 Noriaki Sasai
Noriaki Sasai Michinori Toriyama
Michinori Toriyama Toru Kondo4*
Toru Kondo4*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 08 November 2019
Sec. Stem Cell Research
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.01103
This article is part of the Research Topic Recent Advances of Hedgehog Signaling Pathway in Development and Cancer View all 6 articles
The hedgehog (Hh) family comprises sonic hedgehog (Shh), Indian hedgehog (Ihh), and desert hedgehog (Dhh), which are versatile signaling molecules involved in a wide spectrum of biological events including cell differentiation, proliferation, and survival; establishment of the vertebrate body plan; and aging. These molecules play critical roles from embryogenesis to adult stages; therefore, alterations such as abnormal expression or mutations of the genes involved and their downstream factors cause a variety of genetic disorders at different stages. The Hh family involves many signaling mediators and functions through complex mechanisms, and achieving a comprehensive understanding of the entire signaling system is challenging. This review discusses the signaling mediators of the Hh pathway and their functions at the cellular and organismal levels. We first focus on the roles of Hh signaling mediators in signal transduction at the cellular level and the networks formed by these factors. Then, we analyze the spatiotemporal pattern of expression of Hh pathway molecules in tissues and organs, and describe the phenotypes of mutant mice. Finally, we discuss the genetic disorders caused by malfunction of Hh signaling-related molecules in humans.
In vertebrates, at least three hedgehog (Hh)-related genes have been reported (Ingham and McMahon, 2001). The three Hh genes identified in mice and in humans are sonic hedgehog (Shh), desert hedgehog (Dhh), and Indian hedgehog (Ihh), which are similarly processed and secreted from cells, and evoke common signaling pathways in receiving cells (Ingham and McMahon, 2001).
Once produced, Hh polypeptides are cleaved at the intermediate site, and the amino-terminal part is functional as the signal molecules. After processed by cholesterol and palmitate, the protein is secreted from the producing cells and binds to the membrane protein Patched (Ptch). The signal is received by another membrane protein, smoothened (Smo), and then transduced into the nucleus by the transcription factor Gli. Gli stability and activity are modified by the kinesin-like protein KIF7 and the scaffold protein suppressor of fused (SuFu).
In the first part of this review, we discuss the production and secretion of Hh proteins in detail, and introduce the mediators involved in protein modifications and signaling pathways. The mechanisms are summarized in Figure 1.
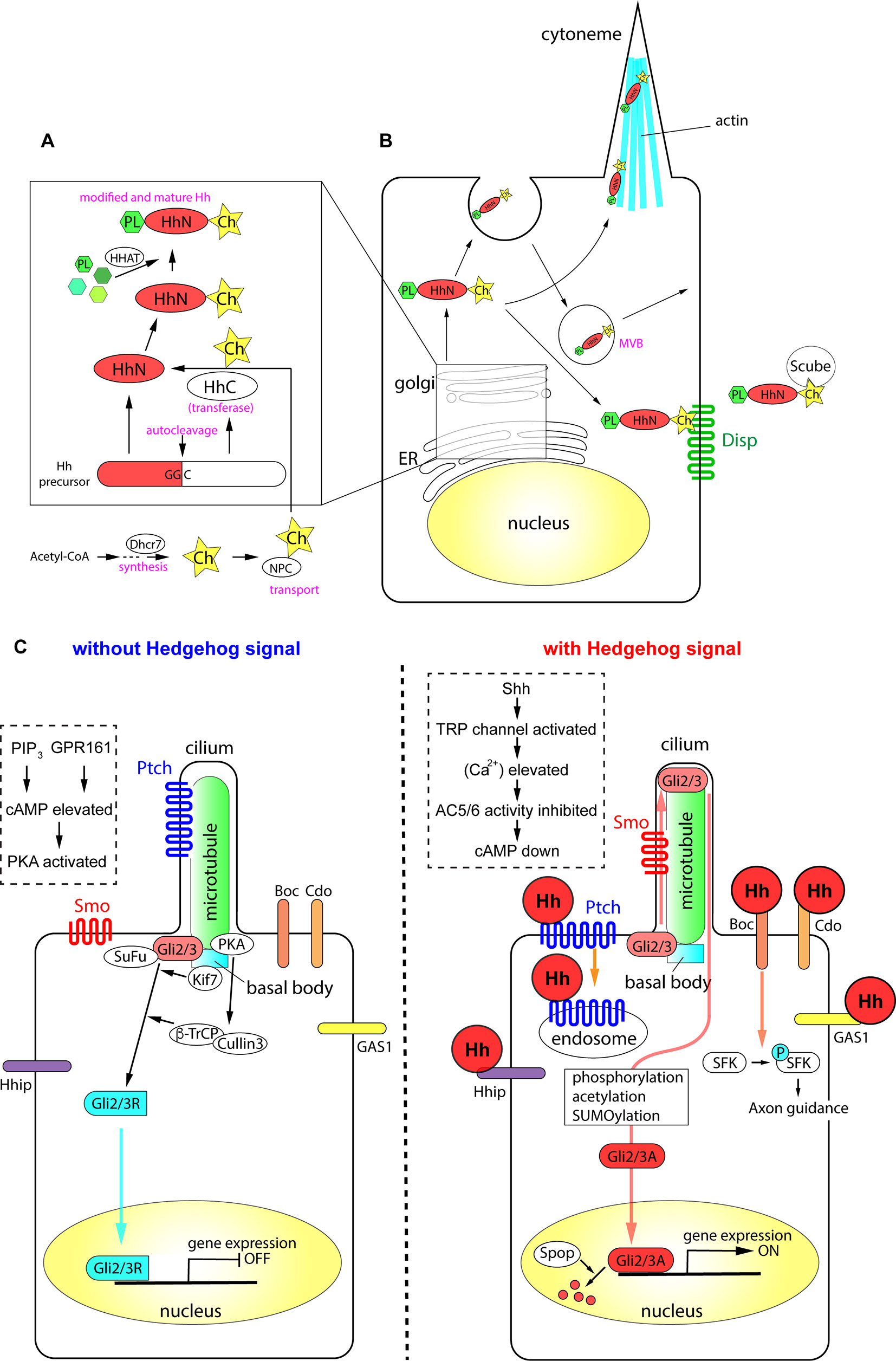
Figure 1 The processing of Hh proteins and their intracellular signaling pathways. (A) Cleavage of Hh polypeptides and their modifications in ER and golgi apparatus. (B) Processing, modifications and transport of Hh proteins in the producing cells. (C) Active signaling pathways in absence and in presence of Hh molecules. Schemas in dotted rectangles represent regulation of cAMP level in the cilia. The figure was made based on the information in (Varjosalo and Taipale, 2008; Balaskas et al., 2012; Moore et al., 2016; Parchure et al., 2018).
All Hh polypeptides produced are transported into the endoplasmic reticulum (ER) and Golgi apparatus, where they undergo autoprocessing (Jeong and McMahon, 2002; Cai and Liu, 2016). The polypeptides are cleaved into two parts, the amino-terminal domain, which functions as a signaling molecule, and the carboxyl-terminal domain, which functions in autoprocessing regulation (Mann and Beachy, 2004). The amino-terminal part is further modified with palmitate and cholesterol (Porter et al., 1996; Chamoun et al., 2001; Lewis et al., 2001; Mann and Beachy, 2004; Chen et al., 2004a; Chen et al., 2011a; Li et al., 2006; Hardy and Resh, 2012). The carboxyl ends of the initial polypeptides act as cholesterol transferases (Porter et al., 1996), and palmitoylation is mediated by skinny Hh (Ski) acyltransferase (also known as Hh acyltransferase or HHAT), a transmembrane acyltransferase located in the ER (Chamoun et al., 2001; Dennis et al., 2012; Konitsiotis et al., 2015). The modified Shh protein was recently purified, and the fatty acids bound to the amino terminus were identified, which revealed that other unsaturated fatty acids in addition to palmitate are involved in the modification (Long et al., 2015). These lipid modifications are essential for the stability of the protein in the extracellular matrix (Callejo et al., 2006) and during long-range transport (Lewis et al., 2001; Zeng et al., 2001). The Niemann-Pick C (NPC1/2) proteins transport cholesterol from the endosome to the ER (Loftus et al., 1997; Tukachinsky et al., 2012; Formichi et al., 2018), and play an essential role in cholesterol modification of Hh. The membrane protein dispatched (Disp) and the secreted protein Scube2 bind to the cholesterol moiety of the modified Hh proteins (Tukachinsky et al., 2012) and release them from the cell membrane.
Some of the Hh proteins at the cell surface can be recycled. After recycling, the Hh proteins are released by lipid bilayer membrane vesicles called exosomes (Vlassov et al., 2012; Parchure et al., 2018). Exosomes are formed at the multivesicular body (MVB) in the cytosol, and bud from the plasma membrane with a size of 30–100 nm containing functional molecules (Parchure et al., 2018). For exosome formation, membrane proteins collectively called endosomal sorting complexes required for transport (ESCRT) are essential (Juan and Furthauer, 2018).
A specialized filopodial structure, the cytoneme, which forms in Hh-producing cells, is important for transmission of the Hh signal. The cytoneme was initially identified in flies (Bischoff et al., 2013; Gonzalez-Mendez et al., 2017), and later reported in the chick embryonic limb bud (Panman and Zeller, 2003; Sanders et al., 2013). The presence of microvesicles and the cytoneme, together with the palmitoylation and cholesterol modification of Hh proteins, mediate the transport of the Hh signal to distant parts in the tissues (Chen et al., 2004a; Sanders et al., 2013; D'Angelo et al., 2015).
Overall, the efficient production and secretion of active Hh proteins involves the following: (i) processing of the polypeptides by autocleavage, (ii) modification of the polypeptides by cholesterol and palmitate, (iii) recycling and packaging of the proteins in the microvesicles, and (iv) the presence of cytosolic structures including cytonemes.
When Hh proteins reach the surface of a responding cell, the signals are introduced into the cell by two membrane proteins, Ptch and Smo. Ptch, a receptor protein that directly interacts with Hh proteins (Marigo et al., 1996; Motoyama et al., 1998b), is a 12-span membrane protein that localizes to the ciliary membrane in the absence of Hh (Rohatgi et al., 2007). Ptch binds to Hh, forming a complex that moves from primary cilia to the plasma membrane. In the case of Shh, the Ptch-Shh complex undergoes endocytosis, which requires the membrane-remodeling GTPase dynamin (Incardona et al., 2000; Ferguson and De Camilli, 2012) and the HECT-domain ubiquitin E3 ligases Smurf1/2 (Yue et al., 2014), and leads to degradation in the lysosome. Once Ptch is cleared from the ciliary membrane, the G protein-coupled receptor (GPCR) Smo is translocated from the plasma membrane to the cilium (Corbit et al., 2005). During this process, Smo is phosphorylated at specific serine residues in the carboxyl intracellular region by casein kinase 1α (CK1α) and G protein-coupled receptor kinase 2 (GRK2) (Chen et al., 2004b; Chen et al., 2011b), and interacts with β-arrestin 2 (Chen et al., 2004b). Smo further forms a complex with the kinesin-like protein Kif3a, and is translocated into primary cilia (Kovacs et al., 2008). β-Arrestin 2 is essential for the translocation of Smo into cilia (Kovacs et al., 2008), and contributes to the dynamin-dependent internalization of Smo (Chen et al., 2004b). The role of β-arrestin 2-mediated internalization of the signaling molecule (Ma and Pei, 2007), together with its involvement in the translocation of Smo, suggests that β-arrestin 2 regulates the activity and the stability of the Smo protein.
The Ptch polypeptide contains a sterol sensing domain (Martin et al., 2001; Strutt et al., 2001) and binds to cholesterol. The interaction of Ptch and cholesterol is necessary for pumping out cholesterol molecules from the endosome or from the plasma membrane (Bidet et al., 2011). In the absence of Hh, Ptch pumps out the cholesterol molecule (Denef et al., 2000; Khaliullina et al., 2009; Blassberg and Jacob, 2017). By contrast, when cells are exposed to Hh, Ptch is transported into the endosome and degraded (Incardona et al., 2000; Yue et al., 2014), and Smo is modified with cholesterol. Modified Smo activates cytosolic phospholipase A2 to release a fatty acid, arachidonic acid, and the interaction between arachidonic acid and Smo promotes the ciliary localization of Smo through allosteric changes (Arensdorf et al., 2017). Altogether, the cholesterol modification of Smo is essential for its complete activity and for the localization of the protein (Blassberg et al., 2016; Huang et al., 2016; Luchetti et al., 2016).
Activation of the Hh signal at the membrane level leads to its transduction into the nucleus by the transcription factors Gli2/3. Gli2/3 are Krüppel-like transcription factors containing zinc-finger DNA-binding domains with dual activity: the amino terminus contains a transcriptional repressor domain and the carboxyl terminus has an activator domain. In the absence of the extracellular Hh ligand, the synthesized Gli2/3 polypeptides form complexes with protein kinase A (PKA), CK1, and glycogen synthase kinase 3 (GSK3), the latter two of which are serine-threonine protein kinases (Tempe et al., 2006). This complex interacts with the scaffold protein SuFu in the absence of the Hh signal. The role of SuFu is to stabilize the Gli2/3 proteins (Chen et al., 2009; Humke et al., 2010); in the absence of SuFu, both Gli2/3 proteins are drastically destabilized and become undetectable by western blotting (Chen et al., 2009; Humke et al., 2010). Activation of the Hh signaling pathway disrupts the interaction between SuFu and Gli (Tukachinsky et al., 2010).
In the absence of an extracellular Hh ligand, the calcium level in the cytosol and in the cilium is relatively high (Moore et al., 2016). Gli2/3 are phosphorylated by PKA and subsequently targeted by the Skp, cullin, F-box (SCF) ubiquitin ligase complex, which is composed of the scaffold protein cullin 3 and the E3-type ubiquitin ligase β-Trcp (Bhatia et al., 2006; Wang and Li, 2006). The Gli2/3 proteins are cleaved at the amino terminus, and the truncated proteins with the transcriptional repressor domains (Gli2/3R; R for "repressive") are translocated into the nucleus to bind to DNA target sites (Svard et al., 2006; Chen et al., 2009).
As Smo couples with Gi-type G proteins (Riobo et al., 2006), the exposure of cells to Hh (i.e., activation of Smo) reduces PKA activity (Moore et al., 2016), which decreases cAMP levels. Thus, the newly synthesized Gli2/3 polypeptides are not cleaved, and the full-length polypeptides are maintained. These Gli2/3 polypeptides are further modified by sumoylation, phosphorylation, and acetylation (Cox et al., 2010; Humke et al., 2010; Coni et al., 2013; Niewiadomski et al., 2014), and translocated into the nucleus to act as transcriptional activators (Gli2/3A; A for “active”).
In the nucleus, the representative DNA sequence for Gli2/3 is GACCACCCA/TGGGTGGTC (Sasaki et al., 1997). However, recent chromatin immunoprecipitation (ChIP) and computational prediction studies identified several other sequences with different affinities (Vokes et al., 2007; Vokes et al., 2008; Oosterveen et al., 2012; Peterson et al., 2012).
Compared with Gli2/3R, Gli2/3A are relatively labile, and are ubiquitinated at the nucleus by the E3 ubiquitin ligase adaptor speckle-type POZ protein (Spop) (Humke et al., 2010; Cai and Liu, 2017).
Target genes of the Hh signaling pathway are cell type-dependent. In most cells responsive to Hh, the common target genes are Ptch and Gli1 (Stamataki et al., 2005; Cohen et al., 2015; Tickle and Towers, 2017). In the developing neural tube, the Nkx6.1, Olig2, Nkx2.2, and FoxA2 genes (Vokes et al., 2007; Kutejova et al., 2016), which confer ventral neural fates, have Gli-binding sites in their regulatory regions (mentioned in detail later).
Additional Hh receptors that act in concert with Ptch and/or Smo have been identified and characterized. Hh-interacting protein (Hhip) is a single-pass membrane glycoprotein with epidermal growth factor (EGF)-repeat domains that binds to the three Hh proteins with similar affinity to Ptch (Chuang and McMahon, 1999). Although the downstream pathway remains to be fully elucidated, Hhip overexpression blocks Shh signaling activity, suggesting that Hhip is a negative regulator of Hh signaling (Bishop et al., 2009; Kwong et al., 2014). However, this effect occurs in a non-cell autonomous manner. The Hhip protein is released from producing cells and directly binds to Shh to block signaling (Kwong et al., 2014). On the other hand, in Shh-producing cells, Hhip gene expression is induced by the Hh signal, and the Hhip protein is internalized and degraded upon Smo activation (Kwong et al., 2014), thereby enabling cell autonomous Hh signaling. This complex regulatory loop may explain the temporal changes in Hh signaling activity (Dessaud et al., 2007; Balaskas et al., 2012). Other Hh receptor proteins identified include the glycosylphosphatidylinositol-anchored cell-surface protein growth arrest-specific 1 (Gas1), the membrane protein cell-adhesion molecule-related, downregulated by oncogenes (Cdo), and brother of Cdo (Boc) (Tenzen et al., 2006; Zhang et al., 2006; Allen et al., 2007; Allen et al., 2011). Cdo and Boc have immunoglobulin and fibronectin type III (FNIII) repeats in the extracellular domain, and possess structural properties similar to those of the axon guidance receptors Robo and deleted in colorectal cancer (Tessier-Lavigne and Goodman, 1996; Okada et al., 2006). Cdo and Boc function in axon guidance (Okada et al., 2006). In the developing chick limb, Cdo and Boc localize to discrete microdomains along extensive filopodial structures in the signal-receiving cells, and these receptor proteins interact with the filopodia (cytonemes) of Shh-producing cells (Sanders et al., 2013).
Hhip, Gas1, Cdo, and Boc bind to Shh directly. However, in contrast to Ptch, which localizes to cilia in the absence of Hh, these membrane proteins localize to the plasma membrane including filopodial structures. The distinct localization of the proteins may account for the variation in temporal and/or spatial Hh signaling activity. In summary, receptors in cilia and the plasma membrane interact with Hh proteins and transduce the signals into the nucleus, where Gli transcription factors act as effectors of Hh signaling.
