
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Ecol. Evol. , 22 March 2021
Sec. Evolutionary and Population Genetics
Volume 9 - 2021 | https://doi.org/10.3389/fevo.2021.646130
This article is part of the Research Topic Life and Death: New Perspectives and Applications in Forensic Science View all 13 articles
 Deidra Jordan1,2
Deidra Jordan1,2 DeEtta Mills1,2*
DeEtta Mills1,2*Forensic DNA analysis has vastly evolved since the first forensic samples were evaluated by restriction fragment length polymorphism (RFLP). Methodologies advanced from gel electrophoresis techniques to capillary electrophoresis and now to next generation sequencing (NGS). Capillary electrophoresis was and still is the standard method used in forensic analysis. However, dependent upon the information needed, there are several different techniques that can be used to type a DNA fragment. Short tandem repeat (STR) fragment analysis, Sanger sequencing, SNapShot, and capillary electrophoresis-single strand conformation polymorphism (CE-SSCP) are a few of the techniques that have been used for the genetic analysis of DNA samples. NGS is the newest and most revolutionary technology and has the potential to be the next standard for genetic analysis. This review briefly encompasses many of the techniques and applications that have been utilized for the analysis of human and nonhuman DNA samples.
Forensic genetics applies genetic tools and scientific methodology to solve criminal and civil litigations (Editorial, 2007). Locard’s Exchange Principle states that every contact leaves a trace, making any evidence a key component in forensic analysis. Biological evidence can comprise of cellular material or cell-free DNA from crime scenes, and as technologies improved, genetic methodologies were expanded to include human and non-human forensic analyses. Although these methodologies can be used for any genome, the prevalence of databases and standard guidelines has allowed human DNA typing to become the gold standard. This review will discuss the historical progression of DNA analysis techniques, strengths and limitations, and their possible forensic applications applied to human and non-human genetics.
“DNA fingerprinting” was serendipitously discovered in 1984 (Jeffreys, 2013). What they found propelled DNA “fingerprinting,” or DNA typing, to the forefront in legal cases to become the “gold standard” for forensic genetics in a court of law. Jeffreys first used restriction enzymes to fragment DNA, a method in which restriction endonucleases (RE) enzymes fragment the genomic DNA, producing restriction fragment length polymorphisms (RFLP) patterns. Since each RE recognizes specific DNA sequences to enzymatically cut the DNA, then inherent differences between gene sequences, due to evolutionary changes, will produce different fragment lengths. If the enzyme site is present in one individual but has changed in a different individual, the fragment lengths, once separated and visualized, will differ. While this technique was useful for some studies, Jeffreys did not find it useful for his particular genetic studies. Subsequently when working with the myoglobin gene in seals, he discovered that a short section of that gene – a minisatellite – was conserved and when isolated and cloned could be used to detect inherited genetic lineages as well as individualize a subject. Fragment length separation by electrophoresis, followed by transfer to Southern blot membranes, hybridized with a specific or non-specific complementary isotopic DNA probe, allowed for DNA fragments visualization (Jeffreys et al., 1985b). Upon careful analysis, Jeffreys determined that the fragments represented different combinations of DNA repetitive elements, unique to each individual, and could be used to better identify individuals or kinship lineages (Jeffreys et al., 1985b). Jeffreys’ technology was used in several subsequent paternity, immigration, and forensic genetics cases (Gill et al., 1985; Jeffreys et al., 1985a; Evans, 2007). This was just the beginning of a whole new era in DNA typing.
After Jeffreys’ discoveries, many DNA analyses methods involving electrophoretic fragment separation were discovered. Many were based on RFLP principles (Botstein et al., 1980), e.g., amplified fragment length polymorphism (AFLP) (Vos et al., 1995), and terminal restriction fragment length polymorphism (TRFLP) (Liu et al., 1997). Others like length heterogeneity- polymerase chain reaction (LH-PCR) (Suzuki et al., 1998) were based on intrinsic insertions and deletions of bases within specific genetic markers. Sanger sequencing (Sanger and Coulson, 1975), and single-strand conformational polymorphism (SSCP) analysis (Orita et al., 1989), while separated by electrophoresis, are theoretically based on single base sequence changes rather than insertions, deletions or RE site differences. While Jeffrey’s DNA fingerprinting method provided a very high power of discrimination, the main limitations were it was very time-consuming and required at least 10–25 ng of DNA to be successful (Wyman and White, 1980). With these limitations, RFLP was not always feasible for forensic cases.
