 Silvian Tan
Silvian Tan Nonhlanhla Khumalo
Nonhlanhla Khumalo Ardeshir Bayat
Ardeshir Bayat- 1Plastic and Reconstructive Surgery Research, Centre for Dermatology Research, NIHR Manchester Biomedical Research Centre, University of Manchester, Manchester, United Kingdom
- 2Hair and Skin Research Laboratory, Department of Dermatology, Groote Schuur Hospital, University of Cape Town, Cape Town, South Africa
Keloids are considered as benign fibroproliferative skin tumors growing beyond the site of the original dermal injury. Although traditionally viewed as a form of skin scarring, keloids display many cancer-like characteristics such as progressive uncontrolled growth, lack of spontaneous regression and extremely high rates of recurrence. Phenotypically, keloids are consistent with non-malignant dermal tumors that are due to the excessive overproduction of collagen which never metastasize. Within the remit of keloid pathobiology, there is increasing evidence for the various interplay of neoplastic-promoting and suppressing factors, which may explain its aggressive clinical behavior. Amongst the most compelling parallels between keloids and cancer are their shared cellular bioenergetics, epigenetic methylation profiles and epithelial-to-mesenchymal transition amongst other disease biological (genotypic and phenotypic) behaviors. This review explores the quasi-neoplastic or cancer-like properties of keloids and highlights areas for future study.
Introduction
Keloids are considered as benign fibroproliferative dermal tumors, which are borne out of abnormal wound healing processes following injury to the skin. They are characterized visually by raised exophytic dermal outgrowths extending beyond the original wound boundary and microscopically by thickened hyalinized collagen bundles (1, 2).
Most individuals affected by keloid disease are aged between 10 and 30 years and are of pigmented skin with high reports in African, Afro-Caribbean, Afro-American, Hispanic, or Asian ancestry (3–6). Keloids may develop months or years after the initial injury and can be accompanied by intense pain, pruritus, and other physical, and psychosocial symptoms (7). Common sites affected are the anterior chest, shoulders, back, and earlobe, with those on the pre-sternum and shoulder regions developing under high tension. Keloids tend to be aggressive in invading adjacent surrounding healthy (normal) skin and can often recur following any form of treatment. In particular, monotherapy with surgical removal alone carries a recurrence rate of up to 100% (8). Phenotypically, keloids are consistent with non-malignant dermal tumors that are due to the excessive overproduction of collagen which never metastasize. However, the morphology and clinically aggressive behavior of keloids can be thought to bear a resemblance to neoplastic dermal tumors.
Different theories exist to explain the etiology of keloids, including elevated skin tension (9, 10), hypoxia (11), chronic inflammation (12), autoimmune (13–15), genetics (16, 17), and vascular factors (18), none of which, however, are independently sufficient to do so. To date, options for keloid treatment are poorly defined, in part due to unsatisfactory outcomes of current treatments and the poor quality of evidence surrounding their use. Compounding keloid research is the lack of animal disease models for testing.
This review aims to explore the salient cancer-like or quasi-neoplastic attributes and features of keloids (Figure 1) and highlights several key areas for future study.
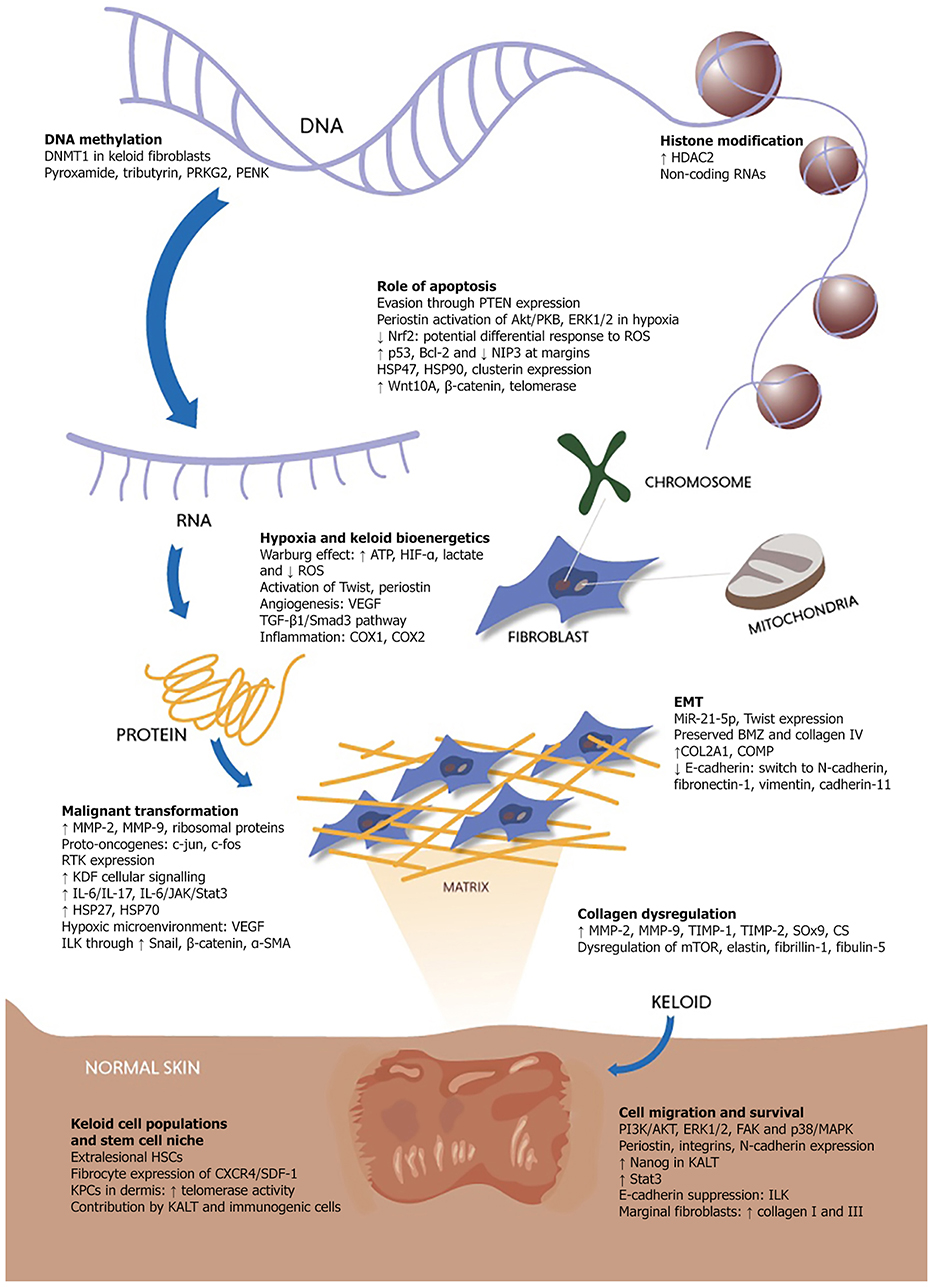
Figure 1. Key processes contributing to the quasi-neoplastic expression of keloid pathobiology.
Keloids: Scars or Cancerous Tumors?
Keloids are traditionally viewed as scars on the spectrum of fibroproliferative dermal diseases. To elucidate the etiopathogenesis of keloids, however, the distinction between scar and disease must be made. A scar is the endpoint of physiological wound healing, preceded by inflammation, fibroplasia and granulation, and manifest as non-functioning fibrotic tissue, which may regress over time (19, 20). Cutaneous scars often undergo changes in properties such as thickness, texture, color, and strength but remain within the confines of the surrounding intact skin (19, 21). In contrast, keloids are aggressive exophytic dermal outgrowths disproportionately grown beyond the boundaries of the original wound, from a source, which remains suspended in “wound healing” and scar maturation. Although keloids are not routinely classified as true neoplasms due to their lack of spontaneous occurrence and absence of metastasis, they exhibit various cancer-like characteristics such as uncontrolled proliferation, invasiveness into surrounding tissue, lack of spontaneous regression and ability to vascularize (22–24). Their disproportionately locally aggressive clinical phenotype suggests possible links with skin or mesenchymal tumors that need to be explored in further detail.
Owing to their similar clinical presentation, keloids have been misdiagnosed as other benign and malignant skin tumors and vice versa (25). Of these, dermatofibrosarcoma protuberans is the most commonly linked to keloids amongst others such as keloidal dermatofibroma (26), keloidal basal cell carcinoma, keloidal atypical fibroxanthoma (27), suggesting a possible overlap between them (28). Based on their pathobiology and clinical phenotype, keloids bear the most similarity between tumors relating to fibrosis, dermal origin, and mesenchymal origin (29). Similarities between keloids and mesenchymal tumors can be found across a range of characteristics from biomarker expression to clinical presentation (30). Seminal work on keloid mesenchymal-related biomarkers suggests that stem-cell like cells identified in keloids deviate from dermal to chondrocytic or osteogenic lineage (31), however research to confirm such hypotheses in this direction is still lacking.
Keloid Morphology: Cellular and Matrix Composition
On gross examination, keloids are well-demarcated, raised cutaneous lesions in different shapes and may be bosselated, nodular, or pendulous (32). They may appear shiny with a discoloration—often erythematous with telangiectasia in individuals with fairer skin and hyperpigmented in those with darker skin (32)—and can reach sizes between several millimeters to many centimeters in diameter (33). Keloids display a heterogenic phenotype in relation to stiffness and range from soft to extremely firm with decreased skin plasticity. The center of keloid tissue often exhibit central hypoxia due to capillary occlusion as a result of exuberant collagen and endothelial cells (34). Keloid margins contain active fibroblasts which invade into surrounding tissue and are well-vascularized through angiogenesis to upkeep the oxygen and nutrient supply required presumably to fuel their invasiveness (24, 35). This is thought to be the result of endothelial cell migration and survival from growth factor activation through kinase signaling pathways such as PI3K/AKT, ERK1/2, FAK, and p38/MAPK (36, 37).
Most of the expansile keloid tissue is in the reticular dermal layer with a thickened overlying epidermis. Keloid tissues are not encapsulated and have a perimeter that advances into the surrounding tissue (2). Marginal fibroblasts are more metabolically active and engage in higher rates of collagen I and III production, as reflected by more erythematous skin overlying these margins (38). Several studies have shown that the outer margins and inner complex of keloids are populated by different types of stem cells. In particular, mesenchymal-like stem cells expressing non-hemapoetic markers constituting the inner complex appear to represent the niche that is vital to sustain keloid growth, surrounded by extralesional hematopoietic stem cells (39, 40). In relation to this, the concentration of homogenous thickened hyalinized collagen in the center surrounded by more typical-looking collagen fibers near the edges is unique to keloids (41), but not always present (2).
In brief, cutaneous wound healing involves the formation and remodeling of collagen matrix over time by fibroblasts and myofibroblasts. Similarly, activated myofibroblasts and keloid fibroblasts represent the main generators of keloid matrix through their synergistic action in increasing keloid tissue stiffness (42, 43). Fibrocytes, thought to be myofibroblast precursors, are bone marrow-derived circulating cells present in keloids and tissues undergoing wound healing (44–46). They appear as a cross between fibroblasts, monocytes, and hematopoietic stem cells (HSCs) due to their fibroblast products, hematopoietic surface markers, myeloid antigen expression, and shared morphological characteristics (47). Their expression of CXCR4 forms part of the CXCR4/SDF-1 axis, which is crucial in skin regulating cutaneous wound healing, systematic lupus erythematosus and angiogenesis of basal cell carcinoma (48).
The keloid matrix consists of different collagens, glycoproteins, and glycosaminoglycans (GAGs) (23, 49, 50). The initial overproduction of type III collagen in keloid matrix is replaced by type I collagen, with an extremely high ratio of type I to type III collagen (17:1) compared to normal scars (6:1) (51). The collagen fibers constituting keloids are larger than those in normal scars and disorderly arranged (2) in loosely cross-linked (51) whorls of thick bundles in the same plane as the epidermis (1). Elevated levels of MMP-2, TIMP-1, and TIMP-2 are observed in keloids (52), the imbalance of which appears to dysregulate collagen production and accumulation, with upregulated MMP-2 and MMP-9 expression additionally linked to cancer invasiveness (53). Overproduction of collagen and matrix is also linked with dysregulated mTOR signaling (54), which plays a crucial role in human cancers (55). Interestingly, elastic fiber deposition is higher in the reticular dermis than the papillary dermis (56, 57), but has been shown to be absent in the keloid extracellular matrix (ECM) alongside deregulated expression of elastic fiber assembly proteins such as elastin, fibrillin-1 and fibulin-5 (58). This is attributed to the over-deposition of chondroitin sulfate (CS) which is 6.9 times higher than that in normal skin. CS is thought to suppress elastic fiber assembly through the dysregulation of fibrillin-1 deposition (58) and its aberrant expression has been found to contribute to tumor metastasis in breast cancer (59). The functions of fibulin-5 and elastin in tumor formation are complex, involving the regulation of metalloproteinases (MMPs) amongst others (60–62). The interplay between these proteoglycan-associated factors in keloid pathogenesis remain to be elucidated.
