 Maxime Montembeault
Maxime Montembeault Simona M. Brambati
Simona M. Brambati Maria Luisa Gorno-Tempini4
Maria Luisa Gorno-Tempini4 Raffaella Migliaccio
Raffaella Migliaccio- 1INSERM U 1127, CNRS UMR 7225, Sorbonne Universités, and Université Pierre et Marie Curie-Paris 6, UMR S 1127, Institut du Cerveau et de la Moelle Épinière (ICM), FrontLab, Paris, France
- 2Centre de Recherche de l'Institut Universitaire de Gériatrie de Montréal, Montréal, QC, Canada
- 3Département de Psychologie, Université de Montréal, Montréal, QC, Canada
- 4Memory and Aging Center, University of California at San Francisco, San Francisco, CA, United States
- 5Department of Neurology, Institut de la Mémoire et de la Maladie d'Alzheimer (IM2A), Paris, France
Primary progressive aphasias (PPA) are neurodegenerative diseases clinically characterized by an early and relatively isolated language impairment. Three main clinical variants, namely the nonfluent/agrammatic variant (nfvPPA), the semantic variant (svPPA), and the logopenic variant (lvPPA) have been described, each with specific linguistic/cognitive deficits, corresponding anatomical and most probable pathological features. Since the discovery and the development of diagnostic criteria for the PPA variants by the experts in the field, significant progress has been made in the understanding of these diseases. This review aims to provide an overview of the literature on each of the PPA variant in terms of their clinical, anatomical and pathological features, with a specific focus on recent findings. In terms of clinical advancements, recent studies have allowed a better characterization and differentiation of PPA patients based on both their linguistic and non-linguistic profiles. In terms of neuroimaging, techniques such as diffusion imaging and resting-state fMRI have allowed a deeper understanding of the impact of PPA on structural and functional connectivity alterations beyond the well-defined pattern of regional gray matter atrophy. Finally, in terms of pathology, despite significant advances, clinico-pathological correspondence in PPA remains far from absolute. Nonetheless, the improved characterization of PPA has the potential to have a positive impact on the management of patients. Improved reliability of diagnoses and the development of reliable in vivo biomarkers for underlying neuropathology will also be increasingly important in the future as trials for etiology-specific treatments become available.
Introduction
In 1892, Arnold Pick (1) first described a patient with a clinical history of progressive and isolated language deficits, along with mild memory impairment and progressive social dysfunction. Around the same time, Paul Sérieux (2) described a woman who presented with a progressive loss of word comprehension and in whom, contrary to Pick's patient, memory and intelligence were initially preserved. When this patient died in 1897, Jules Déjerine examined his brain, discovering neuronal loss and cortical atrophy in bilateral temporal regions (2). More recently, Marsel Mesulam had the opportunity to examine the cell and myelin preparations of Sérieux's patient, finding no evidence of either senile plaques or neurofibrillary tangles (3). For this reason, he considered this patient the closest prototypical example of the syndrome now known as primary progressive aphasia (PPA).
In the modern literature, the first systematic description of a series of PPA cases was published in 1982 by Marsel Mesulam (4). The disorder was characterized as a “slowly progressive aphasia without generalized dementia.” The six reported patients had very heterogeneous linguistic profiles, which did not completely fit the classic vascular aphasia models of Broca and Wernicke, thus suggesting the existence of several variants of PPA.
In the mid-1970s, Warrington (5), followed by Snowden (6), Hodges and their collaborators (7) described a progressive disorder of semantic memory that they termed “semantic dementia.” In 1996, Grossman (8) described a different form of progressive language disorder, termed “progressive nonfluent aphasia.” At the end of the 1990s, Neary and collaborators (9) proposed a classification for frontotemporal dementia (FTD, then used as a clinical term) that included semantic dementia and progressive nonfluent aphasia. However, the definition of progressive nonfluent aphasia was very broad and likely included a variety of clinical syndromes. In 2004, a third subtype of PPA was described by Gorno-Tempini and colleagues (10), the “logopenic” variant of PPA (lvPPA). In 2011, an international group of PPA investigators (11) put forth new criteria that included these three main variants. These criteria are based on clinical features, along with neuroimaging, neuropathological, and genetic data, to allow homogeneous patient classification for research purposes. In this framework, patients must first meet the general PPA criteria proposed by Mesulam, in which a difficulty with language must be (1) the most prominent clinical feature, both at symptom onset and for the initial phases of the diseases, and (2) must be the principal cause of impaired daily living activities (12). The general clinical evaluation of PPA patients aims to identify the speech-language profile, showing impaired vs. spared speech-language skills, in order to identify the variant. In addition to the clinical features, the diagnosis can be further supported by neuroimaging (atrophy, hypometabolism, hypoperfusion), and pathology. Table 1 summarizes the clinical, anatomical, and biological features of the three main variants of PPA.
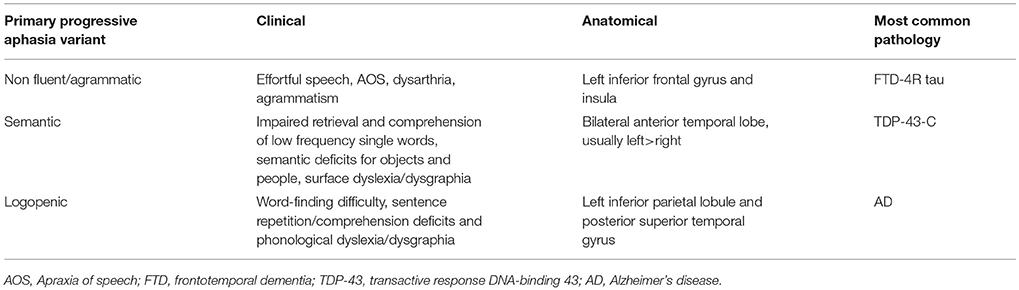
Table 1. Clinical, anatomical, and biological features of the three variants of PPA.
Although these three main variants do not account for all possible presentations of PPA, this classification is thought to capture most patients who do not a have a genetic form of the disease. It should be noted that, while correct clinical characterization allows accurate prediction of anatomical involvement, the correspondence between clinico-anatomical and molecular findings at pathology is only probabilistic. It reflects partially selective vulnerability of certain networks to certain neurodegenerative diseases (13).
Since the discovery and the development of diagnosis criteria for the PPA variants by the experts in the field in 2011, significant progress has been made in the understanding of PPA. While most neuroimaging studies on PPA had focused on regional atrophy, recent neuroimaging studies have focused on the impact of PPA on selective brain networks sustaining different language functions. Furthermore, recent longitudinal studies have allowed a better delineation of clinical and cerebral changes associated with the progression of the disease. This review aims to provide an overview and an update of the literature on each of the PPA variant in terms of their clinical, anatomical and pathological features. More specifically, in addition to reviewing the 2011 criteria, this review will provide an update on recent findings on the linguistic/cognitive manifestations of the disease and on their assessment. The latest neuroimaging studies will be reviewed with a specific focus on disconnection aspects of the disease. Issues and controversies associated with the diagnosis of PPA will be discussed, and possible avenues will be examined in the light of the most recent research.
General Background On Pathology in PPA
From a pathological point of view, each PPA variant seems to correspond to a specific tissue pathology, and in some cases to a gene mutation. However, the clinico-pathological correspondence in PPA remains far from absolute, as will be discussed more specifically for each PPA variant in the following sections (Table 1).
One of the possible causes is the accumulation of pathological aggregates of tau protein. Tau protein is a highly soluble microtubule-associated protein (MAPT) and promotes microtubule polymerization and stabilization. Disorders in which tau pathology is considered the major contributing factor to neurodegeneration are referred to as “primary tauopathies” (e.g., fronto-temporal dementia tau related, FTD tau). Tau protein in the brain is heterogeneous due to alternative splice forms, as well as post-translational modifications, including phosphorylation (14). The terms “3R” and “4R” tau refers to the products of the alternative splicing of the MAPT gene, generating tau species with either three or four conserved ~32 amino acid repeats in the microtubule binding domain of tau protein (15). There is preferential accumulation of 3R or 4R tau in various tauopathies, providing a biochemical subclassification of the tauopathies. The MAPT gene is located on chromosome 17. Mutations in the MAPT gene induce the formation of abnormal normal tau protein inclusions, leading to abnormal functioning of these cells. Changes in tau protein induced by mutations can also decrease its effectiveness or increase its amount, which can lead to disease.
Another frequent pathological substrate is the deposits of transactive response DNA-binding protein 43 (TDP-43) which is a cellular protein encoded by the TARDBP gene. Four subtypes have been described (A, B, C, D), in some cases associated with genes mutations. For instance, type A (TDP-43-A) is associated in a proportion of cases with mutations in the progranulin (PGRN or GRN) gene (16). The PGRN gene is located on chromosome 17 and induces the production of the progranulin protein. Mutations in the PGRN gene reduce the production of progranulin and increase the neural aggregates of TDP-43. The progranulin helps cell growth, and the protein TDP-43 regulates the process of making proteins from DNA (expression). The TARDBP gene on chromosome 1, encoding the TDP-43 protein, is very rarely involved.
Finally, some patients (especially those with lvPPA) have an underlying AD pathology. AD is characterized by intracellular tau-associated neurofibrillary tangles and extracellular amyloid-β (Aβ)–associated plaques in the brain.
Variants of Primary Progressive Aphasia (PPA)
Non Fluent/Agrammatic Variant (nfvPPA)
Clinical Manifestations
NfvPPA is a rare, early-onset neurodegenerative syndrome with a mean age of onset of ~60 years (17). The duration of survival is quite variable, ranging from 2 years in cases associated with amyotrophic lateral sclerosis to about 12 years in cases not associated with any motor disorder (18, 19).
The hallmark clinical features of nfvPPA are effortful speech and agrammatism. Effortful speech is characterized by slow, labored speech production, mainly due to a speech motor planning deficit, i.e., apraxia of speech (AOS) (20). Speech sound errors, consisting of distortions, deletions, substitutions, insertions, or transpositions of speech sounds are present. Distortions are considered as phonetic errors and are caused by AOS, while deletions, substitutions, insertions and transpositions are phonemic errors and can be caused by a motor speech impairment or a phoneme selection deficit (21, 22). However, there are some significant challenges in differentiating these two types of errors clinically, and both studies showing higher rates of phonetic errors (23, 24) or higher rates of phonemic errors (21, 22) in nfvPPA patients have been reported. Prosody is also typically affected in nfvPPA. Dysarthric features often co-occur with AOS, usually with mixed hypophonic and spastic features (20). Agrammatism is characterized by short, simple phrases, and omission of grammatical morphemes. Difficulties are present in language production (e.g., omission of articles, use of incorrect morphological endings), as well as in comprehension (e.g., difficulties in understanding complex syntactic structures, such as passives and relative clauses) (25). Patients with nfvPPA often use fewer verbs compared with healthy controls, in part because verbs play a critical part in syntactically structuring a sentence. They also have difficulty with verb naming and comprehension tasks (26).
