 Zijun Y. Xu-Monette
Zijun Y. Xu-Monette Mingzhi Zhang
Mingzhi Zhang Jianyong Li
Jianyong Li Ken H. Young
Ken H. Young- 1Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Department of Oncology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
- 3Department of Hematology, JiangSu Province Hospital, The First Affiliated Hospital of NanJing Medical University, NanJing, JiangSu Province, China
- 4Graduate School of Biomedical Science, The University of Texas Health Science Center at Houston, Houston, TX, United States
PD-1–PD-L1 interaction is known to drive T cell dysfunction, which can be blocked by anti-PD-1/PD-L1 antibodies. However, studies have also shown that the function of the PD-1–PD-L1 axis is affected by the complex immunologic regulation network, and some CD8+ T cells can enter an irreversible dysfunctional state that cannot be rescued by PD-1/PD-L1 blockade. In most advanced cancers, except Hodgkin lymphoma (which has high PD-L1/L2 expression) and melanoma (which has high tumor mutational burden), the objective response rate with anti-PD-1/PD-L1 monotherapy is only ~20%, and immune-related toxicities and hyperprogression can occur in a small subset of patients during PD-1/PD-L1 blockade therapy. The lack of efficacy in up to 80% of patients was not necessarily associated with negative PD-1 and PD-L1 expression, suggesting that the roles of PD-1/PD-L1 in immune suppression and the mechanisms of action of antibodies remain to be better defined. In addition, important immune regulatory mechanisms within or outside of the PD-1/PD-L1 network need to be discovered and targeted to increase the response rate and to reduce the toxicities of immune checkpoint blockade therapies. This paper reviews the major functional and clinical studies of PD-1/PD-L1, including those with discrepancies in the pathologic and biomarker role of PD-1 and PD-L1 and the effectiveness of PD-1/PD-L1 blockade. The goal is to improve understanding of the efficacy of PD-1/PD-L1 blockade immunotherapy, as well as enhance the development of therapeutic strategies to overcome the resistance mechanisms and unleash the antitumor immune response to combat cancer.
Introduction
It is widely known that ligation of programmed cell death protein 1 (PD-1, also known as CD279) (1) with PD-1 ligand 1 (PD-L1, also called B7-H1 or CD274) (2, 3) activates a critical immune checkpoint leading to T cell dysfunction, exhaustion, and tolerance; high-affinity anti-PD-1 or anti-PD-L1 monoclonal antibodies (mAbs) (4), which block PD-1–PD-L1 interaction, can reverse the immune checkpoint, releasing the brake on T cell responses. However, neither PD-1 nor PD-L1 expression is specific for the reversible T cell dysfunction state, and the effect of PD-1/PD-L1 blockade can be context-dependent. In addition, PD-1 signaling and the mechanism of action of anti-PD-1/L1 mAbs are not completely understood.
Despite these discrepancies and unknowns, PD-1/PD-L1 blockade has achieved great clinical success in combating cancers. Durable response could also be achieved in PD-L1− patients (5, 6). Nonetheless, a large proportion of patients, including those with PD-L1+/PD-1+ expression, do not respond to PD-1/PD-L1 blockade. Some rational combination therapies have shown synergy in vivo or in clinical trials (as well as immune-related toxicities, unfortunately). This article summarizes functional and clinical studies of PD-1/PD-L1 and the resistance mechanisms for PD-1/L1 blockade, and discusses several important questions arising from the disparate data, with the goal of increasing understanding of PD-1, PD-L1, and PD-1/PD-L1 blockade.
PD-1 and PD-1 Expression: Markers of T Cell Exhaustion or Activation
Contrary to the common perception that PD-1 and PD-L1 expression is a marker of T cell dysfunction associated with cancer and chronic viral infection, PD-1 and PD-L1 can also be expressed under normal physiologic conditions. PD-1 is expressed on 40–80% of memory T cells but not on naïve T cells in the peripheral blood of healthy human adults, and PD-1 expression levels do not directly affect the cytokine production function of CD8+ T cells (7).
PD-1 expression may indicate T cell activation, because PD-1 is expressed only on activated T cells in vivo, and not on resting T cells. PD-1 (PDCD1) mRNA is mainly expressed in the thymus in vivo, with additional possible distribution in the spleen and lung (1). PD-1 protein can be detected in normal murine thymus and spleen T cells at low levels (8), but is strongly induced on thymocytes and T cells in the spleen and lymph nodes after stimulation with an anti-CD3 mAb in vitro (9) and increased on T cells in the spleen and liver after tumor cell injection in vivo (10). PD-1 is also expressed on activated B cells in vitro after stimulation with anti-IgM antibodies, but was undetectable on activated macrophages or dendritic cells (9, 11). In human reactive tonsils, PD-1 is expressed primarily on T cells, as well as a small subset of follicular dendritic cells (12).
The association of PD-1 expression with antigen-specific T cells has also been illustrated in cancer patients. PD-1 expression was significantly higher on antigen-specific CD8+ T cells than other CD8+ T cells in metastatic melanoma lesions in the same patients (13). In a melanoma mouse model, compared with tumor-ignorant bystander CD8+ T cells, tumor-specific CD8+ T cells infiltrating the same tumor had significantly higher levels of PD-1, LAG-3, CD69 (activation marker), and 4-1BB (costimulatory molecule) expression and gained 1,414 activation-related (but not exhaustion-related) accessible chromatin regions (14). Adoptive T cell therapy with cells expanded from PD-1+CD8+ tumor-infiltrating lymphocytes (TILs), but not from PD-1− or bulk CD8+ TILs, showed tumor-reactivity and therapeutic benefit in vivo (15).
On the other hand, PD-1 expression is associated with suboptimal costimulation and T cell dysfunction when antigen is presented on non-activated or non-professional antigen-presenting cells (16, 17), and PD-1 expression is often induced by high antigen concentration and prolonged antigen stimulation (18, 19). PD-1 may not be a good T cell activation marker because PD-1 surface expression is not rapidly induced on stimulated CD4+/CD8+ T cells. PD-1 expression has been shown to be increased 24–48 h after stimulation in vivo (20–22), 5–7 days after antigen experience (17), 3–8 days after adoptive transfer of pre-activated antigen-reactive CD8+ T cells (14), and 19 days after immunization in vivo (19), although PDCD1 mRNA expression was shown to be increased at an earlier time point, as was the suppression of T-cell function. An in vivo kinetics study of T cell response to hepatitis B virus infection also showed that after intrahepatic antigen recognition, CD8+ T cells first showed rapid induction and decline of IFN-γ-producing capacity, followed by delayed T cell expansion and an increase in cytolytic activity, and the functional oscillation coincided with strong PD-1 induction on antigen-specific T cells (23).
Furthermore, in a melanoma model, the “exhausted” (showing reduced cytokine production capability) tumor-reactive CD8+ T cells, compared with “non-exhausted” bystander CD8+ T cells, had Pdcd1 upregulation but downregulation of genes involved in CD8+ T cell survival and function (Il7r, Bcl2, Cxcr3, Ifngr1, and Ifngr2) (14). In patients with metastatic melanoma, tumor-infiltrating T cells had high PD-1 expression and decreased functional avidity compared with T cells infiltrating normal tissues, whereas circulating peripheral blood T cells had minimal PD-1 expression comparable with that in healthy donors. Smaller fraction of antigen-specific CD8+ T cells in metastatic melanoma lesions produced IFN-γ compared with those circulating in blood, which was inversely correlated with PD-1 expression (13). Similarly, PD-1 expression gradually increased in TILs with tumor growth but not on spleen T cells in a melanoma tumor model; although a higher percentage of TILs produced IFN-γ after stimulation ex vivo compared with spleen T cells, the amount of IFN-γ produced by TILs was lower, and smaller percentage of TILs produced TNF-α (19). In a colon cancer model, the cellular expression levels of PD-1 on intratumoral T cells inversely correlated with the function of CD8+ T cells (24).
During chronic infection with lymphocytic choriomeningitis virus (LCMV), PDCD1 mRNA levels were upregulated in “exhausted” CD8+ T cells with impaired cytokine production and proliferation, but PDCD1 was not upregulated in functional LCMV-specific memory CD8+ T cells during acute viral infection (25). Paradoxically, PD-1 protein expression was not limited to chronic LCMV infection, and PD-1 protein was also transiently expressed on CD8+ T cells in acute viral infection and downregulated along with LCMV clearance, suggesting that PD-1 protein expression is not a specific marker of exhaustion (25). In fact, during acute infection with rapid control of the viral infection, PD-1lo cells mainly produced antiviral cytokines and PD-1hi cells were the main mediators of cytotoxicity activity (26). Similarly, during chronic mycobacterial infection in vivo, PD-1+ T cells were not functionally exhausted (highly proliferative and could differentiate into cytokine-secreting T cells), and probably critical for antigen-specific T cell responses (27). Moreover, during tumor growth in a mouse model, although increased PD-1 and LAG-3 expression was accompanied by decreased T-cell effector function, enhancing fatty acid catabolism increased PD-1 expression and improved T-cell effector function; conversely, inhibiting fatty acid catabolism decreased PD-1 expression and impaired T-cell function (28).
PD-1hi expression also does not mark T cell exhaustion in patients with autoimmune disease or cancer. In patients with rheumatoid arthritis, PD-1hiCXCR5−CD4+ cells are expanded in pathologically inflamed non-lymphoid tissues and are functionally active (promoting B cell responses) (29). In follicular lymphoma patients, PD-1+ T cells include both functionally “exhausted” (unable to produce cytokines) PD-1lo T cells and PD-1hi “non-exhausted” follicular helper T cells (CXCR5+BCL6+CD4+, supporting the growth and survival of B cells, and secreting IL-21 and IL-4) (30). Increased PD-1+ cells in tumor biopsies have been associated with either favorable prognosis in patients with follicular lymphoma (31, 32), lung cancer (33), ovarian cancer (34), or poor survival in cancer patients (35, 36). Furthermore, in melanoma patients, PD-1+ T cell clones are antigen-specific T cell clonotypes with higher functional avidity and reactivity (IFN-γ and TNF-α production after activation) than PD-1− T cell clones (37), and PD-1 expression can be used as a biomarker for neoantigen-specific T cells in TILs and in the peripheral blood (38–40). The discrepancies in association of PD-1 expression with T-cell function (exhaustion or avidity) may reflect the complex interplay between various driving forces and effectors of the PD-1 pathway, suggesting that factors other than PD-1 are also important for T-cell functionality.
Similar to PD-1, PD-L1 expression can also be a marker of immune activation. PD-L1 is often not expressed in cell lines in vitro but is induced on tumors and in the tumor microenvironment (exceptions include some lymphoma and myeloma cell lines) (10, 41). IFN-γ produced by effector T cells soon after but not before activation of immune response (23), is the major inducer of PD-L1 expression at the transcription level (42). Supporting this, in metastatic melanoma samples, PD-L1+ cell densities were shown to significantly correlate with CD8+ T cell densities in the tumor and at the invasive tumor margin (43). IFN-γ and TLR ligands induce PD-L1 through the JAK/STAT/IRF-1, MEK/ERK, and MyD88/TRAF6 pathways (44–47). JAK2 (46), MEK/ERK, and p38 MAPK (48) signaling pathways were critical for PD-L1 expression in Hodgkin lymphoma cells. Furthermore, PD-L1 expression is also induced on immune cells after immune activation, including dendritic cells, macrophages, B cells (8, 11), T cells (49), and natural killer cells (50), and this is mediated through the cytokine/chemokine and STAT3 pathways (50–52).
Immune responses are not the only processes that can induce PD-L1 expression; tumor-intrinsic oncogenic pathways can also upregulate PD-L1 expression. For example, oncogenic c-Jun (AP-1) and STAT3 signaling (53), and hypoxia-inducible factor HIF-1α (54) upregulate PD-L1 expression transcriptionally; the oncogenic epigenetic writer EZH2 (55) and epigenetic reader BET4 upregulate PD-L1 (56), whereas the epigenetic eraser histone deacetylase downregulates PD-L1 expression (57). In addition, loss of PTEN function and oncogenic activation of the PI3K/AKT/mTOR pathway increase PD-L1 expression posttranscriptionally (58, 59) [however, in vivo PTEN loss did not always affect PD-L1 expression significantly (60)]. Moreover, CSN5, induced by NF-κB p65 (61), and novel CMTM6/4 transmembrane proteins (62, 63) decrease ubiquitination and stabilize PD-L1. EGF signaling induces PD-L1 glycosylation and antagonizes GSK3β-mediated PD-L1 phosphorylation and degradation (64). Enhanced glycolysis and lactate production activate transcriptional coactivator TAZ and induce PD-L1 expression on tumor cells (65). The glycolytic intermediate pyruvate can also metabolically control PD-L1 expression on macrophages through the BMP4/p-SMAD1/5/IRF-1 signaling pathway (66).
Furthermore, PD-L1 is also expressed under normal conditions in both lymphoid and non-lymphoid tissues on human placental trophoblasts, myocardial endothelia cells, and cortical thymic epithelial cells (8, 11, 42), which is involved in peripheral tolerance and immune privilege (67–69). PD-L1 expression has been correlated with either poorer or better survival of cancer patients (70, 71). Taking together, these findings show that, similar to PD-1, PD-L1 expression is not a specific marker for T cell activation or exhaustion.
PD-1 and PD-L1 Expression as Driver or Biomarker of Immune Suppression: Tumor-Driven or Host-Driven Evolution
As mentioned above, PD-L1 expression can be either immunogenic (tumor-extrinsic, driven by the immune system) (72) or oncogenic (tumor cell-intrinsic, driven by intrinsic mechanisms in cancer cells). It has been controversial whether the immunogenic and oncogenic PD-L1 expression on tumor cells or PD-L1 expression on activated host immune cells is essential for immune evasion. Recently, four studies addressed this question in vivo and showed that although all forms of PD-L1 expression contribute to immune suppression in a non-redundant fashion, the relative roles (i.e., predominant or minor) of immunogenic tumor-derived PD-L1 and host-derived PD-L1 expression in suppressing T cell cytotoxicity and infiltration varied depending on the mouse models used, which had different levels of tumor immunogenicity (73–76). PD-L1 gene deletion in highly immunogenic MC38 colorectal adenocarcinoma tumors resulted in loss of protection from T cell cytotoxicity, whereas the growth of MC38 tumors in PD-L1/PD-L2-knockout (PD-L1−/−/L2−/−) mice was as robust as in wild-type mice, which elegantly demonstrated that induced tumor PD-L1 expression directly and sufficiently inhibits antitumor immunity, serving as far more than a marker of an ineffective immune response (74).
Similarly designed experiments demonstrated that oncogenic PD-L1 expression in BRAF.PTEN melanoma tumors only slightly inhibited antitumor immunity (74), whereas immunogenic PD-L1 expression on non-tumor cells was critical for immune evasion. Similarly, in a mouse model of melanoma tumors with low immunogenicity, host PD-L1 and PD-1 expression on non-tumor cells is essential for suppressing antitumor immunity. Therefore, although the prevailing notion is that tumors exploit the PD-1 pathway and evade immune response by actively overexpressing PD-L1, this “adaptive immune resistance mechanism” is largely limited to immunogenic PD-L1 expression (74), which is ultimately driven by the host immune response (72).
Although tumor PD-L1 expression in the MC38 model has a driver role, tumor PD-L1-mediated immune suppression has local limitations, which one study proposed as the “molecular shield” functional model. In this model, PD-L1 forms only a temporal molecular shield to protect PD-L1+ tumor cells, and the cytolytic function of T cells against other PD-L1− tumor cells with the same antigen is not impaired (77), likely because a close proximity between PD-1–PD-L1 and immunologic synapses is required for PD-L1 function to disturb the T-cell receptor (TCR)–major histocompatibility complex (MHC) interaction. This functional mode is somewhat like another mechanistic model, in which PD-1–PD-L1 interaction increases T cell motility through inhibition of TCR-driven “stop signals” (78). Consistent with this functional model, two (73, 74) of the four recent studies mentioned above showed that tumor PD-L1 expression can protect only PD-L1+ tumor cells from cytolytic T cell killing in situ, and not PD-L1− cells in trans, conferring a selective growth advantage on PD-L1+ tumor cells.
However, as shown in mouse models and in cancer patients, immunogenic tumor PD-L1 expression is heterogeneous (76) and transient (75), which does not support the idea that tumor-derived PD-L1 expression is required for tolerance induction and maintenance or that PD-L1+ tumor clones are preferably selected during tumorigenesis. It is postulated that PD-L1− tumor cells escape immune surveillance through alternative mechanisms such as decreased MHC expression, increased PD-L2 expression on PD-L1− tumor cells, stromal remodeling, and epithelial–mesenchymal transition (73), as well as compensatory PD-L1 expression on host cells, including T cells (79–81), antigen-presenting cells, monocytic myeloid-derived suppressor cells (MDSCs), and host tissues (81, 82). The compensatory PD-L1 expression can be both IFN-γ-dependent and IFN-γ-independent (75), and may be able to trigger a vicious cycle of immune suppression in the tumor microenvironment (83). Moreover, PD-1 signaling was recently proposed to affect antigen-presenting cells more than tumor cells owing to the increased CD80/CD86 expression on antigen-presenting cells, given that the CD28 receptor is the primary target for PD-1/SHP2-mediated dephosphorylation, as was newly discovered in that study (84). Therefore, host-derived PD-L1 appears to be indispensable for the inhibitory function of the PD-L1/PD-1 axis. However, whether the minor role of the oncogenic PD-L1 expression in the BRAF.PTEN melanoma model applies to tumor PD-L1 expression upregulated by other tumor-intrinsic mechanisms in different types of cancer is unclear.
Furthermore, the driver role of PD-1 on host T cells in immune suppression is demonstrated by the fact that MC38 tumors were completely cleared in PD-1-knockout (PD-1−/−) mice. TILs from PD-1−/− mice had an increased ratio of CD8+ cells to regulatory T cells (Tregs) and granzyme expression compared with TILs from wild-type mice. In contrast, MC38 tumors (with immunogenic PD-L1 expression) grew similarly robust in PD-L1−/−/L2−/− mice as in wild-type mice; PD-L1−/−/L2−/− mice and wild-type mice had similar CD8/Treg ratios and PD-1, granzyme, and Ki-67 expression levels in TILs (74). In addition, earlier studies also showed that blockade of PD-1, but not PD-L1, by genetic deletion or mAbs cleared the tumor growth in tumor models (10, 74, 85), and PD-L1 knockout in vivo had no effect on PD-1 expression in TILs (74).