Sox9, the master regulator of chondrogenesis (63), is upregulated in keloids (31) and its ectopic expression is linked with the upregulation of COL2A1 and cartilage oligomeric matrix protein (COMP) which culminates in ECM production geared toward chondrogenesis and fibrosis (64). COMP stimulates the assembly of collagen 1 fibrils (65, 66), with an expression level proportional to keloid size and is expressed in scleroderma and other tumors (67–69). Keloids exhibit significantly raised levels of biglycan in the nodular dermis of active keloid lesions alongside decreased decorin expression (70). This is interesting as decorin, which has recently been found to suppress collagen production, is raised in malignant conditions (71, 72). All these proteoglycans have been shown to be differentially regulated by basic fibroblast growth factor, that is thought to be involved in producing the tumor phenotype (73, 74).
Keloids share many similarities with other dermal tumors by virtue of their shared tissue origin. This includes the integral activation of Wnt β-catenin pathway in desmoid tumors (75) and elevated levels of TGF-β, collagen, and GAG in dermatofibrosarcoma protuberans (76). In both of these tumors, trauma has been cited as a precipitating factor (75, 77).
Keloids and the Hallmarks of Cancer
Although keloids exhibit the hallmarks of cancer to a large extent, these characteristics remain to be fully explored in this context (78, 79). There is potentially a key regulator or group of pre-requisite factors, which when activated after skin injury, triggers a cascade of events that culminate in keloid scar formation. The various relationships between tumor-related factors expressed in keloids may be complex and their roles in sustaining keloid growth are still unclear. Table 1 highlights key tumor biomarkers discussed in this review.
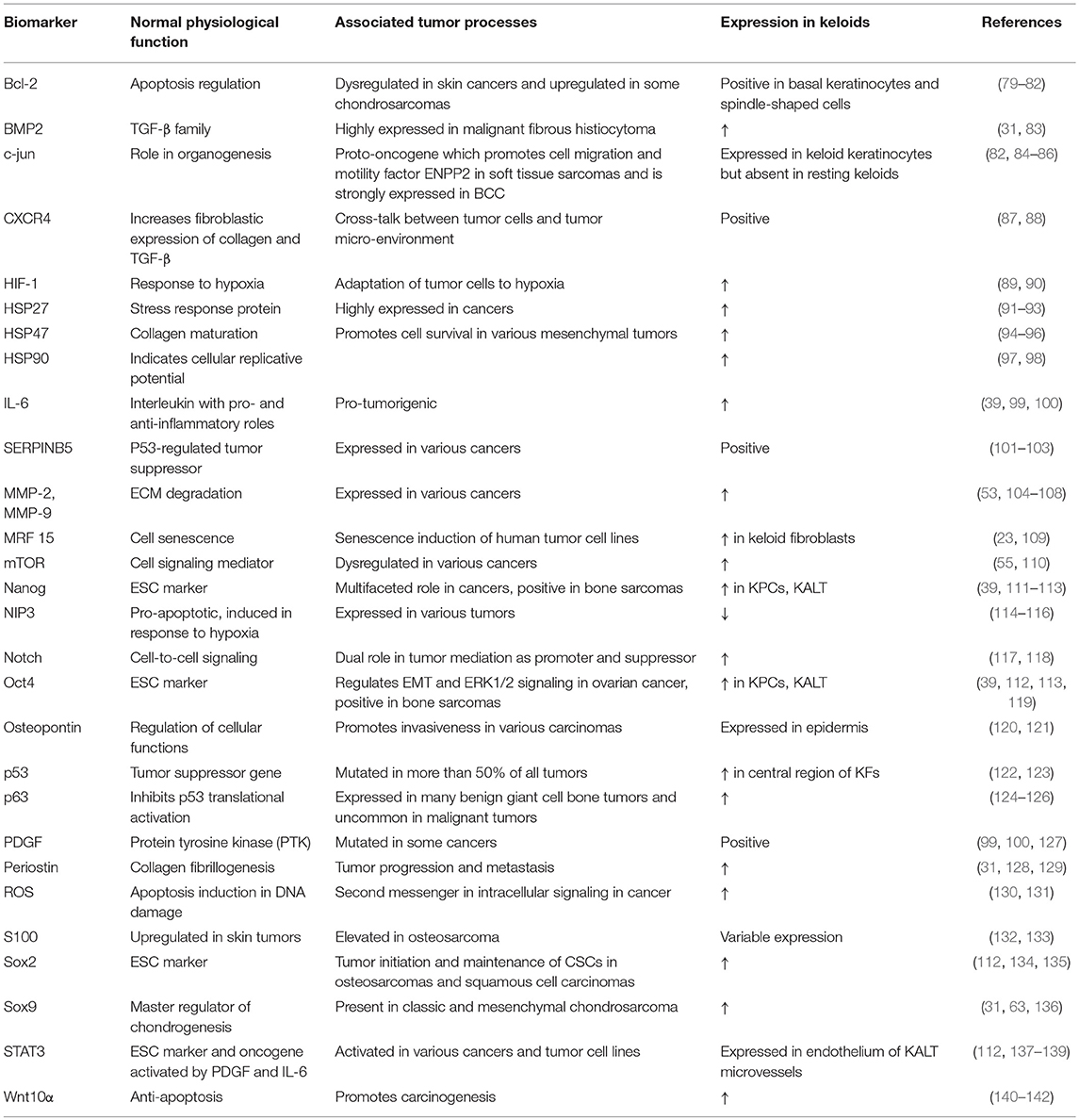
Table 1. Expression of tumor biomarkers in keloids.
Sustaining Proliferative Signaling
Keloid fibroblasts express higher receptor tyrosine kinase signals compared to normal skin derived fibroblasts (143, 144). This increased keloid-derived fibroblast cellular signaling, which may influence cell growth, differentiation, and survival, is linked to cancer development when dysregulated (145). Nanog, a gene which confers self-replication abilities to cells and is elevated in various cancers (111, 146, 147), is absent in somatic cells (148), yet recently found to be upregulated in keloid associated lymphoid tissue (KALT) (112). Zhang et al. (39) described a population of clonogenic, self-renewing keloid-derived precursor cells expressing mesenchymal and embryonic stem cell markers and are driven by IL-6/IL-17 mediated inflammation. IL-6, alongside hepatocyte growth factor and epidermal growth factor, is pro-tumorigenic (39). Of interest, IL-6 and its second messengers JAK/Stat3, both of which are elevated in keloid fibroblasts, form the IL-6/JAK/Stat3 pathway, which contributes toward tumorigenesis within the tumor micro-environment. In particular, Stat3 confers increased cell proliferation and migration within keloid fibroblasts (99, 149).
Transforming growth factor beta (TGF-β) is a well-researched cytokine in keloid pathogenesis due to its pivotal role in directing keloid fibrosis (150, 151). TGF-β1 influences keloid keratinocytes through both Smad-dependent and Smad-independent signaling pathways such as ERK1/2 and p38 to promote collagen accumulation (150, 151). In relation to cancer, TGF-β1 has been shown to inhibit initial tumor formation before accelerating its malignancy in a transgenic mouse skin cancer model (152). Keloid and normal fibroblasts exhibit a differential response to PDGF, EGF, FGF (153), and TGF-β (154), the majority of which bind to protein tyrosine kinase (PTK) family receptors. PTK receptors contribute to cell proliferation, differentiation (155), and carcinogenesis (156).
Satish et al. (23) found the upregulation of ribosomal proteins in keloid fibroblasts, which is also seen in cancer growth, indicating tighter regulations in keloids which prevent the malignant transformation of their precursor cells. Transcriptional activating factors involved include c-jun and c-fos which function as proto-oncogenes. In addition to its role in cell cycle regulation of fibroblasts (84) and Ras-activated human epidermal neoplasia, c-jun is also linked to benign inflammatory conditions like psoriasis and arthritis (157).
Evasion of Growth Suppressors
The mechanisms underlying keloid growth are not straightforward, involving both up- and downregulation of tumor suppressors. The deregulation of p53 forms the hallmark in various cancers; more recently a mechanism by which it is deregulated in keloids has been identified as TRAF4-USP10 interaction that culminates in fibroblast proliferation (158). Interestingly, whilst p53 mutation is prevalent in keloids (159), its expression has also been found to be elevated in the central region of keloid fibroblasts alongside an increase in p63 which inhibits its translation (101, 160). In turn, this leads to downstream effects including elevated SERPINB5 and maspin, the latter of which is also upregulated during keratinocyte senescence (101). The link between TP53 codon 72 polymorphism and keloids remains unclear (124, 161, 162). Other examples include downregulated miR-1224-5p which normally functions through TGF-β1/Smad3, and has a role in cancers (163). Similarly, miR-21-5p plays a key role in PTEN-mediated proliferative and apoptotic mechanisms in keloid fibroblasts (164), causing decreased levels of PTEN which is a tumor suppressor with wide-ranging downstream effects on cellular maintenance and morphology through PI3K/AKT/mTOR (165). Interestingly, elevated levels of tumor suppressors such as PML are also seen in the hypercellular regions of keloids, suggesting senescence as a factor which confers keloids their benign nature (166). It is plausible that the concerted interplay between these factors result in the quasi-neoplastic behavior of keloids.
Induction of Angiogenesis: The Role of Hypoxia in Keloid Bioenergetics
The Warburg effect is a phenomenon in cancer cells in which metabolism is favored and ATP is produced through anaerobic glycolysis in favor of oxidative metabolism. In cancer cells, the rate of glucose uptake is dramatically increased leading to lactate production, despite the presence of fully functioning mitochondria and availability of oxygen. Specifically, this refers to an increased rate of glycolysis, which is followed by a surge of lactic acid fermentation even if there is no lack of oxygen (167, 168). Similarly, keloid fibroblasts have been found to preferentially utilize the glycolytic pathway instead of oxidative phosphorylation, the latter of which is utilized by normal fibroblasts. The ability of keloid fibroblasts to tap into a wider source of energy may explain why keloids thrive in a hypoxic microenvironment. Indeed, the same study reported that keloid fibroblasts have more ATP, are 3.7 times more active than normal fibroblasts and exhibit lower levels of reactive oxygen species (ROS), which is a byproduct of mitochondrial oxidative phosphorylation (22). The switch to aerobic glycolysis in tumors as a means of ATP production is thought to be the result of glycolytic enzymes activated by HIF-α (169), which is also upregulated in keloids (170, 171). These findings are supported by the higher levels of lactate in keloids due to glycolysis and increase in keloid ATP synthesis on exposure to hypoxia (89, 172). As tumors become increasingly reliant on glycolysis with tumor progression (173), it may even be possible to correlate the growth of keloids with their utilization of glycolysis as a form of keloid disease staging.
Hypoxia is thought to promote tumor proliferation and invasiveness in cancer (174). In keloids, local hypoxia within the injury zone accelerates wound healing by stimulating angiogenesis and driving fibroblast proliferation (175), as evidenced by hypoxia-induced vascular endothelial growth factor (VEGF) expression (176) in keloid fibroblasts and a higher density of blood vessels in keloids than normal dermis and scars. In addition to angiogenesis, hypoxia-induced fibroblast to myofibroblast-like conversion has also been observed in keloids and postulated to occur by the TGF-β1/Smad3 pathway (177). Angiogenesis is vital in the development of many cutaneous diseases (178). Hypoxia induces the activation of HIF-α1 in both keloids and cancer (179). This results in the activation of Twist which mediates EMT-related cadherin switching and is a master regulator of morphogenesis and other embryonic processes (180).
Periostin, a cell-adhesion ECM protein (128), is increased in physiological processes involved in skin repair, fibrosis, cell proliferation, and ECM remodeling, and tumor processes such as growth and metastasis (181). Periostin increases angiogenesis by activating ERK1/2 and focal adhesion kinase (FAK) pathways, increasing VEGF and angiopoeitin-1 (Ang-1) expression (182). Specific to keloids, periostin is associated with promoting the ability of keloid fibroblasts to migrate and invade surrounding tissues in hypoxia (183). Periostin expression is upregulated by hypoxia through an HIF-1α-dependent pathway (183). Another group of cell adhesive receptor proteins implicated are integrins which are thought to interact with periostin to facilitate EMT, angiogenesis, and the mobility of chondrocytes, fibroblasts, and cancer cells (184). Periostin promotes angiogenesis in gastric, breast, and ovarian cancers (185, 186).
