 Ivan Zanoni
Ivan Zanoni Francesca Granucci
Francesca Granucci Achille Broggi
Achille Broggi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 November 2017
Sec. Molecular Innate Immunity
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01661
This article is part of the Research Topic Interferon-λs: New Regulators of Inflammatory Processes View all 11 articles
Type III interferons (IFNs) (or IFN-λ) are the latest addition to the IFN family. Even though they share little protein homology with type I IFN, both exhibit remarkable functional similarities: each can be induced in response to viral infections, and both lead to Janus kinases (JAK) and signal transducer and activator of transcription (STAT) activation. The JAK/STAT pathway induces antiviral responses and IFN-stimulated gene transcription. However, despite the similarities in their effector functions with type I IFNs, IFN-λ also has a non-redundant role in protecting barrier organs: epithelial cells preferentially produce IFN-λ rather than type I IFNs; and interferon lambda receptor 1 (IFNLR1), the specific receptor for IFN-λ, is highly expressed on cells of epithelial lineage. Thus far, IFN-λ has been considered mainly as an epithelial cytokine, which restricts viral replication in epithelial cells and constitutes an added layer of protection at mucosal sites. However, it is now increasingly recognized that IFNLR1 is expressed broadly, and that immune cells such as neutrophils and dendritic cells also respond to IFN-λ. Moreover, in many in vivo models, IFN-λ modulates immune cell functions and thereby configures itself less as a cytokine that is only specific to the epithelium, and more as a cytokine that directly controls the inflammatory response at mucosal sites. Here, we critically review the recent literature on immune modulatory roles for IFN-λ, and distinguish between the direct and indirect effects of this IFN on immune cell functions in different inflammatory settings.
First described more than 60 years ago (1) interferons (IFNs) were the first family of cytokines to be discovered. Since then, IFNs have been extensively studied, and their presence is correlated with a number of immunological and biological processes, such as cell proliferation, regulation of cell survival, and modulation of immune functions. IFNs can be divided into three major subfamilies: type I IFNs (comprising mainly IFN-β and over 20 subtypes of IFN-α, -ε, and -ω), type II IFNs (IFN-γ), and the recently identified type III IFNs (IFN-λ) (2, 3) that comprise four members in human (IFN-λ1/IL-29, IFN-λ2, IFN-λ3/IL-28A-B, and IFN-λ4) and two in mice (IFN-λ2/IL-28A and IFN-λ3/IL-28B, while IFN-λ1 is a pseudogene interrupted by a stop codon). IFN-λ2 and IFN-λ3 are highly related and have 96% sequence identity, while IFN-λ1 shares 81% sequence identity with IFN-λ2 and IFN-λ3 (4).
The gene and protein structure of IFN-λ2 and -λ3 share little homology to those of type I IFNs (15%) (4); but they exert remarkably overlapping functions. The heterodimeric receptor for IFN-λ is named IFNLR (or IL-28R), and comprises the specific subunit interferon lambda receptor 1 (IFNLR1, also known as IL-28R1) plus the IL-10R2 subunit that is common to many type II cytokines (such as IL-10, IL-22, IL-24, and IL-26). Once IFNLR is engaged, IFN-λ activate an antiviral response that is very similar to the one triggered by type I IFNs (5). In fact, both engage a similar JAK–STAT pathway, with the only difference that IFN-λ can also use the adaptor JAK2 (6). Both cytokine families also induce IFN-stimulated gene (ISG) transcription, and both confer protection against viral infections (5). This overlap in functions raises the question of why two distinct but similar IFN systems have been maintained throughout evolution, considering that these two systems separated as far back in evolution as did amphibians, reptiles, and birds (7).
The main distinction between the two IFN systems has to do with the tropism between expression of the cytokine and its specific receptors. Myeloid cells at mucosal sites express both type I IFNs and IFN-λ in response to viral as well as bacterial ligands (8–11). However, type I IFN and IFN-λ production are regulated differently. Stimulation of plasma membrane toll-like receptor (TLR) (such as TLR2 and TLR5), both in myeloid and epithelial cells, selectively induces IFN-λ, and not type I IFN, mRNA expression. Moreover, activation of TLR5 has recently been proved to be essential for the induction of IFN-λ upon Salmonella encounter (9). Also, cells of epithelial lineage, both in the gut (12) and in the liver (13), preferentially produce IFN-λ over type I IFNs in response to viral ligands. In particular, while both IFNs are induced downstream of pattern recognition receptor and mitochondrial antiviral signaling protein (MAVS), the production of IFN-λ is favored subsequent to activation of the MAVS that reside in peroxisomes (6, 14). The abundance of peroxisomes in cells of epithelial lineage could explain the tropism of IFN-λ production (13).
Other than the tropism of IFN-λ production, the selective expression of the receptor governs the tropism of IFN-λ response. The receptor for type I IFNs (which comprises receptor subunits IFNAR1 and IFNAR2) is expressed in virtually every cell type, while expression of the IFNLR1 receptor is much more specific, and is believed to be most abundant in cells of epithelial origin that are present at barrier surfaces (15). This pattern of expression, along with the recently documented non-redundant role of IFN-λ in protecting against virus infection at mucosal sites [e.g., at the intestinal barrier (12, 16–18) and in the lung (19)], suggest a model in which IFN-λ represents an epithelial cytokine that protect mucosal surfaces without activating widespread and possibly nocuous immune responses, while type I IFNs represent a more general and potent system that is activated once the mucosal barrier is broken. However, recent findings challenge the view that IFN-λ is primarily an epithelial cytokine, describe IFN-λ’s ability to directly and indirectly modulate immune cell functions and document the expression of IFNLR1 on immune cells; they also document that among immune cells, neutrophils express IFNLR1 and directly respond to IFN-λ, in the setting of viral infections (19) as well as other forms of acute inflammation (20–22). IFN-λ reportedly also interferes with the function of NK cells (23, 24), and favors the skewing of T cell activation toward type I (rather than type II) responses, by modulating DC functions (25). While the study of immunomodulatory effects of IFN-λ is still in its infancy—in part due to a lack of specific tools such as good antibodies against IFNLR1—a new role for IFN-λ in shaping the mucosal immune response is emerging. In this review, we critically examine recent literature on the role of IFN-λ in immune cells, differentiating between a direct IFN-λ effect on specific cell types and possible indirect phenomena; we also evaluate what is known about how IFN-λ participates in the control of mucosal immune responses.