With the progression of the disease, other cognitive deficits may emerge, including a decline in attentional resources and verbal working memory, as well as executive functions, episodic memory, praxis and behavioral symptoms (27, 28). General neurological examination is normal early in the disease course but extrapyramidal signs, and in many cases, a florid progressive supranuclear palsy syndrome (PSP-s) or corticobasal syndrome (CBS), can occur later in the disease course (24, 29–34) when the disease advances to SMA and subcortical regions, which might be due to the underlying FTD-4R pathology (see section Pathology). In cases in which language difficulties are very early accompanied by a clear extrapyramidal syndrome (for example, a generalized rigidity or tremor), the diagnosis of PPA is excluded. In these cases, even though the specific criteria for nfvPPA might be fulfilled, the general PPA criteria proposed by Mesulam are not fulfilled (12), given the predominance of the extrapyramidal syndrome and its impact on daily living activities. Therefore, clinically, these patients are diagnosed with PSP-s or CBS with speech/language features (35, 36). Conversely, mild deficits, such as mild limb apraxia or slowness in fine finger movements, do not exclude a diagnosis of PPA.
The current clinical criteria (11) for nfvPPA include at least one of the following core features: (1) Agrammatism in language production; (2) Effortful, halting speech with inconsistent speech sound errors and distortions (AOS); and at least 2 of 3 of the following other features: (1) Impaired comprehension of syntactically complex sentences; (2) Spared single-word comprehension; (3) Spared object knowledge.
Linguistic/Cognitive Assessment
Spontaneous speech in nfvPPA can be assessed using description of a picture, such as “The cookie theft” (37), “The picnic scene” from the Western Aphasia Battery (38), or a picture story such as “Frog, where are you?” (39). These connected speech samples may yield information about fluency, grammatical competence, and motor speech abilities, amongst others (23, 40). Previous studies have revealed that speech samples in nfvPPA are characterized speech sound errors, as well as slow rate, syntactic errors, and reduced complexity (21–23). Specific tests for the motor speech component, performed by an expert speech pathologist, are also highly recommended. These may include articulatory tasks of increasing difficulty, from simple phonation and production of single syllables (e.g., puh/puh/puh) to more complex diadochokinetic tasks (e.g., puh/tuh/kuh/), repetition of multisyllabic words (e.g., impossibility), and finally, sentence repetition [e.g., (41)].
Motor speech disorders may, in some cases, prevent the accurate assessment of agrammatism. One approach is to use sentence production tasks, such as the Northwestern Anagram Test, which requires to assemble individual word cards into a meaningful sentence (42). Another approach is to assess grammatical processing in comprehension, by asking patients to point to one of two pictures after hearing an auditorily presented sentence. In this task, accurate decoding of the grammatical structure of the sentence is required to select the correct picture. Conversely, in very early stages of disease, written language (such as a written description of a picture) can often reveal early, mild grammatical errors. Specific tests for word-comprehension and object knowledge, functions usually spared in the early phases, should also be systematically administrated.
Neuroimaging Findings
The left inferior frontal gyrus (pars opercularis) is considered as the syndrome-specific epicenter in nfvPPA (43, 44). It is also associated with gray matter (GM) atrophy in the insula, premotor regions, SMA and striatum (Figure 1) (10, 45, 46). Syntactic processing deficits observed in these patients are associated with structural and functional abnormalities in the posterior part of inferior frontal gyrus (47). Some patients can become mute early in the course of the disease. This profile is associated with GM atrophy that is more prominent in the pars opercularis, extending into the left basal ganglia (48).
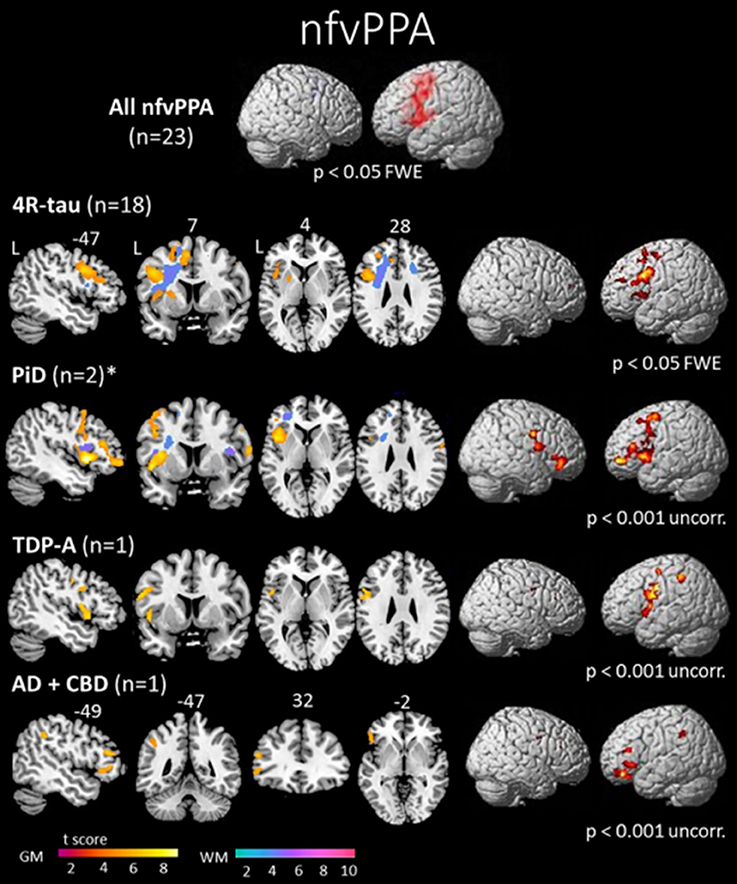
Figure 1. Areas of significant atrophy in a group of patients with nonfluent/agrammatic variant of primary progressive aphasia (nfvPPA) and then subdivided by pathological subgroups. The main areas of atrophy are mainly in the left inferior frontal gyrus, insula and premotor areas. No significant difference was observed in the comparison between nfvPPA-tau and nfvPPA-TDP-43 patients in this study. However, greater white matter damage has been reported in nfvPPA-tau cases (69). FWE, family wise error; 4R-tau, nfvPPA patients with FTD-tau with 4 repeats pathology; PiD, nfvPPA patients with Pick's disease pathology; TDP-A, nfvPPA patients with FTD-TDP-43 depositions type A pathology; AD + CBD, nfvPPA patients with Alzheimer's disease and corticobasal degeneration pathologies; GM, gray matter; WM, white matter; L, left [from (45), Permission to reproduce have been obtained from the copyright holders of this work].
A few studies reported that AOS and agrammatism can occur separately in nfvPPA, affecting different subcomponents of the same brain network (49). In cases in which AOS presents as an isolated symptom, the term of “primary progressive apraxia of speech” has been applied (PPAOS). On one hand, patients with PPAOS, might show focal imaging abnormalities in the premotor cortex. On the other hand, patients with dominant agrammatic deficits show widespread involvement of premotor, prefrontal, temporal, and parietal lobes, as well as in the caudate nucleus and the insula (49). Nevertheless, in the most common presentation of nfvPPA, motor speech deficits are prevalent, but signs of agrammatism are also present. Atrophy in these cases includes premotor and posterior inferior frontal regions, progressing along the aslant tract to the supplementary motor area (SMA) complex and eventually to the basal ganglia and supramarginal gyrus (46). nfvPPA is therefore an example of a network disorder involving the circuit of regions and connections involved in speech production.
Diffusion magnetic resonance imaging (MRI) techniques have shown that the dorsal language pathway of long-range white matter (WM) fibers connecting frontal, subcortical, and parietal areas are primarily involved in neurodegeneration in nfvPPA (46, 50) (Figure 2). This damage appears to be specific to this variant and is not observed in other PPA variants. WM damage in the dorsal pathway (superior longitudinal fasciculus) is also observed (Figure 3) (51–53). Consistently, a recent resting state fMRI study has demonstrated decreased functional connectivity between the left inferior frontal gyrus and the posterior middle temporal gyrus in nfvPPA, even in patients in which the atrophy is not severe (54).
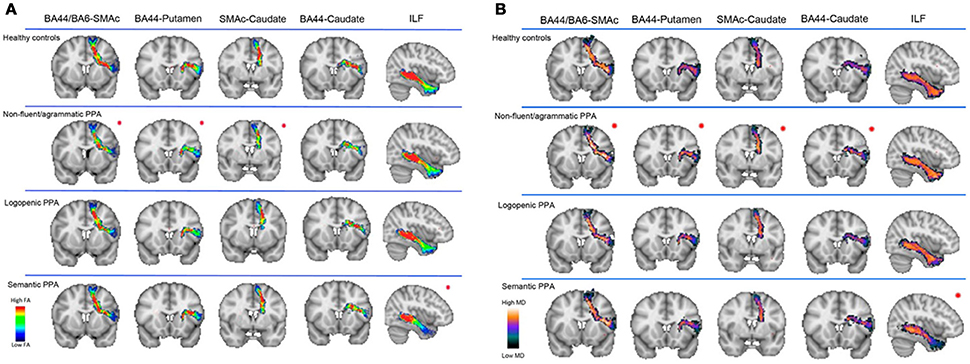
Figure 2. Speech production white matter tracts are shown in healthy controls, nfvPPA, lvPPA, and svPPA patients. Fractional anisotropy (FA) (A) and mean diffusivity (MD) (B) metrics were evaluated in four speech production tracts (BA44-SMAc, BA44-Putamen, SMAc-Caudate, and BA44-Caudate) and in the inferior longitudinal fasciculus (ILF). FA is a measure that quantifies the degree to which the diffusion of water molecules in WM fiber bundles is restricted to a specific direction (a higher FA value is associated with a better structural integrity). MD is a measure that quantifies the average degree of diffusion of water molecules in all directions (a lower MD value is associated with a better structural integrity). These results suggest that the white matter tracts connecting the speech production network are selectively damaged in nfvPPA. BA44, Broadmann Area 44; SMAc, Supplementary motor area complex; ILF, inferior longitudinal fasciculus. [from (46), Permission to reproduce have been obtained from the copyright holders of this work].
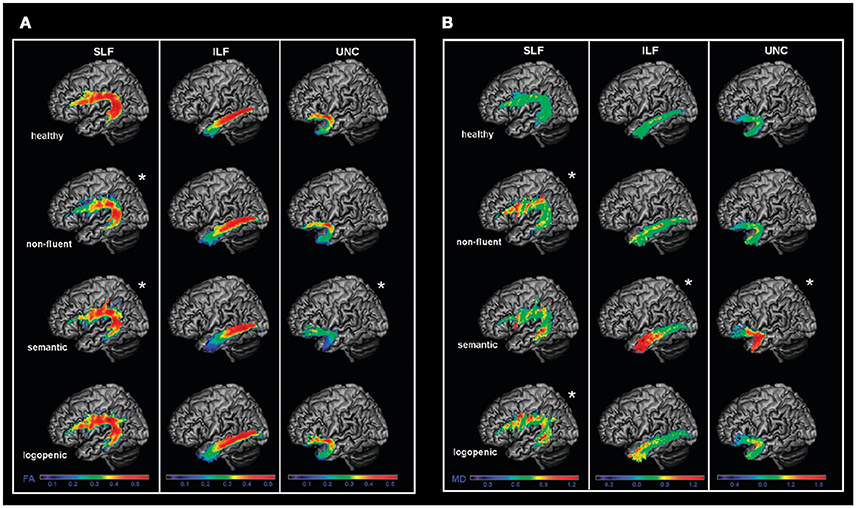
Figure 3. Left language-related white matter tracts are shown in healthy controls, nfvPPA, svPPA, and lvPPA patients. Fractional anisotropy (FA) (A) and mean diffusivity (MD) (B) metrics were evaluated in three tracts: the SLF (dorsal pathway), the ILF and UNC (ventral pathways). FA is a measure that quantifies the degree to which the diffusion of water molecules in WM fiber bundles is restricted to a specific direction (a higher FA value is associated with a better structural integrity). MD is a measure that quantifies the average degree of diffusion of water molecules in all directions (a lower MD value is associated with a better structural integrity). In nfvPPA, the main damage was observed in the dorsal pathway. In svPPA, the two ventral pathways as well as the temporal part of the dorsal pathway was damaged. In lvPPA, only the temporoparietal part of the dorsal pathway was damaged. SLF, Superior longitudinal fasciculus; ILF: inferior longitudinal fasciculus; UNC: uncinated fasciculus; FA, fractional anisotropy; MD, mean diffusivity [from (51), Permission to reproduce have been obtained from the copyright holders of this work].