Hh plays an important role in many biological events, and mutant mice deficient in Hh-related genes therefore show various and severe phenotypes. Mutations, deficiencies, and abnormal expression levels of Hh proteins and related genes cause malformations, hyperplasia, and growth retardation in tissues, especially in the central nervous system (CNS), craniofacial structures, limbs, and skeleton at developmental stages. Conversely, persistent or aberrantly high Hh activation leads to tumorigenesis in organs at postnatal stages. In the next sections, we discuss the tissue distribution of Hh proteins, especially Shh, and the related genes. In addition, we provide data on the critical roles of these molecules in development and tissue homeostasis, which are mostly derived from studies in model mice. The principal gene knockout or mutant phenotypes in mice are summarized in Table 1.
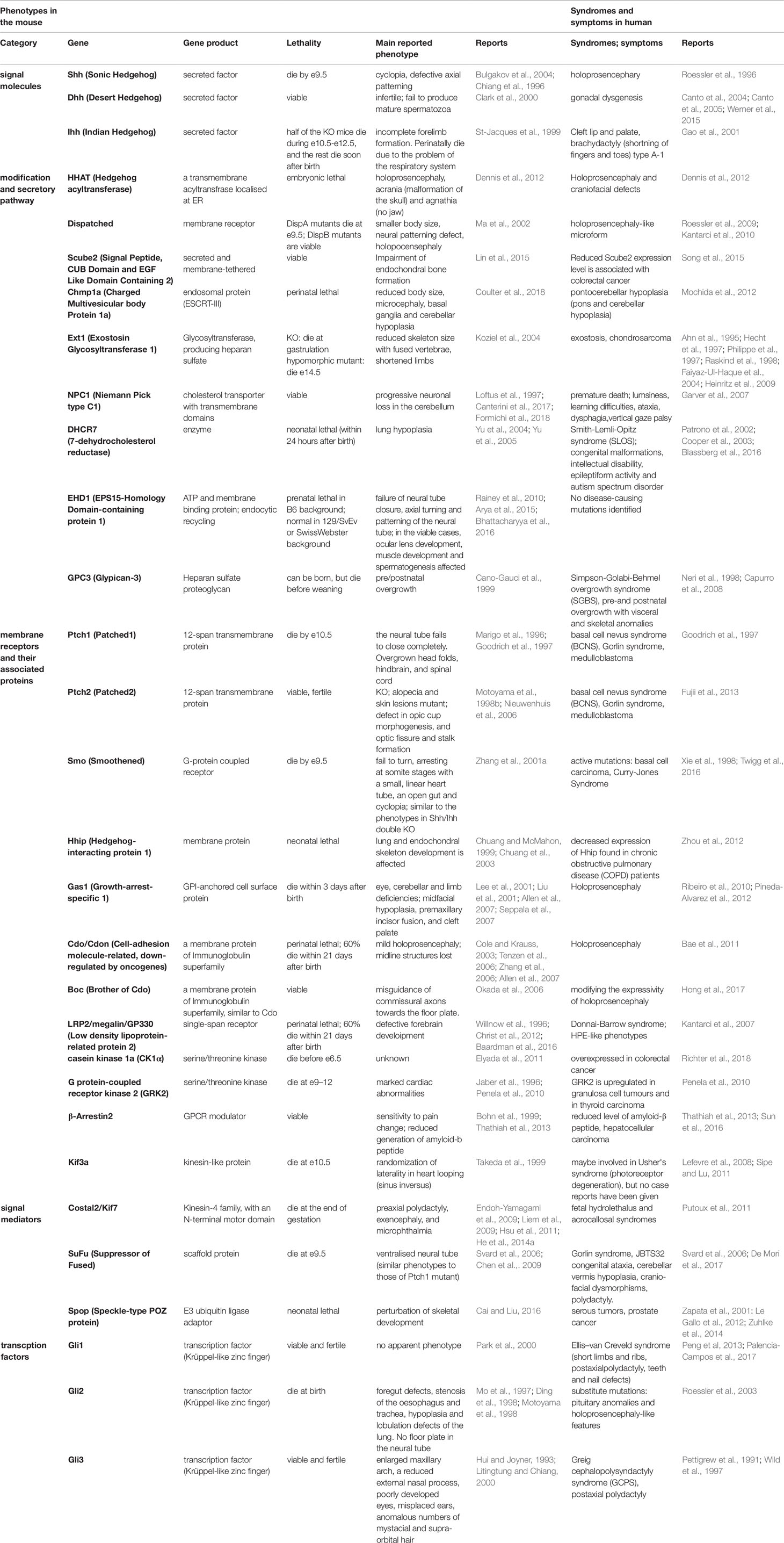
Table 1 Main phenotypes and symptoms caused by the mutations and deficiencies of the Hh-related genes in mice and in humans.
Hh genes are expressed in a variety of tissues throughout developmental and postnatal stages (Petrova and Joyner, 2014), and play critical roles in the development and homeostasis of these tissues. Among the three vertebrate Hh genes, Shh is highly expressed starting at early developmental stages and shows strong activity; therefore, it has been analyzed extensively. Shh expression is initially detected in the axial mesoderm in the head and caudal areas at embryonic day 7.5 (e7.5) (Echelard et al., 1993; Marti et al., 1995; Blaess et al., 2014). At e8.5, Shh expression is detected in the neural tube (i.e., the embryonic CNS). Shh expression in the neural tube is restricted to the ventral region. At the trunk level, Shh is expressed in the floor plate domain, which is located at the ventral-most part of the neural tube. In addition to its expression in the nervous system and the axial mesoderm, Shh is expressed in the zone of polarizing activity (ZPA) of the fore- and hind-limb starting at e9.5 (Echelard et al., 1993). This expression pattern is maintained until late developmental stages (Echelard et al., 1993).
One important characteristic of Shh is that it acts as a morphogen. Morphogens are a group of signaling (secretory) molecules that determine different cell fates in a concentration-dependent manner (Wolpert, 1969; Cohen et al., 2013; Matise, 2013; Briscoe and Small, 2015; Tickle and Towers, 2017). Shh is expressed in the floor plate in the neural tube, and its levels reach a concentration gradient along the dorsal-ventral axis, assigning different neural identities in a concentration-dependent manner (Alaynick et al., 2011). This is experimentally proven by incubating neural progenitor cells with different concentrations of Shh, which results in the production of different neural subtypes after a certain time period (Dessaud et al., 2007; Dessaud et al., 2010). However, the concentration of Shh in the neural tube changes dynamically over time during embryogenesis, and the distribution of the Shh protein does not correspond to intracellular Shh activity or Gli activity (Balaskas et al., 2012). Cells responsive to Shh show a rapid increase in activity followed by a gradual decrease. Multiple mechanisms for this negative feedback (termed "temporal adaptation") (Dessaud et al., 2007; Cohen et al., 2015) have been proposed. One mechanism involves the expression of Ptch (Dessaud et al., 2007); the Ptch gene is induced by Shh, and the accumulation of Ptch in the ciliary membrane blocks the Shh signal (Dessaud et al., 2007). Moreover, Gli2 transcription in the neural tube gradually decreases during development, and this decrease downregulates the Shh signal transduction over time (Cohen et al., 2015). In addition, the new regulator GPR17, which plays an essential role in this temporal adaptation, was recently isolated (Yatsuzuka et al., 2019).
In the limb bud, a classical experiment shows that transplantation of ZPA into the anterior side of the limb bud induces an extra number of fingers, which is called "polydactyly" (Tickle and Towers, 2017). Therefore, the regulation of Shh activity in the limb bud is critical for proper digit formation.
Shh is expressed in the CNS and limb bud starting at early stages, and it plays essential roles in the differentiation and construction of these tissues. Mice deficient in the Shh gene have holoprosencephaly (smaller head) with cyclopia (single eye), and lack ventral cell types within the neural tube of the spinal cord and in most of the ribs. Consequently, knockout mice usually die by e10.5 (Chiang et al., 1996; Bulgakov et al., 2004).
Shh is also expressed in the lung, which is an endoderm-derived organ, and Shh is expressed in the distal epithelium at least from e10.5 to e16.5. Shh is detected in epithelial cells both in peripheral and conducting airways (Miller et al., 2001). Analysis of Shh knockout mice with a mild phenotype shows defects in foregut development and lung branching morphogenesis (Litingtung et al., 1998; Pepicelli et al., 1998).
Although Shh plays essential roles in various developmental stages, it is also expressed and plays important roles at postnatal stages (Alvarez-Buylla and Ihrie, 2014; Petrova and Joyner, 2014), particularly in brain neural stem cells. In the adult brain, two regions undergo continuous neurogenesis: the ventricular subventricular zone (V-SVZ) and the subgranular zone (SGZ) of the dentate gyrus (Ahn and Joyner, 2005; Alvarez-Buylla and Ihrie, 2014). After these regions are established (Ahn and Joyner, 2005), both quiescent and transit-amplifying neural stem cells respond to Shh and start to expand (Ahn and Joyner, 2005; Alvarez-Buylla and Ihrie, 2014). Shh is required for the maintenance of stem cells and migrating newborn neurons (Balordi and Fishell, 2007a). Analysis of tamoxifen-inducible Smo mutants shows that both proliferation and neurogenesis of neural stem cells are compromised (Balordi and Fishell, 2007b). Therefore, Shh is required for the maintenance, proliferation, differentiation, and migration of adult neural stem cells.
Ihh is mainly expressed in the developing skeletal system. The Ihh mRNA can be detected in cartilage at e11.5, and later in prehypertrophic chondrocytes (Bitgood and McMahon, 1995; St-Jacques et al., 1999). In developing chondrocytes, parathyroid hormone-related peptide (PTHrP) is one of the target genes of Ihh. PTHrP in turn antagonizes Ihh and sequesters the Ihh signal, and thereby proceeds differentiation.
Half of the Ihh knockout mice die between e10.5 and e12.5, probably because of circulatory abnormalities. The rest of the mutant mice remain alive until the end of gestation, but die at birth from respiratory failure. The mutant embryos are smaller in size and have short forelimbs with reduced chondrocyte proliferation (St-Jacques et al., 1999; Karp et al., 2000; Levi et al., 2011). PTHrP expression is downregulated in Ihh knockout mice (Karp et al., 2000). However, constitutive expression of PTHrP in the Ihh knockout background cannot rescue all Ihh knockout phenotypes, suggesting the existence of a PTHrP-independent pathway involved in the short-limb phenotype or decreased chondrocyte proliferation.
Although Ihh knockout mice are embryonic-lethal, the postnatal function of Ihh was elucidated by conditional gene disruption in a tamoxifen-inducible system (Zhou et al., 2014). In these conditional knockout mice, surgically induced osteoarthritis (OA) was attenuated, whereas wild-type mice showed OA progression (Zhou et al., 2014). This result provides possible novel therapeutic strategies to prevent OA progression.
In the mouse, Dhh is expressed in Sertoli cells immediately after sex determination at e11.5 (Bitgood et al., 1996) and in Schwann cells, which are a component of the peripheral nervous system, at e14.5 (Bitgood and McMahon, 1995). Dhh expression lasts until the postnatal stages (Bitgood and McMahon, 1995; Bitgood et al., 1996). Dhh-deficient mice are viable, and show sex-specific phenotypes. Female knockout mice do not exhibit an obvious phenotype and are fertile, whereas mutant male mice show defects in spermatogenesis and are therefore infertile (Bitgood et al., 1996; Clark et al., 2000). In the adult testis, Dhh is expressed in Sertoli cells, and Dhh knockout mice lack Ptch1 expression in neighboring Leydig cells, suggesting that Leydig cells are the target of Dhh (Bitgood et al., 1996). In the peripheral nervous system, detailed analysis of Dhh mutant mice showed a thin perineurium and abnormal formation of perineural tight junctions (Parmantier et al., 1999). These findings suggest that Dhh is also important for the development of peripheral nerve sheaths.
Taken together, these studies indicate that Hh signaling is critical not only during embryogenesis, but also for postnatal biological processes.
The acyltransferase HHAT is essential for embryonic development (Dennis et al., 2012). Deficiency of HHAT function perturbs long-range Hh transport in tissues, and affects the activity of other signaling molecules of FGF and BMP as a secondary effect, which results in holoprosencephaly and craniofacial defects (Dennis et al., 2012; Konitsiotis et al., 2015).
Chmp1a is an ESCRT-III component that mediates Shh secretion through the endosome. Shh secretion is impaired in Chmp1a knockout mice, and mutant mice show a small body size and microcephaly (small brain), and die soon after birth (Coulter et al., 2018). This phenotype can be reversed through crossing with a Ptch1 heterozygote. Ptch per se antagonizes the Hh signal by blocking Smo activity, suggesting that Hh signaling is activated in the Ptch mutant (Goodrich et al., 1997). The reversal of the phenotype by the heterozygotic reduction of Ptch expression under the Chmp1a-deficient background suggests that the phenotypes caused by single Chmp1a gene knockout are caused by decreased Hh function (Coulter et al., 2018).
The ESCRT-II subunit Vps25 (Handschuh et al., 2014) does not directly affect Shh secretion. However, degradation of the FGF receptor is perturbed in Vps25 hypomorphic mutants. This increases FGF activity and accelerates the feedback loop between FGF and Shh involved in limb bud development. Deficiency of the Vps25 gene therefore represents aberrant upregulation of Shh, and results in the polydactyly phenotype (Handschuh et al., 2014).
Two proteins of Disp and Scube2 are related to the Hh protein releasing process. DispA is detected from e7.5 throughout the embryo stage (Ma et al., 2002), and DispA mutant embryos die at e9.5 with defects in head structure and abnormal neural patterning (Ma et al., 2002). In mutant mice, the Hh modification is intact, but the secretion process is perturbed (Ma et al., 2002). Scube2 encodes a secreted and membrane-tethered protein with complement C1r/C1s, Uegf, Bmp1 (CUB)- (Bork and Beckmann, 1993) and EGF-like domains, and plays essential roles in chondrocyte differentiation and cell proliferation (Lin et al., 2015). Detailed analysis revealed that Scube2 is a strong promoter of Ihh activity (Lin et al., 2015).
After Hh proteins are released from producing cells, their stability and tissue distribution depend on the extracellular environment. For instance, vitronectin is an extracellular matrix protein that acts synergistically with Shh during motor neuron differentiation (Pons and Marti, 2000). By contrast, the heparan sulfate (HS) chain, synthesized by the glycosyltransferase exostosin1 (Ext1), restricts the spread of Hh (Koziel et al., 2004; Callejo et al., 2006). Ext1 acts in the Golgi apparatus and catalyzes the elongating reaction of the HS tetrasaccharide chain, thereby contributing to the production of HS proteoglycan (HSPG) (Koziel et al., 2004). HSPG binds to Ihh and restricts Ihh signaling in chondrocytes. In Ext1 hypomorphic mutants (mutants in which the Ext1 expression is significantly reduced), HS synthesis is suppressed and hypertrophic chondrocyte differentiation is delayed, presumably because Ihh is spread in the tissue (Koziel et al., 2004). In a similar process, deficiency of the Ext2 gene results in the reduction of HS synthesis and early embryonic lethality (i.e., embryos die around e6.5) (Stickens et al., 2005).
One of the glypican family proteins, glypican-3 (GPC-3), is expressed on the surface of Hh-receiving cells and directly binds to Hh proteins (Capurro et al., 2008), thereby suppressing Hh signaling (Capurro et al., 2008). This activity involves endocytosis and degradation of the GPC-3-Hh complex, and low-density lipoprotein receptor-related protein-1 plays an essential role in this process (Capurro et al., 2012).
The interaction between GPC-3 and Hh competes with the Ptch-Hh interaction. Therefore, the phenotypes of GPC-3 mutant mice are related to excessive Hh signaling, and are characterized by embryonic overgrowth (heavier embryos) with expanded ventral identity in the spinal cord, and renal and lung anomalies (Cano-Gauci et al., 1999; Chiao et al., 2002).; Capurro et al., 2008)
The main function of Ptch1 (Goodrich et al., 1997) and Ptch2 (Alfaro et al., 2014; Zhulyn et al., 2015) in Hh signaling is to inhibit Smo activity, thereby maintaining the quiescence of the Hh signaling pathway. Knockout of the Ptch1 gene causes constitutive activation of Smo and its downstream Hh pathway, and the phenotype is reminiscent of that caused by overactivation of the Hh pathway. For instance, Ptch1 knockout mice, which die by e10.5, show a ventralized neural tube (Goodrich et al., 1997). Ptch2 plays redundant roles during embryogenesis (Motoyama et al., 1998b; Zhulyn et al., 2015), and the phenotypes of Ptch2 mutants are manifested at postnatal stages; the mutation causes alopecia (hair loss) and epidermal hyperplasia (Nieuwenhuis et al., 2006).
Studies of another membrane protein, Hhip, show that Hhip knockout mice die at neonatal stages because of lung and endochondral skeleton defects (Chuang et al., 2003). Conversely, transgenic mice in which Hhip is overexpressed in cartilage show a shortened skeleton, which is similar to the phenotype of Ihh knockout mice (Chuang and McMahon, 1999), suggesting that Hhip is a negative regulator of the Hh signal.
During the translocation of Smo into primary cilia, phosphorylation of the serine residues at the carboxyl-terminal region is essential for its full function. Although CK1α and GRK2 are essential for the phosphorylation and translocation of Smo into cilia, they have a wide variety of substrate proteins, namely, Smo is not the only substrate for these kinases. Therefore, the phenotypes associated with mutations of each gene are more severe or distinct from the ones expected from the deficiency of Hh signaling molecules (Jaber et al., 1996; Elyada et al., 2011).
