore et al., 1993
; Barnes et al., 2000
). After LTP has been induced, the maintenance of LTP can also be measured and divided into an ‘early phase’ lasting one to 3 h and a ‘late phase’ lasting for more than 24 h. Several studies have demonstrated that in the short-term (i.e., over 1 h), LTP decay rates are not different between aged and young rats (e.g. Landfield and Lynch, 1977
; Landfield et al., 1978
), but over a longer time course, significant age-related maintenance deficits do appear (e.g. Barnes, 1979
; Barnes and McNaughton, 1980
; for review see Barnes, 2003
). For example, Barnes and McNaughton (1980)
administered LTP-inducing stimuli at 24-hour intervals on 12 consecutive days, then monitored the evoked field response for several weeks. Although there were no age-related differences in the final levels of LTP induction, LTP decayed nearly twice as fast in the aged rats over several weeks.
Several experiments have also demonstrated a significant correlation between LTP and memory deficits in aged animals (Barnes, 1979
; Barnes and McNaughton, 1980
, 1985
; de Toledo-Morrell and Morrell, 1985
; Bach et al., 1999
). This was first demonstrated by Barnes (1979)
who showed that aged rats took longer path lengths and a greater amount of time to solve a spatial memory task (i.e., the Barnes maze). These deficits in spatial memory performance were significantly correlated with the durability of synaptic enhancement in the dentate gyrus both within and between age groups. In subsequent studies, Barnes et al. (1980)
and Barnes and McNaughton (1985)
also demonstrated a significant correlation between the rates of acquisition and forgetting of spatial memory and the induction and maintenance of LTP. Aged rats were slower to reach asymptotic performance levels and forgot the problem faster than did young rats. Similarly, LTP was induced more slowly and decayed more rapidly in aged rats. In both cases, the rate of decay for the memory of the escape box and LTP was nearly twice as fast for the aged rats. Bach et al. (1999)
also found a significant relationship between LTP decay rates in area CA1 in vitro and performance on the Barnes maze in aged but not young mice. These findings are further supported by observations that old animals with spatial memory deficits on the 8-arm radial maze also show faster decay rates for hippocampal LTP (de Toledo-Morrell and Morrell, 1985
). Taken together, the currently available data indicate that learning and memory deficits observed in aged rats and mice parallel deficits in the induction and maintenance of LTP.
Gene Transcription in Learning and Memory
Early work on the molecular mechanisms of memory highlighted the need for gene transcription and expression to support the maintenance of long-term memory (reviewed in Alberini, 2009
). Operationally defined, IEGs are those genes whose transcription can be induced in the presence of protein synthesis inhibitors, and they represent the earliest genomic response to patterned synaptic activity (Guzowski, 2002
). The intracellular events that trigger IEG transcription begin with NMDA receptor activation and the eventual translocation of ERK to the nucleus where it activates the transcription factors CREB and Elk-1 (Impey et al., 1998
; Roberson et al., 1999
), resulting in changes in IEG transcription. The putative cellular functions of this diverse group of genes (e.g. transcription factors, cellular growth, synaptic modification/structural changes, synaptic homeostasis) make them excellent candidates for critical components of synaptic plasticity processes (reviewed in Lanahan and Worley, 1998
). Evidence supporting a role for IEGs in memory function is abundant, although some particularly compelling evidence exists for Arc, zif268 and bdnf. In the following section we will briefly review some of the relevant background for these specific memory-associated target genes.
In many respects Arc is the prototypical rapidly induced memory-associated gene in the CNS. Arc transcription is induced selectively in the principal cells of the hippocampus and other brain regions by neural activity specifically associated with active information processing that occurs in spatial learning and memory formation (Guzowski et al., 2005
; Miyashita et al., 2008
). When Arc expression is inhibited, there is no significant effect on the induction of LTP, or on acquisition of the Morris swim task; however, there is interference with the maintenance of LTP over several days (Guzowski et al., 2000
). Moreover, rats that had previously learned the location of the hidden platform on the Morris swim task, when treated to eliminate Arc expression 8 h after acquisition of the task, failed to recall the platform’s location upon later testing (Guzowski et al., 2000
). Experiments using Arc knockout mice have revealed similar effects on long-term spatial, taste, and fear memory (McIntyre et al., 2005
; Plath et al., 2006
). Mechanistically, Arc plays a critical role in regulating AMPA receptor trafficking (Chowdhury et al., 2006
) and homeosynaptic scaling of synapses that contain AMPA receptors (Rial Verde et al., 2006
; Shepherd et al., 2006
), both of which are essential for efficient and reliable synaptic plasticity (Malinow and Malenka, 2002
; Bredt and Nicoll, 2003
).