Longitudinal GM atrophy changes in nfvPPA occur, 1 year after the first visit, in the left posterior frontal regions (often comprising inferior middle and superior gyri), supplementary motor area, insula, striatum, inferior parietal regions, and underlying WM (43, 55–57). Atrophy progresses to the supplementary motor complex region through the aslant tract. This tract is involved in the initiation and execution of movements, especially articulation. In nfvPPA, its integrity is associated with the number of distortion errors made by patients in spontaneous speech as well as with performance in a verbal fluency task (46, 58).
Pathology
NfvPPA is most commonly associated with a form of FTD-4R tau (45, 59–65). Other reports indicate TDP-43-A pathology in nfvPPA (45, 59, 66) and in some cases associated with progranulin (PGRN) or chromosome 9 open reading frame 72 (C9ORF72) gene mutations (67). Less frequently, AD pathology has also been reported in nfvPPA (45, 60, 61). However, a recent study investigating PPA patients with discordant amyloid status (i.e., nfvPPA with AD pathology) has suggested that most of these cases actually present mixed pathology (FTD tau pathology as primary pathologic diagnosis and AD pathology as contributing pathologic diagnosis) (68).
While progress has been made in understanding the underlying pathology in nfvPPA, some recent studies have also begun to characterize nfvPPA patients according to their underlying pathology. Clinically, it has been hypothesized that cases with predominant and isolated motor speech disorders would be associated with tau (24), while predominant agrammatism could predict TDP-43-A pathology. While similar GM damage have been observed in nfvPPA-tau and nfvPPA-TDP patients (Figure 1), greater WM damage has been observed in nfvPPA-tau cases (45, 69). Recently, further involvement of temporo-parietal regions beyond GM loss in the frontal lobe has been detected in a clinically heterogeneous group of TDP-43-A cases (64).
NfvPPA patients with underlying FTD-4R tau can also be further divided in nfv-PPA associated with progressive supranuclear palsy (PSP) and with corticobasal degeneration (CBD) anatomopathologies (56). At initial presentation, dysarthria and relatively selective WM atrophy appear typical of nfvPPA-PSP, while greater sentence comprehension deficits appear typical of nfvPPA-CBD. While these speech/language differences dissipate at a one-year follow-up, the progression of atrophy also allow to differentiate the two subgroups (56). In nfvPPA-PSP, atrophy spreads within the subcortical/brainstem motor system, which is consistent with greater oculomotors deficits and swallowing difficulty. In nfvPPA-CBD, atrophy progresses anteriorly in prefrontal regions, which is thought to generate greater working memory impairment and behavioral symptoms.
Semantic Variant (svPPA)
Clinical Manifestations
SvPPA is characterized by a progressive and multimodal loss of semantic knowledge (70). Age at onset is variable, most often between 55 and 70 years (71). The duration of the disease is also variable and can range from 2 to 15 years, although patients typically survive 7–8 years after onset (72, 73). Studies of the prevalence and incidence of svPPA are relatively limited, but a recent epidemiology study estimated the prevalence of FTD at 10.8/100,000, with svPPA accounting for approximately one-third of these cases (74).
These patients progressively lose the meaning of words, and they usually present with severe, progressive anomia, and markedly impaired comprehension of single words (11, 30). In earlier stages, the loss of meaning of words, and subsequently, naming and single-word comprehension deficits, are more prominent for low frequency/familiarity items (e.g., “rhinoceros” vs. “dog”) (75). Often, the patients replace less frequent words with more familiar ones, typically using the superordinate category (e.g., “animal” for “cat”). Another salient aspect of the syndrome is the production of semantic paraphasias in naming (e.g., “brush” for “comb”). Anomia can also be observed in spontaneous speech that is often empty and not very informative (23, 76). In the early stages, inability to comprehend low-familiarity words can be the only symptom accompanying anomia, and patients frequently ask for the meaning of words. The progression of semantic deficits leads to impaired object recognition affecting all sensory modalities, including vision, touch, olfaction, and gustation (7, 70) [e.g., visual agnosia; (73)]. The ability to correctly identify objects is strongly influenced by familiarity with the object (e.g., “fork” is more familiar than “compass”) (77). Additionally, individuals with svPPA appear to have disproportionate difficulty understanding concrete concepts relative to abstract concepts (78–80). Rarely, cases have been described with greater, or even selective, deficits for people (81) and animals (82). Some patients demonstrate impairment in the recognition of faces, which stems from a loss of person knowledge (83) that is also familiarity-dependent (84).
In contrast, episodic memory is relatively preserved in svPPA, especially when tasks with minimal conceptual loading are used (85, 86). The intact performance on traditional non-conceptually loaded episodic memory tasks converges with the performance of svPPA patients on autobiographical memory tasks. Patients typically show relatively preserved recollection of recent autobiographical memory in the context of poorer remote autobiographical memory (known as the reverse temporal gradient or step-function), reflecting increased semanticisation of past events (87, 88). SvPPA patients have also difficulties in episodic future thinking (89, 90).
The loss of word meaning is also apparent in reading. Patients do not recognize words as whole entities, but rather adopt a phonological strategy, deriving pronunciations using letter-sound conversion. As a result, irregular words are pronounced as if they are regular (“yatsht” for “yacht”), a phenomenon called surface dyslexia (91, 92). A similar pattern of selective impairment for irregular words is observed in spelling (surface dysgraphia).
Behavioral abnormalities are typically present in mid-late phases, including disinhibition, irritability, and food taste changes (e.g., preference for sweet foods). Lack of empathy, mental inflexibility, and compulsions - including clockwatching and intense interest in jigsaws—are also frequently noted (93–95). Almost 50% of svPPA patients report experiencing somatic symptom disorder as misidentification and preoccupation with normal bodily sensations such as hunger, bladder filling, borborygmi, rhinorrhea, and reflux; excessive concern over the incompletely understood meaning or source of pain or other symptoms; and Cotard syndrome or the delusion that unidentified somatic symptoms signify death or deterioration (96). This inability to read and name somatic sensations, or “alexisomia,” results in disproportionate and persistent concern about somatic sensations with consequent significant disability (96).
It has been demonstrated that non-right-handedness is overrepresented in svPPA patients, at nearly twice the prevalence of the general population. Left-handedness has been described as a proxy for atypical brain hemispheric lateralization (97).
The current diagnostic guidelines (11) identify anomia and single-word comprehension deficits as core features, both essential for diagnosis. At least 3 of the following other diagnostic features must also be present: (1) impaired object knowledge, particularly for low- frequency or low-familiarity items; (2) surface dyslexia or dysgraphia; (3) spared repetition; and 4. spared speech production (grammar and motor speech).
Linguistic/Cognitive Assessment
Language assessment of svPPA includes tests of confrontation naming, in which the patient is asked to retrieve the word in response to a picture [e.g., the Boston Naming test (98)]. Object and person knowledge are also examined using tests of semantic associations, gesture-object matching, and sound-picture matching tasks. The popular Pyramids and Palm Trees Test (99) is a semantic association task that measures the capacity to access detailed semantic information about words and pictures necessary for the identification of the relationships that conceptually link two perceptually and functionally distinct entities. The loss of concepts can be also tested using other types of stimuli, including sounds, foods, and odors. Famous faces and buildings naming tasks, as well as semantic knowledge tasks are also very sensitive with svPPA patients (100).
Reading and spelling of regular and irregular words are also tested in order to identify surface dyslexia and dysgraphia. Spontaneous speech should also be assessed in svPPA. Differently from nfvPPA, the spontaneous speech should not present AOS and the syntactic structure should be preserved. On the other side, svPPA present increased use of highly familiar words, anomia characterized by long pauses and use of general words (such as “thing”) for identifying the items displayed on the image.
Episodic memory tests based on non-linguistic stimuli (such as Rey Complex Figure) should be administered to exclude the presence of major episodic memory deficits, especially at early stages of the disease. Repetition and syntactic comprehension tests should be evaluated as an exclusion criterion.
Neuroimaging Findings
The anterior temporal lobes show bilateral atrophy and hypoperfusion in svPPA and is considered as the syndrome-specific epicenter (45, 101–104) (Figure 4). This focal anatomical damage makes neuroimaging a complementary tool in the diagnostic process for this PPA variant (105). The damage is usually greater in the left hemisphere at first stages of the disease (30). Typical semantic impairment is associated with greater left-sided anterior temporal atrophy/hypometabolism (106, 107), naming difficulties are correlated with superior portions of the left temporal pole (108), and finally, loss of person knowledge and behavioral changes are associated with more extensive right temporal atrophy (109, 110). Atrophy of the hippocampus has been reported mainly involving the anterior portion, which is connected to the semantic memory system (111). On the other hand, the posterior portion, mainly connected to the episodic memory system, would be relatively spared (112). This pattern of hippocampal atrophy would explain the dissociation between semantic and episodic memory deficits in svPPA population.