Neutrophils are the first line of defense of the immune system: following pathogen invasion or tissue injury, these cells are quickly and massively recruited to barrier sites, where they protect the host by killing invading pathogens via a very rapid release of toxic mediators, independent of de novo protein synthesis (26). At later stages, neutrophils regulate the inflammatory response, either passively by undergoing apoptosis and turning off their toxic potential, or actively by secreting anti-inflammatory cytokines and lipidic mediators (27). The ability of these cells to potently kill bacteria is also accompanied by the necessary evil of tissue damage, since many of the toxic mediator released, such as reactive oxygen species (ROS) and proteases, are unable to discriminate between host and pathogen cells. Given the tropism of IFN-λ production to mucosal sites and the complex crosstalk between epithelial cells and neutrophils at mucosal surfaces (28, 29), it is remarkable that among murine immune cells, neutrophils express IFNLR1 at the highest level (19–21). Murine neutrophils express IFNLR1 at very high levels (19–21, 30) that are comparable to those in colonic epithelial cells (20) and in epithelial cells from the lung (19). Human neutrophils have also been found to express IFNLR1 at higher levels as compared to lymphocytes (30) and upregulate its expression following treatment with pro-inflammatory agents such as LPS (20), or after encounter with Aspergillus fumigatus (30). In addition to the high levels of receptor expression, mouse and human neutrophils also respond to IFN-λ stimulation (19–21, 30), and activate the canonical JAK–STAT pathway, that leads to phosphorylation of STAT1, STAT2, and STAT3 (21, 30) and induces upregulation of ISGs at levels similar to those induced by type I IFNs (19, 20). Surprisingly, in addition to the canonical ISG response induced downstream of the JAK–STAT pathway, IFN-λ also down-modulates tissue-damaging, transcription-independent responses such as production of ROS, granule mobilization (20), release of neutrophil extracellular traps (NETs) (22), and cellular migration (21); while cytokine production in response to inflammatory stimuli, phagocytosis, and apoptosis is not affected by IFN-λ (20).
Irina Udalova and colleagues were the first to report that neutrophils respond to IFN-λ (21), and that treatment of neutrophils with IFN-λ in vitro leads to activation of the JAK–STAT pathway and STAT1 phosphorylation; they also first described the ability of IFN-λ to regulate pro-inflammatory neutrophil functions. In arthritic mice treated with recombinant IFN-λ, they observed a defect in neutrophil migration to the inflamed joint; this defect was attributed to the capacity of IFN-λ to directly inhibit neutrophil migration. Also in an air pouch model of acute inflammation, and when neutrophil migration toward leukotrien B4 was assessed in vitro, the cells exhibited a defect in migration: fewer neutrophils were recovered in the air pouch in the presence of IFN-λ, and a shorter Euclidean distance was traveled by neutrophils treated with IFN-λ in vitro (21).
More recently, we showed that IFN-λs (but not type I IFNs) are able to regulate a non-translational signaling pathway that diminishes ROS production by neutrophils as well as degranulation following activation of the cells with pro-inflammatory stimuli, but that it does not alter cytokine production induced by inflammatory stimuli or phagocytosis (20). We additionally demonstrated that IFN-λ inhibits degranulation and decreases ROS production even when de novo protein synthesis is inhibited with cycloheximide, or when STAT1 or STAT3 are genetically ablated or pharmacologically inhibited. Inhibition of all JAK kinases, or specific inhibition of JAK2, which is involved only in IFN-λ signaling (and not in type I IFN responses) (6, 14) impairs the ability of IFN-λ to inhibit ROS production and degranulation (20). Neutrophils treated with IFN-λ are nevertheless able to phagocytose both opsonized and non-opsonized E. coli, and to produce cytokines in response to LPS. Human neutrophils appear to have similar regulating mechanisms: treatment with IFN-λ reduces the ability of these cells to produce ROS (20), and also impairs their ability to generate NETs in an in vitro model of thromboinflammation, wherein neutrophils are incubated with activated platelets in the presence of IFN-λ (22). IFN-λ treatment also inhibits NET generation in response to platelet-derived inorganic polyphosphate (polyP) and interferes with the ability of polyP to inhibit mTOR activation and induce the autophagy marker LC3, which is a requisite for NET release (31). IFN-λ, thus, profoundly influences neutrophil non-transcriptional functions and engages a pathway that is independent of the canonical JAK–STAT pathway and does not rely on de novo protein synthesis. In contrast to the transcriptional responses, these characteristics are not shared with type I IFNs and seem to specifically target the potent cytotoxic responses that can threaten mucosal integrity.
