 Brent D. Kerger
Brent D. Kerger Robert C. James2
Robert C. James2- 1Cardno ChemRisk, LLC, Aliso Viejo, CA, USA
- 2ToxStrategies, Tallahassee, FL, USA
- 3Cardno ChemRisk, LLC, San Francisco, CA, USA
The diagnosis of mesothelioma is not always straightforward, despite known immunohistochemical markers and other diagnostic techniques. One reason for the difficulty is that extrapleural tumors resembling mesothelioma may have several possible etiologies, especially in cases with no meaningful history of amphibole asbestos exposure. When the diagnosis of mesothelioma is based on histologic features alone, primary mesotheliomas may resemble various primary or metastatic cancers that have directly invaded the serosal membranes. Some of these metastatic malignancies, particularly carcinomas and sarcomas of the pleura, pericardium and peritoneum, may undergo desmoplastic reaction in the pleura, thereby mimicking mesothelioma, rather than the primary tumor. Encasement of the lung by direct spread or metastasis, termed pseudomesotheliomatous spread, occurs with several other primary cancer types, including certain late-stage tumors from genetic cancer syndromes exhibiting chromosomal instability. Although immunohistochemical staining patterns differentiate most carcinomas, lymphomas, and mestastatic sarcomas from mesotheliomas, specific genetic markers in tumor or somatic tissues have been recently identified that may also distinguish these tumor types from asbestos-related mesothelioma. A registry for genetic screening of mesothelioma cases would help lead to improvements in diagnostic criteria, prognostic accuracy and treatment efficacy, as well as improved estimates of primary mesothelioma incidence and of background rates of cancers unrelated to asbestos that might be otherwise mistaken for mesothelioma. This information would also help better define the dose-response relationships for mesothelioma and asbestos exposure, as well as other risk factors for mesothelioma and other mesenchymal or advanced metastatic tumors that may be indistinguishable by histology and staining characteristics.
Introduction
By far, the most extensively investigated cause of mesothelioma is asbestos exposure (Sporn and Roggli, 2004). Asbestos occurs in two mineralogic forms: commercially used amphiboles (e.g., commercially available amosite and crocidolite) and chrysotile. Amosite and crocidolite are considerably more persistent in tissue than is chrysotile, which disappears from lung tissue rapidly (Bernstein et al., 2005, 2010, 2011). This difference in biopersistence is believed to be the one of reasons that mesothelioma incidence rates are much higher in those who used or manufactured products with amosite and crocidolite, when compared to those exposed to only chrysotile (Churg, 1998; Hodgson and Darnton, 2000; Yarborough, 2006).
Although there is some debate regarding the potency gradient between chrysotile and asbestos fiber types, most agree that amphiboles are more toxic on a fiber-to-fiber basis (Hodgson and Darnton, 2000, 2010; Berman and Crump, 2008a,b). Some epidemiologic observations suggest that the incidence of mesothelioma caused by asbestos exposure peaked between about 1990 and 2010, because the highly potent amphibole exposures were largely curtailed in the 1960s and the typical latency period of 20–40 years since first exposure has now transpired (Hemminki and Li, 2003; Hillard et al., 2003; Price and Ware, 2004; Weill et al., 2004; Burdorf et al., 2007; Teta et al., 2008; Harding and Darnton, 2010). Epidemiologic observations also suggest a much lower, if not zero, mesothelioma potency for the more prominent chrysotile exposures from various sources and products in subsequent years (Hodgson and Darnton, 2000, 2010; Berman and Crump, 2003, 2008a,b; Goodman et al., 2004; Laden et al., 2004; Gibbs and Berry, 2008; Sichletidis et al., 2008). If this is true, it suggests that sometime in the near future there may be a substantial decline in the number of asbestos-induced mesothelioma cases. Consequently, there will be a greater need to clinically distinguish and better understand the etiology of mesothelioma cases unrelated to asbestos exposure.
While only perhaps 10–20% of pleural mesothelioma cases in the past decade are reportedly due to causes other than asbestos exposure (Sporn and Roggli, 2004), the risk factors for peritoneal mesothelioma are far more diverse, with less than half of the cases in recent case series explained by heavy amphibole exposures (Neumann et al., 2001; Hillard et al., 2003; Weill et al., 2004; Reid et al., 2005; Bofetta and Stayner, 2006; Bofetta, 2007; Larson et al., 2007; Magnani et al., 2007; Gibbs and Berry, 2008). Pericardial mesothelioma is far less common than the pleural and peritoneal forms and has no strong or consistent association with asbestos exposure (Papi et al., 2005; Luk et al., 2008). In addition, unlike pleural mesothelioma, peritoneal and pericardial mesotheliomas often occur in younger individuals with no known or substantial source of past asbestos exposure (Sporn and Roggli, 2004; Papi et al., 2005; Luk et al., 2008). Recent reviews acknowledge that asbestos is not the sole cause of pleural and peritoneal mesothelioma, with erionite and ionizing radiation being established causes and the relatively low frequency of mesothelioma among more highly exposed individuals indicating that genetic susceptibility factors and/or co-carcinogens may play an important role (Neri et al., 2005; Testa et al., 2011; Jasani and Gibbs, 2012; Jean et al., 2012; Tallet et al., 2013).
Rates of peritoneal mesothelioma have stayed relatively flat in developed countries during the past two decades, in contrast to the dramatic rise in pleural mesothelioma rates from about the 1960s through at least the mid-1990s that was associated with asbestos use in prior decades (Hemminki and Li, 2003; Leigh, 2003; Weill et al., 2004; Burdorf et al., 2007; Teta et al., 2008; Moolgavkar et al., 2009; Harding and Darnton, 2010). This divergence suggests that asbestos is not likely to be among the current major causes of peritoneal mesothelioma. In fact, there is emerging evidence that some of the tumors initially diagnosed as peritoneal mesotheliomas are in actuality a cancer with another origin. Peritoneal tumors may easily be misclassified as they often result from metastatic disease originating in other organs and are typically discovered at late stages when such tumors may exhibit mixed pathological characteristics that do not closely resemble the original tumor cell type (Pass et al., 2005; Burdorf et al., 2007). It has even been suggested that peritoneal mesothelioma in younger individuals (e.g., under age 50) may represent a distinctly different disease entity; one that is more curable and more likely related to gonadal cancers, and so, not plausibly attributed to asbestos exposure (Weill et al., 2004; Hodgson et al., 2005; Larson et al., 2007).
Because mesothelioma lacks a unique and distinctive pattern of histological features, the diagnosis is often tentative and based primarily on a defined pattern of staining with immunohistochemical markers (Ordonez, 1998; Cappello and Barnes, 2001). While most tumor presentations can be distinguished by the presence/absence of these specific immunohistochemical markers, in many cases a definitive staining pattern is lacking. The difficulties sometimes encountered with mesothelioma diagnosis based on only cytology and immunohistochemical staining pattern are summarized in Figure 1.

Figure 1. Difficulties with diagnosis of primary mesothelioma using only cytology and immunohistochemical staining pattern.
Although a highly specific biomarker for mesothelioma has not yet been demonstrated, a variety of case series reports have been used to identify patterns of positive and negative staining characteristics that are “consistent with” malignant mesothelioma. The primary goal in distinguishing epithelioid types of mesothelioma involves a staining pattern that is generally not seen in adenocarcinomas; this includes positive staining for pankeratin, calretinin, keratin 5/6, and WT1 (but not necessarily all of these), and negative staining for CEA, CD15, Ber-EP4 Moc-31, TTF-1, and B72.3 (Ordonez, 1998; Cappello and Barnes, 2001; Pass et al., 2008). But the heterogeneous histology of mesothelioma (e.g., the involvement of epithelial cells, spindle cells or both cell types) can lead to inconsistent or ambiguous staining patterns that overlap with other tumor types, e.g., certain soft tissue sarcomas (Nicholson et al., 1998; Cappello and Barnes, 2001; Miettinen et al., 2001). In addition, the chromosomal instability of metastatic tumors that often leads to more primitive cell types with indistinct staining patterns may in some cases overlap with those of mesothelioma or soft tissue sarcoma (Morin et al., 2005).
To date, searches for genetic aberrations associated with malignant mesothelioma have failed to identify a highly specific chromosomal abnormality, although several associated lesions based on newer techniques like micro-RNA expression, DNA methylation, telomere status, micronucleus formation, and gene expression profiling are under investigation (Bott et al., 2011; Testa et al., 2011; Jean et al., 2012). In contrast to many other types of solid tumor, mesotheliomas generally lack mutations of the fundamental tumor suppressor genes, p53 and RB. Only a few molecular defects have been identified in mesotheliomas thus far. Pisick and Salgia (2005) reviewed the literature and concluded that while there are several different genetic and signal pathway alterations that can occur in malignant mesothelioma cells and cell lines, these changes are heterogeneous, and therefore not clearly useful for diagnostic purposes at this time. These authors noted that transformation occurs either by direct stimulation of pathways activated by receptor tyrosine kinases or by a loss of genetic material containing tumor suppressor genes that prevent unchecked cell proliferation. The more commonly noted losses occur on the short arms of chromosomes 1 (notably 1p21–p22), 3 (notably 3p21.3), and 9 (notably 9p14/p16), and on the long arms of chromosomes 6 (notably 6q14–q21, 6q16.6–q21, 6q21–q23.2, and 6q25), 15 (notably 15q11.1–q15), and both arms of chromosome 22 (Pisick and Salgia, 2005; Pass et al., 2008). Loss of a copy of chromosome 22 is thought to be the single most consistent karyotypic alteration seen in malignant mesothelioma (Pass et al., 2008). Unfortunately, these patterns of genetic loss also represent genetic variations seen in more common forms of cancer, such as non-Hodgkin's lymphoma, breast, ovarian, prostate, lung and colon cancers (Pisick and Salgia, 2005).
A more recent review on molecular changes observed in mesothelioma by Jean et al. (2012) summarized findings of emerging research on possible genetic markers. These authors identified a slightly wider range of associated chromosomal alterations including losses at 1p, 3p, 4q, 6q, 13p, 14q, and 22q, and gains at 1q, 5p, 7p, 8q, and 17q and noted that more specific mutations on chromosome 22 (e.g., deletions at the neurofibromatosis 2 locus, 22q12), chromosome 9 (e.g., INK4 genes, specifically deletions at cyclin-dependent kinase inhibitor genes CDKN2A and CDKN2B at 9p21.3), and chromosome 17 (TP53 gene deletion at 17p13.1, though less common) have been associated with mesothelioma in recent reports. However, these mutations occur with several other cancer types (Jean et al., 2012). They noted a new association between mesotheliomas and increased microRNA expression of certain types (MiR-31, -141, -192, -193, -200a-c, -203, -205, and -429); greater DNA methylation at certain loci, and increases in markers of telomere length maintenance mechanisms were also reportedly associated with mesotheliomas (Ivanov et al., 2010; Jean et al., 2012). While many of these new associations appear promising as adjuncts to current immunohistochemical staining techniques in discerning mesothelioma from other cancer types, further research is needed to determine their specificity and reliability in refining diagnosis, treatment, and prognosis for mesothelioma.