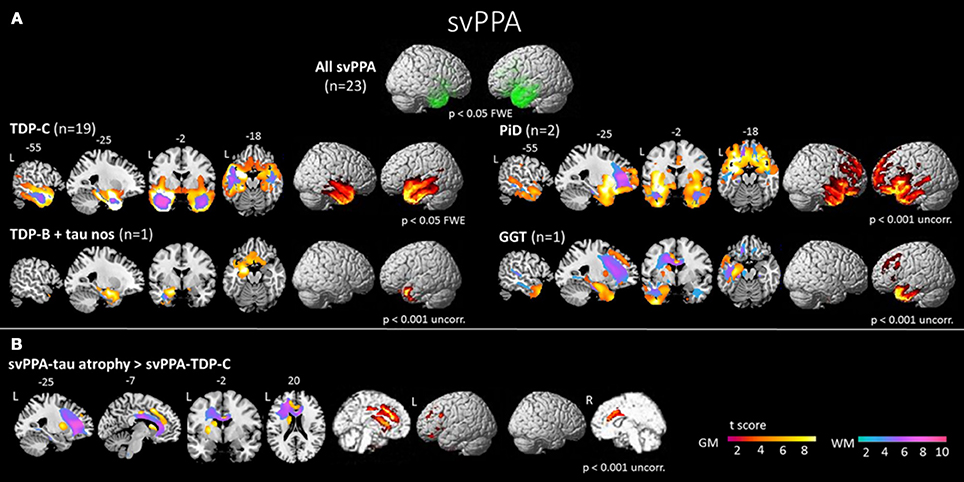
Figure 4. (A) Areas of significant atrophy in a group of patients with semantic variant of primary progressive aphasia (svPPA) and subdivided by pathological subgroups. The main areas of atrophy are in the bilateral anterior temporal lobes, more predominantly in the left hemisphere. (B) Areas of significant atrophy in svPPA patients with FTD tau pathology, in comparison to those with TDP-43 pathology. These patients present greater atrophy of frontotemporal cortex (medial anterior temporal lobe, orbitofrontal cortex, anterior cingulate cortex), basal ganglia and connecting white matter structures. FWE, family wise error; TDP-C, svPPA patients with FTD-TDP-43 depositions type C pathology; TDP-C+ tau nos, svPPA patients with FTD-TDP-43 depositions type C and tau pathology not otherwise specified; PiD, svPPA patients with Pick's disease pathology; GGT, svPPA patients with globular glial tauopathy pathology; L, left; R, right; GM, gray matter; WM, white matter [from (45), Permission to reproduce have been obtained from the copyright holders of this work].
Microstructural studies of WM integrity have shown damage in the ventral tracts that connect the temporal lobe to the occipital lobe and to the orbitofrontal cortex, with left side predominance (Figure 3) (51). The dorsal frontoparietal tracts that do not involve the temporal lobes are spared bilaterally, except for the temporal segment of the dorsal pathway (Figure 3) (51, 113, 114). This pattern is highly left lateralized compared with behavioral variant of fronto-temporal dementia which has a right predominance (115). During the progression of the disease, right-hemisphere WM bundles, in particular the uncinate, are preferentially damaged (116).
At the functional level, svPPA patients manifest extensive reduced anterior temporal lobe connectivity with primary and modality-selective cortices (117, 118). The longitudinal pattern of atrophy can be predicted by functional MRI connectivity between the temporal pole and the rest of the brain following connectional pathways within a large-scale network (119).
As the disease progresses, the atrophy involves the ventral and lateral temporal regions, as well as the contralateral temporal lobe and frontal regions (57, 120, 121). Beyond the specific metabolic signatures, additional dysfunctional patterns in the early stages can predict clinical progression: svPPA patients who present with extended bilateral patterns at baseline eventually develop behavioral disorders and a dysexecutive syndrome at follow-up (122).
Pathology
SvPPA is nearly always associated with underlying TDP-43-C pathological aggregates (75–100% in clinicopathological correlation series), and for the remainder of patients, most often with FTD tau (45, 60, 62, 66, 123–126). Rarely, AD pathology has also been reported in svPPA (60, 126), although a recent study has shown that most svPPA cases with AD pathology also present TDP-43-C pathology (68). In comparison to patients with TDP-43 pathology, those with FTD tau pathology present greater atrophy of frontotemporal cortex (medial anterior temporal lobe, orbitofrontal cortex, anterior cingulate cortex), basal ganglia and connecting WM bundles (Figure 4) (45). Interestingly, svPPA patients and PGRN mutation carriers are both characterized by underlying TDP-43 aggregation. In some cases, patients with PGRN mutations can develop aphasia with semantic deficits (127). However, familial forms of pure svPPA have not been reported. In both svPPA patients and PGRN mutation carriers, an increased prevalence of specific and related autoimmune diseases has been found, suggesting a unique pattern of systemic inflammation (128). Very rarely, mutations of the C9ORF72 gene have also been described in svPPA (129).
Recent studies have tried to identify FTD pathological subtypes with the help of cerebrospinal fluid (CSF) biomarkers. Increased neurofilament light chain (NfL) levels in the CSF, which are associated with neuronal and axonal degeneration, have been reported in patients with neurodegenerative diseases, and more specifically in patients with probable TDP-43 pathology such as svPPA patients (130).
Logopenic Variant (lvPPA)
Clinical Manifestations
LvPPA has been more recently characterized (10, 131) as a distinct form of PPA, and little is known about its age at onset and disease survival. As with the other variants of PPA, the logopenic variant is considered an early onset form of dementia (132).
Patients with lvPPA typically present with word finding difficulty, along with sentence repetition deficits and, as the disease progresses, impaired sentence comprehension. Phonological impairments and, specifically, a phonological short-term memory deficit, have been suggested to be the core of the syndrome (131). In accordance with this interpretation, repetition and comprehension of single words remaining largely spared.
Prior to the current consensus criteria for diagnosis, lvPPA was often diagnosed as nfvPPA (60). Both variants can present with slow speech, frequent word-finding pauses and speech sound errors. However, patients with nfvPPA have slower speech and, conversely, those with lvPPA do not present with severe agrammatism and the distorted, effortful speech production of AOS. Speech sound errors are usually phonemic, but not phonetic (21, 23). Confrontation naming is often impaired, albeit at a lesser degree if compared to svPPA. Phonological paraphasias can occur in spontaneous speech and confrontation naming.
Usually later in the disease, episodic memory impairment (62) is often present, even though the lexical retrieval impairment observed in lvPPA patients contributes to verbal episodic memory performance (133, 134). Longitudinal studies have shown that cognitive decline is faster in lvPPA in comparison to other variants, and that this decline is not restricted to language functioning (135). In lvPPA patients, a more accelerated decline was also observed in visuospatial abilities (136), in memory and in attention (135). This finding has been associated with the underlying AD pathology (see section Pathology). Poor calculation abilities (137) and limb apraxia can also occur (10). Apathy, anxiety, irritability, and agitation are often reported (138).
Recent studies have revealed that in comparison to the general population, PPA patients report higher rates of learning disabilities (and more specifically developmental dyslexia) in their early phases of life (139), and that dyslexia susceptibility genes influence brain atrophy in PPA (140). Further reports suggested that the frequency was specifically higher in lvPPA patients in comparison to the other variants, and that in these patients, learning disability is associated with earlier onset of disease, more isolated language symptoms, and more focal pattern of left posterior temporoparietal atrophy (97). Developmental dyslexia, which is the most common developmental language learning disability, can manifest with phonological disturbances and posterior temporal involvement, similarly to lvPPA. In the framework of network vulnerability hypothesis, learning disability might confer susceptibility of language network to early-onset, focal AD pathology such as lvPPA (97). However, further research is needed to confirm the higher frequency of developmental dyslexia in lvPPA specifically, since another study provided conflicting results (141).
Criteria for lvPPA (11) require that both of the following core features must be present: impaired single-word retrieval in spontaneous speech and naming along with impaired repetition of sentences and phrases. At least 3 of the following other features must be present: (1) phonological errors in spontaneous speech and naming; (2) spared single-word comprehension and object knowledge; (3) spared motor speech; and (4) absence of frank agrammatism.
Linguistic/Cognitive Assessment
The evaluation of spontaneous speech is essential in order to appreciate lvPPA patients' anomia and can be done using description of a picture as previously described. In such tasks, anomia may manifest in phonological paraphasias, hesitations, and frequent pauses for word-finding. Language assessment of lvPPA also includes confrontation naming tasks such as the Boston Naming Test (98). Oral repetition of words, pseudowords, phrases, and sentences, are usually administered in order to show the dissociation between preserved single word repetition, in opposition to the greater impairment for sentences and phrases. Moreover, phonological errors are often appreciated (142). Tests for sentence comprehension consist of matching orally presented sentences to pictures. Patients with nfvPPA also fail on these types of tests because of the effect of grammatical complexity, whereas patients with lvPPA fail because of the effects of length and frequency.
Reading and spelling tests reveal phonological errors as well as difficulty with pseudowords, which rely upon phonological processing (91). LvPPA patients also show difficulties in verbal working memory tests, such as the digit span from the Weschler Adult Intelligence Scale (48, 143). Single-word comprehension, motor speech and agrammatism should be evaluated as exclusion criteria. Recent studies have suggested that non-linguistic features, namely visuospatial functioning (136), episodic memory and emotion processing (144), are also helpful in the differential diagnosis of lvPPA in comparison to the other variants (and specially with nfvPPA).
Neuroimaging Findings
Anatomical damage in lvPPA is typically located in posterior superior temporal and middle temporal gyri as well as the inferior parietal lobule (10, 45). This pattern of atrophy is consistent with the classical anatomical model of the phonological loop (131). This neurodegenerative pattern is very similar to the one observed in early-onset AD (132). Naming difficulties are correlated with the left posterior temporal cortex (108). Recently, a model of the progression of atrophy in lvPPA has been suggested, showing that atrophy progresses from the disease epicenter (left posterior superior and middle temporal gyri) to ipsilateral parietal and frontal lobes and contralateral temporal lobe (145).
WM loss in association tracts in the left hemisphere has been detected (146), mainly in parietal fibers linking the parietal with frontal and posterior temporal regions (Figure 3) (51). A recent longitudinal study also suggested that early WM changes in lvPPA can be observed in the left posterior inferior longitudinal fasciculus, and that these changes become widespread over a year of progression of the disease, also affecting the anterior inferior longitudinal fasciculus, the uncinate fasciculus and the superior longitudinal fasciculus (116).
In terms of functional connectivity, the working memory network (frontal regions, inferior parietal lobule, superior, and middle temporal gyri) and language network (posterior superior temporal gyrus and inferior frontal lobe) have been shown to be altered in lvPPA patients in a resting-state fMRI study (147).
Pathology
LvPPA is most often caused by AD pathology (45, 60, 65), in as many as 95% of cases (68). It is considered one of the possible focal and early onset presentations of AD (62, 107, 132), even if other pathological profiles have been more rarely described, including Lewy body dementia (148), TDP-43, and tau (60).
Cerebro-spinal fluid examination (149) and molecular imaging techniques such as PET with Pittsburgh Compound B (PIB) (107), a ligand for the amyloid, have shown the presence of amyloid in these individuals. However, in comparison to typical AD patients, lvPPA patients with AD pathology show more significant hypoperfusion in the left superior temporal gyrus (Figure 5) (149).
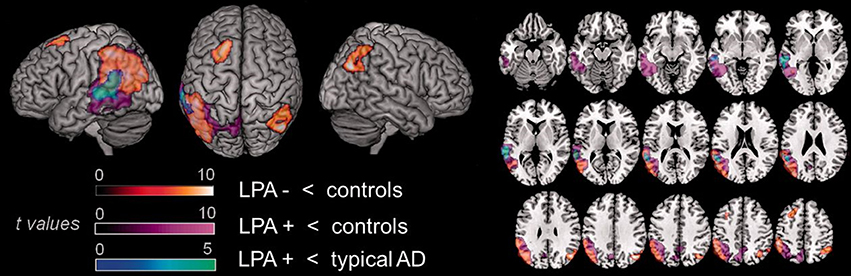
Figure 5. Comparison of areas of hypoperfusion between groups of lvPPA patients with AD pathology, lvPPA patients without AD pathology and typical AD patients. LvPPA patients present significant hypoperfusion in the left temporal-parietal junction, affecting larger portions of the temporal cortex in the lvPPA patients with AD pathology. In comparison to typical AD patients, lvPPA patients with AD pathology show more significant hypoperfusion in the left superior temporal gyrus. LPA, lvPPA; LPA-, lvPPA patients without AD pathology; LPA+, lvPPA patients with AD pathology; AD, Alzheimer's disease [from (149), Permission to reproduce have been obtained from the copyright holders of this work].