As previously described for epithelial cells (2, 3, 5, 32), IFN-λ induces a transcriptional response remarkably similar to that of type I IFNs. So far, no genes have been identified that are selectively upregulated by IFN-λ (and not by type I IFNs), and the upregulation of antiviral ISGs is largely overlapping; however, IFN-λ (as opposed to IFN-α) is unable to directly induce upregulation of pro-inflammatory cytokines, such as TNF, IL-1β, and IL-6, or chemokines, such as CCL2 and CXCL1. The influence of IFN-λ on neutrophils appears, thus, to be anti-inflammatory. Indeed, IFN-λ is able to down-modulate nocuous neutrophil functions—such as the production of toxic mediators or the production of NETs—without interfering with the capacity of these cells to engulf pathogens, or to orchestrate the inflammatory response via cytokine secretion (Figure 1). The importance of such regulation of neutrophil functions has been documented in vivo following viral infections and also in inflammatory pathologies. In fact, when IFNLR1 is depleted specifically in neutrophils, mice are more susceptible to a sublethal dose of influenza virus infection and present a higher viral load, higher number of leukocytes in the BAL, and higher levels of expression of inflammatory cytokines (19). Notably, when low doses of virus are used for infection, IFNLR expression is required both in epithelial cells and in neutrophils to confer maximum protection. In fact, mice with a conditional ablation of IFNLR1 in pulmonary epithelial cells or in neutrophils only partially recapitulate the total knock-out phenotype (19).
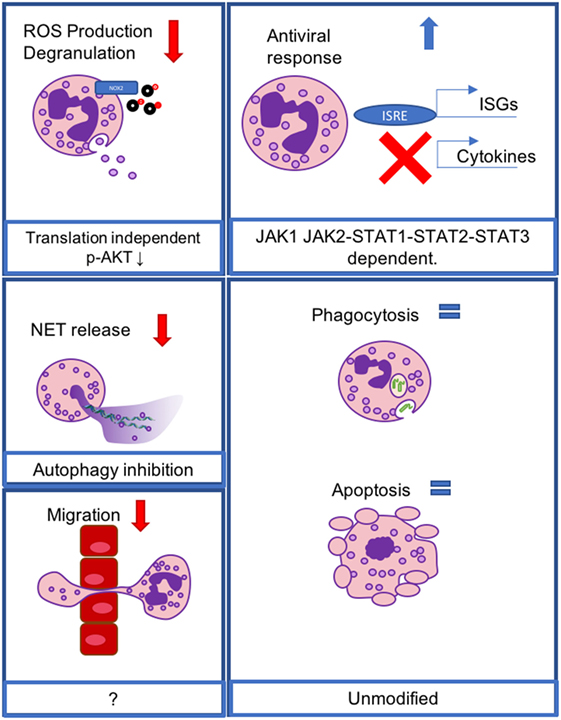
Figure 1. IFN-λ modulates neutrophil functions at the transcriptional and non-transcriptional levels. Reactive oxygen species production and degranulation are regulated at a non-translational level, involving AKT inhibition (upper left), neutrophil extracellular trap release is inhibited via inhibition of autophagy (middle left), and neutrophil migration is inhibited via an unknown mechanism (lower left). Transcriptional antiviral responses lead to the induction of IFN-stimulated genes, but do not mediate cytokine production, and act through a JAK1- and JAK2-dependent, STAT1, -2, -3-dependent mechanism (upper right). Phagocytosis and apoptosis are not affected (lower right).
Interferon-λ also influences neutrophil functions during acute inflammation in the gut mucosa. We and others have described a protective role for IFN-λ in a mouse model of DSS-induced colitis (20, 33, 34). In fact, IFNLR1−/− mice are more susceptible to the induction of colitis than are wild-type mice and present a more severe disease phenotype, which is characterized by shorter colons, greater weight loss, more severe histological damage, and augmented oxidative stress (20). This effect is entirely dependent on the action of IFN-λ on immune cells, because chimeras in which only radio-resistant cells are IFNLR1−/−, and mice that harbor a deletion of IFNLR1 specific to epithelial cells are equally sensitive to DSS administration as are their wild-type counterparts (20).
By contrast, bone marrow chimeras in which IFNLR1 is depleted only in cells of hematopoietic origin, and mice with conditional depletion of IFNLR1 expression restricted to neutrophils, recapitulate the aggravated phenotype of IFNLR1−/− mice. Notably, both chimeras deleted in the hematopoietic compartment, neutrophils specific IFNLR1−/− mice and total IFNLR1−/− mice have a more severe oxidative stress transcriptional signature in the colon epithelium, when compared to their wild-type counterparts. These data strongly suggest that the control exerted by IFN-λ on neutrophil ROS production is pivotal to protect the intestinal mucosa during acute inflammation (20). In the absence of an active viral or bacterial infection, the source of tonic IFN-λ signaling is represented by the commensal virome. In fact, while depletion of intestinal viruses aggravates colitis in wild-type mice (20, 35) as well as in mice that are deficient in type I IFN signaling (20), IFNLR1−/− mice phenocopy WT mice that are depleted of intestinal viruses in that they are insensitive to treatment with antiviral drugs. In particular, alteration of the intestinal virome in humans that are similar to the alteration obtained in mice treated with antiviral drugs is associated with ulcerative colitis and Crohn’s disease (36, 37).
It was recently shown that IFN-λ action on neutrophils can also protect the host during fungal infections (30). In a model of invasive aspergillosis, both IFNLR1−/− and mice bearing neutrophil-specific depletion of IFNLR1 succumb faster after pulmonary infection with A. fumigatus and present an aggravated disease, with higher CFUs recovered from the lungs and more severe invasion as measured by histology. Curiously, neutrophils deficient for IFNLR1 had reduced intracellular ROS levels when stained ex vivo. This phenotype was recapitulated in neutrophils deficient for STAT1 suggesting that, during fungal infections, IFN-λ-dependent STAT1 activation mediates a transcriptional program that protects the host. While early, translation independent, regulation of neutrophil function by IFN-λ suppresses ROS production and degranulation in response to inflammatory stimuli, during fungal infections, STAT1-dependent action is critical for the activation of neutrophil functions in vivo. The apparent contrast between the two mechanisms can be explained by the differential regulation of neutrophil biology in response to different stimuli. Moreover, while immediate responses, such as ROS production and degranulation, are not typically transcriptionally regulated, the optimal expression of NADPH enzymes during neutrophil development could contribute to the protective effect of IFN-λ against fungi. Indeed, in our hands, when neutrophils were stimulated in vitro with C. albicans hyphae, ROS were produced both in the absence and in the presence of recombinant IFN-λ (our unpublished data). Altogether, these data suggest that IFNLR1-stimulation is not necessary to induce ROS production by neutrophils upon fungal encounter in vitro but that, in vivo, IFN-λ can contribute to prime neutrophils during a stage of differentiation that could not be recapitulated in vitro.
