 Emily Jayne Colbeck
Emily Jayne Colbeck Ann Ager
Ann Ager Awen Gallimore
Awen Gallimore Gareth Wyn Jones
Gareth Wyn Jones- 1UCL Cancer Institute, University College London, London, United Kingdom
- 2Division of Infection and Immunity, School of Medicine and Systems Immunity Research Institute, Cardiff University, Cardiff, United Kingdom
Secondary lymphoid organs are integral to initiation and execution of adaptive immune responses. These organs provide a setting for interactions between antigen-specific lymphocytes and antigen-presenting cells recruited from local infected or inflamed tissues. Secondary lymphoid organs develop as a part of a genetically preprogrammed process during embryogenesis. However, organogenesis of secondary lymphoid tissues can also be recapitulated in adulthood during de novo lymphoid neogenesis of tertiary lymphoid structures (TLSs). These ectopic lymphoid-like structures form in the inflamed tissues afflicted by various pathological conditions, including cancer, autoimmunity, infection, or allograft rejection. Studies are beginning to shed light on the function of such structures in different disease settings, raising important questions regarding their contribution to progression or resolution of disease. Data show an association between the tumor-associated TLSs and a favorable prognosis in various types of human cancer, attracting the speculation that TLSs support effective local antitumor immune responses. However, definitive evidence for the role for TLSs in fostering immune responses in vivo are lacking, with current data remaining largely correlative by nature. In fact, some more recent studies have even demonstrated an immunosuppressive, tumor-promoting role for cancer-associated TLSs. In this review, we will discuss what is known about the development of cancer-associated TLSs and the current understanding of their potential role in the antitumor immune response.
Introduction
Secondary lymphoid organs (SLOs) primarily serve to initiate adaptive immune responses to exogenous pathogens. For this, SLOs provide a location for interactions between rare antigen-specific naive lymphocytes and antigen-presenting cells draining from local tissues. The importance of SLOs in mediating homeostatic lymphocyte proliferation and rapid recall responses to returning antigens have long been appreciated (1, 2). Also it is now well recognized that SLOs contribute to peripheral immune tolerance to self-antigens and commensals by regulating trafficking of immunosuppressive Foxp3+ regulatory T cells (Tregs), major cellular mediators of peripheral tolerance, and facilitating the deletion of autoreactive T cells by SLO-resident extrathymic Aire-expressing cells (3, 4).
Canonical SLOs comprise the lymph nodes (LNs), the white pulp of the spleen, the appendix (in humans), and mucosal-associated lymphoid tissues (MALTs) including intestinal Peyer’s patches (PPs) and the tonsils. SLOs are strategically placed at distinct, predetermined sites throughout the body, together forming a sophisticated network that facilitates continual immune surveillance of interstitial areas, epithelial and mucosal surfaces, and the blood. Individual SLOs have highly organized, specialized architecture that is specifically adapted to promote the immune cell interactions necessary for immune response initiation (2, 5, 6).
The development of canonical SLOs is a genetically preprogrammed process initiated during embryogenesis. Although LNs and PPs develop prenatally, the organogenesis of MALT, such as bronchial-associated lymphoid tissue (BALT) and more plastic lymphoid tissues, including cryptopatches and isolated lymphoid follicles in the intestine, occurs postnatally. Thus, there exists a continuum of lymphoid tissues, from canonical, constitutive SLOs preprogrammed during ontogeny to highly plastic, inducible and transient lymphoid structures that form later in life (2, 7).
SLO organogenesis is recapitulated in the de novo development of tertiary lymphoid structures (TLSs) under pathological circumstances (6, 8, 9). TLSs, also termed ectopic lymphoid-like structures or tertiary lymphoid organs, form at the site of infection or chronic inflammation and have been noted in autoimmune disease, allograft rejection, and more recently cancer (2, 6, 10). Crucially, the clinical significance of TLSs is thought to vary from deleterious to protective, emphasizing the need to better understand the formation and function of these structures, which may be contextually different, before clinical targeting.
In this review, we will compare and contrast TLS neogenesis with the development of a prototypic SLO, the LN. Importantly, we will discuss current knowledge surrounding the function of TLSs, specifically within cancer, and consider the implications for the use of next-generation therapeutics.
Composition and Organization of a TLS Compared to a Prototypic SLO: The LN
Lymph nodes comprise an organized collection of immune and stromal cells encapsulated by a fibrous capsule and an underlying subcapsular sinus (SCS; Figure 1) (6, 11, 12). Cells are topologically segregated into a cortex of densely packed B cells and follicular dendritic cells (FDCs) arranged into discrete primary follicles; the paracortex that accommodates less densely packed T cells, dendritic cells (DCs), and fibroblastic reticular cells (FRCs); and the medulla, composed of lymphatic medullary cords, separated by lymph-filled cavities called medullary sinuses. After antigen exposure, B cells proliferate extensively, giving rise to secondary follicles [germinal centers (GCs)] (6). Alongside FDCs and FRCs, marginal reticular cells (MRCs) constitute a third stromal cell network of the LN, situated just under the SCS (13, 14).
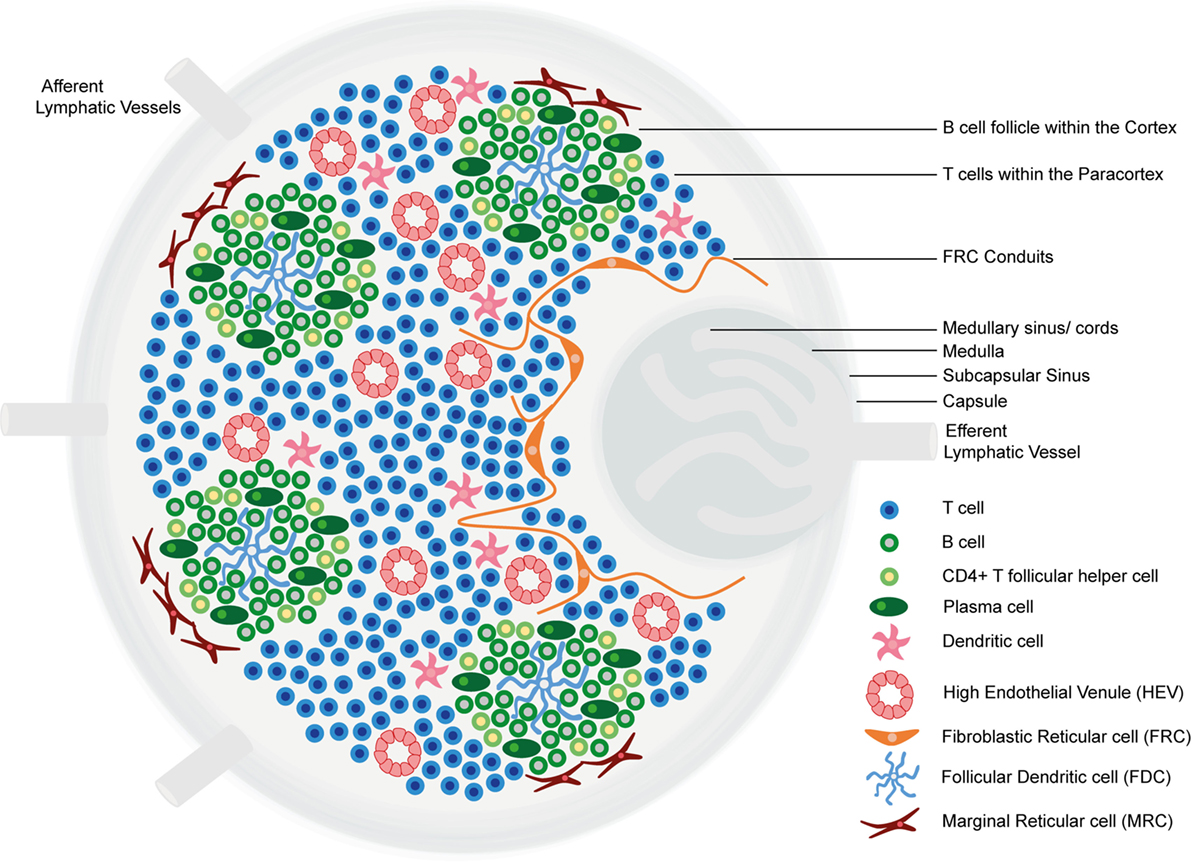
Figure 1. The structure of the lymph node. Lymph nodes comprise a collagen-rich fibrous capsule and an underlying subcapsular sinus (SCS). Cells are segregated into (1) the cortex, consisting of B cells, T follicular helper cells, and follicular dendritic cells (FDCs) arranged in primary follicles, in which B cells survey antigens presented on the FDC stromal network; and (2) the paracortex, which accommodates T cells, dendritic cells (DCs), and fibroblastic reticular cells (FRCs) that form stromal cell networks and reticular fibers, along which T cells and DCs migrate. Upon antigen exposure and stimulation, B cell proliferation within the primary follicle gives rise to germinal centers, containing antibody-producing plasma cells. The inner medulla is composed of lymphatic tissues (medullary cords) separated by medullary sinuses consisting of lymph. FRCs express CCL19 and CCL21, whereas CXCL13 is expressed by FDCs. Marginal reticular cells (MRCs) form a third stromal cell network, situated just under the SCS. Lymph nodes contain lymphatic vasculature and high endothelial venules (HEVs). Afferent lymphatic vessels deliver lymph containing antigen and immune cells, and HEVs are specialized postcapillary venules that primarily deliver naive and central memory lymphocytes.
LNs contain two vasculature systems: lymphatic vasculature and high endothelial venules (HEVs). Afferent lymphatic vessels deliver lymph, containing antigens and immune cells, primarily DCs, to the SCS (11, 15). From the SCS, lymph percolates through cortical and medullary sinuses and leaves the LN via the efferent lymphatic vessel, which delivers lymph to the venous blood (6, 11). HEVs are highly specialized postcapillary venules found in the blood vascular bed within the paracortical region of LNs, the main function of which is homeostatic delivery of naive and central memory lymphocytes from the adjacent bloodstream. Endothelial cells lining HEVs have a distinct plump, cuboidal morphology and express highly specific addressin molecules, collectively termed peripheral node addressins (PNAds) (11, 16). Lymphocytes extravasate through HEV walls according to a multistep adhesion cascade, dictated by the expression of adhesion molecules and chemokines on HEV endothelial cell surfaces (11, 17).
The term “TLS” can refer to structures of varying organization, from simple clusters of lymphocytes, to sophisticated, segregated structures highly reminiscent of SLOs (10, 18–22). TLSs form at localized sites of microbial infection or chronic inflammation and have been noted in many autoimmune diseases and more recently cancer (10). De novo development of TLSs is referred to as ectopic lymphoid neogenesis or lymphoid neoorganogenesis (9, 10).
A notable difference between LNs and TLSs is the fact that the former are encapsulated, while the latter represent a congregation of immune and stromal cells confined within an organ or tissue. Crucially, while SLO development represents a genetically preprogrammed process occurring during ontogeny, TLSs form in response to chronic inflammatory cues. Furthermore, while SLOs form at predetermined specified anatomical locations, TLSs typically form in non-lymphoid organs and exhibit plasticity, as they can present transiently, becoming resolved after the elimination of antigen (2, 6). TLS development has been documented in practically all organ settings under conditions of chronic inflammation, including the heart, kidneys, intestine, vasculature system, central nervous system, and bone marrow. The clinical diseases and experimental models in which TLSs have been documented are extensively reviewed in Ref. (10).
Pathologists use specific criteria to define TLSs. According to these criteria, TLSs contain distinct T and B cell compartments, FRC networks and PNAd+ HEVs within T cell zones, FDCs and evidence for class switching and reactive GCs in B cell zones, and expression of the enzyme activation-induced cytidine deaminase (AID); an enzyme expressed in GC B cells required for the initiation of somatic hypermutation and immunoglobulin gene class switching (18, 23). Like LNs, TLSs have also been shown to contain lymphatic vessels, although the role of these vessels and whether they resemble afferent or efferent lymphatics of LN vasculature is as yet unknown (4–6). A set of criteria to define bona fide TLSs were also recently proposed by Fridman and colleagues (7).