Some authors have demonstrated the coexistence of AD pathology and argyrophilic thorny astrocyte clusters (ATAC), intensely tau immunoreactive, in the fronto-temporoparietal cortex and subcortical regions (150). They suggest that they might represent a marker of a process responsible for the prominent focal clinical manifestations in lvPPA (150). Finally, a clinical syndrome with lvPPA features but also with more global features may be pre-dominantly linked to mutations in the GRN gene (151).
PPA Unclassifiable
In addition to the three most common variants, the 2011 consensus criteria also suggest that a minority of patients might be unclassifiable (11). It might be the case in patients who present for a long time with a single language symptom or in patients who present mixed features. It was further suggested that with the progression of the disease, these patients' profiles might become clearer.
A few studies have directly investigated the extent to which the 2011 consensus criteria cover the diversity of PPA cases. While some of them found that most patients could be classified within the three suggested variants (60, 61, 68, 152), others reported higher percentage (20–41%) of unclassifiable PPA patients (153–155). One of the main difficulty described by these last authors is related to patients who present with a single isolated language symptom, thus fulfilling the root criteria for PPA, but not the criteria for any of the variants. The most often reported cases are the ones who present with isolated anomia, without impaired repetition of single word, sentences and phrases (therefore only partially fulfilling criteria for lvPPA). Another main difficulty reported is related to patients who present a mixed profile, thus fulfilling the criteria for more than one of the variants. Patients presenting both sentence repetition impairments and agrammatism (therefore fulfilling criteria for both lvPPA and nfvPPA), as well as patients presenting both agrammatism and semantic impairments (fulfilling criteria for both nfvPPA and svPPA) have been observed.
Many factors could be associated with a varying proportion of unclassifiable PPA cases. First, the potential inclusion of genetic forms of PPA in studies might lead to higher number of mixed profiles. For example, it has been suggested that PGRN mutation carriers might present with a lvPPA and nfvPPA mixed profile (156). Furthermore, there is likely a high heterogeneity in tests and cutoffs used in the diagnosis of PPA across different clinical and research sites. There is a need for a use of sensitive and specific tests, and some authors have recently published assessment batteries specifically designed for the assessment of PPAs (157, 158). Most importantly, apparently similar symptoms might be due to distinct underlying causes between PPA variants, and this should be considered in tests selection and interpretation. For example, some studies have suggested that repetition deficits might be of different nature in lvPPA and nfvPPA. In lvPPA, they might be due to a disruption of the store component of the phonological loop, while in nfvPPA, impairments in speech motor planning might affect the subvocal rehearsal component of the phonological loop (159). This has also been investigated regarding the overlapping naming impairments in svPPA and lvPPA, which are thought to be respectively due to a semantic impairment vs. a lexical access deficit (160, 161). Finally, more prospective studies investigating the classification of PPA patients are needed, since retrospective studies are more likely to not have had adequate or complete test batteries to apply the current criteria (68). Nonetheless, even though significant progress was made in the recent years in the understanding of PPA, these studies illustrate that there are still controversies in the diagnosis of PPA and the above-mentioned issues will need to be clarified in the next years.
Conclusion
In summary, each variant of PPA (nfv-, sv-, and lv-PPA) is characterized by a prototypic neurolinguistic/neuropsychological, neuroimaging and neuropathological profile. The effervescence of PPA as a research field in the recent years has allowed for key discoveries in each of these domains. In terms of clinical advancements, recent studies have allowed a better characterization and differentiation of PPA patients based on both their linguistic and non-linguistic profiles. In terms of neuroimaging, techniques such as diffusion imaging and resting-state fMRI, as well as multimodal studies, have allowed a deeper understanding of the impact of PPA on structural and functional connectivity alterations beyond the well-defined pattern of regional gray matter atrophy. Finally, in terms of pathology/genetics, despite significant advances, clinico-pathological correspondence is still far to be absolute. The improved characterization of PPA has the potential to have a positive impact on the management of individual patients. It can help to better direct patients toward appropriate therapeutic and behavioral intervention, as well as to provide adequate counseling of families and caregivers (162). Improved reliability of diagnoses and the development of reliable in vivo biomarkers for underlying neuropathology will be increasingly important as trials for etiology-specific treatments become available.
Author Contributions
MLG-T and RM contributed to the conception of the study. RM and MM wrote the first draft of the manuscript, but all authors wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
RM gratefully acknowledges the support from France Alzheimer and Philippe Chatrier Foundations. MLG-T gratefully acknowledges the support from the National Institutes of Health (NINDS R01 NS050915, NIA P50 AG03006, NIA P50 AG023501, NIA P01 AG019724); State of California (DHS04-35516); Alzheimer's Disease Research Centre of California (03–75271 DHS/ADP/ ARCC); Larry L. Hillblom Foundation; John Douglas French Alzheimer's Foundation; Koret Family Foundation; Consortium for Frontotemporal Dementia Research; and McBean Family Foundation and a Career Scientist Award (NFD) from the US Department of Veterans Affairs Clinical Sciences R and D Program. SMB gratefully acknowledges the support of Fonds de recherche du Québec en Santé and Alzheimer Society of Canada. MM gratefully acknowledges the support of Fonds de recherche du Québec en Santé.
Abbreviations
AD, Alzheimer's disease; AOS, apraxia of speech; ATAC, argyrophilic thorny astrocyte clusters; CBD, corticobasal degeneration; CBS, corticobasal syndrome; C9ORF72, chromosome 9 open reading frame 72; FTD, frontotemporal dementia; GM, gray matter; lvPPA, logopenic variant primary progressive aphasia; MAPT, microtubule-associated protein tau; MRI, magnetic resonance imaging; nfvPPA, non-fluent/agrammatic variant primary progressive aphasia; PET, positron emission tomography; PGRN, progranulin; PIB, Pittsburgh Compound B; PPA, primary progressive aphasia; PPAOS, progressive apraxia of speech; PSP, progressive supranuclear palsy; PSP-s, progressive supranuclear palsy syndrome; SMA, supplementary motor area; svPPA, semantic variant primary progressive aphasia; TDP-43, transactive response DNA-binding protein 43; WM, white matter.
References
1. Pick A. Ueber die beziehungen der senilen hirnatrophie zur aphasie. Prager Med Wochenschrift (1892) 17:165–7.
2. Sérieux P, Déjèrine J. Sur un cas de surdité verbale pure terminée par aphasie sensorielle, suivie d'autopsie. Comptes Rendues des Séances de la Soc Biol. (1897) 49:1074–7.
3. Mesulam MM. Primary progressive aphasia: a 25-year retrospective. Alzheimer Dis Assoc Disord. (2007) 21:S8–11. doi: 10.1097/WAD.0b013e31815bf7e1
4. Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol. (1982) 11:592–8. doi: 10.1002/ana.410110607
5. Warrington EK. The selective impairment of semantic memory. Q J Exp Psychol. (1975) 27:635–57. doi: 10.1080/14640747508400525
6. Snowden J, Goulding PJ, Neary D. Semantic dementia: a form of circumscribed cerebral atrophy. Behav Neurol. (1989) 2:167–82.
7. Hodges JR, Patterson K, Oxbury S, Funnell E. Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain (1992) 115(Pt 6):1783–806.
8. Grossman M. Progressive non-fluent aphasia: language, cognitive and PET measures contrasted with probable Alzheimer's disease. J Cogn Neurosci. (1996) 8:135–54.
9. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology (1998) 51:1546–54.
10. Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. (2004) 55:335–46. doi: 10.1002/ana.10825
11. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology (2011) 76:1006–14. doi: 10.1212/WNL.0b013e31821103e6
13. Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron (2009) 62:42–52. doi: 10.1016/j.neuron.2009.03.024
14. Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. (1986) 261:6084–9.
15. Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry (1992) 31:10626–33.
16. Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. (2010) 9:995–1007. doi: 10.1016/S1474-4422(10)70195-2
17. Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. (2005) 62:925–30. doi: 10.1001/archneur.62.6.925
18. Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology (2003) 61:349–54. doi: 10.1212/01.WNL.0000078928.20107.52
19. Josephs KA, Knopman DS, Whitwell JL, Boeve BF, Parisi JE, Petersen RC, et al. Survival in two variants of tau-negative frontotemporal lobar degeneration: FTLD-U vs FTLD-MND. Neurology (2005) 65:645–7. doi: 10.1212/01.wnl.0000173178.67986.7f
20. Ogar JM, Dronkers NF, Brambati SM, Miller BL, Gorno-Tempini ML. Progressive nonfluent aphasia and its characteristic motor speech deficits. Alzheimer Dis Assoc Disord. (2007) 21:S23–30. doi: 10.1097/WAD.0b013e31815d19fe
21. Ash S, Evans E, O'Shea J, Powers J, Boller A, Weinberg D, et al. Differentiating primary progressive aphasias in a brief sample of connected speech. Neurology (2013) 81:329–36. doi: 10.1212/WNL.0b013e31829c5d0e
22. Ash S, McMillan C, Gunawardena D, Avants B, Morgan B, Khan A, et al. Speech errors in progressive non-fluent aphasia. Brain Lang. (2010) 113:13–20. doi: 10.1016/j.bandl.2009.12.001
23. Wilson SM, Henry ML, Besbris M, Ogar JM, Dronkers NF, Jarrold W, et al. Connected speech production in three variants of primary progressive aphasia. Brain (2010) 133(Pt 7):2069–88. doi: 10.1093/brain/awq129
24. Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain (2006) 129(Pt 6):1385–98. doi: 10.1093/brain/awl078
25. Thompson CK, Mack JE. Grammatical Impairments in PPA. Aphasiology (2014) 28:1018–37. doi: 10.1080/02687038.2014.912744
26. Hillis AE, Oh S, Ken L. Deterioration of naming nouns versus verbs in primary progressive aphasia. Ann Neurol. (2004) 55:268–75. doi: 10.1002/ana.10812
27. Rosen HJ, Allison SC, Ogar JM, Amici S, Rose K, Dronkers N, et al. Behavioral features in semantic dementia vs other forms of progressive aphasias. Neurology (2006) 67:1752–6. doi: 10.1212/01.wnl.0000247630.29222.34
28. Rohrer JD, Rossor MN, Warren JD. Syndromes of nonfluent primary progressive aphasia: a clinical and neurolinguistic analysis. Neurology (2010) 75:603–10. doi: 10.1212/WNL.0b013e3181ed9c6b
29. Kertesz A, Martinez-Lage P, Davidson W, Munoz DG. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology (2000) 55:1368–75. doi: 10.1212/WNL.55.9.1368
30. Gorno-Tempini ML, Murray RC, Rankin KP, Weiner MW, Miller BL. Clinical, cognitive and anatomical evolution from nonfluent progressive aphasia to corticobasal syndrome: a case report. Neurocase (2004) 10:426–36. doi: 10.1080/13554790490894011
31. Murray R, Neumann M, Forman MS, Farmer J, Massimo L, Rice A, et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology (2007) 68:1274–83. doi: 10.1212/01.wnl.0000259519.78480.c3
32. Deramecourt V, Lebert F, Debachy B, Mackowiak-Cordoliani MA, Bombois S, Kerdraon O, et al. Prediction of pathology in primary progressive language and speech disorders. Neurology (2010) 74:42–9. doi: 10.1212/WNL.0b013e3181c7198e
33. McMonagle P, Blair M, Kertesz A. Corticobasal degeneration and progressive aphasia. Neurology (2006) 67:1444–51. doi: 10.1212/01.wnl.0000240215.43492.01
34. Rohrer JD, Paviour D, Bronstein AM, O'Sullivan SS, Lees A, Warren JD. Progressive supranuclear palsy syndrome presenting as progressive nonfluent aphasia: a neuropsychological and neuroimaging analysis. Mov Disord. (2010) 25:179–88. doi: 10.1002/mds.22946
35. Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology (2013) 80:496–503. doi: 10.1212/WNL.0b013e31827f0fd1
36. Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. (2017) 32:853–64. doi: 10.1002/mds.26987
37. Goodglass H, Kaplan E. Boston Diagnostic Aphasia Examination (BDAE). Odessa, FL: Lea and Febiger. Distributed by Psychological Assessment Resources (1983).