The polymerase chain reaction (PCR) was discovered by Kary Mullis in 1985 and helped transform all DNA analyses (Mullis et al., 1986). The current standard for human DNA typing is short tandem repeat (STR) analysis (McCord et al., 2019). This method amplifies highly polymorphic, repetitive DNA regions by PCR and separates them by amplicon length using capillary electrophoresis. These inheritable markers are a series of 2–7 bases tandemly repeated at a specific locus, often in non-coding genetic regions. Forensic STRs are commonly tetranucleotide repeats (Goodwin et al., 2011), chosen because of their technical robustness and high variation among individuals (Kim et al., 2015). The combined DNA index system (CODIS) uses 20 core STR loci, expanded in 2017, and several commercial kits are available that contain these STRs (Oostdik et al., 2014; Ludeman et al., 2018). After amplification, different fluorochromes on each primer set allow for visualization of STRs after deconvolution, creating a STR profile consisting of a combination of genotypes (Gill et al., 2015). This method has become the gold standard for human forensics. Its greatest strength is the standardization of loci used by all laboratories and an extremely large searchable database of genetic profiles. However, some limitations and challenges are faced when dealing with highly degraded or low template DNA samples. To overcome these technical challenges, standardized mini-STR kits have been developed which use shorter versions of the core STRs and can be used in the same manner for forensic cases (Butler et al., 2007; Constantinescu et al., 2012). Keep in mind, DNA typing of humans – a single species – is the gold standard because of (a) the concerted scientific effort to standardize loci to analyze, (b) the development of commercial kits that can produce the same results regardless of instrumentation or laboratory performing the work, (c) a compatible and very large database that provides allelic frequencies for all sub-populations of humans, (d) standardized statistical methods used to report the results and (e) many court cases that have accepted human DNA typing evidence in a court of law – setting the precedent for future cases to use DNA typing results.
It was not long before scientists realized that non-human DNA could provide informative genetic evidence in forensic cases. Applications include bioterrorism, wildlife crimes, human identification through skin microorganisms, and so much more (Arenas et al., 2017). Since large quantities of biological materials are frequently not found at crime scenes, successful RFLP analyses were unlikely. Combining restriction enzymes and PCR technology, a process known as AFLP analysis (Vos et al., 1995), became a method for DNA fingerprinting using minute amounts of unknown sourced DNA. REs digest genomic DNA, then ligation of a constructed adapter sequence to the ends of all fragments allows the annealing of primers designed to recognize the adaptor sequences. Subsequent amplification generates many amplicons ranging in length when separated and visualized in an electropherogram or on a gel (Vos et al., 1995; Butler, 2012). AFLP markers for plant forensic DNA typing have been used because it provides high discrimination, requires only small amounts of DNA and the method is reproducible, all forensically important characteristics (Datwyler and Weiblen, 2006). For example, since most cannabis is clonally propagated, subsequent generations will have identical genetic profiles as seen with AFLP (Miller Coyle et al., 2003), providing useful intelligence links back to the source population. But there are significant variation between cultivars and within populations, so not having a standard database representing the species’ diversity for statistical comparisons greatly limits the method’s applicability. Another forensic example of its use is differentiating between marijuana and hemp, two morphologically and genetically similar plants, one an illicit drug while the other is not. In this study, three populations of hemp and one population of marijuana were analyzed with AFLP producing 18 bands that were specific to hemp samples. Additionally, 51.9% of molecular variance occurred within populations indicating these polymorphisms were useful for forensic individualization (Datwyler and Weiblen, 2006).
As a result of the anthrax letter attacks of 2001, microbial forensics came to the forefront (Schmedes et al., 2016), a discipline that combines multiple scientific specialties – microbiology, genetics, forensic science, and analytical chemistry. One method used to compare microbial communities is TRFLP (Liu et al., 1997; Osborn et al., 2000; Butler, 2012). With this method, the DNA is amplified using “universal,” highly conserved primer sequences shared across all organisms of interest, i.e., the 16S rRNA genes in bacteria and Archaea, and then uses REs to fragment the PCR products (Table 1). Separated by capillary electrophoresis, only the fluorescently tagged terminal restricted fragments are visualized (Mrkonjic Fuka et al., 2007), reducing the profile complexity and providing high discrimination. TRFLP has been used to characterize complex microbial communities for forensic applications by linking the similarity of the amplicon patterns generated from the intrinsic soil communities to the evidence from a crime scene (Meyers and Foran, 2008; Habtom et al., 2017). This method does provide a distinct pattern reflective of the microbial community, useful for forensic genetics but the method does not provide any sequence information. Another limitation is no standardization of which primer pairs or REs are used, making direct comparisons between studies difficult. This lack of standardization also hinders the development of a database for species identification. Additionally, the method is time-consuming due to the additional step of restriction digestion and the possibility of incomplete enzymatic digestion can complicate the interpretation of results (Osborn et al., 2000; Moreno et al., 2006).