β-Arrestin 2 also has a variety of partner proteins. In zebrafish, mutation of the Smo gene and knockdown of β-arrestin 2 produce similar phenotypes characterized by defects in muscle and ventral neural tube differentiation. Moreover, the phenotypes caused by the Smo mutation can be rescued by overexpression of β-arrestin 2, suggesting that there are strong epistatic connections between Smo and β-arrestin 2 (Wilbanks et al., 2004). However, β-arrestin 2 mutant mice show different phenotypes involving changes in pain sensitivity (Bohn et al., 1999) and the generation of amyloid β-peptide, which is involved in Alzheimer's disease (AD) (Thathiah et al., 2013), in postnatal stages. Therefore, the dependency on β-arrestin 2 during development may be species-specific, and is partly redundant with other genes in the mouse.
When Smo is translocated into cilia, the endocytic recycling regulatory protein EPS15-homology domain-containing protein 1 (EHD1) (Rainey et al., 2010; Arya et al., 2015; Bhattacharyya et al., 2016) directly interacts with Smo, and is co-trafficked into primary cilia. EHD1-null cells show alterations in the shape of cilia, and the embryonic phenotype is the ventralized neural tube, which is caused by hyperactivation of Hh signaling (Bhattacharyya et al., 2016). This suggests that EHD1 affects the localization or the stability of the Smo protein.
Gas1, Cdo, and Boc are expressed in the dorsal part of the neural tube and in the anterior two-thirds of the forelimb bud in e10.5 mouse embryos, which is complementary to the expression of Shh (Allen et al., 2011). However, overexpression of each of these factors in the neural tube induces the ventralization of the neural tube, suggesting that these factors support Shh signaling (Allen et al., 2011). Cdo and Boc have partially redundant roles in neural tube dorso-ventral pattern formation and digit specification during limb bud development. Cdo/Boc compound mutants show a markedly defective structure, whereas the phenotypes are subtle in each single mutant. By contrast, the Gas1 mutant on its own has a strong phenotype in these tissues (Allen et al., 2011). Triple mutants deficient in the three receptors Gas1, Cdo, and Boc display severe forebrain and cardiovascular defects at e8.5, and later holoprosencephaly, cyclopia, and neural tube pattern defects (Allen et al., 2011), which resemble the Shh/Ihh compound or Smo knockout phenotypes (Zhang et al., 2001b). The mechanisms underlying the coordination of Hh signal reception between Gas1, Cdo, and Boc and the conventional receptor system consisting of Ptch/Smo need to be addressed in the future.
Among mediators of the Hh signaling pathway, SuFu and its modulator Kif7 play critical roles in tissue morphogenesis, and the respective mutants therefore exhibit severe phenotypes.
SuFu mutant mice exhibit a severely ventralized pattern in the neural tube, suggesting that SuFu is a negative regulator of Hh signaling (Svard et al., 2006; Chen et al., 2009).
Kif7, a kinesin-4 family motor protein, localizes to the distal tip of primary cilia in vivo and determines the length of cilia (He et al., 2014a). The Kif7 mutant is characterized by long and twisted cilia (He et al., 2014a).
Kif7 binds to Gli2/3, and Gli activity is aberrantly upregulated in Kif7 mutant cells (Cheung et al., 2009). In addition, ventral domains in the neural tube are expanded and polydactyly is observed in the mutant (Endoh-Yamagami et al., 2009; Liem et al., 2009), suggesting that Kif7 acts as a negative regulator of Hh signaling. In addition, Kif7 mutation partially rescues the phenotype caused by Smo knockout, confirming that Kif7 acts as a negative effector in the Hh pathway downstream of Smo (Liem et al., 2009). However, the effect of combined Kif7 knockout and Ptch1 mutation on rescuing the phenotype indicates that Kif7 can also promote the activity of the Shh pathway (Liem et al., 2009).
Kif7 functions both as a positive and a negative factor in chondrocyte differentiation also in the growth plate (Hsu et al., 2011). Kif7 plays a positive role for the Hh activity by excluding the Gli-SuFu complex from primary cilia, thereby limiting the repressive effect of Gli. However, in the absence of SuFu, Kif7 inhibits Gli transcriptional activity (Hsu et al., 2011). The dual role of Kif7 may be dependent on other partner proteins forming a complex with SuFu and Kif7.
Spop regulates the stability of the Gli2 and Gli3 proteins by ubiquitinating at their carboxyl-terminal domains (Wang et al., 2010). However, the phenotypes in the Spop mutants are more complex than expected. Single Spop deficiency does not severely affect dorsal-ventral pattern formation in the neural tube (Cai and Liu, 2017). On the other hand, Spop and Gli2 compound mutants rescue the loss of ventral neural identity caused by the Gli2 single mutant, suggesting a negative role of Spop in Gli activity (Cai and Liu, 2017). However, during skeletal development, Spop deficiency results in the accumulation of Gli3R, and causes defects in chondrocyte and osteoblast differentiation (Cai and Liu, 2016). Therefore in this context, Spop acts as a positive regulator of Ihh signaling/skeletal development. These findings indicate that the activity of Spop in the Hh signaling pathway is context-dependent.
At the transcriptional level, Gli genes play partially overlapping roles in mediating Shh intracellular signaling (Bai and Joyner, 2001; Lei et al., 2004). However, the three genes have different requirements. Gli1-deficient mice do not show apparent phenotypes and are viable and fertile (Park et al., 2000), whereas Gli2 knockout mice die at birth (Motoyama et al., 1998a). Gli3 mutants are viable and exhibit tumorigenic phenotypes with craniofacial anomalies (Hui and Joyner, 1993; Veistinen et al., 2012). Moreover, Gli2 and Gli3 have partially redundant functions in limb bud development (Mo et al., 1997). The different requirements of Gli proteins may be related to spatial differences in the expression of the three Gli proteins.
Cholesterol biosynthetic enzymes play indirect but important roles in Hh signaling. Cholesterol and fatty acids are synthesized from acetyl-CoA through several steps. The enzyme 7-dehydrocholesterol reductase (DHCR7) converts 7-dehydrocholesterol to cholesterol in the last step of cholesterol synthesis (Yu et al., 2004; Blassberg et al., 2016). Mice deficient in the DHCR7 gene die immediately after birth from lung hypoplasia (Yu et al., 2004). At the molecular level, full activation of Smo requires cholesterol binding at the extracellular cysteine-rich domain (Nedelcu et al., 2013; Luchetti et al., 2016), which induces a conformational change in Smo to its active state (Huang et al., 2016; Myers et al., 2017; Xiao et al., 2017).
The NPC1/2 proteins are also critical for Hh modification. NPC1/2 transport cholesterol from the endosome to the ER/Golgi (Urano et al., 2008), and this transport is blocked in NPC1/2-deficient mice. This results in the accumulation of cholesterol in the lysosome, which prevents cholesterol modification of Hh proteins as well as Ptch and Smo (Orth and Bellosta, 2012) and the production of active proteins. NPC1/2-deficient mice are characterized by increased cholesterol levels and progressive neuronal loss in the cerebellum (Loftus et al., 1997; Canterini et al., 2017; Formichi et al., 2018). In addition, shortened primary cilia are detected in NPC1 mutant cells (Formichi et al., 2018), suggesting that NPC affects the production of sterols in cilia, and Hh signal transduction is perturbed not only at the secretion level but also at the receptor level.
Several target genes are induced by Hh signals. In the spinal cord, the transcription factors that determine neural identity are induced by Shh in a concentration-dependent manner (Dessaud et al., 2008) and form regulatory networks (Briscoe and Small, 2015). The transcription factors FoxA2, Olig2, and Nkx2.2 have Gli-binding sites in their regulatory domains (Vokes et al., 2007; Kutejova et al., 2016). Olig2 and Nkx2.2, which are basic helix-loop-helix and homeobox transcription factors, respectively, are induced by Shh and repress each other (Kutejova et al., 2016). In addition, Nkx2.2 (expressed in the ventral p3 domain) and Pax6 (expressed in the intermediate domains) are mutually repressive (Briscoe et al., 2000). Knockout of each of these components perturbs the transcriptional network and alters pattern formation (Balaskas et al., 2012).
In limb development, Shh induces a set of genes distinct from those involved in neural development (Vokes et al., 2008; Lewandowski et al., 2015). For instance, the transcription factor Blimp1, which is expressed in the posterior forelimb, is a direct target gene of Shh and is essential for the maintenance of ZPA and other mesodermal tissues in mice (Robertson et al., 2007). Gremlin and Hand2, which encode a BMP antagonist and a transcription factor, respectively, are expressed in a region distinct from that of Shh. Nevertheless, they have Gli-binding sites in their regulatory regions and are directly regulated by active Gli (Vokes et al., 2008). As in the neural tube, Shh acts in a long-range and concentration-dependent manner in the limb bud, and target gene expression is regulated both spatially and temporally.
In the Ihh pathway, because PTHrP is a target gene of Ihh signaling, disruption of PTHrP or its receptor PTH/PTHrP-R results in skeletal dysplasia, premature maturation of chondrocytes, and excessive bone formation, and causes a lethal phenotype (Karaplis et al., 1994; Lanske et al., 1996). Overall, the critical roles of Hh molecules are at least partly reflected in the downstream factors and their transcriptional networks.
Although the pathway composed of Ptch, Smo, Gli, and their associated factors plays a major role in Hh function, other pathways that bypass this canonical pathway have been analyzed. Shh functions in axon guidance in commissural axons (Charron et al., 2003; Hammond et al., 2009; Aviles et al., 2013) in collaboration with the long-range diffusible chemoattractant netrin (Kennedy et al., 1994; Charron et al., 2003). Notably, Shh-dependent axon guidance is not mediated by Ptch, but Boc (not Cdo) acts as a receptor for Shh (Okada et al., 2006). Boc knockout mice show misplaced axons invading into the motor column (Okada et al., 2006).
Shh acts as an attractant before commissural axons cross the midline, whereas it acts as a repellent for post-crossing axons. This is related to changes in the protein composition of the growth cone. Accumulation of the adaptor protein 14-3-3 in a time-dependent manner results in repulsive axons (Yam et al., 2012). Because of this change in responsiveness, immediately after crossing the midline the axons are redirected perpendicularly to the rostral direction, where Shh concentration is low (Bourikas et al., 2005). Recently it has been shown that Boc is internalized into early endosomes and this endocytosis is required for the growth-cone turning of neurons (Ferent et al., 2019). Boc is also enriched in ipsilateral retinal ganglion cells (RGCs), and Shh, expressed at the midline, acts as a repellent for the RGCs, which thereby regulates the segregation of ipsilateral and contralateral neurons (Fabre et al., 2010).
In chick, Hhip is transiently expressed in the commissural axon at the time when axons turn into the longitudinal axis (Bourikas et al., 2005). However, this mechanism is not detected in mice, suggesting that the mechanism is species-specific.
Src family kinases (SFKs) are downstream Boc signaling effectors (Yam et al., 2009). While SFK activation is required for axonal guidance, it is not involved in Gli-mediated transcription. In Shh-mediated chemotaxis, cilia are not essential for pathway activation, suggesting that the role of the Shh pathway in chemotaxis and axon guidance is mediated by other factors than Ptch-Smo-Gli (Bijlsma et al., 2012). The extension of the axon correlates with the Wnt signal, supporting a mechanistic difference in the Shh pathway from that mediated by the Ptch-Smo-Gli axis (Aviles et al., 2013).
Mutations or deletions in genomic regions, either in coding or non-coding regions, cause malfunctions of the translated proteins or alterations in gene expression levels, resulting in genetic disorders. The phenotypic penetrance caused is variable in humans compared with that resulting from gene knockout or disease models in mice, as a pure genetic background and rearing under standardized conditions confer sensitivity to genetic mutations. In human mutations, complete removal of genes is rare, as genomic mutations normally encode proteins with incomplete or submaximal levels of activity. Nevertheless, mutations of the indicated Hh-related genes result in severe hereditary disorders in humans, and in most cases, the effects are similar to those in knockout mice (overall symptoms based on the mutations of the Hh-related genes are summarized in Table 1, and the detailed symptoms are shown in Table 2).
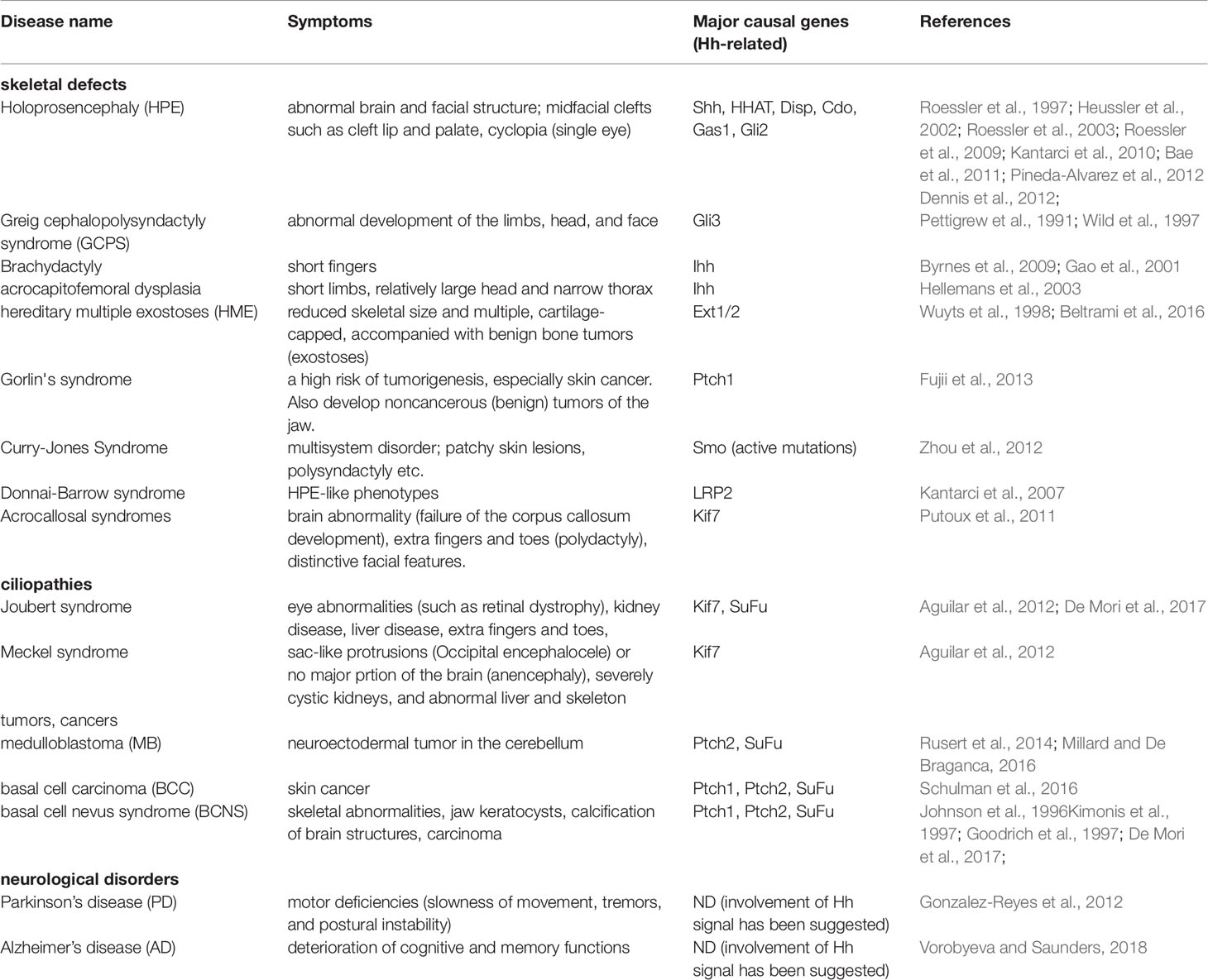
Table 2 Detailed symptoms in the Hh-related disease. ND, not determined.
Among the three Hh genes, Shh, Ihh, and Dhh, mutations in Shh or its abnormal expression produces severe symptoms in humans, including abnormalities in the development of the nervous system, facial structure, and limbs.
Holoprosencephaly (HPE) is a common disorder of brain development with a prevalence of 1 in 250 embryos and in 16,000 live births (Roessler et al., 1997). In HPE, the cerebral hemisphere is not formed, and the patients develop an abnormal brain and facial structure, with frequent midfacial clefts such as cleft lip and palate (Roessler et al., 1997). Severe cases of HPE, which are rare, show cyclopia (single eye) (Sharma et al., 2014). This syndrome is not only associated with mutations in Shh (Roessler et al., 1997; Heussler et al., 2002), but also with those in downstream genes. To date, HHAT (Dennis et al., 2012), Disp (Roessler et al., 2009; Kantarci et al., 2010), Cdo (Bae et al., 2011), Gas1 (Pineda-Alvarez et al., 2012), and Gli2 (Roessler et al., 2003) have been reported as causal genes for HPE.
Greig cephalopolysyndactyly syndrome (GCPS or Greig syndrome), which is characterized by abnormal development of the limbs, head, and face, is considered a genetic developmental disorder. It shows an autosomal dominant inheritance pattern (Pettigrew et al., 1991; Wild et al., 1997), and Gli3 has been recognized as the causative gene for GCPS and its related disorder, Pallister-Hall syndrome (Kang et al., 1997).
In addition Shh-related genes, the genes encoding the transcription factors Zic2, Six3, and TGF-β induced factor homeobox 1 (TGIF 1) are considered major causative genes for HPE (Dubourg et al., 2004; Dubourg et al., 2016). Recent studies using next generation sequencing show that the FGF signaling-related genes FGF8 and FGF receptor 1 are also causal genes (Dubourg et al., 2016).
Mutation of the Ihh gene causes defects in skeletal development, and is associated with brachydactyly (short fingers) (Gao et al., 2001; Byrnes et al., 2009) and acrocapitofemoral dysplasia (short limbs, relatively large head, and narrow thorax) (Hellemans et al., 2003). In all reported cases, the mutations are located in the amino-terminal active fragment of Ihh (Byrnes et al., 2009).