Zif268 functions as an inducible regulatory transcription factor, and it is known to regulate the transcription of at least 20 delayed-response genes (reviewed in Knapska and Kaczmarek, 2004
), and can therefore have a dramatic effect on long-term cellular function. Using a mutant mouse with a targeted disruption of zif268, Jones et al. (2001)
addressed whether zif268 is required for the maintenance of late LTP and for the expression of long-term memory. In this study, mutant mice exhibited early LTP in the dentate gyrus, but late LTP was absent when measured 24 and 48 h after tetanus. Further, on both spatial and non-spatial learning tasks, short-term memory remained intact in the mutant mice, but performance was impaired in tests requiring long-term memory. In addition, subsequent work by Lee et al. (2004)
suggests that zif268 is necessary for hippocampal memory re-consolidation, but not for the initial consolidation of a memory. Although the debate over memory consolidation and re-consolidation processes remains controversial, (see Nader and Hardt, 2009
), these results indicate that zif268 is essential for the stabilization of long-term synaptic plasticity and for the expression of long-term memories.
Bdnf is also a prominent and powerful regulator of synaptic plasticity and memory formation. The rodent bdnf gene consists of nine 5′-exons linked to separate promoters and one 3-exon which codes for the mature bdnf protein (Liu et al., 2006
; Aid et al., 2007
; Pruunsild et al., 2007
). Due to the complexity of the bdnf gene, at least 11 different bdnf transcripts can be generated from the mammalian rodent bdnf gene by alternative splicing (Pruunsild et al., 2007
). The activation of different bdnf promoters is brain-region-specific and depends on the type of stimulus used (Metsis et al., 1993
; Timmusk et al., 1993
; Bishop et al., 1994
; Kokaia et al., 1994
). An abundance of evidence supports the idea that bdnf is critical for learning-related synaptic plasticity and the maintenance of long-term memory. For example, training on a hippocampus-dependent contextual learning task significantly increases bdnf within the hippocampus (Hall et al., 2000
), and when endogenous bdnf is blocked, adult rats experience impairments in spatial learning and memory (Mu et al., 1999
; Heldt et al., 2007
). In addition, the induction of LTP significantly increases bdnf mRNA levels (Patterson et al., 1992
; Castren et al., 1993
), while LTP is impaired in mice that lack the bdnf gene (Korte et al., 1995
, 1996
).
If IEGs are necessary for synaptic plasticity and memory function, and these processes are altered in aged animals, then it would be predicted that the transcription of the IEGs necessary for normal synaptic plasticity and memory function are altered in the aged brain. In fact, decreased IEG expression does accompany the normal aging process. Large-scale microarray investigations have observed a significant down-regulation of zif268, Arc, Narp and Homer1a in the hippocampus of memory-impaired aged rats (Blalock et al., 2003
; Rowe et al., 2007
). In regards to bdnf, a great deal of work indicates that this gene and its receptors undergo significant age-associated down-regulation (reviewed in Tapia-Arancibia et al., 2008
). For example, Hattiangady et al. (2005)
found that bdnf is down-regulated within area CA1, CA3 and the dentate gyrus in middle-aged and aged rats. Taken together, these studies indicate that several genes already known to be necessary for synaptic plasticity and normal memory function are down-regulated within the aged brain, including Arc, zif268 and bdnf. What remains to be determined are the mechanisms that contribute to age-associated changes in gene transcription. We propose that age-related alterations in DNA methylation and histone modifications lead to age-associated disruption of memory-promoting gene transcription.
A Role for DNA Methylation in Memory Function
DNA methylation and histone modifications are two of the most extensively investigated epigenetic mechanisms (Figure 1
). Until recently, it was thought that once laid down, these epigenetic marks remained unchanged for the lifetime of the organism, but recent studies in the nervous system have challenged this view. It is now apparent that epigenetic marks can be modified in response to an organism’s experience, and play a key role in dynamically regulating the gene transcription supporting synaptic plasticity and long-term memory formation (Levenson and Sweatt, 2005
; Graff and Mansuy, 2008
; Jiang et al., 2008
; Liu et al., 2009
; Sweatt, 2009
).