Finally, the inhibitory activity of IFN-λ on neutrophils can also be exploited therapeutically: in fact, IFN-λ administration is protective in pulmonary infections with influenza virus (19), during DSS colitis (20) and in an inflammatory setting such as rheumatoid arthritis (21) or a mouse model of vascular injury (22), where IFN-λ is not produced naturally.
Conventional mouse DCs and human plasmacytoid DCs (pDCs) express low levels of IFNLR1 yet respond to IFN-λ stimulation. In mice, DCs that are derived from the lung express low levels of IFNLR1 (25). Despite these low levels of expression, the central role of DCs at the crossroads between adaptive and innate immunity makes their responses to IFN-λ highly significant. Koltsida and colleagues report that DCs stimulated with IFN-λ, despite responding poorly in terms of ISG induction, are nonetheless able to upregulate T-bet and produce higher levels of IL-12 following LPS stimulation. In the same conditions, they also fail to upregulate OX40L and assume a Th1-polarizing phenotype (25). Indeed, when DCs sorted from the lungs of mice infected with a replication-defective adenovirus expressing IFN-λ under the CMV promoter—or from mice that are treated with recombinant IFN-λ—are used to stimulate T cell polarization in vitro, they favor Th1 skewing. This ability of IFN-λ to induce the skewing of T cell responses is particularly relevant in a model of allergic airway disease (25). In fact, IFNLR1−/− mice present a more severe disease phenotype, with elevated production of type II cytokines, a higher histopathological score, and increased eosinophilic infiltration in the BAL. Moreover, when IFN-λ is administered—either directly or via an IFN-λ-producing adenovirus—mice are protected from allergic airway disease (25). Also, adoptive transfer of DCs purified from mice treated with IFN-λ-producing adenovirus confers protection. Early reports also suggest that when DCs are stimulated with IFN-λ, they acquire a regulatory phenotype and promote FOXP3+ Treg proliferation (38), and that T cell responses can, thus, be skewed toward a Th1 phenotype in vitro (39). These data strongly support a role of IFN-λ-stimulated DCs in skewing T cell responses in vivo, and underscore the need to further investigate how IFN-λ affects DCs (25).
As mentioned above and recently reviewed (40), human pDCs serve an important role in IFN-λ biology. Human pDCs express IFNLR1 and are able to produce as well as respond to IFN-λ (40–42). When stimulated with IFN-λ, they induce the canonical JAK–STAT pathway (43, 44) and upregulate low levels of ISG transcription (43–45). IFN-λ also influences pDC-specific functions: in particular, it can stimulate pDCs to produce type I IFNs and induce the expression of low levels of TNF (44). Moreover, IFN-λ acts synergistically with IL-3 to hyperactivate pDCs and induce higher levels of inflammatory cytokines (45). Treatment of pDCs with IFN-λ also influences the activation status of pDCs, inducing an upregulation of CD80 and CD86. The functional significance of these regulations remains to be determined: while some researchers claim that IFN-λ inhibits the ability of pDCs to activate T cells (42), the enhancement of pDC activation suggests that IFN-λ stimulates pDCs and enhances their capacity to combat viral infections.
While the ability of IFN-λ to influence the activity of DCs is intriguing and could have a substantial effect on how DCs govern innate and adaptive responses, more work is needed to clarify the specific response of DCs to IFN-λ. The discovery of new non-transcriptional pathways induced by IFN-λ should elucidate whether non-transcriptional responses are active in DCs and help reveal additional specific effects of IFN-λ on DCs. But while scattered reports in the literature link IFN-λ to the skewing of T cells toward a Th1 phenotype (46), the expression of IFNLR in T cells and the responsivity of T cells to IFN-λ has not been formally established; this suggests that the influence of this IFN on T cell functions in vivo represents indirect effects that require activation of DCs.
Emerging evidence documents that IFN-λ affects NK cell activity in vivo (23, 24). NK cells are believed to be essential for IFN-λ-mediated protection against influenza virus (24), against tumor growth (23), and in a model of LPS-induced or cecal-ligation puncture (CLP)-induced septic shock (23). However, whether IFN-λ can act directly on NK cells is debated (47–49). Smyth and colleagues (50) report low levels of IFNLR1 expression on mouse NK cells, and to date, there is no evidence of a direct response of NK cells to IFN-λ; in fact, treatment of NK cells with IFN-λ does not activate STAT1 phosphorylation, nor does activate ISG expression (23). However, despite the lack of receptor expression on NK cells and the lack of responsiveness of these cells to IFN-λ in vitro, a model of acute endotoxemia shows that NK cells derived from IFNLR1−/− spleens have defective IFN-γ production, and IFNLR1−/− mice are partially protected from lethal doses of LPS or in a CLP model of sepsis, in a IFN-γ-dependent manner. Together, these observations point to an indirect effect of IFN-λ on NK cells. While NK cells transferred from INFLR1−/− mice into Rag−/− γc−/− mice are also defective in the production of IFN-γ after LPS treatment (23), this does not exclude the possibility that IFNLR1−/− NK cells have defects in differentiation/development. Observations on a recent model of influenza virus infection support this notion: administration of IFN-λ (by continuous overexpression via hydrodynamic gene delivery) protected mice from the viral infection, and influenced NK cell differentiation; indeed, NK cells in these mice exhibited a more mature phenotype and proliferated at a higher rate. However, these authors also claimed that NK cells express extremely low levels of IFNLR1, and they attributed the observed phenotype to the expression of IFNLR1 on myeloid cells. Notably, depletion of phagocytes by administering clodronate liposomes abolishes the protective effect of IFN-λ (24).