However, many groups apply the term TLS to less well-organized structures (18). Here, we will also use the term “TLS” more loosely. The observed heterogeneity in TLS architecture and organization could reflect disease stage at which biopsies are taken and could therefore represent sampling at varying phases of TLS development, maturation, and/or resolution. Furthermore, we believe that the potential functional resemblance that TLS share with canonical SLOs could be more relevant than precise anatomical structure: these structures have been associated with either deleterious or protective clinical outcomes in human patients, leading to the speculation that TLSs can generate functional adaptive immune responses capable of influencing the progression of disease (10). This will be discussed in detail later.
TLSs in Cancer
Tertiary lymphoid structures were initially described in the context of non-neoplastic chronic inflammatory conditions, including autoimmune diseases, infections, and idiopathic diseases (6, 18, 21). Neoplastic malignancies share many features with environments of chronic inflammation, including the chronicity of inflammation itself. However, malignant tumors differ from chronic inflammatory environments in one significant aspect that many would assume may preclude the formation of TLSs: the highly immunosuppressive tumor microenvironment (24, 25). Yet, the occurrence of TLSs of varying degrees of organization has been reported in patients afflicted by multiple types of primary and metastatic cancer (Table 1).
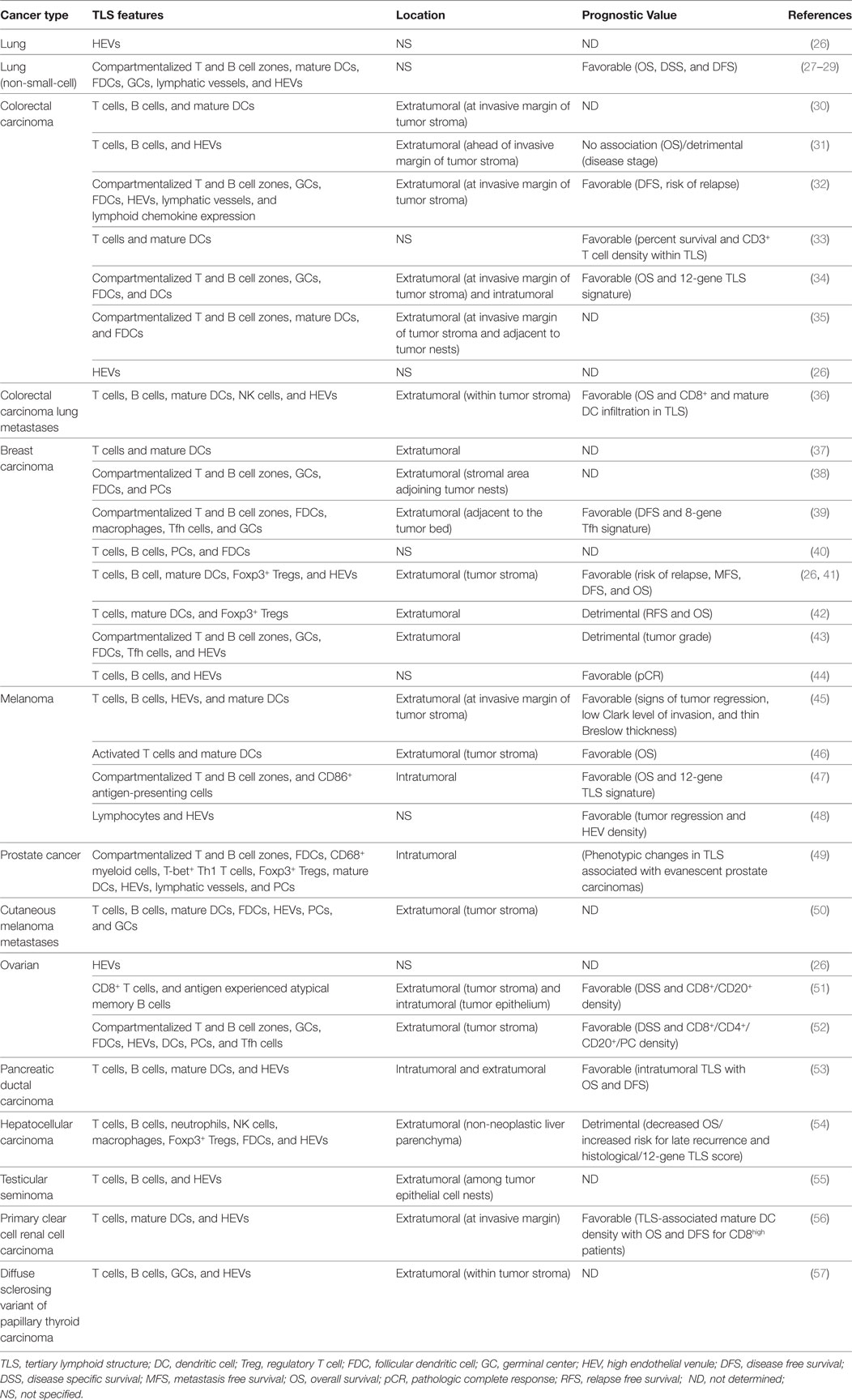
Table 1. Tertiary lymphoid structures and high endothelial venules in human cancer.
Tumor TLSs are largely associated with a favorable clinical prognosis for patients for a number of different solid tumor types. In a retrospective study of 74 early-stage non-small-cell lung cancer (NSCLC) patients, Dieu-Nosjean and colleagues were the first to report the presence of TLSs in patients with lung cancer. The TLSs, referred to as tumor-induced BALT (Ti-BALT), consisted of an organized distribution of DC and T cell clusters and B cell follicles. The researchers demonstrated that the density of mature DC-LAMP (CD208)+ DCs, used as a marker of Ti-BALT, correlated with increased overall, disease-specific and disease-free survival (27). The presence of intratumoral HEVs alone is a strong prognostic marker for various types of human cancer, too. Martinet and colleagues demonstrated a significant correlation between the presence of intratumoral HEVs located within lymphocyte-rich clusters and increased disease-free, metastasis-free, and overall survival rates in a retrospective study of 146 invasive breast cancer patients (26). Furthermore, the same group later found an association between tumor-associated HEVs, infiltrating lymphocytes, and tumor regression in malignant melanoma (45). Multiple other studies have provided evidence for an association between TLSs, or HEVs in the absence of TLSs, in tumors and favorable clinical outcome (Table 1).
However, the relationship between tumor-associated TLSs and patient outcome appears to depend on many parameters, including cancer type and disease stage. In our own studies, the majority of colorectal cancer-associated HEVs are found at the tumor invasive margin, where they are associated with lymphoid aggregates containing CD20+ B cells and CD3+ T cells. However, while extratumoral HEV density correlated significantly with numbers of T cells within the invasive margin, the correlation with T cell densities in the tumor center was weak. In fact, lymphoid aggregates were associated with more advanced (Duke’s C stage) disease and were not associated with a more favorable prognosis; indeed, there was a trend toward higher numbers of lymphoid aggregates in those patients who did not survive >5 years posttumor resection relative to those who did (31). In addition, quantification of TLSs within the non-neoplastic liver parenchyma of 66 patients who had undergone resection for hepatocellular carcinoma (HCC) revealed an increased risk for late tumor recurrence and lower overall survival for patients with a high histological TLS score (54). Furthermore, tumor-associated TLS formation was associated with a higher tumor grade in 290 primary breast carcinoma patients (43). TLSs or isolated PNAd+ HEVs have also been documented in diverse mouse models of cancer, including lung adenocarcinoma, HCC, melanoma, and fibrosarcoma (54, 58–61). As is the case for human cancer, the relationship between TLSs/HEVs and tumor control appears to be variable: data evidencing these disparate roles will be discussed in detail later.
Development of TLSs
Studies of the ontogenic development of SLOs provide a paradigm for understanding TLS formation. Despite structural differences between canonical SLOs and TLSs, we now know that many molecular mechanisms underlying SLOs initiation, development, and maintenance are shared with TLSs formation (10) (Figure 2). For instance, chronically inflamed tissues resident to TLSs are often characterized by the expression of homeostatic chemokines and cytokines reminiscent of SLOs (62). However, a detailed understanding of the exact mechanisms by which TLSs form in various pathogenic circumstances, in particular cancer, is still lacking.
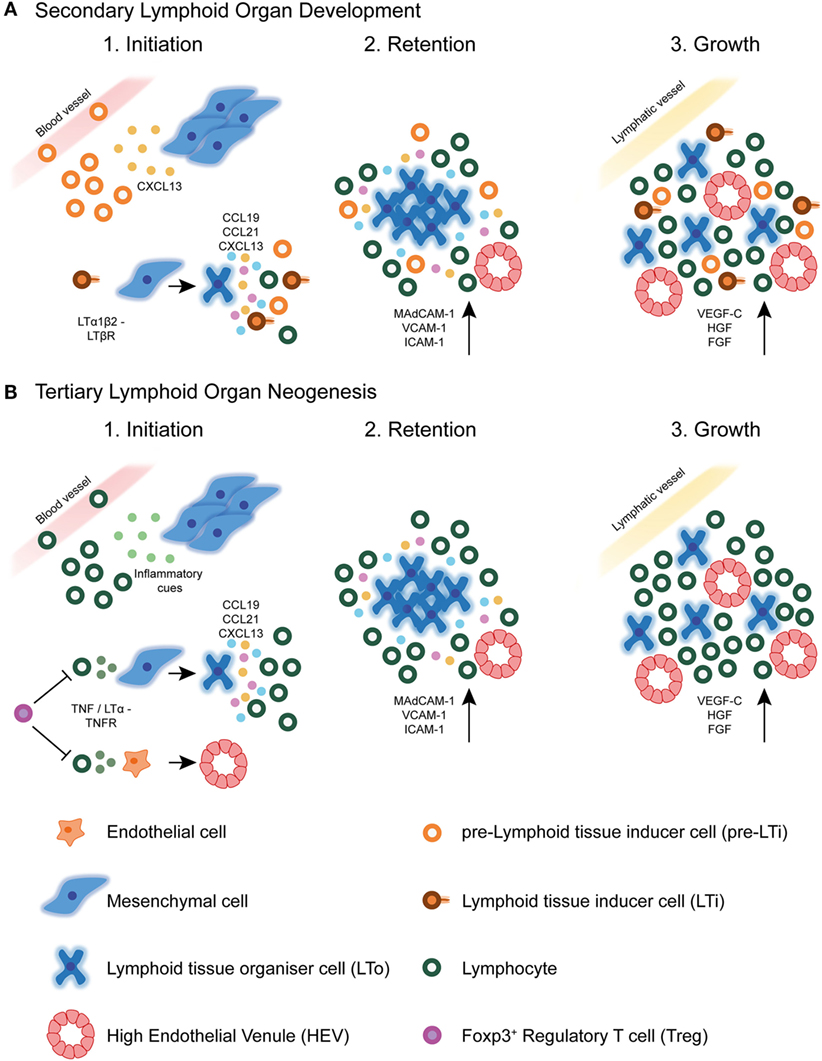
Figure 2. Secondary lymphoid organ and tertiary lymphoid structure development. (A) During secondary lymphoid organ development, precursor lymphoid tissue inducer (pre-LTi) cells are initially attracted to the lymph node anlagen from adjacent blood vessels by CXCL13 secreted by resident mesenchymal cells. Clustering of these first cells facilitates crosstalk leading to their maturation into mature LTi cells, which express surface LTα1β2. Interaction of LTi cells with LTβR expressing stromal cells leads to their differentiation into lymphoid tissue organizer (LTo) cells. Mature LTo cells express chemokines as a result of LTβR triggering, which attract further pre-LTi cells and other hematopoietic cells to the developing lymph node anlagen. Also as a result of LTβR triggering, LTo cells express adhesion molecules, which retain infiltrating hematopoietic cells, eventually leading to lymph node growth. Finally, the developing lymph node fosters formation of high endothelial venules (HEVs), and expression of lymphangiogenic factors aids connection of the lymph node to the surrounding lymphatic vasculature. (B) In tertiary lymphoid structure neogenesis in tumors, the initiating cues are likely to be of inflammatory origin and may differ between different tumors. These cues attract circulating lymphocytic cells, such as T lymphocytes and NK cells, which have been shown to initiate HEV development via secretion of TNFα or LTα3. These cytokines may act directly on TNFR expressing endothelial cells in the tumor microenvironment, causing differentiation of already existing tumor vasculature into specialized HEVs. Whether this event precedes tertiary lymphoid structure neogenesis in tumors is currently unknown. The signals involved in SLO development are also shared with TLS formation, including homeostatic chemokines and adhesion molecules. Little is currently known about the involvement of lymphatic vasculature in TLS development or maintenance. Foxp3+ regulatory T cells (Tregs) exert a negatively regulatory role over HEV/TLS development in tumors, potentially via direct inhibition of initiating hematopoietic cells including T cells.
