 Luisa Iommarini
Luisa Iommarini Anna Maria Porcelli
Anna Maria Porcelli Giuseppe Gasparre
Giuseppe Gasparre Ivana Kurelac
Ivana Kurelac- 1Dipartimento di Farmacia e Biotecnologie, Università di Bologna, Bologna, Italy
- 2Dipartimento di Scienze Mediche e Chirurgiche, Università di Bologna, Bologna, Italy
Hypoxia-inducible factor 1 alpha (HIF-1α) orchestrates cellular adaptation to low oxygen and nutrient-deprived environment and drives progression to malignancy in human solid cancers. Its canonical regulation involves prolyl hydroxylases (PHDs), which in normoxia induce degradation, whereas in hypoxia allow stabilization of HIF-1α. However, in certain circumstances, HIF-1α regulation goes beyond the actual external oxygen levels and involves PHD-independent mechanisms. Here, we gather and discuss the evidence on the non-canonical HIF-1α regulation, focusing in particular on the consequences of mitochondrial respiratory complexes damage on stabilization of this pleiotropic transcription factor.
Hypoxia-inducible factor 1 (HIF-1) is the major orchestrator of cellular adaptation to low oxygen environment (1). In normoxia, prolyl hydroxylases (PHDs) hydroxylate HIF-1α on two proline residues within the oxygen-dependent degradation domain, triggering von Hippel–Lindau (pVHL)-mediated ubiquitination and proteasomal degradation (Figure 1) (2). In parallel, the Factor Inhibiting HIF (FIH), an asparaginyl hydroxylase regulated similarly to PHDs, in an oxygen-dependent manner, suppresses HIF-1 transcriptional activity in normoxia by preventing co-activator recruitment (3, 4). Conversely, hypoxia inhibits PHDs and stabilizes HIF-1α, which then translocates into the nucleus and dimerizes with constitutively expressed HIF-1β, creating active HIF-1 complex and triggering the transcription of genes promoting glycolytic metabolism, angiogenesis, and survival (Figure 1) (5). Activation of HIF-1α is physiological during embryogenesis and in wound-healing processes, whereas in cancer, HIF-1α is associated with malignancy and poor prognosis (6, 7). Abnormal stabilization of HIF-1α and upregulation of its downstream targets have been described in a broad spectrum of solid tumors as they progress to malignancy (8).
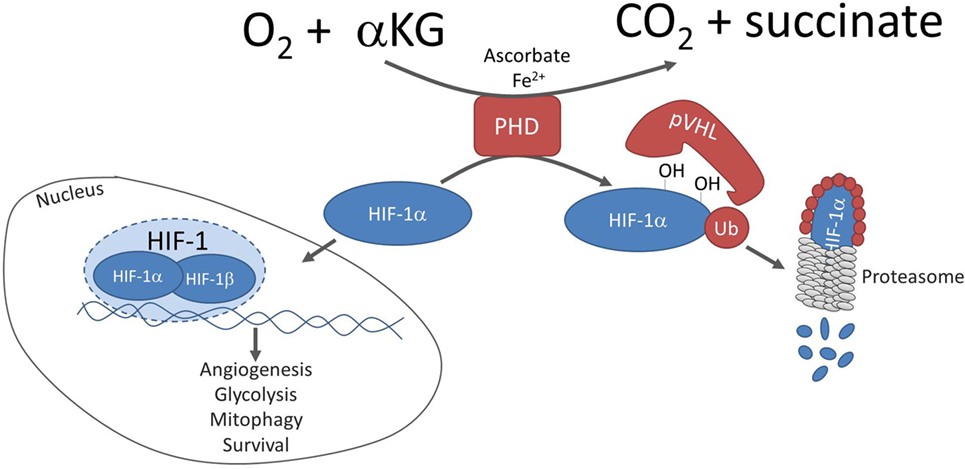
Figure 1. Canonical regulation of HIF-1α stability. In normoxia, prolyl hydroxylases (PHDs) hydroxylate hypoxia-inducible factor 1 alpha (HIF-1α) on two proline residues, triggering pVHL-mediated ubiquitination and proteasomal degradation of hydroxylated HIF-1α. The hydroxylation reaction is coupled to conversion of αKG to succinate and requires co-factors ascorbate and ferrous iron. In hypoxia, hydroxylation is inhibited and HIF-1α dimerizes with constitutively expressed HIF-1β, creating an active HIF-1 complex, which transcribes genes promoting angiogenesis, glycolytic metabolism, mitophagy, and survival.
Since the discovery of HIF-1α and the ingenious oxygen-dependent PHD-mediated regulation, a great number of additional modalities of HIF-1α control has been identified, independently from external oxygen concentrations and acting at the level of its transcription, translation, oxygen-independent stabilization/degradation, translocation from cytoplasm to the nucleus, and even affecting HIF-1 transcriptional activity. Here, we review and discuss the non-canonical regulation of HIF-1α expression and stabilization in cancer cells, focusing on factors which cause pseudohypoxia (HIF-1α stabilization in normoxic conditions) or fail to stabilize HIF-1α in low oxygen atmosphere (pseudonormoxia). Particular attention is given to the discussion of data showing that oxidative phosphorylation (OXPHOS) damage may block HIF-1α stabilization, since this controversial issue has seldom been reviewed elsewhere.