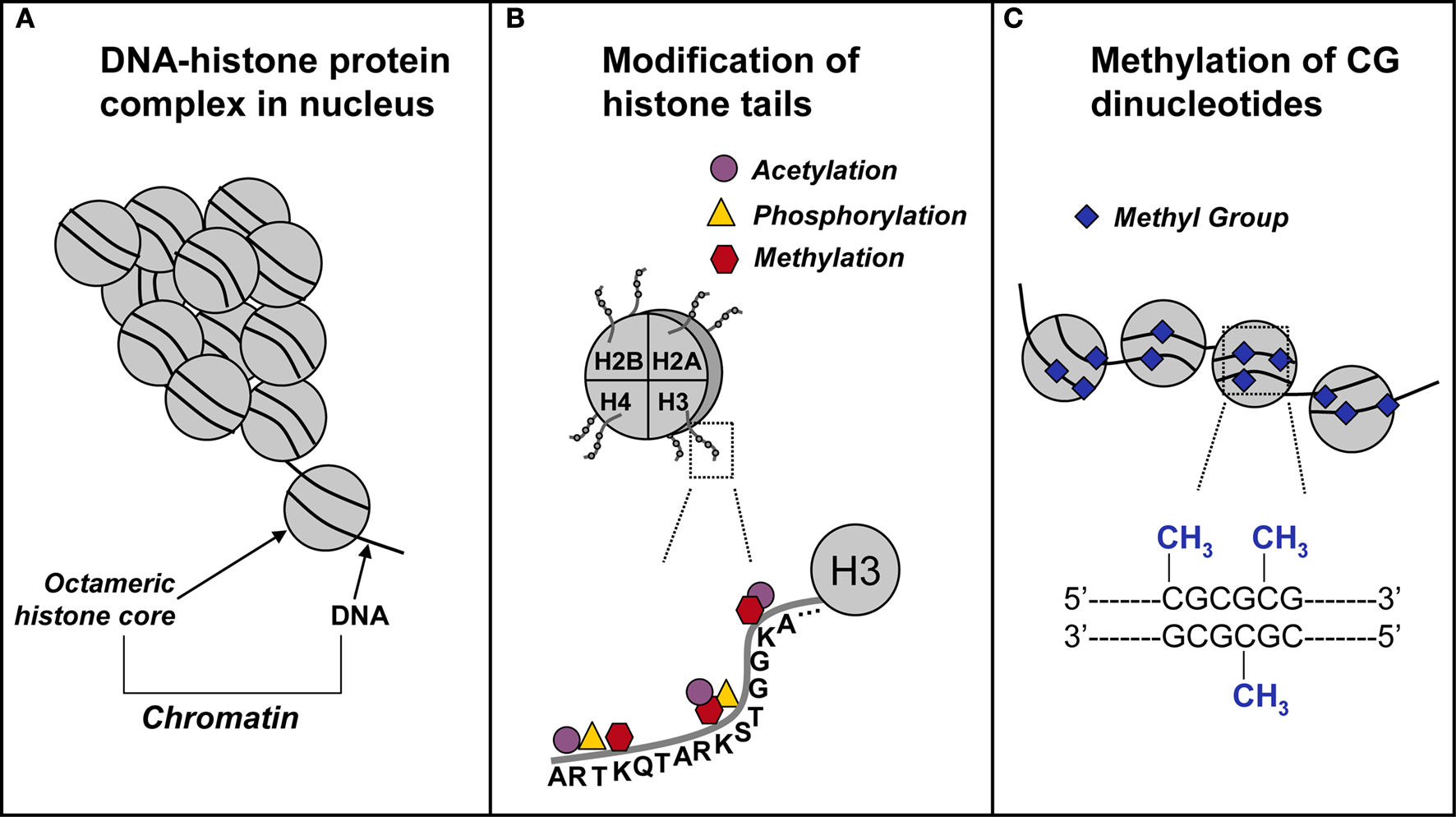
Figure 1. Schematic representation of epigenetic mechanisms. (A) In the nucleus, DNA coils and condenses around histones. Each octameric histone core contains two copies each of histones H2A, H2B, H3, and H4. The DNA–protein complex is referred to as chromatin. (B) The DNA-histone interaction occurs at the N-terminal tail of a histone, where for example on the H3 N-terminal tail, there are several sites for epigenetic marking via acetylation, methylation, and phosphorylation. (C) In and around gene promoters that are rich in cytosine-guanine nucleotides (CpG islands), methyl groups are transferred to CpG sites. This process, called DNA methylation, is catalyzed by a class of enzymes known at DNA methyltransferases.
Much of the work devoted to testing the idea that epigenetic mechanisms regulate gene transcription necessary for memory function has focused on posttranslational histone modifications, especially histone acetylation (reviewed in Barrett and Wood, 2008
; Graff and Mansuy, 2008
; Jiang et al., 2008
; Sweatt, 2009
). (We will return to a review of these studies in the following section.) DNA methylation, the covalent chemical modification of cytosine side-chains, is another epigenetic mechanism important for synaptic plasticity and memory formation. DNA methylation occurs in regions of the genome rich in CpG dinucleotides (i.e. CpG islands), which are often found in the promoter region of genes. The family of enzymes that carry out DNA methylation, the DNA methyltranferases (DNMTs), come in two variants, maintenance DNMTs including DNMT1 and de novo DNMTs, including DNMT3a and 3b. The function of de novo DNMTs is to transfer methyl groups to previously unmethylated CpG sites, whereas the maintenance DNMTs methylate hemi-methylated DNA. DNA methylation has traditionally been associated with transcriptional silencing, but there are also instances in which DNA methylation can have an activating role (Yasui et al., 2007
; Chahrour et al., 2008
). For example, the presence or addition of a methyl group to a stretch of DNA can interefere with the ability of transcription factors to bind to regulatory elements within the DNA sequence, thereby preventing gene transcription from occurring. Conversely, there are several molecules that recognize and preferentially bind to methylated DNA, including methyl CpG-binding domain protein 2 (MeCP2). This activity has the potential to either repress or promote gene transcription and may depend on whether the regulatory region of the gene is heavily methylated or lightly methylated, respectively (Chahrour et al., 2008
; Cohen et al., 2008
), suggesting that the methylation status of the target gene DNA may ultimately determine whether a gene will be transcribed.