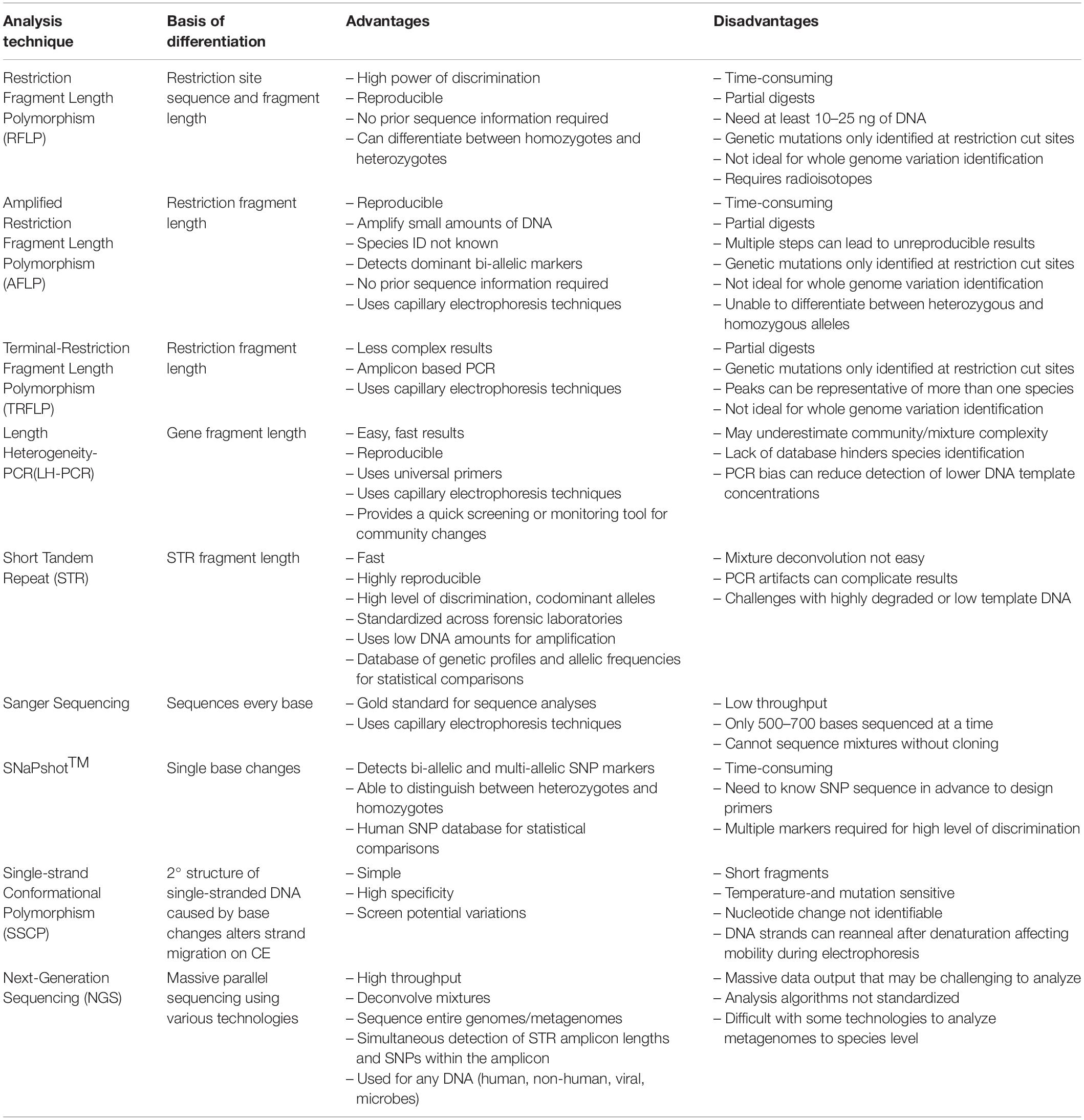
Table 1. The basis of differentiation, advantages, and disadvantages of past and current technologies.
Another methodology has been used to characterize microbial communities is length heterogeneity- polymerase chain reaction (LH-PCR) (Suzuki et al., 1998). Universal primers complementary to highly conserved domains within genomes are used to amplify hypervariable sequences within specific sequence domains. The 16S/18S rRNA genes, the chloroplast genes or Internal Transcribed Spacer (ITS) regions are commonly used. This technique is based on the natural sequence length variation due to insertions and deletions of bases that occur within a domain (Moreno et al., 2006). It has been used to characterize microbial communities for forensic soil applications where a correlation between geographic location and microbial profiles has proven to be more discriminating than elemental soil analysis (Moreno et al., 2006, 2011; Damaso et al., 2018). With LH-PCR, metagenomic DNA extracted from the soil is amplified using fluorescently labeled universal primers with amplicon peaks within the electropherogram representing the minimum diversity within the community. However, specific sequence information is not known as many peaks of the same size could represent more than one species, thereby masking the community’s actual taxonomic diversity. A recent study showed the intrinsic diversity of a microbial mat, masked by LH-PCR, could be further resolved by the inherent sequence differences using capillary electrophoresis-single strand conformational polymorphism (CE-SSCP) analysis (Damaso et al., 2014) and confirmed by sequencing. The advantage of LH-PCR is it is a fast and reproducible method that can correlate geographical areas to microbial patterns with bioinformatics (Damaso et al., 2018); but a soil database would need to be developed to be useful beyond specific geographical areas.