Together, these studies may suggest that immune responses are ultimately regulated by the host rather than the tumor. However, another study showed that continuous antigen encounters and TCR stimulation, rather than factors associated with the tumor microenvironment, induce PD-1 expression and T cell dysfunction (17), which is “imprinted” at the premalignant and early malignant phase and later evolves into a therapeutically irreversible state. In line with the idea of antigen dictation of immune response, increased PD-1 expression in expanded blood CD8+ cells from patients following viral immunotherapy was not necessarily a target for improving the efficacy of viral immunotherapy (86); immunogenic personalized mutanome vaccines have induced durable clinical response in melanoma patients (87, 88). However, resistance to personalized neoantigen vaccines can still be developed through β2M deficiency and other unclear mechanisms in some patients in these personal neoantigen vaccine trials, and patients receiving PD-1 blockade combination therapy achieved complete regression (87, 88). Moreover, in a tumor model, although tumor vaccines increased antigen-specific TILs, they did not decrease PD-1 expression, which impaired the effector function of TILs, nor did they decrease the percentage of MDSCs in the tumor lesions (which accumulated since early-stage and accentuated after immunization) (19). In a clinical trial of immunization in patients with metastatic melanoma, the expansion and function (tested in vivo and in vitro) of stimulated antigen-specific CD8+ T cells by cancer vaccines were also regulated by increased PD-1 expression (89).
The critical role of antigen was also shown in a mouse model with LCMV infection: T cells functioned normally during acute (Armstrong strain) infection with transient PD-1 expression but were exhausted during chronic (clone 13) infection with stable PD-1 expression (25). Although exhausted CD8+ T cells could be reinvigorated by anti-PD-L1 therapy in vivo, T cells became re-exhausted with persistent PD-1 expression if antigen concentration remained high (90). Therefore, persistent tumor antigens appeared to be the dictator for PD-1 expression and T cell re-exhaustion. However, this was not supported by antigen withdrawal in vivo experiment. After antigen clearance, exhausted T cells and anti-PD-L1-treated exhausted T cells failed to downregulate PD-1 expression (or T-bet and Eomes expression) and had poor recall response upon antigen re-challenge (90).
A study assessing changes in chromatin accessibility during viral infection revealed that acute LCMV infection resulted in stable (5–10%) and dynamic (≥25%) changes in accessible chromatin regions in antigen-specific effector and memory CD8+ T cells. In contrast, chronic infection uniquely enriched accessible chromatin regions for NFAT and Nr4a family transcription factors (including enhancers of the PDCD1 locus) but partially lost the accessibility to some regions (such as Satb1 and Il7r loci) in exhausted CD8+ T cells, although exhausted CD8+ T cells and effector CD8+ T cells shared chromatin accessibility at promoter regions of key effector-related genes, including Ifng, Gzma, Gzmk, Fasl, and Prf1, as well at inhibitory receptor genes, including Tim3, Lag3, and Ctla4 (91). Anti-PD-L1 therapy in vivo caused only minimal epigenetic profile changes in exhausted T cells; instead, the T cell reinvigoration by PD-L1 blockade resulted from transcriptional rewiring with different transcription factors (NF-κB, Jun:AP-1, IRFs, and CTCF, instead of “partnerless” NFATc1, NFAT:AP-1, Nr4a1, Nur77, Eomes, and Egr2) in the epigenetic landscape (90). The epigenetic inflexibility is thought to contribute to re-exhaustion with antigen stimulation without memory-like recall response after anti-PD-L1 treatment (90), suggesting the importance of host T cell-intrinsic regulatory factors including PD-1.
Similar to this unsustained therapeutic effect in viral infection models, an anti-PD-L1 mAb was shown to have only transient antitumor effects in a mouse model, in contrast to the complete suppression of myeloma growth by gene knockout of PD-1 (85). Anti-PD-L1 therapy in vivo led to tumor regression with increased antigen-reactive T cell infiltrate and increased IFN-γ and TNF-α production upon antigen stimulation ex vivo. However, PD-L1 blockade had only a moderate effect on gene activation and chromatin accessibility in tumor-infiltrating T cells, including upregulation of a few functionally important genes (including granzyme and serpin genes) and dampened accessibility in limited motifs binding NFAT, NFAT:AP-1, TCF, and bZIP:IRF transcription factors. In contrast, 450 accessible regions (including those accessible for Nr4a and NFAT) were gained in “exhausted” T cells compared with “non-exhausted” T cells before the treatment (14).
Furthermore, in an inducible liver cancer model, dysfunction of antigen-specific T cells lasting for more than 30 days was not rescued either after antigen withdrawal or after a decrease in PD-1 levels in TILs by anti-PD-1/PD-L1 therapy (17), suggesting that the dysfunction state was maintained by multiple factors rather than PD-1 alone. Irreversibility of these TILs, which will be discussed more in later sections, somewhat resembled the unresponsiveness of tolerant/anergic T cells to PD-L1 blockade (92). In these settings, PD-1 appeared to be a biomarker rather than the central driver of immune suppression.
PD-1 and PD-L1: Functionally Dependent or Independent in Driving Immune Suppression
The receptor and ligand relationship between PD-1 and PD-L1 was discovered by Freeman et al. in 2000 (2), and the relationship between PD-1 and PD-1 ligand 2 (PD-L2, also called B7-DC or CD273) was discovered by Latchman et al. in 2001 (93). PD-1 ligation leads to T cell exhaustion (decreased proliferation and effector function) (25), apoptosis (94, 95), or anergy/tolerance (a hyporesponsive state of T cells to a specific antigen that can be induced by lack of costimulation) (96–99). Functional studies have demonstrated that PD-1 receptor ligation is required for PD-1 to prevent T cell activation, and the inhibitory effect of PD-1 ligation depends on TCR strength (21, 22, 42) and co-localization of PD-1 with CD3 and/or CD28 (20, 100).
Molecularly, PD-1 ligation inhibits CD28-mediated costimulation (2, 20, 93); prevents TCR-driven stop signals (78); inhibits TCR signaling in both CD8+ and CD4+ T cells; blocks cell cycle progression in CD4+ T cells; downregulates expression of antiapoptotic molecules and proinflammatory cytokines; and upregulates expression of Cbl-b ubiquitin ligase in CD8+ T cells (20, 93, 100–104). For B cell-derived PD-1 expression, coligation of the PD-1 cytoplasmic region with the B cell receptor (BCR) inhibited BCR signaling in vitro (105). Inhibition of TCR/BCR signaling is mediated by the protein tyrosine phosphatase SHP2, which is recruited to the PD-1 immunoreceptor tyrosine-based switch motif upon PD-1 ligation and dephosphorylates ZAP70 (in T cells), Syk, Igβ, PLCγ2, and ERK (in B/T cells) and other downstream kinases, including PI3K/AKT (20, 93, 102, 105, 106). Although SHP2 can be associated with PD-1 immunoreceptor tyrosine-based switch motif with TCR stimulation in the absence of PD-1 engagement, PD-1 engagement is required to block T cell activation (20).
However, in contrast to these earlier studies, a recent study showed that CD28 and Lck (a kinase associated with CD4/CD8 that phosphorylates CD3/TCR, CD28, and PD-1), but not TCR, were the preferred targets of dephosphorylation by PD-1-bound SHP2 in a biochemical reconstitution system (84). PD-1 co-clustered with CD28 in plasma membrane microclusters in a PD-L1-dependent manner but only partially segregated with TCR in stimulated CD8+ T cells. Furthermore, intact cell assays using Jurkat T cells and Raji B cells confirmed that CD28, but not TCR, was dephosphorylated after PD-1 ligation with PD-L1; however, the dephosphorylation was only transient (84).
The downregulated PI3K/AKT pathway in T cells upon PD-1 ligation is important for the cell cycle, proliferation, survival, apoptosis, and metabolism. PD-1 also inhibits the PI3K/AKT pathway by inhibiting phosphorylation of PTEN in the C-terminal tail, which decreases PTEN stability but increases PTEN phosphatase activity (107). Because the PI3K/AKT/mTOR pathway is critical for metabolic reprogramming, PD-1 expression and ligation has been linked to metabolic dysfunction in T cells. As shown in vitro, ligation of PD-1 on CD4+ T cells inhibited glycolysis (106) and glucose transporter Glut1 as well as transportation and catabolism of glutamine, but augmented lipolysis and fatty acid oxidation (108), which promotes Treg development over that of effector T cells (109, 110). In multiple graft-vs.-host disease (GVHD) models, PD-1 expression was shown to increase levels of reactive oxygen species, which was dependent on oxidative metabolism of fat in both CD4+ and CD8+ T cells, facilitating CD8+ T cell apoptosis (95). Conversely, PD-1/PD-L1 blockade partially decreased the generation of reactive oxygen species and cell death of alloreactive PD-1hi, but not PD-1lo, T cells and increased the severity of GVHD (95). However, in patients with viral infection, exhausted virus-specific CD8+ T cells were dependent on glycolysis with high Glut1 and PD-1 expression and depolarized mitochondria which could be rescued by a signal 3 (111) cytokine IL-12, compared with the non-exhausted CD8+ T cells within the same patients with metabolic flexibility of utilizing mitochondrial oxidative phosphorylation to fuel the effector function (112). A recent study showed that in vivo hypoglycemia and hypoxia metabolic stress caused CD8+ T cell exhaustion (which was independent of the PD-1 pathway however); fatty acid catabolism enhanced in CD8+ T cells (which was also observed in melanoma patients) partially preserved antitumor effector functions of CD8+ TILs but upregulated (possibly indirectly) PD-1 expression; PD-1 blockade synergizes (but did not change) this metabolic reprogramming in inhibiting tumor growth (28). In a B cell leukemia model with increased PD-1 and PD-L1 expression over time in the leukemic microenvironment, impaired T cell metabolism directly contributed to T cell dysfunction, whereas in vivo and in vitro PD-1 blockade was not sufficient to improve T-cell function (113).
Opposite to the PD-1 function in suppressing glycolysis, enhanced glycolysis induces PD-L1 expression (65), which in turn promotes glycolysis in tumor cells and restricts T-cell function by metabolically competing for glucose (114). Of note, PD-1 signaling inhibits the PI3K/AKT/mTOR and MAPK/ERK pathways in T cells but PI3K/AKT and MEK/ERK signaling pathways activate PD-L1 expression in tumor cells. Tumor PD-L1 promotes MTORC1 signaling but inhibits MTORC2 and autophagy (115). Metabolic competition or adaptation between tumor cells and T cells (114) may contribute to these contrasting pathways, and the paradoxical results in transplantation models: alloreactive donor T-cells in PD-L1-deficient GVHD mice had increased aerobic glycolysis and oxidative phosphorylation (116), whereas donor PD-L1-deficient T cells in wild-type mice had reduced aerobic glycolysis, oxidative phosphorylation, fatty acid metabolism, and cytokine production (117).
In line with the requirement of PD-1 ligation for its suppressive function, in follicular lymphoma, which has very low PD-L1 expression, only subsets of PD-1+ T cells have exhausted phenotypes and function (30, 118). However, exhaustion of terminally differentiated PD-1hiCD44intCD8+ T cells during chronic viral infection appeared not to depend on PD-L1 expression, because anti-PD-L1 mAbs could not rescue these PD-1hi T cells from apoptosis or restore the effector function (119). Moreover, PD-1 and PD-L1 expression may be temporally non-overlapping; a kinetics study observed a rapid but transient burst of IFN-γ production at 4 h after adoptive T cell transfer, whereas loss of IFN-γ expression coincided with delayed strong PD-1 induction (23).
PD-L2, the second PD-1 natural ligand, has higher affinity than PD-L1 for PD-1 (120, 121). However, PD-1–PD-L2 interaction is much less functionally significant than the PD-1–PD-L1 interaction owing to the low expression of PD-L2, and PD-1–PD-L1 interaction is sensitive to PD-L2 competition only when PD-L2 levels are very high (120). In sharp contrast to PD-L1, PD-L2 is rarely expressed in lymphohematopoietic and non-hematopoietic tissues (8, 122), except human placental endothelium and medullary thymic epithelial cells (42). PD-L2 can be induced on dendritic cells, macrophages, activated T cells (8, 11, 21, 42), B cells (123–125), and cancer cells by IL-4 through IL-4R/STAT6 in inflammatory macrophages (126), the NF-κB pathway in dendritic cells (8), and IFN-β/IFN-γ in melanoma cells (47). Furthermore, several studies showed that PD-1 and PD-L1, but not PD-L2, induce T cell tolerance and apoptosis, preventing auto/alloimmune responses (16, 67, 97, 116, 127, 128). These data may suggest that PD-1’s suppressive function is largely dependent on PD-L1 but not PD-L2 expression.
In contrast, PD-L1 and PD-L2 can exert inhibitory function independent of PD-1 by binding to B7-1 (CD80) (129) and RGMb (130), respectively. The binding affinity of PD-L1–CD80 is less than that of PD-1–PD-L1 (49). Studies showed that PD-L1–CD80 interaction, but not PD-L1–PD-1 interaction, is responsible for the induction and maintenance of T cell tolerance (131, 132), and that interaction between PD-L1 and PD-1 does not lead to T cell anergy in vitro (77). In contrast, in nonobese diabetic (NOD) mouse models, loss of PD-1, but not PD-L1, on antigen-specific CD4+ T cells resulted in increased proliferation of CD4+ T cells and infiltration of the pancreas during type 1 diabetes (133).
However, early studies showed that similar to the dependence of PD-1 function on receptor ligation (20), the inhibitory activity of PD-L1 and PD-L2 requires the expression of PD-1 (2, 93); in fact, PD-L1 expression in T cells, natural killer cells, and peripheral tissues can have a costimulatory effect with unknown receptors (3, 50, 117, 134–142). PD-L1 expressed on activated CD8+ T cells was shown to promote survival and effector function of CD8+ T cells during the contraction phase following immunization/antigen stimulation (134). PD-L1 expression in pancreatic islet beta cells was shown to accelerate allograft rejection, increase CD8+ T cell proliferation, and promote autoimmune diabetes (135). Likewise, PD-L1 expression induced on donor T cells augmented GVHD lethality (117). A recent study showed that after CD4+ T depletion in hematopoietic cell transplantation in vivo, PD-L1–CD80 interaction augmented survival and expansion of donor CD8+ T cells, resulting in strong graft-vs.-leukemia effects. In contrast, interaction of PD-L1 in recipient tissues with PD-1 on donor CD8+ T cells prevented GVHD (139), suggesting that PD-L1’s inhibitory function depends on PD-1. These contradictory results suggest that PD-L1 interactions with PD-1, CD80, and other unknown receptors have context-dependent functions. Unidentified receptors of PD-L2 with stimulatory function have also been reported (143–145).
PD-1 Blockade and PD-L1 Blockade by Gene Knockout or Antibodies: Efficacies and Limitations
Blocking of the PD-1/PD-L1 pathway by genetic deletion or using anti-PD-1/PD-L1 antibodies has been studied in various preclinical models and the results are quite variable, likely owing to the different roles of PD-1 and PD-L1 in different genetic and immunologic settings. Unlike CTLA-4 germline knockout CTLA-4−/− mice, which spontaneously and rapidly developed fatal lymphoproliferative disease with massive expansion of activated T cells (146, 147), PD-1−/− mice with different genetic backgrounds slowly developed lupus-like proliferative arthritis, glomerulonephritis, splenomegaly, or dilated cardiomyopathy with high-titer autoantibodies in early PD-1 studies (148–150), suggesting that PD-1 can inhibit B cell proliferation and differentiation. In a later study, PD-1 knockout in NOD mice specifically accelerated the onset and frequency of type I diabetes, with strong T helper 1 (Th1) polarization of T cells infiltrating into islets (151). Loss of PD-1, but not PD-L1, was further confirmed to be responsible for the proliferation and infiltration of reactive CD4+ T cells during type 1 diabetes in an adoptive T cell transfer model (133). PD-1 also plays a role in positive and negative selection of T cells, as indicated by the altered thymocyte repertoire in PD-1−/− TCR-transgenic mice (152) and in vitro (104).
In contrast, PD-L1−/− mice appeared normal but were susceptible to experimental autoimmune hepatitis (induced by accumulation of antigen-activated CD8+ T cells in the liver) (153) and experimental autoimmune encephalomyelitis (induced by myelin-reactive CD4+ Th1 cells) (81). PD-L1−/− lupus-susceptible (MRL+/+) mice developed autoimmune myocarditis and pneumonitis with increased PD-1+ macrophage and T cell infiltrates in the heart and lung (154). PD-L2−/− mice exhibited enhanced antigen-specific T cell response and breakdown of oral tolerance compared with wild-type controls (155).
In tumor-formation models, PD-1−/− mice completely suppressed the tumorigenesis of PD-L1+ myeloma cells (85); PD-1 deficiency also inhibited the hematogenous dissemination of poorly immunogenic tumors (which were PD-L1− in vitro) in PD-1−/− mice (10, 85). In viral infection models, both PD-L1−/− (25) and PD-1−/− mice (156) died from immunopathologic damage within a week after being infected with the LCMV clone 13 strain, which causes chronic infections in wild-type mice. However, both PD-L1−/− and PD-1−/− mice exhibited normal T cell responses to acute LCMV infection and controlled the infection as the wild-type mice did (25, 156). The lethal consequence of chronic infection was a result of systemic vascular leakage due to severe perforin-mediated cytolysis with enhanced CD8+ T cell activity (156). These results may suggest that the effectiveness of antiviral immune response is determined by the strain of virus or antigen but not the PD-1/PD-L1 axis, whereas high cytolytic activity due to PD-1/PD-L1 absence results in immunopathologic tissue damage over a prolonged period (23). These results may also suggest that PD-1/PD-L1 interaction has a positive role in generating effective antiviral responses. Indeed, further studies showed that PD-1 deletion in virus-specific CD8+ T cells enhanced T cell proliferation in the acute phase, but overstimulation and robust proliferation lead to increased apoptosis during the contraction phase, as well as accumulation of more cytotoxic but terminally differentiated (Eomeshi cells evolved from T-bethi progenitor cells), “deeply exhausted” CD8+ T cells during chronic LCMV infection (157).
Interestingly, PD-L1 blockade with anti-PD-L1 antibodies during the early-phase (on days 4–6) of systemic LCMV clone 13 infection also caused vascular permeability and ultimately fatal circulatory collapse (156), but anti-PD-L1 therapy on days 23–40 after infection restored the function of exhausted CD8+ T cells (proliferation, cytokine production, degranulation, and viral control) with or without CD4+ T cell depletion (25). Although CD4+ T cell help is critical for sustained CD8+ T cell cytotoxic function during chronic LCMV infection (158), other studies showed that combining PD-L blockade with CD4+ T cell depletion (159) or Treg cell depletion (160) could rescue deeply exhausted CD8+ T cells during the late stage of infection and may result in a significant reduction in viral load.