Control of TLS/HEV Neogenesis by Tregs
A key cellular population recurrently suggested to play a regulatory role in ectopic TLS and/or HEV neogenesis is Foxp3+ Tregs. Tregs are highly immunosuppressive T cells that maintain immune homeostasis and promote immunological tolerance to self-antigens. Tregs prevent autoimmunity by keeping in check the activation and expansion of overreactive immune cells, therefore limiting excessive and harmful immune responses (63, 64).
Regulatory T cells are highly enriched in tumors, where they impinge on antitumor immune responses (65, 66). In line with this negative regulatory role, several seminal studies have demonstrated the prevention of tumor development or regression of established tumors, following efficient and selective depletion of Foxp3+ Tregs (67–69). In our own studies, Treg ablation in the methylcholanthrene (MCA) carcinogen-induced fibrosarcoma mouse model of carcinogenesis resulted in profound activation of Foxp3− CD4+ and CD8+ T cells and an overall highly significant reduction in tumor growth rate. However, the response to Treg loss was highly variable, with a large proportion of Treg-depleted animals displaying no significant alteration in tumor control. Critically, successful tumor control was determined by the extent of T cell infiltration into the tumor, which was in turn dictated by the development of ectopic, isolated PNAd+ HEVs within the tumor mass. HEVs were only ever observed in a proportion of tumors following Treg depletion, and there was an absolute concordance between HEV presence, high numbers of tumor-infiltrating lymphocytes (TILs), and tumor growth control. In our hands, therefore, depletion of immunosuppressive Tregs is a prerequisite for the development of isolated intratumoral HEVs in MCA-induced fibrosarcomas, implying that Tregs can inhibit neogenesis of HEVs, and possibly TLSs, in tumors (61).
Indeed, highly organized inducible BALT (i-BALT) containing PNAd+ HEVs, which normally only develop in response to inflammatory insult in wild-type animals, spontaneously develop in lung tissue of germ-free CCR7−/− mice, which are defective in Treg-mediated immune regulation (70, 71). Adoptive transfer of wild-type but not CCR7−/− Tregs into CCR7- deficient hosts largely interrupted i-BALT formation, and homing of Tregs to peripheral lymphoid organs was essential for their prevention of i-BALT formation (70). A separate study reported the development of spontaneous i-BALT in IL-2-deficient animals, which are devoid of Tregs (72). Furthermore, Foo and colleagues showed that i-BALT that form in response to LPS exposure in the lung was driven by neutrophils and negatively regulated by Tregs (73). Such studies imply a crucial regulatory role for Tregs in the prevention of ectopic lymphoid neogenesis. It will be important in the future to define the mechanisms by which Tregs suppress HEV and/or TLS formation in different scenarios if we wish to develop therapeutic strategies to manipulate these ectopic lymphoid structures.
Lymphotoxin/Tumor Necrosis Factor (TNF) Signaling in SLO Development and TLS Neogenesis
A body of work has demonstrated that the successful development of LNs critically depends on coordinated interactions between stromal lymphoid tissue organizer (LTo) cells and hematopoietic lymphoid tissue inducer (LTi) cells. These interactions occur via signaling between the lymphotoxin (LT) α1β2 ligand, expressed on the surface of LTi cells, and the LTβ receptor (LTβR), expressed on LTo cells [reviewed in Ref. (2, 74, 75)]. Signaling through the LTβR on gp38 (podoplanin) expressing LTo cells results in the expression of lymphoid chemokines CCL19, CCL21, and CXCL13, which attract migrating hematopoietic cells. Also as a result of LTβR signaling, LN stromal cells express adhesion molecules, including vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and mucosal vascular addressin cell adhesion molecule-1 (MAdCAM-1), which retain the newly arriving hematopoietic cells, leading to LN growth [reviewed in Ref. (2)]. Furthermore, LTβR signaling induces the expression of vascular growth factors such as the lymphangiogenic factor, VEGF-C, by LTo cells, which aids connection of the developing LN to the surrounding lymphatic vasculature (76). Finally, the developing LN becomes colonized by infiltrating lymphocytes, which are guided to specific zones by homeostatic chemokine expression, giving rise to a highly organized LN [reviewed in Ref. (2)]. Embryonic venous blood vessels play a key role in lymphoid organogenesis by delivering LTi cells, which initiate development, as well as other cellular subsets that mature and maintain HEVs (77). Interestingly, some recently published data have challenged certain aspects of this long accepted scheme and will be discussed in detail later.
Lymphotoxin signaling also appears to play a central role in TLS neogenesis. Overexpression of Ltα in the kidney and pancreas using the RIP tissue-specific promoter results in chronic inflammation accompanied by highly organized TLS induction. This study by Nancy Ruddle’s group was one of the first to demonstrate the involvement of the same signaling molecules, namely Ltα, that control SLO development in TLS formation (9). The mechanisms by which Ltα directs embryonic lymphoid organogenesis and TLS neogenesis in the context of chronic inflammation seem similar, particularly in regard to the induction of chemokine expression (78).
Studies have also strongly implicated LTβ and LTβR in TLS neogenesis. Pancreatic TLSs in RIPLTαβ transgenic animals displayed more distinct T and B cell zone separation, higher chemokine expression, luminal expression of PNAd on HEVs, and increased infiltration of naive L-selectin+ lymphocytes, relative to RIPLTα mice (79). Furthermore, the loss of LTα1β2-LTβR signaling resulted in reversion of many aspects of pancreatic TLS formation, including HEV dedifferentiation, stromal network disruption, and loss of chemokine expression (80). These and multiple other studies have strongly implicated LTα1β2-LTβR signaling in TLS neogenesis at sites of chronic inflammation, particularly in the development and maintenance of specific TLS structures, namely FDC networks and HEVs (19, 80–86).
Signaling via the LTβR has also been implicated in neogenesis of TLSs and/or HEVs in cancer. Targeting of an antibody-LTα fusion protein directly to mouse melanomas resulted in neogenesis of TLS-like lymphoid aggregates including PNAd+ HEVs (87). Martinet and colleagues demonstrated that DC-LAMP+ DCs were the major producers of LTβ in human breast tumors and that LTβ is overexpressed specifically in tumors displaying a high density of HEVs (41).
The alternative LTβR ligand, LIGHT, is thought to play a redundant role specifically in mesenteric LN development (88). LIGHT has also been implicated in neogenesis of cancer-associated TLSs (89). In breast cancer patients, enhanced expression of LIGHT in ectopically formed TLSs in breast tissue relative to SLOs implies a role for this TNF superfamily member in driving cancer-associated TLS formation (90). Similarly, the expression of LIGHT in a fibrosarcoma cell line resulted in upregulation of CCL21 and MAdCAM-1, facilitating the recruitment of vast numbers of naive CD8+ T lymphocytes, which were then sufficiently activated in situ to facilitate rejection of established tumors (91). Furthermore, a very recent study demonstrated the ability of LIGHT, if targeted to tumor vasculature by the use of a vascular targeting peptide (VTP), to normalize aberrant blood vessels, induce de novo TLS neogenesis, and facilitate influx of endogenous T cells. In combination with checkpoint inhibitor immunotherapy and vaccination, the LIGHT-VTP agent enabled the efficient destruction of tumors previously refractory to immunotherapy (92). These results suggest that, at least in some malignant contexts, LIGHT represents an initiating signal to induce TLS formation in tumors.
Interestingly, studies have demonstrated that TLS neogenesis can be dictated by different signaling circuitries to those classically associated with SLO development. Moyron-Quiroz and colleagues cataloged the presence of primitive TLSs, lacking FDC networks and HEVs, in Ltα-deficient mice, which lack LNs and PPs (93–96). More recent studies of tumor-associated TLSs in mouse models of cancer have also suggested a potential departure from canonical SLO development. Peske and colleagues noted the presence of HEV-like vasculature expressing PNAd and CCL21 within lymphocytic aggregates in several murine tumors growing in different anatomical locations in wild-type mice (60). Surprisingly, the authors found that blockade of LTβR signaling in B16-OVA tumor bearing animals had no effect on PNAd expression on tumor vasculature and concurrent trafficking of naive OT-I cells into tumors was even enhanced, implicating no loss of HEV function. Instead, the researchers found that homotrimeric LTα3, but not TNF, signaling via TNFRs was responsible for the induction of HEV-like vasculature in tumors (60).
Indeed, our own studies of Treg-depleted MCA-induced fibrosarcomas underpin this reliance on TNFR signaling in ectopic HEV neogenesis in tumors. Surprisingly, treatment of animals with an antagonistic LTβR.Fc fusion protein increased intratumoral HEV total area and maintained numbers of infiltrating T cells, suggesting preserved function of the vessels. In striking contrast, administration of a TNFR.Ig fusion protein, anti-LTα monoclonal antibody, or anti-TNFα monoclonal antibody led to a drastic decrease in intratumoral HEV total area, with concomitant effects on T cell infiltration. Therefore, we found that, similar to the case of tumor-associated HEV development in the presence of Treg, TNFR signaling predominates as the governing signaling circuitry modulating HEV development in tumors following Treg depletion (97).
Chemokine Involvement in SLO Development and TLS Neogenesis
Precursor LTi (pre-LTi) cells that seed the LN anlagen are initially attracted by CXCL13 expressed by local stromal cells (74, 98–100). Mice genetically deficient in CXCL13 or its receptor, CXCR5, display failed LTi clustering and an absence of several peripheral LNs. Lymphatic endothelium-derived CCL21 is able to compensate for the lack of CXCL13 expression in the development of certain LNs by attracting the first LTi cellular clusters (99, 101). However, in general, the CCR7/CCL21 ligand receptor pair seems to contribute only an additive affect to the attractive function of CXCL13, as all LNs form in a normal fashion in mice deficient for the CCR7/CCL21 axis but competent in CXCR5/CXCL13 signaling (102).
Chronically inflamed tissues containing TLSs display significantly increased expression levels of homeostatic chemokines involved in SLO development, including CXCL12, CXCL13, CCL19, and CCL21. All four lymphoid chemokines are individually capable of inducing TLS formation when overexpressed using the RIP in the pancreas (80, 103–106). Conversely, the loss of CXCL13, CXCR5, or CCR7 prohibits the formation of TLSs in disease settings (107–109).
However, the TLSs formed following forced expression of these various chemokines differ significantly in terms of size, cellular composition, and structural organization, revealing defined roles for these chemokines in governing organization and thereby the functionality, of TLSs. For instance, while CXCL13 and CCL21 function in terms of segregation of T and B cells into distinct compartments, CCL19 and CXCL12 primarily seem to facilitate lymphocyte recruitment and positioning of various cell types within the organized TLS. These chemokines seem to function upstream of LTα1β2 signaling, as shown by induction of LTα1β2 expression on T or B cells by CCL19, CCL21, or CXCL13 (80, 103). However, the anatomical site of TLS formation and/or specific disease processes may influence the relative involvement of various lymphoid chemokines: while CCL21 overexpression drives TLS formation in the pancreas, ectopic expression of this chemokine in the skin fails to stimulate lymphoid neogenesis (104).
Studies of human cancer patients have additionally implicated chemokines CCL19, CCL21, and CXCL13 in the formation of intratumoral TLSs (24, 28, 110). In human lung carcinoma-associated TLSs, CCL19-expressing cells were found to belong predominantly to the mature DC-LAMP+ DC population and are suggested to contribute toward maintenance of the lymphoid structure via recruitment of CCR7+ immune cells, including naive and central memory T cells, and further, recently activated, DCs. CXCL13 expression in lung carcinoma TLSs was detected specifically on CD21+ FDCs within GC-like areas of the TLS. These CXCL13-expressing FDCs were found to colocalize with CXCR5+ follicular helper T cell-like cells, suggesting that CXCL13 expression in tumor TLSs could contribute toward generation of humoral immune responses via recruitment of CXCR5+ immune cells into the GC. As is true for TLSs in inflammatory settings, the CCL21 expression in lung tumor TLSs was found to be restricted to lymphatic vessels. This invites the attractive speculation that CCL21+ lymphatic vessels in and around tumor TLSs could provide major trafficking systems guiding activated immune cells to local tumor-draining LNs where systemic protection against metastatic dissemination of primary tumor cells could be established (24, 28). However, there is, as of yet, no formal evidence to support this hypothesis. Overexpression of lymphoid chemokines has also been more recently reported in breast carcinomas, where CXCL13-expressing CD4+ T follicular helper cells constitute an important component of breast tumor TLSs. Furthermore, a strong Tfh signature robustly predicted increased patient survival (39).