Oxygen-Independent HIF-1α Stabilization by Oncometabolite-Mediated Regulation of PHDs Activity
The first evidence of an oxygen-independent regulation of HIF-1α stability in vivo was found in tumors harboring succinate dehydrogenase (SDH) and fumarate hydratase mutations (9). Soon after, it was demonstrated that SDH inhibition stabilizes HIF-1α in normoxia due to increased concentrations of succinate, a by-product and allosteric inhibitor of the PHD reaction (10). This finding gave birth to the concept of “oncometabolites,” which initially regarded the accumulation of certain Krebs cycle intermediates, such as succinate and fumarate (11, 12), but may now be extended to any metabolite capable of triggering oncogenic or tumor suppressor signals. In the context of HIF-1α regulation, pyruvate and lactate were suggested to promote pseudohypoxia (13–15), whereas the PHD substrate alpha-ketoglutarate (αKG), as well as PHD co-factors ascorbate and Fe2+, were all shown to confer a dose-dependent HIF-1α destabilization in hypoxia (16) (Figure 2A). For example, αKG increases the PHD affinity for oxygen and thus promotes HIF-1α hydroxylation and degradation even at low oxygen concentrations (17, 18). Accordingly, pseudonormoxia is observed in cells suffering nicotinamide nucleotide transhydrogenase deficiency or severe complex I damage, both conditions leading to NADH accumulation and consequent increase in αKG, due to the slowdown of the Krebs cycle rate (19–22). Conversely, the mitochondrial isocitrate dehydrogenase 3 alpha overexpression decreases αKG concentrations and promotes HIF-1α stability (23). Although mechanisms balancing oncometabolite concentrations represent intriguing therapeutic targets, their successful manipulation to fight cancer is still to be optimized, most likely due to the complexity of oncometabolite-mediated HIF-1α regulation. For instance, hypoxia-induced miR-210 expression was shown to contribute to the succinate accumulation by causing respiratory complex II defects (24, 25). Moreover, whereas (L)-2 hydroxyglutarate promotes HIF-1α stabilization (26), genetic lesions leading to the accumulation of the (R)-2 hydroxyglutarate enantiomer instead activate PHDs (27).
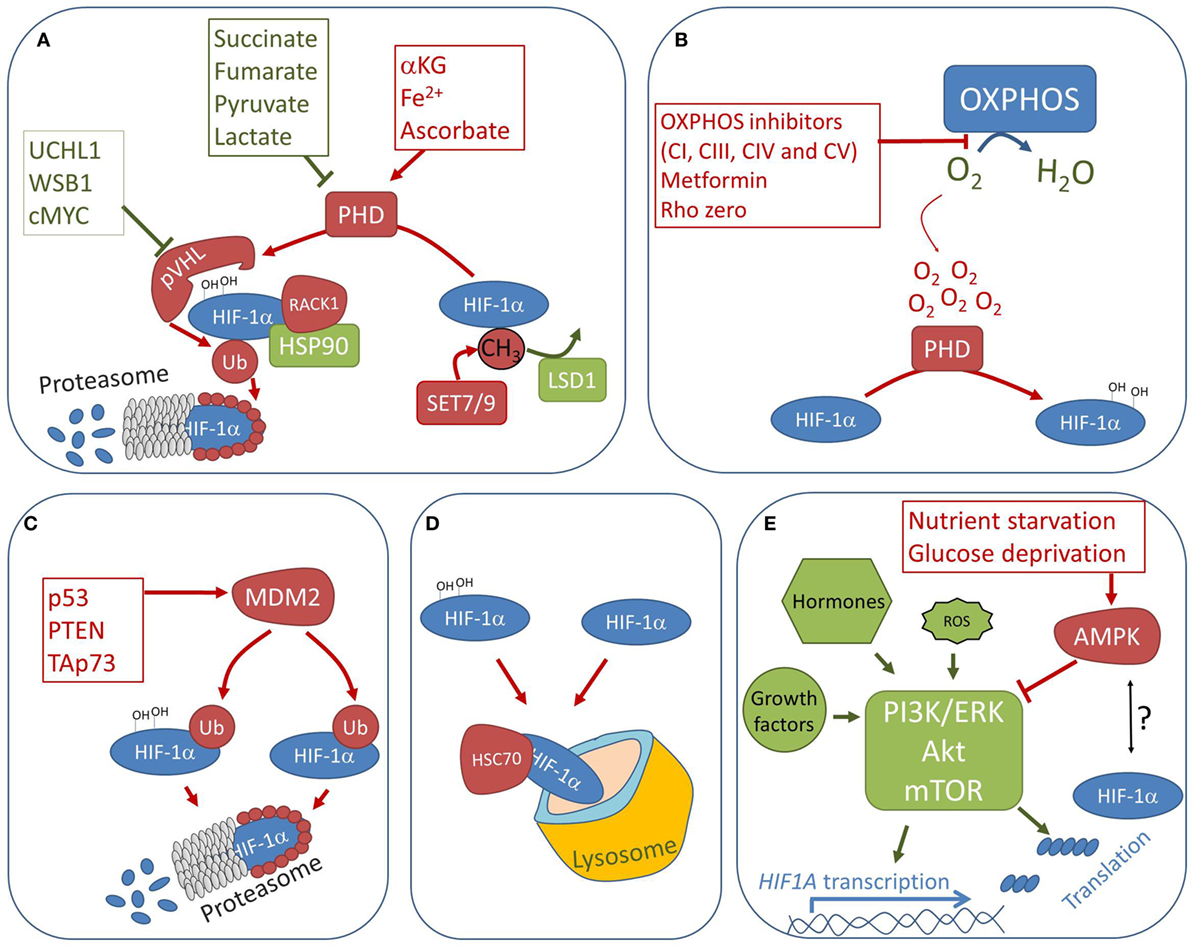
Figure 2. Non-canonical regulation of HIF-1α stability. Factors promoting pseudonormoxia and pseudohypoxia are indicated in red and green, respectively. (A) Prolyl hydroxylase (PHD) activity may be blocked by accumulation of Krebs cycle metabolites succinate and fumarate, whereas αKG, and co-factors ascorbate and iron, boost PHDs activity regardless of oxygen levels. Activation of any factor promoting pVHL downregulation in normoxia will also lead to pseudohypoxic stabilization of HIF-1α. Finally, posttranslational modifications, such as methylation by SET7/9, or interactions with proteins, such as receptor of activated protein C kinase (RACK1) and HSP90, may regulate PHD accessibility to HIF-1α and promote or block hydroxylation regardless of oxygen concentrations. (B) Severe damage or inhibition of oxidative phosphorylation (OXPHOS) complexes I, III, IV, or V, reduces oxygen consumption, which in turn may increase intracellular oxygen concentrations and cause pseudonormoxia. (C) MDM2 is an ubiquitine ligase, which promotes HIF-1α degradation in hypoxic environment when associated with tumor suppressor proteins. (D) Proteasome-independent HIF-1α degradation via chaperone-mediated autophagy is mediated by HSC70. (E) PI3K/Akt/mTOR axis is the major pathway involved in promoting HIF1A transcription and translation, regardless of oxygen concentrations and upon numerous protumorigenic stimuli. For example, elevated reactive oxygen species concentrations were shown to promote HIF1A transcription and translation via Akt signaling. On the other hand, conditions counteracting mTOR pathway, such as nutrient starvation, and possibly adenosine monophosphate kinase (AMPK) activation, may lead to HIF-1α downregulation.