Although the autoimmune diseases against self-antigens were much milder and at later onset in PD-1/PD-L1/L2 deficient mice than in CTLA-4−/− mice, anti-PD-1 mAbs exhibited stronger antitumor effects than anti-CTLA-4 mAbs in tumor models (10, 25). The enhanced antitumor immunity is believed to result from the occupancy of the PD-1 receptor by anti-PD-1 mAbs which prevents PD-1 from interacting with its natural ligands PD-L1/L2. It has been demonstrated that PD-1 blockade with anti-PD-1 mAbs can increase proliferation and cytokine production of antigen-specific T cells (4, 10, 161), expand intratumoral frequencies of CD8+ effector memory T cells (162), enhance the cytotoxicity activity of effector T cells (preferably PD-1+ memory T cells with higher functional avidity) (37), augment recruitment of effector cells into the tumor site (4, 10, 161), decrease T cell mobility and enhance stable T–dendritic cell interaction (78), and promote CD8+ T cell priming (97, 163) [however, some studies showed that PD-1 blockade alone did not affect CD8+ T cell priming and costimulation from CD27 or CD28 may also be required for T cell priming (164, 165)].
In addition, PD-1 expression was found on 64% of freshly isolated natural killer cells from patients with multiple myeloma (166). Anti-PD-1 treatment in vitro with a CT-011 antibody (however, its specificity for PD-1 has been questioned) enhanced natural killer cell trafficking, immune complex formation, and cytotoxicity against PD-L1-bearing multiple myeloma cells (166). In multiple tumor models, IL-18 upregulated PD-1 expression on mature natural killer cells only in lymphoid organs but not in tumors; anti-PD-1 therapy in vivo abrogated IL-18-mediated metastases (167). PD-1 expression was also found on tumor-associated macrophages in patients with colorectal cancer (168) and on tumor-infiltrating myeloid dendritic cells in ovarian cancer patients (169). PD-1/PD-L1 blockade alone or combined with anti-CD47 therapy in vivo increased macrophage phagocytosis but decreased tumor growth and increased survival of mice (168). PD-1 blockade in vitro or in vivo enhanced dendritic cell function, including cytokine (TNF-α and IL-6) release, antigen presentation, and costimulation owing to NF-κB activation. PD-L1 blockade also increased cytokine release although to less extent (169, 170).
Similar to PD-1 blockade, PD-L1 blockade with anti-PD-L1 mAbs was also shown to increase cytokine production of T helper cells, enhance the cytolytic activity of cytotoxic T cells, and lengthen the duration of antigen-driven T cell migration arrest in vitro (78, 100, 171, 172). PD-L1 blockade strongly enhanced proliferation and cytokine production of memory or recently activated T cells from peripheral blood of healthy donors ex vivo, but only slightly enhanced naive T cell activation during a primary response (173). In contrast, a study showed that anti-PD-1/PD-L1 mAbs in vivo enhanced IFN-γ production but inhibited naïve CD4+ T cell proliferation, mediated by IFN-γ from CD4+ T cells and nitric oxide from macrophages (174). In a murine model of chronic colitis induced by adoptive transfer of CD4+CD45RBhi T cells, PD-L1 blockade treatment before (but not after) the onset of severe colitis suppressed T cell expansion and Th1 cytokine production and prevented the development of colitis (141).
However, the effects of PD-1/PD-L1 blockade were contextual in viral infection models. PD-L1 blockade and PD-1 blockade were effective only for exhausted T cells during chronic LCMV infection, and they did not increase virus-specific CD8+ T cells during acute infection (25, 26). Moreover, in a chronic LCMV infection model, PD-L1 blockade rescued only the rescuable subset of exhausted CD8+ T cells and not the more terminally differentiated (PD-1hiCD44int) subset of CD8+ T cells (119). Similarly, adoptive transfer of CXCR5+CD44hi but not CXCR5−CD44loCD8+ T cells (the former had PD-1loTIM-3lo expression and higher effector function) reduced the viral load in mice chronically infected with LCMV; the therapeutic effect was further enhanced with anti-PD-L1 combination (175). T cell terminal differentiation during chronic viral infection was also shown to be associated with the EomeshiPD-1hiBLIMP-1+T-betlo phenotype (converted from T-bethiPD-1int cells) and increased cytotoxicity but decreased co-production of IFN-γ and TNF-α (176); the therapeutic reversibility of EomeshiPD-1hiT-betlo cells compared with T-bethiPD-1int cells was not examined in that study (176). Another study demonstrated opposite results, showing that anti-PD-L1 or anti-PD-1 therapy during chronic viral infection in vivo expanded only the TCF1+ memory-like CD8+ T cells with PD-1hiT-betloEomes+ expression but not terminally differentiated TCF1−CD8+ T cells (177). However, whether the effector functions of expanded TCF1+CD8+ T cells were restored was not shown.
PD-1/PD-L1 blockade also had no effect on established T cell anergy in autoimmune models (92) nor on “non-reversible” dysfunction of T cells in tumor models. In a breast cancer mouse model, the PD-1hi-expressing CD8+ T cell population failed to be rescued by anti-PD-1 therapy, showing increases in the Treg/CD8+ T ratio, in contrast to CD8+ T cells with PD-1lo surface expression, which were sensitive to anti-PD-1 mAb in a colon cancer mouse model (24). Several studies demonstrated that the therapeutic reversibility correlated to the duration of dysfunction. In a tamoxifen-inducible autochthonous liver cancer model, dysfunctional tumor-specific CD8+ T cells could be rescued by PD-1/PD-L1 blockade in the early-phase, but after 30 or more days the dysfunction was irreversible (17). Notably, this timing effect is opposite to that for PD-L1 blockade during systemic LCMV infection [fatal during the early-phase (156) but effective on days 23–40 (25)]. Also, PD-1 blockade at early time points following viral immunotherapy did not improve durable control of metastatic disease in vivo despite the high frequency of PD-1+TIM-3+CD8+ T cells (86).
Transcriptional factors (17) and epigenetic programs may define the function of tumor-specific T cells in TILs and therapeutic reprogrammability (178). Dysfunctional TILs were found to lose access to some intergenic/intragenic regions (probably enhancers), including those in Ifng, Cd5, and Tcf7, but gain access to some NFATC1-binding sites, including those in Pdcd1, Ctla4, Cd38, and Egr1/2. The reprogrammability of dysfunction, as assessed by whether the ability to produce IFN-γ and TNF-α was regained after anti-PD-1/PD-L1 therapy, is associated with the discrete chromatin state of T cells—i.e., the “plastic dysfunctional state” at early tumorigenesis and the “fixed dysfunctional state” after day 14–35—and the differential expression of TCF and NFAT family transcription factors. The chromatin changes associated with the fixed dysfunction state included closed TCF/FOS motifs and opened E2F/ETS/KLF motifs. Antigen exposure in tumors has a pivotal role in determining the chromatin state in T cells, whereas PD-1hi-expressing CD8+ T cells can be in either a plastic or fixed dysfunctional state (178).
However, PD-L1 and B7/CD28 expression in these viral infection models and tumor models, which may be relevant for the therapeutic efficacy, were unclear. For example, terminal differentiated TILs with reduced IFN-γ production may induce very low PD-L1 expression, contributing to the hyporesponsiveness to anti-PD-1/L1 therapy, if pre-existing PD-1–PD-L1 interaction is required for the anti-PD-1/L1 therapy to have a positive effect. It has been shown in vitro that PD-1 engagement with anti-PD-1 mAbs inhibited rather than enhanced CD4+ T cell expansion and cytokine production with optimal ICOS or suboptimal CD28 costimulation (20, 21, 101) and inhibited glycolysis and glutamine catabolism in T cells (106, 108). However, it is unclear why anti-PD-1 mAbs do not activate similar inhibitory signaling in T cells after blocking the PD-1–PD-L1 interaction in PD-L1+ tumors. Also unknown are whether after anti-PD-1 mAbs occupy PD-1, blocked PD-L1 will bind to the alternative CD80 receptor and whether the PD-L1–CD80 interaction in tumors is inhibitory or stimulatory.
In contrast, anti-PD-L1 mAbs, which do not bind to PD-1, should not induce de novo inhibitory signaling in T cells in PD-L1− tumors. In addition, anti-PD-L1 mAbs block both PD-1 and CD80 interaction with PD-L1, suggesting that anti-PD-L1 mAbs may have higher efficacy than anti-PD-1 antibodies in PD-L1+ tumors. However, treatment with anti-PD-L1 mAbs will not block PD-1–PD-L2 interaction or decrease PD-1 expression, and PD-L1 is broadly expressed in normal tissues, which may suggest that anti-PD-L1 mAbs are less efficacious in PD-1+ PD-L2+ scenarios but have more immune-related toxicities than anti-PD-1 mAbs.
In preclinical models, comparison between PD-1 blockade and PD-L1 blockade showed inconsistent or contradictory results. Several studies demonstrated that PD-1 and PD-L1 blockade had similar efficacy in preclinical models with PD-L1+ tumors (19, 77). In tumor-formation mouse models, PD-1 blockade showed striking efficacy in inhibiting hematogenous dissemination of tumor cells with poor immunogenicity, but PD-L1 blockade had no effect (10). However, PD-L1 blockade was more effective than PD-1 blockade in restoring the function of exhausted T cells in PD-L1-expressing mice with chronic viral infection (25). Moreover, an antibody against PD-L1 on myeloid dendritic cells improved T cell antitumor immunity, although it did not block PD-1–PD-L1 interaction (179). PD-L1 blockade had a stronger effect than PD-1 blockade in breaking T cell anergy in vivo in an OT-1 T-cell anergy model. Anergy prevention required early treatment with PD-1 or PD-L1 antibodies after tolerogen exposure, whereas delayed treatment had no effect in preventing T cell anergy (127). The ineffectiveness of PD-1/PD-L1 antibodies in breaking established T cell tolerance, in sharp contrast to the effectiveness in preventing tolerance induction, was also observed in other mouse models (92, 97, 159). In contrast, an anti-PD-L1 mAb, which specifically blocks PD-L1/CD80 but not PD-L1/PD-1 interaction (131), was able to break the pre-established T-cell anergy. However, another study showed that in NOD mice, both PD-L1 and PD-1 blockade enhanced the interactions of tolerized T cells with antigen-bearing dendritic cells, abrogated tolerance, and induced rapid development of autoimmune diabetes, whereas CTLA-4 blockade or anti-CD80 had no such effects (78).
In addition to anti-PD-1 antibodies, small-molecule compounds and peptide antagonists have been reported to inhibit the interaction between PD-1 and PD-L1 (180–183), but their clinical efficacies and dependence on PD-1/PD-L1 expression are currently unknown.
Clinical PD-1 Blockade and PD-L1 Blockade in Cancer Patients: Successes and Failures
Immune checkpoint blockade with anti-CTLA-4, anti-PD-1, and anti-PD-L1 antibodies has changed the paradigm of cancer treatment. Compared with the CTLA-4 antibodies, anti-PD-1/L1 antibodies have the advantage of lower toxicities (184–186). Currently, the US Food and Drug Administration (FDA) has approved two anti-PD-1 mAbs (PD-1 blockade), nivolumab (Opdivo; Bristol-Myers Squibb Co.) and pembrolizumab (KEYTRUDA; Merck and Co., Inc.), and three anti-PD-L1 mAbs (PD-L1 blockade), atezolizumab (TECENTRIQ; Genentech Oncology), avelumab (BAVENCIO; EMD Serono, Inc.), and durvalumab (IMFINZI; AstraZeneca UK Limited), for the treatment of cancer. The approvals were based on a high objective response rate (ORR), durability of response, or improved survival rate as demonstrated in successful clinical trials (Tables 1 and 2).
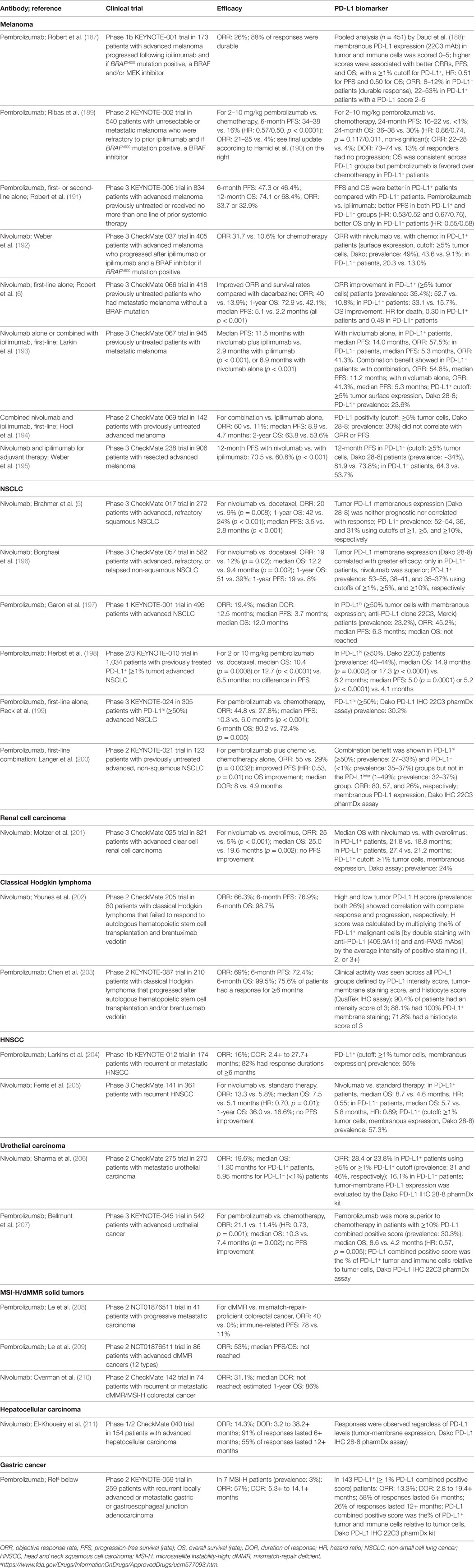
Table 1. Brief summary of the results of anti-PD-1 therapy clinical trials leading to US food and drug administration approval.
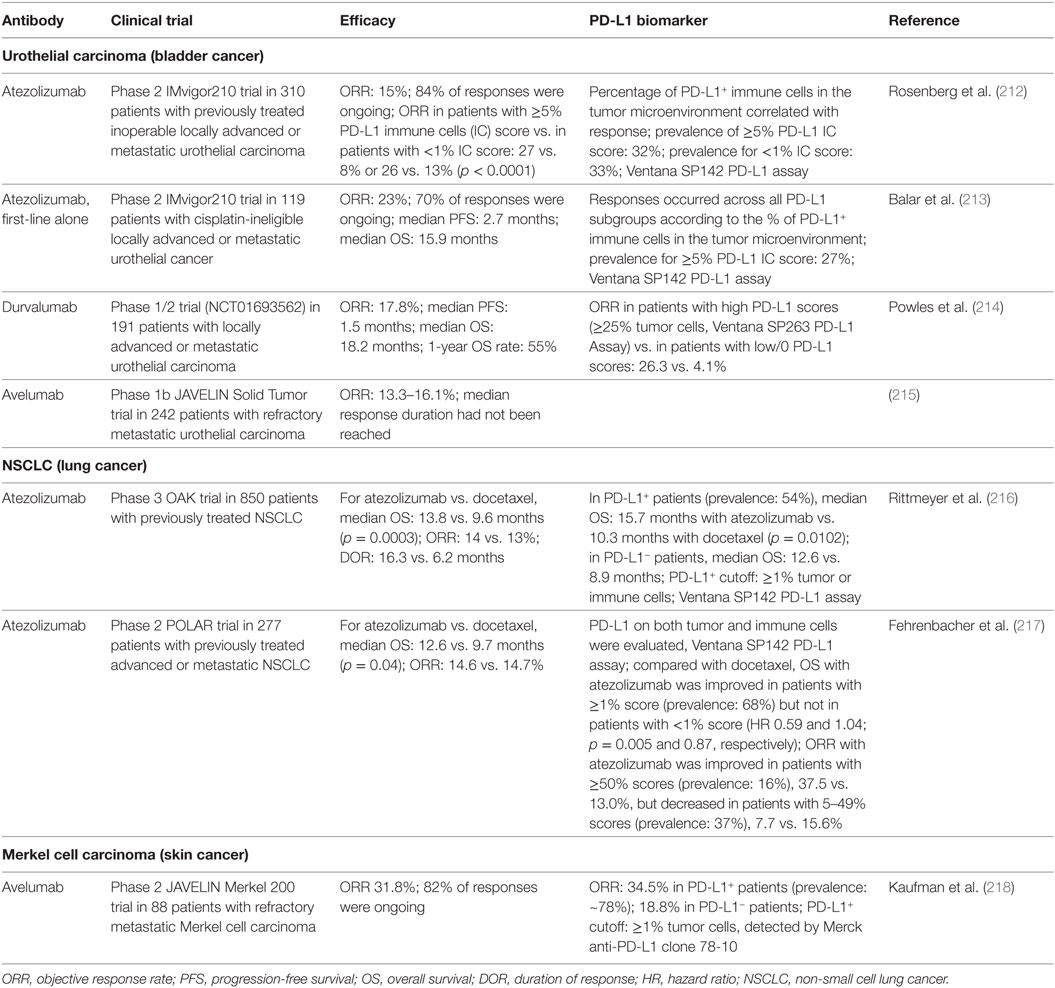
Table 2. Brief summary of the results of anti-PD-L1 therapy clinical trials leading to US food and drug administration approval.
Anti-PD-1 mAbs as single agents or combined with chemotherapy or ipilimumab (anti-CTLA-4 mAb) have been approved for the treatment of the following cancers as first-line, second-line, third-line, or later-line therapies: melanoma (6, 184, 187–195), non-small cell lung cancer (NSCLC) (5, 196–200, 219), classical Hodgkin lymphoma (202, 203, 220), renal cell carcinoma (201), head and neck squamous cell carcinoma (HNSCC) (204), urothelial carcinoma (205–207), microsatellite instability-high (MSI-H) cancers (including colorectal cancer and other solid cancers) (208–210), hepatocellular carcinoma (211), and gastric or gastroesophageal junction adenocarcinoma [approval to pembrolizumab (Table 1); however, only nivolumab phase 3 results are available (221)]. Anti-PD-L1 mAbs as single agents in first-line, second-line, or salvage therapies have been approved in urothelial carcinomas (212–215, 222), NSCLC (216, 217), and Merkel cell carcinoma (218). Many clinical trials in different cancer types or settings are still ongoing and some have shown good results, such as the phase 3 PACIFIC clinical trial for durvalumab as consolidation therapy in patients with stage III NSCLC (223). The ORRs with PD-1/PD-L1 blockade as monotherapy in relapse/recurrence settings largely differ by disease entities; the ORR is close to 70% in classical Hodgkin lymphoma which frequently has 9p24 copy number alterations (202), ~40% in skin cancers, ~20% in lung cancers, ~25% in renal cancer, 13–23% in bladder cancer, and 13–16% in HNSCC. PD-1 blockade and PD-L1 blockade largely showed similar efficacy, although the ORRs were ~5% higher with PD-1 blockade than with PD-L1 blockade in NSCLC, and results of PD-L1 blockade need to be validated in phase 3 studies.