Cellular Initiators of SLO Development and TLS Neogenesis
Lymphoid tissue inducer cells were first described in the mouse as a fetal population of hematopoietic cells, essential for secondary lymphoid organogenesis (111–114). LTi cells are CD45+ CD4+ CD3− c-Kit+ interleukin 7 receptor-α (IL-7Rα)+ ID2+ RORγt+ cells that derive from a common progenitor found in the liver (115, 116). The absolute requirement for LTi cells in LN development was demonstrated by the complete lack of LNs in animals genetically deficient in genes required for LTi development and maturation: Rorc, which encodes the transcription factor retinoic acid receptor-related orphan receptor-γt (RORγt), and Id2, which encodes helix-loop-helix protein inhibitor of DNA binding 2 (ID2) (117–119). The function of LTi cells in the initiation of LN development is absolutely dependent on their expression of LTα1β2, which is induced by TNF-related activation-induced cytokine receptor (TRANCER) signaling (or IL-7Rα signaling in PP development) (116, 120).
Despite the requirement for LTβR signaling between LTi and LTo cells for LN development, LTi clustering proceeds uninterrupted when LTα1β2 expression by LTi cells (76, 118, 120, 121) or LTβR expression by LTo cells (122) is prevented. A more recent study demonstrated the ability of TNFα, if expressed above basal levels, to compensate for the lack of LTi cells in Rorc(γt)−/− animals in driving organogenesis of several peripheral LNs (those draining the skin), including axillary, cervical, inguinal, and brachial, albeit at lower frequency than in wild-type (123). Therefore, this study contradicts the dogma stating that LTi cells are absolutely required for SLO formation. Although these data imply redundancy in the communication networks governing SLO development, for normal development of LN anlagen in early postnatal life, LTβR signaling is absolutely required suggesting that while the identity of the cell providing the signal may not be as important, the signaling pathway driving secondary lymphoid organogenesis must be preserved (123).
Surprisingly, the loss of LTβR expression in CCL19-expressing mesenchymal LTo cells does not impede LN organogenesis, challenging the existing model delineating a crucial role for LTo cells (124). More recently, the same group led by Burkhard Ludewig have elegantly illustrated that lymphatic endothelial cells (LECs), which form a monolayer of cells lining the SCS in mature LNs, in fact act as the first LTo cells by regulating LTi cell migration and retention; activation of mesenchymal LTo cells only occurred following productive crosstalk between LECs and LTi cells in the LN anlagen (125). These results place LECs above mesenchymal LTo cells in the hierarchical contribution by different stromal cell populations in LN organogenesis. Collectively, such studies challenge the widely accepted dogma of a two-cell type crosstalk scheme involving mesenchymal LTo and hematopoietic LTi cells and illustrate the ongoing gap in our knowledge concerning LN organogenesis.
Experimental data gathered from studies utilizing knockout and transgenic mouse models have indicated cooperation between TNF superfamily members and lymphoid chemokines in the process of lymphoid neogenesis, much as in SLO development (6). However, the identity of both a TNF/LT producing LTi-like cell and a TNF/LT responsive stromal LTo-like cell involved in TLS formation still remains elusive. The recent discovery of an equivalent population of RORγt+ LTi cells in the adult, which are members of the innate lymphoid cell (ILC) family, prompted many to speculate that such a population could be directly responsible for TLS formation during lymphoid neogenesis (126, 127). Some studies have even suggested a direct role for adult LTi cells in lymphoid neogenesis. For instance, Meier and colleagues demonstrated that the induction of ectopic lymphoid tissue by overexpression of the gene encoding IL-7 was entirely dependent on the presence of LTi cells, as TLSs failed to develop in the absence of RORγt (128). A separate study, in which adult WT LTi cells were adoptively transferred into neonatal CXCR5−/− mice, demonstrated the ability of these cells to induce de novo formation of TLSs in the intestine (127). More recently, natural cytotoxicity receptor (NCR) expressing ILC3 cells were found closely associated with lymphoid aggregates in NSCLC, where they induced LTαβ and adhesion molecule expression, suggesting a potential role in neogenesis of these structures (129).
However, several studies have now provided evidence supporting the notion that TLSs can develop in the absence of canonical RORγt+ LTi cells and ILC3 cells. Marinkovic and colleagues used a mouse model in which CCL21 overexpression in the thyroid results in TLS formation, to demonstrate the dispensability of conventional RORγt+ cells for ectopic lymphoid neogenesis (106). Deletion of Id2 resulted in the absence of LNs and PPs as expected, but had no effect on the development of TLSs in the thyroid of CCL21 overexpressing animals. Rather, the authors showed that mature CD3+ CD4+ T cells were absolutely required for the molecular program that instructs TLS development and suggested that these cells interact with DCs to initiate such a program (106). TLSs also develop in the colon of Rorc(γt)−/− animals following inflammatory insult (130). Furthermore, i-BALT forms in the lungs of Rorc(γt)−/− animals subjected to pulmonary inflammation (131). Interestingly, TNF/Rorc(γt)−/− mice demonstrate development not only of some SLOs but also of TLSs in the absence of LTi cells, provided TNFα signaling is increased. However, although TNFα compensates for LTi cell loss to a certain degree, Id2 expression as well as LTβR signaling is required for complete secondary lymphoid organogenesis and de novo TLS neogenesis (123).
In the study by Rangel-Moreno and colleagues, the investigators concluded that CD4+ T cell-derived IL-17 was responsible for i-BALT development by inducing LTα-independent CXCL13 expression (131). Other studies have linked IL-17 and/or TH17 cells to TLS neogenesis (132, 133). A recently published study demonstrated the involvement of TH17 cells in synovial ectopic lymphoid structure development in both experimental and clinical rheumatoid arthritis (RA). This process was under the inhibitory control of IL-27, a cytokine that is often elevated in the inflamed synovium and serum of certain RA patients (134). Interestingly, the IL-17-producing capability of LTi cells is one of the features that has recently indicated an ancestral relationship between LTi and TH17 cells (126, 135). However, the identity of a distinct TLS-inducing cell type remains elusive, with B cells and TNF-producing myeloid cells also representing candidates (123, 130).
Dendritic cells, well known for their role in antigen presentation, are also implicated in the modulation and maintenance of the LN HEV phenotype in the adult, via expression of the LTβ ligand. After depletion of CD11c+ DCs in adult mice, cellularity and size of peripheral LNs was significantly reduced, expression of HEV markers was downregulated, and homing of lymphocytes to LNs impaired. HEVs reverted to an immature phenotype as a result of a direct interaction between DCs and endothelial cells, presumably via LT ligands expressed by DCs and endothelial cell-expressed LTβR (136). Similarly, DC-LAMP+ DCs positively correlated with HEV density in breast tumors and were significantly associated with a favorable clinical prognosis (41). The presence of DC-LAMP+ DCs was also shown to correlate with HEV density in primary melanoma (45). However, despite clustering of DCs around HEVs in breast cancer, the majority of DCs are located outside basal laminal layers encapsulating HEVs, as in LNs, and are therefore unlikely to be capable of direct contact with endothelial cells; a likely prerequisite for initiation of HEV neogenesis (41).
In the above-mentioned study by Peske and colleagues, there was a distinctive lack of PNAd+ tumor vasculature in intraperitoneal B16-OVA tumors of gene-targeted animals specifically devoid of CD8+ T cells and RAG2-deficient animals. Furthermore, PNAd-expressing vessels were restored in these tumors to levels observed in WT animals upon adoptive transfer of CD8+ T cells to RAG2-deficient mice. In contrast, NK cells could compensate for the loss of CD8+ T cells in induction of PNAd+ vasculature in subcutaneous B16-OVA tumors, suggesting that these two cellular populations can act in a redundant fashion in particular tumors (60).
Indeed, our own studies of the MCA-induced mouse model of carcinogenesis have shown that conventional CD4+ CD3− IL-7R+ RORγ+ LTi cells are absent from HEV+ tumors of Treg-depleted animals (61). Instead, by selectively depleting different subsets of immune cells by monoclonal antibody treatment, we found that CD8+ T lymphocytes are the primary cell directing neogenesis of HEVs (97). Hence, rather than a reliance on canonical LTi cells, as is the case for ontogenic SLO development, neogenesis of tumor-associated HEVs, and perhaps thereafter TLSs, appears to rely on cytokine-secreting CD8-expressing or NK effector lymphocytes.
While studies have implied a degree of lymphocyte dependency for the development of a mature HEV phenotype in LNs (137), L-selectin-dependent lymphocyte trafficking to peripheral LNs in RAG-1-deficient mice suggests that the development of functional LN HEVs can proceed in the absence of lymphocytes (138). HEV neogenesis in Treg-depleted MCA tumors and Treg replete B16 tumors could therefore represent a significant mechanistic departure from normal LN HEV development and indeed TLS development in non-malignant scenarios of chronic inflammation (60, 97). We hypothesize that robust ongoing antigenic stimulation provided by tumor-associated antigens (TAAs) leads to sufficient activation of intratumoral lymphocytes, which then secrete the required cytokines to induce HEV differentiation of existing tumor vasculature. In the case of strong antigens, such as OVA, this activation of lymphocytes can override Treg-induced immunosuppression to enable a degree of HEV neogenesis in the presence of these potently immunosuppressive cells. However, depletion of Treg, a type of immunotherapy currently being given to cancer patients in multiple clinical trials, appears to unleash the reins on effector lymphocytes, such that their activation leads to prolific HEV-inducing cytokine production in the tumor bed, robust HEV formation, and concomitant tumor control.
Function of TLSs in Cancer: Sentinel Bystanders or Active Proprietors of the Immune Response or Immunosuppression?
It has been suggested that, similar to SLOs, TLSs function primarily to potentiate the local immune response at the site of formation. Accordingly, TLSs would have the potential to exacerbate or control disease, depending on the nature of the pathology. By-in-large, the current consensus suggests that there would be rationale for therapeutically potentiating TLS formation in the contexts of microbial infection and malignancy, where exacerbation of local immunity could lead to clearance of infection or rejection of a tumor, but inhibiting the formation of TLSs in chronic inflammation and autoimmunity, where heightened local immune responses would contribute to disease progression (10).
However, TLSs are often documented in pathology by histological examination of tissues, which precludes the gathering of definitive evidence for their functional consequence. The majority of the current data linking TLSs with prognosis in patients or disease progression in animals is correlative by nature. The prognostic association of tumor-associated TLSs or HEVs in the absence of TLSs may be confounded by the fact that their neogenesis often occurs in the context of a robust immune response; the link between these ectopic lymphoid structures and a favorable clinical outcome may be indirect and simply reflect the presence of effector T cells. Conclusive functional data are significantly lacking, without which we cannot be sure as to the precise role of these ectopic lymphoid structures within pathological foci; a prerequisite for effective therapeutic targeting.
Studies have implied the occurrence of an active immune response within TLSs found in autoimmune conditions in both mice and humans, evidenced by AID activity in TLS GCs of Sjorgen’s syndrome patients’ salivary glands, and T cell priming and epitope spreading within TLSs in a mouse model of multiple sclerosis, for example (139, 140). One particular TLS, i-BALT can initiate both humoral and cellular immune responses to protect against influenza infection independently of SLOs in animals lacking spleen, LNs, and PPs (94). These structures were even shown to foster and support immunological memory (93). However, studies addressing the question of tumor-associated TLS function in disease progression have yet to provide definitive conclusive evidence for their role.