Another Ihh-related growth defect is hereditary multiple exostoses (HME), which is characterized by reduced skeletal size and multiple, cartilage-capped bony outgrowths, accompanied by benign bone tumors (exostoses) in endochondral bones (Wuyts et al., 1998; Beltrami et al., 2016). HME and two other related bone disorders, exostosis and chondrosarcoma, are found in families that have mutations in the Ext1/2 genes (Ahn et al., 1995; Hecht et al., 1997; Philippe et al., 1997; Raskind et al., 1998; Faiyaz-Ul-Haque et al., 2004; Heinritz et al., 2009). Ext1 is essential for HS synthesis and proper Ihh signal distribution (Koziel et al., 2004) in the mouse, and the symptoms of HME are reminiscent of the mutant phenotypes.
Mutations in Dhh are associated with congenital disorders. Dhh mutation was found in a patient with premature female genitalia and an immature uterus, regardless of the XY karyotype (Umehara et al., 2000). This patient also suffered from neuropathy (Umehara et al., 2000), which is reasonable considering that Dhh is expressed in the peripheral nervous system (Parmantier et al., 1999).
Primary cilia act as a cellular "antenna" that receives chemical and mechanical signals from the extracellular environment to transduce them into the cells, and play important roles in development and homeostasis (Sasai and Briscoe, 2012; Wheway et al., 2018). Several transmembrane receptors related to Wnt, Notch, Hippo, and platelet-derived growth factor, as well as the receptors for Hh (i.e., Ptch, Gli; Figure 1) localize to cilia (Sasai and Briscoe, 2012; Malicki and Johnson, 2017; Wheway et al., 2018). Therefore, ciliary dysfunction abrogates signal transduction by these factors, resulting in a group of diseases termed ciliopathies.
The symptoms of ciliopathies are malformation of the body, including abnormal neural development (Waters and Beales, 2011; Guo et al., 2015) and situs inversus (major visceral organs are deployed with right and left sides reversed) (Pennekamp et al., 2015), during development, and retinal degeneration (Wheway et al., 2014; Bujakowska et al., 2017), kidney disease (Hildebrandt et al., 2009; Habbig and Liebau, 2015), and male infertility (Inaba and Mizuno, 2016) in the postnatal stages (Nigg and Raff, 2009; Waters and Beales, 2011; Shaheen et al., 2016). To date, more than 400 proteins have been identified and characterized, and many more unknown genes/proteins are expected to exist (Nigg and Raff, 2009; Goetz and Anderson, 2010; Reiter and Leroux, 2017).
Ciliary defects cause symptoms similar to those found in Hh signaling mutants such as polydactyly, encephalocele, neural tube defects, and cerebellar vermis hypoplasia (Huangfu et al., 2003; Sasai and Briscoe, 2012; Bangs and Anderson, 2017). Dysfunction of Jbts17 (also known as CPLANE1), a gene mutated in Joubert syndrome, Meckel syndrome, and oral-facial-digital syndrome, causes ciliogenesis and ciliary trafficking defects, and results in decreased Shh target gene expression (Damerla et al., 2015; Toriyama et al., 2016). Disruption of the Cep290 gene, which encodes a centrosomal protein, causes severe renal defects (Hynes et al., 2014), and treatment with purmorphamine, an agonist of Hh signaling (Briscoe, 2006), increases the number of ciliated cells. This suggests that Hh signaling is a potential therapeutic target in some ciliopathies. Similarly, mutations in MKS1 (Weatherbee et al., 2009) and MKS3 (Abdelhamed et al., 2013), which encode centrosome and orphan proteins, respectively, cause systemic defects related to ciliogenesis in association with alterations in Shh, and sometimes Wnt signaling.
Blockade of primary cilia normally perturbs the introduction of Hh into cells. However, lack of primary cilia caused by Kif3a mutation increases Hh activity in facial mesenchymal cells and results in craniofacial defects, hypertelorism (abnormal distance between the eyes), and frontonasal dysplasia (FND; developmental disorder with abnormal facial structure such as distant eyes and flat nose) (Wong et al., 2009; Brugmann et al., 2010).
Because the symptoms of ciliopathies are chronic, the patients are normally not easily treatable. Nevertheless, a number of trials have been made to cure these intractable diseases. For instance, gene therapy, where a viral infection of the functional ciliary genes into the body, has been shown to be successful to restore sensory input in the olfactory ciliopathy model mice (Green et al., 2018). In addition, by utilizing the method to make an organoid (a imitated three-dimensional organ differentiated in vitro from stem cells (Rossi et al., 2018)), a three-dimensional retina has been made from the iPS cells derived from the patients of retinitis pigmentosa, a type of ciliopathies. This method is useful because the progress of ciliopathies can be chased in vitro, and gives hints to find the strategies for the treatment (Deng et al., 2018). In addition, a microscopy-based automated system for small molecule screening was designed to search for new drugs that encourage ciliation. This system is expected to facilitate drug screening to find good chemicals to treat ciliopathies (Zhang et al., 2019).
Decreased Hh activity causes systemic tissue hypoplasia; however, overactive and persistent Hh signaling also leads to abnormalities in tissue homeostasis (Petrova and Joyner, 2014).
The representative phenotype induced by persistent activation of the Hh signal is cancer (Gupta et al., 2010). Two major types of cancer associated with Hh signaling have been extensively studied, namely, medulloblastoma (MB) and basal cell carcinoma (BCC) (Teglund and Toftgard, 2010).
MB is a primitive neuroectodermal tumor that most commonly arises from the cerebellum, and is normally diagnosed at a young age, especially in children younger than 10 years (de Robles et al., 2015). Approximately 200–500 cases are diagnosed in the United States every year (https://www.cancer.net/cancer-types/medulloblastoma-childhood/statistics). MB is divided into four types, Wnt-activated, Shh-activated, group 3, and group 4, according to diagnostic symptoms, genetics, and gene expression (Rusert et al., 2014; Millard and De Braganca, 2016). Approximately 30% of all cases, whether sporadic or genetic in nature, that are found in the cerebellar hemispheres (Rusert et al., 2014; Millard and De Braganca, 2016) are the Hh-activated type. In cases of germline mutations (genetic background), the Ptch and SuFu tumor suppressor genes tend to be abnormal (Yin et al., 2019).
BCC is a common skin cancer, accounting for >75% of all skin cancers (Zhang et al., 2001a). BCC is detected in 450–490 per 100,000 individuals in the UK (Epstein, 2008), and some BCC is associated with mutation of the Ptch gene (Gailani et al., 1996; Johnson et al., 1996). Analysis of biopsy samples from human BCC patients identified mutations in the p53 and Ptch1 genes in 30–50% and 20–30% of cases, respectively (Zhang et al., 2001a). Its related syndrome, basal cell nevus syndrome (BCNS), is characterized by skeletal abnormalities, jaw keratocysts, and calcification of brain structures in addition to carcinoma (Kimonis et al., 1997), and mutations in the Ptch1/2 and SuFu genes have been found in BCNS patients (Johnson et al., 1996; Goodrich et al., 1997; Kimonis et al., 1997; De Mori et al., 2017).
Other types of cancer caused by the abnormal Hh expression include glioma (Gupta et al., 2010). Glioma is induced by the aberrant proliferation of glial cells both in the brain and in the spinal cord (Gupta et al., 2010).
A number of studies have shown that Hh, as well as Wnt, BMP, and Notch, pathway acts for the initiation step of glioma, namely gliomagenesis (Palma and Ruiz i Altaba, 2004; Laug et al., 2018). Importantly, human glioma-initiating cells (GICs; also known as glioma stem cells) undergo an indefinite self-renewal and are resistant to conventional anti-cancer drugs (Clement et al., 2007; Takezaki et al., 2011; Auffinger et al., 2015; Phi et al., 2018). On the other hand, it was demonstrated that overexpression of a C-terminal-truncated form of Gli2, exerted an antagonistic effect on the Gli activity and knocking-down of Gli1 and Gli2 expression prevented proliferation of GICs and their tumorigenesis (Clement et al., 2007; Takezaki et al., 2011). Therefore, it would be possible to cure the gliomagenesis by blocking the Shh signaling pathway, and as mentioned above, candidate chemicals are being tested for clinical applications (Pietrobono and Stecca, 2018).
As for therapy of the tumors, Hh antagonist could be used to block the signal. For instance, cyclopamine, which strongly blocks the Hh signal pathway, could have been a candidate chemical to ameliorate the symptoms (Chen et al., 2002). However, poor oral solubility and severe side effects in mice limited further clinical development of cyclopamine (Pietrobono and Stecca, 2018). Several other chemicals, which either bind to the Smo protein or inhibit its ciliary localization, have been developed and are currently under evaluation at preclinical or clinical trial stages (Pietrobono and Stecca, 2018). Among them, two Smo inhibitors, vismodegib and sonidegib, have already been approved by the US Food and Drug Administration (FDA) for treating the advanced BCC (Axelson et al., 2013; Migden et al., 2018; Pietrobono and Stecca, 2018; Niyaz et al., 2019), and clinical trials to apply these drugs for MB are ongoing as of October 2019 (Li et al., 2019). These drugs, especially sonidegib, can cross the blood-brain-barrier (Li et al., 2019) and have the advantage in the accessibility to tumors in the brain. In addition, statins, HMG-CoA reductase inhibitors, have been shown to synergize with Hh pathway inhibitors (e.g., vismodegib) in inhibiting the MB growth (Gordon et al., 2018). As cholesterol biosynthesis is required for Smo activity in the Hh signaling pathway, it is reasonable to decrease the cholesterol level to treat MB.
However, because Hh pathway is involved in many biological events, modification of Smo activity could cause a number of negative side effects. Therefore, a recent study proposed targeting a specific protein located in primary cilia (Bay et al., 2018). Knockdown of the Arl13b gene, which encodes a GTPase expressed in cilia, in MB cell lines suppresses only high level of Shh signaling and decreases cell proliferation. Loss of Arl13b in the Ptch1-null background abolishes the tumorigenic effect of loss of Ptch1 (Goodrich et al., 1997), inhibiting MB formation in the developing cerebellum (Bay et al., 2018).
In the treatment of Hh-related cancers, GPCRs are considered effective targets because they are easily accessible from the cell exterior and could therefore be potential drug targets (He and Lu, 2015). Hh signal transduction is associated with the downregulation of PKA and decreased cAMP levels; therefore, chemicals that modify the levels of these molecules can be developed as an effective anti-cancer treatment. For instance, Gαs-type GPCRs upregulate intracellular cAMP and thus inhibit Hh signaling (He and Lu, 2015). For example, GPR161 overexpression changes the sensitivity of cells to Hh and is considered a potential drug target (Mukhopadhyay et al., 2013; He and Lu, 2015; Rao et al., 2016; Shimada et al., 2018; Pusapati et al., 2018a).
Parkinson's disease (PD) is one of the most common neurodegenerative disorders, with a prevalence of 1–2 in 1,000 people (Collaborators, G.B.D.P.s.D, 2018). PD is characterized by motor deficiencies (slowness of movement, tremors, and postural instability) (Kalia and Lang, 2015), which are caused by degeneration of dopaminergic (DA) neurons (dopamine-producing neurons) in a specific area of the brain called the substantia nigra pars compacta (SNpc). However, the mechanisms underlying neurodegeneration in PD remain unknown (Kalia and Lang, 2015).
In adult mice, Shh is expressed in the SNpc, as well as in other areas including the ventral tegmental area and the retrorubral field in the ventral midbrain (Gonzalez-Reyes et al., 2012; Alvarez-Buylla and Ihrie, 2014). Shh expression in these regions is necessary for long-term maintenance of DA neurons, and ablation of Shh leads to DA neuron degeneration and causes PD (Gonzalez-Reyes et al., 2012). Activation of Hh signaling protects DA neurons, and treatment with the Smo agonist purmorphamine (Briscoe, 2006; Riobo et al., 2006) attenuates inflammatory responses in cells, thereby blocking DA neuron degeneration (Shao et al., 2017). Detailed mechanistic analyses are critical to clarify these effects.
AD is another common neurodegenerative disorder, with an incidence of 1–3% in the population >65 years of age. AD patients show deterioration of cognitive and memory functions caused by loss of neurons and synapses (Masters et al., 2015). Patients with AD show abnormal elevation and accumulation of amyloid-β, a small neurotoxic peptide cleaved from amyloid precursor protein. Amyloid-β interrupts Shh signaling by distorting primary cilia, suggesting that primary cilia-mediated signal transduction, including the Shh signaling pathway, is attenuated in AD patients (Vorobyeva and Saunders, 2018). In a mouse model of AD and in human AD patients, whereas Shh expression is upregulated, Ptch1 and Gli1 signaling is decreased, and impaired neurogenesis occurs (He et al., 2014b). Taken together, these findings suggest that Shh signal plays essential roles for the integrity of the brain.
Since the identification of Hh as a segment polarity gene in a mutant screen in flies by German biologist Christiane Nüsslein-Volhard in 1980 (Nusslein-Volhard and Wieschaus, 1980), more than 8,000 scientific papers related to Hh have been published in a 40 year period according to a simple survey of the PubMed database (https://www.ncbi.nlm.nih.gov/pubmed/). Nevertheless, new functions, mediators, and regulatory mechanisms are continuously being proposed. The Hh signaling pathway continues to attract the attention of researchers not only because of its important roles in biological processes, but also its unique regulatory mechanisms. Hh proteins are cleaved from long polypeptides, and the receptor proteins for Hh are localized in a specific region of the membrane, the cilium. In addition, the bimodality of the transcription factors Gli2/3, which act as repressors in the absence of Hh and activators in the presence of Hh, constitutes an unusual regulatory mechanism. This bimodal regulation involves ubiquitin-mediated cleavage, which further highlights the uniqueness of the transcriptional regulation. In tissues, Hh acts as a morphogen, and target genes are induced in a concentration-dependent manner. Moreover, multiple feedback mechanisms dynamically regulate Hh activity in a temporal manner, which confers variation in cell fates. Distinct target genes are upregulated by the same Hh signal in a context-dependent manner.
The regulatory mechanisms of the Hh pathway are complex, and new mechanisms are continuously being identified. Because each new finding triggers another question, many researchers in various fields including molecular and cell biology, genetics, medicine, biochemistry, protein structure, chemistry, and mathematical biology have chosen Hh as the focus of their research.
Hh proteins are involved in a variety of biological events such as cell differentiation, proliferation, and survival. The fact that multiple processes that are apparently distinct from each other are induced by a single Hh protein should be addressed in the future. Future studies on Hh could focus on the cell type-specific expression levels of each mediator of the Hh signaling pathway. Although there are more than 30 mediators of Hh signaling, the expression levels of these proteins are likely cell type-specific, which may confer variation in the kinetics and responsiveness to the signal. For instance, Hh target genes are expressed within 2 hours in NIH3T3 cells (Humke et al., 2010), whereas expression begins after several hours in neural progenitor cells (Dessaud et al., 2007; Dessaud et al., 2010). Quantitative analyses of the expression levels of Hh signaling mediator genes or proteins may explain the variation in Hh signaling kinetics and its physiological significance.
The mechanisms underlying the cell type specificity of target genes involved in the Hh signaling pathway should be investigated. Although most signaling mediators are common to Shh, Ihh, and Dhh, the downstream genes induced are context-dependent. This variation may be achieved through crosstalk with other signaling molecules, or differences in the transcription factors interacting with Gli or the epigenetic background (chromatin status) of cells. Even in the same neural progenitor cells, early and late progenitor cells show differential responses to the same Shh protein (Sasai et al., 2014). A recent genome-wide loss of function-based screen performed using CRISPR identified positive and negative effectors of Shh signaling (Pusapati et al., 2018b). This screen was performed in NIH3T3 cells, and the potential findings in other cell types are intriguing. Systematic analysis of the expression of Hh target genes in different cells may reveal the mechanisms underlying the diverse roles of Hh signals in different cellular contexts.
Despite extensive research, many mechanisms underlying Hh signaling may remain undiscovered, and cutting-edge approaches, such as chasing single cells or single proteins, computational prediction, and genome-wide functional screens, are warranted to elucidate these mechanisms.
NS undertook the groundwork. MT, an expert of cell biology of cilia and ciliopathies, wrote the genetic disease part. TK, an expert of cancer, wrote the cancer part.
The work and publication of this article are supported by grants-in-aid from Japan Society for the Promotion of Science (17H03684, 17K19399, NS; 17H05003, MT) and from MEXT (19H04781; NS), and the Joint Research Program of the Institute for Genetic Medicine, Hokkaido University (TK, NS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
NS is thankful to Ryo Sekido for expert advice. A part of the disease cases were found by referring to the website OMIM (Online Mendelian Inheritance in Man; https://www.omim.org) and NIH Genetics Home Reference (https://ghr.nlm.nih.gov).