However, anti-PD-1/PD-L1 therapies did not work in all cancers [e.g., chronic lymphocytic leukemia (224)]. Although most of responses were more durable than traditional therapies, some patients who initially responded to checkpoint blockade experienced relapse [acquired resistance; however, a small subset of relapsed patients could still respond to continuing blockade therapy; the rate was 3.6% in urothelial carcinoma patients treated with atezolizumab (225)]. Moreover, recently five phase 3 studies have failed to meet the endpoints [first-line nivolumab alone or durvalumab plus tremelimumab compared with chemotherapy; nivolumab, pembrolizumab, or atezolizumab as a later-line therapy compared with chemotherapy or standard treatment (226, 227), Table 3], even though blockade has shown clinical activity in phase 1/2 trials (212, 228–231). Two phase 3 clinical trials of pembrolizumab in multiple myeloma have been placed on full clinical hold owing to increased risk of death.
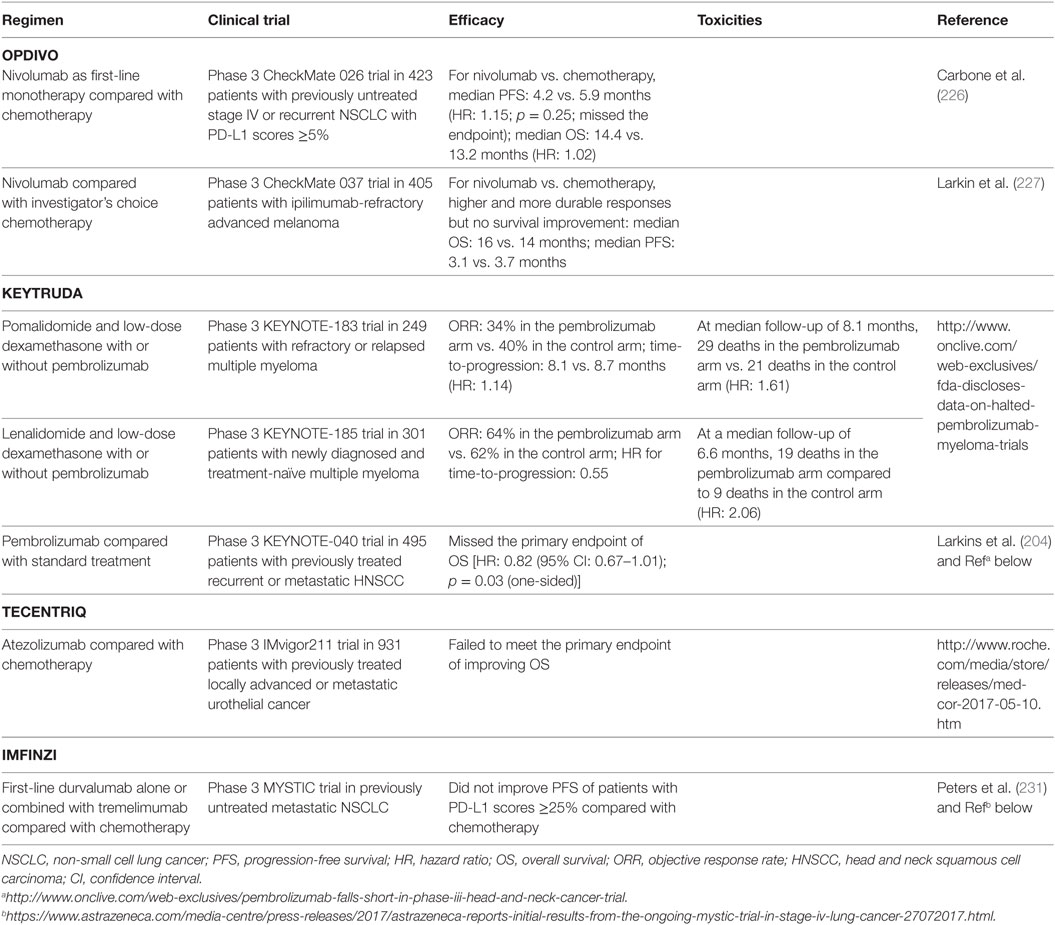
Table 3. Examples of anti-PD-1/L1 clinical trials that missed the endpoint or were discontinued owing to increased risk of death.
In addition, hyperprogression, a new pattern of disease progression after anti-PD-1/PD-L1 monotherapy, that is associated with elderly age and worse overall survival but not specific tumor types, has been identified in ~9% of cancer patients (232, 233). A higher rate of hyperprogression (regional recurrence in most cases without any cases of pseudoprogression), 29%, was retrospectively identified in patients with HNSCC (234). The different rates may result from differences in hyperprogression definition and size of the cohorts, since in the HNSCC cohort, hyperprogression was significantly associated with shorter progression-free survival but not with overall survival. These unexpected clinical observations may reflect our incomplete understanding of the PD-1/PD-L1 pathway and immune regulation mechanisms.
Molecular Determinants and Predictive Biomarkers for PD-1/PD-L1 Blockade Immunotherapy: PD-L1+, Tumor Mutational Load, T Cell Functional State, or Other Host Factors
Given the high cost and potential toxicities of the treatment, efforts have been made to identify predictive biomarkers for selecting patients who are most likely to benefit from anti-PD-1 immunotherapy. PD-L1 is the first and most studied biomarker for PD-1 blockade (188, 235). Theoretically, PD-1 blockade should work only in PD-1+ PD-L+ patients and not in PD-1− patients (4) or PD-L− patients (most PD-L1− cases are PD-L−) (Figure 1), because PD-1 ligation is indispensable for PD-1-mediated suppression, and in the absence of PD-1 natural ligand, anti-PD-1 mAbs can act as PD-1 agonists to inhibit rather than enhance PD-1+CD4+ T-cell function (20, 21, 101). However, in multiple clinical trials, PD-L1 negativity was not found as an excluding factor for patient selection (Table 1). Durable clinical response to PD-1 blockade was also observed in some PD-L1− patients with unknown PD-L2 status (although with a lower response rate in most studies). Furthermore, in some studies of squamous NSCLC and renal cell carcinoma, the efficacy of PD-1 blockade (response rate or survival outcome) in PD-L1− patients was similar to or even better than that in PD-L1+ patients (5, 201). The predictive values of the percentage and cellular levels of PD-1 expression in correlation with PD-L1 expression were unclear in these clinical studies.
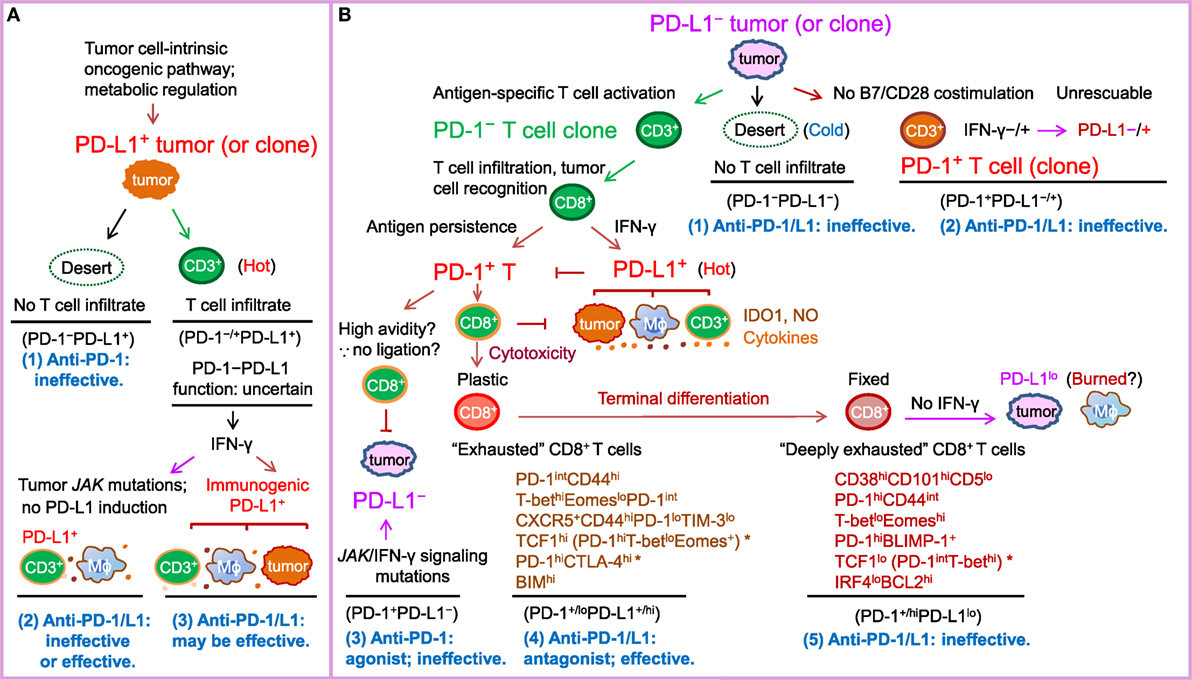
Figure 1. Schematic illustration of PD-1/PD-L1 expression in the tumor setting as a marker of T cell activation and driver of T cell dysfunction, as well as a predictive biomarker for response to PD-1/PD-L1 blockade in PD-L1− and PD-L1+ tumors according to the prevailing notion. PD-L2, which is infrequently expressed and potentially has PD-1-independent positive function, is not depicted in the figure for clarity. The PD-L1–CD80 axis is also not illustrated because its role and significance in the cancer setting is unclear. (A) In tumors (or tumor clones) with cell-intrinsic PD-L1 expression driven by the oncogenic pathways, whether anti-PD-1/PD-L1 is effective may depend on the activity of the PD-1–PD-L1 axis. If T-cell infiltration is lacking (a “desert”-like immune landscape, or “cold” tumors), or PD-1 is not expressed on T cells, anti-PD-1 therapy will not elicit a de novo T cell response. If the tumor is infiltrated with immune cells (“hot” tumor) and the oncogenic or immunogenic PD-L1 expression suppresses T cell activation by binding to PD-1 within the T-cell receptor microclusters, anti-PD-1/PD-L1 therapy can be effective. IDO1, NO (nitric oxide), and suppressive cytokines in the tumor microenvironment may contribute to resistance to PD-1/PD-L1 blockade therapy. (B) In tumors without cell-intrinsic PD-L1 expression, tumors (or tumor clones) with low immunogenicity (“cold” tumors) or costimulation may not respond to anti-PD-1/PD-L1 therapy, whereas tumors (or tumor clones) with a high neoantigen load elicit antitumor T cell responses (“hot”) but their response to anti-PD-1/PD-L1 therapy varies. Antigen-specific CD8+ T cells secrete IFN-γ, which may turn PD-L1− tumors into PD-L1+ tumors infiltrated with PD-L1+ macrophages, dendritic cells, and T cells. However, if tumors do not have IFN-γ receptors or have JAK2 mutations, tumors may remain PD-L1− and not respond to anti-PD-1/PD-L1 treatment or respond if PD-L1 is induced on non-tumor immune cells. In PD-L1+ tumors, prolonged antigen stimulation gradually induces PD-1 expression on antigen-specific T cells. PD-1 ligation with PD-L1 induced on tumors, antigen-presenting cells, and T cells in hot tumors in turn suppresses antitumor function of effector T cells, leading to T cell “exhaustion” (a term initially used for T cell dysfunction during chronic viral infection). Early-phase T cell “exhaustion” is plastic and can be reversed by PD-1/PD-L1 blockade; in contrast, if T cell dysfunction is fixed after terminal differentiation, “deeply exhausted” T cells cannot be rescued by PD-1/L1 blockade. Inflexibility in transcriptional and epigenetic programs may contribute to the therapeutic irreversibility of deeply exhausted T cells. Potential markers suggested by studies in tumor models, viral infection models, and cancer patients are summarized below the labels for these two different dysfunctional stages of PD-1+CD8+ T cells. * indicates disparities in PD-1 levels in the literature (please refer to the text for details).
Unlike tumor PD-L1 expression, which has shown predictive value for the efficacy of anti-PD-1 therapy in most studies (Table 1), PD-L1 expression on immune cells in the tumor microenvironment was more correlated with treatment response to anti-PD-L1 therapy (212, 236) (Table 2). However, the correlation of PD-L1 expression with response was absent in other studies, with similar ORRs or improved survival rates occurring across all PD-L1 subgroups (213, 216). Even in studies showing correlations, some patients who lacked both tumor and immune cell expression of PD-L1 still responded to anti-PD-L1 therapy (212, 218). The mechanisms of response to anti-PD-1/L1 therapy in these PD-L1− patients are unknown, posing intriguing questions. One plausible explanation would be failure to detect PD-L1 expression owing to technical reasons or temporal and spatial expression (for example, clustered PD-L1 expression in the early time course of T cell activation and the dynamic PD-L1 expression on circulating T cells).
Efforts have been made to improve the prediction accuracy, by standardizing the detection antibodies and immunohistochemistry assays (Tables 1 and 2), separately assessing PD-L1 expression on tumor cells and PD-L1 expression on non-tumor cells, and optimizing PD-L1 cutoffs. Because most studies have used low PD-L1 cutoffs (≥1% or ≥5%), and PD-L1 function is limited to local inhibition, acting as the “molecular shield” of PD-L1+ cells, we would postulate that the association of PD-L1 expression with immune activation status, rather than the correlation with immune suppression strength, may underlie the predictive value of PD-L1 expression for anti-PD-L1 therapy. Interestingly, in NSCLC, anti-PD-1 therapy (pembrolizumab) demonstrated superiority over chemotherapy in patients with ≥50% or <1% tumor PD-L1 scores, but this benefit was absent in patients with 1–49% tumor PD-L1 scores (200). This non-linear correlation reappeared in an anti-PD-L1 study in NSCLC, in which atezolizumab compared with docetaxel was associated with improved ORR in the ≥50% PD-L1+ group but decreased ORR in the 1–49% group (217). The predictive 50% cutoff of PD-L1 expression has been included in the FDA indication for pembrolizumab in metastatic NSCLC tumors as frontline therapy (199).
A recent biomarker study using longitudinal tumor samples from patients with metastatic melanoma showed that expression of PD-1, LAG-3, and PD-L1 in early on-treatment (median: 1.4 months after initiation of treatment), but not in pre-treatment (median: 3 months prior to treatment), biopsies was highly predictive for response to PD-1 blockade, suggesting the inability to accurately predict the clinical response before anti-PD-1 therapy (237). In this study, some responders had no PD-1/PD-L1 expression in pre-treatment samples but had high immune marker expression in on-treatment samples; conversely, many non-responders had high PD-L1 expression in pre-treatment samples but had low PD-L1 expression in on-treatment samples. The observation that PD-L1− patients turned into PD-L1+ patients appeared to suggest that immunogenic PD-L1 expression was induced after anti-PD-1 therapy. However, results from in vitro experiments (20, 21, 101) suggest that in PD-L1− patients, binding of anti-PD-1 mAbs to PD-1 will inhibit IFN-γ production, and therefore, the baseline PD-L1− status should not be changed after anti-PD-1 therapy. In contrast to these discrepancies, hyperprogression after anti-PD-1/L1 therapy tended to be associated with PD-L1 negativity (232, 233).
Because PD-L1 expression in on-treatment tumors predicted response to anti-PD-1 treatment (237), one would postulate that inducibility of PD-L1 expression can predict effectiveness of PD-1 blockade. Consistently, JAK2/STAT1 signaling is increased in classical Hodgkin lymphoma (238) which showed high ORRs [(220) and Table 1] to PD-1 blockade. Conversely, JAK1/2 and APLNR loss-of-function mutations, which result in non-inducibility of tumor PD-L1 expression by IFN-γ, have been associated with primary or acquired resistance to PD-1 blockade in solid tumors; PD-1 blockade was ineffective for these patients even if their mutational load was high (239–241). However, PD-L1 should still be inducible on nonmalignant immune cells, which did not harbor JAK1/2 and APLNR mutations as tumors did, suggesting that other immune escape/suppressive mechanisms may also contribute to the treatment resistance in these patients. Indeed, JAK1/2 or IFN-γ pathway gene mutations were not always found to be associated with clinical response (242, 243).
Microsatellite instability arising from mismatch-repair deficiency is the second predictive biomarker (208, 244) approved by FDA (245). MSI-H tumors have high levels of neoantigens associated with a strong local and systemic immune response (246). In addition, MSI-H tumors were shown to display upregulation of multiple immune checkpoints, including PD-1, which may limit the vigorous immune microenvironment (247), making PD-1 blockade a rational treatment approach. In metastatic colorectal cancer, the ORR with pembrolizumab was 40% in MSI-H patients compared with 0% in mismatch-repair-proficient patients (208). In an expanded study of advanced mismatch-repair-deficient cancers across 12 different tumor types, the objective radiographic response rate was 53% and the complete response rate was 21% (209).
High tumor mutational burden and neoantigen load, which are fairly common across cancer types compared with the uncommon MSI-H (248), have also been correlated with sensitivity to PD-1 blockade (higher ORR and/or prolonged survival) in melanoma, NSCLC, glioma (243, 249–252), and likely across types of solid cancers (252). In addition, high numbers of indel mutations were found in renal cell carcinomas, and frameshift indel count was associated with response to PD-1 blockade in melanoma patients (253). Conversely, high copy number loss burden was associated with resistance to checkpoint blockade (242). However, classical Hodgkin lymphoma has a high ORR but not a high mutational burden. Some gene mutations may correlate with treatment resistance (such as JAK2 and B2M). Although a study showed that neoantigen load correlated with T-cell infiltration in colorectal cancers (254), another study showed that the density of immunogenic antigens did not correlated with T-cell infiltration and local immunity in melanoma (255). To reduce whole-exome sequencing and enhance the clinical applicability of tumor mutational burden, targeted comprehensive genomic profiling (248) and small next generation sequencing panels (256) have been developed. Progress has been made in understanding the association of response with particular gene (such as DNA repair genes BRCA2 and POLE; potentially also PMS2, MSH2/6, and MLH1) mutations and clonal neoantigens, as well as T cell clones responding to PD-1/L1 blockade (243, 248, 250, 256–259). POLE mutations have been shown to be associated with not only higher mutational burden (248) but also immune signatures and lymphocytic infiltration independent of MSI-H status in endometrial cancer (260). However, particular gene mutations and alterations (such as loss of PTEN and CDKN2A) and mutational burden showed inconsistent significance in studies (60, 242, 243, 258). Tumor mutation load and clonal mutation load (less heterogeneity) were associated with overall survival and response to nivolumab in ipilimumab-naive patients but not in patients who had previously progressed on ipilimumab (243). In the latter group of patients, response to PD-1 blockade was inconsistently associated with T cell clonality (242, 243).