Particular TLS features are indicative of the ability to support an immune response to antigen. For instance, HEVs found within human and mouse tumors, either in association with defined TLSs or not, express the same adhesion molecules and chemokines as LN HEVs (PNAd, MAdCAM-1, CCL21, and ICAM-1), presumably endowing them with the ability to interact with and promote the egress of lymphocytes of the naive and central memory phenotype from the bloodstream (28, 55, 57, 60, 61). In a preclinical mouse model of colon carcinogenesis, Di Caro and colleagues were able to demonstrate the migration of GFP-labeled splenocytes to TLSs in the colonic mucosa, suggesting active recruitment of lymphocytes (32). Furthermore, the presence of thin walled vessels expressing typical LN lymphatic markers such as LYVE-1 and podoplanin has been documented in cancer-associated TLSs, indicating a means for the entrance of antigen-presenting cells (28, 32). However, live in vivo imaging studies of lymphocytes entering via HEVs and antigen-presenting cells entering via lymphatic vessels are required to solidify the functional consequence of the presence of such vascular structures.
The presence of tumor-associated TLSs in NSCLC, for which mature DCs serve as a reliable marker, was found to shape the T cell infiltrate toward an activated, Th1 and cytotoxic orientation (29). Interestingly, patients with a high infiltration of CD8+ T cells in combination with a high density of TLSs demonstrated significantly improved survival relative to patients with high CD8+ T cell infiltration in the absence of TLSs, suggesting TLSs actively license the prognostic value of intratumoral cytotoxic T cells. Similarly, the presence of plasma cells expressing markers of antigen-specific responses within TLSs in ovarian cancer was associated with increased responses of tumor-infiltrating CD8+ T cells (52). While such studies of gene expression analysis in humans can only ever offer suggestive evidence for a functional role for tumor-associated TLSs, they do propose that TLSs may educate tumor-infiltrating lymphocytes to control tumors better.
Studies attempting to address the question of whether TLSs are capable of supporting an antigen-specific response to endogenous antigen in vivo in animal models have yielded intriguing results. Expression of LIGHT in a fibrosarcoma cell line resulted in upregulation of CCL21 and MAdCAM-1 on tumor vasculature, facilitating recruitment of vast numbers of naive CD8+ T lymphocytes, which appeared to then be sufficiently activated in situ to facilitate rejection of established tumors (91). Furthermore, TLS aggregates induced in mouse melanoma lesions by targeted LTα expression by tumor cells seem to be able to foster an active antitumor immune response in the absence of all canonical SLOs in vivo: Schrama and colleagues documented retarded tumor growth and even tumor regression in splenectomized LTα−/− animals. Furthermore, endogenous CD8+ T cells specific for the melanoma-associated antigen TRP-2 were detected by in situ tetramer staining only within tumors of LTα−/− animals in which TLSs had been induced (141). While previous studies had demonstrated the ability of B16 melanoma tumors to foster T cell priming in the absence of SLOs in LTα−/− mice (142), the study by Schrama and colleagues was one of the first studies to specifically associate this capability with the presence of intratumoral TLSs.
However, it is possible that tumor-associated TLSs are not essential for in situ T cell priming in all cases. The absence of fully formed TLSs in subcutaneous tumors that support HEV neogenesis does not appear to influence the ability of these tumors to facilitate in situ priming and activation of tumor-specific naive T lymphocytes (60, 61, 97). It is possible that once lymphocytes have successfully accessed the tumor site, via lymphoid-like vasculature, priming and initiation of an in situ immune response can occur without the support of an organized lymphoid structure. However, this may require activating signals that are not present in every tumor microenvironment: in the case of LIGHT expressing fibrosarcomas, the dual role of LIGHT as a potent costimulatory molecule for T cells in combination with tumor-associated antigens could suffice for robust T cell priming and expansion (91).
Coronella and colleagues described the presence of sophisticated TLSs within human infiltrating ductal carcinoma of the breast, including segregated T and B cell zones with GCs and interdigitating FDCs (38). What is more, the researchers demonstrated a preponderance of clonal intratumoral B cells, relative to peripheral B cells, by sequencing IgG1 heavy chains isolated from three tumors. Analysis of somatic hypermutation levels and patterns were suggestive of affinity maturation occurring within TLS GCs. These findings were later supported by another independent study of breast carcinoma (40) and a separate study of metastatic melanoma (50). Indeed, GCs in LNs are indicative of an active immune response, and studies have demonstrated a humoral immune response associated with TLS GCs in human lung cancer (143) and a correlation between patient survival and TLS GC makers in breast cancer patients (39). These data indicate that tumor-associated TLSs have the capacity to support in situ oligoclonal B cell responses driven by tumor tissue associated antigens.
The cellular composition, organization, and localization of tumor-associated TLSs may dictate whether these structures confer an advantageous or deleterious outcome for disease progression. Following on from the observation that the density of DC-LAMP+ mature DCs, as a marker of TLSs, correlated with long-term survival in NSCLC patients (27), Germain and colleagues went on to show that a high tumor follicular B cell density correlates with increased survival and that this prognostic value is enhanced when follicular B cells are present in combination with high mature DC densities (143). However, these analyses were conducted across the entire tumor area and not restricted to tumor-associated TLSs, precluding any inference regarding immune responses specifically ongoing within TLSs. In a separate study, García-Hernández and colleagues found a dramatic change in the cellular composition of prostate cancer-associated TLSs in patients that experienced spontaneous tumor regression: tumor-associated TLSs in patients with evanescent prostate cancer consisted of lower frequencies of Tregs and greater frequencies of T-bet+ Th1 T cells than those in patients with more advanced disease (49). This dichotomy in TLS composition between immunostimulatory and immunosuppressive components could be crucial in dictating the immunological outcome of these tumor-associated structures.
In accordance with their well-defined immunosuppressive role, the recruitment of Foxp3+ Tregs and myeloid derived suppressor cells (MDSCs) to TLSs within B16 melanomas engineered to express CCL21 led to promotion of tumor growth (59). In addition, high numbers of Foxp3+ Tregs within lymphoid aggregates surrounding primary breast tumors was indicative of an increased risk of disease relapse and death (42). Furthermore, Tregs within tumor-associated TLSs actively suppressed antitumor immune responses in a mouse model of lung adenocarcinoma (58). These studies demonstrate that tumor-associated TLSs are sometimes associated with immunosuppression rather than immune activation. It is possible, therefore, that in the absence of a strong stimulus provided by tumor neoantigens or without adoptive transfer of transgenic T cells targeted to known tumor antigens, these ectopic lymphoid structures can foster immunosuppression and support rather than limit tumor growth. Indeed, in a mouse model of chronic hepatitis driven by constitutive IKK-NFkB signaling in hepatocytes, which develop aggressive malignant HCC (IKKβ(EE)Hep mice), researchers noted the development of TLSs highly reminiscent of human hepatic TLSs associated with HCC (54). Importantly, hepatic TLSs were found to foster HCC progenitor cells in the mouse model and depletion of TLSs by ablation of adaptive immunity via crossing IKKβ(EE)Hep mice to lymphocyte deficient Rag1−/− mice substantially attenuated hepatocarcinogenesis. Hence, in certain cancers, tumor-associated TLSs may even serve as immunological microniches promoting the generation of progenitor cancer cells, rather than an effective antitumor immune response.
Importantly, in the study by Gobert and colleagues, the specific location of Treg infiltration in primary breast tumors proved critical to the prognostic value of this observation; Treg presence within the tumor bed itself did not influence disease evolution. However, Tregs present within tumor-associated TLSs displayed a highly activated phenotype, suggestive of their in situ activation in response to TAA presented within the TLSs (42). Collectively, these data are indicative of local suppression of T cell responses by activated Tregs within tumor-associated lymphoid structures rather than within the tumor mass itself, supporting the idea that TLSs are active sites of immune responses.
Crucially, depletion of Tregs within TLSs of mouse lung adenocarcinomas led to enhanced costimulatory capacity of DCs, T cell proliferation, and protective antitumor immune responses leading to tumor regression (58). Not only does this study support the developing hypothesis that intratumoral TLSs can represent sites of active local adaptive immunity against tumor but it also highlights a potential requirement to overcome the profound immunosuppression within the tumor microenvironment to license an effective antitumor response fostered by TLSs. Indeed, in our own studies, HEVs that develop in the absence of Tregs in MCA-induced fibrosarcomas are associated with significantly higher intratumoral T lymphocyte frequencies and reduced tumor growth rates (61, 97). While HEVs have been documented in other mouse models of cancer in the presence of Tregs, amplification protocols are required to visualize HEVs and tumor growth control is negligible despite adoptive transfer of high numbers of transgenic T cells reactive to tumor-expressed antigen (60). It is possible therefore that in the absence of strong antigenic stimulation (which are provided in transgenic T cell models of cancer) or immune activation, HEVs and tumor-associated TLSs could foster immunosuppression over antitumor immunity.
Indeed, the precise location of TLSs in regard to the tumor mass may have important implications for the prognostic value of these structures. Tumor-associated TLSs and/or HEVs can be extratumoral, positioned at or outside the tumor invasive margin, or intratumoral, situated within the true tumor mass or tumor nests. In colorectal carcinoma (CRC), PNAd+ HEVs are rarely found within the tumor stroma or epithelium and are instead mainly situated in the surrounding extratumoral area (31). The lack of an association between HEVs and prognosis in CRC could suggest that extratumoral lymphoid neogenesis may be indicative of an immune response driven by tumor and one therefore in support of cancer progression and immune evasion. Other studies documenting HEVs truly embedded within the tumor stroma have found a positive prognostic value associated with these structures (26, 45). These discrepancies could also be reconciled by the importance of TLSs/HEVs in enabling infiltration of T cells into the tumor; extratumoral HEVs were not associated with increased TIL frequencies (31), whereas intratumoral HEVs were (26, 45). Similarly, while extratumoral TLS density was not a prognostic marker in pancreatic cancer patients, intratumoral TLSs functioned as an independent favorable prognosticator (53). Furthermore, in support of the idea that the function and therefore prognostic significance of TLSs/HEVs differs with disease stage also, there is no association between extratumoral HEVs in advanced (Dukes’ C, or stage III) CRC tumors and prognosis while lymphoid aggregates in stage II CRC (with no LN involvement) are associated with a favorable prognosis (31, 32). Hence, tumor-associated TLSs/HEVs may only function in the antitumor response during early disease stages, a capability that may be lost during cancer progression in parallel to the establishment of an immunosuppressive tumor microenvironment and loss of tumor immunogenicity. Finally, the primary tumor origin of distal metastases appears to significantly influence the clinical impact, and hence presumably the immune response, of metastasis associated TLSs: T cell and DC infiltration in TLSs of lung metastases of colorectal carcinomas is a predictor of longer overall survival but appears to correlate with poor survival in lung metastases of renal cell carcinoma (RCC) (36, 56). Collectively, these observations indicate that TLS/HEV location in relation to the tumor mass, disease stage, and tumor origin may all be absolutely crucial in dictating the resulting immune response to tumor.
These data underpin the importance of assessing the functional consequences of TLSs and/or HEVs in the absence of TLSs in different cancers prior to therapeutic intervention. It appears that tumor-associated lymphoid structures are capable of supporting an effective immune response in certain contexts. However, a word of caution is issued by studies demonstrating an immune evasive and even disease-promoting role for TLSs (Figure 3). It could be that robust immune system activation, for instance via Treg depletion, is required for the beneficial role of intratumoral lymphoid aggregates and vasculature to be unmasked, and encouraging ectopic lymphoid-like vasculature in combination with such therapies may induce an effective immune response to eradicate tumor.