Abdelhamed, Z. A., Wheway, G., Szymanska, K., Natarajan, S., Toomes, C., Inglehearn, C., et al. (2013). Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum. Mol. Genet. 22, 1358–1372. doi: 10.1093/hmg/dds546
Aguilar, A., Meunier, A., Strehl, L., Martinovic, J., Bonniere, M., Attie-Bitach, T., Encha-Razavi, F., Spassky, N. (2012). Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. Ed. Anderson, K. V. (New York: Sloan–Kettering Institute). p. 16951-16956. doi: 10.1073/Pnas.1201408109
Ahn, J., Ludecke, H. J., Lindow, S., Horton, W. A., Lee, B., Wagner, M. J., et al. (1995). Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat. Genet. 11, 137–143. doi: 10.1038/ng1095-137
Ahn, S., Joyner, A. L. (2005). In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature 437, 894–897. doi: 10.1038/nature03994
Alaynick, W. A., Jessell, T. M., Pfaff, S. L. (2011). SnapShot: spinal cord development. Cell 146, 178–178 e171. doi: 10.1016/j.cell.2011.06.038
Alfaro, A. C., Roberts, B., Kwong, L., Bijlsma, M. F., Roelink, H. (2014). Ptch2 mediates the Shh response in Ptch1-/- cells. Development 141, 3331–3339. doi: 10.1242/dev.110056
Allen, B. L., Song, J. Y., Izzi, L., Althaus, I. W., Kang, J. S., Charron, F., et al. (2011). Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev. Cell 20, 775–787. doi: 10.1016/j.devcel.2011.04.018
Allen, B. L., Tenzen, T., McMahon, A. P. (2007). The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 21, 1244–1257. doi: 10.1101/gad.1543607
Alvarez-Buylla, A., Ihrie, R. A. (2014). Sonic hedgehog signaling in the postnatal brain. Semin. Cell Dev. Biol. 33, 105–111. doi: 10.1016/j.semcdb.2014.05.008
Arensdorf, A. M., Dillard, M. E., Menke, J. M., Frank, M. W., Rock, C. O., Ogden, S. K. (2017). Sonic hedgehog activates phospholipase A2 to enhance smoothened ciliary translocation. Cell Rep. 19, 2074–2087. doi: 10.1016/j.celrep.2017.05.033
Arya, P., Rainey, M. A., Bhattacharyya, S., Mohapatra, B. C., George, M., Kuracha, M. R., et al. (2015). The endocytic recycling regulatory protein EHD1 Is required for ocular lens development. Dev. Biol. 408, 41–55. doi: 10.1016/j.ydbio.2015.10.005
Auffinger, B., Spencer, D., Pytel, P., Ahmed, A. U., Lesniak, M. S. (2015). The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 15, 741–752. doi: 10.1586/14737175.2015.1051968
Aviles, E. C., Wilson, N. H., Stoeckli, E. T. (2013). Sonic hedgehog and Wnt: antagonists in morphogenesis but collaborators in axon guidance. Front. Cell. Neurosci. 7, 86. doi: 10.3389/fncel.2013.00086
Axelson, M., Liu, K., Jiang, X., He, K., Wang, J., Zhao, H., et al. (2013). U.S. food and drug administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin. Cancer Res. 19, 2289–2293. doi: 10.1158/1078-0432.CCR-12-1956
Baardman, M. E., Zwier, M. V., Wisse, L. J., Gittenberger-de Groot, A. C., Kerstjens-Frederikse, W. S., Hofstra, R. M., et al . (2016). Common arterial trunk and ventricular non-compaction in Lrp2 knockout mice indicate a crucial role of LRP2 in cardiac development. Dis. Models Mech. 9, 413–425. doi: 10.1242/dmm.022053
Bae, G. U., Domene, S., Roessler, E., Schachter, K., Kang, J. S., Muenke, M., et al. (2011). Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am. J. Hum. Genet. 89, 231–240. doi: 10.1016/j.ajhg.2011.07.001
Bai, C. B., Joyner, A. L. (2001). Gli1 can rescue the in vivo function of Gli2. Development 128, 5161–5172.
Balaskas, N., Ribeiro, A., Panovska, J., Dessaud, E., Sasai, N., Page, K. M., et al. (2012). Gene regulatory logic for reading the Sonic Hedgehog signaling gradient in the vertebrate neural tube. Cell 148, 273–284. doi: 10.1016/j.cell.2011.10.047
Balordi, F., Fishell, G. (2007a). Hedgehog signaling in the subventricular zone is required for both the maintenance of stem cells and the migration of newborn neurons. J. Neurosci.: Off. J. Soc. Neurosci. 27, 5936–5947. doi: 10.1523/JNEUROSCI.1040-07.2007
Balordi, F., Fishell, G. (2007b). Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J. Neurosci.: Off. J. Soc. Neurosci. 27, 14248–14259. doi: 10.1523/JNEUROSCI.4531-07.2007
Bangs, F., Anderson, K. V. (2017). Primary cilia and mammalian hedgehog signaling. Cold Spring Harbor Perspect. Biol. 9. doi: 10.1101/cshperspect.a028175
Bay, S. N., Long, A. B., Caspary, T. (2018). Disruption of the ciliary GTPase Arl13b suppresses Sonic hedgehog overactivation and inhibits medulloblastoma formation. Proc. Natl. Acad. Sci. U.S.A. 115, 1570–1575. doi: 10.1073/pnas.1706977115
Beltrami, G., Ristori, G., Scoccianti, G., Tamburini, A., Capanna, R. (2016). Hereditary multiple exostoses: a review of clinical appearance and metabolic pattern. Clin. Cases Miner. Bone Metabol.: Off. J. Ital. Soc. Osteoporosis Miner. Metabol. Skeletal Dis. 13, 110–118. doi: 10.11138/ccmbm/2016.13.2.110
Bhatia, N., Thiyagarajan, S., Elcheva, I., Saleem, M., Dlugosz, A., Mukhtar, H., et al. (2006). Gli2 is targeted for ubiquitination and degradation by beta-TrCP ubiquitin ligase. J. Biol. Chem. 281, 19320–19326. doi: 10.1074/jbc.M513203200
Bhattacharyya, S., Rainey, M. A., Arya, P., Mohapatra, B. C., Mushtaq, I., Dutta, S., et al. (2016). Endocytic recycling protein EHD1 regulates primary cilia morphogenesis and SHH signaling during neural tube development. Sci. Rep. 6, 20727. doi: 10.1038/srep20727
Bidet, M., Joubert, O., Lacombe, B., Ciantar, M., Nehme, R., Mollat, P., et al. (2011). The hedgehog receptor patched is involved in cholesterol transport. PloS One 6, e23834. doi: 10.1371/journal.pone.0023834
Bijlsma, M. F., Damhofer, H., Roelink, H. (2012). Hedgehog-stimulated chemotaxis is mediated by smoothened located outside the primary cilium. Sci. Signaling 5, ra60. doi: 10.1126/scisignal.2002798
Bischoff, M., Gradilla, A. C., Seijo, I., Andres, G., Rodriguez-Navas, C., Gonzalez-Mendez, L., et al. (2013). Cytonemes are required for the establishment of a normal Hedgehog morphogen gradient in Drosophila epithelia. Nat. Cell Biol. 15, 1269–1281. doi: 10.1038/ncb2856
Bishop, B., Aricescu, A. R., Harlos, K., O'Callaghan, C. A., Jones, E. Y., Siebold, C. (2009). Structural insights into hedgehog ligand sequestration by the human hedgehog-interacting protein HHIP. Nat. Struct. Mol. Biol. 16, 698–703. doi: 10.1038/nsmb.1607
Bitgood, M. J., McMahon, A. P. (1995). Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 172, 126–138. doi: 10.1006/dbio.1995.0010
Bitgood, M. J., Shen, L., McMahon, A. P. (1996). Sertoli cell signaling by desert hedgehog regulates the male germline. Curr. Biol.: CB 6, 298–304. doi: 10.1016/S0960-9822(02)00480-3
Blaess, S., Szabo, N., Haddad-Tovolli, R., Zhou, X., Alvarez-Bolado, G. (2014). Sonic hedgehog signaling in the development of the mouse hypothalamus. Front. Neuroanat. 8, 156. doi: 10.3389/fnana.2014.00156
Blassberg, R., Jacob, J. (2017). Lipid metabolism fattens up hedgehog signaling. BMC Biol. 15, 95. doi: 10.1186/s12915-017-0442-y
Blassberg, R., Macrae, J. I., Briscoe, J., Jacob, J. (2016). Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome. Hum. Mol. Genet. 25, 693–705. doi: 10.1093/hmg/ddv507
Bohn, L. M., Lefkowitz, R. J., Gainetdinov, R. R., Peppel, K., Caron, M. G., Lin, F. T. (1999). Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–2498. doi: 10.1126/science.286.5449.2495
Bork, P., Beckmann, G. (1993). The CUB domain. a widespread module in developmentally regulated proteins. J. Mol. Biol. 231, 539–545. doi: 10.1006/jmbi.1993.1305
Bourikas, D., Pekarik, V., Baeriswyl, T., Grunditz, A., Sadhu, R., Nardo, M., et al. (2005). Sonic hedgehog guides commissural axons along the longitudinal axis of the spinal cord. Nat. Neurosci. 8, 297–304. doi: 10.1038/nn1396
Briscoe, J., Pierani, A., Jessell, T. M., Ericson, J. (2000). A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101, 435–445. doi: 10.1016/S0092-8674(00)80853-3
Briscoe, J., Small, S. (2015). Morphogen rules: design principles of gradient-mediated embryo patterning. Development 142, 3996–4009. doi: 10.1242/dev.129452
Brugmann, S. A., Allen, N. C., James, A. W., Mekonnen, Z., Madan, E., Helms, J. A. (2010). A primary cilia-dependent etiology for midline facial disorders. Hum. Mol. Genet. 19, 1577–1592. doi: 10.1093/hmg/ddq030
Bujakowska, K. M., Liu, Q., Pierce, E. A. (2017). Photoreceptor cilia and retinal ciliopathies. Cold Spring Harbor Perspect. Biol. 9. doi: 10.1101/cshperspect.a028274
Bulgakov, O. V., Eggenschwiler, J. T., Hong, D. H., Anderson, K. V., Li, T. (2004). FKBP8 is a negative regulator of mouse sonic hedgehog signaling in neural tissues. Development 131, 2149–2159. doi: 10.1242/dev.01122
Byrnes, A. M., Racacho, L., Grimsey, A., Hudgins, L., Kwan, A. C., Sangalli, M., et al. (2009). Brachydactyly A-1 mutations restricted to the central region of the N-terminal active fragment of Indian Hedgehog. Eur. J. Hum. Genet.: EJHG 17, 1112–1120. doi: 10.1038/ejhg.2009.18
Cai, H., Liu, A. (2016). Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc. Natl. Acad. Sci. U.S.A. 113, 14751–14756. doi: 10.1073/pnas.1612520114
Cai, H., Liu, A. (2017). Spop regulates Gli3 activity and Shh signaling in dorsoventral patterning of the mouse spinal cord. Dev. Biol. 432, 72–85. doi: 10.1016/j.ydbio.2017.04.002
Callejo, A., Torroja, C., Quijada, L., Guerrero, I. (2006). Hedgehog lipid modifications are required for Hedgehog stabilization in the extracellular matrix. Development 133, 471–483. doi: 10.1242/dev.02217
Cano-Gauci, D. F., Song, H. H., Yang, H., McKerlie, C., Choo, B., Shi, W., et al. (1999). Glypican-3-deficient mice exhibit developmental overgrowth and some of the abnormalities typical of Simpson-Golabi-Behmel syndrome. J. Cell Biol. 146, 255–264. doi: 10.1083/jcb.146.1.255
Canterini, S., Dragotto, J., Dardis, A., Zampieri, S., De Stefano, M. E., Mangia, F., et al. (2017). Shortened primary cilium length and dysregulated Sonic hedgehog signaling in Niemann-Pick C1 disease. Hum. Mol. Genet. 26, 2277–2289. doi: 10.1093/hmg/ddx118
Canto, P., Soderlund, D., Reyes, E., Mendez, J. P. (2004). Mutations in the desert hedgehog (DHH) gene in patients with 46,XY complete pure gonadal dysgenesis. J. Clin. Endocrinol. Metabol. 89, 4480–4483. doi: 10.1210/jc.2004-0863
Canto, P., Vilchis, F., Soderlund, D., Reyes, E., Mendez, J. P. (2005). A heterozygous mutation in the desert hedgehog gene in patients with mixed gonadal dysgenesis. Mol. Hum. Reprod. 11, 833–836. doi: 10.1093/molehr/gah216
Capurro, M. I., Shi, W., Filmus, J. (2012). LRP1 mediates Hedgehog-induced endocytosis of the GPC3-Hedgehog complex. J. Cell Sci. 125, 3380–3389. doi: 10.1242/jcs.098889
Capurro, M. I., Xu, P., Shi, W., Li, F., Jia, A., Filmus, J. (2008). Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev. Cell 14, 700–711. doi: 10.1016/j.devcel.2008.03.006
Chamoun, Z., Mann, R. K., Nellen, D., von Kessler, D. P., Bellotto, M., Beachy, P. A., et al. (2001). Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science 293, 2080–2084. doi: 10.1126/science.1064437
Charron, F., Stein, E., Jeong, J., McMahon, A. P., Tessier-Lavigne, M. (2003). The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 113, 11–23. doi: 10.1016/S0092-8674(03)00199-5
Chen, J. K., Taipale, J., Cooper, M. K., Beachy, P. A. (2002). Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 16, 2743–2748. doi: 10.1101/gad.1025302
Chen, M. H., Li, Y. J., Kawakami, T., Xu, S. M., Chuang, P. T. (2004a). Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 18, 641–659. doi: 10.1101/gad.1185804
Chen, M. H., Wilson, C. W., Li, Y. J., Law, K. K., Lu, C. S., Gacayan, R., et al. (2009). Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 23, 1910–1928. doi: 10.1101/gad.1794109
Chen, W., Ren, X. R., Nelson, C. D., Barak, L. S., Chen, J. K., Beachy, P. A., et al. (2004b). Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science 306, 2257–2260. doi: 10.1126/science.1104135
Chen, X., Tukachinsky, H., Huang, C. H., Jao, C., Chu, Y. R., Tang, H. Y., et al. (2011a). Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J. Cell Biol. 192, 825–838. doi: 10.1083/jcb.201008090
Chen, Y., Sasai, N., Ma, G., Yue, T., Jia, J., Briscoe, J., et al. (2011b). Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PloS Biol. 9, e1001083. doi: 10.1371/journal.pbio.1001083
Cheung, H. O., Zhang, X., Ribeiro, A., Mo, R., Makino, S., Puviindran, V., et al. (2009). The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signaling 2, ra29. doi: 10.1126/scisignal.2000405
Chiang, C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L., Westphal, H., et al. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407–413. doi: 10.1038/383407a0
Chiao, E., Fisher, P., Crisponi, L., Deiana, M., Dragatsis, I., Schlessinger, D., et al. (2002). Overgrowth of a mouse model of the Simpson-Golabi-Behmel syndrome is independent of IGF signaling. Dev. Biol. 243, 185–206. doi: 10.1006/dbio.2001.0554
Christ, A., Christa, A., Kur, E., Lioubinski, O., Bachmann, S., Willnow, T. E., et al. (2012). LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev. Cell 22, 268–278. doi: 10.1016/j.devcel.2011.11.023
Chuang, P. T., Kawcak, T., McMahon, A. P. (2003). Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 17, 342–347. doi: 10.1101/gad.1026303
Chuang, P. T., McMahon, A. P. (1999). Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 397, 617–621. doi: 10.1038/17611
Clark, A. M., Garland, K. K., Russell, L. D. (2000). Desert hedgehog (Dhh) gene is required in the mouse testis for formation of adult-type Leydig cells and normal development of peritubular cells and seminiferous tubules. Biol. Reprod. 63, 1825–1838. doi: 10.1095/biolreprod63.6.1825
Clement, V., Sanchez, P., de Tribolet, N., Radovanovic, I., Ruiz i Altaba, A. (2007). HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol.: CB 17, 165–172. doi: 10.1016/j.cub.2006.11.033
Cohen, M., Briscoe, J., Blassberg, R. (2013). Morphogen interpretation: the transcriptional logic of neural tube patterning. Curr. Opin. Genet. Dev. 23, 423–428. doi: 10.1016/j.gde.2013.04.003
Cohen, M., Kicheva, A., Ribeiro, A., Blassberg, R., Page, K. M., Barnes, C. P., et al. (2015). Ptch1 and Gli regulate Shh signalling dynamics via multiple mechanisms. Nat. Commun. 6, 6709. doi: 10.1038/ncomms7709
Cole, F., Krauss, R. S. (2003). Microform holoprosencephaly in mice that lack the Ig superfamily member Cdon. Curr. Biol.: CB 13, 411–415. doi: 10.1016/S0960-9822(03)00088-5
Collaborators, G.B.D.P.s.D. (2018). Global, regional, and national burden of Parkinson’s disease, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 17, 939–953. doi: 10.1016/S1474-4422(18)30295-3
Coni, S., Antonucci, L., D'Amico, D., Di Magno, L., Infante, P., De Smaele, E., et al. (2013). Gli2 Acetylation at Lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PloS One 8. doi: 10.1371/journal.pone.0065718
Cooper, M. K., Wassif, C. A., Krakowiak, P. A., Taipale, J., Gong, R., Kelley, R. I., et al. (2003). A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 33, 508–513. doi: 10.1038/ng1134
Corbit, K. C., Aanstad, P., Singla, V., Norman, A. R., Stainier, D. Y., Reiter, J. F. (2005). Vertebrate smoothened functions at the primary cilium. Nature 437, 1018–1021. doi: 10.1038/nature04117
Coulter, M. E., Dorobantu, C. M., Lodewijk, G. A., Delalande, F., Cianferani, S., Ganesh, V. S., et al. (2018). The ESCRT-III Protein CHMP1A mediates secretion of sonic hedgehog on a distinctive subtype of extracellular vesicles. Cell Rep. 24, 973–986 e978. doi: 10.1016/j.celrep.2018.06.100
Cox, B., Briscoe, J., Ulloa, F. (2010). SUMOylation by Pias1 regulates the activity of the Hedgehog dependent Gli transcription factors. PloS One 5, e11996. doi: 10.1371/journal.pone.0011996
D'Angelo, G., Matusek, T., Pizette, S., Therond, P. P. (2015). Endocytosis of Hedgehog through dispatched regulates long-range signaling. Dev. Cell 32, 290–303. doi: 10.1016/j.devcel.2014.12.004
Damerla, R. R., Cui, C., Gabriel, G. C., Liu, X., Craige, B., Gibbs, B. C., et al. (2015). Novel Jbts17 mutant mouse model of Joubert syndrome with cilia transition zone defects and cerebellar and other ciliopathy related anomalies. Hum. Mol. Genet. 24, 3994–4005. doi: 10.1093/hmg/ddv137
De Mori, R., Romani, M., D'Arrigo, S., Zaki, M. S., Lorefice, E., Tardivo, S., et al. (2017). Hypomorphic recessive variants in SUFU Impair the Sonic Hedgehog pathway and cause joubert syndrome with cranio-facial and skeletal defects. Am. J. Hum. Genet. 101, 552–563. doi: 10.1016/j.ajhg.2017.08.017
de Robles, P., Fiest, K. M., Frolkis, A. D., Pringsheim, T., Atta, C., Germaine-Smith, C., et al. (2015). The worldwide incidence and prevalence of primary brain tumors: a systematic review and meta-analysis. Neuro-oncology 17, 776–783. doi: 10.1093/neuonc/nou283
Denef, N., Neubuser, D., Perez, L., Cohen, S. M. (2000). Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102, 521–531. doi: 10.1016/S0092-8674(00)00056-8
Deng, W. L., Gao, M. L., Lei, X. L., Lv, J. N., Zhao, H., He, K. W., et al. (2018). Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Rep. 10, 1267–1281. doi: 10.1016/j.stemcr.2018.02.003
Dennis, J. F., Kurosaka, H., Iulianella, A., Pace, J., Thomas, N., Beckham, S., et al. (2012). Mutations in Hedgehog acyltransferase (Hhat) perturb Hedgehog signaling, resulting in severe acrania-holoprosencephaly-agnathia craniofacial defects. PloS Genet. 8, e1002927. doi: 10.1371/journal.pgen.1002927
Dessaud, E., McMahon, A. P., Briscoe, J. (2008). Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development 135, 2489–2503. doi: 10.1242/dev.009324
Dessaud, E., Ribes, V., Balaskas, N., Yang, L. L., Pierani, A., Kicheva, A., et al. (2010). Dynamic assignment and maintenance of positional identity in the ventral neural tube by the morphogen sonic hedgehog. PloS Biol. 8, e1000382. doi: 10.1371/journal.pbio.1000382
Dessaud, E., Yang, L. L., Hill, K., Cox, B., Ulloa, F., Ribeiro, A., et al. (2007). Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450, 717–720. doi: 10.1038/nature06347
Ding, Q., Motoyama, J., Gasca, S., Mo, R., Sasaki, H., Rossant, J., et al. (1998). Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125, 2533–2543.