Other recent observations on mesothelioma genetic markers have focused on mesenchymal membrane receptor tyrosine kinases that dirve downstream cell signaling of cell proliferation, cell cycle control, survival and differentiation (Lemmon and Schlessinger, 2010). First, there is recent evidence suggesting that telomere status and telomere maintenance mechanisms tied to P53, ATRX and DAXX mutations may be helpful in distinguishing more aggressive mesotheliomas (Durant, 2012; Gocha et al., 2013; Tallet et al., 2013). Positive telomerase activity is observed in 91–100% of pleural mesotheliomas in two studies (Dhaene et al., 1998; Au et al., 2011), and telomerase-independent pathways for telomere lengthening through DNA damage repair mechanisms (ALT positive cells) are more commonly observed in sarcomatoid tumors (Heaphy et al., 2011; Durant, 2012; Hu et al., 2012) and persons with multiple endocrine neoplasia type 1 (Gocha et al., 2013). Second, the mutations leading to telomere lengthening and survival of clonally expanding mesothelioma cells using ALT mechanisms are reported to form ALT-associated promyelocytic bodies that may be akin to micronuclei observed in tumor cells and blood polynucleated lymphocytes with exposure to asbestos (Dopp et al., 1995; Bolognesi et al., 2005; Martini et al., 2011). Third, soluble mesothelin-related peptides are overexpressed in persons with mesothelioma (Robinson et al., 2003; Scherpereel et al., 2006) and have been associated with increased micronuclei in peripheral lymphocytes (Martini et al., 2011). And fourth, germline mutations in nuclear deubiquitinase BRCA1-associated protein 1 (BAP1) have been observed in familial clusters of mesothelioma not necessarily linked to asbestos (Testa et al., 2011) and in a fraction of pleural mesotheliomas in other case series (Bott et al., 2011; Jean et al., 2012; Tallet et al., 2013). These more recent associations are all tied into loss of normal cell signaling that occurs in a variety of cancer types, but further investigations may provide more specific genetic markers to help better distinguish mesothelioma diagnosis, treatment, and prognoses in the future (Bott et al., 2011; Jean et al., 2012; Gocha et al., 2013).
Given the preceding issues, it is important to collect and more thoroughly evaluate genetic evidence for mesothelioma cases, particularly in younger cases or those with little or no known amphibole asbestos exposure. Genetic screening in conjunction with assessment of histology and immunohistochemical staining patterns in these cases may result in more definitive identification of the true tissue/cell-type of origin, thereby avoiding misdiagnosis of mesothelioma. This, in turn, would allow for a better identification of tumor-specific risk factors and thereby increase the possibility of a more effective treatment regimen specific for those cancers that are not a true mesothelioma. Screening for genetic aberrations corresponding to tumor types known to mimic mesothelioma would seem to be particularly important in cases where no occupational asbestos exposure has been identified, in cases involving an individual under 50 years old, and in cases with no evidence of pleural plaques, asbestosis, or elevated lung asbestos fiber counts.
Genetic testing of suspected mesothelioma cases with non-distinct staining patterns is apparently an infrequent medical consideration despite the potential value to the patient and to medical science in the form of learning more about the genetics underlying mesothelioma and other tumors types with histological resemblance1. Additional factors that can provide important evidence reflecting on the likelihood of misdiagnosis and/or unlikely association with asbestos exposure for tentatively diagnosed mesothelioma cases are provided in Figure 2.

Figure 2. Additional factors to consider in evaluating suspected asbestos-related mesothelioma.
The purpose of this paper is to set out the scientific basis for a registry-based research protocol for the collection of medical information and tissue samples to refine medical knowledge on mesothelioma diagnosis, prognosis, and rates. The basis for genetic testing that may distinguish specific cancer types probably unrelated to asbestos exposure is reviewed, and the utility of a registry-based research program to enhance medical knowledge regarding the etiology of general and specific subtypes of mesothelioma is explored.
Basis for Genetic Testing of Mesothelioma and Histologically Similar Tumors
Germ Cell Tumors and Mesothelioma
Advanced stage extragonadal germ cell tumors (GCTs) are known to commonly occur in, or metastasize to, tissues along the body midline from the pineal gland to the sacrum. Due to their multi-potential nature they can exhibit features, both for histology and immunohistochemical markers, of primary pleural or peritoneal malignant mesothelioma. Mesothelioma of the tunica vaginalis is an example of this occurring within the testicle, yet little attention has been given to potential metastatic forms of this tumor and/or to extragonadal GCTs that originate from mutated embryonic cells of the urogenital ridge that may be mistakenly diagnosed as a mesothelioma of the peritoneum or pleura. The occurrence of mesothelioma in children and adolescents without significant or identifiable asbestos exposure (Fraire et al., 1988; Coffin and Dehner, 1992; Niggli et al., 1994; Hubbard, 1997) could plausibly represent a GCT or other congenital/genetic mutations affecting mesenchymal tissues, especially given the much higher background incidence of GCTs relative to that of mesothelioma.
GCTs can arise from germ cell tissues in the testes, the prostate, and other tissue locations (extragonadal GCT) as a result of a mutation and abnormal migration of germ cells during embryonic development (Blossom et al., 1997; Sarma et al., 2006). Blossom et al. (1997) have explained that extragonadal GCTs occur along the body's midline from the presacral areas and retroperitoneum up to the cranium (pineal gland), a pattern that corresponds to the embryologic urogenital ridge extending along the mediastinum and peritoneum from vertebrae C-6 to L-4. Extragonadal GCTs result from malignant transformation of germinal elements that are displaced to extragonadal sites in the absence of any gonadal primary tumor (Blossom et al., 1997). There is no plausible etiologic role of asbestos exposure in the development of extragonadal GCTs.
Blossom et al. (1997) have also noted that approximately 90% of malignant extragonadal GCT occur in men, most of which are diagnosed in the third decade of life or later. While the lifetime probability of developing a GCT (most of which originate from the testes) is only about 0.2% in Caucasian males in the United States, this lifetime risk is far greater than that of mesothelioma. Thus, potential misdiagnosis of these tumors might represent a substantial portion of the tumors mistakenly diagnosed as pleural or peritoneal mesothelioma in males, and may falsely increase the estimated background incidence of mesothelioma. The cause of these tumors is unknown but they are most likely explained by genetic mutation events in early embryonic development (Bosl et al., 1997). Individuals with a history of cryptorchidism are known to exhibit a significantly increased risk of testicular GCT (Bosl et al., 1997; Motzer and Bosl, 2005), as are brothers of individuals with GCTs (Kumar et al., 2005a; Motzer and Bosl, 2005).
The multi-potential nature of GCTs leads to a diverse pathological appearance including combined epithelial and spindle cell presentations that might be mistaken for mesothelioma (Blossom et al., 1997; Bosl et al., 1997; Motzer and Bosl, 2005). Due to this diversity of GCTs, and especially for advanced stage metastatic tumors, common stains and immunohistochemical markers may be unhelpful in identifying the origins of extragonadal GCTs. Fortunately, there are characteristic DNA changes in some tumors affecting chromosome 12 (iso-12p) that may help one to diagnose tumors originating from germ cells from gonadal or extragonadal sites (Bosl et al., 1997; Kumar et al., 2005a; Sarma et al., 2006). Since GCTs are not inhibited in their lines of differentiation, most presentations include two or more histologic patterns (e.g., mixed sarcomatoid and epithelioid as seen with mesothelioma), and males with a history of cryptorchid and dysgenetic testes show familial clustering and a higher frequency of intratubular GCTs (Kumar et al., 2005a).
Mullerian Tissue Tumors and Mesothelioma
Mullerian tissue tumors, of both breast and ovarian/adnexal origins, include certain mesenchymal subtypes that can spread to either the pleura or peritoneum in females. Breast cancer is a common metastatic tumor affecting the pleura in females (Winston et al., 2000; Kolschmann et al., 2005), and ovarian/adnexal cancers are also reported to spread to both the pleura and the peritoneum at advanced stages (Cormio et al., 2003). Breast cancer risk factors such as BRCA phenotype are now known to carry risks for certain female gonadal cancers that mimic mesothelioma (Hankinson and Danforth, 2006; Cannistra et al., 2008). More specifically, primary papillary serous carcinomas are among the most common forms of aggressive cancer types that originate in female gonadal tissues (i.e., ovarian surface epithelium, fallopian tube epithelium and fimbria, or mullerian tissue gonadal remnants) and can be difficult to distinguish from advanced forms of mesothelioma when the immunohistochemical staining pattern is ambiguous (Kannerstein et al., 1977; Bannatyne and Russell, 1981; Raju et al., 1989; Altaras et al., 1991; Fox, 1993; Alvarado-Cabrero et al., 1997; Crum et al., 2007). The ovarian surface epithelium is comprised of a specialized type of mesothelial cell forming embryonic mesoderm tissue that is a direct descendent of the coelomic epithelium of the embryonic gonadal ridge and of the mullerian duct system (Lauchlan, 1972; Parmley and Woodruff, 1974; Kannerstein et al., 1977; Fox, 1993; Katabuchi and Okamura, 2003; Piek et al., 2004).
Altaras et al. (1991) explained some of the history behind papillary serous carcinomas (PSC) being ultimately distinguished as separate clinical entities from mesotheliomas. Specifically, the peritoneal location of PSC is explained by the common coelomic ancestry of the mullerian duct epithelium and the peritoneal mesothelium. Mullerian differentiation can occur in a variety of metaplastic and neoplastic ways in diverse tissue locations, such as PSC, ovarian tumors of common epithelial origin, endometriosis, endosalpingiosis, and epithelial inclusions of the ovary and lymph nodes (Altaras et al., 1991). Diffuse PSC of the peritoneum without overt involvement of the ovary was initially described as malignant mesothelioma, until Kannerstein et al. (1977) identified histological criteria that distinguished PSC from mesotheliomas and from all extraovarian epithelial tumors. However, PSC of peritoneal and ovarian origin remain indistinguishable from each other by histopathology (Altaras et al., 1991).
More advanced-stage tumors and metastases from mullerian cells may become more primitive (undifferentiated) such that the tumor cell histology and immunohistochemical markers of the ovarian surface epithelial cells cannot be readily distinguished from less specialized mesothelial tumors that may originate in the peritoneal lining tissue (Hernandez et al., 1984; August et al., 1985; Cramer et al., 1987; Lund et al., 1991; Sakamoto et al., 1994; Gilks et al., 2008).
Some key tumor immunohistochemical markers (e.g., positive calretinin, CK7, and WT-1, with negative CEA) and cellular characteristics (epithelioid, poorly differentiated) are commonly seen for both mesothelioma and PSC (Fox, 1993; Schorge et al., 2000; Shimizu et al., 2000; Al-Hussaini et al., 2004; McCoubrey et al., 2005; Kobel et al., 2008). Indeed, PSC in many ways can mimic epithelioid mesothelioma, but unlike mesothelioma, PSC is linked to hereditary and sporadic mutations of two breast cancer susceptibility genes (BRCA-1 and BRCA-2). Primary epithelial cancers of the fallopian tubes (particularly the fimbria) appear to be linked to more aggressive and early-onset PSC in women with the BRCA-1 mutation (Sobol et al., 2000; Zweemer et al., 2000; Aziz et al., 2001; Paley et al., 2001; Levine et al., 2003; Piek et al., 2003; Finch et al., 2006; Medeiros et al., 2006; Kindelberger et al., 2007), while ovarian surface epithelium tumors have been linked to both BRCA-1 and BRCA-2 mutations (Werness et al., 2000; Colgan et al., 2001, 2002; Piura et al., 2001; Risch et al., 2001; Agoff et al., 2002; Brose et al., 2002; Geisler et al., 2002; Hilton et al., 2002; Cass et al., 2005). More advanced PSC tumors originating from ovarian surface epithelium or tubal fimbria epithelium are subject to genomic instability, loss of WT-1 gene expression, and the presence of both BRCA and p53 mutations, sometimes making the site or origin difficult to distinguish in more advanced stages of the disease (Schorge et al., 2000; Piek et al., 2004; Kindelberger et al., 2007; Lee et al., 2007; Gilks et al., 2008).