While the above findings unequivocally establish that NK cell functions are modified by IFN-λ in vivo, they also strongly suggest that NK cells can be instructed by other cell types that directly respond to IFN-λ stimulation. DCs and neutrophils—the two cell types that do express IFNLR1 and respond to IFN-λ—can influence NK cell functionality in vivo. In fact, DCs activate NK cells by secreting cytokines, such as IL-2, IL-18, and IL-12; and DCs also present IL-15 to NK cells in an IFN-β-dependent manner (51–56). It will be important to test in the future the hypothesis that, similarly to type I IFNs (19), IFN-λ could also directly induce low levels of IL-15 that are presented to NK cells. In the same model of airway allergic inflammation that revealed IFN-λs ability to influence DC-mediated skewing of the immune response, it was shown that NK cells preferentially produced IFN-γ and that they were protective against airway inflammation (57). While a direct activity of IFN-λ on NK cells for the observed protection cannot be excluded, the striking similarity of the two models implicates DCs in both skewing NK cell activation and inducing IFN-γ production.
Neutrophils also profoundly influence the functions of NK cells. Consistent with the model in which IFN-λ regulates NK cell maturation, defects in NK cell terminal differentiation and survival were observed in congenitally neutropenic mice and in mice depleted of neutrophils, as well as in patients with neutropenia (57). Also, ROS produced by human neutrophils inhibit NK cell functions in vitro (58). The ability of IFN-λ to suppress ROS production and to counteract this inhibition feedback can potentially explain the increased activation of NK cells in the presence of IFN-λs. Some early in vivo studies support the hypothesis of a crosstalk between neutrophils and NK cells that governs the antitumoral activity of IFN-λ. In fact, when IFN-λ is administered via retroviral transduction into a mouse fibrosarcoma cell line, it is effective in controlling tumor growth, but this protective effect is lost when either NK cells or neutrophils are depleted (59). While IFN-λ undeniably influences NK cell functions in vivo, the phenotypes observed might be ascribed to unexplored modulation of NK cell functions by neutrophils or DCs. However, the emergence of non-transcriptional roles for IFN-λ on neutrophils opens up the possibility that similar overlooked non-transcriptional pathways are active in NK cells.
Reports of other cell types expressing IFNLR1 and responding to IFN-λ stimulation exist in the literature. In particular, human B cells have been shown to express IFNLR1 (43, 60, 61) and respond to IFN-λ by upregulating ISGs (61). While the functional role of IFN-λ in B cells is still open for investigations, early pieces of evidence suggest that, similar to type I IFNs, IFN-λ augments TLR-mediated activation of B cells.
Scattered reports describing a role of IFN-λ in human macrophage activation also exist. In particular, IFN-λ can protect human monocyte-derived macrophages from HIV infection (62, 63) and treatment of human monocyte-derived macrophages with IFN-λ augments the production of pro-inflammatory cytokines following stimulation with LPS or R848 (64).
Historically, IFN-λ has been recognized as an epithelium-specific cytokine that affects antiviral responses in epithelial cells; however, a growing body of literature supports a critical role for these IFNs in influencing the modulation of immune responses. The action of IFN-λ on immune cells is now configured in a model wherein this cytokine represents the first line of defense of mucosal surfaces. In fact, IFN-λ has non-redundant functions in conditions such as low viral loads (19), or when the epithelial layer is preferentially affected (12): under these conditions, IFN-λ acts directly on epithelial cells to exert local antiviral activity and on DCs to skew the T cell response toward an antiviral Th1 response; IFN-λ also acts directly or indirectly on NK cells to potentiate their activation and protect against viruses. At the same time, IFN-λ also serves important functions in neutrophils, inhibiting tissue-damaging events, such as ROS production, degranulation, and NET formation, without impairing cytokine production or pathogen engulfment. Indeed, IFN-λ activity on neutrophils does not impair, but enhances, responses to pathogenic fungi (30). This modulation of neutrophil activities is pivotal for protecting the mucosae from excessive damage and for maintaining the integrity and barrier functions of epithelia at mucosal sites. IFN-λ is, thus, deemed to be a mucosal cytokine whose evolutionary role is to precede activation of type I IFN, eliminate invading pathogens at mucosal sites without compromising their barrier functions, and limit dissemination of the pathogen (Figure 2). If the pathogen spreads and reaches the underlying tissues, a more potent inflammatory response orchestrated by type I IFNs is needed, but comes at the cost of extensive tissue damage. Such protective activity is also relevant in the absence of a viral infection: tonic IFN-λ production induced by commensal viruses protects the colon mucosa during experimental colitis by dampening neutrophil responses, and administration of IFN-λ is protective in a number of inflammatory settings such as allergic airway diseases, or arthritis. Such evidence of immunomodulatory roles for IFN-λ in vivo highlights that these cytokines have additional, as yet unexplored roles in the stimulation of immune cells.