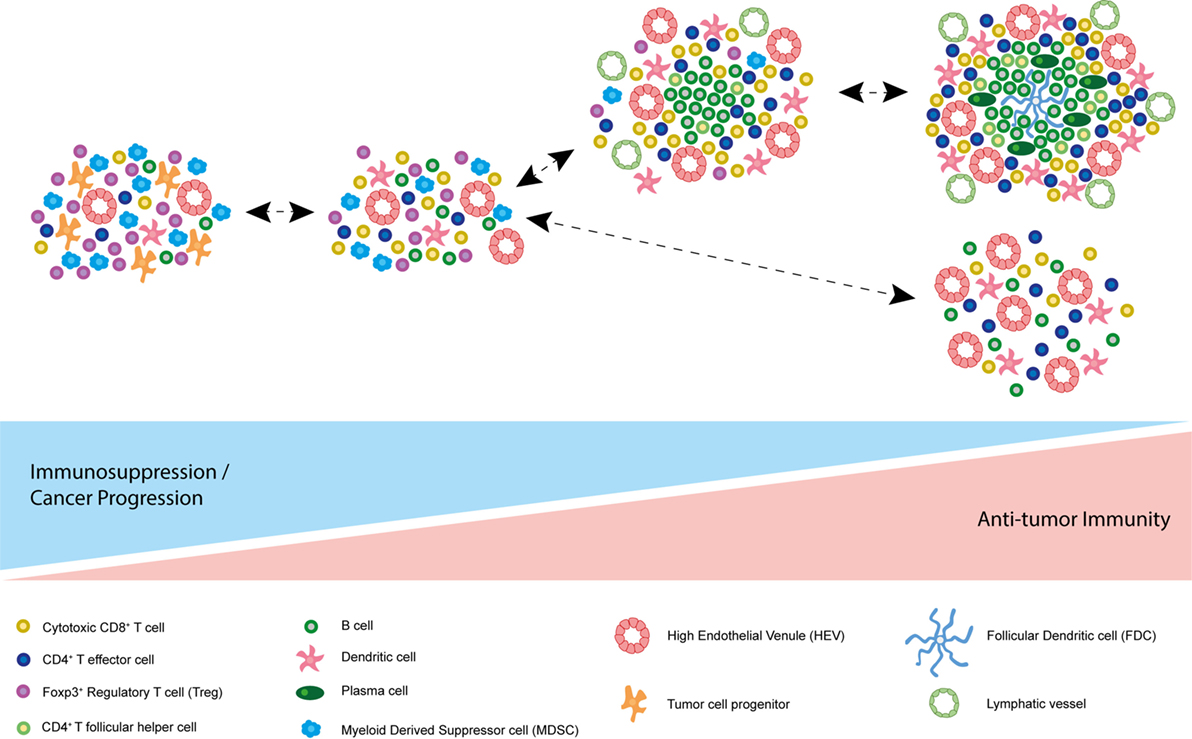
Figure 3. Model of spectrum of tumor-associated tertiary lymphoid structures (TLSs). Based on current literature, it is possible that the function of different tumor-associated TLSs differs due to their composition. At one end of the spectrum, accumulation of immunosuppressive Foxp3+ regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) may promote immune tolerance to the tumor. In some cases, TLSs have been described as immunological microniches, which foster malignant progenitor cells, thereby driving cancer progression. At the other end of the spectrum, many cancer-associated TLSs appear to correlate with improved survival and disease regression in multiple human malignancies and animal models. TLS characteristics associated with a good prognosis and an antitumor immune response in addition to cytotoxic CD8+ T cells and CD4+ effector T cells include plasma cells, T follicular helper cells, B cells, follicular dendritic cells (FDCs), mature DC-LAMP+ dendritic cells, and PNAd+ high endothelial venules (HEVs). Of note, these differing states of tertiary lymphoid neogensis are not mutually exclusive and can co-exist in the same tumor at the same point in time.
Concluding Remarks: Toward Therapeutic Targeting of TLS or HEV Neogenesis in Cancer
Observed correlations between cancer-associated TLSs or HEVs in the absence of TLSs and favorable prognosis in several human malignancies have provided the rationale for therapeutically targeting TLSs in an attempt to drive an effective antitumor immune response. Agents directed against key signaling molecules now known to be involved in TLS development have already entered the clinical arena. For instance, clinical trials are investigating the efficacy of intratumoral injections of autologous DCs transduced to express CCL21 in stage IIIB and IV and recurrent NSCLC patients (NCT00601094 and NCT01574222). Although these trials’ primary aim is to determine safety, best dose, and side effects of treatment, secondary objectives are to monitor changes in the infiltrating immune cell populations by immunohistochemistry, which may provide insights into TLS neogenesis within tumors before and after treatment, and whether this will in turn correlate with improved antitumor immunity.
However, as we have learnt, many of the signaling circuitries are overlapping with those essential for SLO development and maintenance, making selective targeting challenging. One signaling pathway that our group and others have identified as playing a major role in HEV neogenesis in tumors but little role in canonical SLO formation is the TNFR signaling pathway (60, 95, 97, 144, 145). There is evidence to suggest that anti-TNF therapy can reverse aspects of TLS neogenesis in RA patients (146), which could account, at least in part, for the therapeutic efficacy of this treatment. Therefore, this pathway represents a potentially selective targetable axis via which HEV neogenesis, and possibly TLS development, could be encouraged in tumors. Targeting of TNFα to the tumor vasculature has already been shown to upregulate adhesion molecule expression on the surface of endothelial cells and subsequently enhance CD8+ cytotoxic T cell infiltration (147). It will be intriguing to determine whether targeting of TNFα to the tumor, or otherwise stimulating TNFR signaling at this site, may encourage differentiation of tumor vessels toward an HEV-like phenotype, and whether this will in turn enhance T cell infiltration further, and/or antitumor responses.
This review aims to tell a cautionary tale, however: it is becoming clear that lymphoid neogenesis is a highly complex process, which may have wide ranging implications, from antitumor immunity-promoting to immunosuppression. Recent studies have even highlighted an active disease-promoting role for intratumoral TLSs (54). It appears that the function of tumor-associated lymphoid structures may be dictated by their cellular composition and the surrounding immune contexture. It is obvious that we must learn more about the function of these ectopic structures in different human malignancies before we attempt to induce their formation in people. It could be that lymphoid structures within tumors may only serve to promote the antitumor immune response in the context of profound immune activation, a state that can be induced by depletion of immunosuppressive cells such as Tregs and MDSCs. Treg-depleting therapies such as low-dose cyclophosphamide, PI3Kδ inhibitors, and IDO inhibitors are already being used in the clinic for the treatment of human cancers or progressing through clinical trials (148–152) (NCT00567931 and NCT01042535). It is of utmost importance for us to establish whether these treatments alone can induce HEVs or TLSs in tumors. It could be that combining immune-modulating agents with HEV/TLS-targeted therapies may represent the recipe for ultimate effective immune-mediated tumor control.
Author Contributions
All authors contributed to the conceptualization and design of the work. EJC wrote the article and designed the figures. All authors made significant contributions to the editing and reviewing of the manuscript at multiple stages.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank all members of the Gallimore, Ager and Jones labs for stimulating discussions around Tertiary Lymphoid Structures in Cancer, and the Funding bodies who supported this work.
Funding
EJC is supported by a PhD studentship funded by Cancer Research UK. AG is supported by a programme grant from Cancer Research UK (C16731/A21200). AA is supported by programme grants from the Wellcome Trust (094511/Z/10/Z) and Medical Research Council UK (MR/L008742/1). GWJ is supported by an Arthritis Research UK Career Development Fellowship (grant number 20305).
Abbreviations
SLO, secondary lymphoid organ; TLS, tertiary lymphoid structure; MALT, mucosal-associated lymphoid tissue; LN, lymph node; PP, Peyer’s patch; NALT, nasal-associated lymphoid tissue; BALT, bronchial-associated lymphoid tissue; CCL, CC motif chemokine ligand; CCR, CC-chemokine receptor; CXCL, CXC motif chemokine ligand; CXCR, CXC-chemokine receptor; FDC, follicular dendritic cell; HEV, high endothelial venule.
References
1. Andrian von UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol (2003) 3:867–78. doi:10.1038/nri1222
2. van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol (2010) 10:664–74. doi:10.1038/nri2832
3. Gardner JM, Devoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X, et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science (2008) 321:843–7. doi:10.1126/science.1159407
4. Lee J-W, Epardaud M, Sun J, Becker JE, Cheng AC, Yonekura A-R, et al. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol (2007) 8:181–90. doi:10.1038/ni1427
5. Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Annu Rev Immunol (2008) 26:627–50. doi:10.1146/annurev.immunol.26.021607.090257
6. Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol (2006) 7:344–53. doi:10.1038/ni1330
7. Dieu-Nosjean M-C, Goc J, Giraldo NA, Sautès-Fridman C, Fridman W-H. Tertiary lymphoid structures in cancer and beyond. Trends Immunol (2014) 35:571–80. doi:10.1016/j.it.2014.09.006
8. Picker LJ, Butcher EC. Physiological and molecular mechanisms of lymphocyte homing. Annu Rev Immunol (1992) 10:561–91. doi:10.1146/annurev.iy.10.040192.003021
9. Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med (1996) 183:1461–72. doi:10.1084/jem.183.4.1461
10. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol (2014) 14:447–62. doi:10.1038/nri3700
11. Girard J-P, Moussion C, Förster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol (2012) 12:762–73. doi:10.1038/nri3298
12. Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol (2009) 183:2205–12. doi:10.4049/jimmunol.0804324
13. Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, et al. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity (2009) 30:264–76. doi:10.1016/j.immuni.2008.12.014
14. Katakai T, Suto H, Sugai M, Gonda H, Togawa A, Suematsu S, et al. Organizer-like reticular stromal cell layer common to adult secondary lymphoid organs. J Immunol (2008) 181:6189–200. doi:10.4049/jimmunol.181.9.6189
15. Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol (2005) 5:617–28. doi:10.1038/nri1670
16. Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol (2004) 22:129–56. doi:10.1146/annurev.immunol.21.090501.080131
17. Ager A, May MJ. Understanding high endothelial venules: lessons for cancer immunology. Oncoimmunology (2015) 4:e1008791. doi:10.1080/2162402X.2015.1008791
18. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol (2012) 33:297–305. doi:10.1016/j.it.2012.04.006
19. Lu TT, Browning JL. Role of the lymphotoxin/LIGHT system in the development and maintenance of reticular networks and vasculature in lymphoid tissues. Front Immunol (2014) 5:47. doi:10.3389/fimmu.2014.00047
20. Stranford S, Ruddle NH. Follicular dendritic cells, conduits, lymphatic vessels, and high endothelial venules in tertiary lymphoid organs: parallels with lymph node stroma. Front Immunol (2012) 3:350. doi:10.3389/fimmu.2012.00350
21. Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol (2006) 6:205–17. doi:10.1038/nri1786
22. Carragher DM, Rangel-Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immunol (2008) 20:26–42. doi:10.1016/j.smim.2007.12.004
23. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102:553–63. doi:10.1016/S0092-8674(00)00078-7
24. Goc J, Fridman W-H, Sautès-Fridman C, Dieu-Nosjean M-C. Characteristics of tertiary lymphoid structures in primary cancers. Oncoimmunology (2013) 2:e26836. doi:10.4161/onci.26836
25. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348:74–80. doi:10.1126/science.aaa6204
26. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie J-J, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res (2011) 71:5678–87. doi:10.1158/0008-5472.CAN-11-0431
27. Dieu-Nosjean M-C, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol (2008) 26:4410–7. doi:10.1200/JCO.2007.15.0284
28. de Chaisemartin L, Goc J, Damotte D, Validire P, Magdeleinat P, Alifano M, et al. Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer. Cancer Res (2011) 71:6391–9. doi:10.1158/0008-5472.CAN-11-0952
29. Goc J, Germain C, Vo-Bourgais TKD, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res (2014) 74:705–15. doi:10.1158/0008-5472.CAN-13-1342
30. Suzuki A, Masuda A, Nagata H, Kameoka S, Kikawada Y, Yamakawa M, et al. Mature dendritic cells make clusters with T cells in the invasive margin of colorectal carcinoma. J Pathol (2002) 196:37–43. doi:10.1002/path.1018
31. Bento DC, Jones E, Junaid S, Tull J, Williams GT, Godkin A, et al. High endothelial venules are rare in colorectal cancers but accumulate in extra-tumoral areas with disease progression. Oncoimmunology (2015) 4:e974374. doi:10.4161/2162402X.2014.974374
32. Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A, et al. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin Cancer Res (2014) 20:2147–58. doi:10.1158/1078-0432.CCR-13-2590
33. McMullen TPW, Lai R, Dabbagh L, Wallace TM, de Gara CJ. Survival in rectal cancer is predicted by T cell infiltration of tumour-associated lymphoid nodules. Clin Exp Immunol (2010) 161:81–8. doi:10.1111/j.1365-2249.2010.04147.x
34. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol (2011) 179:37–45. doi:10.1016/j.ajpath.2011.03.007
35. Bergomas F, Grizzi F, Doni A, Pesce S, Laghi L, Allavena P, et al. Tertiary intratumor lymphoid tissue in colo-rectal cancer. Cancers (Basel) (2011) 4:1–10. doi:10.3390/cancers4010001
36. Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean M-C, Riquet M, et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res (2013) 19:4079–91. doi:10.1158/1078-0432.CCR-12-3847
37. Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med (1999) 190:1417–26. doi:10.1084/jem.190.10.1417
38. Coronella JA, Spier C, Welch M, Trevor KT, Stopeck AT, Villar H, et al. Antigen-driven oligoclonal expansion of tumor-infiltrating B cells in infiltrating ductal carcinoma of the breast. J Immunol (2002) 169:1829–36. doi:10.4049/jimmunol.169.4.1829
39. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4? follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest (2013) 123:2873–92. doi:10.1172/JCI67428
40. Nzula S, Going JJ, Stott DI. Antigen-driven clonal proliferation, somatic hypermutation, and selection of B lymphocytes infiltrating human ductal breast carcinomas. Cancer Res (2003) 63:3275–80.