Activating Invasion and Metastasis
There is a growing body of evidence recognizing the importance of EMT in keloid pathophysiology. Whilst EMT is known to promote the migratory behavior of metastatic cells, the benign nature of keloids makes it unclear whether they are the result of type II fibrotic EMT or suspended type III metastatic EMT (187). There is no evidence of significant epidermal-dermal basement membrane zone (BMZ) breakdown or disrupted collagen IV expression in the BMZ of keloids (188), which may explain why they do not metastasize. Investigating other cell motility factors in keloids may reveal unique key agents in metastatic prevention.
E-cadherin, a component of epithelial adherens junction, is encoded by CDH1 and becomes lost in cancer cells undergoing EMT (189). Although CDH1 gene expression levels are similar between normal and keloid keratinocytes, keloids express decreased protein levels of E-cadherin (188, 190), in line with cadherin switching from E-cadherin to mesenchymal markers such as N-cadherin (191), fibronectin-1, vimentin, and cadherin-11. Indeed, vimentin, fibronectin-1, and cadherin-11 have been identified in keloids, with overexpression of fibronectin-1 (23, 49). Vimentin, which is commonly expressed in soft tissue tumors, is highly expressed in keloid keratinocytes (190) and associated with changes in shape, motility and adhesion properties during EMT (192). N-cadherin, which is linked to increased tumor cell mobility (193) and progression (194), remains to be studied in keloids. Integrin-linked kinase (ILK), which is normally involved in regulating ECM signaling (195, 196), leads to E-cadherin suppression and promotion of tumor invasiveness (197–200) alongside a fibroblastic phenotype when aberrantly upregulated in EMT. In basal cell carcinoma, ILK expression leads to increased tumor invasiveness and EMT markers through upregulated Snail, β-catenin and α-SMA (197). β-catenin is elevated in the nucleus and cytoplasm of cells undergoing EMT. Similarly, a higher level of β-catenin protein activity has been found in keloid keratinocytes compared to normal keratinocytes, and is inversely proportional to E-cadherin expression despite no significant difference at transcription level (140).
Upon injury, epidermal keratinocytes induce dermal fibroblasts to produce keloid matrix features by expressing genes involved in epithelial-to-mesenchymal transition (EMT) (201) and upregulating the expression of inflammatory mediators such as COX1 and COX2 in keloid fibroblasts, endothelial cells, and inflammatory cells (202). The fibroblast-like phenotype in these keloid keratinocytes is potentially perpetuated by the local hypoxic environment in keloids which increase the invasiveness of these keratinocytes, thereby leading to excessive keloid growth (202).
MicroRNAs are known to facilitate oncogenesis and metastasis by regulating post-transcriptional and translational gene expression in cell proliferation, EMT and cancer stem cells (203, 204). This may explain several observations in keloids, for example, the decreased elastic fiber density in keloids despite normal elastin expression coding for elastic fibers (205). MiR-21-5p which mediates PTEN in keloids, also contributes to EMT in keloids (188, 206), thereby suggesting EMT phenotype maintenance in keloid keratinocytes through exertion of their stem-cell like effects (206). This is significant, as MiR-21-5p has been claimed to be overexpressed in most cancers and various fibrotic disorders such as those involving the skin, kidneys and cardiopulmonary systems (206, 207).
Additionally, insulin-like growth factor-I receptor, which promotes fibroblast invasiveness, is highly upregulated in keloid fibroblasts compared to normal fibroblasts (208). Osteopontin is a major cytokine, which promotes matricellular interaction, tumor progression, angiogenesis, and resistance to apoptosis in malignancies (120). It is expressed in keloids, skin, and mesenchymal tumors. S100 influences the chondroid metaplasia of fibroblasts to fibrocartilage cells (209) and is expressed in desmoplastic melanoma (160), however it has only been found in low levels in keloids (132).
Resisting Cell Death
Keloid myofibroblasts are thought to sustain extended “healing” by eluding apoptosis (210) despite prolonged hypoxia. Tumor cells may escape apoptosis through periostin-activated Akt/PKB pathway in hypoxia (211). In keloids, periostin activation is linked with downstream activation of ERK1/2 (182) which is known to regulate the functions of Fra-1 and ZEB1/2 in tumorigenesis (212). There is no accepted consensus on the apoptosis levels in keloids. It was previously thought that keloids elude apoptosis by sustaining lower levels of oxidative stress (213). More recent evidence contradicts this; the discovery of increased ROS (125) and downregulated Nrf2 (214) is consistent with other papers suggesting increased rates of apoptosis in keloids (215, 216). Nrf2 is protective against oxidative stresses and various diseases including cancer (217, 218), whereas ROS is thought to act both as a second messenger in intracellular signaling (130) to facilitate cancer progression and as an apoptotic agent by promoting cell senescence (219). This suggests the possibility of differential response to ROS by different cell populations.
Up to eight apoptosis genes were found to be down-regulated amongst 64 which were studied in keloids, compared to normal skin by Sayah et al. (114). Amongst these, NIP3 is known pro-apoptotic, induced in response to hypoxia by HIF-1α (115). The distribution of apoptotic cells was mapped and found to be equal across the normal tissues but were fewer and concentrated at the margins of keloid tissues. From a regional perspective, Luo et al. (220) described in detail the heterogeneous nature of the microenvironment and subcellular regions in keloids with regard to apoptosis. Within the keloid tissue itself, apoptotic cells are concentrated in hypercellular areas at the edges and lacking in the collagen-abundant center (221), corroborating with previous findings of p53 and Bcl-2 localizing to the same regions (79). The concerted up- and down- regulation of various apoptotic-related genes in keloid tissues suggest an interplay of these factors in the tumorigenic capabilities of keloids.
Keloids express several heat shock proteins (HSPs) which are responsible for increasing cell survivability through interprotein interactions in response to stresses (97, 222). HSP47, which is vital in collagen synthesis and maturation (223), is highly expressed in carcinomas of the head, neck (224), and pancreas (225). HSP90 increases in response to cellular stresses to increase cell survival (97) and is upregulated in keloids (98). Similarly, clusterin is increased in cellular stresses (226, 227), and invasive tumors (228).
A study by Yu et al. (140) found increased expression of Wnt10A, β-catenin, and telomerase in keloids. Increased Wnt signaling in keloids promotes cell growth by minimizing apoptosis through upregulation of β-catenin and telomerase activity (140). In normal cells, β-catenin is inhibited by Sox9 to promote chondrogenesis (229, 230). It is unclear whether these two molecules are simultaneously upregulated and if so, the effects they have on keloid growth. Telomerase activity, which is linked with disease aggressiveness of bone and soft tissue tumors, is found in 10% of benign and 44% of malignant tumors (231). Keloid precursor cells have been shown to demonstrate telomerase activity which may mediate telomere lengthening by interacting with IL-6 (39). There are reports, however, of decreased telomere length (131) and suppressed telomerase activity in keloids as it is thought that telomerase ceases to function after the initial stages of keloid formation (232). Unsurprisingly, telomere length is negatively correlated with ROS levels (131, 233).
It is postulated that a reciprocal protective relationship exists between tumor cells and their stem cell niche, as shown by the upregulation of various genes and proteins in endothelial cells to increase survivability in response to cytotoxic conditions (234, 235). The role of micro-environmental signals (17, 236) which promote chemokine-related changes in keloid fibroblasts (237) has been investigated recently following similar studies on epigenetics in cancer development (238, 239). Studies on DNA methylation profiles of keloid tissue and healthy skin suggest that DNA methylation may be a key driver in the pathology of keloids. These include the discovery of DNMT1 in keloid fibroblasts (240) and master regulator genes such as pyroxamide, tributyrin, PRKG2, and PENK in keloid tissue (241–243). Other studies found evidence for histone modification such as upregulated HDAC2 in keloid tissue (244) and expression of non-coding RNAs in keloids (245). These discoveries are significant as they represent known biomarkers of cancer. HSPs such as HSP27, HSP47, and HSP70 are also found to be overexpressed in keloids (246), with HSP27 and HSP70 having links to cancer (247).
Enabling Replicative Immortality: The Role of a Keloid Stem Cell Niche
Stem cells perform various roles in cutaneous wound healing (248). Distinct populations of several stem or stem-like cells (249) have been identified in keloid tissues, including hematopoietic stem cells, mesenchymal-like stem cells, and keloid progenitor cells (40). The fate of stem cells dictated by signals from the stem cell niche microenvironment (250) represents a tightly regulated process in keloids (251).
A population of keloid precursor stem cells (KPCs) was described in the dermis which harbor the capacity for multi-lineage differentiation and self-renewal, the latter of which may explain the high recurrence rates in keloids (39). Their telomerase activity is higher than normal skin precursor cells but lower than cancers. The significance of this is their correlation with tumor or cancer stem cells (252) which are thought to originate from adult stem cells mutated under the influence of a deregulated stem cell niche, following which they become capable of uncontrolled growth, perpetual self-renewal and multi-lineage differentiation, eventually leading to cancer. They may also have a role in conferring resistance to treatment (253, 254). The role of the cancer stem cell niche is outlined in further detail by Borovski (255).
As part of the wound healing process, orchestrated upregulation and interactions between neutrophils, eosinophils, T-cells, B-cells, macrophages, and mast cells have been demonstrated in keloid tissue, and more recently along with keloid-associated lymphoid tissue (KALT) (41, 256). Within KALT, primitive cells within the sub-epidermal micro-vessel endothelium have been recently found to express embryonic stem cell (ESC) markers Oct4, Sox2, pSTAT3, and Nanog (112), suggesting a possible relationship between the immunogenicity and presence of stem cells in keloids.
Responsiveness to Cancer Treatments
High quality evidence on keloid treatment is limited (257, 258). Larger keloids often require surgical excision (259) which carry recurrence rates of up to 100% as monotherapy (8). Radiation, which is another long-established modality of treatment, has been shown to produce varying results in the literature. Primary radiation given to a sample of 84 unresectable keloids over 5 weeks resulted in 97% significant regression in the course of 18 months (260). Another study found that post-operative radiotherapy in the form of beta radiation at 400cGy twice a week totaling up to 16Gy was found to result in flattened scars in 67% of patients, but not without side effects such as pain and atrophy, and carries a recurrence rate of 21.2% (258, 261). Brachytherapy has also yielded favorable results, with a recurrence rate of 3.1% at 33.6 months in a recent study (262). Combined with surgical excision and application of platelet-rich plasma, post-operative superficial radiotherapy up to three fractions has been demonstrated to result in low disease recurrence (263).
Chemotherapy and targeted therapy agents have also shown promising results. Sorafenib, a tyrosine kinase inhibitor, has been found to be effective in suppressing keloid activity by targeting the TGF-β/Smad and MAPK/ERK pathways which are involved in keloid fibroblast growth and cell cycle processes (264). A recent systematic review identified post-excision 5-fluorouracil in combination with triamcinolone acetonide as a favorable treatment with up to 92% non-recurrence (265) in what is termed as “combination therapy.” In comparison, monotherapy using intra-lesional corticosteroids, the first-line treatment for keloids (8) has been shown to yield positive results in only 77% of patients (266). The recurrence rates in triple therapy incorporating surgical excision, intra-lesional steroids, and silicone sheet have been reported at 4.6% (263), 9.1% (267), and 12.5% (268). Keloids are also responsive to recombinant adenovirus-mediated double suicide gene therapy using CDglyTK which is used in cancer (269).
The use of photodynamic therapy (PDT) in keloids led to the suppression of blood flow and collagen levels alongside improved tissue flexibility (270). This is a cancer treatment used in Bowen's, basal cell carcinoma and actinic keratosis, thought to suppress fibroproliferation through protoporphyrin 9 (PpIX)-mediated activation of ROS, which is in turn cytotoxic (271, 272). Although keloids are not metastatic, their responsiveness to multimodal cancer treatment suggests similar underlying disease mechanisms with tumorigenesis.
Conclusion
The idea that keloids behave like non-malignant locally aggressive cutaneous cancers is not new and this is evident in both its phenotypical and genetic properties. The biomarker expression profile in these diseases highlights the striking parallels between keloids and both benign and malignant mesenchymal tumors across transcriptional, translational, cellular, and tissue levels. Keloids also exhibit characteristics displayed by cancer cells to some degree, in particular the Warburg effect which confers increased survival to keloid cells in hypoxia. Furthermore, signaling pathways common to these diseases have been found to mold the matrix composition of keloids with the inclusion of chondrogenic signatures. This, in turn, is perpetuated by key cells such as melanocytes, keratinocytes, fibroblasts, and those from the fibrocytic family, which participate in EMT and possess stem cell-like properties. A keloid stem cell niche has also been postulated to support this. Many important tumor-related factors, which have been shown to contribute to the overall pathogenesis by influencing cellular processes such as apoptosis. To this point in time, the use of cancer treatments in keloids has shown encouraging results, further diminishing the fine line between keloids and cancerous tumors. Taken together, this poses many questions in relation to keloid pathobiology which need to be answered, in order to understand how the cascade encompassing prolonged, dysregulated wound healing culminates in the cancer-like or quasi-neoplastic processes which result in keloid formation, progression, persistence and recurrence. This is vital due to the implications it may have for the future therapy and further investigative research of this elusive disease and those with which it shares similar properties.