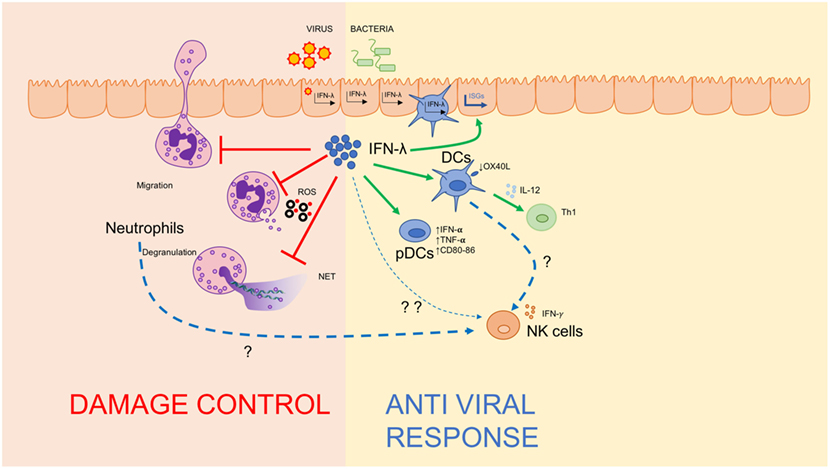
Figure 2. IFN-λ regulates the mucosal inflammatory process. Schematic depiction of IFN-λ’s ability to regulate immunity at mucosal sites by amplifying the antiviral response via directly stimulating dendritic cells and plasmacytoid DCs (right), and dampening damage-inducing neutrophil functions to maintain mucosal integrity (left).
However, support for a direct role for IFN-λ in the modulation of immune functions is fragmented. This is in part due to the lack of biological tools such as specific antibodies against IFNLR1 and the existence of a splicing variant of IFNLR1 in humans that gives rise to a secreted protein with decoy functions (65), which further complicate the correlation of IFNLR1 expression and IFN-λ responsiveness. The translation of findings based on mouse models to human biology is further complicated by the apparent different pattern of expression of the IFNLR1. Indeed, while pDCs and B cells express IFNLR1 and respond to IFN-λ stimulation in humans, the same cell types are not responsive to IFN-λ in mice. Also, while both murine and human neutrophil express the IFNLR1, it is still a matter of discussion if and how inflammatory stimuli and differentiation status of these cells can influence IFNLR1 expression. Despite these confounds, recent reports have uncovered the immune-modulating properties of IFN-λ, as well as new specific non-translational pathways that further differentiate its action from that of type I IFNs. These new insights will pave the way toward an in-depth understanding of the physiological role of these cytokines and will help in exploring the unappreciated functions of IFN-λ in the context of immune cells.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
IZ is supported by NIH grant 1R01AI121066-01A1, 1R01DK115217, P30 DK034854, CCFA Senior Research Awards (412708), and the Cariplo Foundation. FG is supported by Associazione Italiana per la Ricerca sul Cancro, Cariplo Foundation, ARISLA, and Fondazione Regionale per la Ricerca Biomedica (FRRB).
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci (1957) 147:258–67. doi:10.1098/rspb.1957.0049
2. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4:69–77. doi:10.1038/ni875
3. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol (2003) 4:63–8. doi:10.1038/ni873
4. Fox BA, Sheppard PO, O’Hara PJ. The role of genomic data in the discovery, annotation and evolutionary interpretation of the interferon-lambda family. PLoS One (2009) 4:e4933. doi:10.1371/journal.pone.0004933
6. Odendall C, Kagan JC. The unique regulation and functions of type III interferons in antiviral immunity. Curr Opin Virol (2015) 12:47–52. doi:10.1016/j.coviro.2015.02.003
7. Kotenko SV, Durbin JE. Contribution of type III interferons to antiviral immunity: location, location, location. J Biol Chem (2017) 292:7295–303. doi:10.1074/jbc.R117.777102
8. Lazear HM, Nice TJ, Diamond MS. Interferon-lambda: immune functions at barrier surfaces and beyond. Immunity (2015) 43:15–28. doi:10.1016/j.immuni.2015.07.001
9. Odendall C, Voak AA, Kagan JC. Type III IFNs are commonly induced by bacteria-sensing TLRs and reinforce epithelial barriers during infection. J Immunol (2017) 199:3270–9. doi:10.4049/jimmunol.1700250
10. Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, et al. Viral infection and toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol (2004) 34:796–805. doi:10.1002/eji.200324610
11. Bierne H, Travier L, Mahlakõiv T, Tailleux L, Subtil A, Lebreton A, et al. Activation of type III interferon genes by pathogenic bacteria in infected epithelial cells and mouse placenta. PLoS One (2012) 7:e39080. doi:10.1371/journal.pone.0039080
12. Mahlakõiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog (2015) 11:e1004782. doi:10.1371/journal.ppat.1004782
13. Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity (2015) 42:123–32. doi:10.1016/j.immuni.2014.12.016
14. Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol (2014) 15:717–26. doi:10.1038/ni.2915
15. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog (2008) 4:e1000017. doi:10.1371/journal.ppat.1000017
16. Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, et al. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of IFN-lambda against norovirus and reovirus. J Virol (2017) 13:91(7). doi:10.1128/JVI.02079-16
17. Lin J-D, Feng N, Sen A, Balan M, Tseng H-C, McElrath C, et al. Distinct roles of type I and type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog (2016) 12:e1005600. doi:10.1371/journal.ppat.1005600
18. Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, et al. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science (2015) 347:269–73. doi:10.1126/science.1258100
19. Galani IE, Triantafyllia V, Eleminiadou E-E, Koltsida O, Stavropoulos A, Manioudaki M, et al. Interferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity (2017) 46:875–90.e6. doi:10.1016/j.immuni.2017.04.025