41. Martinet L, Filleron T, Le Guellec S, Rochaix P, Garrido I, Girard J-P. High endothelial venule blood vessels for tumor-infiltrating lymphocytes are associated with lymphotoxin β-producing dendritic cells in human breast cancer. J Immunol (2013) 191:2001–8. doi:10.4049/jimmunol.1300872
42. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res (2009) 69:2000–9. doi:10.1158/0008-5472.CAN-08-2360
43. Figenschau SL, Fismen S, Fenton KA, Fenton C, Mortensen ES. Tertiary lymphoid structures are associated with higher tumor grade in primary operable breast cancer patients. BMC Cancer (2015) 15:101. doi:10.1186/s12885-015-1116-1
44. Song IH, Heo S-H, Bang WS, Park HS, Park IA, Kim Y-A, et al. Predictive value of tertiary lymphoid structures assessed by high endothelial venule counts in the neoadjuvant setting of triple-negative breast cancer. Cancer Res Treat (2017) 49:399–407. doi:10.4143/crt.2016.215
45. Martinet L, Le Guellec S, Filleron T, Lamant L, Meyer N, Rochaix P, et al. High endothelial venules (HEVs) in human melanoma lesions: major gateways for tumor-infiltrating lymphocytes. Oncoimmunology (2012) 1:829–39. doi:10.4161/onci.20492
46. Ladányi A, Kiss J, Somlai B, Gilde K, Fejos Z, Mohos A, et al. Density of DC-LAMP(+) mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol Immunother (2007) 56:1459–69. doi:10.1007/s00262-007-0286-3
47. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep (2012) 2:765. doi:10.1038/srep00765
48. Avram G, Sánchez-Sendra B, Martín JM, Terrádez L, Ramos D, Monteagudo C. The density and type of MECA-79-positive high endothelial venules correlate with lymphocytic infiltration and tumour regression in primary cutaneous melanoma. Histopathology (2013) 63:852–61. doi:10.1111/his.12235
49. García-Hernández ML, Uribe-Uribe NO, Espinosa-González R, Kast WM, Khader SA, Rangel-Moreno J. A unique cellular and molecular microenvironment is present in tertiary lymphoid organs of patients with spontaneous prostate cancer regression. Front Immunol (2017) 8:563. doi:10.3389/fimmu.2017.00563
50. Cipponi A, Mercier M, Seremet T, Baurain J-F, Théate I, van den Oord J, et al. Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res (2012) 72:3997–4007. doi:10.1158/0008-5472.CAN-12-1377
51. Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, et al. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res (2012) 18:3281–92. doi:10.1158/1078-0432.CCR-12-0234
52. Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin Cancer Res (2016) 22:3005–15. doi:10.1158/1078-0432.CCR-15-2762
53. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer (2015) 112:1782–90. doi:10.1038/bjc.2015.145
54. Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol (2015) 16:1235–44. doi:10.1038/ni.3290
55. Sakai Y, Hoshino H, Kitazawa R, Kobayashi M. High endothelial venule-like vessels and lymphocyte recruitment in testicular seminoma. Andrology (2014) 2:282–9. doi:10.1111/j.2047-2927.2014.00192.x
56. Giraldo NA, Becht E, Pagès F, Skliris G, Verkarre V, Vano Y, et al. Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin Cancer Res (2015) 21:3031–40. doi:10.1158/1078-0432.CCR-14-2926
57. Low S, Sakai Y, Hoshino H, Hirokawa M, Kawashima H, Higuchi K, et al. High endothelial venule-like vessels and lymphocyte recruitment in diffuse sclerosing variant of papillary thyroid carcinoma. Pathology (2016) 48:666–74. doi:10.1016/j.pathol.2016.08.002
58. Joshi NS, Akama-Garren EH, Lu Y, Lee D-Y, Chang GP, Li A, et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity (2015) 43:579–90. doi:10.1016/j.immuni.2015.08.006
59. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science (2010) 328:749–52. doi:10.1126/science.1185837
60. Peske JD, Thompson ED, Gemta L, Baylis RA, Fu Y-X, Engelhard VH. Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun (2015) 6:7114. doi:10.1038/ncomms8114
61. Hindley JP, Jones E, Smart K, Bridgeman H, Lauder SN, Ondondo B, et al. T-cell trafficking facilitated by high endothelial venules is required for tumor control after regulatory T-cell depletion. Cancer Res (2012) 72:5473–82. doi:10.1158/0008-5472.CAN-12-1912
62. Buckley CD, Barone F, Nayar S, Bénézech C, Caamaño J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol (2015) 33:715–45. doi:10.1146/annurev-immunol-032713-120252
63. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
64. Attridge K, Walker LSK. Homeostasis and function of regulatory T cells (Tregs) in vivo: lessons from TCR-transgenic Tregs. Immunol Rev (2014) 259:23–39. doi:10.1111/imr.12165
65. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer (2010) 127:759–67. doi:10.1002/ijc.25429
66. Gallimore A, Godkin A. Regulatory T cells and tumour immunity – observations in mice and men. Immunology (2008) 123:157–63. doi:10.1111/j.1365-2567.2007.02748.x
67. Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MWL, Ngiow SF, et al. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res (2010) 70:7788–99. doi:10.1158/0008-5472.CAN-10-1736
68. Li X, Kostareli E, Suffner J, Garbi N, Hämmerling GJ. Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur J Immunol (2010) 40:3325–35. doi:10.1002/eji.201041093
69. Teng MWL, Ngiow SF, Scheidt von B, McLaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res (2010) 70:7800–9. doi:10.1158/0008-5472.CAN-10-1681
70. Kocks JR, Davalos-Misslitz ACM, Hintzen G, Ohl L, Förster R. Regulatory T cells interfere with the development of bronchus-associated lymphoid tissue. J Exp Med (2007) 204:723–34. doi:10.1084/jem.20061424
71. Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med (2007) 204:735–45. doi:10.1084/jem.20061405
72. Contractor NV, Bassiri H, Reya T, Park AY, Baumgart DC, Wasik MA, et al. Lymphoid hyperplasia, autoimmunity, and compromised intestinal intraepithelial lymphocyte development in colitis-free gnotobiotic IL-2-deficient mice. J Immunol (1998) 160:385–94.
73. Foo SY, Zhang V, Lalwani A, Lynch JP, Zhuang A, Lam CE, et al. Regulatory T cells prevent inducible BALT formation by dampening neutrophilic inflammation. J Immunol (2015) 194:4567–76. doi:10.4049/jimmunol.1400909
74. van de Pavert SA, Mebius RE. Development of secondary lymphoid organs in relation to lymphatic vasculature. Adv Anat Embryol Cell Biol (2014) 214:81–91. doi:10.1007/978-3-7091-1646-3_7
75. Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol (2003) 3:292–303. doi:10.1038/nri1054
76. Vondenhoff MF, Greuter M, Goverse G, Elewaut D, Dewint P, Ware CF, et al. LTbetaR signaling induces cytokine expression and up-regulates lymphangiogenic factors in lymph node anlagen. J Immunol (2009) 182:5439–45. doi:10.4049/jimmunol.0801165
77. Ager A. High endothelial venules and other blood vessels: critical regulators of lymphoid organ development and function. Front Immunol (2017) 8:45. doi:10.3389/fimmu.2017.00045
78. Hjelmström P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am J Pathol (2000) 156:1133–8. doi:10.1016/S0002-9440(10)64981-4
79. Drayton DL, Ying X, Lee J, Lesslauer W, Ruddle NH. Ectopic LT alpha beta directs lymphoid organ neogenesis with concomitant expression of peripheral node addressin and a HEV-restricted sulfotransferase. J Exp Med (2003) 197:1153–63. doi:10.1084/jem.20021761
80. Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity (2000) 12:471–81. doi:10.1016/S1074-7613(00)80199-5
81. Fava RA, Kennedy SM, Wood SG, Bolstad AI, Bienkowska J, Papandile A, et al. Lymphotoxin-beta receptor blockade reduces CXCL13 in lacrimal glands and improves corneal integrity in the NOD model of Sjögren’s syndrome. Arthritis Res Ther (2011) 13:R182. doi:10.1186/ar3507
82. Gatumu MK, Skarstein K, Papandile A, Browning JL, Fava RA, Bolstad AI. Blockade of lymphotoxin-beta receptor signaling reduces aspects of Sjögren’s syndrome in salivary glands of non-obese diabetic mice. Arthritis Res Ther (2009) 11:R24. doi:10.1186/ar2617
83. Gräbner R, Lötzer K, Döpping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE-/- mice. J Exp Med (2009) 206:233–48. doi:10.1084/jem.20080752
84. Seleznik GM, Reding T, Romrig F, Saito Y, Mildner A, Segerer S, et al. Lymphotoxin β receptor signaling promotes development of autoimmune pancreatitis. Gastroenterology (2012) 143:1361–74. doi:10.1053/j.gastro.2012.07.112
85. Motallebzadeh R, Rehakova S, Conlon TM, Win TS, Callaghan CJ, Goddard M, et al. Blocking lymphotoxin signaling abrogates the development of ectopic lymphoid tissue within cardiac allografts and inhibits effector antibody responses. FASEB J (2012) 26:51–62. doi:10.1096/fj.11-186973
86. Furtado GC, Marinkovic T, Martin AP, Garin A, Hoch B, Hubner W, et al. Lymphotoxin beta receptor signaling is required for inflammatory lymphangiogenesis in the thyroid. Proc Natl Acad Sci U S A (2007) 104:5026–31. doi:10.1073/pnas.0606697104
87. Schrama D, thor Straten P, Fischer WH, McLellan AD, Bröcker EB, Reisfeld RA, et al. Targeting of lymphotoxin-alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity (2001) 14:111–21. doi:10.1016/S1074-7613(01)00094-2
88. Scheu S, Alferink J, Pötzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med (2002) 195:1613–24. doi:10.1084/jem.20020215
89. Weinstein AM, Storkus WJ. Therapeutic lymphoid organogenesis in the tumor microenvironment. Adv Cancer Res (2015) 128:197–233. doi:10.1016/bs.acr.2015.04.003
90. Gantsev SK, Umezawa K, Islamgulov DV, Khusnutdinova EK, Ishmuratova RS, Frolova VY, et al. The role of inflammatory chemokines in lymphoid neoorganogenesis in breast cancer. Biomed Pharmacother (2013) 67:363–6. doi:10.1016/j.biopha.2013.03.017
91. Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol (2004) 5:141–9. doi:10.1038/ni1029
92. Johansson-Percival A, He B, Li Z-J, Kjellén A, Russell K, Li J, et al. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat Immunol (2017) 18:1207–17. doi:10.1038/ni.3836
93. Moyron-Quiroz JE, Rangel-Moreno J, Hartson L, Kusser K, Tighe MP, Klonowski KD, et al. Persistence and responsiveness of immunologic memory in the absence of secondary lymphoid organs. Immunity (2006) 25:643–54. doi:10.1016/j.immuni.2006.08.022
94. Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, et al. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med (2004) 10:927–34. doi:10.1038/nm1091
95. De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science (1994) 264:703–7. doi:10.1126/science.8171322
96. Banks TA, Rouse BT, Kerley MK, Blair PJ, Godfrey VL, Kuklin NA, et al. Lymphotoxin-alpha-deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J Immunol (1995) 155:1685–93.