Dubourg, C., Carre, W., Hamdi-Roze, H., Mouden, C., Roume, J., Abdelmajid, B., et al. (2016). Mutational spectrum in holoprosencephaly shows That FGF is a new major signaling pathway. Hum. Mutation 37, 1329–1339. doi: 10.1002/humu.23038
Dubourg, C., Lazaro, L., Pasquier, L., Bendavid, C., Blayau, M., Le Duff, F., et al. (2004). Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: mutation review and genotype-phenotype correlations. Hum. Mutation 24, 43–51. doi: 10.1002/humu.20056
Echelard, Y., Epstein, D. J., St-Jacques, B., Shen, L., Mohler, J., McMahon, J. A., et al. (1993). Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75, 1417–1430. doi: 10.1016/0092-8674(93)90627-3
Elyada, E., Pribluda, A., Goldstein, R. E., Morgenstern, Y., Brachya, G., Cojocaru, G., et al. (2011). CKIalpha ablation highlights a critical role for p53 in invasiveness control. Nature 470, 409–413. doi: 10.1038/nature09673
Endoh-Yamagami, S., Evangelista, M., Wilson, D., Wen, X., Theunissen, J. W., Phamluong, K., et al. (2009). The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol.: CB 19, 1320–1326. doi: 10.1016/j.cub.2009.06.046
Epstein, E. H. (2008). Basal cell carcinomas: attack of the hedgehog. Nat. Rev. Cancer 8, 743–754. doi: 10.1038/nrc2503
Fabre, P. J., Shimogori, T., Charron, F. (2010). Segregation of ipsilateral retinal ganglion cell axons at the optic chiasm requires the Shh receptor Boc. J. Neurosci.: Off. J. Soc. Neurosci. 30, 266–275. doi: 10.1523/JNEUROSCI.3778-09.2010
Faiyaz-Ul-Haque, M., Ahmad, W., Zaidi, S. H., Hussain, S., Haque, S., Ahmad, M., et al. (2004). Novel mutations in the EXT1 gene in two consanguineous families affected with multiple hereditary exostoses (familial osteochondromatosis). Clin. Genet. 66, 144–151. doi: 10.1111/j.1399-0004.2004.00275.x
Ferent, J., Giguere, F., Jolicoeur, C., Morin, S., Michaud, J. F., Makihara, S., et al. (2019). Boc Acts via numb as a Shh-dependent endocytic platform for Ptch1 internalization and Shh-mediated axon guidance. Neuron 102, 1157–1171 e1155. doi: 10.1016/j.neuron.2019.04.003
Ferguson, S. M., De Camilli, P. (2012). Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13, 75–88. doi: 10.1038/nrm3266
Formichi, P., Battisti, C., De Santi, M. M., Guazzo, R., Tripodi, S. A., Radi, E., et al. (2018). Primary cilium alterations and expression changes of Patched1 proteins in niemann-pick type C disease. J. Cell. Physiol. 233, 663–672. doi: 10.1002/jcp.25926
Fujii, K., Ohashi, H., Suzuki, M., Hatsuse, H., Shiohama, T., Uchikawa, H., et al. (2013). Frameshift mutation in the PTCH2 gene can cause nevoid basal cell carcinoma syndrome. Familial Cancer 12, 611–614. doi: 10.1007/s10689-013-9623-1
Gailani, M. R., Stahle-Backdahl, M., Leffell, D. J., Glynn, M., Zaphiropoulos, P. G., Pressman, C., et al. (1996). The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 14, 78–81. doi: 10.1038/ng0996-78
Gao, B., Guo, J., She, C., Shu, A., Yang, M., Tan, Z., et al. (2001). Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat. Genet. 28, 386–388. doi: 10.1038/ng577
Garver, W. S., Francis, G. A., Jelinek, D., Shepherd, G., Flynn, J., Castro, G., et al. (2007). The National Niemann-Pick C1 disease database: report of clinical features and health problems. Am. J. Med. Genet. Part A 143A, 1204–1211. doi: 10.1002/ajmg.a.31735
Goetz, S. C., Anderson, K. V. (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344. doi: 10.1038/nrg2774
Gonzalez-Mendez, L., Seijo-Barandiaran, I., Guerrero, I. (2017). Cytoneme-mediated cell-cell contacts for Hedgehog reception. eLife 6. doi: 10.7554/eLife.24045
Gonzalez-Reyes, L. E., Verbitsky, M., Blesa, J., Jackson-Lewis, V., Paredes, D., Tillack, K., et al. (2012). Sonic hedgehog maintains cellular and neurochemical homeostasis in the adult nigrostriatal circuit. Neuron 75, 306–319. doi: 10.1016/j.neuron.2012.05.018
Goodrich, L. V., Milenkovic, L., Higgins, K. M., Scott, M. P. (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113. doi: 10.1126/science.277.5329.1109
Gordon, R. E., Zhang, L., Peri, S., Kuo, Y. M., Du, F., Egleston, B. L., et al. (2018). Statins synergize with hedgehog pathway inhibitors for treatment of medulloblastoma. Clin. Cancer Res. 24, 1375–1388. doi: 10.1158/1078-0432.CCR-17-2923
Green, W. W., Uytingco, C. R., Ukhanov, K., Kolb, Z., Moretta, J., McIntyre, J. C., et al. (2018). Peripheral gene therapeutic rescue of an olfactory ciliopathy restores sensory input, axonal pathfinding, and odor-guided behavior. J. Neurosci.: Off. J. Soc. Neurosci. 38, 7462–7475. doi: 10.1523/JNEUROSCI.0084-18.2018
Guo, J., Higginbotham, H., Li, J., Nichols, J., Hirt, J., Ghukasyan, V., et al. (2015). Developmental disruptions underlying brain abnormalities in ciliopathies. Nat. Commun. 6, 7857. doi: 10.1038/ncomms8857
Gupta, S., Takebe, N., Lorusso, P. (2010). Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2, 237–250. doi: 10.1177/1758834010366430
Habbig, S., Liebau, M. C. (2015). Ciliopathies - from rare inherited cystic kidney diseases to basic cellular function. Mol. Cell. Pediatr. 2, 8. doi: 10.1186/s40348-015-0019-1
Hammond, R., Blaess, S., Abeliovich, A. (2009). Sonic hedgehog is a chemoattractant for midbrain dopaminergic axons. PloS One 4, e7007. doi: 10.1371/journal.pone.0007007
Handschuh, K., Feenstra, J., Koss, M., Ferretti, E., Risolino, M., Zewdu, R., et al. (2014). ESCRT-II/Vps25 constrains digit number by endosome-mediated selective modulation of FGF-SHH signaling. Cell Rep. 9, 674–687. doi: 10.1016/j.celrep.2014.09.019
Hardy, R. Y., Resh, M. D. (2012). Identification of N-terminal residues of Sonic Hedgehog important for palmitoylation by Hedgehog acyltransferase. J. Biol. Chem. 287, 42881–42889. doi: 10.1074/jbc.M112.426833
He, M., Subramanian, R., Bangs, F., Omelchenko, T., Liem, K. F., Kapoor, T. M., et al. (2014a). The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol. 16, 663–672. doi: 10.1038/ncb2988
He, P., Staufenbiel, M., Li, R., Shen, Y. (2014b). Deficiency of patched 1-induced Gli1 signal transduction results in astrogenesis in Swedish mutated APP transgenic mice. Hum. Mol. Genet. 23, 6512–6527. doi: 10.1093/hmg/ddu370
He, X., Lu, Q. R. (2015). G-Protein Galphas controls medulloblastoma initiation by suppressing sonic hedgehog signaling. Mol. Cell. Oncol. 2, e975070. doi: 10.4161/23723556.2014.975070
Hecht, J. T., Hogue, D., Wang, Y., Blanton, S. H., Wagner, M., Strong, L. C., et al. (1997). Hereditary multiple exostoses (EXT): mutational studies of familial EXT1 cases and EXT-associated malignancies. Am. J. Hum. Genet. 60, 80–86.
Heinritz, W., Huffmeier, U., Strenge, S., Miterski, B., Zweier, C., Leinung, S., et al. (2009). New mutations of EXT1 and EXT2 genes in German patients with Multiple Osteochondromas. Ann. Hum. Genet. 73, 283–291. doi: 10.1111/j.1469-1809.2009.00508.x
Hellemans, J., Coucke, P. J., Giedion, A., De Paepe, A., Kramer, P., Beemer, F., et al. (2003). Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am. J. Hum. Genet. 72, 1040–1046. doi: 10.1086/374318
Heussler, H. S., Suri, M., Young, I. D., Muenke, M. (2002). Extreme variability of expression of a Sonic Hedgehog mutation: attention difficulties and holoprosencephaly. Arch. Dis. Childhood 86, 293–296. doi: 10.1136/adc.86.4.293
Hildebrandt, F., Attanasio, M., Otto, E. (2009). Nephronophthisis: disease mechanisms of a ciliopathy. J. Am. Soc. Nephrol: JASN 20, 23–35. doi: 10.1681/ASN.2008050456
Hong, M., Srivastava, K., Kim, S., Allen, B. L., Leahy, D. J., Hu, P., et al. (2017). BOC is a modifier gene in holoprosencephaly. Hum. Mutation 38, 1464–1470. doi: 10.1002/humu.23286
Hsu, S. H., Zhang, X., Yu, C., Li, Z. J., Wunder, J. S., Hui, C. C., et al. (2011). Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 138, 3791–3801. doi: 10.1242/dev.069492
Huang, P., Nedelcu, D., Watanabe, M., Jao, C., Kim, Y., Liu, J., et al. (2016). Cellular cholesterol directly activates smoothened in hedgehog signaling. Cell 166, 1176–1187 e1114. doi: 10.1016/j.cell.2016.08.003
Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L., Anderson, K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87. doi: 10.1038/nature02061
Hui, C. C., Joyner, A. L. (1993). A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 3, 241–246. doi: 10.1038/ng0393-241
Humke, E. W., Dorn, K. V., Milenkovic, L., Scott, M. P., Rohatgi, R. (2010). The output of Hedgehog signaling is controlled by the dynamic association between suppressor of fused and the Gli proteins. Genes Dev. 24, 670–682. doi: 10.1101/gad.1902910
Hynes, A. M., Giles, R. H., Srivastava, S., Eley, L., Whitehead, J., Danilenko, M., et al. (2014). Murine Joubert syndrome reveals Hedgehog signaling defects as a potential therapeutic target for nephronophthisis. Proc. Natl. Acad. Sci. U.S.A. 111, 9893–9898. doi: 10.1073/pnas.1322373111
Inaba, K., Mizuno, K. (2016). Sperm dysfunction and ciliopathy. Reprod. Med. Biol. 15, 77–94. doi: 10.1007/s12522-015-0225-5
Incardona, J. P., Lee, J. H., Robertson, C. P., Enga, K., Kapur, R. P., Roelink, H. (2000). Receptor-mediated endocytosis of soluble and membrane-tethered Sonic hedgehog by Patched-1. Proc. Natl. Acad. Sci. U.S.A. 97, 12044–12049. doi: 10.1073/pnas.220251997
Ingham, P. W., McMahon, A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087. doi: 10.1101/gad.938601
Jaber, M., Koch, W. J., Rockman, H., Smith, B., Bond, R. A., Sulik, K. K., et al. (1996). Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc. Natl. Acad. Sci. U.S.A. 93, 12974–12979. doi: 10.1073/pnas.93.23.12974
Jeong, J., McMahon, A. P. (2002). Cholesterol modification of Hedgehog family proteins. J. Clin. Invest. 110, 591–596. doi: 10.1172/JCI16506
Johnson, R. L., Rothman, A. L., Xie, J., Goodrich, L. V., Bare, J. W., Bonifas, J. M., et al. (1996). Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272, 1668–1671. doi: 10.1126/science.272.5268.1668
Juan, T., Furthauer, M. (2018). Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin. Cell Dev. Biol. 74, 66–77. doi: 10.1016/j.semcdb.2017.08.022
Kalia, L. V., Lang, A. E. (2015). Parkinson's disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kang, S., Graham, J. M., Olney, A. H., Biesecker, L. G. (1997). GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat. Genet. 15, 266–268. doi: 10.1038/ng0397-266
Kantarci, S., Ackerman, K. G., Russell, M. K., Longoni, M., Sougnez, C., Noonan, K. M., et al. (2010). Characterization of the chromosome 1q41q42.12 region, and the candidate gene DISP1, in patients with CDH. Am. J. Med. Genet. Part A 152A, 2493–2504. doi: 10.1002/ajmg.a.33618
Kantarci, S., Al-Gazali, L., Hill, R. S., Donnai, D., Black, G. C., Bieth, E., et al. (2007). Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat. Genet. 39, 957–959. doi: 10.1038/ng2063
Karaplis, A. C., Luz, A., Glowacki, J., Bronson, R. T., Tybulewicz, V. L., Kronenberg, H. M., et al. (1994). Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 8, 277–289. doi: 10.1101/gad.8.3.277
Karp, S. J., Schipani, E., St-Jacques, B., Hunzelman, J., Kronenberg, H., McMahon, A. P. (2000). Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development 127, 543–548.