Author Contributions
The review article was originally conceived and structured by AB. ST performed literature search and wrote the article. NK contributed by reading and editing. AB edited the overall article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Jumper N, Paus R, Bayat A. Functional histopathology of keloid disease. Histol Histopathol. (2015) 30:1033–57. doi: 10.14670/HH-11-624
2. Lee JY, Yang CC, Chao SC, Wong TW. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. (2004) 26:379–84. doi: 10.1097/00000372-200410000-00006
3. Ketchum LD, Cohen IK, Masters FW. Hypertrophic scars and keloids. A collective review. Plastic Reconstr Surg. (1974) 53:140–54. doi: 10.1097/00006534-197402000-00004
4. Lu W-s, Zheng X-d, Yao X-h, Zhang L-f. Clinical and epidemiological analysis of keloids in Chinese patients. Arch Dermatol Res. (2015) 307:109–14. doi: 10.1007/s00403-014-1507-1
5. English RS, Shenefelt PD. Keloids and hypertrophic scars. Dermatol Surg. (1999) 25:631–8. doi: 10.1046/j.1524-4725.1999.98257.x
6. Ramakrishnan KM, Thomas KP, Sundararajan CR. Study of 1,000 patients with keloids in South India. Plastic Reconstr Surg. (1974) 53:276–80. doi: 10.1097/00006534-197403000-00004
7. Ud-Din S, Bayat A. New insights on keloids, hypertrophic scars, and striae. Dermatol Clin. (2014) 32:193–209. doi: 10.1016/j.det.2013.11.002
8. Mustoe TA, Cooter RD, Gold MH, Hobbs FD, Ramelet AA, Shakespeare PG, et al. International clinical recommendations on scar management. Plastic Reconstr Surg. (2002) 110:560–71. doi: 10.1097/00006534-200208000-00031
9. Chipev CC, Simon M. Phenotypic differences between dermal fibroblasts from different body sites determine their responses to tension and TGFbeta1. BMC Dermatol. (2002) 2:13. doi: 10.1186/1471-5945-2-13
10. Suarez E, Syed F, Rasgado TA, Walmsley A, Mandal P, Bayat A. Skin equivalent tensional force alters keloid fibroblast behavior and phenotype. Wound Repair Regen. (2014) 22:557–68. doi: 10.1111/wrr.12215
11. Kischer CW, Shetlar MR, Chvapil M. Hypertrophic scars and keloids: a review and new concept concerning their origin. Scan Electron Microsc. (1982) (Pt 4):1699–713.
12. Ogawa R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int J Mol Sci. (2017) 18:606. doi: 10.3390/ijms18030606
13. Kazeem AA. The immunological aspects of keloid tumor formation. J Surg Oncol. (1988) 38:16–8. doi: 10.1002/jso.2930380106
14. Placik OJ, Lewis VL Jr. Immunologic associations of keloids. Surg Gynecol Obstet. (1992) 175:185–93.
15. McCarty SM, Syed F, Bayat A. Influence of the human leukocyte antigen complex on the development of cutaneous fibrosis: an immunogenetic perspective. Acta Derm Venereol. (2010) 90:563–74. doi: 10.2340/00015555-0975
16. Bayat A, Arscott G, Ollier WE, McGrouther DA, Ferguson MW. Keloid disease: clinical relevance of single versus multiple site scars. Br J Plastic Surg. (2005) 58:28–37. doi: 10.1016/j.bjps.2004.04.024
17. Shih B, Bayat A. Genetics of keloid scarring. Arch Dermatol Res. (2010) 302:319–39. doi: 10.1007/s00403-009-1014-y
18. Ogawa R, Akaishi S. Endothelial dysfunction may play a key role in keloid and hypertrophic scar pathogenesis—Keloids and hypertrophic scars may be vascular disorders. Med Hypotheses. (2006) 96:51–60. doi: 10.1016/j.mehy.2016.09.024
19. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. (2008) 453:314–21. doi: 10.1038/nature07039
20. Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. (1999) 341:738–46. doi: 10.1056/NEJM199909023411006
21. Ferguson MW, Whitby DJ, Shah M, Armstrong J, Siebert JW, Longaker MT. Scar formation: the spectral nature of fetal and adult wound repair. Plastic Reconstr Surg. (1996) 97:854–60. doi: 10.1097/00006534-199604000-00029
22. Vincent AS, Phan TT, Mukhopadhyay A, Lim HY, Halliwell B, Wong KP. Human skin keloid fibroblasts display bioenergetics of cancer cells. J Invest Dermatol. (2008) 128:702–9. doi: 10.1038/sj.jid.5701107
23. Satish L, Lyons-Weiler J, Hebda PA, Wells A. Gene expression patterns in isolated keloid fibroblasts. Wound Repair Regen. (2006) 14:463–70. doi: 10.1111/j.1743-6109.2006.00135.x
24. Niessen FB, Spauwen PH, Schalkwijk J, Kon M. On the nature of hypertrophic scars and keloids: a review. Plastic Reconstr Surg. (1999) 104:1435–58. doi: 10.1097/00006534-199910000-00031
25. Kimura K, Inadomi T, Yamauchi W, Yoshida Y, Kashimura T, Terui T. Dermatofibrosarcoma protuberans on the chest with a variety of clinical features masquerading as a keloid: is the disease really protuberant? Ann Dermatol. (2014) 26:643–5. doi: 10.5021/ad.2014.26.5.643
26. Kanitakis J. Keloidal dermatofibroma: report of a rare dermatofibroma variant in a young white woman. Am J Dermatopathol. (2013) 35:400–1. doi: 10.1097/DAD.0b013e31825d9d30
27. Tongdee E, Touloei K, Shitabata PK, Shareef S, Maranda EL. Keloidal atypical fibroxanthoma: case and review of the literature. Case Rep Dermatol. (2016) 8:156–63. doi: 10.1159/000446343
28. Canady J, Karrer S, Fleck M, Bosserhoff AK. Fibrosing connective tissue disorders of the skin: molecular similarities and distinctions. J Dermatol Sci. (2013) 70:151–8. doi: 10.1016/j.jdermsci.2013.03.005
29. Bienias W, Miȩkoś-Zydek B, Kaszuba A. Current views on the etiopathogenesis of keloids. Postepy Dermatol Alergol. (2011) 28:467–75.
30. Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F. WHO Classification of Tumours. 4th ed. IARC (2013).
31. Naitoh M, Kubota H, Ikeda M, Tanaka T, Shirane H, Suzuki S, et al. Gene expression in human keloids is altered from dermal to chondrocytic and osteogenic lineage. Genes Cells. (2005) 10:1081–91. doi: 10.1111/j.1365-2443.2005.00902.x
32. Bayat A, Arscott G, Ollier WE, Ferguson MW, Mc Grouther DA. Description of site-specific morphology of keloid phenotypes in an Afrocaribbean population. Br J Plastic Surg. (2004) 57:122–33. doi: 10.1016/j.bjps.2003.11.009
33. Crockett DJ. Regional keloid susceptibility. Br J Plastic Surg. (1964) 17:245–53. doi: 10.1016/S0007-1226(64)80040-0
34. Kischer CW, Thies AC, Chvapil M. Perivascular myofibroblasts and microvascular occlusion in hypertrophic scars and keloids. Hum Pathol. (1982) 13:819–24. doi: 10.1016/S0046-8177(82)80078-6
35. Bran GM, Goessler UR, Hormann K, Riedel F, Sadick H. Keloids: current concepts of pathogenesis (review). Int J Mol Med. (2009) 24:283–93. doi: 10.3892/ijmm_00000231
36. Powazniak Y, Kempfer AC, de la Paz Dominguez M, Farias C, Keller L, Calderazzo JC, et al. Effect of estradiol, progesterone and testosterone on apoptosis- and proliferation-induced MAPK signaling in human umbilical vein endothelial cells. Mol Med Rep. (2009) 2:441–7. doi: 10.3892/mmr_00000119
37. Esfahanian N, Shakiba Y, Nikbin B, Soraya H, Maleki-Dizaji N, Ghazi-Khansari M, et al. Effect of metformin on the proliferation, migration, and MMP-2 and−9 expression of human umbilical vein endothelial cells. Mol Med Rep. (2012) 5:1068–74. doi: 10.3892/mmr.2012.753
38. Syed F, Ahmadi E, Iqbal SA, Singh S, McGrouther DA, Bayat A. Fibroblasts from the growing margin of keloid scars produce higher levels of collagen I and III compared with intralesional and extralesional sites: clinical implications for lesional site-directed therapy. Br J Dermatol. (2011) 164:83–96. doi: 10.1111/j.1365-2133.2010.10048.x
39. Zhang Q, Yamaza T, Kelly AP, Shi S, Wang S, Brown J, et al. Tumor-like stem cells derived from human keloid are governed by the inflammatory niche driven by IL-17/IL-6 axis. PLoS ONE. (2009) 4:e7798. doi: 10.1371/journal.pone.0007798
40. Iqbal SA, Syed F, McGrouther DA, Paus R, Bayat A. Differential distribution of haematopoietic and nonhaematopoietic progenitor cells in intralesional and extralesional keloid: do keloid scars provide a niche for nonhaematopoietic mesenchymal stem cells? Br J Dermatol. (2010) 162:1377–83. doi: 10.1111/j.1365-2133.2010.09738.x
41. Huang C, Akaishi S, Hyakusoku H, Ogawa R. Are keloid and hypertrophic scar different forms of the same disorder? A fibroproliferative skin disorder hypothesis based on keloid findings. Int Wound J. (2014) 11:517–22. doi: 10.1111/j.1742-481X.2012.01118.x
42. Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J. Keloids: The paradigm of skin fibrosis–pathomechanisms and treatment. Matrix Biol. (2016) 51:37–46. doi: 10.1016/j.matbio.2016.01.013
43. Ashcroft KJ, Syed F, Bayat A. Site-specific keloid fibroblasts alter the behaviour of normal skin and normal scar fibroblasts through paracrine signalling. PLoS ONE. (2013) 8:e75600. doi: 10.1371/journal.pone.0075600
44. Bellini A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. (2007) 87:858–70. doi: 10.1038/labinvest.3700654
45. Mori L, Bellini A, Stacey MA, Schmidt M, Mattoli S. Fibrocytes contribute to the myofibroblast population in wounded skin and originate from the bone marrow. Exp Cell Res. (2005) 304:81–90. doi: 10.1016/j.yexcr.2004.11.011
46. Iqbal SA, Sidgwick GP, Bayat A. Identification of fibrocytes from mesenchymal stem cells in keloid tissue: a potential source of abnormal fibroblasts in keloid scarring. Arch Dermatol Res. (2012) 304:665–71. doi: 10.1007/s00403-012-1225-5
47. Metz CN. Fibrocytes: a unique cell population implicated in wound healing. Cell Mol Life Sci. (2003) 60:1342–50. doi: 10.1007/s00018-003-2328-0
48. Bollag WB, Hill WD. CXCR4 in epidermal keratinocytes: crosstalk within the skin. J Invest Dermatol. (2013) 133:2505–8. doi: 10.1038/jid.2013.271
49. Chen W, Fu X, Sun X, Sun T, Zhao Z, Sheng Z. Analysis of differentially expressed genes in keloids and normal skin with cDNA microarray. J Surg Res. (2003) 113:208–16. doi: 10.1016/S0022-4804(03)00188-4
50. Bux S, Madaree A. Keloids show regional distribution of proliferative and degenerate connective tissue elements. Cells Tissues Organs. (2010) 191:213–34. doi: 10.1159/000231899
51. Verhaegen PD, van Zuijlen PP, Pennings NM, van Marle J, Niessen FB, van der Horst CM, et al. Differences in collagen architecture between keloid, hypertrophic scar, normotrophic scar, and normal skin: an objective histopathological analysis. Wound Repair Regen. (2009) 17:649–56. doi: 10.1111/j.1524-475X.2009.00533.x
52. Ulrich D, Ulrich F, Unglaub F, Piatkowski A, Pallua N. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with different types of scars and keloids. J Plast Reconstr Aesthet Surg. (2010) 63:1015–21. doi: 10.1016/j.bjps.2009.04.021
53. Roomi MW, Monterrey JC, Kalinovsky T, Rath M, Niedzwiecki A. Patterns of MMP-2 and MMP-9 expression in human cancer cell lines. Oncol Rep. (2009) 21:1323–33. doi: 10.3892/or_00000358