Non-Canonical Oxygen-Dependent Regulation of PHDs by Redistribution of Intracellular Oxygen Following OXPHOS Damage
As a solid cancer progresses, transformed cells usually activate HIF-1-mediated adaptations to hypoxic stress, which include downregulation of mitochondrial respiration to decrease the cells’ requirement for oxygen (24, 28, 29). However, several xenograft studies, and a few examples from human tumors, demonstrate that severe OXPHOS damage induces a series of metabolic and molecular anti-tumorigenic events which, among other, include destabilization of HIF-1α (20, 21, 30–34). The anti-tumorigenic consequences of OXPHOS damage leading to HIF-1α destabilization come as a paradox to the known role of HIF-1 in promoting mitophagy and downregulation of OXPHOS genes (24, 28, 29) and are, therefore, discussed here in more detail. Hagen and colleagues pioneered in demonstrating that decreased oxygen consumption, due to OXPHOS inhibition in cancer cell lines, may result in redistribution of intracellular oxygen from respiratory enzymes to the PHDs, so that the latter become unable to sense external hypoxia (35, 36). As a result, HIF-1α is destabilized in cells with severe mitochondrial respiration damage, despite the outer hypoxic environment (Figure 2B). The association between mitochondrial respiration damage and HIF-1α inactivation despite hypoxia has also been observed in Rho zero cells and diverse cancer cell types, in which OXPHOS complexes I, III, IV, or V were pharmacologically inhibited (37–39). In accordance, by using a phosphorescent probe quenched by oxygen, a recent study showed that increasing concentrations of complex I inhibitor rotenone decrease intracellular hypoxia in a dose-dependent manner in a prostate cancer cell line (40). The conditions applied in these studies usually consisted of 3–6 h culture in the presence of 1–3% oxygen. On the other hand, studies applying 0.1–1% oxygen concentrations, reported that HIF-1α stabilizes in Rho zero cancer cells or upon rotenone treatment (41, 42), and Gong and Agani demonstrated that, in near-anoxic conditions, HIF-1α is stabilized despite OXPHOS damage (43). Therefore, OXPHOS damage does not seem to irreversibly prevent, but may rather attenuate HIF-1α stabilization, suggesting that the increased intracellular oxygen concentrations, caused by the lower oxygen consumption, may rapidly equilibrate with the extracellular tensions. Such equilibration probably depends on the cellular membrane permeability to molecular oxygen, which among other is influenced by cholesterol levels and, therefore, lipid metabolism, which is conditioned by the OXPHOS status (44).
Notably, because of the short HIF-1α half-life (<5 min) in well oxygenated atmosphere, changes in ambient oxygen concentrations and variations of oxygen diffusion in the culture medium have a strong impact on HIF-1α stabilization when working in vitro. Therefore, precautions must be applied during cellular extraction and during cell washing, to avoid making biased conclusions regarding HIF-1α regulation. Moreover, for the time being, experimental limits prevent precise dissection of oxygen distribution in a growing tumor. Indeed, it must be noted that, to the best of our knowledge, the formal demonstration of the mechanism linking OXPHOS deficiency and HIF-1α destabilization in vivo, where selective pressures and microenvironment are radically different from in vitro conditions, has yet to be reported. Based on our data from complex I-deficient models, we hypothesize that more than one factor is involved in HIF-1α destabilization in OXPHOS-deficient tumors, since, if compared to counterpart controls, they display not only increased intracellular oxygen concentrations (unpublished data) but also higher αKG levels (20–22) and iron accumulation (unpublished data), all factors known to promote PHD-mediated HIF-1α hydroxylation.
To add complexity, OXPHOS damage is a known source of reactive oxygen species (ROS), which were suggested to promote HIF-1α stability in hypoxia and normoxia, although their role in HIF-1α regulation is still controversial (45, 46). Brunell and colleagues suggested that oxygen sensing in OXPHOS does not depend on oxygen consumption in human fibroblasts, but rather on ROS production deriving from decreased activity of complexes III and IV (47). On the other hand, by working on cancer cells, Chua and colleagues report that HIF-1α stabilization in hypoxia is not dependent on ROS and that re-establishing oxygen consumption in complex III-repressed cells is sufficient to induce HIF-1α stabilization, most likely due to a decrease of intracellular oxygen (48). The role of ROS in oxygen sensing has extensively been reviewed elsewhere (46, 49–51), and we discuss the role of ROS in promoting HIF1A transcription in the next paragraph. Still, it is interesting to note that OXPHOS damage leading to elevated ROS was suggested to promote HIF-1α stabilization (45), whereas severe respiratory deficiency associated to a decreased consumption of NADH results in pseudonormoxia. These apparently opposite effects may be explained by the fact that particularly severe damage, at least in the context of certain complex I mutations (20, 21), could destroy ROS-generating sites of respiratory multi-enzymes, resulting in unchanged or even decreased ROS concentrations. In this context, it is not surprising that mitochondrial DNA (mtDNA) mutations, not infrequent modifiers of tumorigenesis, may have opposing consequences on cancer progression, depending on the type of damage they induce (20). For example, mtDNA mutations increasing ROS production have been suggested to promote tumorigenesis and metastases, whereas those causing severe damage, such as complex I disassembly, compromise tumor progression (20, 21).