40. Boschi V, Catricalà, E Consonni, M, Chesi, C, Moro, A, Cappa SF. Connected speech in neurodegenerative language disorders: a review. Front Psychol. (2017) 8:269. doi: 10.3389/fpsyg.2017.00269
41. Wertz RT, LaPointe LL, Rosenbek JC. Apraxia of Speech in Adults: The Disorder and its Management. Indiana: Singular (1984).
42. Weintraub S, Mesulam MM, Wieneke C, Rademaker A, Rogalski EJ, Thompson CK. The northwestern anagram test: measuring sentence production in primary progressive aphasia. Am J Alzheimers Dis Other Demen. (2009) 24:408–16. doi: 10.1177/1533317509343104
43. Mandelli ML, Vilaplana E, Brown JA, Hubbard HI, Binney RJ, Attygalle S, et al. Healthy brain connectivity predicts atrophy progression in non-fluent variant of primary progressive aphasia. Brain (2016) 139(Pt 10):2778–91. doi: 10.1093/brain/aww195
44. Mandelli ML, Vitali P, Santos M, Henry M, Gola K, Rosenberg L, et al. Two insular regions are differentially involved in behavioral variant FTD and nonfluent/agrammatic variant PPA. Cortex (2016) 74:149–57. doi: 10.1016/j.cortex.2015.10.012
45. Spinelli EG, Mandelli ML, Miller ZA, Santos-Santos MA, Wilson SM, Agosta F, et al. Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol. (2017) 81:430–43. doi: 10.1002/ana.24885
46. Mandelli ML, Caverzasi E, Binney RJ, Henry ML, Lobach I, Block N, et al. Frontal white matter tracts sustaining speech production in primary progressive aphasia. J Neurosci. (2014) 34:9754–67. doi: 10.1523/JNEUROSCI.3464-13.2014
47. Wilson SM, Dronkers NF, Ogar JM, Jang J, Growdon ME, Agosta F, et al. Neural correlates of syntactic processing in the nonfluent variant of primary progressive aphasia. J Neurosci. (2010) 30:16845–54. doi: 10.1523/JNEUROSCI.2547-10.2010
48. Gorno-Tempini ML, Ogar JM, Brambati SM, Wang P, Jeong JH, Rankin KP, et al. Anatomical correlates of early mutism in progressive nonfluent aphasia. Neurology (2006) 67:1849–51. doi: 10.1212/01.wnl.0000237038.55627.5b
49. Josephs KA, Duffy JR, Strand EA, Machulda MM, Senjem ML, Lowe VJ, et al. Syndromes dominated by apraxia of speech show distinct characteristics from agrammatic PPA. Neurology (2013) 81:337–45. doi: 10.1212/WNL.0b013e31829c5ed5
50. Wilson SM, Galantucci S, Tartaglia MC, Rising K, Patterson DK, Henry ML, et al. Syntactic processing depends on dorsal language tracts. Neuron (2011) 72:397–403. doi: 10.1016/j.neuron.2011.09.014
51. Galantucci S, Tartaglia MC, Wilson SM, Henry ML, Filippi M, Agosta F, et al. White matter damage in primary progressive aphasias: a diffusion tensor tractography study. Brain (2011) 134(Pt 10):3011–29. doi: 10.1093/brain/awr099
52. Grossman M, Powers J, Ash S, McMillan C, Burkholder L, Irwin D, et al. Disruption of large-scale neural networks in non-fluent/agrammatic variant primary progressive aphasia associated with frontotemporal degeneration pathology. Brain Lang. (2013) 127:106–20. doi: 10.1016/j.bandl.2012.10.005
53. Grossman M. The non-fluent/agrammatic variant of primary progressive aphasia. Lancet Neurol. (2012) 11:545–55. doi: 10.1016/S1474-4422(12)70099-6
54. Bonakdarpour B, Rogalski EJ, Wang A, Sridhar J, Mesulam MM, Hurley RS. Functional Connectivity is Reduced in Early-stage Primary Progressive Aphasia When Atrophy is not Prominent. Alzheimer Dis Assoc Disord. (2017) 31:101–6. doi: 10.1097/wad.0000000000000193
55. Rogalski E, Cobia D, Harrison TM, Wieneke C, Weintraub S, Mesulam MM. Progression of language decline and cortical atrophy in subtypes of primary progressive aphasia. Neurology (2011) 76:1804–10. doi: 10.1212/WNL.0b013e31821ccd3c
56. Santos-Santos MA, Mandelli ML, Binney RJ, Ogar J, Wilson SM, Henry ML, et al. Features of patients with nonfluent/agrammatic primary progressive aphasia with underlying progressive supranuclear palsy pathology or corticobasal degeneration. JAMA Neurol. (2016) 73:733–42. doi: 10.1001/jamaneurol.2016.0412
57. Brambati SM, Amici S, Racine CA, Neuhaus J, Miller Z, Ogar J, et al. Longitudinal gray matter contraction in three variants of primary progressive aphasia: a tenser-based morphometry study. Neuroimage Clin. (2015) 8:345–55. doi: 10.1016/j.nicl.2015.01.011
58. Catani M, Mesulam MM, Jakobsen E, Malik F, Martersteck A, Wieneke C, et al. A novel frontal pathway underlies verbal fluency in primary progressive aphasia. Brain (2013) 136(Pt 8):2619–28. doi: 10.1093/brain/awt163
59. Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. (2011) 122:137–53. doi: 10.1007/s00401-011-0839-6
60. Chare L, Hodges JR, Leyton CE, McGinley C, Tan RH, Kril JJ, et al. New criteria for frontotemporal dementia syndromes: clinical and pathological diagnostic implications. J Neurol Neurosurg Psychiatry (2014) 85:865–70. doi: 10.1136/jnnp-2013-306948
61. Harris JM, Gall C, Thompson JC, Richardson AM, Neary D, du Plessis D, et al. Classification and pathology of primary progressive aphasia. Neurology (2013) 81:1832–9. doi: 10.1212/01.wnl.0000436070.28137.7b
62. Mesulam M, Wicklund A, Johnson N, Rogalski E, Leger GC, Rademaker A, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol. (2008) 63:709–19. doi: 10.1002/ana.21388
63. Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer's and frontotemporal pathology in primary progressive aphasia. Brain (2014) 137(Pt 4):1176–92. doi: 10.1093/brain/awu024
64. Rohrer JD, Lashley T, Schott JM, Warren JE, Mead S, Isaacs AM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain (2011) 134(Pt 9):2565–81. doi: 10.1093/brain/awr198
65. Rohrer JD, Rossor MN, Warren JD. Alzheimer's pathology in primary progressive aphasia. Neurobiol Aging (2012) 33:744–52. doi: 10.1016/j.neurobiolaging.2010.05.020
66. Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. (2007) 114:31–8. doi: 10.1007/s00401-007-0236-3
67. Cioffi SM, Galimberti D, Barocco F, Spallazzi M, Fenoglio C, Serpente M, et al. Non fluent variant of primary progressive aphasia due to the novel GRN g.9543delA(IVS3-2delA) mutation. J Alzheimers Dis. (2016) 54:717–21. doi: 10.3233/jad-160185
68. Santos-Santos MA, Rabinovici GD, Iaccarino L, et al. Rates of amyloid imaging positivity in patients with primary progressive aphasia. JAMA Neurol. (2018) 75:342–52. doi: 10.1001/jamaneurol.2017.4309
69. Caso F, Mandelli ML, Henry M, Gesierich B, Bettcher BM, Ogar J, et al. In vivo signatures of nonfluent/agrammatic primary progressive aphasia caused by FTLD pathology. Neurology (2014) 82:239–47. doi: 10.1212/WNL.0000000000000031
70. Snowden J. Semantic dementia: a form of circumscribed cerebral atrophy. Behav Neurol. (1989) 2:167–82
71. Hodges JR, Patterson K. Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol. (2007) 6:1004–14. doi: 10.1016/S1474-4422(07)70266-1
72. Kertesz A, Blair M, McMonagle P, Munoz DG. The diagnosis and course of frontotemporal dementia. Alzheimer Dis Assoc Disord. (2007) 21:155–63. doi: 10.1097/WAD.0b013e31806547eb
73. Kertesz A, Jesso S, Harciarek M, Blair M, McMonagle P. What is semantic dementia?: a cohort study of diagnostic features and clinical boundaries. Arch Neurol. (2010) 67:483–9. doi: 10.1001/archneurol.2010.55
74. Coyle-Gilchrist IT, Dick KM, Patterson K, Vazquez Rodriquez P, Wehmann E, Wilcox A, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology (2016) 86:1736–43. doi: 10.1212/wnl.0000000000002638
75. Caine D, Breen N, Patterson K. Emergence and progression of ‘non-semantic' deficits in semantic dementia. Cortex (2009) 45:483–94. doi: 10.1016/j.cortex.2007.07.005
76. Garrard P, Rentoumi V, Gesierich B, Miller B, Gorno-Tempini ML. Machine learning approaches to diagnosis and laterality effects in semantic dementia discourse. Cortex (2014) 55:122–9. doi: 10.1016/j.cortex.2013.05.008
77. Mayberry EJ, Sage K, Ralph MA. At the edge of semantic space: the breakdown of coherent concepts in semantic dementia is constrained by typicality and severity but not modality. J Cogn Neurosci. (2011) 23:2240–51. doi: 10.1162/jocn.2010.21582
78. Hoffman P, Lambon Ralph MA. Reverse concreteness effects are not a typical feature of semantic dementia: evidence for the hub-and-spoke model of conceptual representation. Cereb Cortex (2011) 21:2103–12. doi: 10.1093/cercor/bhq288.