Some T cell-derived biomarkers have also been found to be predictive of response to PD-1 blockade in patients with advanced melanoma; these biomarkers include high baseline CD8+ and PD-1+ density at the invasive tumor margin and inside the tumor, proximity between PD-1+ and PD-L1+ cells, clonal TCR repertoire (43, 242), BIM expression in tumor-reactive PD-1+CD8+ T cells (261, 262), and higher proportion of PD-1hiCTLA-4hi cells with a partially exhausted T cell phenotype (capable of producing IFN-γ but lost the ability to produce TNF-α and IL-2) within CD8+ TILs (263). Baseline PDCD1 mRNA expression was also associated with progression-free survival after anti-PD-1 therapy in a pooled cohort of cancer patients (264). However, the findings that PD-1hiCTLA-4hi TILs that were preferably expanded after anti-PD-1 therapy in melanoma patients (263) counters the findings in preclinical models [PD-1hi T cells were irreversible (178) and anti-PD-1 therapy was effective only in tumors with low frequencies of PD-1+ T cells (24)].
In addition, in preclinical models, low levels of CD38, CD101, and CD30L whereas high levels of CD5 surface expression (178), low to intermediate levels of PD-1 expression on CD8+ T cells (24), as well as high TCF1 (177) and IRF4 nuclear expression were associated with T cell plastic dysfunctional state whereas high BCL2 expression in CD8+ T cells was associated with fixed dysfunctional state (178). The potential of these biomarkers may be clarified in future anti-PD-1/L1 clinical trials.
Moreover, several non-T host factors, including absolute lymphocyte count, relative eosinophil count, ≤2.5-fold elevation of serum lactate dehydrogenase, and the absence of metastasis other than soft-tissue/lung metastasis, have also been associated with improved overall survival in melanoma patients treated with pembrolizumab (265). However, efficacy comparison with controlled arms (anti-PD-1 therapy compared with traditional therapy) will be more informative (266). Also notably, a retrospective analysis found a five-factor {serum lactate dehydrogenase elevation, age <65 years, female sex, previous ipilimumab treatment [however, this factor was non-significant in the earliest pembrolizumab trial (184)], and liver metastasis} prediction scale was associated with lower ORRs to anti-PD-1 therapy (267). Although studies have shown that response to anti-PD-L1 therapy was associated with a Th1 gene signature in on-treatment samples (236), a recent study found that early decrease of IL-8 (a Th1-associated cytokine) levels in the serum 2-3 weeks after anti-PD-1 therapy was predictive of response in melanoma and NSCLC patients, including rare cases [0.6–4% (268, 269)] with pseudoprogression (270). A prospective trial in melanoma patients found that response to anti-PD-1 therapy induced genomic contraction, which was associated with pronounced pre-existing immune signatures in pre-treatment samples, including TCR/PD-1/IFN-γ/IL-2/PI3K signaling signatures as well as MHC class II and other genes resembling a macrophage signature (243).
The gut microbiome in cancer patients has been shown to influence PD-1 blockade efficacy. Clinical responses to anti-PD-1 immunotherapy were associated with high diversity and relative abundance of Ruminococcaceae bacteria in prospectively collected microbiome samples from patients with metastatic melanoma (271) and relative abundance of A. muciniphila in patients with NSCLC, renal cell carcinoma, or urothelial carcinoma (272). In addition, commensal Bifidobacterium was shown to confer improved anti-PD-L1 efficacy in vivo (273). Mechanisms accounting for the favorable prognosis may include increased tumor infiltration of CD8+ T cells, more effector T cells than Tregs in systemic circulation, dendritic cell function, IL-12 secretion, anabolic metabolism, and systemic inflammation (271–273), but the mechanistic links for these immunomodulatory effects remain unknown. PD-1 also regulates the gut microbiota and the function and survival of IgA-producing plasma B cells, but this effect can be abrogated by PD-1 blockade, as was shown in vivo (274).
Overcoming Resistance to PD-1/PD-L1 Blockade: Various Combination Strategies
Like a tug-of-war, the actions of immune response and tumor development resist each other. PD-1 blockade may have antitumor effects in cancer patients (275) but this is not always sufficient for a clinical response. Resistance mechanisms may come from either the immune system or the tumor. The ratio of immunologic reinvigoration to tumor burden, but not the magnitude of reinvigoration alone, was found to be predictive of response to pembrolizumab and overall survival in patients with advanced melanoma (276). Maximized innate and adaptive responses, achieved through combination therapies, were capable to eliminate large, advanced, poorly immunogenic tumors in mice (277).
Multiple tumor- or immune-driven resistance mechanisms have been identified and targeted in combination with PD-1 blockade. First, absence of “signal 1” and T cell activation leads to ineffectiveness of anti-PD-1/L1 monotherapy (278). Studies have shown that B2M mutations, deletions, or loss of heterozygosity, which leads to loss of MHC class I expression and failure of antigen recognition, is a potential mechanism for immune escape and resistance to PD-1 blockade in patients with melanoma (239, 279). Clinical outcome of anti-PD-1/PD-L1 therapy was shown to correlate with MHC class II positivity in a unique subset of melanoma cells (typically MHC class II is expressed only on immune cells in solid tumors), as well as increased CD4+ and CD8+ TILs in melanoma patients (280).
However, a surprisingly high frequencies of decreased or absent expression of β2M/MHC class I (79% overall; 92% in PD-L1/L2 amplified cases) and MHC class II (67%) were found in 108 patients newly diagnosed with classical Hodgkin lymphoma (88% of patients had nodular sclerosis Hodgkin lymphoma; 82.5% were negative for Epstein-Barr virus) (281). High frequencies of abnormal MHC expression were also observed in another 233 patients with Epstein–Barr virus-negative classical Hodgkin lymphoma (83.2% for MHC class I and 46.8% for MHC class II) (282). Because classical Hodgkin lymphoma has a high ORR to PD-1 blockade, these data may suggest that non-T responses also play important roles in the effect of PD-1 blockade, which is supported by a study showing that after PD-1 blockade, genes implicated in cytolysis and natural killer cell function were upregulated in patients (283). In addition to natural killer cells whose antitumor function is MHC-independent, invariant natural killer T cells can be activated by signals from a lipid–CD1d complex (284), and alloreactive CD8 T cells demonstrated cytotoxicity effector function against MHC class I-deficient allogeneic cells in a TCR-independent manner (285). To enhance antigen recognition and T cell response, chimeric antigen receptor T cell therapies, bispecific T-cell engagers, oncolytic viruses, vaccination, and intratumoral IL-12 plasmid electroporation have been combined with PD-1/PD-L1 blockade (86, 286–290) but the clinical results are currently unavailable.
Second, because the absence of costimulation (“signal 2”) can result in T cell anergy (278), impaired costimulation could lead to ineffectiveness of PD-1/PD-L1 blockade. This is supported by recent studies showing that rescue of exhausted CD8+ T cells with PD-1 blockade requires CD28/B7 costimulation in a mouse model with chronic viral infection (291) and that response to PD-1 blockade requires the presence of both CD4+ and CD8+ T cells as well as CD28 and CD80/CD86 costimulation in a murine melanoma tumor model with low mutational load (165). However, an earlier study showed that PD-1 blockade in vivo leads to accelerated rejection of heart allografts only in the absence of CD28 costimulation, accompanied by expansion of alloreactive T cells and enhanced generation of effector T cells (292).
Although PD-1 is expressed only after T cell activation, which requires costimulation (9), it has been shown that PD-1 can be induced without CD28 costimulation (11); in fact, lack of costimulation leads to upregulation of PD-1 (16). In one study of patients with early-stage lung cancer, 10–80% of tumor-infiltrating CD8+ T cells were CD28− (291). CD28 could be lost during aging, with repeated antigenic stimulation, and after exposure to some cytokines (293). Therefore, insufficient CD28 costimulation could be an important resistance mechanism for PD-1 blockade. Consistent with the high efficacy of PD-1 blockade in Hodgkin lymphoma, CD28 is strongly or moderately expressed on T cells surrounding CD80/CD86hi-expressing Reed-Sternberg cells (294–296). In contrast, chronic lymphocytic leukemia has no or low levels of CD80/CD86 expression on leukemia cells (297–299) with immunologic synapse formation defects (300) and is resistant to pembrolizumab in a clinical trial (224).
In addition to the CD28 pathway, the CD40–CD40L costimulatory pathway has been shown to be required for the ameliorative effects of anti-PD-L1 therapy and plays a critical role in rescue of exhausted CD8 T cells (301). Anti-CD40 agonists, which alone could effectively reverse cytotoxic T cell exhaustion by activating the mTORC1 pathway in vivo, significantly enhanced action of PD-1 antagonists in chronic infection in vivo (302). In addition, combining PD-1/PD-L1 blockade with costimulatory agonist antibodies to CD27 (164), CD137 (4-1BB) (303, 304), TLR3/7/9 (305–307) [TLR3 is also a safe vaccine adjuvant (308)], GITR (309), STING (310), or OX40 [the synergy to restore function of exhausted CD8+ T cells was only observed under helpless (no CD4+ T cell) condition (311)] have demonstrated enhanced antitumor effects in preclinical models. However, sequential (delayed anti-PD-1) but not concurrent anti-OX40 and anti-PD-1 treatment (combination) in vivo resulted in increased efficacy which required both CD4+ and CD8+ T cells (312).
Third, although anti-PD-1/PD-L1 antibodies block PD-1–PD-L1 interaction, they do not affect PD-1/L1 expression. Studies have demonstrated that expanded exhausted CD8+ T cells reactive to anti-PD-1/PD-L1 therapy in vivo retain high PD-1 expression (25); PD-1/PD-L1 blockade was shown to enhance IFN-γ and PD-L1 expression (42, 72) and increase tumor-infiltrating PD-1+ T cell frequencies (14). One preclinical study showed that the antitumor effect of anti-PD-1 therapy required the presence of PD-1loCD8+ T cells before treatment and decreased frequencies of tumor-infiltrating PD-1+CD8+ T cells below a threshold after the anti-PD-1 therapy (24). However, clinical studies showed that PD-1hi expression before treatment (263) or on treatment correlated with response to PD-1 blockade in melanoma patients (237).
High PD-1 expression as resistance mechanism is probably more relevant for anti-PD-L1 therapy, which only blocks PD-1–PD-L1 interaction by modulating cytosolic signaling pathways and does not reduce PD-1 expression. In a chronic LCMV infection model and a melanoma tumor model, anti-PD-L1 therapy did neither downregulate the PDCD1 gene in treated T cells nor did reprogram the epigenetic landscape, including chromatin accessibility to Nr4a and NFAT transcription factors (14, 90).
Strategies to modulate the transcriptional (including epigenetic) and posttranscriptional regulation of PD-1/PD-L1 expression may lead to a more durable response in patients. The transcription factors and pathways positively regulating PD-1 expression include BLIMP-1 (although conflicting results were also reported) (313, 314), IFN-α–IRF9 (315), TGFβ–SMAD3 (316), NFATc1 (317), STAT3/4/NFATc1/CTCF (318), the Notch signaling pathway (319), FOXP1 (320), c-FOS (321), STAT1/2 (322), and NF-κB (323). In contrast, T-bet (324), trimethylation (37, 325, 326), and a chromatin organizer SATB1 (327) negatively regulate PDCD1 expression. Chromatin accessibility to PDCD1 enhancers (including the −23.8 kb enhancer) is important for PD-1 expression in exhausted T cells (328).
Fourth, insufficient antitumor activity may result from multiple T cell subtypes and subclones (including those with “fixed” T cell dysfunction) that are not responsive to PD-1/L1 blockade. Dysfunction of these T cell subclones may lead to tumor evolution of subclonal neoantigens, which were associated with primary and acquired resistance to checkpoint blockade in patients (250, 258). In a cancer model, “fixed” dysfunction of driver-antigen-specific T cells was associated with PD-1, TIM-3, LAG-3, and 2B4 expression (17). Although PD-1 has a uniquely critical role in immune suppression, co-expression of multiple immune checkpoint receptors on T cells resulted in greater T cell exhaustion (329).
Multiple blockade combinations have shown synergetic effects in releasing adaptive immune resistance in preclinical models (330), as well as combination strategies targeting the transcriptional program (17). Histone deacetylase inhibitors have been shown to increase expression of multiple T cell chemokine (paradoxically also PD-L1 expression) and enhance the response to PD-1 blockade in vivo (57, 331). EZH2 and DNMT1 inhibitors increased Th1-type chemokines and T-cell infiltration, and augmented the efficacy of PD-L1 blockade therapy in vivo (332). Simultaneous blockade of PD-1 and LAG-3 synergistically improved viral control and tumor eradication (329, 333, 334). Combined TIGIT and PD-1 blockade (335), or combined PD-1, TIM-3 (336), and BLTA blockade (337), increased the expansion and effector function of antigen-specific CD8+ T cells from melanoma patients ex vivo.
The combination of PD-1 blockade and CTLA-4 blockade, which has distinct immunologic effect and activates different T cell populations in vivo (283, 338), demonstrated greater antitumor effects than the use of either antibody alone (339, 340). Furthermore, clinical trials have demonstrated remarkable efficacy of combined nivolumab and ipilimumab therapy in melanoma (ORR: ~60%) (194, 341), although combined durvalumab (anti-PD-L1) and tremelimumab (anti-CTLA-4) in NSCLC was not successful in a recent phase 3 study (Table 3). Sequential use of nivolumab followed by ipilimumab or in reverse sequence did not reduce the toxicities resulting from concurrent (combination) therapy with nivolumab and ipilimumab, as found in a phase 2 study; nivolumab followed by ipilimumab showed higher response and survival rates but also higher toxicities compared with sequential use of ipilimumab followed by nivolumab, in which the synergistic effect was lost (342).
Fifth, the immunosuppressive tumor microenvironment may contribute to the in effectiveness of anti-PD-1/L1 treatment. Tregs, MDSCs, M2 macrophages, and their associated cytokines, chemokines, and other soluble factors are well-recognized inhibitory mechanisms orchestrated to suppress antitumor immunity (72). Depletion of tumor-infiltrating Tregs was shown to synergize with PD-1 blockade to eradicate established tumors in vivo (343). However, the clinical significance of Tregs was inconsistent in different studies, likely due to the differential function of Treg subsets (344). Moreover, as shown in vivo, the suppressive function of NRP1+/+ Tregs could be lost and converted to antitumor immunity in the presence of IFN-γ produced by HIF-1αhi NRP1−/− Tregs. This functional fragility signaled through the IFN-γ receptor was required for the effectiveness of PD-1 blockade in vivo (345).
Increased MDSCs have been shown to be associated with poor prognosis (346), whereas decrease in macrophages after anti-PD-1 therapy was associated with clinical response in melanoma patients (243). Combination of PD-1/PD-L1 blockade with tumor vaccines only partially restored the effector function of TILs stimulated by immunization and decreased Treg infiltration, but had little effect on the frequencies of MDSCs in the tumor lesions in vivo (19). Anti-PD-L1 blocking mAb augmented IFN-γ-mediated nitric oxide production by macrophages which inhibited CD4+ T cell proliferation; nitric oxide synthase inhibitor L-NMMA abrogated the inhibition and increased cytokine production (174). Indoleamine 2,3-dioxygenase (IDO) expression in tumor-associated macrophages and MDSCs induced by IFN-γ during CD8+ T cell response, can cause tryptophan deficiency and “metabolic checkpoint” in T cells (347, 348). Combining IDO inhibitors with anti-PD-1 therapy was shown to increase effector T-cell infiltration in vivo (349), and this combination has shown promising results in ongoing clinical trials (350). In addition, upregulation of IL10 and macrophage/monocyte chemotactic genes was associated with resistance to anti-PD-1 therapy (259). Combination of PD-1 blockade with IL-10 neutralization in vivo resulted in reduced tumor burden and improved murine survival, accompanied by augmented antitumor function of T cells and decreased infiltration of MDSCs (351). However, recent clinical trials demonstrated that pegylated recombinant IL-10 combined with PD-1 blockade therapy enhanced the antitumor effect (352).
Moreover, a study showed that in vivo PD-1− tumor-associated macrophages removed anti-PD-1 mAbs from the surface of PD-1+CD8+ T cells, mediated by the interaction between FcγII/III receptors and the anti-PD-1 Fc domain glycan (353). Therapeutic inhibition of FcγR interaction enhanced anti-PD-1 efficacy in vivo. Also, nivolumab was transferred from human CD8+ T cells to macrophages in an in vitro coculture system (353), although the IgG4 constant region sequences of nivolumab are designed to contain an S228P mutation to prevent antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity (4). It is unknown whether pembrolizumab, which binds to PD-1 at a completely different region than does nivolumab (354), can also be transferred by this FcγR–mediated mechanism. Unlike anti-PD-1 mAbs, selective depletion of Tregs, dependent on activating Fcγ receptors expressed by macrophages, is essential for the activity of anti-CTLA-4 therapy in vivo (355, 356).
Sixth, systemic immunity is critical for tumor eradication and protection against new tumors; in the immune network, dendritic cell function and T cell infiltration play an important role (357). Gut dysbiosis (loss of microbial diversity) and antibiotic treatment were associated with shorter progression-free and/or overall survival in cancer patients receiving anti-PD-1 immunotherapy (271, 272). Conversely, improving the gut microbiome may lower the cancer-immune set point and circumvent resistance to PD-1 blockade (272). Peritumoral injection of LCMV alone or combined with PD-1 blockade has also been shown to induce immune surveillance and tumor regression in vivo (358).
Concluding Remarks
Although the complexity of the PD-1/PD-L1 pathway has been revealed, our current understanding of the rejuvenation potential of T cells is only the tip of the iceberg. Accumulating evidence has demonstrated that PD-1 ligation suppresses the effector function of activated T cells; PD-L1 can directly cause tumor immune evasion; and anti-PD-1/PD-L1 mAbs that prevent PD-1–PD-L1 interaction can restore T-cell effector function. However, tumor PD-L1 expression through cell-intrinsic mechanisms may not have a significant role in driving immune suppression; PD-L1 and PD-L2 may also have costimulatory functions; and PD-1/PD-L1 blockade did not always elicit an effective antitumor response in preclinical studies. Moreover, although many anti-PD-1/PD-L1 clinical trials were remarkably successful which have revolutionized the treatment of cancer, some failed to reach the endpoint or resulted in an increased risk of death. In the setting of advanced cancers except Hodgkin lymphoma (likely also MSI-H tumors), only the minority of cancer patients had durable response to PD-1/PD-L1 blockade monotherapy, and some patients even had disease hyperprogression. Classical Hodgkin lymphoma, which does not have a high mutational burden or MHC class I expression, demonstrated a high response rate to PD-1 blockade therapy.
In addition to summarizing these paradoxical results in studies of PD-1/PD-L1 and PD-1/PD-L1 blockade, this review discussed a few open questions from mechanistic and clinical perspectives. As discussed, both PD-1 and PD-L1 are often (but not always) associated with T cell dysfunction; PD-1+ and PD-L1+ expression can also indicate T cell activation although PD-L1 and PD-1 may be expressed in different stages of immune response; markers to distinguish PD-1+ T cells with high functional avidity from exhausted PD-1+ T cells are unclear. Both PD-1 and PD-L1 can either dependently or independently drive immune suppression. Whether tumor or host factors dictate immunity remains to be determined. Mechanisms that are not completely understood also include those governing PD-1 expression, molecular pathways underlying PD-1/PD-L1 blockade, the difference in PD-1 signaling upon PD-L1 binding and upon anti-PD-1 mAb binding, metabolic crosstalk between tumor cells and T cells, and functional relationship (causal, consequential, or independent) between PD-1/PD-L1 expression and cell metabolism. Molecular delineation and critical node identification may also help clarify the inconsistent preclinical results of blocking PD-1 compared with blocking PD-L1.