97. Colbeck EJ, Jones E, Hindley JP, Smart K, Schulz R, Browne M, et al. Treg depletion licenses T cell-driven HEV neogenesis and promotes tumor destruction. Cancer Immunol Res (2017) 5(11):1005–15. doi:10.1158/2326-6066.CIR-17-0131
98. Ansel KM, Ngo VN, Hyman PL, Luther SA, Förster R, Sedgwick JD, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature (2000) 406:309–14. doi:10.1038/35018581
99. Luther SA, Ansel KM, Cyster JG. Overlapping roles of CXCL13, interleukin 7 receptor alpha, and CCR7 ligands in lymph node development. J Exp Med (2003) 197:1191–8. doi:10.1084/jem.20021294
100. van de Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol (2009) 10:1193–9. doi:10.1038/ni.1789
101. Ohl L, Henning G, Krautwald S, Lipp M, Hardtke S, Bernhardt G, et al. Cooperating mechanisms of CXCR5 and CCR7 in development and organization of secondary lymphoid organs. J Exp Med (2003) 197:1199–204. doi:10.1084/jem.20030169
102. Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell (1999) 99:23–33. doi:10.1016/S0092-8674(00)80059-8
103. Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol (2002) 169:424–33. doi:10.4049/jimmunol.169.1.424
104. Chen S-C, Vassileva G, Kinsley D, Holzmann S, Manfra D, Wiekowski MT, et al. Ectopic expression of the murine chemokines CCL21a and CCL21b induces the formation of lymph node-like structures in pancreas, but not skin, of transgenic mice. J Immunol (2002) 168:1001–8. doi:10.4049/jimmunol.168.3.1001
105. Fan L, Reilly CR, Luo Y, Dorf ME, Lo D. Cutting edge: ectopic expression of the chemokine TCA4/SLC is sufficient to trigger lymphoid neogenesis. J Immunol (2000) 164:3955–9. doi:10.4049/jimmunol.164.8.3955
106. Marinkovic T, Garin A, Yokota Y, Fu Y-X, Ruddle NH, Furtado GC, et al. Interaction of mature CD3+CD4+ T cells with dendritic cells triggers the development of tertiary lymphoid structures in the thyroid. J Clin Invest (2006) 116:2622–32. doi:10.1172/JCI28993
107. Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Kusser K, Randall TD. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc Natl Acad Sci U S A (2007) 104:10577–82. doi:10.1073/pnas.0700591104
108. Wengner AM, Höpken UE, Petrow PK, Hartmann S, Schurigt U, Bräuer R, et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum (2007) 56:3271–83. doi:10.1002/art.22939
109. Winter S, Loddenkemper C, Aebischer A, Räbel K, Hoffmann K, Meyer TF, et al. The chemokine receptor CXCR5 is pivotal for ectopic mucosa-associated lymphoid tissue neogenesis in chronic Helicobacter pylori-induced inflammation. J Mol Med (2010) 88:1169–80. doi:10.1007/s00109-010-0658-6
110. Hamanishi J, Mandai M, Matsumura N, Baba T, Yamaguchi K, Fujii S, et al. Activated local immunity by CC chemokine ligand 19-transduced embryonic endothelial progenitor cells suppresses metastasis of murine ovarian cancer. Stem Cells (2010) 28:164–73. doi:10.1002/stem.256
111. Mebius RE, Streeter PR, Michie S, Butcher EC, Weissman IL. A developmental switch in lymphocyte homing receptor and endothelial vascular addressin expression regulates lymphocyte homing and permits CD4+ CD3- cells to colonize lymph nodes. Proc Natl Acad Sci U S A (1996) 93:11019–24. doi:10.1073/pnas.93.20.11019
112. Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity (1997) 7:493–504.
113. Adachi S, Yoshida H, Kataoka H, Nishikawa S. Three distinctive steps in Peyer’s patch formation of murine embryo. Int Immunol (1997) 9:507–14. doi:10.1093/intimm/9.4.507
114. Yoshida H, Honda K, Shinkura R, Adachi S, Nishikawa S, Maki K, et al. IL-7 receptor alpha+ CD3(-) cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int Immunol (1999) 11:643–55. doi:10.1093/intimm/11.5.643
115. Mebius RE, Miyamoto T, Christensen J, Domen J, Cupedo T, Weissman IL, et al. The fetal liver counterpart of adult common lymphoid progenitors gives rise to all lymphoid lineages, CD45+CD4+CD3- cells, as well as macrophages. J Immunol (2001) 166:6593–601. doi:10.4049/jimmunol.166.11.6593
116. Yoshida H, Kawamoto H, Santee SM, Hashi H, Honda K, Nishikawa S, et al. Expression of alpha(4)beta(7) integrin defines a distinct pathway of lymphoid progenitors committed to T cells, fetal intestinal lymphotoxin producer, NK, and dendritic cells. J Immunol (2001) 167:2511–21. doi:10.4049/jimmunol.167.5.2511
117. Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science (2000) 288:2369–73. doi:10.1126/science.288.5475.2369
118. Eberl G, Marmon S, Sunshine M-J, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol (2004) 5:64–73. doi:10.1038/ni1022
119. Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature (1999) 397:702–6. doi:10.1038/17812
120. Yoshida H, Naito A, Inoue J-I, Satoh M, Santee-Cooper SM, Ware CF, et al. Different cytokines induce surface lymphotoxin-alphabeta on IL-7 receptor-alpha cells that differentially engender lymph nodes and Peyer’s patches. Immunity (2002) 17:823–33. doi:10.1016/S1074-7613(02)00479-X
121. Coles MC, Veiga-Fernandes H, Foster KE, Norton T, Pagakis SN, Seddon B, et al. Role of T and NK cells and IL7/IL7r interactions during neonatal maturation of lymph nodes. Proc Natl Acad Sci U S A (2006) 103:13457–62. doi:10.1073/pnas.0604183103
122. Bénézech C, White A, Mader E, Serre K, Parnell S, Pfeffer K, et al. Ontogeny of stromal organizer cells during lymph node development. J Immunol (2010) 184:4521–30. doi:10.4049/jimmunol.0903113
123. Furtado GC, Pacer ME, Bongers G, Bénézech C, He Z, Chen L, et al. TNFα-dependent development of lymphoid tissue in the absence of RORγt+ lymphoid tissue inducer cells. Mucosal Immunol (2014) 7:602–14. doi:10.1038/mi.2013.79
124. Chai Q, Onder L, Scandella E, Gil-Cruz C, Perez-Shibayama C, Cupovic J, et al. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity (2013) 38:1013–24. doi:10.1016/j.immuni.2013.03.012
125. Onder L, Mörbe U, Pikor N, Novkovic M, Cheng H-W, Hehlgans T, et al. Lymphatic endothelial cells control initiation of lymph node organogenesis. Immunity (2017) 47:80–92.e4. doi:10.1016/j.immuni.2017.05.008
126. Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science (2010) 330:665–9. doi:10.1126/science.1194597
127. Schmutz S, Bosco N, Chappaz S, Boyman O, Acha-Orbea H, Ceredig R, et al. Cutting edge: IL-7 regulates the peripheral pool of adult ROR gamma+ lymphoid tissue inducer cells. J Immunol (2009) 183:2217–21. doi:10.4049/jimmunol.0802911
128. Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, et al. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity (2007) 26:643–54. doi:10.1016/j.immuni.2007.04.009
129. Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun (2015) 6:8280. doi:10.1038/ncomms9280
130. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med (2011) 208:125–34. doi:10.1084/jem.20100052
131. Rangel-Moreno J, Carragher DM, la Luz Garcia-Hernandez de M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol (2011) 12:639–46. doi:10.1038/ni.2053
132. Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol (2010) 184:5344–51. doi:10.4049/jimmunol.0902999
133. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity (2011) 35:986–96. doi:10.1016/j.immuni.2011.10.015
134. Jones GW, Bombardieri M, Greenhill CJ, McLeod L, Nerviani A, Rocher-Ros V, et al. Interleukin-27 inhibits ectopic lymphoid-like structure development in early inflammatory arthritis. J Exp Med (2015) 212:1793. doi:10.1084/jem.20132307
135. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med (2009) 206:35–41. doi:10.1084/jem.20072713
136. Moussion C, Girard J-P. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature (2011) 479:542–6. doi:10.1038/nature10540
137. Liao S, Ruddle NH. Synchrony of high endothelial venules and lymphatic vessels revealed by immunization. J Immunol (2006) 177:3369–79. doi:10.4049/jimmunol.177.5.3369
138. Galkina E, Tanousis K, Preece G, Tolaini M, Kioussis D, Florey O, et al. L-selectin shedding does not regulate constitutive T cell trafficking but controls the migration pathways of antigen-activated T lymphocytes. J Exp Med (2003) 198:1323–35. doi:10.1084/jem.20030485
139. Bombardieri M, Barone F, Humby F, Kelly S, McGurk M, Morgan P, et al. Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjögren’s syndrome. J Immunol (2007) 179:4929–38. doi:10.4049/jimmunol.179.7.4929
140. McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med (2005) 11:335–9. doi:10.1038/nm1202
141. Schrama D, Voigt H, Eggert AO, Xiang R, Zhou H, Schumacher TNM, et al. Immunological tumor destruction in a murine melanoma model by targeted LTalpha independent of secondary lymphoid tissue. Cancer Immunol Immunother (2008) 57:85–95. doi:10.1007/s00262-007-0352-x
142. Kirk CJ, Hartigan-O’Connor D, Mulé JJ. The dynamics of the T-cell antitumor response: chemokine-secreting dendritic cells can prime tumor-reactive T cells extranodally. Cancer Res (2001) 61:8794–802.
143. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med (2014) 189:832–44. doi:10.1164/rccm.201309-1611OC
144. Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, et al. Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity (2005) 23:539–50. doi:10.1016/j.immuni.2005.10.002
145. Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity (1998) 9:71–9. doi:10.1016/S1074-7613(00)80589-0
146. Cañete JD, Celis R, Moll C, Izquierdo E, Marsal S, Sanmartí R, et al. Clinical significance of synovial lymphoid neogenesis and its reversal after anti-tumour necrosis factor alpha therapy in rheumatoid arthritis. Ann Rheum Dis (2009) 68:751–6. doi:10.1136/ard.2008.089284
147. Calcinotto A, Grioni M, Jachetti E, Curnis F, Mondino A, Parmiani G, et al. Targeting TNF-α to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J Immunol (2012) 188:2687–94. doi:10.4049/jimmunol.1101877
148. Scurr M, Bloom A, Pembroke T, Srinivasan R, Brown C, Smart K, et al. Escalating regulation of 5T4-specific IFN-γ(+) CD4(+) T cells distinguishes colorectal cancer patients from healthy controls and provides a target for in vivo therapy. Cancer Immunol Res (2013) 1:416–25. doi:10.1158/2326-6066.CIR-13-0035
149. Vermeij R, Leffers N, Hoogeboom B-N, Hamming ILE, Wolf R, Reyners AKL, et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: a single-arm phase II study. Int J Cancer (2012) 131:E670–80. doi:10.1002/ijc.27388
150. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med (2012) 18:1254–61. doi:10.1038/nm.2883
151. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med (2014) 370:997–1007. doi:10.1056/NEJMoa1315226
Keywords: tertiary lymphoid structures, cancer immunotherapy, high endothelial venules, lymphoid neogenesis, tumor microenvironment
Citation: Colbeck EJ, Ager A, Gallimore A and Jones GW (2017) Tertiary Lymphoid Structures in Cancer: Drivers of Antitumor Immunity, Immunosuppression, or Bystander Sentinels in Disease? Front. Immunol. 8:1830. doi: 10.3389/fimmu.2017.01830
Received: 29 September 2017; Accepted: 04 December 2017;
Published: 19 December 2017
Edited by:
David Robert Kroeger, British Columbia Cancer Agency, CanadaReviewed by:
Cara Haymaker, University of Texas MD Anderson Cancer Center, United StatesKawaljit Kaur, University of California, Los Angeles, United States
Copyright: © 2017 Colbeck, Ager, Gallimore and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emily Jayne Colbeck, e.colbeck@ucl.ac.uk