Since PSC can originate as an occult mullerian neoplasm that commonly proliferates at other peritoneal sites (Colgan et al., 2001, 2002; Cass et al., 2005; Powell et al., 2005; Finch et al., 2006; Chand et al., 2007), the pathological (histology and staining) evaluation of PSC often does not identify the original tumor site and should be augmented with detailed analysis of the patient's clinical and family history and BRCA phenotype in order to assign the final diagnosis with reasonable medical certainty (Risch et al., 2001; Piek et al., 2004; Kindelberger et al., 2007; Gilks et al., 2008). BRCA-positive families are noted to have higher cancer rates affecting the colon, prostate, pancreas and peritoneum, as well as melanoma (Struewing et al., 1997; Frank, 2001; Paley et al., 2001; Al-Hussaini et al., 2004). Three familial genetic syndromes associated with excess ovarian cancer risk have been identified: certain BRCA-1 or BRCA-2 mutations result in Breast Ovarian Cancer Syndrome, Site-Specific Ovarian Cancer Syndrome, and Hereditary Non-Polyposis Colon Cancer (or Lynch II Syndrome) which are associated with mutations in DNA mismatch repair genes in affected families (Muto, 2005).
In contrast, PSC has not been specifically associated with asbestos exposure (Fox, 1993), and peritoneal mesothelioma are predominantly associated with relatively high exposures to primarily amphibole forms of asbestos (Sporn and Roggli, 2004; Bofetta and Stayner, 2006; Yarborough, 2007; Gibbs and Berry, 2008).
Congenital Heart Disease and Pericardial Mesothelioma
Pericardial mesothelioma is estimated to comprise only 0.7–1% of all mesotheliomas (Papi et al., 2005; Molina Garrido et al., 2006) and lacks a specific immunohistochemical staining pattern that would distinguish this lesion from mesotheliomas originating at other sites (Reis-Filho et al., 2002; Papi et al., 2005). In contrast to pleural mesothelioma, the association of asbestos exposure with pericardial mesothelioma is unclear (Eren and Akar, 2002; Val-Bernal et al., 2002; Lagrotteria et al., 2005; Papi et al., 2005; Luk et al., 2008). Although a few reported cases have occurred in individuals with occupational asbestos exposure (Kahn et al., 1980; Beck et al., 1982; Thomason et al., 1994; Fujiwara et al., 2005), it appears that the vast majority of recently reported cases had no known asbestos exposure (Quinn et al., 2000; Hirano et al., 2002; Reis-Filho et al., 2002; Val-Bernal et al., 2002; Susman et al., 2004; Yakirevich et al., 2004; Erdogan et al., 2005; Lagrotteria et al., 2005; Maruyama et al., 2006; Molina Garrido et al., 2006; Doval et al., 2007; Vornicu et al., 2007; Akamoto et al., 2008; Santos et al., 2008), and many were reported in younger individuals (e.g., age 19–50 years). Malignant mesothelioma of the pleural cavity was also reported to occur in a 15-year old boy with rheumatic heart disease and a prosthetic aortic valve implant; this child was reported to have no history of asbestos or chemical exposure (Tewari et al., 1989).
As reviewed by Kumar et al. (2005b), the heart is one of the first organs to form and function during early embryonic development. Developmental errors in mesenchymal tissue migration are just one example of genetic errors leading to certain types of congenital heart disease, e.g., involving various anomalies of the outflow tract, some due to failure of fusion and others due to failure of septation. Parallel to the genesis of some extragonadal GCTs from abnormal germ cell migration along the embryological urogenital ridge, some congenital heart defects may be caused by abnormal development of neural crest-derived cells, whose migration into the heart is required for proper formation of the outflow tracts. Many congenital heart defects are related to trisomies of chromosomes 13, 15, 18, and 21, and deletions in chromosome 22 (22q11.2) are thought to play a major role in congenital heart disorders of the conotruncus and the brachial arches (Kumar et al., 2005b). Accordingly, it seems appropriate that identification of a case of malignant mesothelioma involving congenital heart disease would initiate genetic screening to help define the common congenital defects that may relate to the onset of both diseases in the same individual. Further research is also recommended to examine the incidence of malignant mesothelioma among individuals with various forms and severity of congenital heart disease, and perhaps also in relation to rheumatic heart disease.
Synovial Sarcomas Presenting as Mesothelioma
There is no demonstrated association between synovial sarcoma and asbestos exposure, although synovial sarcoma of the pleura is difficult to distinguish from sarcomatoid mesothelioma without the use of genetic screening (Nicholson et al., 1998; Cappello and Barnes, 2001; Colwell et al., 2002; Weinbreck et al., 2007). These tumors are thought to originate from primitive pluripotent mesenchyme cells capable of sarcomatous differentiation. This tumor can be highly aggressive, involving metastases to distant locations (Siegel et al., 2007; Eilber and Dry, 2008), and consistent with this they have been observed to occur in tissues with little or no associated synovium. Indeed, synovial sarcoma does not arise from or differentiate toward synovium—it is an unfortunate misnomer since the vast majority arise in deep soft tissue of extremities, especially around the knee. Because of this it has been proposed that synovial sarcoma be renamed as carcinosarcoma or spindle cell carcinoma of soft tissue due to its immunoreactivity to cytokeratins and EMA. Relevant to mistaken pleural mesotheliomas, synovial sarcomas have been prominently noted to occur in the pleural cavity (Gaertner et al., 1996; Nicholson et al., 1998; Caliandro et al., 2000; Cappello and Barnes, 2001; Colwell et al., 2002; Vohra et al., 2004; Lee et al., 2006; Tsukioka et al., 2006; Galetta et al., 2007; Iwata et al., 2007; Mukhopadhyay and Aubry, 2007; Satoh et al., 2007; Weinbreck et al., 2007), as well as in the lung (Hosono et al., 2005; Taylor et al., 2005). Other reported sites include the peritoneal cavity (Vera et al., 2006; Gofman et al., 2007), and the stomach (Makhlouf et al., 2008).
Synovial sarcoma is a relatively rare malignancy that typically occurs in adolescents and young adults between the ages of 15 and 50 years of age and most commonly affects the extremities in the vicinity of large joints such as the knee or the thigh (Cadman et al., 1965; Cordon-Cardo, 1997; Nicholson et al., 1998; Cappello and Barnes, 2001; Kumar et al., 2005c) and the hands or feet (Michal et al., 2006). Because synovial sarcoma can be difficult to distinguish from reactive mesothelial proliferation and sarcomatoid mesothelioma by use of histology and immunohistochemical markers alone (Shiraki et al., 1989; Moran et al., 1992; Nicholson et al., 1998; Miettinen et al., 2001; Carbone et al., 2002; Gladish et al., 2002; Vohra et al., 2004; Taylor et al., 2005; Michal et al., 2006; Rdzanek et al., 2006), it represents another alternative carcinoma to consider when evaluating suspected sarcomatoid mesothelioma cases. This has been particularly true since the discovery of highly specific genetic lesions [t(x;18), SYT-SSX1, SSX2, and SSX4 fusion genes] in synovial sarcoma tumors that clinically distinguish this particular this tumor type (Colwell et al., 2002; Amary et al., 2007; Weinbreck et al., 2007). Indeed, Sandberg (2002) provides a summary of specific chromosomal translocations corresponding to a variety of soft tissue sarcomas including synovial sarcoma where—the translocation is the sole cytogenetic anomaly, indicating the probable causative role of this translocation in the genesis of these tumors. In synovial sarcoma, the normal SYT gene encodes a transcription factor whereas the SSX1 and SSX2 genes produce proteins that are transcription inhibitors. The available evidence suggests that the specific type of translocation may be related to the aggressiveness of the synovial sarcoma and its prognosis. The most common sites of metastases are the lung/mediastinum, the skeleton, and regional lymph nodes (Hosono et al., 2005; Kumar et al., 2005c; Michal et al., 2006).
Two additional soft tissue sarcomas should be mentioned here because they have the potential to occur as poorly differentiated metastatic tumors with staining characteristics that are difficult to distinguish from sarcomatoid mesothelioma, but they can be positively identified by their unusual chromosomal alterations (Kumar et al., 2005a). Desmoplastic small round cell tumor, a tumor of unknown hisotogenesis in the peritoneum, shows a specific translocation [t(11;22)(p13;q12)], involving the EWS-WT-1 gene fusion product in 90% of cases. Endometrial stromal sarcoma, a tumor that may occur as distant metastases decades after hysterectomy, also shows a specific translocation [t(7;17)(p15;q21)], involving JAZF1-JJAZ1 gene fusion product in about 65% of cases. These tumors are relatively rare sarcomas that have sufficiently unique markers to distinguish the origin of poorly differentiated spindle cell tumors that may be mistaken as mesotheliomas.
Inherited Cancer Susceptibility Syndromes and Mesothelioma
Certain inherited disorders produce multiple cancers that are prone to develop chromosomal instability and present at a late stage with histological features that mimic disseminated mesothelioma (Hawley and Pandolfi, 2008). Two examples of such inherited disorders are Multiple Endocrine Neoplasia Syndrome-1 (MEN-1) and Hereditary Non-Polyposis Colon Cancer (HNPCC). Both syndromes are known to have diagnostic genetic mutations in somatic cells that identify the syndrome and overt clinical outcome which consists of multiple, but often survivable, cancers in affected subjects and their blood relatives.
MEN-1 involves a germline mutation at the 11q13 (a tumor suppressor gene) which encodes for a protein menin and is associated with increased risk of parathyroid, endocrine (thyroid, prostate, testicular, breast, and ovarian), pancreas, and pituitary neoplasia (mostly adenomas) (Mayer, 2005; Morin et al., 2005). MEN-1 is known to occur in conjunction with Zollinger-Ellison Syndrome (ZES) in both sporadic and heritable forms, with an estimated 50% penetration among blood relatives. ZES leads to the formation of gastrinomas in the pancreas and/or duodenum that lead to hypergastrinemia and intractable peptic ulcer disease, however the most common and earliest manifestation is usually primary hyperparathyroidism. Clinical markers for ZES are elevated fasting gastrin (>150 pg/ml) and sometimes hyperparathyroidism resulting in elevated blood calcium, parathyroid hyperplasia, and other sequelae (Goyal, 2005). The late stages of MEN-1 are associated with increasing chromosomal instability among tumors that may have metastasized to other tissues decades earlier, or involving proliferation from surgical scars of the stomach, since the only way to resolve duodenal hypergastrinemia is with distal gastrectomy. Late stage tumors of MEN-1 patients have been associated with chromosomal instability that may plausibly lead to local tissue invasion and conversion to mixed neoplastic cell types including soft tissue sarcomas that may mimic mesotheliomas. Thus, it is suggested that persons with characteristic prior cancers and/or ZES and distal gastrectomy who present with apparent mesothelioma should be tested for the somatic cell mutation (11q13) of the MEN-1 tumor suppressor gene, in addition to testing for other translocations that may be diagnostic for various types of soft tissue sarcoma that may be relevant to the tumor/site (Sandberg, 2002).
Similarly, HNPCC involves germline mutations at certain loci (2p16, 3p21.3, and 7p22) that are associated with the formation of carcinoid tumors of the duodenum and ascending colon (Mayer, 2005; Morin et al., 2005; Hawley and Pandolfi, 2008). As with MEN-1, this germline mutation is thought to have a high penetration rate, and often involves multiple additional tumor sites including endometrial, ovarian, stomach, small bowel, and ureter carcinoma. Also, late stage tumors in HNPCC patients are associated with chromosomal instability that may plausibly lead to local tissue invasion and conversion to mixed neoplastic cell types including soft tissue sarcomas that may mimic mesotheliomas. Thus, persons with characteristic prior cancers who present with apparent mesothelioma should be tested for the somatic cell mutations of HNPCC, in addition to testing for other translocations that may be diagnostic for various types of soft tissue sarcoma that may be relevant to the tumor/site (Sandberg, 2002).