The idea that dynamic changes in DNA methylation may contribute to synaptic plasticity and memory function in the adult CNS is still a relatively novel one, and as such, a limited number of studies have been directed at testing this hypothesis. Work by Levenson et al. (2006)
was the first to demonstrate that general inhibitors of DNMT activity alter DNA methylation in the adult brain, block hippocampal LTP in vitro, and alter the DNA methylation status of the plasticity-promoting genes Reln (Reelin) and bdnf. Additional studies demonstrated that de novo DNMT expression is upregulated in the adult rat hippocampus after contextual fear conditioning and that blocking DNMT activity blocked contextual fear conditioning (Miller and Sweatt, 2007
). In addition, fear conditioning is associated with rapid methylation and transcriptional silencing of the memory suppressor gene Protein Phosphatase 1 (PP1) and demethylation and transcriptional activation of the synaptic plasticity gene Reln. These findings have the surprising implication that both active DNA methylation and active demethylation might be involved in long-term memory consolidation in the adult CNS.
More recent work from Sweatt and colleagues has investigated the mechanistic interplay between histone acetylation and DNA methylation (Miller et al., 2008
). Inhibition of DNMT activity was found to block histone acetylation, along with memory consolidation and LTP, as previously reported (Levenson et al., 2006
; Miller and Sweatt, 2007
). These deficits, however, were rescued by pharmacologically increasing histone acetylation prior to DNMT inhibition. In another recent study on epigenetic regulation of the bdnf gene during fear memory formation, it has also been found that contextual- fear conditioning evokes differential methylation and demethylation of the various bdnf promoters (Lubin et al., 2008
). In animals with a learned fear association, these modifications are associated with localized histone modifications at specific bdnf promoters and upregulation of bdnf transcription (Lubin et al., 2008
). Finally, it has been shown that pharmacologically disrupting DNA methylation prevents formation of fear memory (Lubin et al., 2008
). All together, these findings lend strong support to the idea that DNA methylation and histone modifications work together to regulate plasticity and memory formation in the adult CNS.
Whether these processes also contribute to the age-related changes in synaptic plasticity and memory that have been observed in aged, cognitively impaired humans and animal models is the focus of on-going studies. For example, work by Siegmund et al. (2007)
demonstrates significant increases and decreases in DNA methylation within the human brain during both normal and pathological aging that corresponds to a concomitant change in mRNA levels. In addition, recent work by Penner et al. (2010)
demonstrates that dynamic DNA methylation of the Arc gene is altered in the hippocampus of memory-impaired aged rats compared to adult rats. Future studies will be directed at attempts to ameliorate these changes in an effort to restore memory function.
A Histone Code in Memory?
The general hypothesis that there is an epigenetic “histone code” for memory formation is new and still quite speculative (Chwang et al., 2006
, 2007
; Wood et al., 2006
; Borrelli et al., 2008
), and derives from an earlier idea proposed by Allis and colleagues. The overall concept is that multiple post-translational histone modifications may be integrated together, combinatorially driving gene expression patterns by remodeling the structure of chromatin or by recruiting signaling complexes (Jenuwein and Allis, 2001
). As different histone modifications may be driven by different upstream signaling pathways, multiple signals might thus converge on the nucleus, controlling gene readout through regulating chromatin structure. This would result in a mapping of multiple histone alteration states onto subsets of genes that are transcribed as a result of those changes.
Preliminary evidence already exists that supports the idea of a histone code for memory. For example, the ERK/MAPK cascade has been found to regulate chromatin structure in the hippocampus, and protein kinase MSK1 has been shown to be a mediator of the effects of ERK (Chwang et al., 2006
, 2007
). Interestingly, histone H3 phosphorylation at Ser 10 and acetylation at Lys 14, both transcriptional activation markers, are significantly increased with fear conditioning training; however, histone H4 acetylation at Lys 5/8/12/16, also an indicator for activation, is unchanged. Moreover, Histone H4 acetylation at those same sites is increased with latent inhibition training. At a minimum, these observations show that patterns of histone regulation in the hippocampus are specific for the type of modification as well as the residue on which it occurs, and can differ with different types of learning. To begin to try to explain this complexity of chromatin regulation in memory, we and others hypothesized that regulation of histone phosphorylation and acetylation is part of a “histone code” that is read out as a pattern of gene transcription specific to different types of hippocampus-dependent memory (Wood et al., 2006
; Borrelli et al., 2008
).