79. Yi HA, Moore P, Grossman M. Reversal of the concreteness effect for verbs in patients with semantic dementia. Neuropsychology (2007) 21:9–19. doi: 10.1037/0894-4105.21.1.9
80. Joubert S, Vallet GT, Montembeault M, Boukadi M, Wilson MA, Laforce RJ, et al. Comprehension of concrete and abstract words in semantic variant primary progressive aphasia and Alzheimer's disease: a behavioral and neuroimaging study. Brain Lang. (2017) 170:93–102. doi: 10.1016/j.bandl.2017.04.004
81. Gainotti G. Different patterns of famous people recognition disorders in patients with right and left anterior temporal lesions: a systematic review. Neuropsychologia (2007) 45:1591–607. doi: 10.1016/j.neuropsychologia.2006.12.013
82. Mendez MF, Kremen SA, Tsai PH, Shapira JS. Interhemispheric differences in knowledge of animals among patients with semantic dementia. Cogn Behav Neurol. (2010) 23:240–6. doi: 10.1097/WNN.0b013e3181f22448
83. Josephs KA, Whitwell JL, Vemuri P, Senjem ML, Boeve BF, Knopman DS, et al. The anatomic correlate of prosopagnosia in semantic dementia. Neurology (2008) 71:1628–33. doi: 10.1212/01.wnl.0000334756.18558.92
84. Mondini S, Semenza C. How Berlusconi keeps his face: a neuropsychological study in a case of semantic dementia. Cortex (2006) 42:332–5. doi: 10.1016/S0010-9452(08)70359-9
85. Irish M, Bunk S, Tu S, Kamminga J, Hodges JR, Hornberger M, et al. Preservation of episodic memory in semantic dementia: the importance of regions beyond the medial temporal lobes. Neuropsychologia (2016) 81:50–60. doi: 10.1016/j.neuropsychologia.2015.12.005
86. Adlam AL, Patterson K, Hodges JR. “I remember it as if it were yesterday”: memory for recent events in patients with semantic dementia. Neuropsychologia (2009) 47:1344–51. doi: 10.1016/j.neuropsychologia.2009.01.029
87. Irish M, Hornberger M, Lah S, Miller L, Pengas G, Nestor PJ, et al. Profiles of recent autobiographical memory retrieval in semantic dementia, behavioural-variant frontotemporal dementia, and Alzheimer's disease. Neuropsychologia (2011) 49:2694–702. doi: 10.1016/j.neuropsychologia.2011.05.017
88. Viard A, Desgranges B, Matuszewski V, Lebreton K, Belliard S, de La Sayette V, et al. Autobiographical memory in semantic dementia: new insights from two patients using fMRI. Neuropsychologia (2013) 51:2620–32. doi: 10.1016/j.neuropsychologia.2013.08.007
89. Irish M, Addis DR, Hodges JR, Piguet O. Considering the role of semantic memory in episodic future thinking: evidence from semantic dementia. Brain (2012) 135(Pt 7):2178–91. doi: 10.1093/brain/aws119
90. La Corte V, Piolino P. On the Role of Personal semantic memory and temporal distance in episodic future thinking: the TEDIFT model. Front Hum Neurosci. (2016) 10:385. doi: 10.3389/fnhum.2016.00385
91. Brambati SM, Ogar J, Neuhaus J, Miller BL, Gorno-Tempini ML. Reading disorders in primary progressive aphasia: a behavioral and neuroimaging study. Neuropsychologia (2009) 47:1893–900. doi: 10.1016/j.neuropsychologia.2009.02.033
92. Wilson SM, Brambati SM, Henry RG, Handwerker DA, Agosta F, Miller BL, et al. The neural basis of surface dyslexia in semantic dementia. Brain (2009) 132(Pt 1):71–86. doi: 10.1093/brain/awn300
93. Irish M, Hodges JR, Piguet O. Right anterior temporal lobe dysfunction underlies theory of mind impairments in semantic dementia. Brain (2014) 137(Pt 4):1241–53. doi: 10.1093/brain/awu003
94. Seeley WW, Bauer AM, Miller BL, Gorno-Tempini ML, Kramer JH, Weiner M, et al. The natural history of temporal variant frontotemporal dementia. Neurology (2005) 64:1384–90. doi: 10.1212/01.WNL.0000158425.46019.5C
95. Kumfor F, Miller L, Lah S, Hsieh S, Savage S, Hodges JR, et al. Are you really angry? The effect of intensity on facial emotion recognition in frontotemporal dementia. Soc Neurosci. (2011) 6:502–14. doi: 10.1080/17470919.2011.620779
96. Gan JJ, Lin A, Samimi MS, Mendez MF. Somatic symptom disorder in semantic dementia: the role of alexisomia. Psychosomatics (2016) 57:598–604. doi: 10.1016/j.psym.2016.08.002
97. Miller ZA, Mandelli ML, Rankin KP, Henry ML, Babiak MC, Frazier DT, et al. Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain (2013) 136(Pt 11):3461–73. doi: 10.1093/brain/awt242
98. Kaplan E, Goodglass H, Weintraub S. Boston Naming Test: Philadelphia, PA: Lea & Febiger. (1983).
99. Howard D, Patterson K. The Pyramids and Palm Trees Test. A Test of Semantic Access From Words and Pictures. Bury St. Edmunds: Thames Valley Company (1992).
100. Montembeault M, Brambati SM, Joubert S, Boukadi M, Chapleau M, Laforce RJ, et al. Naming unique entities in the semantic variant of primary progressive aphasia and Alzheimer's disease: Towards a better understanding of the semantic impairment. Neuropsychologia (2017) 95:11–20. doi: 10.1016/j.neuropsychologia.2016.12.009
101. Mummery CJ, Patterson K, Price CJ, Ashburner J, Frackowiak RS, Hodges JR. A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol. (2000) 47:36–45. doi: 10.1002/1531-8249(200001)47:1<36::AID-ANA8>3.0.CO;2-L
102. Galton CJ, Patterson K, Graham K, Lambon-Ralph MA, Williams G, Antoun N, et al. Differing patterns of temporal atrophy in Alzheimer's disease and semantic dementia. Neurology (2001) 57:216–25. doi: 10.1212/WNL.57.2.216
103. Mesulam M, Wieneke C, Rogalski E, Cobia D, Thompson C, Weintraub S. Quantitative template for subtyping primary progressive aphasia. Arch Neurol. (2009) 66:1545–51. doi: 10.1001/archneurol.2009.288
104. Rosen HJ, Kramer JH, Gorno-Tempini ML, Schuff N, Weiner M, Miller BL. Patterns of cerebral atrophy in primary progressive aphasia. Am J Geriatr Psychiatry (2002) 10:89–97
105. Yang J, Pan P, Song W, Shang HF. Quantitative meta-analysis of gray matter abnormalities in semantic dementia. J Alzheimers Dis. (2012) 31:827–33. doi: 10.3233/jad-2012-120736
106. Kato T, Inui Y, Nakamura A, Ito K. Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Res Rev. (2016) 30:73–84. doi: 10.1016/j.arr.2016.02.003
107. Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, et al. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. (2008) 64:388–401. doi: 10.1002/ana.21451
108. Migliaccio R, Boutet C, Valabregue R, Ferrieux S, Nogues M, Lehéricy S, et al. The brain network of naming: a lesson from primary progressive aphasia. PLoS ONE (2016) 11:e0148707. doi: 10.1371/journal.pone.0148707
109. Thompson SA, Patterson K, Hodges JR. Left/right asymmetry of atrophy in semantic dementia: behavioral-cognitive implications. Neurology (2003) 61:1196–203. doi: 10.1212/01.WNL.0000091868.28557.B8
110. Henry ML, Wilson SM, Ogar JM, Sidhu MS, Rankin KP, Cattaruzza T, et al. Neuropsychological, behavioral, and anatomical evolution in right temporal variant frontotemporal dementia: a longitudinal and post-mortem single case analysis. Neurocase (2012) 20:100–9. doi: 10.1080/13554794.2012.732089
111. La Joie R, Landeau B, Perrotin A, Bejanin A, Egret S, Pelerin A, et al. Intrinsic connectivity identifies the hippocampus as a main crossroad between Alzheimer's and semantic dementia-targeted networks. Neuron (2014) 81:1417–28. doi: 10.1016/j.neuron.2014.01.026
112. Chapleau M, Aldebert J, Montembeault M, Brambati SM. Atrophy in Alzheimer's disease and semantic dementia: an ALE meta-analysis of voxel-based morphometry studies. J Alzheimers Dis. (2016) 54:941–55. doi: 10.3233/jad-160382
113. Agosta F, Henry RG, Migliaccio R, Neuhaus J, Miller BL, Dronkers NF, et al. Language networks in semantic dementia. Brain (2010) 133(Pt 1):286–99. doi: 10.1093/brain/awp233
114. Acosta-Cabronero J, Patterson K, Fryer TD, Hodges JR, Pengas G, Williams GB, et al. Atrophy, hypometabolism and white matter abnormalities in semantic dementia tell a coherent story. Brain (2011) 134(Pt 7):2025–35. doi: 10.1093/brain/awr119
115. Meijboom R, Steketee RM, Ham LS, van der Lugt A, van Swieten JC, Smits M. Differential hemispheric predilection of microstructural white matter and functional connectivity abnormalities between respectively semantic and behavioral variant frontotemporal dementia. J Alzheimers Dis. (2017) 56:789–804. doi: 10.3233/jad-160564
116. Tu S, Leyton CE, Hodges JR, Piguet O, Hornberger M. Divergent longitudinal propagation of white matter degradation in logopenic and semantic variants of primary progressive aphasia. J Alzheimers Dis. (2016) 49:853–61. doi: 10.3233/jad-150626
117. Guo CC, Gorno-Tempini ML, Gesierich B, Henry M, Trujillo A, Shany-Ur T, et al. Anterior temporal lobe degeneration produces widespread network-driven dysfunction. Brain (2013) 136(Pt 10):2979–91. doi: 10.1093/brain/awt222
118. Agosta F, Galantucci S, Valsasina P, Canu E, Meani A, Marcone A, et al. Disrupted brain connectome in semantic variant of primary progressive aphasia. Neurobiol Aging (2014) 35:2646–55. doi: 10.1016/j.neurobiolaging.2014.05.017
119. Collins JA, Montal V, Hochberg D, Quimby M, Mandelli ML, Makris N, et al. Focal temporal pole atrophy and network degeneration in semantic variant primary progressive aphasia. Brain (2017) 140(Pt 2):457–71. doi: 10.1093/brain/aww313
120. Brambati SM, Rankin KP, Narvid J, Seeley WW, Dean D, Rosen HJ, et al. Atrophy progression in semantic dementia with asymmetric temporal involvement: a tensor-based morphometry study. Neurobiol Aging (2009) 30:103–11. doi: 10.1016/j.neurobiolaging.2007.05.014
121. Kumfor F, Landin-Romero R, Devenney E, Hutchings R, Grasso R, Hodges JR, et al. On the right side? A longitudinal study of left- versus right-lateralized semantic dementia. Brain (2016) 139(Pt 3):986–98. doi: 10.1093/brain/awv387
122. Cerami C, Dodich A, Greco L, Iannaccone S, Magnani G, Marcone A, et al. The role of single-subject brain metabolic patterns in the early differential diagnosis of primary progressive aphasias and in prediction of progression to dementia. J Alzheimers Dis. (2016) 55:183–97. doi: 10.3233/JAD-160682
123. Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol. (2004) 56:399–406. doi: 10.1002/ana.20203
124. Davies RR, Hodges JR, Kril JJ, Patterson K, Halliday GM, Xuereb JH. The pathological basis of semantic dementia. Brain (2005) 128(Pt 9):1984–95. doi: 10.1093/brain/awh582
125. Grossman M, Wood EM, Moore P, Neumann M, Kwong L, Forman MS, et al. TDP-43 pathologic lesions and clinical phenotype in frontotemporal lobar degeneration with ubiquitin-positive inclusions. Arch Neurol. (2007) 64:1449–54. doi: 10.1001/archneur.64.10.1449
126. Hodges JR, Mitchell J, Dawson K, Spillantini MG, Xuereb JH, McMonagle P, et al. Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain (2010) 133(Pt 1):300–6. doi: 10.1093/brain/awp248
127. Mesulam M, Johnson N, Krefft TA, Gass JM, Cannon AD, Adamson JL, et al. Progranulin mutations in primary progressive aphasia: the PPA1 and PPA3 families. Arch Neurol. (2007) 64:43–7. doi: 10.1001/archneur.64.1.43
128. Miller ZA, Rankin KP, Graff-Radford NR, Takada LT, Sturm VE, Cleveland CM, et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J Neurol Neurosurg Psychiatry (2013) 84:956–62. doi: 10.1136/jnnp-2012-304644
129. Le Ber I, Camuzat, A, Guillot-Noel L, Hannequin D, Lacomblez L, Golfier V, et al. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow-chart for genetic testing. J Alzheimers Dis. (2013) 34:485–99. doi: 10.3233/jad-121456
130. Scherling CS, Hall T, Berisha F, Klepac K, Karydas A, Coppola G, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. (2014) 75:116–26. doi: 10.1002/ana.24052
131. Gorno-Tempini ML, Brambati SM, Ginex V, Ogar J, Dronkers NF, Marcone A, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology (2008) 71:1227–34. doi: 10.1212/01.wnl.0000320506.79811.da
132. Migliaccio R, Agosta F, Rascovsky K, Karydas A, Bonasera S, Rabinovici GD, et al. Clinical syndromes associated with posterior atrophy: early age at onset AD spectrum. Neurology (2009) 73:1571–8. doi: 10.1212/WNL.0b013e3181c0d427
133. Win KT, Pluta J, Yushkevich P, Irwin DJ, McMillan CT, Rascovsky K, et al. Neural correlates of verbal episodic memory and lexical retrieval in logopenic variant primary progressive aphasia. Front Neurosci. (2017) 11:330. doi: 10.3389/fnins.2017.00330
134. Win KT, Pluta J, Yushkevich P, Irwin DJ, McMillan CT, Rascovsky K, et al. Neural correlates of verbal episodic memory and lexical retrieval in logopenic variant primary progressive aphasia. Front Neurosci. (2017) 11:330. doi: 10.3389/fnins.2017.00330.