Discussion
Based on the preceding research summary, a preliminary genetic screening matrix has been outlined in Table 1 that could be used as a starting point for further characterizing and understanding the clinical and genetic features of malignant mesothelioma and other primary cancers that may sometimes mimic asbestos-related mesothelioma. As noted in the Introduction, there are many additional candidate features that could be added to Table 1 as the associations between mesothelioma and various genetic markers are further clarified. This preliminary matrix could assist in developing future research plans and incorporating other clinical observations that may help to fulfill the goal of obtaining unambiguous diagnostic criteria for various forms of malignant mesothelioma and tumors that may mimic mesothelioma.
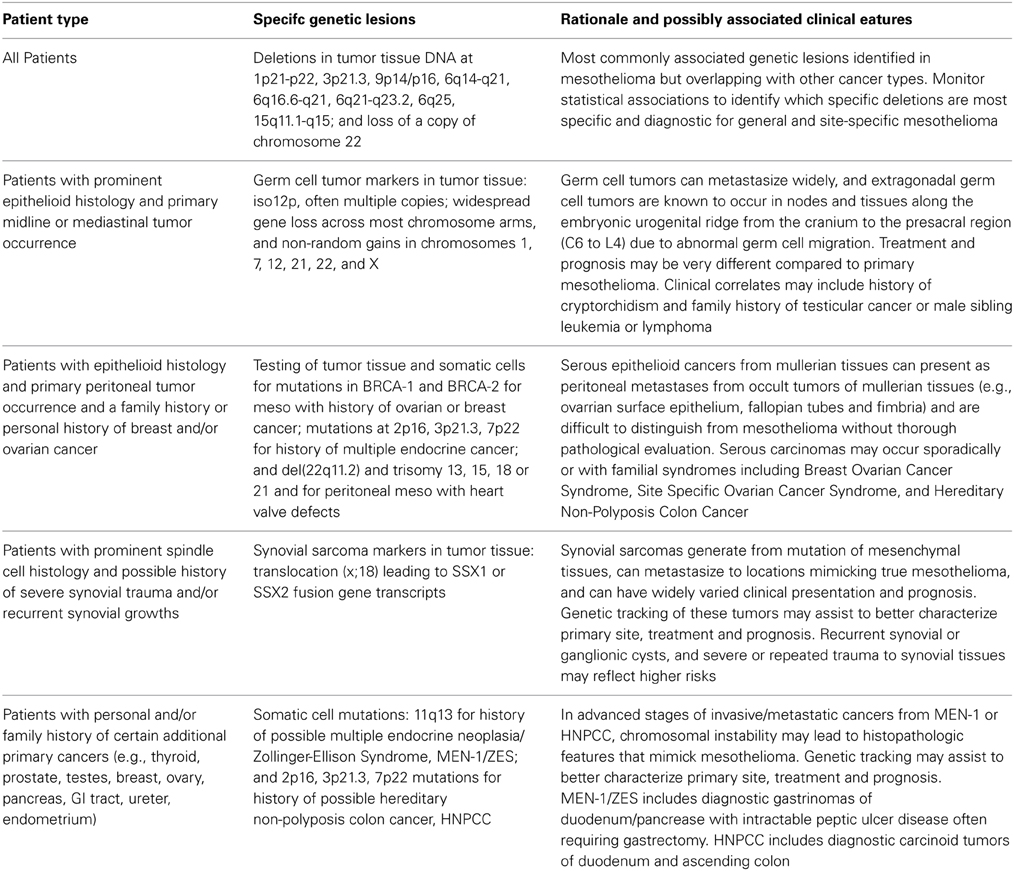
Table 1. Suggested matrix for genetic screening of suspected malignant mesothelioma cases.
Diagnosing a tumor as mesothelioma based on histologic and immunohistochemical staining characteristics alone can lump together several mesenchymal tumor types of different origins that should be treated as distinct clinical entities with unique risk factors, prognosis, and best treatment modalities. Several mesenchymal tumors that are difficult to distinguish based on histology and staining characteristics are already known to be distinguished by genetic markers in somatic or tumor cells.
The number of mullerian or germ cell tumors, advanced metastatic tumors, or mistaken synovial sarcomas that may have no plausible connection to asbestos exposure will remain obscure without appropriate research that supports a more definitive, evidence- based decision matrix for correct diagnosis. While the prognosis of any given individual's cancer can be difficult to predict, this research may identify genetic and/or clinical characteristics that presently, or in the future, will aid in more accurate prognostic predictions and perhaps more tumor-specific and efficacious treatments. For example, some forms of synovial sarcoma that remain circumscribed and relatively indolent are survivable without the aggressive clinical interventions typical of the treatment of malignant mesothelioma. Similarly, the confirmation of germ cell tumor origins would call for more focused clinical investigations and interventions that greatly differ from mesothelioma treatments. By adopting better diagnostic criteria for those mesenchymal tumor subtypes that may be mistaken as mesothelioma, physicians will improve epidemiologic estimates of the incidence and prevalence of the tumor types discussed above. Thus, this approach would ultimately improve dose-response relationships in the low dose region for asbestos and mesothelioma as well as that of other apparent risk factors for this deadly disease.
As discussed earlier, the proportion of future mesothelioma cases that can be credibly attributed to asbestos is likely to lessen because the highly potent amphibole exposures were largely curtailed in the 1960s and the typical latency period of 20–40 years since first exposure has now transpired. The incidence of pleural mesotheliomas that are more specifically associated with asbestos is projected to continue declining, while peritoneal mesothelioma incidence (associated primarily with non-asbestos causes) has been essentially flat for decades (Teta et al., 2008; Moolgavkar et al., 2009). Parallel to mesothelioma risk trends, asbestosis is a fibrotic interstitial lung disease with a comparable latency period that has also generally diminished in both incidence and clinical severity in the past two decades. This trend has raised questions about how many valid asbestosis cases arise from the many claims that today are often based solely on equivocal radiographic findings and questionable occupational history of asbestos exposure (Bang et al., 2008; Mizell et al., 2009; Harding and Darnton, 2010).
Physicians are urged to recognize that the proportion of mesothelioma cases unrelated to asbestos will likely increase in coming decades, and that better vigilance will be needed for proper diagnosis, treatment, and prevention. This will undoubtedly involve the development and use of more definitive diagnostic tools like genetic screening that can help differentiate asbestos-related mesothelioma from other mesenchymal tumor subtypes or advanced metastatic tumors that can mimic the histopathological presentation of mesothelioma.
A national or international registry-based mesothelioma research program may be an achievable and appropriate means for augmenting the knowledge base on clinical features and genetic markers for distinguishing specific forms of mesothelioma and tumors that may mimic mesothelioma. Since the annual number of mesothelioma cases is relatively small, the scope of research each year may be reasonably defined. Incident case detection could be linked to existing state and/or national cancer registries, with the registry research program overseen by a steering committee with outside peer review resources. A working group could be tasked with developing appropriate research protocols for collection of needed clinical and family history information in addition to samples of tumor tissue and somatic cells for genetic screening. The scope of analysis and quality control procedures of the working group should be clearly defined and transparent, with assurance of evidence-based data analysis and outside peer review of findings in a manner that also assures objectivity and patient privacy.
In conclusion, the identification of relatively specific clinical features and genetic markers that may avoid misdiagnosis of mesothelioma and other mesenchymal tumor subtypes or advanced metastatic tumors is an important future research need. Currently, the diagnosis of mesothelioma rests largely on histology and staining patterns that are sometimes inconclusive. It is recommended that further research be directed at identifying those genetic and clinical features unique to malignant mesothelioma and the cancers that may mimic its histopathology. This research could be fostered through a national or international mesothelioma registry with requisite medical history questionnaire and tumor/somatic tissue submission, hopefully leading to more specific diagnostic tools, better disease classification and incidence data, and a uniform and enhanced database for understanding the natural history and prognosis of various mesothelioma subtypes.
Conflict of Interest Statement
The authors are research scientists employed by scientific consulting firms that are paid to conduct research for private clients relating to questions raised in regulatory and legal arenas. Each of the authors has conducted such research on behalf of clients with alleged asbestos liabilities, including expert witness activities. This work was funded solely by the authors and their institutions, without financial or technical assistance from any client. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank David Garabrant, Mark Burton, Fionna Mowat, and Jane Teta for their peer review and insights. The authors are independent consulting scientists who have researched asbestos on behalf of private clients in regulatory and litigation settings. This manuscript was funded solely by the authors and their employers, and no client participated in the creation or editing of this work.
Footnotes
- ^Each of the research categories proposed in this manuscript is based on a compilation of notes from actual mesothelioma claims with findings that supported the alternative diagnosis. However, current privacy laws in the United States prevent reporting specific details of these cases here. Establishing a registry-based research program as proposed in this paper would generate the epidemiologic and clinical information for the improvement of diagnosis and identification of risk factors for this cancer in a manner that protects patient privacy. A registry would provide a neutral setting outside of the litigation arena where legal maneuvering would not hamper necessary data collection and analysis. Unfortunately, it has been the experience of one of the authors (BDK) that information that could improve disease causation analysis for mesothelioma cases has been blocked when a legal case has been filed. These attorneys may not realize that a more definitive diagnosis might lead to a more effective treatment and a better prognosis for the affected individual.
References
Agoff, S. N., Mendelin, J. E., Grieco, V. S., and Garcia, R. L. (2002). Unexpected gynecologic neoplasms in patients with proven or suspected BRCA-1 or -2 mutations: implications for gross examination, cytology, and clinical follow-up. Am. J. Surg. Pathol. 26, 171–178. doi: 10.1097/00000478-200202000-00003
Akamoto, S., Ono, Y., Ota, K., Suzaki, N., Sasaki, A., Matsuo, Y., et al. (2008). Localized malignant mesothelioma in the middle madiastinum. Surg. Today 38, 635–638. doi: 10.1007/s00595-007-3679-1
Al-Hussaini, M., Stockman, A., Foster, H., and McCluggage, W. G. (2004). WT-1 assists in distinguishing ovarian from uterine serous carcinoma and in distinguishing between serous and endometrioid ovarian carcinoma. Histopathol 44, 109–115. doi: 10.1111/j.1365-2559.2004.01787.x
Altaras, M. M., Aviram, R., Cohen, I., Cordoba, M., Weiss, E., and Beyth, Y. (1991). Primary peritoneal papillary serous adenocarcinoma: clinical and management aspects. Gynecol. Oncol. 40, 230–236. doi: 10.1016/0090-8258(90)90283-Q
Alvarado-Cabrero, I., Navani, S. S., Young, R. H., and Scully, R. E. (1997). Tumors of the fimbriated end of the fallopian tube: a clinicopathologic analysis of 20 cases, including nine carcinomas. Int. J. Gynecol. Pathol. 16, 189–196. doi: 10.1097/00004347-199707000-00001
Amary, M. F. Berisha, F., Bernardi Fdel, C., Herbert, A., James, M., Reis-Filho, J. S., et al. (2007). Detection of SS18-SSX fusion transcripts in formalin-fixed paraffin-embedded neoplasms: analysis of conventional RT-PCR, qRT-PCR and dual color FISH as diagnostic tools for synovial sarcoma. Mod. Pathol. 20, 482–496. doi: 10.1038/modpathol.3800761
Au, A. Y. M., Hackl, T., Yeager, T. R., Cohen, S. B., Pass, H. I., Harris, C. C., et al. (2011). Telomerase activity in pleural malignant mesotheliomas. Lung Cancer 73, 283–288. doi: 10.1016/j.lungcan.2010.12.023
August, C. Z., Murad, T. M., and Newton, M. (1985). Multiple focal extraovarian serous carcinoma. Int. J. Gynecol. Pathol. 4, 11–23. doi: 10.1097/00004347-198501000-00002
Aziz, S., Kuperstein, G., Rosen, B., Cole, D., Nedelcu, R., McLaughlin, J., et al. (2001). A genetic epidemiological study of carcinoma of the fallopian tube. Gynecol. Oncol. 80, 341–345. doi: 10.1006/gyno.2000.6095
Bang, K. M., Mazurek, J. M., Syamlal, G., and Wood, J. M. (2008). Asbestosis mortality surveillance in the United States, 1970–2004. Int. J. Occup. Environ. Health 14, 161–169. doi: 10.1179/oeh.2008.14.3.161
Bannatyne, P., and Russell, P. (1981). Early adenocarcinoma of the fallopian tubes. Diag. Gynecol. Obstet. 3, 49–60. doi: 10.1007/s00595-007-3679-1
Beck, B., Konetzke, G., Ludwig, V., Rothig, W., and Sturm, W. (1982). Malignant pericardial mesotheliomas and asbestos exposure: a case report. Am. J. Ind. Med. 3, 149–159. doi: 10.1002/ajim.4700030205
Berman, D. W., and Crump, K. S. (2003). Final Draft: Technical Support Document for a Protocol To Assess Asbestos-Related Risk. Washington, DC: Office of Solid Waste and Emergency Response, U.S. Environmental Protection Agency.