Regardless of the validity of the histone code hypothesis, these results also support the idea that chromatin acts as a platform for signal integration in memory formation (Schreiber and Bernstein, 2002
). Each of the core histones contains N-terminal tails on which the majority of histone modifications take place (Peterson and Laniel, 2004
). Each modification may be targeted by separate, independent signaling pathways that converge after reaching the nucleus, and chromatin itself may serve as a scaffold on which disparate signals are integrated. We and others have shown that phosphorylation and acetylation of histone H3 are targeted by ERK and MSK1 in hippocampal neurons (Chwang et al., 2007
); however, studies in cultured embryonic fibroblasts have also implicated the p38 MAPK, SAPK2 and other signal transduction pathways in targeting histone phosphorylation and acetylation (Thomson et al., 1999
; see Figure 2
). Thus, multiple signal transduction pathways may target histones independently, and their effects on transcriptional output may be additive, redundant, or even antagonistic. Additional work will be needed to determine precisely what other signaling pathways may be involved in histone regulation in memory, how their relative contributions in histone regulation are integrated to produce a coordinated transcriptional output, and whether aging results in disruption of baseline chromatin states and alterations of memory-associated histone modification patterns.
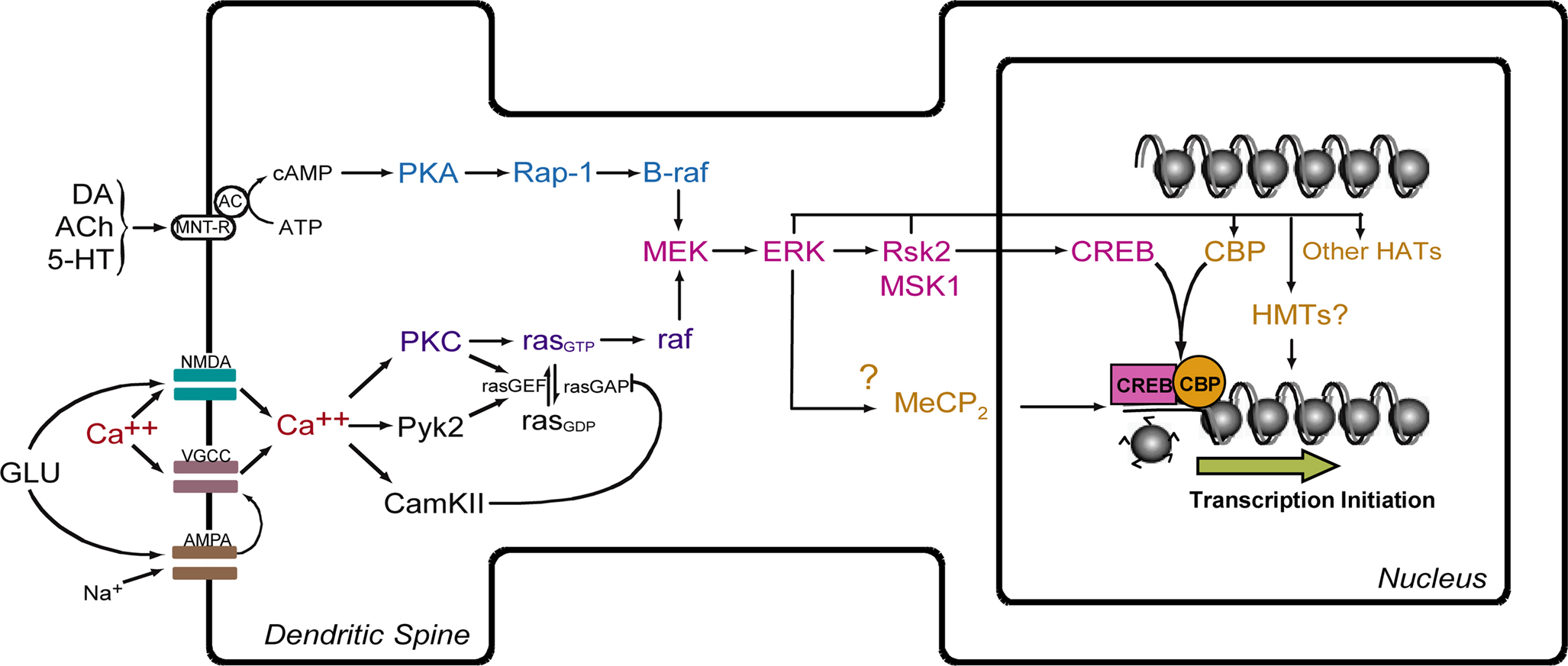
Figure 2. Model for ERK-mediated regulation of histone modification and gene transcription. Activation of NMDA receptors and voltage-gated Ca++ channels (VGCC) leads to influx of Ca++ and activation of the ras-MEK-ERK signaling cascade. This leads to activation of CREB-mediated transcription via action of Rsk2 and MSK1. CREB is postulated to facilitate transcription through interaction with CREB-binding protein (CBP) and acetylation of histones. MeCP2, a methyl-binding domain protein, regulates gene activity by binding to methylated CpG dinucleotides. HAT = histone acetyltransferase. HMT = histone methyltransferase. MNT-R = modulatory neurotransmitter receptors.