Kennedy, T. E., Serafini, T., de la Torre, J. R., Tessier-Lavigne, M. (1994). Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell 78, 425–435. doi: 10.1016/0092-8674(94)90421-9
Khaliullina, H., Panakova, D., Eugster, C., Riedel, F., Carvalho, M., Eaton, S. (2009). Patched regulates smoothened trafficking using lipoprotein-derived lipids. Development 136, 4111–4121. doi: 10.1242/dev.041392
Kimonis, V. E., Goldstein, A. M., Pastakia, B., Yang, M. L., Kase, R., DiGiovanna, J. J., et al. (1997). Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am. J. Med. Genet. 69, 299–308. doi: 10.1002/(SICI)1096-8628(19970331)69:3<299::AID-AJMG16>3.0.CO;2-M
Konitsiotis, A. D., Jovanovic, B., Ciepla, P., Spitaler, M., Lanyon-Hogg, T., Tate, E. W., et al. (2015). Topological analysis of Hedgehog acyltransferase, a multipalmitoylated transmembrane protein. J. Biol. Chem. 290, 3293–3307. doi: 10.1074/jbc.M114.614578
Kovacs, J. J., Whalen, E. J., Liu, R., Xiao, K., Kim, J., Chen, M., et al. (2008). Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 320, 1777–1781. doi: 10.1126/science.1157983
Koziel, L., Kunath, M., Kelly, O. G., Vortkamp, A. (2004). Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev. Cell 6, 801–813. doi: 10.1016/j.devcel.2004.05.009
Kutejova, E., Sasai, N., Shah, A., Gouti, M., Briscoe, J. (2016). Neural progenitors adopt specific identities by directly repressing all alternative progenitor transcriptional programs. Dev. Cell 36, 639–653. doi: 10.1016/j.devcel.2016.02.013
Kwong, L., Bijlsma, M. F., Roelink, H. (2014). Shh-mediated degradation of Hhip allows cell autonomous and non-cell autonomous Shh signalling. Nat. Commun. 5, 4849. doi: 10.1038/ncomms5849
Lanske, B., Karaplis, A. C., Lee, K., Luz, A., Vortkamp, A., Pirro, A., et al. (1996). PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 273, 663–666. doi: 10.1126/science.273.5275.663
Laug, D., Glasgow, S. M., Deneen, B. (2018). A glial blueprint for gliomagenesis. Nat. Rev. Neurosci. 19, 393–403. doi: 10.1038/s41583-018-0014-3
Le Gallo, M., O'Hara, A. J., Rudd, M. L., Urick, M. E., Hansen, N. F., O'Neil, N. J., et al. (2012). Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 44, 1310–1315. doi: 10.1038/ng.2455
Lee, C. S., Buttitta, L., Fan, C. M. (2001). Evidence that the WNT-inducible growth arrest-specific gene 1 encodes an antagonist of sonic hedgehog signaling in the somite. Proc. Natl. Acad. Sci. U.S.A. 98, 11347–11352. doi: 10.1073/pnas.201418298
Lefevre, G., Michel, V., Weil, D., Lepelletier, L., Bizard, E., Wolfrum, U., et al. (2008). A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development 135, 1427–1437. doi: 10.1242/dev.012922
Lei, Q., Zelman, A. K., Kuang, E., Li, S., Matise, M. P. (2004). Transduction of graded Hedgehog signaling by a combination of Gli2 and Gli3 activator functions in the developing spinal cord. Development 131, 3593–3604. doi: 10.1242/dev.01230
Levi, B., James, A. W., Nelson, E. R., Brugmann, S. A., Sorkin, M., Manu, A., et al. (2011). Role of Indian hedgehog signaling in palatal osteogenesis. Plast. Reconstructive Surg. 127, 1182–1190. doi: 10.1097/PRS.0b013e3182043a07
Lewandowski, J. P., Du, F., Zhang, S., Powell, M. B., Falkenstein, K. N., Ji, H., et al. (2015). Spatiotemporal regulation of GLI target genes in the mammalian limb bud. Dev. Biol. 406, 92–103. doi: 10.1016/j.ydbio.2015.07.022
Lewis, P. M., Dunn, M. P., McMahon, J. A., Logan, M., Martin, J. F., St-Jacques, B., et al. (2001). Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell 105, 599–612. doi: 10.1016/S0092-8674(01)00369-5
Li, Y., Song, Q., Day, B. W. (2019). Phase I and phase II sonidegib and vismodegib clinical trials for the treatment of paediatric and adult MB patients: a systemic review and meta-analysis. Acta Neuropathol. Commun. 7, 123. doi: 10.1186/s40478-019-0773-8
Li, Y., Zhang, H., Litingtung, Y., Chiang, C. (2006). Cholesterol modification restricts the spread of Shh gradient in the limb bud. Proc. Natl. Acad. Sci. U.S.A. 103, 6548–6553. doi: 10.1073/pnas.0600124103
Liem, K. F., He, M., Ocbina, P. J., Anderson, K. V. (2009). Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 13377–13382. doi: 10.1073/pnas.0906944106
Lin, Y. C., Roffler, S. R., Yan, Y. T., Yang, R. B. (2015). Disruption of Scube2 Impairs Endochondral Bone Formation. J. Bone Miner. Res.: Off. J. Am. Soc. Bone Miner. Res. 30, 1255–1267. doi: 10.1002/jbmr.2451
Litingtung, Y., Chiang, C. (2000). Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat. Neurosci. 3, 979–985. doi: 10.1038/79916
Litingtung, Y., Lei, L., Westphal, H., Chiang, C. (1998). Sonic hedgehog is essential to foregut development. Nat. Genet. 20, 58–61. doi: 10.1038/1717
Liu, Y., May, N. R., Fan, C. M. (2001). Growth arrest specific gene 1 is a positive growth regulator for the cerebellum. Dev. Biol. 236, 30–45. doi: 10.1006/dbio.2000.0146
Loftus, S. K., Morris, J. A., Carstea, E. D., Gu, J. Z., Cummings, C., Brown, A., et al. (1997). Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 277, 232–235. doi: 10.1126/science.277.5323.232
Long, J., Tokhunts, R., Old, W. M., Houel, S., Rodgriguez-Blanco, J., Singh, S., et al. (2015). Identification of a family of fatty-acid-speciated sonic hedgehog proteins, whose members display differential biological properties. Cell Rep. 10, 1280–1287. doi: 10.1016/j.celrep.2015.01.058
Luchetti, G., Sircar, R., Kong, J. H., Nachtergaele, S., Sagner, A., Byrne, E. F., et al. (2016). Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife 5. doi: 10.7554/eLife.20304
Ma, L., Pei, G. (2007). Beta-arrestin signaling and regulation of transcription. J. Cell Sci. 120, 213–218. doi: 10.1242/jcs.03338
Ma, Y., Erkner, A., Gong, R., Yao, S., Taipale, J., Basler, K., et al. (2002). Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of dispatched. Cell 111, 63–75. doi: 10.1016/S0092-8674(02)00977-7
Malicki, J. J., Johnson, C. A. (2017). The cilium: cellular antenna and central processing unit. Trends Cell Biol. 27, 126–140. doi: 10.1016/j.tcb.2016.08.002
Mann, R. K., Beachy, P. A. (2004). Novel lipid modifications of secreted protein signals. Annu. Rev. Biochem. 73, 891–923. doi: 10.1146/annurev.biochem.73.011303.073933
Marigo, V., Davey, R. A., Zuo, Y., Cunningham, J. M., Tabin, C. J. (1996). Biochemical evidence that patched is the Hedgehog receptor. Nature 384, 176–179. doi: 10.1038/384176a0
Marti, E., Takada, R., Bumcrot, D. A., Sasaki, H., McMahon, A. P. (1995). Distribution of Sonic hedgehog peptides in the developing chick and mouse embryo. Development 121, 2537–2547.
Martin, V., Carrillo, G., Torroja, C., Guerrero, I. (2001). The sterol-sensing domain of Patched protein seems to control Smoothened activity through Patched vesicular trafficking. Curr. Biol.: CB 11, 601–607. doi: 10.1016/S0960-9822(01)00178-6
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., Cummings, J. L. (2015). Alzheimer's disease. Nat. Rev. Dis. Primers 1, 15056. doi: 10.1038/nrdp.2015.56
Matise, M. P. (2013). Molecular genetic control of cell patterning and fate determination in the developing ventral spinal cord. Wiley Interdiscip. Rev. Dev. Biol. 2, 419–425. doi: 10.1002/wdev.83
Migden, M. R., Chang, A. L. S., Dirix, L., Stratigos, A. J., Lear, J. T. (2018). Emerging trends in the treatment of advanced basal cell carcinoma. Cancer Treat. Rev. 64, 1–10. doi: 10.1016/j.ctrv.2017.12.009
Millard, N. E., De Braganca, K. C. (2016). Medulloblastoma. J. Child Neurol. 31, 1341–1353. doi: 10.1177/0883073815600866
Miller, L. A., Wert, S. E., Whitsett, J. A. (2001). Immunolocalization of sonic hedgehog (Shh) in developing mouse lung. J. Histochem. Cytochem.: Off. J. Histochem. Soc. 49, 1593–1604. doi: 10.1177/002215540104901213
Mo, R., Freer, A. M., Zinyk, D. L., Crackower, M. A., Michaud, J., Heng, H. H., et al. (1997). Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123.
Mochida, G. H., Ganesh, V. S., de Michelena, M. I., Dias, H., Atabay, K. D., Kathrein, K. L., et al. (2012). CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat. Genet. 44, 1260–1264. doi: 10.1038/ng.2425
Moore, B. S., Stepanchick, A. N., Tewson, P. H., Hartle, C. M., Zhang, J., Quinn, A. M., et al. (2016). Cilia have high cAMP levels that are inhibited by Sonic Hedgehog-regulated calcium dynamics. Proc. Natl. Acad. Sci. U.S.A. 113, 13069–13074. doi: 10.1073/pnas.1602393113
Motoyama, J., Liu, J., Mo, R., Ding, Q., Post, M., Hui, C. C. (1998a). Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat. Genet. 20, 54–57. doi: 10.1038/1711
Motoyama, J., Takabatake, T., Takeshima, K., Hui, C. (1998b). Ptch2, a second mouse Patched gene is co-expressed with Sonic hedgehog. Nat. Genet. 18, 104–106. doi: 10.1038/ng0298-104
Mukhopadhyay, S., Wen, X., Ratti, N., Loktev, A., Rangell, L., Scales, S. J., et al. (2013). The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210–223. doi: 10.1016/j.cell.2012.12.026
Myers, B. R., Neahring, L., Zhang, Y., Roberts, K. J., Beachy, P. A. (2017). Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl. Acad. Sci. U.S.A. 114, E11141–E11150. doi: 10.1073/pnas.1717891115
Nedelcu, D., Liu, J., Xu, Y., Jao, C., Salic, A. (2013). Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat. Chem. Biol. 9, 557–564. doi: 10.1038/nchembio.1290
Neri, G., Gurrieri, F., Zanni, G., Lin, A. (1998). Clinical and molecular aspects of the Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. 79, 279–283. doi: 10.1002/(SICI)1096-8628(19981002)79:4<279::AID-AJMG9>3.0.CO;2-H
Nieuwenhuis, E., Motoyama, J., Barnfield, P. C., Yoshikawa, Y., Zhang, X., Mo, R., et al. (2006). Mice with a targeted mutation of patched2 are viable but develop alopecia and epidermal hyperplasia. Mol. Cell. Biol. 26, 6609–6622. doi: 10.1128/MCB.00295-06
Niewiadomski, P., Kong, J. H., Ahrends, R., Ma, Y., Humke, E. W., Khan, S., et al. (2014). Gli protein activity is controlled by multisite phosphorylation in vertebrate hedgehog signaling. Cell Rep. 6, 168–181. doi: 10.1016/j.celrep.2013.12.003
Nigg, E. A., Raff, J. W. (2009). Centrioles, centrosomes, and cilia in health and disease. Cell 139, 663–678. doi: 10.1016/j.cell.2009.10.036
Niyaz, M., Khan, M. S., Mudassar, S. (2019). Hedgehog signaling: an Achilles' heel in cancer. Transl. Oncol. 12, 1334–1344. doi: 10.1016/j.tranon.2019.07.004
Nusslein-Volhard, C., Wieschaus, E. (1980). Mutations affecting segment number and polarity in Drosophila. Nature 287, 795–801. doi: 10.1038/287795a0
Okada, A., Charron, F., Morin, S., Shin, D. S., Wong, K., Fabre, P. J., et al. (2006). Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature 444, 369–373. doi: 10.1038/nature05246
Oosterveen, T., Kurdija, S., Alekseenko, Z., Uhde, C. W., Bergsland, M., Sandberg, M., et al. (2012). Mechanistic differences in the transcriptional interpretation of local and long-range Shh morphogen signaling. Dev. Cell 23, 1006–1019. doi: 10.1016/j.devcel.2012.09.015
Orth, M., Bellosta, S. (2012). Cholesterol: its regulation and role in central nervous system disorders. Cholesterol 2012, 292598. doi: 10.1155/2012/292598
Palencia-Campos, A., Ullah, A., Nevado, J., Yildirim, R., Unal, E., Ciorraga, M., et al. (2017). GLI1 inactivation is associated with developmental phenotypes overlapping with Ellis-van Creveld syndrome. Hum. Mol. Genet. 26, 4556–4571. doi: 10.1093/hmg/ddx335
Palma, V., Ruiz i Altaba, A. (2004). Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development 131, 337–345. doi: 10.1242/dev.00930
Panman, L., Zeller, R. (2003). Patterning the limb before and after SHH signalling. J. Anat. 202, 3–12. doi: 10.1046/j.1469-7580.2003.00138.x
Parchure, A., Vyas, N., Mayor, S. (2018). Wnt and Hedgehog: secretion of lipid-modified morphogens. Trends Cell Biol. 28, 157–170. doi: 10.1016/j.tcb.2017.10.003
Park, H. L., Bai, C., Platt, K. A., Matise, M. P., Beeghly, A., Hui, C. C., et al. (2000). Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127, 1593–1605.
Parmantier, E., Lynn, B., Lawson, D., Turmaine, M., Namini, S. S., Chakrabarti, L., et al. (1999). Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron 23, 713–724. doi: 10.1016/S0896-6273(01)80030-1
Patrono, C., Dionisi-Vici, C., Giannotti, A., Bembi, B., Digilio, M. C., Rizzo, C., et al. (2002). Two novel mutations of the human delta7-sterol reductase (DHCR7) gene in children with Smith-Lemli-Opitz syndrome. Mol. Cell. Probes 16, 315–318. doi: 10.1006/mcpr.2002.0426
Penela, P., Murga, C., Ribas, C., Lafarga, V., Mayor, F., Jr. (2010). The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br. J. Pharmacol. 160, 821–832. doi: 10.1111/j.1476-5381.2010.00727.x
Peng, T., Tian, Y., Boogerd, C. J., Lu, M. M., Kadzik, R. S., Stewart, K. M., et al. (2013). Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature 500, 589–592. doi: 10.1038/nature12358
Pennekamp, P., Menchen, T., Dworniczak, B., Hamada, H. (2015). Situs inversus and ciliary abnormalities: 20 years later, what is the connection? Cilia 4, 1. doi: 10.1186/s13630-014-0010-9
Pepicelli, C. V., Lewis, P. M., McMahon, A. P. (1998). Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr. Biol.: CB 8, 1083–1086. doi: 10.1016/S0960-9822(98)70446-4
Peterson, K. A., Nishi, Y., Ma, W., Vedenko, A., Shokri, L., Zhang, X., et al. (2012). Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning. Genes Dev. 26, 2802–2816. doi: 10.1101/gad.207142.112
Petrova, R., Joyner, A. L. (2014). Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 141, 3445–3457. doi: 10.1242/dev.083691
Pettigrew, A. L., Greenberg, F., Caskey, C. T., Ledbetter, D. H. (1991). Greig syndrome associated with an interstitial deletion of 7p: confirmation of the localization of Greig syndrome to 7p13. Hum. Genet. 87, 452–456. doi: 10.1007/BF00197167
Phi, L. T. H., Sari, I. N., Yang, Y. G., Lee, S. H., Jun, N., Kim, K. S., et al. (2018). Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 5416923. doi: 10.1155/2018/5416923
Philippe, C., Porter, D. E., Emerton, M. E., Wells, D. E., Simpson, A. H., Monaco, A. P. (1997). Mutation screening of the EXT1 and EXT2 genes in patients with hereditary multiple exostoses. Am. J. Hum. Genet. 61, 520–528. doi: 10.1086/515505
Pietrobono, S., Stecca, B. (2018). Targeting the oncoprotein smoothened by small molecules: focus on novel acylguanidine derivatives as potent smoothened inhibitors. Cells 7. doi: 10.3390/cells7120272
Pineda-Alvarez, D. E., Roessler, E., Hu, P., Srivastava, K., Solomon, B. D., Siple, C. E., et al. (2012). Missense substitutions in the GAS1 protein present in holoprosencephaly patients reduce the affinity for its ligand, SHH. Hum. Genet. 131, 301–310. doi: 10.1007/s00439-011-1078-6
Pons, S., Marti, E. (2000). Sonic hedgehog synergizes with the extracellular matrix protein vitronectin to induce spinal motor neuron differentiation. Development 127, 333–342.
Porter, J. A., Young, K. E., Beachy, P. A. (1996). Cholesterol modification of hedgehog signaling proteins in animal development. Science 274, 255–259. doi: 10.1126/science.274.5285.255
Pusapati, G. V., Kong, J. H., Patel, B. B., Gouti, M., Sagner, A., Sircar, R., et al. (2018a). G protein-coupled receptors control the sensitivity of cells to the morphogen Sonic Hedgehog. Sci. Signaling 11. doi: 10.1126/scisignal.aao5749
Pusapati, G. V., Kong, J. H., Patel, B. B., Krishnan, A., Sagner, A., Kinnebrew, M., et al. (2018b). CRISPR Screens Uncover Genes that Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Dev. Cell 44, 113–129 e118. doi: 10.1016/j.devcel.2017.12.003
Putoux, A., Thomas, S., Coene, K. L., Davis, E. E., Alanay, Y., Ogur, G., et al. (2011). KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 43, 601–606. doi: 10.1038/ng.826
Rainey, M. A., George, M., Ying, G., Akakura, R., Burgess, D. J., Siefker, E., et al. (2010). The endocytic recycling regulator EHD1 is essential for spermatogenesis and male fertility in mice. BMC Dev. Biol. 10, 37. doi: 10.1186/1471-213X-10-37
Rao, R., Salloum, R., Xin, M., Lu, Q. R. (2016). The G protein Galphas acts as a tumor suppressor in sonic hedgehog signaling-driven tumorigenesis. Cell Cycle 15, 1325–1330. doi: 10.1080/15384101.2016.1164371
Raskind, W. H., Conrad, E. U., Matsushita, M., Wijsman, E. M., Wells, D. E., Chapman, N., et al. (1998). Evaluation of locus heterogeneity and EXT1 mutations in 34 families with hereditary multiple exostoses. Hum. Mutation 11, 231–239. doi: 10.1002/(SICI)1098-1004(1998)11:3<231::AID-HUMU8>3.0.CO;2-K
Reiter, J. F., Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 18, 533–547. doi: 10.1038/nrm.2017.60
Ribeiro, L. A., Quiezi, R. G., Nascimento, A., Bertolacini, C. P., Richieri-Costa, A. (2010). Holoprosencephaly and holoprosencephaly-like phenotype and GAS1 DNA sequence changes: report of four Brazilian patients. Am. J. Med. Genet. Part A 152A, 1688–1694. doi: 10.1002/ajmg.a.33466
Richter, J., Kretz, A. L., Lemke, J., Fauler, M., Werner, J. U., Paschke, S., et al. (2018). CK1alpha overexpression correlates with poor survival in colorectal cancer. BMC Cancer 18, 140. doi: 10.1186/s12885-018-4019-0
Riobo, N. A., Saucy, B., Dilizio, C., Manning, D. R. (2006). Activation of heterotrimeric G proteins by Smoothened. Proc. Natl. Acad. Sci. U.S.A. 103, 12607–12612. doi: 10.1073/pnas.0600880103
Robertson, E. J., Charatsi, I., Joyner, C. J., Koonce, C. H., Morgan, M., Islam, A., et al. (2007). Blimp1 regulates development of the posterior forelimb, caudal pharyngeal arches, heart and sensory vibrissae in mice. Development 134, 4335–4345. doi: 10.1242/dev.012047
Roessler, E., Belloni, E., Gaudenz, K., Jay, P., Berta, P., Scherer, S. W., et al. (1996). Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 14, 357–360. doi: 10.1038/ng1196-357
Roessler, E., Belloni, E., Gaudenz, K., Vargas, F., Scherer, S. W., Tsui, L. C., et al. (1997). Mutations in the C-terminal domain of Sonic Hedgehog cause holoprosencephaly. Hum. Mol. Genet. 6, 1847–1853. doi: 10.1093/hmg/6.11.1847
Roessler, E., Du, Y. Z., Mullor, J. L., Casas, E., Allen, W. P., Gillessen-Kaesbach, G., et al. (2003). Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc. Natl. Acad. Sci. U.S.A. 100, 13424–13429. doi: 10.1073/pnas.2235734100
Roessler, E., Ma, Y., Ouspenskaia, M. V., Lacbawan, F., Bendavid, C., Dubourg, C., et al. (2009). Truncating loss-of-function mutations of DISP1 contribute to holoprosencephaly-like microform features in humans. Hum. Genet. 125, 393–400. doi: 10.1007/s00439-009-0628-7
Rohatgi, R., Milenkovic, L., Scott, M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376. doi: 10.1126/science.1139740
Rossi, G., Manfrin, A., Lutolf, M. P. (2018). Progress and potential in organoid research. Nat. Rev. Genet. 19, 671–687. doi: 10.1038/s41576-018-0051-9
Rusert, J. M., Wu, X., Eberhart, C. G., Taylor, M. D., Wechsler-Reya, R. J. (2014). SnapShot: medulloblastoma. Cancer Cell 26, 940–940 e941. doi: 10.1016/j.ccell.2014.11.015
Sanders, T. A., Llagostera, E., Barna, M. (2013). Specialized filopodia direct long-range transport of SHH during vertebrate tissue patterning. Nature 497, 628–632. doi: 10.1038/nature12157
Sasai, N., Briscoe, J. (2012). Primary cilia and graded Sonic Hedgehog signaling. wiley interdisciplinary reviews. Dev. Biol. 1, 753–772. doi: 10.1002/wdev.43
Sasai, N., Kutejova, E., Briscoe, J. (2014). Integration of signals along orthogonal axes of the vertebrate neural tube controls progenitor competence and increases cell diversity. PloS Biol. 12, e1001907. doi: 10.1371/journal.pbio.1001907
Sasaki, H., Hui, C., Nakafuku, M., Kondoh, H. (1997). A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124, 1313–1322.