135. Leyton CE, Hsieh S, Mioshi E, Hodges JR. Cognitive decline in logopenic aphasia: more than losing words. Neurology (2013) 80:897–903. doi: 10.1212/WNL.0b013e318285c15b
136. Watson CL, Possin K, Allen IE, Hubbard HI, Meyer M, Welch AE, et al. Visuospatial functioning in the primary progressive aphasias. J Int Neuropsychol Soc. (2017):24:259–68. doi: 10.1017/S1355617717000984.
137. Rohrer JD, Ridgway GR, Crutch SJ, Hailstone J, Goll JC, Clarkson MJ, et al. Progressive logopenic/phonological aphasia: erosion of the language network. Neuroimage (2010) 49:984–93. doi: 10.1016/j.neuroimage.2009.08.002
138. Rohrer JD, Warren JD. Phenomenology and anatomy of abnormal behaviours in primary progressive aphasia. J Neurol Sci. (2010) 293:35–8. doi: 10.1016/j.jns.2010.03.012
139. Rogalski E, Johnson N, Weintraub S, Mesulam M. Increased frequency of learning disability in patients with primary progressive aphasia and their first-degree relatives. Arch Neurol. (2008) 65:244–8. doi: 10.1001/archneurol.2007.34
140. Paternicó D, Premi E, Alberici A, Archetti S, Bonomi E, Gualeni V, et al. Dyslexia susceptibility genes influence brain atrophy in frontotemporal dementia. Neurol Genet (2015) 1:e24. doi: 10.1212/NXG.0000000000000024
141. Rogalski E, Rademaker A, Wieneke C, Bigio E, Weintraub S, Mesulam MM. The prevalence of learning disabilities in primary progressive aphasia is not segregated by pathology or subtype. JAMA Neurol. (2014) 71:1576–7. doi: 10.1001/jamaneurol.2014.2805
142. Leyton CE, Ballard KJ, Piguet O, Hodges JR. Phonologic errors as a clinical marker of the logopenic variant of PPA. Neurology (2014) 82:1620–7. doi: 10.1212/wnl.0000000000000387
143. Wechsler D. The Wechsler Adult Intelligence Scale–Revised. San Antonio, TX: Psychological Corporation (1981).
144. Piguet O, Leyton CE, Gleeson LD, Hoon C, Hodges JR. Memory and emotion processing performance contributes to the diagnosis of non-semantic primary progressive aphasia syndromes. J Alzheimers Dis. (2015) 44:541–7. doi: 10.3233/jad-141854
145. Phillips JS, Da Re F, Dratch L, Xie SX, Irwin DJ, McMillan CT, et al. Neocortical origin and progression of gray matter atrophy in nonamnestic Alzheimer's disease. Neurobiol Aging (2018) 63:75–87. doi: 10.1016/j.neurobiolaging.2017.11.008
146. Migliaccio R, Agosta F, Possin KL, Rabinovici GD, Miller BL, Gorno-Tempini ML. White matter atrophy in Alzheimer's disease variants. Alzheimer's Dement. (2012) 8(Suppl. 5):S78–87e1-2. doi: 10.1016/j.jalz.2012.04.010
147. Whitwell JL, Jones DT, Duffy JR, Strand EA, Machulda MM, Przybelski SA, et al. Working memory and language network dysfunctions in logopenic aphasia: a task-free fMRI comparison with Alzheimer's dementia. Neurobiol Aging (2015) 36:1245–52. doi: 10.1016/j.neurobiolaging.2014.12.013
148. Teichmann M, Migliaccio R, Kas A, Dubois B. Logopenic progressive aphasia beyond Alzheimer's–an evolution towards dementia with Lewy bodies. J Neurol Neurosurg Psychiatry (2013) 84:113–4. doi: 10.1136/jnnp-2012-302638
149. Teichmann M, Kas A, Boutet C, Ferrieux S, Nogues M, Samri D, et al. Deciphering logopenic primary progressive aphasia: a clinical, imaging and biomarker investigation. Brain (2013) 136(Pt 11):3474–88. doi: 10.1093/brain/awt266
150. Munoz DG, Woulfe J, Kertesz A. Argyrophilic thorny astrocyte clusters in association with Alzheimer's disease pathology in possible primary progressive aphasia. Acta Neuropathol. (2007) 114:347–57. doi: 10.1007/s00401-007-0266-x
151. Rohrer JD, Warren JD, Omar R, Mead S, Beck J, Revesz T, et al. Pariet al lobe deficits in frontotemporal lobar degeneration caused by a mutation in the progranulin gene. Arch Neurol. (2008) 65:506–13. doi: 10.1001/archneur.65.4.506
152. Leyton CE, Villemagne VL, Savage S, Pike KE, Ballard KJ, Piguet O, et al. Subtypes of progressive aphasia: application of the International Consensus Criteria and validation using beta-amyloid imaging. Brain (2011) 134(Pt 10):3030–43. doi: 10.1093/brain/awr216
153. Mesulam MM, Wieneke C, Thompson C, Rogalski E, Weintraub S. Quantitative classification of primary progressive aphasia at early and mild impairment stages. Brain (2012) 135(Pt 5):1537–53. doi: 10.1093/brain/aws080
154. Sajjadi SA, Patterson K, Arnold RJ, Watson PC, Nestor PJ. Primary progressive aphasia: a tale of two syndromes and the rest. Neurology (2012) 78:1670–7. doi: 10.1212/WNL.0b013e3182574f79
155. Wicklund MR, Duffy JR, Strand EA, Machulda MM, Whitwell JL, Josephs KA. Quantitative application of the primary progressive aphasia consensus criteria. Neurology (2014) 82:1119–26. doi: 10.1212/wnl.0000000000000261
156. Rohrer JD, Crutch SJ, Warrington EK, Warren JD. Progranulin-associated primary progressive aphasia: a distinct phenotype? Neuropsychologia (2010) 48:288–97. doi: 10.1016/j.neuropsychologia.2009.09.017
157. Catricalà E, Gobbi E, Battista P, Miozzo A, Polito C, Boschi V, et al. SAND: a Screening for aphasia in neurodegeneration. development and normative data. Neurol Sci. (2017) 38:1469–83. doi: 10.1007/s10072-017-3001-y
158. Savage S, Hsieh S, Leslie F, Foxe D, Piguet O, Hodges JR. Distinguishing subtypes in primary progressive aphasia: application of the Sydney language battery. Dement Geriatr Cogn Disord. (2013) 35:208–18. doi: 10.1159/000346389
159. Leyton CE, Savage S, Irish M, Schubert S, Piguet O, Ballard KJ, et al. Verbal repetition in primary progressive aphasia and Alzheimer's disease. J Alzheimers Dis. (2014) 41:575–85. doi: 10.3233/jad-132468
160. Leyton CE, Hodges JR, Piguet O, Ballard KJ. Common and divergent neural correlates of anomia in amnestic and logopenic presentations of Alzheimer's disease. Cortex (2017) 86:45–54. doi: 10.1016/j.cortex.2016.10.019
161. Reilly J, Peelle JE, Antonucci SM, Grossman M. Anomia as a marker of distinct semantic memory impairments in Alzheimer's disease and semantic dementia. Neuropsychology (2011) 25:413–26. doi: 10.1037/a0022738
Keywords: primary progressive aphasia, nonfluent/agrammatic variant, semantic variant, logopenic variant, language, brain connectivity
Citation: Montembeault M, Brambati SM, Gorno-Tempini ML and Migliaccio R (2018) Clinical, Anatomical, and Pathological Features in the Three Variants of Primary Progressive Aphasia: A Review. Front. Neurol. 9:692. doi: 10.3389/fneur.2018.00692
Received: 30 April 2018; Accepted: 31 July 2018;
Published: 21 August 2018.
Edited by:
Raymond Scott Turner, Georgetown University, United StatesReviewed by:
Jed A. Meltzer, Baycrest Hospital, CanadaCristian E. Leyton, University of Sydney, Australia
Copyright © 2018 Montembeault, Brambati, Gorno-Tempini and Migliaccio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raffaella Migliaccio, bGFyYS5taWdsaWFjY2lvQGdtYWlsLmNvbQ==