20. Broggi A, Tan Y, Granucci F, Zanoni I. IFN-λ suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol (2017) 18:1084–93. doi:10.1038/ni.3821
21. Blazek K, Eames HL, Weiss M, Byrne AJ, Perocheau D, Pease JE, et al. IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J Exp Med (2015) 212:845–53. doi:10.1084/jem.20140995
22. Chrysanthopoulou A, Kambas K, Stakos D, Mitroulis I, Mitsios A, Vidali V, et al. Interferon lambda1/IL-29 and inorganic polyphosphate are novel regulators of neutrophil-driven thromboinflammation. J Pathol (2017) 243:111–22. doi:10.1002/path.4935
23. Lasfar A, de laTorre A, Abushahba W, Cohen-Solal KA, Castaneda I, Yuan Y, et al. Concerted action of IFN-α and IFN-λ induces local NK cell immunity and halts cancer growth. Oncotarget (2016) 7:49259–67. doi:10.18632/oncotarget.10272
24. Wang Y, Li T, Chen Y, Wei H, Sun R, Tian Z. Involvement of NK cells in IL-28B-mediated immunity against influenza virus infection. J Immunol (2017) 199:1012–20. doi:10.4049/jimmunol.1601430
25. Koltsida O, Hausding M, Stavropoulos A, Koch S, Tzelepis G, Ubel C, et al. IL-28A (IFN-λ2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol Med (2011) 3:348–61. doi:10.1002/emmm.201100142
26. Mócsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med (2013) 210:1283–99. doi:10.1084/jem.20122220
27. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11:519–31. doi:10.1038/nri3024
28. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity (2014) 40:66–77. doi:10.1016/j.immuni.2013.11.020
29. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol (2012) 5:354–66. doi:10.1038/mi.2012.24
30. Espinosa V, Dutta O, McElrath C, Du P, Chang Y-J, Cicciarelli B, et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci Immunol (2017) 2:eaan5357. doi:10.1126/sciimmunol.aan5357
31. Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One (2012) 7:e45427. doi:10.1371/journal.pone.0045427
32. Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine (2005) 31:109–18. doi:10.1016/j.cyto.2005.04.003
33. Rauch I, Rosebrock F, Hainzl E, Heider S, Majoros A, Wienerroither S, et al. Noncanonical effects of IRF9 in intestinal inflammation: more than type I and type III interferons. Mol Cell Biol (2015) 35:2332–43. doi:10.1128/MCB.01498-14
34. Chiriac M, Günther C, Becker C, Siebler J, Neurath M. IL-28-dependent activation of epithelial STAT1 drives epithelial wound healing in inflammatory bowel disease. Gastroenterology (2017) 152:S90. doi:10.1016/S0016-5085(17)30650-9
35. Yang J-Y, Kim M-S, Kim E, Cheon JH, Lee Y-S, Kim Y, et al. Enteric viruses ameliorate gut inflammation via toll-like receptor 3 and toll-like receptor 7-mediated interferon-β production. Immunity (2016) 44:889–900. doi:10.1016/j.immuni.2016.03.009
36. Sun L, Nava GM, Stappenbeck TS. Host genetic susceptibility, dysbiosis and viral triggers in IBD. Curr Opin Gastroenterol (2011) 27:321–7. doi:10.1097/MOG.0b013e32834661b4
37. Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell (2015) 160:447–60. doi:10.1016/j.cell.2015.01.002
38. Mennechet FJD, Uzé G. Interferon-λ-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood (2006) 107:4417–23. doi:10.1182/blood-2005-10-4129
39. Jordan WJ, Eskdale J, Srinivas S, Pekarek V. Human interferon lambda-1 (IFN-1/IL-29) modulates the Th1/Th2 response. Genes Immun (2007) 8:254–61. doi:10.1038/sj.gene.6364382
40. Finotti G, Tamassia N, Cassatella MA. Interferon-λs and plasmacytoid dendritic cells: a close relationship. Front Immunol (2017) 8:e74. doi:10.3389/fimmu.2017.01015
41. Megjugorac NJ, Gallagher GE, Gallagher G. IL-4 enhances IFN-λ1 (IL-29) production by plasmacytoid DCs via monocyte secretion of IL-1Ra. Blood (2010) 15:4185–90. doi:10.1182/blood-2009-09-246157
42. Liu F, Liu C, Hu X, Shang Y, Wu L. MicroRNA-21: a positive regulator for optimal production of type I and type III interferon by plasmacytoid dendritic cells. Front Immunol (2017) 8:947. doi:10.3389/fimmu.2017.00947
43. Kelly A, Robinson MW, Roche G, Biron CA, O’Farrelly C, Ryan EJ. Immune cell profiling of IFN-λ response shows pDCs express highest level of IFN-λR1 and are directly responsive via the JAK-STAT pathway. J Interferon Cytokine Res (2016) 36:671–80. doi:10.1089/jir.2015.0169
44. Finotti G, Tamassia N, Calzetti F, Fattovich G, Cassatella MA. Endogenously produced TNF-α contributes to the expression of CXCL10/IP-10 in IFN-λ3-activated plasmacytoid dendritic cells. J Leukoc Biol (2016) 99:107–19. doi:10.1189/jlb.3VMA0415-144R
45. Finotti G, Tamassia N, Cassatella MA. Synergistic production of TNFα and IFNα by human pDCs incubated with IFNλ3 and IL-3. Cytokine (2016) 86:124–31. doi:10.1016/j.cyto.2016.08.005
46. Morrow MP, Pankhong P, Laddy DJ, Schoenly KA, Yan J, Cisper N, et al. Comparative ability of IL-12 and IL-28B to regulate Treg populations and enhance adaptive cellular immunity. Blood (2009) 113:5868–77. doi:10.1182/blood-2008-11-190520