Taken together, the effects of OXPHOS deficiency on HIF-1α will depend on the type of damage inflicted, probably through different mechanisms depending on the mitochondrial respiratory complex involved. Nevertheless, while the downregulation of mitochondrial respiration by HIF-1 is certainly a valid mechanism for adaptation of cancer cells to low oxygen tension, the block of OXPHOS may not be severe, since this would lead to HIF-1α destabilization. The latter is supported by studies such as the recent Hamanaka’s work in epidermal keratinocytes, where the knock-out of mtDNA replication and transcription factor TFAM caused reduction of HIF-1α protein levels (52), indicating that HIF-1α destabilization in cells suffering mitochondrial respiratory damage seems to be a rather general phenomenon.
Interestingly, since severe OXPHOS damage seems to prevent cancer cells from experiencing hypoxia, they should be exempted from the need to adapt to low oxygen environment. Nevertheless, the growth of OXPHOS-deficient tumors is still challenged, as seen in complex I-deficient xenograft models (20, 21, 30, 31, 34) and in oncocytoma patients, who develop slowly proliferating masses, which rarely progress to malignancy (33). On one hand, this may be explained by the metabolic insufficiency, such as the recently described deficit in nucleotide biosynthesis, caused by aspartate shortage upon complex I inhibition (53). However, the consequences of the lack of HIF-1α in such tumors is not to be neglected, especially in the light of studies demonstrating that inhibition of HIF-1α is sufficient to block tumor growth (54, 55). In this context, it is intriguing to hypothesize that, in certain cancers, hypoxia may be advantageous, rather than a drawback for growing tumors, since the survival signals promoted by HIF-1 may actually be a requirement for malignant progression.
PHD-Independent Pathways Regulating HIF-1α Stabilization
While PHDs control the oxygen-dependent HIF-1α stability, many other proteins are emerging as additional mediators of HIF-1α regulation, which act in an oxygen-independent manner and, therefore, regardless of the HIF-1α hydroxylation status. For example, several factors modulate pVHL activity (Figure 2A), such as WD repeat and SOCS box-containing protein 1 (WSB1), which was found to promote HIF-1α stabilization and metastases via ubiquitination and degradation of pVHL in renal carcinoma, breast cancer, and melanoma models (56). Similarly, ubiquitin C-terminal hydrolase-L1 was described to abrogate the pVHL-mediated ubiquitination of HIF-1α in mouse models of pulmonary metastasis (57), and c-Myc has been shown to weaken HIF-1α binding to pVHL complex, eventually leading to normoxic HIF-1α stabilization in breast cancer cells (58). Besides pVHL, E3 ubiquitin-protein ligase MDM2 was also found to ubiquitinate HIF-1α, but in a hydroxylation-independent manner, promoting its destabilization despite hypoxic atmosphere (Figure 2C). MDM2-mediated oxygen-independent HIF-1α degradation seems to occur upon binding with tumor suppressor proteins, such as TAp73 (59) or p53 (60). On a similar note, it has recently been shown that PTEN and PI3K inhibitors promote HIF-1α destabilization by preventing MDM2 phosphorylation and subsequent translocation in the nucleus, suggesting that cytoplasmic MDM2 is then able to ubiquitinate HIF-1α and promote its degradation in hypoxia (61). Therefore, in cancers carrying mutations in tumor suppressor proteins such as TP53, MDM2-mediated HIF-1α degradation would be suspended, leading to synergic promotion of cancer progression, through blockage of the p53 pro-apoptotic stimuli and activation of the survival pathways upregulated by HIF-1α. Conversely, p53-independent binding of MDM2 to HIF-1α was associated with the increase in HIF-1α protein content (62), warning that the role of MDM2 in HIF-1α regulation might be more ambiguous than initially described. Further examples of oxygen-independent HIF-1α regulation involve factors, which may act either as promoters of HIF-1α degradation (Figure 2A), such as receptor of activated protein C kinase (RACK1), or as protectors from pVHL-mediated ubiquitination, such as heat shock protein (Hsp90) or Sentrin/SUMO-specific protease 1 (SENP1) (63–65). Inhibition of Hsp90 promotes the proteasome-mediated degradation of HIF-1α even in hypoxia or when functional pVHL is lacking (66). Moreover, it has been reported that gamma rays stimulate the mTOR-dependent synthesis of Hsp90 leading to HIF-1α stabilization and radiotherapy resistance of lung cancer cells (64). The mechanism of RACK1/Hsp90 competition in enhancing/decreasing HIF-1α-pVHL binding has already been reviewed (67), but it is interesting to note that, among other, calcium may influence RACK1 activity. For instance, calcium-activated phosphatase calcineurin prevented RACK1 dimerization and subsequent HIF-1α degradation in Hek293 and renal carcinoma RCC4 cells (68). Other studies also report a role for calcium in HIF-1α regulation (69, 70), suggesting that HIF-1α is not only an oxygen and nutrient sensor but may also promote adaptive responses to changes in cellular calcium homeostasis. It is probably due to its pleiotropic function that we find such intricate and multilayered control of HIF-1α, as testified by its numerous posttranslational modifications (1, 71, 72). Recently, SET7/9-mediated methylation of the HIF-1α lysine 32 residue was identified to destabilize HIF-1α, and promote its proteasomal degradation even in hypoxia (73). This reaction is contrasted by LSD1-mediated demethylation, which stabilizes HIF-1α, protecting it from ubiquitination (73). Furthermore, deacetylation of HIF-1α at lysine residue 709 by SIRT2 enhances PHD recognition of hydroxylating residues, promoting pseudonormoxia (74). It is interesting that, apart from proteasomal degradation, the mechanism of lysosomal digestion of HIF-1α has been described (Figure 2D). In particular, HIF-1α was first found to interact and co-localize with lysosome-associated membrane protein type 2A in HK2 human kidney and RCC4 renal cancer cells (75). The authors showed that the lysosomal digestion of HIF-1α is slower and less pronounced than its proteasomal degradation, but suggested it may become more important in circumstances where pVHL pathway is not working. Later, it was demonstrated that lysosomal degradation of HIF-1α is mediated by heat shock cognate 70-kDa protein (HSC70) via chaperone-mediated autophagy, which specifically targets individual proteins (76).