54. Ong CT, Khoo YT, Mukhopadhyay A, Do DV, Lim IJ, Aalami O, et al. mTOR as a potential therapeutic target for treatment of keloids and excessive scars. Exp Dermatol. (2007) 16:394–404. doi: 10.1111/j.1600-0625.2007.00550.x
55. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. (2007) 12:9–22. doi: 10.1016/j.ccr.2007.05.008
56. Chen J, Zhuo S, Jiang X, Zhu X, Zheng L, Xie S, et al. Multiphoton microscopy study of the morphological and quantity changes of collagen and elastic fiber components in keloid disease. J Biomed Opt. (2011) 16:051305. doi: 10.1117/1.3569617
57. Amadeu TP, Braune AS, Porto LC, Desmoulière A, Costa AM. Fibrillin-1 and elastin are differentially expressed in hypertrophic scars and keloids. Wound Repair Regen. (2004) 12:169–74. doi: 10.1111/j.1067-1927.2004.012209.x
58. Ikeda M, Naitoh M, Kubota H, Ishiko T, Yoshikawa K, Yamawaki S, et al. Elastic fiber assembly is disrupted by excessive accumulation of chondroitin sulfate in the human dermal fibrotic disease, keloid. Biochem Biophys Res Commun. (2009) 390:1221–8. doi: 10.1016/j.bbrc.2009.10.125
59. Cooney CA, Jousheghany F, Yao-Borengasser A, Phanavanh B, Gomes T, Kieber-Emmons AM, et al. Chondroitin sulfates play a major role in breast cancer metastasis: a role for CSPG4 and CHST11 gene expression in forming surface P-selectin ligands in aggressive breast cancer cells. Breast Cancer Res. (2011) 13:R58. doi: 10.1186/bcr2895
60. Yanagisawa H, Schluterman MK, Brekken RA. Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J Cell Commun Signal. (2009) 3:337–47. doi: 10.1007/s12079-009-0065-3
61. Lee YH, Albig AR, Regner M, Schiemann BJ, Schiemann WP. Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis. (2008) 29:2243–51. doi: 10.1093/carcin/bgn199
62. Donet M, Brassart-Pasco S, Salesse S, Maquart FX, Brassart B. Elastin peptides regulate HT-1080 fibrosarcoma cell migration and invasion through an Hsp90-dependent mechanism. Br J Cancer. (2014) 111:139–48. doi: 10.1038/bjc.2014.239
63. Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. (1999) 22:85–9. doi: 10.1038/8792
64. Hanley KP, Oakley F, Sugden S, Wilson DI, Mann DA, Hanley NA. Ectopic SOX9 mediates extracellular matrix deposition characteristic of organ fibrosis. J Biol Chem. (2008) 283:14063–71. doi: 10.1074/jbc.M707390200
65. Inui S, Shono F, Nakajima T, Hosokawa K, Itami S. Identification and characterization of cartilage oligomeric matrix protein as a novel pathogenic factor in keloids. Am J Pathol. (2011) 179:1951–60. doi: 10.1016/j.ajpath.2011.06.034
66. Agarwal P, Schulz JN, Blumbach K, Andreasson K, Heinegard D, Paulsson M, et al. Enhanced deposition of cartilage oligomeric matrix protein is a common feature in fibrotic skin pathologies. Matrix Biol. (2013) 32:325–31. doi: 10.1016/j.matbio.2013.02.010
67. Yamamoto M, Takahashi H, Suzuki C, Naishiro Y, Yamamoto H, Imai K, et al. Cartilage oligomeric matrix protein in systemic sclerosis. Rheumatology. (2007) 46:1858–9. doi: 10.1093/rheumatology/kem254
68. Englund E, Bartoschek M, Reitsma B, Jacobsson L, Escudero-Esparza A, Orimo A, et al. Cartilage oligomeric matrix protein contributes to the development and metastasis of breast cancer. Oncogene. (2016) 35:5585–96. doi: 10.1038/onc.2016.98
69. Liu TT, Liu XS, Zhang M, Liu XN, Zhu FX, Zhu FM, et al. Cartilage oligomeric matrix protein is a prognostic factor and biomarker of colon cancer and promotes cell proliferation by activating the Akt pathway. J Cancer Res Clin Oncol. (2018) 144:1049–63. doi: 10.1007/s00432-018-2626-4
70. Hunzelmann N, Anders S, Sollberg S, Schonherr E, Krieg T. Co-ordinate induction of collagen type I and biglycan expression in keloids. Br J Dermatol. (1996) 135:394–9. doi: 10.1111/j.1365-2133.1996.tb01502.x
71. Adany R, Heimer R, Caterson B, Sorrell JM, Iozzo RV. Altered expression of chondroitin sulfate proteoglycan in the stroma of human colon carcinoma. Hypomethylation of PG-40 gene correlates with increased PG-40 content and mRNA levels. J Biol Chem. (1990) 265:11389–96.
72. Hunzelmann N, Schonherr E, Bonnekoh B, Hartmann C, Kresse H, Krieg T. Altered immunohistochemical expression of small proteoglycans in the tumor tissue and stroma of basal cell carcinoma. J Invest Dermatol. (1995) 104:509–13. doi: 10.1111/1523-1747.ep12605979
73. Tan EM, Hoffren J, Rouda S, Greenbaum S, Fox JWt, Moore JH Jr, et al. Decorin, versican, and biglycan gene expression by keloid and normal dermal fibroblasts: differential regulation by basic fibroblast growth factor. Exp Cell Res. (1993) 209:200–7. doi: 10.1006/excr.1993.1302
74. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. (2010) 10:116–29. doi: 10.1038/nrc2780
75. Skubitz KM. Biology and treatment of aggressive fibromatosis or desmoid tumor. Mayo Clin Proc. (2017) 92:947–64. doi: 10.1016/j.mayocp.2017.02.012
76. Locci P, Balducci C, Lilli C, Marinucci L, Becchetti E, Dolci C, et al. Desmoid and fibroma tumors differently respond to TGFbeta(1) stimulus and ECM macromolecule accumulation. Biomed Pharmacother. (2007) 61:131–6. doi: 10.1016/j.biopha.2006.09.011
77. Stivala A, Lombardo GA, Pompili G, Tarico MS, Fraggetta F, Perrotta RE. Dermatofibrosarcoma protuberans: our experience of 59 cases. Oncol Lett. (2012) 4:1047–55. doi: 10.3892/ol.2012.887
78. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
79. Ladin DA, Hou Z, Patel D, McPhail M, Olson JC, Saed GM, et al. p53 and apoptosis alterations in keloids and keloid fibroblasts. Wound Repair Regen. (1998) 6:28–37. doi: 10.1046/j.1524-475X.1998.60106.x
80. Nys K, Agostinis P. Bcl-2 family members: essential players in skin cancer. Cancer Lett. (2012) 320:1–13. doi: 10.1016/j.canlet.2012.01.031
81. Bovee JV, van den Broek LJ, Cleton-Jansen AM, Hogendoorn PC. Up-regulation of PTHrP and Bcl-2 expression characterizes the progression of osteochondroma towards peripheral chondrosarcoma and is a late event in central chondrosarcoma. Lab Invest. (2000) 80:1925–34. doi: 10.1038/labinvest.3780202
82. Teofoli P, Barduagni S, Ribuffo M, Campanella A, De Pita O, Puddu P. Expression of Bcl-2, p53, c-jun and c-fos protooncogenes in keloids and hypertrophic scars. J Dermatol Sci. (1999) 22:31–7. doi: 10.1016/S0923-1811(99)00040-7
83. Asano N, Yamakazi T, Seto M, Matsumine A, Yoshikawa H, Uchida A. The expression and prognostic significance of bone morphogenetic protein-2 in patients with malignant fibrous histiocytoma. J Bone Joint Surg B. (2004) 86:607–12. doi: 10.1302/0301-620X.86B4.14484
84. Wilkinson DG, Bhatt S, Ryseck RP, Bravo R. Tissue-specific expression of c-jun and junB during organogenesis in the mouse. Development. (1989) 106:465–71.
85. Sioletic S, Czaplinski J, Hu L, Fletcher JA, Fletcher CD, Wagner AJ, et al. c-Jun promotes cell migration and drives expression of the motility factor ENPP2 in soft tissue sarcomas. J Pathol. (2014) 234:190–202. doi: 10.1002/path.4379
86. Laner-Plamberger S, Kaser A, Paulischta M, Hauser-Kronberger C, Eichberger T, Frischauf AM. Cooperation between GLI and JUN enhances transcription of JUN and selected GLI target genes. Oncogene. (2009) 28:1639–51. doi: 10.1038/onc.2009.10
87. Burger JA, Kipps TJ. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood. (2006) 107:1761–7. doi: 10.1182/blood-2005-08-3182
88. Shin JU, Kim SH, Kim H, Noh JY, Jin S, Park CO, et al. TSLP is a potential initiator of collagen synthesis and an activator of CXCR4/SDF-1 axis in keloid pathogenesis. J Invest Dermatol. (2016) 136:507–15. doi: 10.1016/j.jid.2015.11.008
89. Zhang Q, Oh CK, Messadi DV, Duong HS, Kelly AP, Soo C, et al. Hypoxia-induced HIF-1 alpha accumulation is augmented in a co-culture of keloid fibroblasts and human mast cells: involvement of ERK1/2 and PI-3K/Akt. Exp Cell Res. (2006) 312:145–55. doi: 10.1016/j.yexcr.2005.10.006
90. Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. (2004) 9(Suppl 5):10–7. doi: 10.1634/theoncologist.9-90005-10
91. Cohly HHP, Scott H, Ndebele2 K, Jenkins JK, Angel MF. Differential gene expression of fibroblasts: keloid versus normal. Int J Mol Sci. (2002) 3:1162–76. doi: 10.3390/i3111162
92. Mehlen P, Schulze-Osthoff K, Arrigo AP. Small stress proteins as novel regulators of apoptosis. Heat shock protein 27 blocks Fas/APO-1- and staurosporine-induced cell death. J Biol Chem. (1996) 271:16510–4. doi: 10.1074/jbc.271.28.16510
93. Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. (2006) 5:2592–601. doi: 10.4161/cc.5.22.3448
94. Kuroda K, Tajima S. Proliferation of HSP47-positive skin fibroblasts in dermatofibroma. J Cutan Pathol. (2008) 35:21–6. doi: 10.1111/j.1600-0560.2007.00768.x
95. Uozaki H, Ishida T, Kakiuchi C, Horiuchi H, Gotoh T, Iijima T, et al. Expression of heat shock proteins in osteosarcoma and its relationship to prognosis. Pathol Res Pract. (2000) 196:665–73. doi: 10.1016/S0344-0338(00)80118-1
96. Naitoh M, Hosokawa N, Kubota H, Tanaka T, Shirane H, Sawada M, et al. Upregulation of HSP47 and collagen type III in the dermal fibrotic disease, keloid. Biochem Biophys Res Commun. (2001) 280:1316–22. doi: 10.1006/bbrc.2001.4257
97. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. (2005) 5:761–72. doi: 10.1038/nrc1716
98. Lee WJ, Lee JH, Ahn HM, Song SY, Kim YO, Lew DH, et al. Heat shock protein 90 inhibitor decreases collagen synthesis of keloid fibroblasts and attenuates the extracellular matrix on the keloid spheroid model. Plastic Reconstr Surg. (2015) 136:328e−37e. doi: 10.1097/PRS.0000000000001538
99. Lim CP, Phan TT, Lim IJ, Cao X. Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and migration. Oncogene. (2006) 25:5416–25. doi: 10.1038/sj.onc.1209531
100. Ghazizadeh M, Tosa M, Shimizu H, Hyakusoku H, Kawanami O. Functional implications of the IL-6 signaling pathway in keloid pathogenesis. J Invest Dermatol. (2007) 127:98–105. doi: 10.1038/sj.jid.5700564
101. Ong CT, Khoo YT, Mukhopadhyay A, Masilamani J, Do DV, Lim IJ, et al. Comparative proteomic analysis between normal skin and keloid scar. Br J Dermatol. (2010) 162:1302–15. doi: 10.1111/j.1365-2133.2010.09660.x
102. Nickoloff BJ, Lingen MW, Chang BD, Shen M, Swift M, Curry J, et al. Tumor suppressor maspin is up-regulated during keratinocyte senescence, exerting a paracrine antiangiogenic activity. Cancer Res. (2004) 64:2956–61. doi: 10.1158/0008-5472.CAN-03-2388
103. Zou Z, Gao C, Nagaich AK, Connell T, Saito S, Moul JW, et al. p53 regulates the expression of the tumor suppressor gene maspin. J Biol Chem. (2000) 275:6051–4. doi: 10.1074/jbc.275.9.6051
104. Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol. (1993) 9:541–73. doi: 10.1146/annurev.cb.09.110193.002545
105. Curran S, Murray GI. Matrix metalloproteinases: molecular aspects of their roles in tumour invasion and metastasis. Eur J Cancer. (2000) 36:1621–30. doi: 10.1016/S0959-8049(00)00156-8
106. Yang HK, Jeong KC, Kim YK, Jung ST. Role of matrix metalloproteinase (MMP) 2 and MMP-9 in soft tissue sarcoma. Clin Orthop Surg. (2014) 6:443–54. doi: 10.4055/cios.2014.6.4.443
107. Tanriverdi-Akhisaroglu S, Menderes A, Oktay G. Matrix metalloproteinase-2 and−9 activities in human keloids, hypertrophic and atrophic scars: a pilot study. Cell Biochem Funct. (2009) 27:81–7. doi: 10.1002/cbf.1537
108. Li H, Nahas Z, Feng F, Elisseeff JH, Boahene K. Tissue engineering for in vitro analysis of matrix metalloproteinases in the pathogenesis of keloid lesions. JAMA Facial Plast Surg. (2013) 15:448–56. doi: 10.1001/jamafacial.2013.1211
109. Pardo PS, Leung JK, Lucchesi JC, Pereira-Smith OM. MRG15, a novel chromodomain protein, is present in two distinct multiprotein complexes involved in transcriptional activation. J Biol Chem. (2002) 277:50860–6. doi: 10.1074/jbc.M203839200
110. Syed F, Sherris D, Paus R, Varmeh S, Singh S, Pandolfi PP, et al. Keloid disease can be inhibited by antagonizing excessive mTOR signaling with a novel dual TORC1/2 inhibitor. Am J Pathol. (2012) 181:1642–58. doi: 10.1016/j.ajpath.2012.08.006
111. Jeter CR, Yang T, Wang J, Chao HP, Tang DG. Concise review: NANOG in cancer stem cells and tumor development: an update and outstanding questions. Stem Cells. (2015) 33:2381–90. doi: 10.1002/stem.2007
112. Grant C, Chudakova DA, Itinteang T, Chibnall AM, Brasch HD, Davis PF, et al. Expression of embryonic stem cell markers in keloid-associated lymphoid tissue. J Clin Pathol. (2016) 69:643–6. doi: 10.1136/jclinpath-2015-203483
113. Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell. (2006) 9:261–72. doi: 10.1016/j.ccr.2006.03.010
114. Sayah DN, Soo C, Shaw WW, Watson J, Messadi D, Longaker MT, et al. Downregulation of apoptosis-related genes in keloid tissues. J Surg Res. (1999) 87:209–16. doi: 10.1006/jsre.1999.5761
115. Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA. (2000) 97:9082–7. doi: 10.1073/pnas.97.16.9082
116. Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. (2001) 61:6669–73. Available online at: http://cancerres.aacrjournals.org/content/61/18/6669
117. Dotto GP. Notch tumor suppressor function. Oncogene. (2008) 27:5115–23. doi: 10.1038/onc.2008.225
118. Syed F, Bayat A. Notch signaling pathway in keloid disease: enhanced fibroblast activity in a Jagged-1 peptide-dependent manner in lesional vs. extralesional fibroblasts. Wound Repair Regen. (2012) 20:688–706. doi: 10.1111/j.1524-475X.2012.00823.x
119. Liu L, Zhang J, Fang C, Zhang Z, Feng Y, Xi X. OCT4 mediates FSH-induced epithelial-mesenchymal transition and invasion through the ERK1/2 signaling pathway in epithelial ovarian cancer. Biochem Biophys Res Commun. (2015) 461:525–32. doi: 10.1016/j.bbrc.2015.04.061
120. Shevde LA, Samant RS. Role of osteopontin in the pathophysiology of cancer. Matrix Biol. (2014) 37:131–41. doi: 10.1016/j.matbio.2014.03.001
121. Miragliotta V, Pirone A, Donadio E, Abramo F, Ricciardi MP, Theoret CL. Osteopontin expression in healing wounds of horses and in human keloids. Equine Vet J. (2016) 48:72–7. doi: 10.1111/evj.12372
122. Lu F, Gao J, Ogawa R, Hyakusoku H, Ou C. Biological differences between fibroblasts derived from peripheral and central areas of keloid tissues. Plastic Reconstr Surg. (2007) 120:625–30. doi: 10.1097/01.prs.0000270293.93612.7b
123. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. (1991) 253:49–53. doi: 10.1126/science.1905840
124. Yang A, McKeon F. P63 and P73: P53 mimics, menaces and more. Nat Rev Mol Cell Biol. (2000) 1:199–207. doi: 10.1038/35043127
125. De Felice B, Garbi C, Santoriello M, Santillo A, Wilson RR. Differential apoptosis markers in human keloids and hypertrophic scars fibroblasts. Mol Cell Biochem. (2009) 327:191–201. doi: 10.1007/s11010-009-0057-x
126. Maues De Paula A, Vasiljevic A, Giorgi R, Gomez-Brouchet A, Aubert S, Leroy X, et al. A diagnosis of giant cell-rich tumour of bone is supported by p63 immunohistochemistry, when more than 50% of cells is stained. Virchows Arch. (2014) 465:487–94. doi: 10.1007/s00428-014-1637-z
127. Demoulin JB, Essaghir A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev. (2014) 25:273–83. doi: 10.1016/j.cytogfr.2014.03.003
128. Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem. (2007) 101:695–711. doi: 10.1002/jcb.21224
129. Fukuda K, Sugihara E, Ohta S, Izuhara K, Funakoshi T, Amagai M, et al. Periostin is a key niche component for wound metastasis of melanoma. PLoS ONE. (2015) 10:e0129704. doi: 10.1371/journal.pone.0129704
130. Baeuerle PA, Rupec RA, Pahl HL. Reactive oxygen intermediates as second messengers of a general pathogen response. Pathol Biol. (1996) 44:29–35.
131. De Felice B, Wilson RR, Nacca M. Telomere shortening may be associated with human keloids. BMC Med Genet. (2009) 10:110. doi: 10.1186/1471-2350-10-110
132. Hunasgi S, Koneru A, Vanishree M, Shamala R. Keloid: acase report and review of pathophysiology and differences between keloid and hypertrophic scars. J Oral Maxillofac Pathol. (2013) 17:116–20. doi: 10.4103/0973-029X.110701
133. Gebhardt C, Riehl A, Durchdewald M, Nemeth J, Furstenberger G, Muller-Decker K, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. (2008) 205:275–85. doi: 10.1084/jem.20070679
134. Basu-Roy U, Seo E, Ramanathapuram L, Rapp TB, Perry JA, Orkin SH, et al. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene. (2012) 31:2270–82. doi: 10.1038/onc.2011.405
135. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. (2014) 511:246–50. doi: 10.1038/nature13305
136. Wehrli BM, Huang W, De Crombrugghe B, Ayala AG, Czerniak B. Sox9, a master regulator of chondrogenesis, distinguishes mesenchymal chondrosarcoma from other small blue round cell tumors. Hum Pathol. (2003) 34:263–9. doi: 10.1053/hupa.2003.41
137. Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. (2000) 19:2474–88. doi: 10.1038/sj.onc.1203527
138. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. (1999) 98:295–303. doi: 10.1016/S0092-8674(00)81959-5
139. Xue H, McCauley RL, Zhang W. Elevated interleukin-6 expression in keloid fibroblasts. J Surg Res. (2000) 89:74–7. doi: 10.1006/jsre.1999.5805
140. Yu D, Shang Y, Yuan J, Ding S, Luo S, Hao L. Wnt/β-catenin signaling exacerbates keloid cell proliferation by regulating telomerase. Cell Physiol Biochem. (2016) 39:2001–13. doi: 10.1159/000447896
141. Igota S, Tosa M, Murakami M, Egawa S, Shimizu H, Hyakusoku H, et al. Identification and characterization of Wnt signaling pathway in keloid pathogenesis. Int J Med Sci. (2013) 10:344–54. doi: 10.7150/ijms.5349
142. Kirikoshi H, Inoue S, Sekihara H, Katoh M. Expression of WNT10A in human cancer. Int J Oncol. (2001) 19:997–1001. doi: 10.3892/ijo.19.5.997
143. Chin GS, Liu W, Peled Z, Lee TY, Steinbrech DS, Hsu M. Differential expression of transforming growth factor-beta receptors I and II and activation of Smad 3 in keloid fibroblasts. Plast Reconstr Surg. (2001) 108:423–9. doi: 10.1097/00006534-200108000-00022
144. Chin GS, Liu W, Steinbrech D, Hsu M, Levinson H, Longaker MT. Cellular signaling by tyrosine phosphorylation in keloid and normal human dermal fibroblasts. Plastic Reconstr Surg. (2000) 106:1532–40. doi: 10.1097/00006534-200012000-00014
145. Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Receptor Tyrosine Kinases and Ras. Molecular Cell Biology. 4th ed. New York, NY: W. H. Freeman (2000).
146. Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. (2003) 113:643–55. doi: 10.1016/S0092-8674(03)00392-1
147. Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. (2003) 113:631–42. doi: 10.1016/S0092-8674(03)00393-3
148. Chambers I. The molecular basis of pluripotency in mouse embryonic stem cells. Cloning Stem Cells. (2004) 6:386–91. doi: 10.1089/clo.2004.6.386
149. Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. (2013) 15:848–62. doi: 10.1593/neo.13706
150. Bettinger DA, Yager DR, Diegelmann RF, Cohen IK. The effect of TGF-beta on keloid fibroblast proliferation and collagen synthesis. Plastic Reconstr Surg. (1996) 98:827–33. doi: 10.1097/00006534-199610000-00012
151. Jagadeesan J, Bayat A. Transforming growth factor beta (TGFbeta) and keloid disease. Int J Surg. (2007) 5:278–85. doi: 10.1016/j.ijsu.2006.04.007
152. Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, et al. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. (1996) 86:531–42. doi: 10.1016/S0092-8674(00)80127-0
153. Haisa M, Okochi H, Grotendorst GR. Elevated levels of PDGF alpha receptors in keloid fibroblasts contribute to an enhanced response to PDGF. J Invest Dermatol. (1994) 103:560–3. doi: 10.1111/1523-1747.ep12396856
154. Babu M, Diegelmann R, Oliver N. Keloid fibroblasts exhibit an altered response to TGF-beta. J Invest Dermatol. (1992) 99:650–5. doi: 10.1111/1523-1747.ep12668146
155. Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. (1990) 61:203–12. doi: 10.1016/0092-8674(90)90801-K
156. Kolibaba KS, Druker BJ. Protein tyrosine kinases and cancer. Biochim Biophys Acta. (1997) 1333:F217–48. doi: 10.1016/S0304-419X(97)00022-X
157. Zenz R, Efer R, Kenner L, Florin L, Hummerich L, Mehic D, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. (2005) 437:369–75. doi: 10.1038/nature03963
158. Deng CC, Zhu DH, Chen YJ, Huang TY, Peng Y, Liu SY, et al. TRAF4 promotes fibroblast proliferation in keloids by destabilizing p53 via interacting with the deubiquitinase USP10. J Invest Dermatol. (2019). doi: 10.1016/j.jid.2019.03.1136
159. Saed GM, Ladin D, Olson J, Han X, Hou Z, Fivenson D. Analysis of p53 gene mutations in keloids using polymerase chain reaction-based single-strand conformational polymorphism and DNA sequencing. Arch Dermatol. (1998) 134:963–7. doi: 10.1001/archderm.134.8.963
160. Kaneishi NK, Cockerell CJ. Histologic differentiation of desmoplastic melanoma from cicatrices. Am J Dermatopathol. (1998) 20:128–34. doi: 10.1097/00000372-199804000-00004
161. Yan L, Lü XY, Wang CM, Cao R, Yin YH, Jia CS, et al. Association between p53 gene codon 72 polymorphism and keloid in Chinese population. Zhonghua Zheng Xing Wai Ke Za Zhi. (2007) 23:428−30.
162. Zhuo Y, Gao JH, Luo SQ, Zeng WS, Hu ZQ, Lu F, et al. p53 gene codon 72 polymorphism and susceptibility to keloid in Chinese population. Zhonghua Zheng Xing Wai Ke Za Zhi. (2005) 19:28–30.