Berman, D. W., and Crump, K. S. (2008a). A meta-analysis of asbestos-related cancer risk that addresses fiber size and mineral type. Crit. Rev. Toxicol. 38, 49–73. doi: 10.1080/10408440802273156
Berman, D. W., and Crump, K. S. (2008b). Update of potency factors for asbestos-related lung cancer and mesothelioma. Crit. Rev. Toxicol. 38, 1–47. doi: 10.1080/10408440802276167
Bernstein, D. M., Rogers, R. A., Sepulveda, R., Donladson, K., Schuler, D., Gaering, S., et al. (2010). The pathological response and fate in the lung and pleura of chrysotile in combination with fine particles compared to amosite asbestos following short-term inhalation exposure: interim results. Inhal. Toxicol. 22, 937–962. doi: 10.3109/08958378.2010.497818
Bernstein, D. M., Rogers, R. A., Sepulveda, R., Donaldson, K., Schuler, D., Gaering, S., et al. (2011). Quantification of the pathological response and fate in the lung and pleura of chrysotile in combination with fine particles compared to amosite-asbestos following short-term inhalation exposure. Inhal. Toxicol. 23, 372–391. doi: 10.3109/08958378.2011.575413
Bernstein, D. M., Rogers, R., and Smith, P. (2005). The biopersistence of Canadian chrysotile asbestos following inhalation: final results through 1 year after cessation of exposure. Inhal. Toxicol. 17, 1–14. doi: 10.1080/08958370590885663
Blossom, G. B., Steiger, Z., and Stephenson, L. W. (1997). “Neoplasms of the mediastinum,” in Cancer, Principles and Practice of Oncology, 5th Edn., eds. V. T. DeVita, S. Hellman, and S. A.Rosenberg (Philadelphia, PA: Lippincott-Raven), 951–969.
Bofetta, P. (2007). Epidemiology of peritoneal mesothelioma: a review. Ann. Oncol. 18, 985–990. doi: 10.1093/annonc/mdl345
Bofetta, P., and Stayner, L. T. (2006). “Pleural and peritoneal neoplasms,” in Cancer Epidemiology and Prevention, 3rd Edn., eds D. Schottenfeld, and J. F. Fraumeni, Jr (London: Oxford University Press), 659–673. doi: 10.1093/acprof:oso/9780195149616.003.0034
Bolognesi, C., Martini, F., Tognon, M., Filiberti, R., Neri, M., Perrone, E., et al. (2005). A moleculr epidemiology case control study on pleural malignant mesothelioma. Cancer Epidemiol. Biomark. Prev. 14, 1741–1746. doi: 10.1158/1055-9965.EPI-04-0903
Bosl, G. J., Sheinfeld, J., Bajorin, D. F., and Motzer, R. J. (1997). “Cancer of the testis,” in Cancer: Principles and Practice of Oncology, 5th Edn., eds V. T. DeVita, S. Hellman, and S. A. Rosenberg (Philadelphia: Lippincott-Raven), 1397–1425.
Bott, M., Brevet, M., Taylor, B. S., Shimizu, S., Ito, T., Wang, L., et al. (2011). The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet 43, 668–672. doi: 10.1038/ng.855
Brose, M. S., Rebbeck, T. R., Calzone, K. A., Stopfer, J. E., Nathanson, K. L., and Weber, B. L. (2002). Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J. Natl. Cancer Inst. 94, 1365–1372. doi: 10.1093/jnci/94.18.1365
Burdorf, A., Jarvholm, B., and Siesling, S. (2007). Asbestos exposure and differences in occurrence of peritoneal mesothelioma in the Netherlands and Sweden. Occup. Environ. Med. 64, 839–842. doi: 10.1136/oem.2006.031724
Cadman, M. L., Soule, E. H., and Kelly, P. J. (1965). Synovial sarcoma: an analysis of 134 tumors. Cancer 18, 613–627.
Caliandro, R., Terrier, P., Regnard, J. F., De Montpréville, V., and Ruffié, P. (2000). Primary biphasic synovial sarcoma of the pleura. Rev. Mal. Respir. 17, 498–502.
Cannistra, S. A., Gershenson, D. M., and Recht, A. (2008). “Ovarian cancer, fallopian tube carcinoma, and peritoneal carcinoma,” in Cancer: Principles and Practice of Oncology, 8th Edn., eds V. T. DeVita, T. S. Lawrence, and S. A. Rosenberg Lippincott (Philadelphia: Williams and Wilkins), 1568–1594.
Cappello, F., and Barnes, L. (2001). Synovial sarcoma and malignant mesothelioma of the pleura: review, differential diagnosis and possible role of apoptosis. Pathology 33, 142–148. doi: 10.1080/00313020120038728
Carbone, M., Rizzo, P., Powers, A., Bocchetta, M., Fresco, R., and Krausz, T. (2002). Molecular analyses, morphology and immunohistochemistry together differentiate pleural synovial sarcomas from mesotheliomas: clinical implications. Anticancer Res. 22, 3443–3448.
Cass, I., Holschneider, C., Datta, N., Barbuto, D., Walts, A. E., and Karlan, B. Y. (2005). BRCA- mutation—associated fallopian tube carcinoma: a distinct clinical phenotype? Obstet. Gynecol. 106, 1327–1334. doi: 10.1097/01.AOG.0000187892.78392.3f
Chand, M., Moore, P. J., Clarke, A. D., Nash, G. F., and Hickisk, T. (2007). A diagnostic dilemma following risk-reducing surgery for BRCA1 mutation—a case report of primary papillary serous carcinoma presenting as sigmoid cancer. World J. Surg. Oncol. 5, 102. doi: 10.1186/1477-7819-5-102
Churg, A. (1998). “Nonneoplastic diseases caused by asbestos,” in Pathology of Occupational Lung Disease, 2nd Edn., eds A. Churg and F. H. Y. Green (Baltimore: Williams and Wilkins), 277–339.
Coffin, C. M., and Dehner, L. P. (1992). Mesothelial and related neoplasms in children and adolescents: a clinicopathologic and immunohistochemical analysis of eight cases. Pediatr. Pathol. 12, 333–347. doi: 10.3109/15513819209023314
Colgan, T. J., Boerner, S. L., Murphy, J., Cole, D. E., Narod, S., and Rosen, B. (2002). Peritoneal lavage cytology: an assessment of its value during prophylactic oophorectomy. Gynecol. Oncol. 85, 397–403. doi: 10.1006/gyno.2002.6638
Colgan, T. J., Murphy, J., Cole, D. E., Narod, S., and Rosen, B. (2001). Occult carcinoma in Prophylactic Oophorectomy specimens: prevalence and association with BRCA germline mutation status. Am. J. Surg. Pathol. 25, 1283–1289. doi: 10.1097/00000478-200110000-00009
Colwell, A. S., O'Cunha, J., Vargas, S. O., Parker, B., Dal Cin, P., and Maddaus, M. A. (2002). Synovial sarcoma of the pleura: a clinical and pathologic study of three cases. J. Thoracic Cardiovasc. Surg. 124, 828–832. doi: 10.1067/mtc.2002.124242
Cordon-Cardo, C. (1997). Molecular biology of sarcomas, In: Cancer, principles and practice of oncology, 7th Edn., eds V. T. DeVita, S. Hellman, and S. A. Rosenberg (Philadelphia, PA: Lippincott-Raven), 1731–1737.
Cormio, G., Rossi, C., Cazzolla, A., Resta, L., Loverro, G., Greco, P., et al. (2003). Distant metastases in ovarian carcinoma. Int. J. Gynecol. Cancer 13, 125–129. doi: 10.1046/j.1525-1438.2003.13054.x
Cramer, S. F., Roth, L. M., Ulbright, T. M., Mazur, M. T., Nunez, C. A., Gersell, D. J., et al. (1987). Evaluation of the reproducibility of the World Health Organization classification of common ovarian cancers. Arch. Pathol. Lab. Med. 111, 819–829.
Crum, C. P., Drapkin, R., Kindelberger, D., Medeiros, F., Miron, A., and Lee, Y. (2007). Lessons from BRCA: the tubal fimbria emerges as an origin for pelvic serous cancer. Clin. Med. Res. 5, 35–44. doi: 10.3121/cmr.2007.702
Dhaene, K., Hubner, R., Kumar-Singh, S., Weyn, B., and Van Marck, E. (1998). Telomerase activity in human pleural mesothelioma. Thorax 53, 195–918. doi: 10.1136/thx.53.11.915
Dopp, E., Saedler, J., Stopper, H., Weiss, D. G., and Schiffmann, D. (1995). Mitotic disturbances and micronucleus induction in Syrian hamster embryo fibroblast cells caused by asbestos fibers. Environ. Health Perspect. 103, 268–271.