Regulation of HIF-1α on Transcriptional and Translational Level
Besides the regulation of its protein stability and half-life, HIF-1α may also be regulated in a more conventional manner, via mRNA transcription and protein synthesis, in response not only to hypoxia itself but also to the stimulation by growth factors, cytokines and hormones, heat shock, irradiation, and nutrient availability. In this context, three major pro-survival pathways, namely ERK/MAPK, JAK/STAT, and PI3K/Akt/mTOR, concur to increase transcription and translation of HIF1A, especially in cancer (77). MAPK signaling via ERK1/2 was mainly associated with regulation of HIF-1 transactivation through phosphorylation of p300/CPB cofactors. On the other hand, JAK/STAT pathway triggers Akt-mediated HIF1A transcription via STAT3 (78, 79). The PI3K/Akt/mTOR signaling cascade directly increases HIF1A transcription and translation (80–82). Therefore, any aberrant stimulation of this pathway, which in cancer often occurs through growth factors, hormones, or oncogenes/tumor suppressor mutations, leads to the activation of HIF-1α, even in normoxic conditions (83–85). Concordantly, elevated ROS production caused by OXPHOS deficiency (86), and several other conditions leading to elevated ROS and reactive nitrogen species, including mtDNA mutations (87), chemical toxicants (88), intermittent hypoxia (89), and treatment with pro-inflammatory factors (90), have been associated with PI3K/Akt/mTOR-mediated increase of HIF1A transcription and translation (Figure 2E). Moreover, Akt pathway boosts HIF-1α-mediated response by stabilization and transactivation regardless of oxygen levels (91). For example, the ERK-PI3K/Akt mediate HIF-1α levels by stimulating protein synthesis of the molecular chaperone Hsp90, which in turn is able to stabilize HIF-1α in an oxygen-independent fashion (66, 92).
The PI3K/Akt-mediated activation of mTOR is antagonized by the 5′-adenosine monophosphate kinase (AMPK), the major sensor of cellular energy charge (93). In the context of a progressing cancer cell, PI3K/Akt/mTOR promotes survival and proliferation when conditions are fertile for cell proliferation, whereas AMPK serves as a sensor of nutrient starvation and ensures optimization of energetic sources when a cancer cell requires saving energy. Thus, it is intuitive to hypothesize that AMPK would counteract the effects of Akt-mediated increase of HIF-1α signaling. Indeed, an anticorrelation between active AMPK and HIF-1α has been confirmed by a recent system biology analysis (94) and, concordantly, by in vitro studies showing HIF-1α destabilization in hypoxia under glucose deprivation, suggesting that starvation dampens HIF-1α translation (95–97). However, the relationship between AMPK and HIF-1α is still unclear. On one hand, the lack of AMPK in MEFs stimulates HIF-1α expression in normoxia (98, 99), and mTORC1 activation and increased ROS production have been appointed for the normoxic stabilization of HIF-1α in AMPK-defective MEFs (99, 100). On the other hand, it has been reported that oxidative stress may induce AMPK activation leading to a reduction in HIF-1α degradation (101) and active AMPK was shown to stimulate ROS-mediated increase of HIF-1α (102). It seems that the AMPK control of HIF-1α may be dependent on the contexts and phases of tumor progression, concordantly to the recently reviewed double-edged role of this energy sensor (103).
Concluding Remarks
Taken together, studies we discuss here show that, even though PHD-mediated hydroxylation of HIF-1α seems an impeccable mechanism to control its stability, many novel regulators of HIF-1α are emerging, especially in the context of cancer, where the selective pressures to activate this protumorigenic protein are particularly strong. Unraveling the complexity of HIF-1α regulation might lead to development of more precise anticancer treatments. In particular, considering the heterogeneous OXPHOS activity in different cancers, a better understanding of the mechanisms by which HIF-1α and mitochondrial respiratory chain complexes control oxygen sensing, may identify means for optimization of targeting HIF-1α, possibly based on the OXPHOS status of tumors. For example, therapies targeting HIF-1α could be avoided in tumors suffering OXPHOS deficiency, whereas targeting complex I could be adopted as a strategy to block HIF-1α in tumors which rely on the activity of this pleiotropic transcription factor.
Author Contributions
IK designed the work. LI and IK wrote the manuscript. GG and AMP critically revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC) grant TOUch ME—IG 17387 to AMP and by Italian Ministry of Health grant DISCO TRIP GR-2013-02356666 to GG.