163. Yao X, Cui X, Wu X, Xu P, Zhu W, Chen X, et al. Tumor suppressive role of miR-1224-5p in keloid proliferation, apoptosis and invasion via the TGF-β1/Smad3 signaling pathway. Biochem Biophys Res Commun. (2018) 495:713–20. doi: 10.1016/j.bbrc.2017.10.070
164. Liu Y, Wang X, Yang D, Xiao Z, Chen X. MicroRNA-21 affects proliferation and apoptosis by regulating expression of PTEN in human keloid fibroblasts. Plastic Reconstr Surg. (2014) 134:561e−73e. doi: 10.1097/PRS.0000000000000577
165. Chen C-Y, Chen J, He L, Stiles BL. PTEN: Tumor suppressor and metabolic regulator. Front Endocrinol. (2018) 9:338. doi: 10.3389/fendo.2018.00338
166. Varmeh S, Egia A, McGrouther D, Tahan SR, Bayat A, Pandolfi PP. Cellular senescence as a possible mechanism for halting progression of keloid lesions. Genes Cancer. (2011) 2:1061–6. doi: 10.1177/1947601912440877
167. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
168. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
169. Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Ann Rev Cell Dev Biol. (1999) 15:551–78. doi: 10.1146/annurev.cellbio.15.1.551
170. Zhang Q, Wu Y, Ann DK, Messadi DV, Tuan TL, Kelly AP, et al. Mechanisms of hypoxic regulation of plasminogen activator inhibitor-1 gene expression in keloid fibroblasts. J Invest Dermatol. (2003) 121:1005–12. doi: 10.1046/j.1523-1747.2003.12564.x
171. Wu Y, Zhang Q, Ann DK, Akhondzadeh A, Duong HS, Messadi DV, et al. Increased vascular endothelial growth factor may account for elevated level of plasminogen activator inhibitor-1 via activating ERK1/2 in keloid fibroblasts. Am J Physiol Cell Physiol. (2004) 286:C905–12. doi: 10.1152/ajpcell.00200.2003
172. Hoopes JE, Su CT, Im MJ. Enzyme activities in hypertrophic scars and keloids. Plast Reconstr Surg. (1971) 47:132–7. doi: 10.1097/00006534-197102000-00006
173. Ramanathan A, Wang C, Schreiber SL. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci USA. (2005) 102:5992–7. doi: 10.1073/pnas.0502267102
174. Mucaj V, Shay JE, Simon MC. Effects of hypoxia and HIFs on cancer metabolism. Int J Hematol. (2012) 95:464–70. doi: 10.1007/s12185-012-1070-5
175. Lokmic Z, Musyoka J, Hewitson TD, Darby IA. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int Rev Cell Mol Biol. (2012) 296:139–85. doi: 10.1016/B978-0-12-394307-1.00003-5
176. Steinbrech DS, Mehrara BJ, Chau D, Rowe NM, Chin G, Lee T, et al. Hypoxia upregulates VEGF production in keloid fibroblasts. Ann Plast Surg. (1999) 42:514–9. doi: 10.1097/00000637-199905000-00009
177. Zhao B, Guan H, Liu JQ, Zheng Z, Zhou Q, Zhang J, et al. Hypoxia drives the transition of human dermal fibroblasts to a myofibroblast-like phenotype via the TGF-beta1/Smad3 pathway. Int J Mol Med. (2017) 39:153–9. doi: 10.3892/ijmm.2016.2816
178. Laquer V, Hoang V, Nguyen A, Kelly KM. Angiogenesis in cutaneous disease: part II. J Am Acad Dermatol. (2000) 61:945–58. doi: 10.1016/j.jaad.2009.05.053
179. O'Connell MP, Weeraratna AT. Change is in the air: the hypoxic induction of phenotype switching in melanoma. J Invest Dermatol. (2013) 133:2316–7. doi: 10.1038/jid.2013.208
180. Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1): Implications in metastasis and development. Cell Cycle. (2008) 7:2090–6. doi: 10.4161/cc.7.14.6324
181. Zhou HM, Wang J, Elliott C, Wen W, Hamilton DW, Conway SJ. Spatiotemporal expression of periostin during skin development and incisional wound healing: lessons for human fibrotic scar formation. J Cell Commun Signal. (2010) 4:99–107. doi: 10.1007/s12079-010-0090-2
182. Zhang Z, Nie F, Chen X, Qin Z, Kang C, Chen B, et al. Upregulated periostin promotes angiogenesis in keloids through activation of the ERK 1/2 and focal adhesion kinase pathways, as well as the upregulated expression of VEGF and angiopoietin1. Mol Med Rep. (2015) 11:857–64. doi: 10.3892/mmr.2014.2827
183. Zhang Z, Nie F, Kang C, Chen B, Qin Z, Ma J, et al. Increased periostin expression affects the proliferation, collagen synthesis, migration and invasion of keloid fibroblasts under hypoxic conditions. Int J Mol Med. (2014) 34:253–61. doi: 10.3892/ijmm.2014.1760
184. Ruan K, Bao S, Ouyang G. The multifaceted role of periostin in tumorigenesis. Cell Mol Life Sci. (2009) 66:2219–30. doi: 10.1007/s00018-009-0013-7
185. Zhu M, Fejzo MS, Anderson L, Dering J, Ginther C, Ramos L, et al. Periostin promotes ovarian cancer angiogenesis and metastasis. Gynecol Oncol. (2010) 119:337–44. doi: 10.1016/j.ygyno.2010.07.008
186. Qiu F, Shi CH, Zheng J, Liu YB. Periostin mediates the increased pro-angiogenic activity of gastric cancer cells under hypoxic conditions. J Biochem Mol Toxicol. (2013) 27:364–9. doi: 10.1002/jbt.21498
187. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
188. Hahn JM, McFarland KL, Combs KA, Supp DM. Partial epithelial-mesenchymal transition in keloid scars: regulation of keloid keratinocyte gene expression by transforming growth factor-β1. Burns Trauma. (2016) 4:30. doi: 10.1186/s41038-016-0055-7
189. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. (2009) 119:1429–37. doi: 10.1172/JCI36183
190. Yan L, Cao R, Wang L, Liu Y, Pan B, Yin Y. Epithelial-mesenchymal transition (EMT) in keloid tissues and TGF-beta1-induced hair follicle outer root sheath keratinocytes. Wound Repair Regen. (2015) 23:601–10. doi: 10.1111/wrr.12320
191. Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci. (2008) 121:727–35. doi: 10.1242/jcs.000455
192. Mendez MG, Kojima SI, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. (2010) 24:1838–51. doi: 10.1096/fj.09-151639
193. Hulit J, Suyama K, Chung S, Keren R, Agiostratidou G, Shan W, et al. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. (2007) 67:3106–16. doi: 10.1158/0008-5472.CAN-06-3401
194. Hazan RB, Qiao R, Keren R, Badano I, Suyama K. Cadherin switch in tumor progression. Ann N Y Acad Sci. (2004) 1014:155–63. doi: 10.1196/annals.1294.016
195. Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. (1996) 379:91–6. doi: 10.1038/379091a0
196. Dedhar S. Cell-substrate interactions and signaling through ILK. Curr Opin Cell Biol. (2000) 12:250–6. doi: 10.1016/S0955-0674(99)00083-6
197. Papanikolaou S, Bravou V, Gyftopoulos K, Nakas D, Repanti M, Papadaki H. ILK expression in human basal cell carcinoma correlates with epithelial-mesenchymal transition markers and tumour invasion. Histopathology. (2010) 56:799–809. doi: 10.1111/j.1365-2559.2010.03556.x
198. Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, Del Barrio MG, et al. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. (2000) 2:76–83. doi: 10.1038/35000025
199. Poser I, Dominguez D, de Herreros AG, Varnai A, Buettner R, Bosserhoff AK. Loss of E-cadherin expression in melanoma cells involves up-regulation of the transcriptional repressor Snail. J Biol Chem. (2001) 276:24661–6. doi: 10.1074/jbc.M011224200
200. Cheng CW, Wu PE, Yu JC, Huang CS, Yue CT, Wu CW, et al. Mechanisms of inactivation of E-cadherin in breast carcinoma: modification of the two-hit hypothesis of tumor suppressor gene. Oncogene. (2001) 20:3814–23. doi: 10.1038/sj.onc.1204505
201. Hahn JM, Glaser K, McFarland KL, Aronow BJ, Boyce ST, Supp DM. Keloid-derived keratinocytes exhibit an abnormal gene expression profile consistent with a distinct causal role in keloid pathology. Wound Repair Regen. (2013) 21:530–44. doi: 10.1111/wrr.12060
202. Abdou AG, Maraee AH, Abd-Elsattar Saif HF. Immunohistochemical evaluation of cox-1 and cox-2 expression in keloid and hypertrophic scar. Am J Dermatopathol. (2014) 36:311–7. doi: 10.1097/DAD.0b013e3182a27b83
203. Sanchez-Diaz PC, Hsiao TH, Chang JC, Yue D, Tan MC, Chen HI, et al. De-regulated microRNAs in pediatric cancer stem cells target pathways involved in cell proliferation, cell cycle and development. PLoS ONE. (2013) 8:e61622. doi: 10.1371/journal.pone.0061622
204. Hao J, Zhang Y, Deng M, Ye R, Zhao S, Wang Y, et al. MicroRNA control of epithelial-mesenchymal transition in cancer stem cells. Int J Cancer. (2014) 135:1019–27. doi: 10.1002/ijc.28761
205. Liu Y, Yang D, Xiao Z, Zhang M. MiRNA expression profiles in keloid tissue and corresponding normal skin tissue. Aesthetic Plast Surg. (2012) 36:193–201. doi: 10.1007/s00266-011-9773-1
206. Yan L, Cao R, Liu Y, Wang L, Pan B, Lv X, et al. MiR-21-5p links epithelial-mesenchymal transition phenotype with stem-like cell signatures via AKT signaling in keloid keratinocytes. Sci Rep. (2016) 6:28281. doi: 10.1038/srep28281
207. Kumarswamy R, Volkmann I, Thum T. Regulation and function of miRNA-21 in health and disease. RNA Biol. (2011) 8:706–13. doi: 10.4161/rna.8.5.16154
208. Yoshimoto H, Ishihara H, Ohtsuru A, Akino K, Murakami R, Kuroda H, et al. Overexpression of insulin-like growth factor-1 (IGF-I) receptor and the invasiveness of cultured keloid fibroblasts. Am J Pathol. (1999) 154:883–9. doi: 10.1016/S0002-9440(10)65335-7
209. Narama I, Masuoka-Nishiyama M, Matsuura T, Ozaki K, Nagatani M, Morishima T. Morphogenesis of degenerative changes predisposing dogs to rupture of the cranial cruciate ligament. J Vet Med Sci. (1996) 58:1091–7. doi: 10.1292/jvms.58.11_1091
210. Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. (2005) 13:7–12. doi: 10.1111/j.1067-1927.2005.130102.x
211. Bao S, Ouyang G, Bai X, Huang Z, Ma C, Liu M, et al. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell. (2004) 5:329–39. doi: 10.1016/S1535-6108(04)00081-9
212. Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell. (2010) 38:114–27. doi: 10.1016/j.molcel.2010.02.020
213. Calderon M, Lawrence WT, Banes AJ. Increased proliferation in keloid fibroblasts wounded in vitro. J Surg Res. (1996) 61:343–7. doi: 10.1006/jsre.1996.0127
214. Lee YJ, Kwon SB, Kim CH, Cho HD, Nam HS, Lee SH, et al. Oxidative damage and nuclear factor erythroid 2-related factor 2 protein expression in normal skin and keloid tissue. Ann Dermatol. (2015) 27:507–16. doi: 10.5021/ad.2015.27.5.507
215. Akasaka Y, Ishikawa Y, Ono I, Fujita K, Masuda T, Asuwa N, et al. Enhanced expression of caspase-3 in hypertrophic scars and keloid: induction of caspase-3 and apoptosis in keloid fibroblasts in vitro. Lab Invest. (2000) 80:345–57. doi: 10.1038/labinvest.3780039
216. Appleton I, Brown NJ, Willoughby DA. Apoptosis, necrosis, and proliferation: possible implications in the etiology of keloids. Am J Pathol. (1996) 149:1441–7.
217. Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. (2004) 10:549–57. doi: 10.1016/j.molmed.2004.09.003
218. Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. (2008) 58:262–70. doi: 10.1016/j.phrs.2008.09.003
219. Chen QM, Bartholomew JC, Campisi J, Acosta M, Reagan JD, Ames BN. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem J. (1998) 332(Pt 1):43–50. doi: 10.1042/bj3320043
220. Luo S, Benathan M, Raffoul W, Panizzon RG, Egloff DV. Abnormal balance between proliferation and apoptotic cell death in fibroblasts derived from keloid lesions. Plastic Reconstr Surg. (2001) 107:87–96. doi: 10.1097/00006534-200101000-00014
221. Akasaka Y, Fujita K, Ishikawa Y, Asuwa N, Inuzuka K, Ishihara M, et al. Detection of apoptosis in keloids and a comparative study on apoptosis between keloids, hypertrophic scars, normal healed flat scars, and dermatofibroma. Wound Repair Regen. (2001) 9:501–6. doi: 10.1046/j.1524-475x.2001.00501.x
222. Beere HM. “The stress of dying”: the role of heat shock proteins in the regulation of apoptosis. J Cell Sci. (2004) 117(Pt 13):2641–51. doi: 10.1242/jcs.01284
223. Sauk JJ, Nikitakis N, Siavash H. Hsp47 a novel collagen binding serpin chaperone, autoantigen and therapeutic target. Front Biosci. (2005) 10:107–18. doi: 10.2741/1513
224. Welkoborsky HJ, Bernauer HS, Riazimand HS, Jacob R, Mann WJ, Hinni ML. Patterns of chromosomal aberrations in metastasizing and nonmetastasizing squamous cell carcinomas of the oropharynx and hypopharynx. Ann Otol Rhinol Laryngol. (2000) 109:401–10. doi: 10.1177/000348940010900411
225. Maitra A, Iacobuzio-Donahue C, Rahman A, Sohn TA, Argani P, Meyer R, et al. Immunohistochemical validation of a novel epithelial and a novel stromal marker of pancreatic ductal adenocarcinoma identified by global expression microarrays: sea urchin fascin homolog and heat shock protein 47. Am J Clin Pathol. (2002) 118:52–9. doi: 10.1309/3PAM-P5WL-2LV0-R4EG
226. Djeu JY, Wei S. Clusterin and chemoresistance. Adv Cancer Res. (2009) 105:77–92. doi: 10.1016/S0065-230X(09)05005-2
227. Shannan B, Seifert M, Leskov K, Willis J, Boothman D, Tilgen W, et al. Challenge and promise: roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. (2006) 13:12–9. doi: 10.1038/sj.cdd.4401779
228. Trougakos IP, Djeu JY, Gonos ES, Boothman DA. Advances and challenges in basic and translational research on clusterin. Cancer Res. (2009) 69:403–6. doi: 10.1158/0008-5472.CAN-08-2912
229. Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. (2004) 18:1072–87. doi: 10.1101/gad.1171104
230. Yano F, Kugimiya F, Ohba S, Ikeda T, Chikuda H, Ogasawara T, et al. The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem Biophys Res Commun. (2005) 333:1300–8. doi: 10.1016/j.bbrc.2005.06.041
231. Umehara N, Ozaki T, Sugihara S, Kunisada T, Morimoto Y, Kawai A, et al. Influence of telomerase activity on bone and soft tissue tumors. J Cancer Res Clin Oncol. (2004) 130:411–6. doi: 10.1007/s00432-004-0553-z
232. Granick M, Kimura M, Kim S, Daniali L, Cao X, Herbig U, et al. Telomere dynamics in keloids. Eplasty. (2011) 11:e15.
233. Coluzzi E, Colamartino M, Cozzi R, Leone S, Meneghini C, O'Callaghan N, et al. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE. (2014) 9:e110963. doi: 10.1371/journal.pone.0110963
234. Ezhilarasan R, Mohanam I, Govindarajan K, Mohanam S. Glioma cells suppress hypoxia-induced endothelial cell apoptosis and promote the angiogenic process. Int J Oncol. (2007) 30:701–7. doi: 10.3892/ijo.30.3.701
235. Brown CK, Khodarev NN, Yu J, Moo-Young T, Labay E, Darga TE, et al. Glioblastoma cells block radiation-induced programmed cell death of endothelial cells. FEBS Lett. (2004) 565:167–70. doi: 10.1016/j.febslet.2004.03.099
236. Nirodi CS, Devalaraja R, Nanney LB, Arrindell S, Russell S, Trupin J, et al. Chemokine and chemokine receptor expression in keloid and normal fibroblasts. Wound Repair Regen. (2000) 8:371–82. doi: 10.1111/j.1524-475X.2000.00371.x
237. Russell SB, Russell JD, Trupin KM, Gayden AE, Opalenik SR, Nanney LB, et al. Epigenetically altered wound healing in keloid fibroblasts. J Invest Dermatol. (2010) 130:2489–96. doi: 10.1038/jid.2010.162
238. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. (2010) 31:27–36. doi: 10.1093/carcin/bgp220
239. Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. (2016) 17:284–99. doi: 10.1038/nrg.2016.13
240. E Y, Qipa Z, Hengshu Z. The expression of DNMT1 in pathologic scar fibroblasts and the effect of 5-aza-2-deoxycytidine on cytokines of pathologic scar fibroblasts. Wounds. (2014) 26:139–46.
241. Jones LR, Young W, Divine G, Datta I, Chen KM, Ozog D, et al. Genome-wide scan for methylation profiles in keloids. Dis Markers. (2015) 2015:943176. doi: 10.1155/2015/943176
242. Jones LR, Greene J, Chen KM, Divine G, Chitale D, Shah V, et al. Biological significance of genome-wide DNA methylation profiles in keloids. Laryngoscope. (2017) 127:70–8. doi: 10.1002/lary.26063
243. Garcia-Rodriguez L, Jones L, Chen KM, Datta I, Divine G, Worsham MJ. Causal network analysis of head and neck keloid tissue identifies potential master regulators. Laryngoscope. (2016) 126:E319–24. doi: 10.1002/lary.25958
244. Fitzgerald O'Connor EJ, Badshah II, Addae LY, Kundasamy P, Thanabalasingam S, Abioye D, et al. Histone deacetylase 2 is upregulated in normal and keloid scars. J Invest Dermatol. (2012) 132:1293–6. doi: 10.1038/jid.2011.432
245. He Y, Deng Z, Alghamdi M, Lu L, Fear MW, He L. From genetics to epigenetics: new insights into keloid scarring. Cell Prolif. (2017) 50:e12326. doi: 10.1111/cpr.12326
246. Totan S, Echo A, Yuksel E. Heat shock proteins modulate keloid formation. Eplasty. (2011) 11:e21.
247. Lianos GD, Alexiou GA, Mangano A, Rausei S, Boni L, Dionigi G, et al. The role of heat shock proteins in cancer. Cancer Lett. (2015) 360:114–8. doi: 10.1016/j.canlet.2015.02.026
248. Lau K, Paus R, Tiede S, Day P, Bayat A. Exploring the role of stem cells in cutaneous wound healing. Exp Dermatol. (2009) 18:921–33. doi: 10.1111/j.1600-0625.2009.00942.x
249. Wang DL, Zhu JJ, Deng CL, Wang B, Yu LM. Identification of biological characteristics of human keloid-derived stem cells. Zhonghua Shao Shang Za Zhi. (2011) 27:210–4. doi: 10.3760/cma.j.issn.1009-2587.2011.03.016
250. Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. (2006) 66:4553–7. doi: 10.1158/0008-5472.CAN-05-3986
251. Moon JH, Kwak SS, Park G, Jung HY, Yoon BS, Park J, et al. Isolation and characterization of multipotent human keloid-derived mesenchymal-like stem cells. Stem Cells Dev. (2008) 17:713–24. doi: 10.1089/scd.2007.0210
252. Guo W, Lasky JL 3rd, Wu H. Cancer stem cells. Pediatr Res. (2006) 59(4 Pt 2):59R−64R. doi: 10.1203/01.pdr.0000203592.04530.06
253. Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. (2007) 1:389–402. doi: 10.1016/j.stem.2007.08.001
254. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444:756–60. doi: 10.1038/nature05236
255. Borovski T, De Sousa EMF, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. (2011) 71:634–9. doi: 10.1158/0008-5472.CAN-10-3220
256. Bagabir R, Byers RJ, Chaudhry IH, Muller W, Paus R, Bayat A. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br J Dermatol. (2012) 167:1053–66. doi: 10.1111/j.1365-2133.2012.11190.x
257. Durani P, Bayat A. Levels of evidence for the treatment of keloid disease. J Plastic Reconstr Aesth Surg. (2008) 61:4–17. doi: 10.1016/j.bjps.2007.05.007
258. Reish RG, Eriksson E. Scars: a review of emerging and currently available therapies. Plastic Reconstr Surg. (2008) 122:1068–78. doi: 10.1097/PRS.0b013e318185d38f
259. Juckett G, Hartman-Adams H. Management of keloids and hypertrophic scars. Am Fam Phys. (2009) 80:253–60.
260. Malaker K, Vijayraghavan K, Hodson I, Yafi TA. Retrospective analysis of treatment of unresectable keloids with primary radiation over 25 years. Clin Oncol. (2004) 16:290–8. doi: 10.1016/j.clon.2004.03.005
261. Darzi MA, Chowdri NA, Kaul SK, Khan M. Evaluation of various methods of treating keloids and hypertrophic scars: a 10-year follow-up study. Br J Plastic Surg. (1992) 45:374–9. doi: 10.1016/0007-1226(92)90008-L
262. van Leeuwen MC, Stokmans SC, Bulstra AE, Meijer OW, van Leeuwen PA, Niessen FB. High-dose-rate brachytherapy for the treatment of recalcitrant keloids: a unique, effective treatment protocol. Plastic Reconstr Surg. (2014) 134:527–34. doi: 10.1097/PRS.0000000000000415
263. Jones ME, Hardy C, Ridgway J. Keloid management: a retrospective case review on a new approach using surgical excision, platelet-rich plasma, and in-office superficial photon X-ray radiation therapy. Adv Skin Wound Care. (2016) 29:303–7. doi: 10.1097/01.ASW.0000482993.64811.74
264. Wang W, Qu M, Xu L, Wu X, Gao Z, Gu T, et al. Sorafenib exerts an anti-keloid activity by antagonizing TGF-β/Smad and MAPK/ERK signaling pathways. J Mol Med. (2016) 94:1181–94. doi: 10.1007/s00109-016-1430-3
265. Bijlard E, Steltenpool S, Niessen FB. Intralesional 5-fluorouracil in keloid treatment: a systematic review. Acta Derm Venereol. (2015) 95:778–82. doi: 10.2340/00015555-2106
266. Ud-Din S, Bowring A, Derbyshire B, Morris J, Bayat A. Identification of steroid sensitive responders versus non-responders in the treatment of keloid disease. Arch Dermatol Res. (2013) 305:423–32. doi: 10.1007/s00403-013-1328-7
267. De Sousa RF, Chakravarty B, Sharma A, Parwaz MA, Malik A. Efficacy of triple therapy in auricular keloids. J Cutan Aesthet Surg. (2014) 7:98–102. doi: 10.4103/0974-2077.138347
268. Agbenorku P. Triple keloid therapy: a combination of steroids, surgery and silicone gel strip/sheet for keloid treatment. Eur J Plast Surg. (2000) 23:150–1. doi: 10.1007/s002380050236
269. Xu B, Liu ZZ, Zhu GY, Yang JF, Zhao JP, Wang JC, et al. Efficacy of recombinant adenovirus-mediated double suicide gene therapy in human keloid fibroblasts. Clin Exp Dermatol. (2008) 33:322–8. doi: 10.1111/j.1365-2230.2007.02615.x
270. Ud-Din S, Thomas G, Morris J, Bayat A. Photodynamic therapy: an innovative approach to the treatment of keloid disease evaluated using subjective and objective non-invasive tools. Arch Dermatol Res. (2013) 305:205–14. doi: 10.1007/s00403-012-1295-4
271. Calzavara-Pinton PG, Venturini M, Sala R. Photodynamic therapy: update 2006–part 1: photochemistry and photobiology. J Eur Acad Dermatol. (2007) 21:293–302. doi: 10.1111/j.1468-3083.2006.01902.x
Keywords: keloid, skin scarring, quasi-neoplastic, tumor, cancer, bioenergetics, fibroproliferative, epigenetics
Citation: Tan S, Khumalo N and Bayat A (2019) Understanding Keloid Pathobiology From a Quasi-Neoplastic Perspective: Less of a Scar and More of a Chronic Inflammatory Disease With Cancer-Like Tendencies. Front. Immunol. 10:1810. doi: 10.3389/fimmu.2019.01810
Received: 03 September 2018; Accepted: 17 July 2019;
Published: 07 August 2019.
Edited by:
Miriam Wittmann, University of Leeds, United KingdomReviewed by:
Alexandar Tzankov, University Hospital of Basel, SwitzerlandEva Turley, University of Western Ontario, Canada
Copyright © 2019 Tan, Khumalo and Bayat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ardeshir Bayat, YXJkZXNoaXIuYmF5YXQmI3gwMDA0MDttYW5jaGVzdGVyLmFjLnVr