The basis of genomic differentiation is the intrinsic order of base pairs within a region that can be evaluated by sequencing. Sanger sequencing has been the gold standard since the 1970s (Sanger and Coulson, 1975). Sanger sequencing was termed the gold standard because of the ability for single base pair resolution allowing for full sequence information to be determined. Robust and extensive databases are also readily available for comparison, i.e., GenBank, to identify an organism. However, it does have some limitations such as the short length (<500–700 bp) and it cannot sequence mixtures of organisms, for example, without cloning, so it would not be useful for sequencing complex microbial communities without intense time, effort and cost.
Other approaches use the ability to identify intrinsic single base sequence variation using single nucleotide polymorphisms (SNPs) within four forensically relevant SNP classes: identity-testing, ancestry informative, phenotype informative, and lineage informative. SNPs are particularly useful when typing degraded DNA or increasing the amount of genetic information retrieved from a sample (Budowle and van Daal, 2008; Goodwin et al., 2011). SNaPshotTM is a commercially available SNP kit that can identify known SNPs using single base extension (SBE) technology (Daniel et al., 2015; Fondevila et al., 2017). Wildlife forensics has used SNaPshotTM to identify endangered or trafficked species that are illegally poached to support criminal prosecutions. Elephant species identification from ivory and ivory products (Kitpipit et al., 2017) or differentiating wolf species from dog subspecies (Jiang et al., 2020) are both examples of SNaPshotTM assays developed for wildlife forensics. By using species-specific SNPs, the samples could be identified. But yet again, the limitation becomes the need for species-specific reference databases and the monumental task of developing a robust database for each species. Human SNPs databases with allele frequencies, as seen in dbSNP, however, are available making their forensic application more feasible in some cases.
Massively parallel sequencing (MPS) or next-generation sequencing (NGS) allows for mixtures of genomes of any species to be sequenced in one analysis (Ansorge, 2009). This technology can sequence thousands of genomic regions simultaneously, allowing for whole-genome, metagenomic sequencing or targeted amplicon sequencing (Gettings et al., 2016). Various NGS technologies are available each using slightly different technologies to sequence DNA (Heather and Chain, 2016). Verogen has developed kits explicitly for human forensic genomics using Illumina’s MiSeq FGx system (Guo et al., 2017; Moreno et al., 2018). The FBI recently approved DNA profiles generated by Verogen forensic technology to be uploaded into the National DNA Index System (NDIS) (SWGDAM, 2019), making it the first NGS technology approved for NDIS.
Short tandem repeat mixture deconvolution, degraded, low template samples, and even microbial community samples are just a few of the potential NGS applications for forensic genomics and metagenomics (Borsting and Morling, 2015). In human STR analyses, the greatest challenge is mixture deconvolution. NGS technology presents an increased power of discrimination of STR alleles using the intrinsic SNPs genetic microhaplotypes – a combination of 2–4 closely linked SNPs within an allele (Kidd et al., 2014; Pang et al., 2020). However, the acceptance of analyses programs to deconvolve mixtures has not been standardized to the same level as it has for STRs.
Microbes are the first responders to changes in any environment because they are rapidly affected by the availability of nutrients and their intrinsic habitats. This makes them excellent indicators for studies investigating post-mortem interval (PMI) or as an indicator of soil geographical provenance (Giampaoli et al., 2014; Finley et al., 2015). In decaying organisms, shifts in epinecrotic communities or the thanatomicrobiome are becoming increasingly critical components in investigating PMI (Javan et al., 2016). Sequencing of the thanatomicrobiome revealed the Clostridium spp. varied during different stages human decomposition, the “Postmortem Clostridium Effect” (PCE), providing a time signature of the thanatomicrobiome, which could only have been uncovered through NGS (Javan et al., 2017). However, the lack of consensus in analyses techniques must be addressed before NGS methodologies can be introduced into the justice system (Table 1).
Forensic DNA typing has progressed quickly within a short timeframe (Figure 1), which can be attributed to the many advancements in molecular biology technologies. As these techniques advance, forensic scientists will analyze more atypical forms of evidence to answer questions deemed unresolvable with traditional DNA analyses. For example, epigenetics and DNA methylation markers have been proposed to estimate age, determine the tissue type, and even differentiate between monozygotic twins (Vidaki and Kayser, 2018). However, since epigenetic patterns are also influenced by environmental factors, they can be dynamic, and a number of confounding factors have the potential to affect predictions and must be taken into account when preparing prediction models (i.e., age estimation). Additionally, phenotype informative SNPs across the genome can infer physical characteristics like eye, hair, and skin color, even age, from an unknown source of DNA retrieved from a crime scene. But this technology could pose an “implicit bias” toward minorities, especially in “societies where racism and xenophobia are now on the rise” (Schneider et al., 2019) if not ethically and judicially implemented. With the increased sensitivity of NGS, low biomass samples from environmental DNA (eDNA) – DNA from soil, water, air – can complement and enhance intelligence gathering or provenance in criminal cases. Pollen and dust are two types of eDNA recently explored for their future forensic potential (Alotaibi et al., 2020; Young and Linacre, 2021). However, if used in criminal investigations where the eDNA collected has had interaction with other environments, there must be some protocol or quality control established to account for variability that is likely to occur. This makes the prudent validation of this type of DNA analysis, essential. Limitations also arise due to lack of a database for comparison of samples and statistical analyses to evaluate the strength of a match like in the analysis of human STR profiles.