Manipulating the Epigenome to Improve Memory Function
The efficacy of many transcription factors in modulating transcription depends critically upon the recruitment and activation of a number of co-activators of transcription, one example of which is the HAT CREB-binding protein (CBP). Thus, regulation of CRE-dependent transcription by CREB depends upon the activity of HATs. The effects of HATs are reversed by HDACs and by this mechanism HDACs are also critical controllers of histone acetylation and transcriptional efficacy. In an important breakthrough in the last few years several groups discovered that broad-spectrum HDAC inhibitors enhance LTP in vitro and augment memory formation in vivo in normal rodents, in a neurodegeneration model, or both (Alarcón et al., 2004
; Korzus et al., 2004
; Levenson et al., 2004
; Chwang et al., 2006
, 2007
; Bredy et al., 2007
; Fischer et al., 2007
; Lattal et al., 2007
; Lubin and Sweatt, 2007
; Miller and Sweatt, 2007
; Vecsey et al., 2007
; Miller et al., 2008
). These discoveries open up the possibility that HDAC inhibitors might be useful clinically in treating aging-related memory dysfunction.
To date, there are 11 known HDAC isoforms in the rat (see Chuang et al., 2009
). A variety of isoform-nonselective and selective HDAC inhibitors have been developed (see Table 1
), but the unique contributions of each of the isoforms in memory formation are just beginning to be characterized. Recent work by Guan et al. (2009)
indicates that HDAC2 overexpression in mice can decrease dendritic spine density and synapse numbers, and significantly interfere with both LTP and hippocampal-dependent memory formation. This effect is not observed in mice overexpressing HDAC1. In contrast, Hdac2-deficiency increases synapse number and enhances hippocampal-dependent memory formation, an effect that closely mimics chronic treatment with HDAC inhibitors. In addition, these authors found that HDAC2, but not HDAC1 is enriched at several gene promoter regions including Egr1 (as known as zif268), Bdnf, Fos, and Creb, suggesting that HDAC2 may suppress the expression of these memory-associated genes by binding to their regulatory elements. Interestingly, the promoter region of the memory-promoting gene Arc was predominately associated with HDAC1, suggesting that this isoform also regulates the expression of genes necessary for normal memory function. Overall, these findings indicate that HDAC2 functions as a negative regulator of memory formation, synaptic plasticity, and gene expression, while the precise role of HDAC1 in memory function remains an open question. Nevertheless, the work by Guan et al. (2009)
underscores the importance of indentifying the relative contributions of specific HDAC isoforms to synaptic plasticity and memory function. Moreover, in terms of neurodegenerative and pathological aging, HDAC inhibitors have shown some promise ameliorating symptoms in models of Huntington’s disease, Parkinson’s disease, stroke, amyotrophic lateral sclerosis, spinal muscular atrophy, and Alzheimer’s disease (see Chuang et al., 2009
). One issue that is still of considerable concern is the toxicity and side-effects associated with existing HDAC inhibitors, which often appear after relatively short-term treatment in cancer patients (Bruserud et al., 2007
).

Table 1. Classical HDAC isoforms and some examples of their targeted HDAC inhibitors.
Studies of the potential applicability of HDAC inhibitors to cognitive aging is at such an early stage that, to our knowledge, no studies directly addressing this topic in humans are currently available. Thus, specific human studies of clinically available HDAC inhibitors (e.g. valproate) have not yet been undertaken. In considering the potential for these types of studies, it is important to keep in mind that valproate and sodium butyrate are non-specific inhibitors of HDACs that effect several additional targets (e.g., protein kinase C; Manji and Lenox, 1999
; Seung Kim et al., 2001
). Clearly, additional selective agents acting at specific HDAC isoforms (see Table 1
) are needed to begin to address the question of the applicability of HDAC inhibitors for therapeutics of human cognitive dysfunction. Should improved, isoform-selective HDAC inhibitors become available over the next few years, it will be interesting to determine their efficacy in ameliorating aging-related memory dysfunction in laboratory animal studies. This is not simply a hypothetical consideration, since the development of novel isoform-selective HDAC inhibitors is an active area of drug development at present.
Summary
In this review, we hypothesize that dysregulation of epigenetic control mechanisms and accumulation of aberrant epigenetic “marks” (i.e., the chemical additions to DNA and histone proteins) are a driver for aging-related cognitive dysfunction. Specifically, because transcription of key memory-promoting genes, including Arc, zif268 and bdnf, are known to change during aging (Blalock et al., 2003
; Small et al., 2004
; Rowe et al., 2007
), we propose that these changes are regulated by aberrant epigenetic marks (e.g. DNA methylation or histone acetylation, phosphorylation or deacetylation) and control mechanisms within brain regions particularly vulnerable to the aging process (i.e. hippocampus and prefrontal cortex), which together result in age-related cognitive deficits.
Overall, we feel that our hypothesis will not only fuel new discoveries concerning how memory works at the molecular level, but will work in concert with recent findings from about a dozen laboratories illuminating the high potential of using insights gained from chromatin molecular biology to design potential new routes of therapy for aging-related memory dysfunction. The significance of the hypothesis is 2-fold. One, results will target validation of this new approach to alleviate cognitive aging. Two, results will generate new insights into whether disruption of chromatin regulation and DNA methylation might be a component of the basic molecular biology of aging-associated memory dysfunction.