Doval, D. C., Pande, S. B., Sharma, J. B., Rao, S. A., Prakash, N., and Vaid, A. K. (2007). Report of a case of pericardial mesothelioma with liver metastases responding well to pemetrexed and platinum-based chemotherapy. J. Thorac. Oncol. 2, 780–781. doi: 10.1097/JTO.0b013e31811f3acd
Durant, S. T. (2012). Telomerase-independent paths to immortality in predictable cancer subtypes. J. Cancer 2012, 3, 67–82. doi: 10.7150/jca.3965
Eilber, F. C., and Dry, S. M. (2008). Diagnosis and management of synovial sarcoma. J. Surg. Oncol. 97, 314–320. doi: 10.1002/jso.20974
Erdogan, E., Demirkazik, F. B., Gulsun, M., Ariyurek, M., Emri, S., and Sak, S. D. (2005). Incidental localized (solitary) mediastinal malignant mesothelioma. Br. J. Radiol. 78, 858–861. doi: 10.1259/bjr/19513813
Eren, N. T., and Akar, A. R. (2002). Primary pericardial mesothelioma. Curr. Treat. Options Oncol. 3, 369–373. doi: 10.1007/s11864-002-0002-7
Finch, A., Shaw, P., Rosen, B., Murphy, J., Narod, S. A., and Colgan, T. J. (2006). Clinical and pathologic findings of prophylactic salpingo-oophorectomies in 159 BRCA1 and BRCA2 carriers. Gynecol. Oncol. 100, 58–64. doi: 10.1016/j.ygyno.2005.06.065
Fox, H. (1993). Primary neoplasia of the female peritoneum. Histopathol. 23, 103–110. doi: 10.1111/j.1365-2559.1993.tb00467.x
Fraire, A. E., Cooper, S., Greenberg, S. D., Buffler, P., and Langston, C. (1988). Mesothelioma of childhood. Cancer 62, 838–847. doi: 10.1002/1097-0142(19880815)62:4<838::AID-CNCR2820620433>3.0.CO;2-9
Fujiwara, H., Kamimori, T., Morinaga, K., Takeda, Y., Kohyama, N., Miki, Y., et al. (2005). An autopsy case of primary pericardial mesothelioma in arc cutter exposed to asbestos through talc pencils. Indust. Health 43, 346–350. doi: 10.2486/indhealth.43.346
Gaertner, E., Zeren, E. H., Fleming, M. V., Colby, T. V., and Travis, W. D. (1996). Biphasic synovial sarcoma arising in the pleural cavity. A clinicopathologic study of five cases. Am. J. Surg. Pathol. 20, 36–45. doi: 10.1097/00000478-199601000-00004
Galetta, D., Pelosi, G., Leo, F., Solli, P., Veronesi, G., Borri, A., et al. (2007). Primary thoracic synovial sarcoma: factors affecting long-term survival. J. Thoracic Cardiovasc. Surg. 134, 808–809. doi: 10.1016/j.jtcvs.2007.05.036
Geisler, J. P., Hatterman-Zogg, M. A., Rathe, J. A., and Buller, R. E. (2002). Frequency of BRCA1 dysfunction in ovarian cancer. J. Natl. Cancer. Inst. 94, 61–67. doi: 10.1093/jnci/94.1.61
Gibbs, G. W., and Berry, G. (2008). Mesothelioma and asbestos. Regul. Toxicol. Pharmacol. 52, S223–S231. doi: 10.1016/j.yrtph.2007.10.003
Gilks, C. B., Ionescu, D. N., Kalloger, S. E., Kobel, M., Irving, J., Clarke, B., et al. (2008). Tumor cell type can be reproducibly diagnosed and is of independent prognostic significance in patients with maximally debulked ovarian carcinoma. Human Pathol. 39, 1239–1251. doi: 10.1016/j.humpath.2008.01.003
Gladish, G. W., Sabloff, B. M., Munden, R. F., Truong, M. T., Erasmus, J. J., and Chasen, M. H. (2002). Primary thoracic sarcomas. Radiographics 22, 621–637. doi: 10.1148/radiographics.22.3.g02ma17621
Gocha, A. R. S., Harris, J., and Groden, J. (2013). Alternative mechanisms of telomere lengthening: permissive mutations, DNA repair proteins, and tumorigenic progression. Mutat. Res. 743–744, 142–150. doi: 10.1016/j.mrfmmm.2012.11.006
Gofman, A., Issakov, J., Kollender, Y., Soyfer, V., Dadia, S., Jiveliouk, I., et al. (2007). Synovial sarcoma of the extremities and trunk: a long-lasting disease. Oncol. Rep. 18, 1577–1581. doi: 10.3892/or.18.6.1577
Goodman, M., Teta, M. J., Hessel, P. A., Garabrant, D. H., Craven, V. A., Scrafford, C. G., et al. (2004). Mesothelioma and lung cancer among motor vehicle mechanics: a meta analysis. Ann. Occup. Hyg. 48, 309–326. doi: 10.1093/annhyg/meh022
Goyal, R. K. (2005). “Diseases of the esophagus,” in Harrison's Principles of Internal Medicine, 16th Edn., eds D. L. Kasper (McGraw-Hill, New York), 1739–1762.
Hankinson, S. E., and Danforth, K. N. (2006). “Ovarian Cancer,” in Cancer Epidemiology and Prevention, 3rd Edn., eds D. Schottenfeld and J. F. Fraumeni, Jr (New York, NY: Oxford University Press). 1013–1026. doi: 10.1093/acprof:oso/9780195149616.003.0052
Harding, A. H., and Darnton, A. J. (2010). Asbetsosis and mesothelioma among British asbestos workers (1971–2005). Am. J. Ind. Med. 53, 1070–1080. doi: 10.1002/ajim.20844
Hawley, A. T., and Pandolfi, P. P. (2008). “Etiology of cancer: cancer susceptibility syndromes,” in Cancer, Principles and Practices of Oncology, 8th Edn., eds. V. T.DeVita, T. S. Lawrence, S. A., and Rosenberg (Philadelphia; Lippincott, Williams and Wilkins), 157–168.
Heaphy, C. M., de Wilde, R. F., Jiao, Y., Klein, A. P., Edil, B. H., and Shi, C. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science 333:425. doi: 10.1126/science.1207313
Hemminki, K., and Li, X. (2003). Time trends and occupational risk factors for peritoneal mesothelioma in Sweden. J. Occup. Environ. Med. 45, 451–455. doi: 10.1097/01.jom.0000052960.59271.d4
Hernandez, E., Bhagavan, B. S., Parmley, T. H., and Rosenshein, N. B. (1984). Interobserver variability in the interpretation of epithelial ovarian cancer. Gynecol. Oncol. 17, 117–123. doi: 10.1016/0090-8258(84)90065-9
Hillard, A. K., Lovett, J. K., and McGavin, C. R. (2003). The rise and fall in incidence of malignant mesothelioma from a British naval dockyards, 1979–1999. Occup. Med 53, 209–212. doi: 10.1093/occmed/kqg051
Hilton, J. L., Geisler, J. P., Rathe, J. A., Hattermann-Zogg, M. A., DeYoung, B., and Buller, R. E. (2002). Inactivation of BRCA1 and BRCA2 in ovarian cancer. J. Natl. Cancer Inst. 94, 1396–1406. doi: 10.1093/jnci/94.18.1396
Hirano, H., Maeda, T., Tsuji, M., Ito, Y., Kizaki, T., Yoshii, Y., et al. (2002). Malignant mesothelioma of the pericardium: case reports and immunohistochemical studies including Ki-67 expression. Pathol. Int. 52, 669–676. doi: 10.1046/j.1440-1827.2002.01404.x
Hodgson, J. T., and Darnton, A. (2000). The quantitative risks of mesothelioma and lung cancer in relation to asbestos exposure. Ann. Occup. Hyg. 44, 565–601. doi: 10.1093/annhyg/44.8.565
Hodgson, J. T., and Darnton, A. (2010). Mesothelioma from chrysotile. Occup. Environ. Med. 67, 432. doi: 10.1136/oem.2009.052860
Hodgson, J. T., McElvenny, D. M., Darnton, A. J., Price, M. J., and Peto, J. (2005). The expected burden of mesothelioma mortality in Great Britian from 2002 to 2050. Brit. J. Cancer 92, 587–593. doi: 10.1038/sj.bjc.6602307
Hosono, T., Hironaka, M., Kobayashi, A., Yamasawa, H., Bando, M., Ohno, S., et al. (2005). Primary pulmonary synovial sarcoma confirmed by molecular detection of SYT-SSX1 fusion gene transcripts: a case report and review of the literature. Jpn. J. Clin. Oncol. 35, 274–279. doi: 10.1093/jjco/hyi073
Hu, J., Hwang, S. S., Liesa, M., Gan, B., Sahin, E., Jasekiloff, M., et al (2012). Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 148, 651–663. doi: 10.1016/j.cell.2011.12.028
Hubbard, R. (1997). The aetiology of mesothelioma: are risk factors other than asbestos exposure important? Thorax 52, 496–497. doi: 10.1136/thx.52.6.496
Ivanov, S. V., Goparaju, C. M. V., Lopez, P., Zavadil, J., Toren-Haritan, G., Rosenwald, S., et al. (2010). Pro-tumorigenic effects of miR-31 loss in mesothelioma. J. Biol. Chem. 285, 22809–22817. doi: 10.1074/jbc.M110.100354
Iwata, T., Nishiyama, N., Izumi, N., Tsukioka, T., and Suehiro, S. (2007). Metastatic monophasic synovial sarcoma of the pleura. Ann. Thoracic Cardiovasc. Surg. 13, 258–261.
Jasani, B., and Gibbs, A. (2012). Mesothelioma no associated with asbestos exposure. Arch. Pathol. Lab. Med. 136, 262–267. doi: 10.5858/arpa.2011-0039-RA
Jean, D., Daubriac, J., Le Pimpec-Barthes, F., Galateau-Salle, F., and Jaurand, M-C. (2012). Molecular changes in mesothelioma with an impact on prognosis and treatment. Arch. Pathol. Lab. Med. 136, 277–293. doi: 10.5858/arpa.2011-0215-RA
Kahn, E. I., Rohl, A., Barrett, E. W., Suzuki, Y., and Suzuki, Y. (1980). Primary pericardial mesothelioma following exposure to asbestos. Environ. Res. 23, 270–281. doi: 10.1016/0013-9351(80)90061-4
Kannerstein, M., Churg, J., McCaughey, W. T., and Hill, D. P. (1977). Papillary tumors of the peritoneum in women: mesothelioma or papillary carcinoma. Am. J. Obstet. Gynecol. 127, 306–314.
Katabuchi, H., and Okamura, H. (2003). Cell biology of human ovarian surface epithelial cells and ovarian carcinogenesis. Med. Electron Microsc. 36, 74–86.
Kindelberger, D. W., Lee, Y., Miron, A., Hirsch, M. S., Feltmate, C., Medeiros, F., et al. (2007). Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am. J. Surg. Pathol. 31, 161–169. doi: 10.1097/01.pas.0000213335.40358.47
Kobel, M., Kalloger, S. E., Boyd, N., McKinney, S., Mehl, E., Palmer, C., et al. (2008). Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 5:e232. doi: 10.1371/journal.pmed.0050232
Kolschmann, S., Ballin, A., and Gillissen, A. (2005). Thoracoscopic talc pleurodesis in malignant pleural effusions. Chest 128, 1431–1437. doi: 10.1378/chest.128.3.1431
Kumar, V., Abbas, A. K., and Fausto, N. (2005a). “Germ cell tumors,” in Robbins and Cotran Pathologic Basis of Disease, 7th Edn., eds V. Kumar, A. K. Abbas, and N. Fausto (Philadelphia: Elsevier Saunders), 1040–1048.
Kumar, V., Abbas, A. K., and Fausto, N. (2005b). “Congenital heart defects,” in Robbins and Cotran Pathologic Basis of Disease, 7th Edn., eds V. Kumar, A. K. Abbas, and N. Fausto (Philadelphia: Elsevier Saunders), 564–571.
Kumar, V., Abbas, A. K., and Fausto, N. (2005c). “Synovial sarcoma,” in Robbins and Cotran Pathologic Basis of Disease, 7th Edn., eds V. Kumar, A. K. Abbas, and N. Fausto (Philadelphia: Elsevier Saunders), 1323–1324.
Laden, F., Stampfer, M. J., and Walker, A. M. (2004). Lung cancer and mesothelioma among male automobile mechanics: a review. Rev. Environ. Health 19, 39–61. doi: 10.1515/REVEH.2004.19.1.39
Lagrotteria, D. D., Tsang, B., Elavathil, L. J., and Tomlinson, C. W. (2005). A case of primary malignant pericardial mesothelioma. Can. J. Cardiol. 21, 185–187.