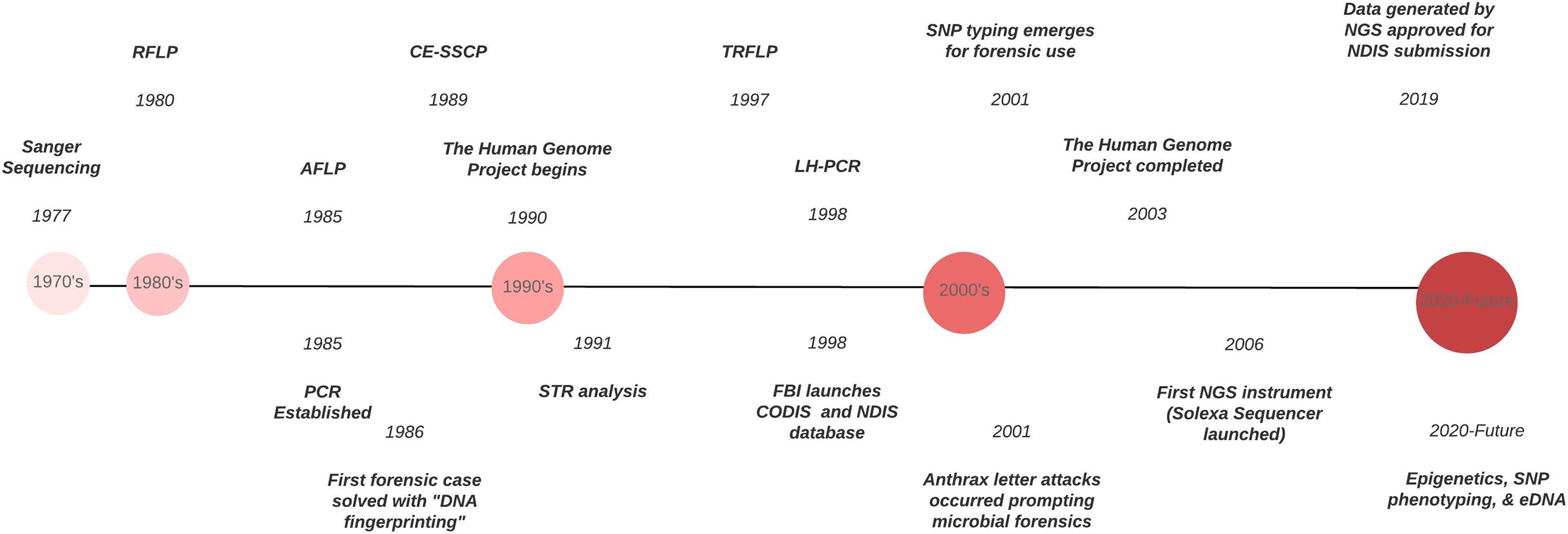
Figure 1. Timeline of the evolution of DNA typing technologies from the 1970’s to the present.
DNA has long been the gold standard in human forensic analysis because of the standardization of DNA markers, databases and statistical analyses. It has laid the foundation for these promising new technologies that will significantly enhance intelligence gathering and species identification – human and non-human – in forensic cases. In order for these methodologies to be useful in criminal investigations, they must adhere to the legal standards such as the Frye or Daubert Standards which determines if an expert testimony or evidence is admissible in court. A method can be deemed acceptable if it follows forensic guidelines set by organizations such as NIST’s Organization Scientific Area Committees (OSAC), Society for Wildlife Forensic Sciences (SWFS), Scientific Working Group on DNA Analysis Methods (SWGDAM), and the International Society for Forensic Genetics (ISFG) (Linacre et al., 2011) just to name a few. These committees provide the guidelines for validation, interpretation, and quality assurance, all necessary components for DNA analysis. The US Fish and Wildlife forensic laboratory has standardized protocols for crimes against federally endangered or threatened species1. However, the more common limiting factors in the development of standard guidelines of non-human forensic genetic analyses across different state laboratories are the lack of consensus in methodologies, supporting allelic databases and standardized statistical analyses. Addressing those issues could lay the foundation for non-human analyses to be on par with human analyses.
DJ designed and wrote the manuscript. DM edited and contributed to the writing of the manuscript. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to acknowledge the invitation by the editors to contribute to this special edition. DJ was supported by the Florida Education Fund’s McKnight Doctoral Fellowship.