The ideas presented here are also relevant to a broader literature concerning a more general epigenetic hypotheses of longevity and cellular senescence (Bandyopadhyay and Medrano, 2003
; Longo and Kennedy, 2006
; Blasco, 2007
; Fraga and Esteller, 2007
; Ramírez et al., 2007
; Sedivy et al., 2008
; Liu et al., 2009
). There is an emerging understanding that chromatin can be altered in a dynamic way and is subject to extensive experience- and age-associated remodeling (Bandyopadhyay and Medrano, 2003
; Sedivy et al., 2008
). Dynamic changes in chromatin structure over the lifespan could potentially either counter aging and age-associated diseases in some cases, or contribute to accelerated aging and age-related dysfunction in other cases (Ramírez et al., 2007
; Sedivy et al., 2008
). For example, global loss of DNA methylation in aging or promoter hypermethylation of genes with a role in progeria have been proposed to control aging and longevity (Fraga and Esteller, 2007
). Moreover, the sirtuins, a family of NAD-dependent histone deacetylases, are now recognized to be a link between chromatin regulation, cellular transformation and lifespan (Longo and Kennedy, 2006
). Chromatin modifications also are now known to be important regulators of mammalian telomeres, and the dysfunction of epigenetic regulators such as histone methyltransferases and DNA methyltransferases, correlates with loss of control of telomere-length control, a mechanism well-known to contribute to the aging process (Blasco, 2007
). Overall, a new unifying hypothesis is emerging that suggests that the accumulation of aberrant epigenetic marks, including histone modification and DNA methylation and demethylation, may be a driver of aging-related cellular and physiologic changes. Here, we propose that these ideas be extended to the specific context of aging-related memory dysfunction.
There are a number of unanswered questions that remain critical to understand concerning the potential roles and regulation of epigenetic molecular mechanisms in the hippocampus and in hippocampus-dependent memory formation in the aging CNS. Among these include: (1) Is aging associated with accumulation of aberrant epigenetic marks in the hippocampus? (2) Is aging associated with disruption of memory-associated epigenomic signaling? (3) Can one restore normal memory function in the aged CNS using pharmacologic agents that manipulate the epigenome? and (4) Can one identify a cellular mechanism, specifically disruption of hippocampal synaptic plasticity, that is a locus for epigenetic disruption of memory with aging? These are four critically important areas for future investigation in order to test the hypothesis of an epigenetic basis for cognitive aging.
Question 1
Do epigenetic changes at key memory gene loci occurring during the aging process? There is a substantial prior literature using rats in basic studies of aging-related memory decline, and this model system presents a very tractable system for addressing the prediction of the hypothesis that aging should be associated with aberrant accumulation of epigenetic marks in memory-associated regions of the CNS. Thus, it will be important to compare younger control animals with both cognitive-healthy aged animals and cognitive-impaired aged animals, and assess whether aging results in alterations in CNS DNA methylation, histone modifications, and associated changes in memory-driven gene transcription.
Question 2
Is aging associated with disruption of memory-driven epigenetic signaling? Previous studies have shown that in younger adult animals memory formation is associated with (and requires) increased gene transcription, altered histone acetylation, and changes in DNA methylation. An important prediction of our hypothesis is that aging will result in disruption of these specific types of experience-driven memory-associated epigenomic signaling mechanisms. These types of studies should provide insights into whether aging results in a disruption of experience-dependent epigenomic signaling.
Question 3
Can aging-related memory dysfunction be ameliorated by pharmacologic intervention that affects epigenetic mechanisms? Currently there are several different pharmacologic means of manipulating epigenetic mechanisms in the CNS, principally but not exclusively through the use of HDAC inhibitors and DNMT inhibitors. One compelling experimental course of action at this point is to undertake preclinical proof-of-principle studies and evaluate if HDAC inhibitors can ameliorate the memory and plasticity deficits exhibited by aged animals. These are exciting studies that will provide an initial validation (or refutation) of the idea of regulation of chromatin structure as a “drugable” target for novel therapeutics in aging-related cognitive dysfunction. We feel that proceeding quickly with the preclinical studies is justified for several reasons. First, the capacity of HDAC inhibitors to augment memory and synaptic plasticity is very well-established for a variety of normal memory behaviors in animals (Alarcón et al., 2004
; Korzus et al., 2004
; Levenson et al., 2004
; Chwang et al., 2006
, 2007
; Bredy et al., 2007
; Lattal et al., 2007
; Lubin and Sweatt, 2007
; Miller and Sweatt, 2007
; Vecsey et al., 2007
; Miller et al., 2008
). (112–122). Second, the Tsai lab has already demonstrated the efficacy of HDAC inhibition to improve memory formation in a mouse model with inducible neurodegeneration (Fischer et al., 2007
). Third, the development of new valid targets for therapeutics related to aging-related memory dysfunction is a pressing societal and biomedical issue and thus must proceed quickly. Finally, the tools and procedures necessary to undertake these studies are all in place and routinely used in a large number of laboratories interested in aging research.