Larson, T., Melinkova, N., Davis, S. I., and Jamison, P. (2007). Incidence and descriptive epidemiology of mesothelioma in the United states, 1999–2002. Int. J. Occup. Environ. Health 13, 398–403. doi: 10.1179/oeh.2007.13.4.398
Lauchlan, S. C. (1972). The secondary mullerian system. Obstet. Gynecol. Surv. 27, 133–146. doi: 10.1097/00006254-197203000-00001
Lee, H. K., Kwon, H. J., Lee, H. B., Jin, G. Y., Chung, M. J., and Lee, Y. C. (2006). Radiofrequency thermal ablation of primary pleural synovial sarcoma. Respiration 73, 250–252. doi: 10.1159/000087153
Lee, Y., Miron, A., Drapkin, R., Nucci, M. R., Medeiros, F., Saleemuddin, A., et al. (2007). A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 211, 26–35. doi: 10.1002/path.2091
Leigh, J. (2003). Letter to USEPA from J. Leigh with Subject: 1986 EPA “Gold Book” Guidance for Preventing Asbestos-Related Disease Among Auto Mechanics. Request for correction, available at #12, University of Sydney.
Lemmon, M. A., and Schlessinger, J. (2010). Cell signaling by receptor tyrosine kinases. Cell 14, 1117–1134. doi: 10.1016/j.cell.2010.06.011
Levine, D. A., Argenta, P. A., Yee, C. J., Marshall, D. S., Olvera, N., Bogomolniy, F., et al. (2003). Fallopian tube and primary peritoneal carcinomas associated with BRCA mutations. J. Clin. Oncol. 21, 4222–4227. doi: 10.1200/JCO.2003.04.131
Luk, A., Ahn, E., Vaideeswar, P., and Butany, J. W. (2008). Pericardial tumors. Semin. Diagnos. Pathol. 25, 47–53. doi: 10.1053/j.semdp.2007.12.001
Lund, B., Thomsen, H. K., and Olsen, J. (1991). Reproducibility of histopathological evaluation in epithelial ovarian carcinoma. Clinical implications. APMIS 99, 353–358. doi: 10.1111/j.1699-0463.1991.tb05161.x
Magnani, C., Ferrante, D., Barone-Adesi, F., Bertolotti, M., Mirabelli, D., and Terracini, B. (2007). Cancer risk after cessation of asbestos exposure, a cohort study of Italian asbestos cement workers. Occup. Environ. Med. 31, 16–22. doi: 10.1136/oem.2007.032847
Makhlouf, H. R., Ahrens, W., Agarwal, B., Dow, N., Marshalleck, J. J., Lee, E. L., et al. (2008). Synovial sarcoma of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 10 cases. Am. J. Surg. Pathol. 37, 275–281. doi: 10.1097/PAS.0b013e31812e6a58
Martini, V., Michelazzi, L., Cioe, A., Fucile, C., Spigno, F., and Robbiano, L. (2011). Exposure to asbestos: correlation between blood levels of mesothelin and frequency of micronuclei in peripheral blood lymphocytes. Mutat. Res. 721, 114–117. doi: 10.1016/j.mrgentox.2010.12.014
Maruyama, R., Sakai, M., Nakamura, T., Suemitsu, R., Okamoto, T., Wataya, H., et al. (2006). Triplet chemotherapy for malignant pericardial mesothelioma: a case report. Jpn. J. Clin. Oncol. 36, 245–248. doi: 10.1093/jjco/hyl003
Mayer, R. L. (2005). “Gastrointestinal tract cancer,” in Harrison's Principles of Internal Medicine, 16th Edn., eds D. L. Kasper (New York, NY: McGraw-Hill), 523–533.
McCoubrey, A., Houghton, O., McCallion, K., and McCluggage, W. G. (2005). Serous adenocarcinoma of the sigmoid mesentery arising in cystic endosalpingiosis. J. Clin. Pathol. 58, 1221–1223. doi: 10.1136/jcp.2005.027946
Medeiros, F., Muto, M. G., Lee, Y., Elvin, J. A., Callahan, M. J., Feltmate, C., et al. (2006). The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am. J. Surg. Pathol. 30, 230–236. doi: 10.1097/01.pas.0000180854.28831.77
Michal, M., Fanburg-Smith, J. C., Lasota, J., Fetsch, J. F., Lichy, J., and Mettinen, M. (2006). Minute synovial sarcomas of the hands and feet: a clinicopathologic study of 21 tumors less than 1 cm. Am. J. Surg. Pathol. 30, 721–726. doi: 10.1097/00000478-200606000-00007
Miettinen, M., Limon, J., Niezabitowski, A., and Lasota, J. (2001). Calretinin and other mesothelioma markers in synovial sarcoma: analysis of antigentic similarities and differences with malignant mesothelioma. Am. J. Surg. Pathol. 25, 610–617. doi: 10.1097/00000478-200105000-00007
Mizell, K. N., Morris, C. G., and Carter, J. E. (2009). Antemortem diagnosis of asbestosis by screening chest radiograph correlated with postmortem histologic features of asbestosis: a study of 273 cases. J. Occup. Med. Toxicol. 4:14. doi: 10.1186/1745-6673-4-14
Molina Garrido, M. J., Rufete, A. M., Rodriguez-Lescure, A., Cascón Pérez, J. D., Guillén Ponce, C., and Carrato Mena, A. (2006). Recurrent pericardial effusion as initial manifestation of primary diffuse pericardial malignant mesothelioma. Clin. Transl. Oncol. 8, 694–696. doi: 10.1007/s12094-006-0042-8
Moolgavkar, S. H., Mesa, R., and Turim, J. (2009). Pleural and peritoneal mesotheliomas in SEER: age effects and temporal trends, 1973–2005. Cancer Causes Control 20, 935–944. doi: 10.1007/s10552-009-9328-9
Moran, C. A., Suster, S., and Koss, M. N. (1992). The spectrum of histologic growth patterns in benign and malignant fibrous tumors of the pleura. Semin. Diagnost. Pathol. 9, 169–180.
Morin, P. J., Trent, J. M., Collins, F. S., and Vogelstein, B. (2005). “Cancer genetics,” in Harrison's Principles of Internal Medicine, 16th Edn., eds D. L. Kasper et al. (New York, NY: McGraw-Hill), 447–453.
Motzer, R. J., and Bosl, G. J. (2005). “Testicular Cancer,” in Harrison's Principles of Internal Medicine, 16th Edn., eds D. L. Kasper, E. Braunwald, S. Hauser, D. Longo, J. L. Jameson, and A. S. Fauci (New York, NY: McGraw-Hill), 550–553.
Mukhopadhyay, S., and Aubry, M. C. (2007). Recurrent primary synovial sarcoma of the chest wall. J. Thorac. Oncol. 2, 660–661. doi: 10.1097/JTO.0b013e31807a2f99
Muto, M. G. (2005). “The patient at risk of ovarian cancer,” in Diagnostic gynecologic and Obstetric Pathology, eds C. P. Crum and K. R. Lee (New York, NY: Elsevier Saunders), 811–819.
Neri, M., Filiberti, R., Taioli, E., Garte, S., Paracchini, V., Bolognesi, C., et al. (2005). Pleural malignant mesothelioma, genetic susceptibility and asbestos exposure. Mutat. Res. 592, 36–44. doi: 10.1016/j.mrfmmm.2005.06.003
Neumann, V., Gunther, S., Muller, K.-M., and Fischer, M. (2001). Malignant mesothelioma—German mesothelioma register 1987–1999. Int. Arch. Occup. Environ. Health 74, 383–395. doi: 10.1007/s004200100240
Nicholson, A. G., Goldstraw, P., and Fisher, C. (1998). Synovial sarcoma of the pleura and its differentiation from other primary pleural tumours: a clinicopathological and imuunohistochemical review of three cases. Histopathology 33, 508–513. doi: 10.1046/j.1365-2559.1998.00565.x
Niggli, F. K., Gray, T. J., Raafat, F., and Stevens, M. C. (1994). Spectrum of peritoneal mesothelioma in childhood: clinical and histopathologic features, including DNA cytometery. Pediatr. Hematol. Oncol. 11, 399–408. doi: 10.3109/08880019409140539
Ordonez, N. G. (1998). In search of a positive immunohistochemical marker for mesothelioma: an update. Adv. Anat. Pathol. 5, 53–60. doi: 10.1097/00125480-199801000-00051
Paley, P. J., Swisher, E. M., Garcia, R. L., Agoff, S. N., Greer, B. E., et al. (2001). Occult cancer of the fallopian tube in BRCA-1 germline mutation carriers at prophylactic oophorectomy: a case for recommending hysterectomy at surgical prophylaxis. Gynecol. Oncol. 80, 176–180. doi: 10.1006/gyno.2000.6071
Papi, M., Genestreti, G., Tassinari, D., Lorenzini, P., Serra, S., Ricci, M., et al. (2005). Malignant pericardial mesothelioma. Report of two cases, review of the literature and differential diagnosis. Tumori 91, 276–279.
Parmley, T. H., and Woodruff, J. D. (1974). The ovarian mesothelioma. Am. J. Obstet. Gynecol. 120, 234–241.
Pass, H. I., Hahn, S. M., Vogelzang, N. J., and Carbone, M. (2005). “Benign and malignant mesothelioma,” in Cancer, Principles and Practice of Oncology, 8th Edn., eds V. T. DeVita, T. S. Lawrence, and S. A. Rosenberg (Philadelphia: Lippincottt, Williams and Wilkins), 1687–1750. doi: 10.1007/0-387-28274-2
Pass, H. I., Vogelzang, N. T., Hahn, S. M., and Carbone, M. (2008). “Benign and malignant mesothelioma,” in Cancer, Principles And Practice of Oncology, 8th Edn., eds. V. T. DeVita, T. S.Lawrence, and S. A.Rosenberg (Philadelphia: Lippincottt, Williams and Wilkins), 1835–1862.