Alotaibi, S. S., Sayed, S. M., Alosaimi, M., Alharthi, R., Banjar, A., Abdulqader, N., et al. (2020). Pollen molecular biology: Applications in the forensic palynology and future prospects: A review. Saudi J. Biol. Sci. 27, 1185–1190. doi: 10.1016/j.sjbs.2020.02.019
Ansorge, W. J. (2009). Next-generation DNA sequencing techniques. N. Biotechnol. 25, 195–203. doi: 10.1016/j.nbt.2008.12.009
Arenas, M., Pereira, F., Oliveira, M., Pinto, N., Lopes, A. M., Gomes, V., et al. (2017). Forensic genetics and genomics: Much more than just a human affair. PLoS Genet. 13:e1006960. doi: 10.1371/journal.pgen.1006960
Borsting, C., and Morling, N. (2015). Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. Genet. 18, 78–89. doi: 10.1016/j.fsigen.2015.02.002
Botstein, D., White, R. L., Skolnick, M., and Davis, R. W. (1980). Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331.
Budowle, B., and van Daal, A. (2008). Forensically relevant SNP classes. Biotechniques 60:610. doi: 10.2144/000112806
Butler, J. M. (2012). “Non-human DNA,” in Advanced Topics in Forensic DNA Typing, ed. J. M. Butler (San Diego: Academic Press), 473–495.
Butler, J. M., Coble, M. D., and Vallone, P. M. (2007). STRs vs. SNPs: thoughts on the future of forensic DNA testing. Forensic Sci. Med. Pathol. 3, 200–205. doi: 10.1007/s12024-007-0018-1
Constantinescu, C. M., Barbarii, L. E., Iancu, C. B., Constantinescu, A., Iancu, D., and Girbea, G. (2012). Challenging DNA samples solved with MiniSTR analysis. Brief overview. Rom. J. Leg. Med. 20, 51–56. doi: 10.4323/rjlm.2012.51
Damaso, N., Martin, L., Kushwaha, P., and Mills, D. (2014). F-108 polymer and capillary electrophoresis easily resolves complex environmental DNA mixtures and SNPs. Electrophoresis 35, 3208–3211. doi: 10.1002/elps.201400069
Damaso, N., Mendel, J., Mendoza, M., von Wettberg, E. J., Narasimhan, G., and Mills, D. (2018). Bioinformatics Approach to Assess the Biogeographical Patterns of Soil Communities: The Utility for Soil Provenance. J. Forensic. Sci. 63, 1033–1042. doi: 10.1111/1556-4029.13741
Daniel, R., Santos, C., Phillips, C., Fondevila, M., van Oorschot, R. A., Carracedo, A., et al. (2015). A SNaPshot of next generation sequencing for forensic SNP analysis. Forensic Sci. Int. Genet. 14, 50–60. doi: 10.1016/j.fsigen.2014.08.013
Datwyler, S. L., and Weiblen, G. D. (2006). Genetic variation in hemp and marijuana (Cannabis sativa L.) according to amplified fragment length polymorphisms. J. Forensic Sci. 51, 371–375. doi: 10.1111/j.1556-4029.2006.00061.x
Editorial. (2007). Launching Forensic Science International daughter journal in 2007: Forensic Science International: Genetics. Forensic Sci. Int. Genet. 1, 1–2. doi: 10.1016/j.fsigen.2006.10.001
Evans, C. (2007). The Casebook of Forensic Detection: How Science Solved 100 of the World’s Most Baffling Crimes. New York, NY: Berkley Books.
Finley, S. J., Benbow, M. E., and Javan, G. T. (2015). Potential applications of soil microbial ecology and next-generation sequencing in criminal investigations. Appl. Soil. Ecol. 88, 69–78. doi: 10.1016/j.apsoil.2015.01.001
Fondevila, M., Borsting, C., Phillips, C., de la Puente, M., Consortium, E. N., Carracedo, A., et al. (2017). Forensic SNP genotyping with SNaPshot: Technical considerations for the development and optimization of multiplexed SNP assays. Forensic Sci. Rev. 29, 57–76.
Gettings, K. B., Kiesler, K. M., Faith, S. A., Montano, E., Baker, C. H., Young, B. A., et al. (2016). Sequence variation of 22 autosomal STR loci detected by next generation sequencing. Forensic Sci. Int. Genet. 21, 15–21. doi: 10.1016/j.fsigen.2015.11.005
Giampaoli, S., Berti, A., Di Maggio, R. M., Pilli, E., Valentini, A., Valeriani, F., et al. (2014). The environmental biological signature: NGS profiling for forensic comparison of soils. Forensic Sci. Int. 240, 41–47. doi: 10.1016/j.forsciint.2014.02.028
Gill, P., Haned, H., Bleka, O., Hansson, O., Dorum, G., and Egeland, T. (2015). Genotyping and interpretation of STR-DNA: Low-template, mixtures and database matches-Twenty years of research and development. Forensic Sci. Int. Genet. 18, 100–117. doi: 10.1016/j.fsigen.2015.03.014
Gill, P., Jeffreys, A. J., and Werrett, D. J. (1985). Forensic application of DNA ‘fingerprints’. Nature 318, 577–579. doi: 10.1038/318577a0
Goodwin, W., Linacre, A., and Hadi, S. (2011). “An introduction to forensic genetics.” 2nd ed. West Sussex, UK: Wiley-Blackwell, 53–62.