47. Krämer B, Eisenhardt M, Glässner A. Do λ-IFNs IL28A and IL28B act on human natural killer cells? Proc Natl Acad Sci U S A (2011) 108:E519–20. doi:10.1073/pnas.1108850108
48. Gardiner CM, Morrison MH, Dring MM. Reply to Krämer et al.: Human natural killer (NK) cell inhibition by IL28A. Proc Natl Acad Sci U S A (2011) 108:E521–2. doi:10.1073/pnas.1109288108
49. Morrison MH, Keane C, Quinn LM, Kelly A, O’Farrelly C, Bergin C, et al. IFNL cytokines do not modulate human or murine NK cell functions. Hum Immunol (2014) 75:996–1000. doi:10.1016/j.humimm.2014.06.016
50. Souza-Fonseca-Guimaraes F, Young A, Mittal D, Martinet L, Bruedigam C, Takeda K, et al. NK cells require IL-28R for optimal in vivo activity. Proc Natl Acad Sci U S A (2015) 112:E2376. doi:10.1073/pnas.1424241112
51. Granucci F, Zanoni I, Pavelka N, van Dommelen SLH, Andoniou CE, Belardelli F, et al. A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J Exp Med (2004) 200:287–95. doi:10.1084/jem.20040370
52. Granucci F, Zanoni I, Ricciardi-Castagnoli P. Central role of dendritic cells in the regulation and deregulation of immune responses. Cell Mol Life Sci (2008) 65:1683–97. doi:10.1007/s00018-008-8009-2
53. Zanoni I, Spreafico R, Bodio C, Di Gioia M, Cigni C, Broggi A, et al. IL-15 cis presentation is required for optimal NK cell activation in lipopolysaccharide-mediated inflammatory conditions. Cell Rep (2013) 4:1235–49. doi:10.1016/j.celrep.2013.08.021
54. Mingozzi F, Spreafico R, Gorletta T, Cigni C, Di Gioia M, Caccia M, et al. Prolonged contact with dendritic cells turns lymph node-resident NK cells into anti-tumor effectors. EMBO Mol Med (2016) 8:1039–51. doi:10.15252/emmm.201506164
55. Ferlazzo G, Pack M, Thomas D, Paludan C, Schmid D, Strowig T, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci U S A (2004) 101:16606–11. doi:10.1073/pnas.0407522101
56. Ferlazzo G, Morandi B, D’Agostino A, Meazza R, Melioli G, Moretta A, et al. The interaction between NK cells and dendritic cells in bacterial infections results in rapid induction of NK cell activation and in the lysis of uninfected dendritic cells. Eur J Immunol (2003) 33:306–13. doi:10.1002/immu.200310004
57. Jaeger BN, Donadieu J, Cognet C, Bernat C, Ordoñez-Rueda D, Barlogis V, et al. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J Exp Med (2012) 209:565–80. doi:10.1084/jem.20111908
58. Costantini C, Cassatella MA. The defensive alliance between neutrophils and NK cells as a novel arm of innate immunity. J Leukoc Biol (2011) 89:221–33. doi:10.1189/jlb.0510250
59. Muneo N, Tagawa M, Iwata F, Suzuki T, Nakamura A, Okada M, et al. IL-28 elicits antitumor responses against murine fibrosarcoma. J Immunol (2007) 178:5086–98. doi:10.4049/jimmunol.178.8.5086
60. Zhang S, Kodys K, Li K, Szabo G. Human type 2 myeloid dendritic cells produce interferon-λ and amplify interferon-α in response to hepatitis C virus infection. Gastroenterology (2013) 144:414–25.e7. doi:10.1053/j.gastro.2012.10.034
61. de Groen RA, Groothuismink ZMA, Liu BS, Boonstra A. IFN-λ is able to augment TLR-mediated activation and subsequent function of primary human B cells. J Leukoc Biol (2015) 98:623–30. doi:10.1189/jlb.3A0215-041RR
62. Wang X, Wang H, Liu M-Q, Li J-L, Zhou R-H, Zhou Y, et al. IFN-λ inhibits drug-resistant HIV infection of macrophages. Front Immunol (2017) 8:210. doi:10.3389/fimmu.2017.00210
63. Hou W, Wang X, Ye L, Zhou L, Yang ZQ, Riedel E, et al. Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J Virol (2009) 83:3834–42. doi:10.1128/JVI.01773-08
64. Liu B-S, Janssen HLA, Boonstra A. IL-29 and IFNα differ in their ability to modulate IL-12 production by TLR-activated human macrophages and exhibit differential regulation of the IFNγ receptor expression. Blood (2011) 117:2385–95. doi:10.1182/blood-2010-07-298976
65. Witte K, Gruetz G, Volk H-D, Looman AC, Asadullah K, Sterry W, et al. Despite IFN-λ receptor expression, blood immune cells, but not keratinocytes or melanocytes, have an impaired response to type III interferons: implications for therapeutic applications of these cytokines. Genes Immun (2009) 10:702–14. doi:10.1038/gene.2009.72
Keywords: interferon lambda, dendritic cells, neutrophils, natural killer cells, type III interferon, viral infection, bacterial infections, fungal infection
Citation: Zanoni I, Granucci F and Broggi A (2017) Interferon (IFN)-λ Takes the Helm: Immunomodulatory Roles of Type III IFNs. Front. Immunol. 8:1661. doi: 10.3389/fimmu.2017.01661
Received: 13 October 2017; Accepted: 13 November 2017;
Published: 28 November 2017
Edited by:
Renato Ostuni, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Nicola Tamassia, University of Verona, ItalyCopyright: © 2017 Zanoni, Granucci and Broggi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivan Zanoni, aXZhbi56YW5vbmlAY2hpbGRyZW5zLmhhcnZhcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.