It is unclear whether PD-1 blockade has different action (antagonist or agonist) in PD-L1+ and PD-L1− patients. Also uncertain is whether this and other differences between PD-L1 and PD-1 (for example, the association of PD-L1 expression with earlier stage of immune activation, the more dynamic PD-1 expression, or other factors which are critical for immune response but differentially associated with PD-1 and PD-L1 expression) underlie the better predictive value of PD-L1 over PD-1 expression as a biomarker for clinical response. Tumor mutational burden has also emerged as a promising biomarker; however, our understanding of clonal mutations, T cell clonality, and neoantigen-reactive TIL clones responsive to PD-1/PD-L1 blockade may be still in its infancy. In addition, infiltration of immune cells, tumor immunogenicity, strength of TCR signaling and costimulation/co-inhibition, T cell differentiation stage and chromatin flexibility, immune cells and soluble factors in the tumor microenvironment, pharmacologic kinetics of antibodies, and systemic immunity may all affect the efficacy of PD-1 blockade. Future studies in the fast advancing field of immunotherapy may shed light on these intriguing questions, develop algorithms to accurately predict the blockade efficacy, and pave the way for a new era of combination immunotherapy.
Author Contributions
ZX-M conceptualized and wrote the manuscript and created the figure. KY contributed to the conception and writing. MZ and JL revised the manuscript. All authors read and approved the final manuscript. The authors thank Erica A. Goodoff from the Department of Scientific Publications, MD Anderson Cancer Center, for her edition of the manuscript.
Conflict of Interest Statement
KY receives research support from Roche Molecular System, Gilead Sciences Pharmaceutical, Seattle Genetics, Dai Sanyo Pharmaceutical, Adaptive Biotechnology, Incyte Pharmaceutical, and HTG Molecular Diagnostics.
Funding
This work was supported by National Cancer Institute/National Institutes of Health grants R01CA138688, R01CA187415, and 1RC1CA146299 to KY, and was also partially supported by National Cancer Institute and National Institutes of Health grants P50CA136411 and P50CA142509. The University of Texas MD Anderson Cancer Center is supported in part by the National Institutes of Health through Cancer Center Support Grant P30CA016672. KY is also supported by The University of Texas MD Anderson Cancer Center Institutional Research and Development Fund, an Institutional Research Grant Award, and MD Anderson Cancer Center Lymphoma and Myeloma Specialized Programs on Research Excellence (SPORE) Research Development Program Award, Hagemeister Lymphoma Foundation Award, Gundersen Lutheran Medical Foundation Award, and the University Cancer Foundation via the Sister institution network fund at The University of Texas MD Anderson Cancer Center.
References
1. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95.
2. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. doi:10.1084/jem.192.7.1027
3. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med (1999) 5(12):1365–9. doi:10.1038/70932
4. Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res (2014) 2(9):846–56. doi:10.1158/2326-6066.CIR-14-0040
5. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med (2015) 373(2):123–35. doi:10.1056/NEJMoa1504627
6. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med (2015) 372(4):320–30. doi:10.1056/NEJMoa1412082
7. Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol (2011) 186(7):4200–12. doi:10.4049/jimmunol.1001783
8. Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol (2003) 33(10):2706–16. doi:10.1002/eji.200324228
9. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol (1996) 8(5):765–72. doi:10.1093/intimm/8.5.765
10. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol (2005) 17(2):133–44. doi:10.1093/intimm/dxh194
11. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol (2002) 169(10):5538–45. doi:10.4049/jimmunol.169.10.5538
12. Iwai Y, Okazaki T, Nishimura H, Kawasaki A, Yagita H, Honjo T. Microanatomical localization of PD-1 in human tonsils. Immunol Lett (2002) 83(3):215–20. doi:10.1016/S0165-2478(02)00088-3
13. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114(8):1537–44. doi:10.1182/blood-2008-12-195792
14. Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc Natl Acad Sci U S A (2017) 114(13):E2776–85. doi:10.1073/pnas.1620498114
15. Fernandez-Poma SM, Salas-Benito D, Lozano T, Casares N, Riezu-Boj JI, Mancheno U, et al. Expansion of tumor-infiltrating CD8+ T cells expressing PD-1 improves the efficacy of adoptive T-cell therapy. Cancer Res (2017) 77(13):3672–84. doi:10.1158/0008-5472.CAN-17-0236
16. Tewalt EF, Cohen JN, Rouhani SJ, Guidi CJ, Qiao H, Fahl SP, et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood (2012) 120(24):4772–82. doi:10.1182/blood-2012-04-427013
17. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity (2016) 45(2):389–401. doi:10.1016/j.immuni.2016.07.011
18. Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol (2009) 182(11):6659–69. doi:10.4049/jimmunol.0804211
19. Zhou Q, Xiao H, Liu Y, Peng Y, Hong Y, Yagita H, et al. Blockade of programmed death-1 pathway rescues the effector function of tumor-infiltrating T cells and enhances the antitumor efficacy of lentivector immunization. J Immunol (2010) 185(9):5082–92. doi:10.4049/jimmunol.1001821
20. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173(2):945–54. doi:10.4049/jimmunol.173.2.945
21. Bennett F, Luxenberg D, Ling V, Wang IM, Marquette K, Lowe D, et al. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J Immunol (2003) 170(2):711–8. doi:10.4049/jimmunol.170.2.711
22. Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol (2002) 32(3):634–43. doi:10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9
23. Isogawa M, Furuichi Y, Chisari FV. Oscillating CD8(+) T cell effector functions after antigen recognition in the liver. Immunity (2005) 23(1):53–63. doi:10.1016/j.immuni.2005.05.005
24. Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, et al. A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res (2015) 75(18):3800–11. doi:10.1158/0008-5472.CAN-15-1082
25. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439(7077):682–7. doi:10.1038/nature04444
26. Zelinskyy G, Myers L, Dietze KK, Gibbert K, Roggendorf M, Liu J, et al. Virus-specific CD8+ T cells upregulate programmed death-1 expression during acute friend retrovirus infection but are highly cytotoxic and control virus replication. J Immunol (2011) 187(7):3730–7. doi:10.4049/jimmunol.1101612
27. Reiley WW, Shafiani S, Wittmer ST, Tucker-Heard G, Moon JJ, Jenkins MK, et al. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A (2010) 107(45):19408–13. doi:10.1073/pnas.1006298107
28. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32(3):377–91.e9. doi:10.1016/j.ccell.2017.08.004
29. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542(7639):110–4. doi:10.1038/nature20810
30. Yang ZZ, Grote DM, Ziesmer SC, Xiu B, Novak AJ, Ansell SM. PD-1 expression defines two distinct T-cell sub-populations in follicular lymphoma that differentially impact patient survival. Blood Cancer J (2015) 5:e281. doi:10.1038/bcj.2015.1
31. Carreras J, Lopez-Guillermo A, Roncador G, Villamor N, Colomo L, Martinez A, et al. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J Clin Oncol (2009) 27(9):1470–6. doi:10.1200/JCO.2008.18.0513
32. Muenst S, Hoeller S, Willi N, Dirnhofera S, Tzankov A. Diagnostic and prognostic utility of PD-1 in B cell lymphomas. Dis Markers (2010) 29(1):47–53. doi:10.3233/DMA-2010-0725
33. Paulsen EE, Kilvaer TK, Khanehkenari MR, Al-Saad S, Hald SM, Andersen S, et al. Assessing PDL-1 and PD-1 in non-small cell lung cancer: a novel immunoscore approach. Clin Lung Cancer (2017) 18(2):220–33.e8. doi:10.1016/j.cllc.2016.09.009
34. Webb JR, Milne K, Nelson BH. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol Res (2015) 3(8):926–35. doi:10.1158/2326-6066.CIR-14-0239
35. Kollmann D, Schweiger T, Schwarz S, Ignatova D, Chang YT, Lewik G, et al. PD1-positive tumor-infiltrating lymphocytes are associated with poor clinical outcome after pulmonary metastasectomy for colorectal cancer. Oncoimmunology (2017) 6(9):e1331194. doi:10.1080/2162402X.2017.1331194
36. Eto S, Yoshikawa K, Nishi M, Higashijima J, Tokunaga T, Nakao T, et al. Programmed cell death protein 1 expression is an independent prognostic factor in gastric cancer after curative resection. Gastric Cancer (2016) 19(2):466–71. doi:10.1007/s10120-015-0519-7
37. Simon S, Vignard V, Florenceau L, Dreno B, Khammari A, Lang F, et al. PD-1 expression conditions T cell avidity within an antigen-specific repertoire. Oncoimmunology (2016) 5(1):e1104448. doi:10.1080/2162402X.2015.1104448
38. Inozume T, Hanada K, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother (2010) 33(9):956–64. doi:10.1097/CJI.0b013e3181fad2b0
39. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest (2014) 124(5):2246–59. doi:10.1172/JCI73639
40. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med (2016) 22(4):433–8. doi:10.1038/nm.4051
41. Andorsky DJ, Yamada RE, Said J, Pinkus GS, Betting DJ, Timmerman JM. Programmed death ligand 1 is expressed by non-hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin Cancer Res (2011) 17(13):4232–44. doi:10.1158/1078-0432.ccr-10-2660
42. Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol (2003) 170(3):1257–66. doi:10.4049/jimmunol.170.3.1257
43. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515(7528):568–71. doi:10.1038/nature13954
44. Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett (2006) 580(3):755–62. doi:10.1016/j.febslet.2005.12.093
45. Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood (2007) 110(1):296–304. doi:10.1182/blood-2006-10-051482
46. Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res (2014) 20(10):2674–83. doi:10.1158/1078-0432.CCR-13-3007
47. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep (2017) 19(6):1189–201. doi:10.1016/j.celrep.2017.04.031
48. Yamamoto R, Nishikori M, Tashima M, Sakai T, Ichinohe T, Takaori-Kondo A, et al. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer Sci (2009) 100(11):2093–100. doi:10.1111/j.1349-7006.2009.01302.x
49. Butte MJ, Pena-Cruz V, Kim MJ, Freeman GJ, Sharpe AH. Interaction of human PD-L1 and B7-1. Mol Immunol (2008) 45(13):3567–72. doi:10.1016/j.molimm.2008.05.014
50. Saudemont A, Jouy N, Hetuin D, Quesnel B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood (2005) 105(6):2428–35. doi:10.1182/blood-2004-09-3458
51. Matta BM, Raimondi G, Rosborough BR, Sumpter TL, Thomson AW. IL-27 production and STAT3-dependent upregulation of B7-H1 mediate immune regulatory functions of liver plasmacytoid dendritic cells. J Immunol (2012) 188(11):5227–37. doi:10.4049/jimmunol.1103382
52. Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C, et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol (2011) 41(2):413–24. doi:10.1002/eji.201040979
53. Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res (2013) 19(3):598–609. doi:10.1158/1078-0432.CCR-12-2731
54. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1 alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi:10.1084/jem.20131916
55. Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep (2017) 20(4):854–67. doi:10.1016/j.celrep.2017.07.007
56. Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep (2017) 18(9):2162–74. doi:10.1016/j.celrep.2017.02.011
57. Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res (2015) 3(12):1375–85. doi:10.1158/2326-6066.CIR-15-0077-T
58. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2007) 13(1):84–8. doi:10.1038/nm1517
59. Lastwika KJ, Wilson W III, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res (2016) 76(2):227–38. doi:10.1158/0008-5472.CAN-14-3362
60. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov (2016) 6(2):202–16. doi:10.1158/2159-8290.CD-15-0283
61. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell (2016) 30(6):925–39. doi:10.1016/j.ccell.2016.10.010
62. Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature (2017) 549(7670):106–10. doi:10.1038/nature23669
63. Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature (2017) 549(7670):101–5. doi:10.1038/nature23643
64. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun (2016) 7:12632. doi:10.1038/ncomms12632
65. Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B, et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene (2017) 36(42):5829–39. doi:10.1038/onc.2017.188
66. Watanabe R, Shirai T, Namkoong H, Zhang H, Berry GJ, Wallis BB, et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J Clin Invest (2017) 127(7):2725–38. doi:10.1172/JCI92167
67. Hori J, Wang M, Miyashita M, Tanemoto K, Takahashi H, Takemori T, et al. B7-H1-induced apoptosis as a mechanism of immune privilege of corneal allografts. J Immunol (2006) 177(9):5928–35. doi:10.4049/jimmunol.177.9.5928
68. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol (2007) 8(3):239–45. doi:10.1038/ni1443
69. Cheng X, Dai H, Wan N, Moore Y, Vankayalapati R, Dai Z. Interaction of programmed death-1 and programmed death-1 ligand-1 contributes to testicular immune privilege. Transplantation (2009) 87(12):1778–86. doi:10.1097/TP.0b013e3181a75633
70. Gabrielson A, Wu Y, Wang H, Jiang J, Kallakury B, Gatalica Z, et al. Intratumoral CD3 and CD8 T-cell densities associated with relapse-free survival in HCC. Cancer Immunol Res (2016) 4(5):419–30. doi:10.1158/2326-6066.CIR-15-0110
71. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A (2007) 104(9):3360–5. doi:10.1073/pnas.0611533104
72. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med (2013) 5(200):200ra116. doi:10.1126/scitranslmed.3006504
73. Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun (2017) 8:14572. doi:10.1038/ncomms14572
74. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med (2017) 214(4):895–904. doi:10.1084/jem.20160801
75. Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res (2017) 5(2):106–17. doi:10.1158/2326-6066.CIR-16-0391
76. Kleinovink JW, Marijt KA, Schoonderwoerd MJA, van Hall T, Ossendorp F, Fransen MF. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology (2017) 6(4):e1294299. doi:10.1080/2162402X.2017.1294299
77. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res (2005) 65(3):1089–96.
78. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol (2009) 10(11):1185–92. doi:10.1038/ni.1790
79. Seo SK, Seo HM, Jeong HY, Choi IW, Park YM, Yagita H, et al. Co-inhibitory role of T-cell-associated B7-H1 and B7-DC in the T-cell immune response. Immunol Lett (2006) 102(2):222–8. doi:10.1016/j.imlet.2005.09.007
80. Yang ZZ, Novak AJ, Stenson MJ, Witzig TE, Ansell SM. Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood (2006) 107(9):3639–46. doi:10.1182/blood-2005-08-3376
81. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A (2004) 101(29):10691–6. doi:10.1073/pnas.0307252101
82. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med (2006) 203(4):883–95. doi:10.1084/jem.20051776
83. Flies DB, Chen L. The new B7s: playing a pivotal role in tumor immunity. J Immunother (2007) 30(3):251–60. doi:10.1097/CJI.0b013e31802e085a
84. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355(6332):1428–33. doi:10.1126/science.aaf1292
85. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A (2002) 99(19):12293–7. doi:10.1073/pnas.192461099
86. Shim KG, Zaidi S, Thompson J, Kottke T, Evgin L, Rajani KR, et al. Inhibitory receptors induced by VSV viroimmunotherapy are not necessarily targets for improving treatment efficacy. Mol Ther (2017) 25(4):962–75. doi:10.1016/j.ymthe.2017.01.023
87. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547(7662):217–21. doi:10.1038/nature22991
88. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547(7662):222–6. doi:10.1038/nature23003
89. Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8(+) T cells induced by melanoma vaccines. Cancer Res (2014) 74(4):1045–55. doi:10.1158/0008-5472.CAN-13-2908
90. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (2016) 354(6316):1160–5. doi:10.1126/science.aaf2807
91. Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, et al. Dynamic changes in chromatin accessibility occur in CD8+ T cells responding to viral infection. Immunity (2016) 45(6):1327–40. doi:10.1016/j.immuni.2016.10.028
92. Pauken KE, Nelson CE, Martinov T, Spanier JA, Heffernan JR, Sahli NL, et al. Cutting edge: identification of autoreactive CD4+ and CD8+ T cell subsets resistant to PD-1 pathway blockade. J Immunol (2015) 194(8):3551–5. doi:10.4049/jimmunol.1402262
93. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2(3):261–8. doi:10.1038/85330
94. Deng R, Cassady K, Li X, Yao S, Zhang M, Racine J, et al. B7H1/CD80 interaction augments PD-1-dependent T cell apoptosis and ameliorates graft-versus-host disease. J Immunol (2015) 194(2):560–74. doi:10.4049/jimmunol.1402157
95. Tkachev V, Goodell S, Opipari AW, Hao LY, Franchi L, Glick GD, et al. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J Immunol (2015) 194(12):5789–800. doi:10.4049/jimmunol.1402180
96. Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol (2005) 6(3):280–6. doi:10.1038/ni1165
97. Goldberg MV, Maris CH, Hipkiss EL, Flies AS, Zhen L, Tuder RM, et al. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood (2007) 110(1):186–92. doi:10.1182/blood-2006-12-062422
98. Keir ME, Freeman GJ, Sharpe AH. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J Immunol (2007) 179(8):5064–70. doi:10.4049/jimmunol.179.8.5064
99. Martin-Orozco N, Wang YH, Yagita H, Dong C. Cutting edge: programmed death (PD) ligand-1/PD-1 interaction is required for CD8+ T cell tolerance to tissue antigens. J Immunol (2006) 177(12):8291–5. doi:10.4049/jimmunol.177.12.8291
100. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med (2012) 209(6):1201–17. doi:10.1084/jem.20112741
101. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal (2012) 5(230):ra46. doi:10.1126/scisignal.2002796
102. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett (2004) 574(1–3):37–41. doi:10.1016/j.febslet.2004.07.083
103. Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med (2011) 3(10):581–92. doi:10.1002/emmm.201100165
104. Keir ME, Latchman YE, Freeman GJ, Sharpe AH. Programmed death-1 (PD-1):PD-ligand 1 interactions inhibit TCR-mediated positive selection of thymocytes. J Immunol (2005) 175(11):7372–9. doi:10.4049/jimmunol.175.11.7372
105. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A (2001) 98(24):13866–71. doi:10.1073/pnas.231486598
106. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
107. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol (2013) 33(16):3091–8. doi:10.1128/MCB.00319-13
108. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
109. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi:10.4049/jimmunol.1003613
110. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi:10.1016/j.cmet.2014.05.004
111. Valenzuela J, Schmidt C, Mescher M. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J Immunol (2002) 169(12):6842–9. doi:10.4049/jimmunol.169.12.6842
112. Schurich A, Pallett LJ, Jajbhay D, Wijngaarden J, Otano I, Gill US, et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep (2016) 16(5):1243–52. doi:10.1016/j.celrep.2016.06.078
113. Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, et al. Suppression of Glut1 and glucose metabolism by decreased Akt/mTORC1 signaling drives T cell impairment in B cell leukemia. J Immunol (2016) 197(6):2532–40. doi:10.4049/jimmunol.1502464
114. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi:10.1016/j.cell.2015.08.016
115. Clark CA, Gupta HB, Curiel TJ. Tumor cell-intrinsic CD274/PD-L1: a novel metabolic balancing act with clinical potential. Autophagy (2017) 13(5):987–8. doi:10.1080/15548627.2017.1280223
116. Saha A, Aoyama K, Taylor PA, Koehn BH, Veenstra RG, Panoskaltsis-Mortari A, et al. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood (2013) 122(17):3062–73. doi:10.1182/blood-2013-05-500801
117. Saha A, O’Connor RS, Thangavelu G, Lovitch SB, Dandamudi DB, Wilson CB, et al. Programmed death ligand-1 expression on donor T cells drives graft-versus-host disease lethality. J Clin Invest (2016) 126(7):2642–60. doi:10.1172/JCI85796
118. Smeltzer JP, Jones JM, Ziesmer SC, Grote DM, Xiu B, Ristow KM, et al. Pattern of CD14+ follicular dendritic cells and PD1+ T cells independently predicts time to transformation in follicular lymphoma. Clin Cancer Res (2014) 20(11):2862–72. doi:10.1158/1078-0432.CCR-13-2367
119. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alpha PD-L1 blockade. Proc Natl Acad Sci U S A (2008) 105(39):15016–21. doi:10.1073/pnas.0801497105
120. Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, et al. Structure and interactions of the human programmed cell death 1 receptor. J Biol Chem (2013) 288(17):11771–85. doi:10.1074/jbc.M112.448126
121. Youngnak P, Kozono Y, Kozono H, Iwai H, Otsuki N, Jin H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun (2003) 307(3):672–7. doi:10.1016/S0006-291X(03)01257-9
122. Ishida M, Iwai Y, Tanaka Y, Okazaki T, Freeman GJ, Minato N, et al. Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol Lett (2002) 84(1):57–62. doi:10.1016/S0165-2478(02)00142-6
123. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol (2014) 15(7):631–7. doi:10.1038/ni.2914
124. Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur J Immunol (2007) 37(9):2405–10. doi:10.1002/eji.200737461
125. Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol (2010) 11(6):535–42. doi:10.1038/ni.1877
126. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A (2003) 100(9):5336–41. doi:10.1073/pnas.0931259100
127. Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H, et al. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood (2007) 110(1):180–5. doi:10.1182/blood-2006-11-060087
128. Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med (2003) 198(1):63–9. doi:10.1084/jem.20022125
129. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27(1):111–22. doi:10.1016/j.immuni.2007.05.016
130. Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med (2014) 211(5):943–59. doi:10.1084/jem.20130790
131. Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood (2010) 116(8):1291–8. doi:10.1182/blood-2010-01-265975
132. Sandner SE, Clarkson MR, Salama AD, Sanchez-Fueyo A, Domenig C, Habicht A, et al. Role of the programmed death-1 pathway in regulation of alloimmune responses in vivo. J Immunol (2005) 174(6):3408–15. doi:10.4049/jimmunol.174.6.3408
133. Pauken KE, Jenkins MK, Azuma M, Fife BT. PD-1, but not PD-L1, expressed by islet-reactive CD4+ T cells suppresses infiltration of the pancreas during type 1 diabetes. Diabetes (2013) 62(8):2859–69. doi:10.2337/db12-1475
134. Pulko V, Harris KJ, Liu X, Gibbons RM, Harrington SM, Krco CJ, et al. B7-h1 expressed by activated CD8 T cells is essential for their survival. J Immunol (2011) 187(11):5606–14. doi:10.4049/jimmunol.1003976
135. Subudhi SK, Zhou P, Yerian LM, Chin RK, Lo JC, Anders RA, et al. Local expression of B7-H1 promotes organ-specific autoimmunity and transplant rejection. J Clin Invest (2004) 113(5):694–700. doi:10.1172/JCI19210
136. Talay O, Shen CH, Chen L, Chen J. B7-H1 (PD-L1) on T cells is required for T-cell-mediated conditioning of dendritic cell maturation. Proc Natl Acad Sci U S A (2009) 106(8):2741–6. doi:10.1073/pnas.0813367106
137. Tamura H, Dong H, Zhu G, Sica GL, Flies DB, Tamada K, et al. B7-H1 costimulation preferentially enhances CD28-independent T-helper cell function. Blood (2001) 97(6):1809–16. doi:10.1182/blood.V97.6.1809
138. Munir Ahmad S, Martinenaite E, Hansen M, Junker N, Borch TH, Met O, et al. PD-L1 peptide co-stimulation increases immunogenicity of a dendritic cell-based cancer vaccine. Oncoimmunology (2016) 5(8):e1202391. doi:10.1080/2162402X.2016.1202391
139. Ni X, Song Q, Cassady K, Deng R, Jin H, Zhang M, et al. PD-L1 interacts with CD80 to regulate graft-versus-leukemia activity of donor CD8+ T cells. J Clin Invest (2017) 127(5):1960–77. doi:10.1172/JCI91138
140. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi:10.1038/nm730
141. Kanai T, Totsuka T, Uraushihara K, Makita S, Nakamura T, Koganei K, et al. Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J Immunol (2003) 171(8):4156–63. doi:10.4049/jimmunol.171.8.4156
142. Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med (2003) 197(9):1083–91. doi:10.1084/jem.20021752
143. Shin T, Kennedy G, Gorski K, Tsuchiya H, Koseki H, Azuma M, et al. Cooperative B7-1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. J Exp Med (2003) 198(1):31–8. doi:10.1084/jem.20030242
144. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med (2001) 193(7):839–46. doi:10.1084/jem.193.7.839
145. Liu X, Gao JX, Wen J, Yin L, Li O, Zuo T, et al. B7DC/PDL2 promotes tumor immunity by a PD-1-independent mechanism. J Exp Med (2003) 197(12):1721–30. doi:10.1084/jem.20022089
146. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi:10.1016/1074-7613(95)90125-6
147. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in CTLA-4. Science (1995) 270(5238):985–8. doi:10.1126/science.270.5238.985
148. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi:10.1016/S1074-7613(00)80089-8
149. Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol (1998) 10(10):1563–72. doi:10.1093/intimm/10.10.1563
150. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291(5502):319–22. doi:10.1126/science.291.5502.319
151. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A (2005) 102(33):11823–8. doi:10.1073/pnas.0505497102
152. Nishimura H, Honjo T, Minato N. Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J Exp Med (2000) 191(5):891–8. doi:10.1084/jem.191.5.891
153. Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity (2004) 20(3):327–36. doi:10.1016/S1074-7613(04)00050-0
154. Lucas JA, Menke J, Rabacal WA, Schoen FJ, Sharpe AH, Kelley VR. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J Immunol (2008) 181(4):2513–21. doi:10.4049/jimmunol.181.4.2513
155. Zhang Y, Chung Y, Bishop C, Daugherty B, Chute H, Holst P, et al. Regulation of T cell activation and tolerance by PDL2. Proc Natl Acad Sci U S A (2006) 103(31):11695–700. doi:10.1073/pnas.0601347103
156. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med (2012) 209(13):2485–99. doi:10.1084/jem.20121015
157. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med (2015) 212(7):1125–37. doi:10.1084/jem.20142237
158. Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol (1994) 68(12):8056–63.
159. Penaloza-MacMaster P, Provine NM, Blass E, Barouch DH. CD4 T cell depletion substantially augments the rescue potential of PD-L1 blockade for deeply exhausted CD8 T cells. J Immunol (2015) 195(3):1054–63. doi:10.4049/jimmunol.1403237
160. Penaloza-MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, West EE, et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med (2014) 211(9):1905–18. doi:10.1084/jem.20132577
161. Wong RM, Scotland RR, Lau RL, Wang C, Korman AJ, Kast WM, et al. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int Immunol (2007) 19(10):1223–34. doi:10.1093/intimm/dxm091
162. Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res (2016) 4(3):194–203. doi:10.1158/2326-6066.CIR-15-0210
163. Fuse S, Tsai CY, Molloy MJ, Allie SR, Zhang W, Yagita H, et al. Recall responses by helpless memory CD8+ T cells are restricted by the up-regulation of PD-1. J Immunol (2009) 182(7):4244–54. doi:10.4049/jimmunol.0802041
164. Ahrends T, Babala N, Xiao Y, Yagita H, van Eenennaam H, Borst J. CD27 agonism plus PD-1 blockade recapitulates CD4+ T-cell help in therapeutic anticancer vaccination. Cancer Res (2016) 76(10):2921–31. doi:10.1158/0008-5472.CAN-15-3130
165. Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol Res (2016) 4(10):845–57. doi:10.1158/2326-6066.CIR-16-0060
166. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116(13):2286–94. doi:10.1182/blood-2010-02-271874
167. Terme M, Ullrich E, Aymeric L, Meinhardt K, Desbois M, Delahaye N, et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res (2011) 71(16):5393–9. doi:10.1158/0008-5472.CAN-11-0993
168. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545(7655):495–9. doi:10.1038/nature22396
169. Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM, et al. PD-1 blunts the function of ovarian tumor-infiltrating dendritic cells by inactivating NF-kappaB. Cancer Res (2016) 76(2):239–50. doi:10.1158/0008-5472.CAN-15-0748
170. Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, et al. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol (2011) 186(12):6905–13. doi:10.4049/jimmunol.1100274
171. Blank C, Kuball J, Voelkl S, Wiendl H, Becker B, Walter B, et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer (2006) 119(2):317–27. doi:10.1002/ijc.21775
172. Honda T, Egen JG, Lammermann T, Kastenmuller W, Torabi-Parizi P, Germain RN. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity (2014) 40(2):235–47. doi:10.1016/j.immuni.2013.11.017
173. Cai G, Karni A, Oliveira EM, Weiner HL, Hafler DA, Freeman GJ. PD-1 ligands, negative regulators for activation of naive, memory, and recently activated human CD4+ T cells. Cell Immunol (2004) 230(2):89–98. doi:10.1016/j.cellimm.2004.09.004
174. Yamazaki T, Akiba H, Koyanagi A, Azuma M, Yagita H, Okumura K. Blockade of B7-H1 on macrophages suppresses CD4+ T cell proliferation by augmenting IFN-gamma-induced nitric oxide production. J Immunol (2005) 175(3):1586–92. doi:10.4049/jimmunol.175.3.1586
175. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature (2016) 537(7620):412–28. doi:10.1038/nature19317
176. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science (2012) 338(6111):1220–5. doi:10.1126/science.1229620
177. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T cell factor 1-expressing memory-like CD8(+) t cells sustain the immune response to chronic viral infections. Immunity (2016) 45(2):415–27. doi:10.1016/j.immuni.2016.07.021
178. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature (2017) 545(7655):452–6. doi:10.1038/nature22367
179. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9(5):562–7. doi:10.1038/nm863
180. Abdel-Magid AF. Inhibitors of the PD-1/PD-L1 pathway can mobilize the immune system: an innovative potential therapy for cancer and chronic infections. ACS Med Chem Lett (2015) 6(5):489–90. doi:10.1021/acsmedchemlett.5b00148
181. Chang HN, Liu BY, Qi YK, Zhou Y, Chen YP, Pan KM, et al. Blocking of the PD-1/PD-L1 interaction by a D-peptide antagonist for cancer immunotherapy. Angew Chem Int Ed Engl (2015) 54(40):11760–4. doi:10.1002/anie.201506225
182. Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. Anti-PD-L1 peptide improves survival in sepsis. J Surg Res (2017) 208:33–9. doi:10.1016/j.jss.2016.08.099
183. Weinmann H. Cancer immunotherapy: selected targets and small-molecule modulators. ChemMedChem (2016) 11(5):450–66. doi:10.1002/cmdc.201500566
184. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369(2):134–44. doi:10.1056/NEJMoa1305133
185. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
186. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
187. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet (2014) 384(9948):1109–17. doi:10.1016/S0140-6736(14)60958-2
188. Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, Ribas A, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol (2016) 34(34):4102–9. doi:10.1200/JCO.2016.67.2477
189. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol (2015) 16(8):908–18. doi:10.1016/S1470-2045(15)00083-2
190. Hamid O, Puzanov I, Dummer R, Schachter J, Daud A, Schadendorf D, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer (2017) 86:37–45. doi:10.1016/j.ejca.2017.07.022
191. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372(26):2521–32. doi:10.1056/NEJMoa1503093
192. Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2015) 16(4):375–84. doi:10.1016/S1470-2045(15)70076-8
193. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi:10.1056/NEJMoa1504030
194. Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol (2016) 17(11):1558–68. doi:10.1016/S1470-2045(16)30366-7
195. Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med (2017) 377(19):1824–35. doi:10.1056/NEJMoa1709030
196. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med (2015) 373(17):1627–39. doi:10.1056/NEJMoa1507643
197. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi:10.1056/NEJMoa1501824
198. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (2016) 387(10027):1540–50. doi:10.1016/S0140-6736(15)01281-7
199. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med (2016) 375(19):1823–33. doi:10.1056/NEJMoa1606774
200. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol (2016) 17(11):1497–508. doi:10.1016/S1470-2045(16)30498-3
201. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373(19):1803–13. doi:10.1056/NEJMoa1510665
202. Younes A, Santoro A, Shipp M, Zinzani PL, Timmerman JM, Ansell S, et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol (2016) 17(9):1283–94. doi:10.1016/S1470-2045(16)30167-X
203. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol (2017) 35(19):2125–32. doi:10.1200/JCO.2016.72.1316
204. Larkins E, Blumenthal GM, Yuan W, He K, Sridhara R, Subramaniam S, et al. FDA approval summary: pembrolizumab for the treatment of recurrent or metastatic head and neck squamous cell carcinoma with disease progression on or after platinum-containing chemotherapy. Oncologist (2017) 22(7):873–8. doi:10.1634/theoncologist.2016-0496
205. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med (2016) 375(19):1856–67. doi:10.1056/NEJMoa1602252
206. Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol (2017) 18(3):312–22. doi:10.1016/S1470-2045(17)30065-7
207. Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med (2017) 376(11):1015–26. doi:10.1056/NEJMoa1613683
208. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi:10.1056/NEJMoa1500596
209. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357(6349):409–13. doi:10.1126/science.aan6733
210. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol (2017) 18(9):1182–91. doi:10.1016/S1470-2045(17)30422-9
211. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389(10088):2492–502. doi:10.1016/S0140-6736(17)31046-2
212. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet (2016) 387(10031):1909–20. doi:10.1016/S0140-6736(16)00561-4
213. Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet (2017) 389(10064):67–76. doi:10.1016/S0140-6736(16)32455-2
214. Powles T, O’Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open-label study. JAMA Oncol (2017) 3(9):e172411. doi:10.1001/jamaoncol.2017.2411
215. Three drugs approved for urothelial carcinoma by FDA. Cancer Discov (2017) 7(7):659–60. doi:10.1158/2159-8290.CD-NB2017-071
216. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (2017) 389(10066):255–65. doi:10.1016/S0140-6736(16)32517-X
217. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet (2016) 387(10030):1837–46. doi:10.1016/S0140-6736(16)00587-0
218. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol (2016) 17(10):1374–85. doi:10.1016/S1470-2045(16)30364-3
219. Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol (2015) 16(3):257–65. doi:10.1016/S1470-2045(15)70054-9
220. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) 372(4):311–9. doi:10.1056/NEJMoa1411087
221. Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2017). doi:10.1016/S0140-6736(17)31827-5
222. Apolo AB, Infante JR, Balmanoukian A, Patel MR, Wang D, Kelly K, et al. Avelumab, an anti-programmed death-ligand 1 antibody, in patients with refractory metastatic urothelial carcinoma: results from a multicenter, phase Ib study. J Clin Oncol (2017) 35(19):2117–24. doi:10.1200/JCO.2016.71.6795
223. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med (2017). doi:10.1056/NEJMoa1709937
224. Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood (2017) 129(26):3419–27. doi:10.1182/blood-2017-02-765685
225. Necchi A, Joseph RW, Loriot Y, Hoffman-Censits J, Perez-Gracia JL, Petrylak DP, et al. Atezolizumab in platinum-treated locally advanced or metastatic urothelial carcinoma: post-progression outcomes from the phase II IMvigor210 study. Ann Oncol (2017). doi:10.1093/annonc/mdx518
226. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med (2017) 376(25):2415–26. doi:10.1056/NEJMoa1613493
227. Larkin J, Minor D, D’Angelo S, Neyns B, Smylie M, Miller WH Jr, et al. Overall survival in patients with advanced melanoma who received nivolumab versus investigator’s choice chemotherapy in CheckMate 037: a randomized, controlled, open-label phase III trial. J Clin Oncol (2017). doi:10.1200/JCO.2016.71.8023
228. Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, et al. Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol (2016) 34(25):2980–7. doi:10.1200/JCO.2016.66.9929
229. Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol (2016) 17(7):883–95. doi:10.1016/S1470-2045(16)30098-5
230. Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, phase II study. J Clin Oncol (2017) 35(14):1542–9. doi:10.1200/JCO.2016.70.1524
231. Peters S, Antonia S, Goldberg SB, Heymach JV, Kim ES, Nakagawa K, et al. 191TiP: MYSTIC: a global, phase 3 study of durvalumab (MEDI4736) plus tremelimumab combination therapy or durvalumab monotherapy versus platinum-based chemotherapy (CT) in the first-line treatment of patients (pts) with advanced stage IV NSCLC. J Thorac Oncol (2016) 11(4 Suppl):S139–40. doi:10.1016/S1556-0864(16)30300-8
232. Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res (2017) 23(8):1920–8. doi:10.1158/1078-0432.CCR-16-1741
233. Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res (2017) 23(15):4242–50. doi:10.1158/1078-0432.CCR-16-3133
234. Saada-Bouzid E, Defaucheux C, Karabajakian A, Coloma VP, Servois V, Paoletti X, et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol (2017) 28(7):1605–11. doi:10.1093/annonc/mdx178
235. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
236. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515(7528):563–7. doi:10.1038/nature14011
237. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov (2016) 6(8):827–37. doi:10.1158/2159-8290.CD-15-1545
238. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood (2010) 116(17):3268–77. doi:10.1182/blood-2010-05-282780
239. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375(9):819–29. doi:10.1056/NEJMoa1604958
240. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov (2017) 7(2):188–201. doi:10.1158/2159-8290.CD-16-1223
241. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature (2017) 548(7669):537–42. doi:10.1038/nature23477
242. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med (2017) 9(379). doi:10.1126/scitranslmed.aah3560
243. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell (2017) 171(4):934–49. doi:10.1016/j.cell.2017.09.028
244. Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res (2016) 22(4):813–20. doi:10.1158/1078-0432.CCR-15-1678
245. First tissue-agnostic drug approval issued. Cancer Discov (2017) 7(7):656. doi:10.1158/2159-8290.CD-NB2017-078
246. Kloor M, Michel S, von Knebel M. Doeberitz: immune evasion of microsatellite unstable colorectal cancers. Int J Cancer (2010) 127(5):1001–10. doi:10.1002/ijc.25283
247. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov (2015) 5(1):43–51. doi:10.1158/2159-8290.CD-14-0863
248. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9(1):34. doi:10.1186/s13073-017-0424-2
249. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi:10.1126/science.aaa1348
250. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (2016) 351(6280):1463–9. doi:10.1126/science.aaf1490
251. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol (2017) 19(8):1047–57. doi:10.1093/neuonc/nox026
252. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther (2017) 16(11):2598–608. doi:10.1158/1535-7163.MCT-17-0386
253. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol (2017) 18(8):1009–21. doi:10.1016/S1470-2045(17)30516-8
254. Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep (2016) 15(4):757–65. doi:10.1016/j.celrep.2016.03.075
255. Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A (2016) 113(48):E7759–68. doi:10.1073/pnas.1609376113
256. Roszik J, Haydu LE, Hess KR, Oba J, Joon AY, Siroy AE, et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med (2016) 14(1):168. doi:10.1186/s12916-016-0705-4
257. Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science (2017) 356(6334):200–5. doi:10.1126/science.aak9510
258. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov (2017) 7(3):264–76. doi:10.1158/2159-8290.CD-16-0828
259. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165(1):35–44. doi:10.1016/j.cell.2016.02.065
260. Mehnert JM, Panda A, Zhong H, Hirshfield K, Damare S, Lane K, et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Invest (2016) 126(6):2334–40. doi:10.1172/JCI84940
261. Dronca RS, Mansfield AS, Park SS, Dong H. BCL-2-interacting mediator of cell death (Bim) is a novel biomarker for response to anti-PD-1 therapy in patients with advanced melanoma. Immunotherapy (2016) 8(12):1351–3. doi:10.2217/imt-2016-0100
262. Dronca RS, Liu X, Harrington SM, Chen L, Cao S, Kottschade LA, et al. T cell Bim levels reflect responses to anti-PD-1 cancer therapy. JCI Insight (2016) 1(6):e86014. doi:10.1172/jci.insight.86014
263. Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest (2016) 126(9):3447–52. doi:10.1172/JCI87324
264. Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, et al. Immune-related gene expression profiling after PD-1 blockade in non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res (2017) 77(13):3540–50. doi:10.1158/0008-5472.CAN-16-3556
265. Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res (2016) 22(22):5487–96. doi:10.1158/1078-0432.CCR-16-0127
266. Friedman CF, Postow MA. Emerging tissue and blood-based biomarkers that may predict response to immune checkpoint inhibition. Curr Oncol Rep (2016) 18(4):21. doi:10.1007/s11912-016-0509-x
267. Nosrati A, Tsai KK, Goldinger SM, Tumeh P, Grimes B, Loo K, et al. Evaluation of clinicopathological factors in PD-1 response: derivation and validation of a prediction scale for response to PD-1 monotherapy. Br J Cancer (2017) 116(9):1141–7. doi:10.1038/bjc.2017.70
268. Nishino M, Dahlberg SE, Adeni AE, Lydon CA, Hatabu H, Janne PA, et al. Tumor response dynamics of advanced non-small cell lung cancer patients treated with PD-1 inhibitors: imaging markers for treatment outcome. Clin Cancer Res (2017) 23(19):5737–44. doi:10.1158/1078-0432.CCR-17-1434
269. Nishino M, Giobbie-Hurder A, Manos MP, Bailey N, Buchbinder EI, Ott PA, et al. Immune-related tumor response dynamics in melanoma patients treated with pembrolizumab: identifying markers for clinical outcome and treatment decisions. Clin Cancer Res (2017) 23(16):4671–9. doi:10.1158/1078-0432.CCR-17-0114
270. Sanmamed MF, Perez-Gracia JL, Schalper KA, Fusco JP, Gonzalez A, Rodriguez-Ruiz ME, et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann Oncol (2017) 28(8):1988–95. doi:10.1093/annonc/mdx190
271. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (2017). doi:10.1126/science.aan4236
272. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (2017). doi:10.1126/science.aan3706
273. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (2015) 350(6264):1084–9. doi:10.1126/science.aac4255
274. Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science (2012) 336(6080):485–9. doi:10.1126/science.1217718
275. Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A (2017) 114(19):4993–8. doi:10.1073/pnas.1705327114
276. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature (2017) 545(7652):60–5. doi:10.1038/nature22079
277. Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med (2016) 22(12):1402–10. doi:10.1038/nm.4200
278. Guinan EC, Gribben JG, Boussiotis VA, Freeman GJ, Nadler LM. Pivotal role of the B7:CD28 pathway in transplantation tolerance and tumor immunity. Blood (1994) 84(10):3261–82.
279. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun (2017) 8(1):1136. doi:10.1038/s41467-017-01062-w
280. Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun (2016) 7:10582. doi:10.1038/ncomms10582
281. Roemer MG, Advani RH, Redd RA, Pinkus GS, Natkunam Y, Ligon AH, et al. Classical Hodgkin lymphoma with reduced beta2M/MHC class I expression is associated with inferior outcome independent of 9p24.1 status. Cancer Immunol Res (2016) 4(11):910–6. doi:10.1158/2326-6066.CIR-16-0201
282. Nijland M, Veenstra RN, Visser L, Xu C, Kushekhar K, van Imhoff GW, et al. HLA dependent immune escape mechanisms in B-cell lymphomas: implications for immune checkpoint inhibitor therapy? Oncoimmunology (2017) 6(4):e1295202. doi:10.1080/2162402X.2017.1295202
283. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol (2015) 194(3):950–9. doi:10.4049/jimmunol.1401686
284. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13(2):101–17. doi:10.1038/nri3369
285. Haspot F, Li HW, Lucas CL, Fehr T, Beyaz S, Sykes M. Allospecific rejection of MHC class I-deficient bone marrow by CD8 T cells. Am J Transplant (2014) 14(1):49–58. doi:10.1111/ajt.12525
286. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126(8):3130–44. doi:10.1172/JCI83092
287. Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T, et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia (2016) 30(2):484–91. doi:10.1038/leu.2015.214
288. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res (2013) 73(23):6900–12. doi:10.1158/0008-5472.CAN-13-1550
289. Canton DA, Shirley S, Wright J, Connolly R, Burkart C, Mukhopadhyay A, et al. Melanoma treatment with intratumoral electroporation of tavokinogene telseplasmid (pIL-12, tavokinogene telseplasmid). Immunotherapy (2017). doi:10.2217/imt-2017-0096
290. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi:10.1038/nature13988
291. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science (2017) 355(6332):1423–7. doi:10.1126/science.aaf0683
292. Ito T, Ueno T, Clarkson MR, Yuan X, Jurewicz MM, Yagita H, et al. Analysis of the role of negative T cell costimulatory pathways in CD4 and CD8 T cell-mediated alloimmune responses in vivo. J Immunol (2005) 174(11):6648–56. doi:10.4049/jimmunol.174.11.6648
293. Weng NP, Akbar AN, Goronzy J. CD28(-) T cells: their role in the age-associated decline of immune function. Trends Immunol (2009) 30(7):306–12. doi:10.1016/j.it.2009.03.013
294. Nozawa Y, Wakasa H, Abe M. Costimulatory molecules (CD80 and CD86) on Reed-Sternberg cells are associated with the proliferation of background T cells in Hodgkin’s disease. Pathol Int (1998) 48(1):10–4. doi:10.1111/j.1440-1827.1998.tb03821.x
295. Munro JM, Freedman AS, Aster JC, Gribben JG, Lee NC, Rhynhart KK, et al. In vivo expression of the B7 costimulatory molecule by subsets of antigen-presenting cells and the malignant cells of Hodgkin’s disease. Blood (1994) 83(3):793–8.
296. Delabie J, Ceuppens JL, Vandenberghe P, de Boer M, Coorevits L, De Wolf-Peeters C. The B7/BB1 antigen is expressed by Reed-Sternberg cells of Hodgkin’s disease and contributes to the stimulating capacity of Hodgkin’s disease-derived cell lines. Blood (1993) 82(9):2845–52.
297. Dorfman DM, Schultze JL, Shahsafaei A, Michalak S, Gribben JG, Freeman GJ, et al. In vivo expression of B7-1 and B7-2 by follicular lymphoma cells can prevent induction of T-cell anergy but is insufficient to induce significant T-cell proliferation. Blood (1997) 90(11):4297–306.
298. Van Gool SW, Delabie J, Vandenberghe P, Coorevits L, De Wolf-Peeters C, Ceuppens JL. Expression of B7-2 (CD86) molecules by Reed-Sternberg cells of Hodgkin’s disease. Leukemia (1997) 11(6):846–51. doi:10.1038/sj.leu.2400683
299. Dakappagari N, Ho SN, Gascoyne RD, Ranuio J, Weng AP, Tangri S. CD80 (B7.1) is expressed on both malignant B cells and nonmalignant stromal cells in non-Hodgkin lymphoma. Cytometry B Clin Cytom (2012) 82(2):112–9. doi:10.1002/cyto.b.20631
300. Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest (2008) 118(7):2427–37. doi:10.1172/JCI35017
301. Bhadra R, Gigley JP, Khan IA. Cutting edge: CD40-CD40 ligand pathway plays a critical CD8-intrinsic and -extrinsic role during rescue of exhausted CD8 T cells. J Immunol (2011) 187(9):4421–5. doi:10.4049/jimmunol.1102319
302. Xu A, Wang R, Freywald A, Stewart K, Tikoo S, Xu J, et al. CD40 agonist converting CTL exhaustion via the activation of the mTORC1 pathway enhances PD-1 antagonist action in rescuing exhausted CTLs in chronic infection. Biochem Biophys Res Commun (2017) 484(3):662–7. doi:10.1016/j.bbrc.2017.01.172
303. Chen S, Lee LF, Fisher TS, Jessen B, Elliott M, Evering W, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res (2015) 3(2):149–60. doi:10.1158/2326-6066.CIR-14-0118
304. Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, Gajewski TF. The EGR2 targets LAG-3 and 4-1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J Exp Med (2017) 214(2):381–400. doi:10.1084/jem.20160485
305. Boes M, Meyer-Wentrup F. TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett (2015) 361(1):49–56. doi:10.1016/j.canlet.2015.02.027
306. Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight (2017) 2(18). doi:10.1172/jci.insight.93397
307. Wang S, Campos J, Gallotta M, Gong M, Crain C, Naik E, et al. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci U S A (2016) 113(46):E7240–9. doi:10.1073/pnas.1608555113
308. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep (2017) 19(9):1874–87. doi:10.1016/j.celrep.2017.05.015
309. Leyland R, Watkins A, Mulgrew KA, Holoweckyj N, Bamber L, Tigue NJ, et al. A novel murine GITR ligand fusion protein induces antitumor activity as a monotherapy that is further enhanced in combination with an OX40 agonist. Clin Cancer Res (2017) 23(13):3416–27. doi:10.1158/1078-0432.CCR-16-2000
310. Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res (2017) 5(8):676–84. doi:10.1158/2326-6066.CIR-17-0049
311. Buchan S, Manzo T, Flutter B, Rogel A, Edwards N, Zhang L, et al. OX40- and CD27-mediated costimulation synergizes with anti-PD-L1 blockade by forcing exhausted CD8+ T cells to exit quiescence. J Immunol (2015) 194(1):125–33. doi:10.4049/jimmunol.1401644
312. Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. Clin Cancer Res (2017) 23(20):6165–77. doi:10.1158/1078-0432.CCR-16-2677
313. Lu P, Youngblood BA, Austin JW, Mohammed AU, Butler R, Ahmed R, et al. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med (2014) 211(3):515–27. doi:10.1084/jem.20130208
314. Zhu L, Kong Y, Zhang J, Claxton DF, Ehmann WC, Rybka WB, et al. Blimp-1 impairs T cell function via upregulation of TIGIT and PD-1 in patients with acute myeloid leukemia. J Hematol Oncol (2017) 10(1):124. doi:10.1186/s13045-017-0486-z
315. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol (2011) 186(5):2772–9. doi:10.4049/jimmunol.1003208
316. Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, et al. TGFbeta1-mediated SMAD3 enhances PD-1 expression on antigen-specific T cells in cancer. Cancer Discov (2016) 6(12):1366–81. doi:10.1158/2159-8290.CD-15-1347
317. Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol (2008) 181(7):4832–9. doi:10.4049/jimmunol.181.7.4832
318. Austin JW, Lu P, Majumder P, Ahmed R, Boss JM. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J Immunol (2014) 192(10):4876–86. doi:10.4049/jimmunol.1302750
319. Mathieu M, Cotta-Grand N, Daudelin JF, Thebault P, Labrecque N. Notch signaling regulates PD-1 expression during CD8(+) T-cell activation. Immunol Cell Biol (2013) 91(1):82–8. doi:10.1038/icb.2012.53
320. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity (2014) 41(5):802–14. doi:10.1016/j.immuni.2014.10.013
321. Xiao G, Deng A, Liu H, Ge G, Liu X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc Natl Acad Sci U S A (2012) 109(38):15419–24. doi:10.1073/pnas.1206370109
322. Cho HY, Lee SW, Seo SK, Choi IW, Choi I, Lee SW. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-alpha-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim Biophys Acta (2008) 1779(12):811–9. doi:10.1016/j.bbagrm.2008.08.003
323. Bally AP, Lu P, Tang Y, Austin JW, Scharer CD, Ahmed R, et al. NF-kappaB regulates PD-1 expression in macrophages. J Immunol (2015) 194(9):4545–54. doi:10.4049/jimmunol.1402550
324. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol (2011) 12(7):663–71. doi:10.1038/ni.2046
325. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity (2011) 35(3):400–12. doi:10.1016/j.immuni.2011.06.015
326. Ahn E, Youngblood B, Lee J, Lee J, Sarkar S, Ahmed R. Demethylation of the PD-1 promoter is imprinted during the effector phase of CD8 T cell exhaustion. J Virol (2016) 90(19):8934–46. doi:10.1128/JVI.00798-16
327. Stephen TL, Payne KK, Chaurio RA, Allegrezza MJ, Zhu H, Perez-Sanz J, et al. SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T cells. Immunity (2017) 46(1):51–64. doi:10.1016/j.immuni.2016.12.015
328. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (2016) 354(6316):1165–9. doi:10.1126/science.aae0491
329. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. doi:10.1038/ni.1679
330. Stecher C, Battin C, Leitner J, Zettl M, Grabmeier-Pfistershammer K, Holler C, et al. PD-1 blockade promotes emerging checkpoint inhibitors in enhancing T cell responses to allogeneic dendritic cells. Front Immunol (2017) 8:572. doi:10.3389/fimmu.2017.00572
331. Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res (2016) 22(16):4119–32. doi:10.1158/1078-0432.CCR-15-2584
332. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi:10.1038/nature15520
333. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A (2010) 107(17):7875–80. doi:10.1073/pnas.1003345107
334. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72(4):917–27. doi:10.1158/0008-5472.CAN-11-1620
335. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest (2015) 125(5):2046–58. doi:10.1172/JCI80445
336. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207(10):2175–86. doi:10.1084/jem.20100637
337. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res (2012) 72(4):887–96. doi:10.1158/0008-5472.CAN-11-2637
338. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell (2017) 170(6):1120–33.e17. doi:10.1016/j.cell.2017.07.024
339. Ma SD, Xu X, Jones R, Delecluse HJ, Zumwalde NA, Sharma A, et al. PD-1/CTLA-4 blockade inhibits Epstein-Barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog (2016) 12(5):e1005642. doi:10.1371/journal.ppat.1005642
340. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A (2010) 107(9):4275–80. doi:10.1073/pnas.0915174107
341. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med (2017) 377(14):1345–56. doi:10.1056/NEJMoa1709684
342. Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL Jr, Lawrence DP, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol (2016) 17(7):943–55. doi:10.1016/S1470-2045(16)30126-7
343. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity (2017) 46(4):577–86. doi:10.1016/j.immuni.2017.03.013
344. Kotsakis A, Koinis F, Katsarou A, Gioulbasani M, Aggouraki D, Kentepozidis N, et al. Prognostic value of circulating regulatory T cell subsets in untreated non-small cell lung cancer patients. Sci Rep (2016) 6:39247. doi:10.1038/srep39247
345. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-gamma drives Treg fragility to promote anti-tumor immunity. Cell (2017) 169(6):1130–41.e11. doi:10.1016/j.cell.2017.05.005
346. Vetsika EK, Koinis F, Gioulbasani M, Aggouraki D, Koutoulaki A, Skalidaki E, et al. A circulating subpopulation of monocytic myeloid-derived suppressor cells as an independent prognostic/predictive factor in untreated non-small lung cancer patients. J Immunol Res (2014) 2014:659294. doi:10.1155/2014/659294
347. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189(9):1363–72. doi:10.1084/jem.189.9.1363
348. Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov (2012) 2(8):722–35. doi:10.1158/2159-8290.CD-12-0014
349. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med (2013) 210(7):1389–402. doi:10.1084/jem.20130066
350. Epacadostat shows value in two SCCHN Trials. Cancer Discov (2017) 7(9):OF2. doi:10.1158/2159-8290.CD-NB2017-100
351. Lamichhane P, Karyampudi L, Shreeder B, Krempski J, Bahr D, Daum J, et al. IL-10 release upon PD-1 blockade sustains immunosuppression in ovarian cancer. Cancer Res (2017). doi:10.1158/0008-5472.CAN-17-0740
352. Durable responses achieved with AM0010. Cancer Discov (2016) 6(12):OF4. doi:10.1158/2159-8290.CD-NB2016-125
353. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med (2017) 9(389). doi:10.1126/scitranslmed.aal3604
354. Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat Commun (2017) 8:14369. doi:10.1038/ncomms14369
355. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med (2013) 210(9):1685–93. doi:10.1084/jem.20130573
356. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi:10.1084/jem.20130579
357. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic immunity is required for effective cancer immunotherapy. Cell (2017) 168(3):487–502.e15. doi:10.1016/j.cell.2016.12.022
Keywords: PD-1, PD-L1, immune checkpoint blockade, biomarker, MSI, TMB, resistance mechanism, combination immunotherapy
Citation: Xu-Monette ZY, Zhang M, Li J and Young KH (2017) PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front. Immunol. 8:1597. doi: 10.3389/fimmu.2017.01597
Received: 18 September 2017; Accepted: 06 November 2017;
Published: 04 December 2017
Edited by:
Jose A. Garcia-Sanz, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Ekaterina Jordanova, Center for Gynaecologic Oncology Amsterdam, NetherlandsTanja Denise De Gruijl, VU University Medical Center, Netherlands
Copyright: © 2017 Xu-Monette, Zhang, Li and Young. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ken H. Young, a2h5b3VuZyYjeDAwMDQwO21kYW5kZXJzb24ub3Jn