Guo, F., Yu, J., Zhang, L., and Li, J. (2017). Massively parallel sequencing of forensic STRs and SNPs using the Illumina((R)) ForenSeq DNA Signature Prep Kit on the MiSeq FGx Forensic Genomics System. Forensic Sci. Int. Genet. 31, 135–148. doi: 10.1016/j.fsigen.2017.09.003
Habtom, H., Demaneche, S., Dawson, L., Azulay, C., Matan, O., Robe, P., et al. (2017). Soil characterisation by bacterial community analysis for forensic applications: A quantitative comparison of environmental technologies. Forensic Sci. Int. Genet. 26, 21–29. doi: 10.1016/j.fsigen.2016.10.005
Heather, J. M., and Chain, B. (2016). The sequence of sequencers: The history of sequencing DNA. Genomics 107, 1–8. doi: 10.1016/j.ygeno.2015.11.003
Javan, G. T., Finley, S. J., Abidin, Z., and Mulle, J. G. (2016). The Thanatomicrobiome: A Missing Piece of the Microbial Puzzle of Death. Front. Microbiol. 7:225. doi: 10.3389/fmicb.2016.00225
Javan, G. T., Finley, S. J., Smith, T., Miller, J., and Wilkinson, J. E. (2017). Cadaver Thanatomicrobiome Signatures: The Ubiquitous Nature of Clostridium Species in Human Decomposition. Front. Microbiol. 8:2096. doi: 10.3389/fmicb.2017.02096
Jeffreys, A. J. (2013). The man behind the DNA fingerprints: an interview with Professor Sir Alec Jeffreys. Investig. Genet. 4:21. doi: 10.1186/2041-2223-4-21
Jeffreys, A. J., Brookfield, J. F., and Semeonoff, R. (1985a). Positive identification of an immigration test-case using human DNA fingerprints. Nature 317, 818–819. doi: 10.1038/317818a0
Jeffreys, A. J., Wilson, V., and Thein, S. L. (1985b). Hypervariable ‘minisatellite’ regions in human DNA. Nature 314, 67–73. doi: 10.1038/314067a0
Jiang, H. H., Li, B., Ma, Y., Bai, S. Y., Dahmer, T. D., Linacre, A., et al. (2020). Forensic validation of a panel of 12 SNPs for identification of Mongolian wolf and dog. Sci. Rep. 10:13249. doi: 10.1038/s41598-020-70225-5
Kidd, K. K., Pakstis, A. J., Speed, W. C., Lagace, R., Chang, J., Wootton, S., et al. (2014). Current sequencing technology makes microhaplotypes a powerful new type of genetic marker for forensics. Forensic Sci. Int. Genet. 12, 215–224. doi: 10.1016/j.fsigen.2014.06.014
Kim, Y. T., Heo, H. Y., Oh, S. H., Lee, S. H., Kim, D. H., and Seo, T. S. (2015). Microchip-based forensic short tandem repeat genotyping. Electrophoresis 36, 1728–1737. doi: 10.1002/elps.201400477
Kitpipit, T., Thongjued, K., Penchart, K., Ouithavon, K., and Chotigeat, W. (2017). Mini-SNaPshot multiplex assays authenticate elephant ivory and simultaneously identify the species origin. Forensic Sci. Int. Genet. 27, 106–115. doi: 10.1016/j.fsigen.2016.12.007
Linacre, A., Gusmão, L., Hecht, W., Hellmann, A. P., Mayr, W. R., Parson, W., et al. (2011). ISFG: Recommendations regarding the use of non-human (animal) DNA in forensic genetic investigations. Forensic Sci. Int. Genet. 5, 501–505. doi: 10.1016/j.fsigen.2010.10.017
Liu, W. T., Marsh, T. L., Cheng, H., and Forney, L. J. (1997). Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microbiol. 63, 4516–4522. doi: 10.1128/AEM.63.11.4516-4522.1997
Ludeman, M. J., Zhong, C., Mulero, J. J., Lagace, R. E., Hennessy, L. K., Short, M. L., et al. (2018). Developmental validation of GlobalFiler PCR amplification kit: a 6-dye multiplex assay designed for amplification of casework samples. Int. J. Legal. Med. 132, 1555–1573. doi: 10.1007/s00414-018-1817-5
McCord, B. R., Gauthier, Q., Cho, S., Roig, M. N., Gibson-Daw, G. C., Young, B., et al. (2019). Forensic DNA Analysis. Anal. Chem. 91, 673–688. doi: 10.1021/acs.analchem.8b05318
Meyers, M. S., and Foran, D. R. (2008). Spatial and temporal influences on bacterial profiling of forensic soil samples. J. Forensic Sci. 53, 652–660. doi: 10.1111/j.1556-4029.2008.00728.x
Miller Coyle, H., Palmbach, T., Juliano, N., Ladd, C., and Lee, H. C. (2003). An overview of DNA methods for the identification and individualization of marijuana. Croat Med. J. 44, 315–321.