References
1. Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol (2014) 49:1–15. doi:10.3109/10409238.2013.838205
2. Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) (2004) 19:176–82. doi:10.1152/physiol.00001.2004
3. Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev (2001) 15:2675–86. doi:10.1101/gad.924501
4. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science (2002) 295:858–61. doi:10.1126/science.1068592
5. Ruas JL, Poellinger L. Hypoxia-dependent activation of HIF into a transcriptional regulator. Semin Cell Dev Biol (2005) 16:514–22. doi:10.1016/j.semcdb.2005.04.001
6. Minet E, Michel G, Remacle J, Michiels C. Role of HIF-1 as a transcription factor involved in embryonic development, cancer progression and apoptosis (review). Int J Mol Med (2000) 5:253–9. doi:10.3892/ijmm.5.3.253
7. Hickey MM, Simon MC. Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr Top Dev Biol (2006) 76:217–57. doi:10.1016/S0070-2153(06)76007-0
8. Schito L, Semenza GL. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer (2016) 2:758–70. doi:10.1016/j.trecan.2016.10.016
9. Morris MR, Maina E, Morgan NV, Gentle D, Astuti D, Moch H, et al. Molecular genetic analysis of FIH-1, FH, and SDHB candidate tumour suppressor genes in renal cell carcinoma. J Clin Pathol (2004) 57:706–11. doi:10.1136/jcp.2003.011767
10. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell (2005) 7:77–85. doi:10.1016/j.ccr.2004.11.022
11. Frezza C, Pollard PJ, Gottlieb E. Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) (2011) 89:213–20. doi:10.1007/s00109-011-0728-4
12. Adam J, Yang M, Soga T, Pollard PJ. Rare insights into cancer biology. Oncogene (2014) 33:2547–56. doi:10.1038/onc.2013.222
13. Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One (2012) 7:e33418. doi:10.1371/journal.pone.0033418
14. Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem (2002) 277:23111–5. doi:10.1074/jbc.M202487200
15. Jung SY, Song HS, Park SY, Chung SH, Kim YJ. Pyruvate promotes tumor angiogenesis through HIF-1-dependent PAI-1 expression. Int J Oncol (2011) 38:571–6. doi:10.3892/ijo.2010.859
16. Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol (2007) 27:912–25. doi:10.1128/MCB.01223-06
17. Tennant DA, Frezza C, MacKenzie ED, Nguyen QD, Zheng L, Selak MA, et al. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene (2009) 28:4009–21. doi:10.1038/onc.2009.250
18. MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, et al. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol (2007) 27:3282–9. doi:10.1128/MCB.01927-06
19. Ho HY, Lin YT, Lin G, Wu PR, Cheng ML. Nicotinamide nucleotide transhydrogenase (NNT) deficiency dysregulates mitochondrial retrograde signaling and impedes proliferation. Redox Biol (2017) 12:916–28. doi:10.1016/j.redox.2017.04.035
20. Iommarini L, Kurelac I, Capristo M, Calvaruso MA, Giorgio V, Bergamini C, et al. Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum Mol Genet (2014) 23:1453–66. doi:10.1093/hmg/ddt533
21. Gasparre G, Kurelac I, Capristo M, Iommarini L, Ghelli A, Ceccarelli C, et al. A mutation threshold distinguishes the antitumorigenic effects of the mitochondrial gene MTND1, an oncojanus function. Cancer Res (2011) 71:6220–9. doi:10.1158/0008-5472.CAN-11-1042
22. Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab (2013) 1:11. doi:10.1186/2049-3002-1-11
23. Zeng L, Morinibu A, Kobayashi M, Zhu Y, Wang X, Goto Y, et al. Aberrant IDH3alpha expression promotes malignant tumor growth by inducing HIF-1-mediated metabolic reprogramming and angiogenesis. Oncogene (2015) 34:4758–66. doi:10.1038/onc.2014.411
24. Puissegur MP, Mazure NM, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ (2011) 18:465–78. doi:10.1038/cdd.2010.119
25. Grosso S, Doyen J, Parks SK, Bertero T, Paye A, Cardinaud B, et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis (2013) 4:e544. doi:10.1038/cddis.2013.71
26. Burr SP, Costa AS, Grice GL, Timms RT, Lobb IT, Freisinger P, et al. Mitochondrial protein lipoylation and the 2-oxoglutarate dehydrogenase complex controls HIF1alpha stability in aerobic conditions. Cell Metab (2016) 24:740–52. doi:10.1016/j.cmet.2016.09.015
27. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature (2012) 483:484–8. doi:10.1038/nature10898
28. Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem (2008) 283:10892–903. doi:10.1074/jbc.M800102200
29. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3:187–97. doi:10.1016/j.cmet.2006.01.012
30. Zhou X, Chen J, Yi G, Deng M, Liu H, Liang M, et al. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget (2016) 7:873–84. doi:10.18632/oncotarget.6418
31. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife (2014) 3:e02242. doi:10.7554/eLife.02242
32. Porcelli AM, Ghelli A, Ceccarelli C, Lang M, Cenacchi G, Capristo M, et al. The genetic and metabolic signature of oncocytic transformation implicates HIF1alpha destabilization. Hum Mol Genet (2010) 19:1019–32. doi:10.1093/hmg/ddp566
33. Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: why choose inefficient mitochondria? Biochim Biophys Acta (2011) 1807:633–42. doi:10.1016/j.bbabio.2010.08.006
34. Ellinghaus P, Heisler I, Unterschemmann K, Haerter M, Beck H, Greschat S, et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med (2013) 2:611–24. doi:10.1002/cam4.112
35. Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science (2003) 302:1975–8. doi:10.1126/science.1088805
36. Hagen T, D’Amico G, Quintero M, Palacios-Callender M, Hollis V, Lam F, et al. Inhibition of mitochondrial respiration by the anticancer agent 2-methoxyestradiol. Biochem Biophys Res Commun (2004) 322:923–9. doi:10.1016/j.bbrc.2004.07.204
37. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A (1998) 95:11715–20. doi:10.1073/pnas.95.20.11715
38. Doege K, Heine S, Jensen I, Jelkmann W, Metzen E. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood (2005) 106:2311–7. doi:10.1182/blood-2005-03-1138
39. Agani FH, Pichiule P, Chavez JC, LaManna JC. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J Biol Chem (2000) 275:35863–7. doi:10.1074/jbc.M005643200
40. Prior S, Kim A, Yoshihara T, Tobita S, Takeuchi T, Higuchi M. Mitochondrial respiratory function induces endogenous hypoxia. PLoS One (2014) 9:e88911. doi:10.1371/journal.pone.0088911
41. Vaux EC, Metzen E, Yeates KM, Ratcliffe PJ. Regulation of hypoxia-inducible factor is preserved in the absence of a functioning mitochondrial respiratory chain. Blood (2001) 98:296–302. doi:10.1182/blood.V98.2.296
42. Srinivas V, Leshchinsky I, Sang N, King MP, Minchenko A, Caro J. Oxygen sensing and HIF-1 activation does not require an active mitochondrial respiratory chain electron-transfer pathway. J Biol Chem (2001) 276:21995–8. doi:10.1074/jbc.C100177200