Schulman, J. M., Oh, D. H., Sanborn, J. Z., Pincus, L., McCalmont, T. H., Cho, R. J. (2016). Multiple hereditary infundibulocystic basal cell carcinoma syndrome associated with a germline SUFU mutation. JAMA Dermatol. 152, 323–327. doi: 10.1001/jamadermatol.2015.4233
Seppala, M., Depew, M. J., Martinelli, D. C., Fan, C. M., Sharpe, P. T., Cobourne, M. T. (2007). Gas1 is a modifier for holoprosencephaly and genetically interacts with sonic hedgehog. J. Clin. Invest. 117, 1575–1584. doi: 10.1172/JCI32032
Shaheen, R., Szymanska, K., Basu, B., Patel, N., Ewida, N., Faqeih, E., et al. (2016). Characterizing the morbid genome of ciliopathies. Genome Biol. 17, 242. doi: 10.1186/s13059-016-1099-5
Shao, S., Wang, G. L., Raymond, C., Deng, X. H., Zhu, X. L., Wang, D., et al. (2017). Activation of Sonic hedgehog signal by Purmorphamine, in a mouse model of Parkinson's disease, protects dopaminergic neurons and attenuates inflammatory response by mediating PI3K/AKt signaling pathway. Mol. Med. Rep. 16, 1269–1277. doi: 10.3892/mmr.2017.6751
Sharma, D., Yadav, J., Garg, E. (2014). Cyclopia syndrome. BMJ Case Rep. 2014. doi: 10.1155/2014/501530
Shimada, I. S., Hwang, S. H., Somatilaka, B. N., Wang, X., Skowron, P., Kim, J., et al. (2018). Basal suppression of the Sonic Hedgehog pathway by the G-Protein-Coupled receptor Gpr161 restricts medulloblastoma pathogenesis. Cell Rep. 22, 1169–1184. doi: 10.1016/j.celrep.2018.01.018
Sipe, C. W., Lu, X. (2011). Kif3a regulates planar polarization of auditory hair cells through both ciliary and non-ciliary mechanisms. Development 138, 3441–3449. doi: 10.1242/dev.065961
Song, Q., Li, C., Feng, X., Yu, A., Tang, H., Peng, Z., et al. (2015). Decreased expression of SCUBE2 is associated with progression and prognosis in colorectal cancer. Oncol. Rep. 33, 1956–1964. doi: 10.3892/or.2015.3790
St-Jacques, B., Hammerschmidt, M., McMahon, A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086. doi: 10.1101/gad.13.16.2072
Stamataki, D., Ulloa, F., Tsoni, S. V., Mynett, A., Briscoe, J. (2005). A gradient of Gli activity mediates graded Sonic Hedgehog signaling in the neural tube. Genes Dev. 19, 626–641. doi: 10.1101/gad.325905
Stickens, D., Zak, B. M., Rougier, N., Esko, J. D., Werb, Z. (2005). Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development 132, 5055–5068. doi: 10.1242/dev.02088
Strutt, H., Thomas, C., Nakano, Y., Stark, D., Neave, B., Taylor, A. M., et al. (2001). Mutations in the sterol-sensing domain of Patched suggest a role for vesicular trafficking in Smoothened regulation. Curr. Biol.: CB 11, 608–613. doi: 10.1016/S0960-9822(01)00179-8
Sun, W. Y., Hu, S. S., Wu, J. J., Huang, Q., Ma, Y., Wang, Q. T., et al. (2016). Down-regulation of beta-arrestin2 promotes tumour invasion and indicates poor prognosis of hepatocellular carcinoma. Sci. Rep. 6, 35609. doi: 10.1038/srep35609
Svard, J., Heby-Henricson, K., Persson-Lek, M., Rozell, B., Lauth, M., Bergstrom, A., et al. (2006). Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell 10, 187–197. doi: 10.1016/j.devcel.2005.12.013
Takeda, S., Yonekawa, Y., Tanaka, Y., Okada, Y., Nonaka, S., Hirokawa, N. (1999). Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A-/- mice analysis. J. Cell Biol. 145, 825–836. doi: 10.1083/jcb.145.4.825
Takezaki, T., Hide, T., Takanaga, H., Nakamura, H., Kuratsu, J., Kondo, T. (2011). Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci. 102, 1306–1312. doi: 10.1111/j.1349-7006.2011.01943.x
Teglund, S., Toftgard, R. (2010). Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 1805, 181–208. doi: 10.1016/j.bbcan.2010.01.003
Tempe, D., Casas, M., Karaz, S., Blanchet-Tournier, M. F., Concordet, J. P. (2006). Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol. Cell. Biol. 26, 4316–4326. doi: 10.1128/MCB.02183-05
Tenzen, T., Allen, B. L., Cole, F., Kang, J. S., Krauss, R. S., McMahon, A. P. (2006). The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev. Cell 10, 647–656. doi: 10.1016/j.devcel.2006.04.004
Tessier-Lavigne, M., Goodman, C. S. (1996). The molecular biology of axon guidance. Science 274, 1123–1133. doi: 10.1126/science.274.5290.1123
Thathiah, A., Horre, K., Snellinx, A., Vandewyer, E., Huang, Y., Ciesielska, M., et al. (2013). beta-arrestin 2 regulates Abeta generation and gamma-secretase activity in Alzheimer's disease. Nat. Med. 19, 43–49. doi: 10.1038/nm.3023
Tickle, C., Towers, M. (2017). Sonic Hedgehog signaling in limb development. Front. Cell Dev. Biol. 5, 14. doi: 10.3389/fcell.2017.00014
Toriyama, M., Lee, C., Taylor, S. P., Duran, I., Cohn, D. H., Bruel, A. L., et al. (2016). The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat. Genet. 48, 648–656. doi: 10.1038/ng.3558
Tukachinsky, H., Kuzmickas, R. P., Jao, C. Y., Liu, J., Salic, A. (2012). Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2, 308–320. doi: 10.1016/j.celrep.2012.07.010
Tukachinsky, H., Lopez, L. V., Salic, A. (2010). A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 191, 415–428. doi: 10.1083/jcb.201004108
Twigg, S. R. F., Hufnagel, R. B., Miller, K. A., Zhou, Y., McGowan, S. J., Taylor, J., et al. (2016). A recurrent mosaic mutation in SMO, encoding the Hedgehog signal transducer smoothened, is the major cause of curry-jones syndrome. Am. J. Hum. Genet. 98, 1256–1265. doi: 10.1016/j.ajhg.2016.04.007
Umehara, F., Tate, G., Itoh, K., Yamaguchi, N., Douchi, T., Mitsuya, T., et al. (2000). A novel mutation of desert hedgehog in a patient with 46,XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am. J. Hum. Genet. 67, 1302–1305. doi: 10.1016/S0002-9297(07)62958-9
Urano, Y., Watanabe, H., Murphy, S. R., Shibuya, Y., Geng, Y., Peden, A. A., et al. (2008). Transport of LDL-derived cholesterol from the NPC1 compartment to the ER involves the trans-Golgi network and the SNARE protein complex. Proc. Natl. Acad. Sci. U.S.A. 105, 16513–16518. doi: 10.1073/pnas.0807450105
Varjosalo, M., Taipale, J. (2008). Hedgehog: functions and mechanisms. Genes Dev. 22, 2454–2472. doi: 10.1101/gad.1693608
Veistinen, L., Takatalo, M., Tanimoto, Y., Kesper, D. A., Vortkamp, A., Rice, D. P. (2012). Loss-of-function of Gli3 in mice causes abnormal frontal bone morphology and premature synostosis of the interfrontal suture. Front. Physiol. 3, 121. doi: 10.3389/fphys.2012.00121
Vlassov, A. V., Magdaleno, S., Setterquist, R., Conrad, R. (2012). Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 1820, 940–948. doi: 10.1016/j.bbagen.2012.03.017
Vokes, S. A., Ji, H., McCuine, S., Tenzen, T., Giles, S., Zhong, S., et al. (2007). Genomic characterization of Gli-activator targets in sonic hedgehog-mediated neural patterning. Development 134, 1977–1989. doi: 10.1242/dev.001966
Vokes, S. A., Ji, H., Wong, W. H., McMahon, A. P. (2008). A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 22, 2651–2663. doi: 10.1101/gad.1693008
Vorobyeva, A. G., Saunders, A. J. (2018). Amyloid-beta interrupts canonical Sonic hedgehog signaling by distorting primary cilia structure. Cilia 7, 5. doi: 10.1186/s13630-018-0059-y
Wang, B., Li, Y. (2006). Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. U.S.A. 103, 33–38. doi: 10.1073/pnas.0509927103
Wang, C., Pan, Y., Wang, B. (2010). Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 137, 2001–2009. doi: 10.1242/dev.052126
Waters, A. M., Beales, P. L. (2011). Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 26, 1039–1056. doi: 10.1007/s00467-010-1731-7
Weatherbee, S. D., Niswander, L. A., Anderson, K. V. (2009). A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum. Mol. Genet. 18, 4565–4575. doi: 10.1093/hmg/ddp422
Werner, R., Merz, H., Birnbaum, W., Marshall, L., Schroder, T., Reiz, B., et al. (2015). 46,XY gonadal dysgenesis due to a homozygous mutation in desert hedgehog (DHH) Identified by exome sequencing. J. Clin. Endocrinol. Metabol. 100, E1022–E1029. doi: 10.1210/jc.2015-1314
Wheway, G., Nazlamova, L., Hancock, J. T. (2018). Signaling through the primary cilium. Front. Cell Dev. Biol. 6, 8. doi: 10.3389/fcell.2018.00008
Wheway, G., Parry, D. A., Johnson, C. A. (2014). The role of primary cilia in the development and disease of the retina. Organogenesis 10, 69–85. doi: 10.4161/org.26710
Wilbanks, A. M., Fralish, G. B., Kirby, M. L., Barak, L. S., Li, Y. X., Caron, M. G. (2004). Beta-arrestin 2 regulates zebrafish development through the hedgehog signaling pathway. Science 306, 2264–2267. doi: 10.1126/science.1104193
Wild, A., Kalff-Suske, M., Vortkamp, A., Bornholdt, D., Konig, R., Grzeschik, K. H. (1997). Point mutations in human GLI3 cause Greig syndrome. Hum. Mol. Genet. 6, 1979–1984. doi: 10.1093/hmg/6.11.1979
Willnow, T. E., Hilpert, J., Armstrong, S. A., Rohlmann, A., Hammer, R. E., Burns, D. K., et al. (1996). Defective forebrain development in mice lacking gp330/megalin. Proc. Natl. Acad. Sci. U.S.A. 93, 8460–8464. doi: 10.1073/pnas.93.16.8460
Wolpert, L. (1969). Positional information and the spatial pattern of cellular differentiation. J. Theor. Biol. 25, 1–47. doi: 10.1016/S0022-5193(69)80016-0
Wong, S. Y., Seol, A. D., So, P. L., Ermilov, A. N., Bichakjian, C. K., Epstein, E. H., et al. (2009). Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 15, 1055–1061. doi: 10.1038/nm.2011
Wuyts, W., Van Hul, W., De Boulle, K., Hendrickx, J., Bakker, E., Vanhoenacker, F., et al. (1998). Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet. 62, 346–354. doi: 10.1086/301726
Xiao, X., Tang, J. J., Peng, C., Wang, Y., Fu, L., Qiu, Z. P., et al. (2017). Cholesterol modification of smoothened is required for hedgehog signaling. Mol. Cell 66, 154–162 e110. doi: 10.1016/j.molcel.2017.02.015
Xie, J., Murone, M., Luoh, S. M., Ryan, A., Gu, Q., Zhang, C., et al. (1998). Activating smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92. doi: 10.1038/34201
Yam, P. T., Kent, C. B., Morin, S., Farmer, W. T., Alchini, R., Lepelletier, L., et al. (2012). 14-3-3 proteins regulate a cell-intrinsic switch from sonic hedgehog-mediated commissural axon attraction to repulsion after midline crossing. Neuron 76, 735–749. doi: 10.1016/j.neuron.2012.09.017
Yam, P. T., Langlois, S. D., Morin, S., Charron, F. (2009). Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron 62, 349–362. doi: 10.1016/j.neuron.2009.03.022
Yatsuzuka, A., Hori, A., Kadoya, M., Matsuo-Takasaki, M., Kondo, T., Sasai, N. (2019). GPR17 is an essential regulator for the temporal adaptation of Sonic hedgehog signalling in neural tube development. Development. doi: 10.1101/424598
Yin, W. C., Satkunendran, T., Mo, R., Morrissy, S., Zhang, X., Huang, E. S., et al. (2019). Dual regulatory functions of SUFU and Targetome of GLI2 in SHH subgroup medulloblastoma. Dev. Cell 48, 167–183 e165. doi: 10.1016/j.devcel.2018.11.015
Yu, H., Wessels, A., Chen, J., Phelps, A. L., Oatis, J., Tint, G. S., et al. (2004). Late gestational lung hypoplasia in a mouse model of the Smith-Lemli-Opitz syndrome. BMC Dev. Biol. 4, 1. doi: 10.1186/1471-213X-4-1
Yu, H., Wessels, A., Tint, G. S., Patel, S. B. (2005). Partial rescue of neonatal lethality of Dhcr7 null mice by a nestin promoter-driven DHCR7 transgene expression. Brain Res. Dev. Brain Res. 156, 46–60. doi: 10.1016/j.devbrainres.2005.01.012
Yue, S., Tang, L. Y., Tang, Y., Tang, Y., Shen, Q. H., Ding, J., et al. (2014). Requirement of Smurf-mediated endocytosis of Patched1 in sonic hedgehog signal reception. eLife 3. doi: 10.7554/eLife.02555
Zapata, J. M., Pawlowski, K., Haas, E., Ware, C. F., Godzik, A., Reed, J. C. (2001). A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J. Biol. Chem. 276, 24242–24252. doi: 10.1074/jbc.M100354200
Zeng, X., Goetz, J. A., Suber, L. M., Scott, W. J., Schreiner, C. M., Robbins, D. J. (2001). A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature 411, 716–720. doi: 10.1038/35079648
Zhang, H., Ping, X. L., Lee, P. K., Wu, X. L., Yao, Y. J., Zhang, M. J., et al. (2001a). Role of PTCH and p53 genes in early-onset basal cell carcinoma. Am. J. Pathol. 158, 381–385. doi: 10.1016/S0002-9440(10)63980-6
Zhang, P., Kiseleva, A. A., Korobeynikov, V., Liu, H., Einarson, M. B., Golemis, E. A. (2019). Microscopy-based automated live cell screening for small molecules that affect ciliation. Front. Genet. 10, 75. doi: 10.3389/fgene.2019.00075
Zhang, W., Kang, J. S., Cole, F., Yi, M. J., Krauss, R. S. (2006). Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev. Cell 10, 657–665. doi: 10.1016/j.devcel.2006.04.005
Zhang, X. M., Ramalho-Santos, M., McMahon, A. P. (2001b). Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell 105, 781–792. doi: 10.1016/S0092-8674(01)00385-3
Zhou, J., Chen, Q., Lanske, B., Fleming, B. C., Terek, R., Wei, X., et al. (2014). Disrupting the Indian hedgehog signaling pathway in vivo attenuates surgically induced osteoarthritis progression in Col2a1-CreERT2; Ihhfl/fl mice. Arthritis Res. Ther. 16, R11. doi: 10.1186/ar4437
Zhou, X., Baron, R. M., Hardin, M., Cho, M. H., Zielinski, J., Hawrylkiewicz, I., et al. (2012). Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum. Mol. Genet. 21, 1325–1335. doi: 10.1093/hmg/ddr569
Zhulyn, O., Nieuwenhuis, E., Liu, Y. C., Angers, S., Hui, C. C. (2015). Ptch2 shares overlapping functions with Ptch1 in Smo regulation and limb development. Dev. Biol. 397, 191–202. doi: 10.1016/j.ydbio.2014.10.023
Keywords: sonic hedgehog (Shh), development, genetic disease, mouse model, ciliopathies, cancer
Citation: Sasai N, Toriyama M and Kondo T (2019) Hedgehog Signal and Genetic Disorders. Front. Genet. 10:1103. doi: 10.3389/fgene.2019.01103
Received: 15 April 2019; Accepted: 11 October 2019;
Published: 08 November 2019.
Edited by:
William Cho, Queen Elizabeth Hospital (QEH), Hong KongReviewed by:
Alain-Pierre Gadeau, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2019 Sasai, Toriyama and Kondo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noriaki Sasai, bm9yaWFraXNhc2FpQGJzLm5haXN0Lmpw; Michinori Toriyama, dG9yaXlhbWFAa3dhbnNlaS5hYy5qcA==; Toru Kondo, dGtvbmRvQGlnbS5ob2t1ZGFpLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.