Piek, J. M., Kenemans, P., and Verheijen, R. H. M. (2004). Intraperitoneal serous adenocarcinoma: a critical appraisal of three hypotheses on its cause. Am. J. Obstet. Gynecol. 191, 718–732. doi: 10.1016/j.ajog.2004.02.067
Piek, J. M., Torrenga, B., Hermsen, B., Verheijen, R. H., Zweemer, R. P., Gille, J. J., et al. (2003). Histopathological characteristics of BRCA1- and BRCA2—associated intraperitoneal cancer: a clinic—based study. Fam. Cancer 2, 73–78. doi: 10.1023/A:1025700807451
Pisick, E., and Salgia, R. (2005). Molecular biology of malignant mesothelioma: a review. Hematol. Oncol. Clin. North Am. 19, 997–1023. doi: 10.1016/j.hoc.2005.09.012
Piura, B., Rabinovich, A., and Yanai-Inbar, I. (2001). Three primary malignancies related to BRCA mutation successively occurring in a BRCA1 185delAG mutation carrier. Eur. J. Obstet. Oncol. Gynecol. Reprd. Biol. 97, 241–244. doi: 10.1016/S0301-2115(00)00521-2
Powell, C. B., Kenley, E., Chen, L. M., Crawford, B., McLennan, J., Zaloudek, C., et al. (2005). Risk—reducing Salpingo—Oophorectomy in BRCA mutation carriers: role of serial sectioning in the detection of occult malignancy. J. Clin. Oncol. 23, 127–132. doi: 10.1200/JCO.2005.04.109
Price, B., and Ware, A. (2004). Mesothelioma trends in the United States: an update based on surveillance, epidemiology, and end results program data for 1973 through 2003. Am. J. Epidemiol. 159, 107–112. doi: 10.1093/aje/kwh025
Quinn, D. W., Qureshi, F., and Mitchell, I. M. (2000). Pericardial mesothelioma: the diagnostic dilemma of misleading images. Ann. Thoracic Surg. 69, 1926–1927. doi: 10.1016/S0003-4975(00)01204-2
Raju, U., Fine, G., Greenawald, K. A., and Ohorodnik, J. M. (1989). Primary papillary serous neoplasia of the peritoneum: a clinicopathologic and ultrastructural study of eight cases. Human Pathol. 20, 426–436. doi: 10.1016/0046-8177(89)90006-3
Rdzanek, M., Fresco, R., Pass, H. I., and Carbone, M. (2006). Spindle cell tumors of the pleura: differential diagnosis. Semin. Diagnost. Pathol. 23, 44–55. doi: 10.1053/j.semdp.2006.06.002
Reid, A., de Klerk, N., Ambrosini, G., Olsen, N., Pang, S. C., and Musk, A. W. (2005). The additional risk of malignant mesothelioma in former workers and residents of Witenoom with benign pleural disease or asbestosis. Occup. Environ. Med 62, 665–669. doi: 10.1136/oem.2004.018531
Reis-Filho, J. S., Pope, L. Z. B., Milanezi, F., Balderrama, C. M., Serapião, M. J., and Schmitt, F. C. (2002). Primary epithelial malignant mesothelioma of the pericardium with deciduoid features: cytohistologic and immunohistochemical. Diagnost. Cytopathol. 26, 117–122. doi: 10.1002/dc.10068
Risch, H. A., McLaughlin, J. R., Cole, D. E., Rosen, B., Bradley, L., Kwan, E., et al. (2001). Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am. J. Hum. Genet. 68, 700–710. doi: 10.1086/318787
Robinson, B. W. S., Creaney, J., Lake, R., Nowak, A., Musk, A. W., de Klerk, N., et al. (2003). Mesothelin-family proteins and diagnosis of mesothelioma. Lancet 362, 1612–1616. doi: 10.1016/S0140-6736(03)14794-0
Sakamoto, A., Sasaki, H., Furusato, M., Suzuki, M., Hirai, Y., Tsugane, S., et al. (1994). Observer disagreement in histological classification of ovarian tumors in Japan. Gynecol. Oncol. 54, 54-58. doi: 10.1006/gyno.1994.1165
Sandberg, A. A. (2002). Cytogenetics and molecular genetics of bone and soft tissue tumors. Am. J. Med. Genet. 115, 189–193. doi: 10.1002/ajmg.10691
Santos, C., Montesinos, J., Castaner, E., Sole, J. M., and Baga, R. (2008). Primary pericardial mesothelioma. Lung Cancer 60, 291–293. doi: 10.1016/j.lungcan.2007.08.029
Sarma, A. V., McLaughlin, J. C., and Schottenfeld, D. (2006). “Testicular cancer,” in Cancer Epidemiology and Prevention, 3rd Edn., eds D. Schottenfeld and J. F. Fraumeni (New York, NY: Oxford University Press), 1151–1165. doi: 10.1093/acprof:oso/9780195149616.003.0060
Satoh, H., Ohara, G., and Hizawa, N. (2007). Primary synovial sarcoma of the chest wall. J. Thorac. Oncol. 2:1060. doi: 10.1097/JTO.0b013e318158ef37
Scherpereel, A., Grigoriu, B., Conti, M., Gey, T., Gregoire, M., Copin, M.-C, et al. (2006). Soluble mesothelin-related peptides in the diagnosis of malignant pleural mesothelioma. Am. J. Respir. Crit. Care Med. 173, 1155–1160. doi: 10.1164/rccm.200511-1789OC
Schorge, J. O., Miller, Y. B., Qi, L. J., Muto, M. G., Welch, W. R., Berkowitz, R. S., et al. (2000). Genetic alterations of the WT1 gene in papillary serous carcinoma of the peritoneum. Gynecol. Oncol. 76, 369–372. doi: 10.1006/gyno.1999.5711
Shimizu, M., Toki, T., Takagi, Y., Konishi, I., and Fuji, S. (2000). Immunohistochemical detection of the Wilms' tumor gene (WT1) in epithelial ovarian tumors. Int. J. Gynecol. Pathol. 19, 158–163. doi: 10.1097/00004347-200004000-00010
Shiraki, M., Enterline, H. T., Brooks, J. J., Cooper, N. S., Hirschi, S., Roth, J. A., et al. (1989). Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 64, 484–490. doi: 10.1002/1097-0142(19890715)64:2<484::AID-CNCR2820640223>3.0.CO;2-T
Sichletidis, L., Chloros, D., Spyratos, D., Haidich, A. B., Fourkiotou, I., Kakoura, M., et al. (2008). Mortality from occupational exposure to relatively pure chrysotile: a 39-year study. Respiration 78, 63–68. doi: 10.1159/000163443
Siegel, H. J., Sessions, W., Casillas, M. A., Said-Al-Naief, N., Lander, P. H., and Lopez-Ben, R. (2007). Synovial sarcoma: clinicopathologic features, treatment, and prognosis. Orthopedics 30, 1020–1025. Available online at: http://www.healio.com/orthopedics/oncology/journals/ortho/%7Bcfaffd56-2438-49b0-918e-551612fc369a%7D/synovial-sarcoma-clinicopathologic-features-treatment-and-prognosis
Sobol, H., Jacquemier, J., Bonaiti, C., Dauplat, J., Birnbaum, D., and Eisinger, F. (2000). Fallopian tube cancer as a feature of BRCA1-associated syndromes. Gynecol. Oncol. 78, 263–266. doi: 10.1006/gyno.2000.5897
Sporn, T. A., and Roggli, V. L. (2004). “Mesothelioma,” in Pathology of Asbestos-Associated Diseases, 2nd Edn., eds. V. L. Roggli, T. D. Oury, and T. A.Sporn (New York, NY: Springer), 104–168. doi: 10.1007/0-387-21819-X_5
Struewing, J. P., Harge, P., Wacholder, S., Baker, S. M., Berlin, M., McAdams, M., et al. (1997). The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. New Eng. J. Med. 336, 1401–1408. doi: 10.1056/NEJM199705153362001
Susman, S., Schofield, P., and Large, S. (2004). Primary pericardial mesothelioma presenting as pericardial constriction: a case report. Heart 90:e4. doi: 10.1136/heart.90.1.e4
Tallet, A., Nault, J-C., Renier, A., Hysi, I., Galateau-Salle, F., Cazes, A., et al. (2013). Overexpression and promoter mutation of the TERT gene in malignant pleural mesothelioma. Oncogene. doi: 10.1038/onc.2013.351. [Epub ahead of print].
Taylor, C. A., Barnhart, A., Pettenati, M. J., and Geisinger, K. R. (2005). Primary pleuropulmonary synovial sarcoma diagnosed by fine needle aspiration with cytogenetic confirmation: a case report. Acta Cytol. 49, 673–676. doi: 10.1159/000326260
Testa, J. R., Cheung, M., Pei, J., Below, J. E., Tan, Y., Sementino, E., et al. (2011). Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genetics 43, 1022–1026. doi: 10.1038/ng.912
Teta, M. J., Mink, P. J., Lau, E., Sceurman, B. K., and Foster, E. D. (2008). U.S. mesothelioma patterns 1973–2002: indicators of change and insights into background rates. Eur. J. Cancer Prev. 17, 525–534. doi: 10.1097/CEJ.0b013e3282f0c0a2
Tewari, S. C., Kurian, G., Jayaswal, R., Chakravorty, S., Chadha, S. K., and Chauhan, M. S. (1989). Malignant mesothelioma in the young (with prosthetic aortic valve, an unusual association). J. Assoc. Phys. India 37, 187–189.
Thomason, R., Schlegel, W., Lucca, M., Cummings, S., and Lee, S. (1994). Primary malignant mesothelioma of the pericardium. Case report and literature review. Tex. Heart Inst. J. 21, 170–174.
Tsukioka, T., Inoue, K., Iwata, T., Mizuguchi, S., Morita, R., and Suehiro, S. (2006). Resected case of synovial sarcoma in the pleural cavity. Jpn. J. Thorac. Cardiovasc. Surg. 54, 263–266. doi: 10.1007/PL00022249
Val-Bernal, J. F., Figols, J., and Gomez-Roman, J. J. (2002). Incidental localized (solitary) epithelial mesothelioma of the pericardium case report and literature review. Cardiovasc. Pathol. 11, 181–185. doi: 10.1016/S1054-8807(02)00097-2
Vera, J., Garcia, M. D., Marigil, M., Abascal, M., Lopez, J. I., and Ligorred, L. (2006). Biphasic synovial sarcoma of the abdominal wall. Virchows Arch. 449, 367–372. doi: 10.1007/s00428-005-0076-2
Vohra, H. A., Davies, S., Vohra, H., Rosin, M. D., and Snead, D. R. (2004). Primary synovial sarcoma of the pleura: beware of misdiagnosis. Eur. J. Intern. Med. 15, 465–466. doi: 10.1016/j.ejim.2004.08.004
Vornicu, M., Arora, S., and Achilleos, A. (2007). Primary pericardial mesothelioma: a rare cardiac malignancy. Intern. Med. J. 37, 576–577. doi: 10.1111/j.1445-5994.2007.01409.x
Weill, H., Hughes, J. M., and Churg, A. M. (2004). Changing trends in US mesothelioma incidence. Occup. Environ. Med. 61, 438–441. doi: 10.1136/oem.2003.010165
Weinbreck, N., Vignaud, J. M., Begueret, H., Burke, L., Benhattar, J., Guillou, L., et al. (2007). SYT-SSX fusion is absent in sarcomatoid mesothelioma allowing its distinction from synovial sarcoma of the pleura. Modern Pathol. 20, 617–621. doi: 10.1038/modpathol.3800775
Werness, B. A., Ramus, S. J., Whittemore, A. S., Garlinghouse-Jones, K., Oakley-Girvan, I., Dicioccio, R. A., et al. (2000). Histopathology of familial ovarian tumors in women from families with and without germline BRCA1 mutations. Hum. Pathol. 31, 1420–1424. doi: 10.1016/S0046-8177(00)80014-3
Winston, C. B., Hadar, O., Teitcher, J. B., Caravelli, J. F., Sklarin, N. T., Panicek, D. M., et al. (2000). Metastatic lobular carcinoma of the breast: patterns of spread in the chest, abdomen, and pelvis on CT. Am. J. Roentgenol. 175, 795–800. doi: 10.2214/ajr.175.3.1750795
Yakirevich, E., Sova, Y., Drumea, K., Bergman, I., Quitt, M., and Resnick, M. B. (2004). Peripheral lymphadenopathy as the initial manifestation of pericardial mesothelioma: a case report. Int. J. Surg. Pathol. 12, 403–405. doi: 10.1177/106689690401200415
Yarborough, C. M. (2006). Chrysotile as a cause of mesothelioma: an assessment based on epidemiology. Off. Rev. Toxicol. 36, 165–187. doi: 10.1080/10408440500534248
Yarborough, C. M. (2007). The risk of mesothelioma from exposure to chrysotile asbestos. Curr. Opin. Pulm. Med. 13, 334–338. doi: 10.1097/MCP.0b013e328121446c
Keywords: germ cell tumors, synovial sarcoma, pericardial mesothelioma, mullerian tissue cancers, chromosomal instability, human, asbestos
Citation: Kerger BD, James RC and Galbraith DA (2014) Tumors that mimic asbestos-related mesothelioma: time to consider a genetics-based tumor registry? Front. Genet. 5:151. doi: 10.3389/fgene.2014.00151
Received: 03 February 2014; Accepted: 08 May 2014;
Published online: 30 May 2014.
Edited by:
David Azorsa, Ron Matricaria Institute of Molecular Medicine at Phoenix Children's Hospital, USAReviewed by:
Paola Parrella, IRCCS Casa Sollievo Della Sofferenza, ItalyParvin Mehdipour, Tehran University of Medical Sciences, Iran
Copyright © 2014 Kerger, James and Galbraith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brent D. Kerger, Cardno ChemRisk LLC, 130 Vantis, Suite 170, Aliso Viejo, CA 92656, USA e-mail:YnJlbnQua2VyZ2VyQGNhcmRuby5jb20=