43. Gong Y, Agani FH. Oligomycin inhibits HIF-1alpha expression in hypoxic tumor cells. Am J Physiol Cell Physiol (2005) 288:C1023–9. doi:10.1152/ajpcell.00443.2004
44. Dotson RJ, Smith CR, Bueche K, Angles G, Pias SC. Influence of cholesterol on the oxygen permeability of membranes: insight from atomistic simulations. Biophys J (2017) 112:2336–47. doi:10.1016/j.bpj.2017.04.046
45. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem (2000) 275:25130–8. doi:10.1074/jbc.M001914200
46. Movafagh S, Crook S, Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem (2015) 116:696–703. doi:10.1002/jcb.25074
47. Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab (2005) 1:409–14. doi:10.1016/j.cmet.2005.05.002
48. Chua YL, Dufour E, Dassa EP, Rustin P, Jacobs HT, Taylor CT, et al. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem (2010) 285:31277–84. doi:10.1074/jbc.M110.158485
49. Pouyssegur J, Mechta-Grigoriou F. Redox regulation of the hypoxia-inducible factor. Biol Chem (2006) 387:1337–46. doi:10.1515/BC.2006.167
50. Galanis A, Pappa A, Giannakakis A, Lanitis E, Dangaj D, Sandaltzopoulos R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett (2008) 266:12–20. doi:10.1016/j.canlet.2008.02.028
51. Bell EL, Chandel NS. Mitochondrial oxygen sensing: regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem (2007) 43:17–27. doi:10.1042/bse0430017
52. Hamanaka RB, Weinberg SE, Reczek CR, Chandel NS. The mitochondrial respiratory chain is required for organismal adaptation to hypoxia. Cell Rep (2016) 15:451–9. doi:10.1016/j.celrep.2016.03.044
53. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell (2015) 162:540–51. doi:10.1016/j.cell.2015.07.016
54. Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res (2000) 60:4010–5.
55. Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res (2007) 67:563–72. doi:10.1158/0008-5472.CAN-06-2701
56. Kim JJ, Lee SB, Jang J, Yi SY, Kim SH, Han SA, et al. WSB1 promotes tumor metastasis by inducing pVHL degradation. Genes Dev (2015) 29:2244–57. doi:10.1101/gad.268128.115
57. Goto Y, Zeng L, Yeom CJ, Zhu Y, Morinibu A, Shinomiya K, et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1alpha. Nat Commun (2015) 6:6153. doi:10.1038/ncomms7153
58. Doe MR, Ascano JM, Kaur M, Cole MD. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res (2012) 72:949–57. doi:10.1158/0008-5472.CAN-11-2371
59. Amelio I, Inoue S, Markert EK, Levine AJ, Knight RA, Mak TW, et al. TAp73 opposes tumor angiogenesis by promoting hypoxia-inducible factor 1alpha degradation. Proc Natl Acad Sci U S A (2015) 112:226–31. doi:10.1073/pnas.1410609111
60. Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev (2000) 14:34–44.
61. Joshi S, Singh AR, Durden DL. MDM2 regulates hypoxic hypoxia-inducible factor 1alpha stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J Biol Chem (2014) 289:22785–97. doi:10.1074/jbc.M114.587493
62. Nieminen AL, Qanungo S, Schneider EA, Jiang BH, Agani FH. Mdm2 and HIF-1alpha interaction in tumor cells during hypoxia. J Cell Physiol (2005) 204:364–9. doi:10.1002/jcp.20406
63. Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell (2007) 25:207–17. doi:10.1016/j.molcel.2007.01.001
64. Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell (2007) 131:584–95. doi:10.1016/j.cell.2007.08.045
65. Baek JH, Liu YV, McDonald KR, Wesley JB, Zhang H, Semenza GL. Spermidine/spermine N(1)-acetyltransferase-1 binds to hypoxia-inducible factor-1alpha (HIF-1alpha) and RACK1 and promotes ubiquitination and degradation of HIF-1alpha. J Biol Chem (2007) 282:33358–66. doi:10.1074/jbc.M705627200
66. Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem (2002) 277:29936–44. doi:10.1074/jbc.M204733200
67. Liu YV, Semenza GL. RACK1 vs. HSP90: competition for HIF-1 alpha degradation vs. stabilization. Cell Cycle (2007) 6:656–9. doi:10.4161/cc.6.6.3981
68. Liu YV, Hubbi ME, Pan F, McDonald KR, Mansharamani M, Cole RN, et al. Calcineurin promotes hypoxia-inducible factor 1alpha expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J Biol Chem (2007) 282:37064–73. doi:10.1074/jbc.M705015200
69. Kim KH, Kim D, Park JY, Jung HJ, Cho YH, Kim HK, et al. NNC 55-0396, a T-type Ca2+ channel inhibitor, inhibits angiogenesis via suppression of hypoxia-inducible factor-1alpha signal transduction. J Mol Med (Berl) (2015) 93:499–509. doi:10.1007/s00109-014-1235-1
70. Chen SJ, Hoffman NE, Shanmughapriya S, Bao L, Keefer K, Conrad K, et al. A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2alpha. J Biol Chem (2014) 289:36284–302. doi:10.1074/jbc.M114.620922
71. Yee Koh M, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci (2008) 33:526–34. doi:10.1016/j.tibs.2008.08.002
72. Lendahl U, Lee KL, Yang H, Poellinger L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat Rev Genet (2009) 10:821–32. doi:10.1038/nrg2665
73. Kim Y, Nam HJ, Lee J, Park DY, Kim C, Yu YS, et al. Methylation-dependent regulation of HIF-1alpha stability restricts retinal and tumour angiogenesis. Nat Commun (2016) 7:10347. doi:10.1038/ncomms10347
74. Seo KS, Park JH, Heo JY, Jing K, Han J, Min KN, et al. SIRT2 regulates tumour hypoxia response by promoting HIF-1alpha hydroxylation. Oncogene (2015) 34:1354–62. doi:10.1038/onc.2014.76
75. Olmos G, Arenas MI, Bienes R, Calzada MJ, Aragones J, Garcia-Bermejo ML, et al. 15-deoxy-delta(12,14)-prostaglandin-J(2) reveals a new pVHL-independent, lysosomal-dependent mechanism of HIF-1alpha degradation. Cell Mol Life Sci (2009) 66:2167–80. doi:10.1007/s00018-009-0039-x
76. Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J Biol Chem (2013) 288:10703–14. doi:10.1074/jbc.M112.414771
77. Kietzmann T, Mennerich D, Dimova EY. Hypoxia-inducible factors (HIFs) and phosphorylation: impact on stability, localization, and transactivity. Front Cell Dev Biol (2016) 4:11. doi:10.3389/fcell.2016.00011
78. Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene (2005) 24:5552–60. doi:10.1038/sj.onc.1208719
79. Niu G, Briggs J, Deng J, Ma Y, Lee H, Kortylewski M, et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol Cancer Res (2008) 6:1099–105. doi:10.1158/1541-7786.MCR-07-2177
80. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest (2013) 123:3664–71. doi:10.1172/JCI67230
81. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science (2016) 352:175–80. doi:10.1126/science.aaf4405
82. Dery MA, Michaud MD, Richard DE. Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. Int J Biochem Cell Biol (2005) 37:535–40. doi:10.1016/j.biocel.2004.08.012
83. Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev (2000) 14:391–6.
84. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res (2000) 60:1541–5.
85. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol (2001) 21:3995–4004. doi:10.1128/MCB.21.12.3995-4004.2001
86. Page EL, Robitaille GA, Pouyssegur J, Richard DE. Induction of hypoxia-inducible factor-1alpha by transcriptional and translational mechanisms. J Biol Chem (2002) 277:48403–9. doi:10.1074/jbc.M209114200
87. Koshikawa N, Hayashi J, Nakagawara A, Takenaga K. Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1alpha gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. J Biol Chem (2009) 284:33185–94. doi:10.1074/jbc.M109.054221
88. Gao N, Ding M, Zheng JZ, Zhang Z, Leonard SS, Liu KJ, et al. Vanadate-induced expression of hypoxia-inducible factor 1 alpha and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J Biol Chem (2002) 277:31963–71. doi:10.1074/jbc.M200082200
89. Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol (2008) 217:674–85. doi:10.1002/jcp.21537
90. Haddad JJ, Saade NE, Safieh-Garabedian B. Redox regulation of TNF-alpha biosynthesis: augmentation by irreversible inhibition of gamma-glutamylcysteine synthetase and the involvement of an IkappaB-alpha/NF-kappaB-independent pathway in alveolar epithelial cells. Cell Signal (2002) 14:211–8. doi:10.1016/S0898-6568(01)00233-9
91. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol (2002) 22:7004–14. doi:10.1128/MCB.22.20.7004-7014.2002
92. Kim WY, Oh SH, Woo JK, Hong WK, Lee HY. Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1alpha. Cancer Res (2009) 69:1624–32. doi:10.1158/0008-5472.CAN-08-0505
93. Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans (2011) 39:1–13. doi:10.1042/BST0390001
94. Yu L, Lu M, Jia D, Ma J, Ben-Jacob E, Levine H, et al. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res (2017) 77:1564–74. doi:10.1158/0008-5472.CAN-16-2074
95. Vordermark D, Kraft P, Katzer A, Bolling T, Willner J, Flentje M. Glucose requirement for hypoxic accumulation of hypoxia-inducible factor-1alpha (HIF-1alpha). Cancer Lett (2005) 230:122–33. doi:10.1016/j.canlet.2004.12.040
96. Osada-Oka M, Hashiba Y, Akiba S, Imaoka S, Sato T. Glucose is necessary for stabilization of hypoxia-inducible factor-1alpha under hypoxia: contribution of the pentose phosphate pathway to this stabilization. FEBS Lett (2010) 584:3073–9. doi:10.1016/j.febslet.2010.05.046
97. Karuppagounder SS, Basso M, Sleiman SF, Ma TC, Speer RE, Smirnova NA, et al. In vitro ischemia suppresses hypoxic induction of hypoxia-inducible factor-1alpha by inhibition of synthesis and not enhanced degradation. J Neurosci Res (2013) 91:1066–75. doi:10.1002/jnr.23204
98. Shackelford DB, Vasquez DS, Corbeil J, Wu S, Leblanc M, Wu CL, et al. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci U S A (2009) 106:11137–42. doi:10.1073/pnas.0900465106
99. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab (2013) 17:113–24. doi:10.1016/j.cmet.2012.12.001
100. Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep (2017) 21:1–9. doi:10.1016/j.celrep.2017.09.026
101. Jung SN, Yang WK, Kim J, Kim HS, Kim EJ, Yun H, et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis (2008) 29:713–21. doi:10.1093/carcin/bgn032
102. Yan M, Gingras MC, Dunlop EA, Nouet Y, Dupuy F, Jalali Z, et al. The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J Clin Invest (2014) 124:2640–50. doi:10.1172/JCI71749
Keywords: hypoxia-inducible factor 1 alpha, cancer, mitochondria, oxidative phosphorylation, electron transport chain, prolyl hydroxylases, pseudohypoxia, pseudonormoxia
Citation: Iommarini L, Porcelli AM, Gasparre G and Kurelac I (2017) Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 7:286. doi: 10.3389/fonc.2017.00286
Received: 18 October 2017; Accepted: 13 November 2017;
Published: 27 November 2017
Edited by:
Sergio Giannattasio, Istituto di Biomembrane, Bioenergetica e Biotecnologie Molecolari (IBIOM), ItalyReviewed by:
Jacques Pouyssegur, Université Côte d’Azur, FranceMichael Breitenbach, University of Salzburg, Austria
Copyright: © 2017 Iommarini, Porcelli, Gasparre and Kurelac. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivana Kurelac, aXZhbmEua3VyZWxhYyYjeDAwMDQwO3VuaWJvLml0