Moreno, L. I., Galusha, M. B., and Just, R. (2018). A closer look at Verogen’s Forenseq DNA Signature Prep kit autosomal and Y-STR data for streamlined analysis of routine reference samples. Electrophoresis 39, 2685–2693. doi: 10.1002/elps.201800087
Moreno, L. I., Mills, D. K., Entry, J., Sautter, R. T., and Mathee, K. (2006). Microbial metagenome profiling using amplicon length heterogeneity-polymerase chain reaction proves more effective than elemental analysis in discriminating soil specimens. J. Forensic Sci. 51, 1315–1322. doi: 10.1111/j.1556-4029.2006.00264.x
Moreno, L. I., Mills, D., Fetscher, J., John-Williams, K., Meadows-Jantz, L., and McCord, B. (2011). The application of amplicon length heterogeneity PCR (LH-PCR) for monitoring the dynamics of soil microbial communities associated with cadaver decomposition. J. Microbiol. Methods 84, 388–393. doi: 10.1016/j.mimet.2010.11.023
Mrkonjic Fuka, M., Gesche Braker, S. H., and Philippot, L. (2007). “Molecular Tools to Assess the Diversity and Density of Denitrifying Bacteria in Their Habitats,” in Biology of the Nitrogen Cycle, eds H. Bothe, S. J. Ferguson, and W. E. Newton (Amsterdam: Elsevier), 313–330.
Mullis, K., Faloona, F., Scharf, S., Saiki, R., Horn, G., and Erlich, H. (1986). Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 1, 263–273. doi: 10.1101/sqb.1986.051.01.032
Oostdik, K., Lenz, K., Nye, J., Schelling, K., Yet, D., Bruski, S., et al. (2014). Developmental validation of the PowerPlex((R)) Fusion System for analysis of casework and reference samples: A 24-locus multiplex for new database standards. Forensic Sci. Int. Genet. 12, 69–76. doi: 10.1016/j.fsigen.2014.04.013
Orita, M., Iwahana, H., Kanazawa, H., Hayashi, K., and Sekiya, T. (1989). Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc. Natl. Acad. Sci. U S A 86, 2766–2770.
Osborn, A. M., Moore, E. R., and Timmis, K. N. (2000). An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol. 2, 39–50. doi: 10.1046/j.1462-2920.2000.00081.x
Pang, J. B., Rao, M., Chen, Q. F., Ji, A. Q., Zhang, C., Kang, K. L., et al. (2020). A 124-plex Microhaplotype Panel Based on Next-generation Sequencing Developed for Forensic Applications. Sci. Rep. 10:1945. doi: 10.1038/s41598-020-58980-x
Sanger, F., and Coulson, A. R. (1975). A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 94, 441–448. doi: 10.1016/0022-2836(75)90213-2
Schmedes, S. E., Sajantila, A., and Budowle, B. (2016). Expansion of Microbial Forensics. J. Clin. Microbiol. 54, 1964–1974. doi: 10.1128/JCM.00046-16
Schneider, P. M., Prainsack, B., and Kayser, M. (2019). The Use of Forensic DNA Phenotyping in Predicting Appearance and Biogeographic Ancestry. Dtsch Arztebl. Int. 52, 873–880. doi: 10.3238/arztebl.2019.0873
Suzuki, M., Rappe, M. S., and Giovannoni, S. J. (1998). Kinetic bias in estimates of coastal picoplankton community structure obtained by measurements of small-subunit rRNA gene PCR amplicon length heterogeneity. Appl. Environ. Microbiol. 64, 4522–4529.
SWGDAM (2019). Addendum to SWGDAM Autosomal Interpretation Guidelines for NGS.Swgdam. Available online at: https://www.swgdam.org/publications
Vidaki, A., and Kayser, M. (2018). Recent progress, methods and perspectives in forensic epigenetics. Forensic Sci. Int. Genet. 37, 180–195. doi: 10.1016/j.fsigen.2018.08.008
Vos, P., Hogers, R., Bleeker, M., Reijans, M., van de Lee, T., Hornes, M., et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23, 4407–4414. doi: 10.1093/nar/23.21.4407
Wyman, A. R., and White, R. (1980). A highly polymorphic locus in human DNA. Proc. Natl. Acad. Sci. U S A 77, 6754–6758. doi: 10.1073/pnas.77.11.6754
Keywords: forensic genetics, DNA typing, metabarcoding, soil, microbes, minisatellites, next-generation sequencing
Citation: Jordan D and Mills D (2021) Past, Present, and Future of DNA Typing for Analyzing Human and Non-Human Forensic Samples. Front. Ecol. Evol. 9:646130. doi: 10.3389/fevo.2021.646130
Received: 25 December 2020; Accepted: 02 March 2021;
Published: 22 March 2021.
Edited by:
Gulnaz T. Javan, Alabama State University, United StatesReviewed by:
Carla Santos, University of Minho, PortugalCopyright © 2021 Jordan and Mills. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: DeEtta Mills, bWlsbHNkQGZpdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.