Question 4
Is disruption of hippocampal LTP a cellular basis for epigenetics-driven cognitive decline in aging? It is well-documented that aging is associated with deficits in LTP, a long-lasting form of synaptic potentiation that is strongly implicated as a cellular mechanism underlying long-term memory. An important question to address in the future is whether pharmacologically manipulating epigenomic signaling can restore hippocampal synaptic plasticity in aged animals. These studies would provide a strong cellular correlate to the behavioral and molecular studies described in Questions 1–3, and also provide a specific candidate cellular mechanism for any cognitive improvements that might be observed in response to HDAC inhibition.
In this article we propose that the dysregulation of epigenetic control mechanisms and the accumulation of aberrant epigenetic marks provide a molecular basis for the cognitive dysfunction associated with aging. The data demonstrating aberrant gene transcription in memory-impaired aged animals was reviewed, as well as the data implicating epigenetic regulation of neural plasticity and memory formation. Furthermore, data concerning how drugs that can promote or reverse epigenetic changes have been highlighted as holding promise for alleviating cognitive dysfunction. The continued study of epigenetic marks in the regulation of cognition promises a future where a viable epigenetic therapy treatment may be found that alleviates the growing prevalence of memory dysfunction in society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank Felecia Hester for her assistance in preparing this article. This work was funded by grants from the National Institutes of Health, the National Alliance for Research on Schizophrenia and Depression, Civitan International, the Rotary Clubs CART fund, and the Evelyn F. McKnight Brain Research Foundation.
Bach, M. E., Barad, M., Son, H., Zhuo, M., Lu, Y. F., Shih, R., Mansuy, I., Hawkins, R. D., and Kandel, E. R. (1999). Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 5280–5285.
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., Heine-Suñer, D., Cigudosa, J. C., Urioste, M., Benitez, J., Boix-Chornet, M., Sanchez-Aguilera, A., Ling, C., Carlsson, E., Poulsen, P., Vaag, A., Stephan, Z., Spector, T. D., Wu, Y. Z., Plass, C., and Esteller, M. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609.
French, P. J., O’Connor, V., Jones, M. W., Davis, S., Errington, M. L., Voss, K., Truchet, B., Wotjak, C., Stean, T., Doyère, V., Maroun, M., Laroche, S., and Bliss, T. V. (2001). Subfield-specific immediate early gene expression associated with hippocampal long-term potentiation in vivo. Eur. J. Neurosci. 13, 968–976.
Guzowski, J. F., Lyford, G. L., Stevenson, G. D., Houston, F. P., McGaugh, J. L., Worley, P. F., and Barnes, C. A. (2000). Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J. Neurosci. 20, 3993–4001.
Hattiangady, B., Rao, M. S., Shetty, G. A., and Shetty, A. K. (2005). Brain-derived neurotrophic factor, phosphorylated cyclic AMP response element binding protein and neuropeptide Y decline as early as middle age in the dentate gyrus and CA1 and CA3 subfields of the hippocampus. Exp. Neurol. 195, 353–371.
Plassman, B. L., Langa, K. M., Fisher, G. G., Heeringa, S. G., Weir, D. R., Ofstedal, M. B., Burke, J. R., Hurd, M. D., Potter, G. G., Rodgers, W. L., Steffens, D. C., Willis, R. J., and Wallace, R. B. (2007). Prevalence of Dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 29, 125–132.
Plath, N., Ohana, O., Dammermann, B., Errington, M. L., Schmitz, D., Gross, C., Mao, X., Engelsberg, A., Mahlke, C., Welzl, H., Kobalz, U., Stawrakakis, A., Fernandez, E., Waltereit, R., Bick-Sander, A., Therstappen, E., Cooke, S. F., Blanquet, V., Wurst, W., Salmen, B., Bösl, M. R., Lipp, H. P., Grant, S. G., Bliss, T. V., Wolfer, D. P., and Kuhl, D. (2006). Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444.
Rowe, W. B., Blalock, E. M., Chen, K. C., Kadish, I., Wang, D., Barrett, J. E., Thibault, O., Porter, N. M., Rose, G. M., and Landfield, P. W. (2007). Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J. Neurosci. 27, 3098–3110.
Siegmund, K. D., Connor, C. M., Campan, M., Long, T. I., Weisenberger, D. J., Biniszkiewicz, D., Jaenisch, R., Laird, P. W., and Akbarian, S. (2007). DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE 2, e895. doi: 10.1371/journal.pone.0000895.
Vecsey, C. G., Hawk, J. D., Lattal, K. M., Stein, J. M., Fabian, S. A., Attner, M. A., Cabrera, S. M., McDonough, C. B., Brindle, P. K., Abel, T., and Wood, M. A. (2007). Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB: CBP-dependent transcriptional activation. J. Neurosci. 27, 6128–6140.
Yasui, D. H., Peddada, S., Bieda, M. C., Vallero, R. O., Hogart, A., Nagarajan, R. P., Thatcher, K. N., Farnham, P. J., and Lasalle, J. M. (2007). Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. U.S.A. 104, 19416–19421.