 Takumi Motoya1,2Koo Nagasawa3Yuki Matsushima4Noriko Nagata1
Takumi Motoya1,2Koo Nagasawa3Yuki Matsushima4Noriko Nagata1 Akihide Ryo5
Akihide Ryo5 Tsuyoshi Sekizuka6
Tsuyoshi Sekizuka6 Akifumi Yamashita6
Akifumi Yamashita6 Makoto Kuroda6
Makoto Kuroda6 Yukio Morita7
Yukio Morita7 Yoshiyuki Suzuki8Nobuya Sasaki2Kazuhiko Katayama9*
Yoshiyuki Suzuki8Nobuya Sasaki2Kazuhiko Katayama9* Hirokazu Kimura3,5,10*
Hirokazu Kimura3,5,10*- 1Ibaraki Prefectural Institute of Public Health, Mito, Japan
- 2Laboratory of Laboratory Animal Science and Medicine, Faculty of Veterinary Medicine, Kitasato University, Towada, Japan
- 3Infectious Disease Surveillance Center, National Institute of Infectious Diseases, Musashimurayama, Japan
- 4Division of Virology, Kawasaki City Institute for Public Health, Kawasaki, Japan
- 5Department of Microbiology, Yokohama City University Graduate School of Medicine, Yokohama, Japan
- 6Pathogen Genomics Center, National Institute of Infectious Diseases, Musashimurayama, Japan
- 7Department of Food and Nutrition, Tokyo Kasei University, Itabashi-ku, Japan
- 8Graduate School of Natural Sciences, Nagoya City University, Nagoya, Japan
- 9Laboratory of Viral Infection I, Kitasato Institute for Life Sciences, Kitasato University, Minato-ku, Japan
- 10School of Medical Technology, Faculty of Health Sciences, Gunma Paz University, Takasaki, Japan
Human norovirus (HuNoV) is a leading cause of viral gastroenteritis worldwide, of which GII.4 is the most predominant genotype. Unlike other genotypes, GII.4 has created various variants that escaped from previously acquired immunity of the host and caused repeated epidemics. However, the molecular evolutionary differences among all GII.4 variants, including recently discovered strains, have not been elucidated. Thus, we conducted a series of bioinformatic analyses using numerous, globally collected, full-length GII.4 major capsid (VP1) gene sequences (466 strains) to compare the evolutionary patterns among GII.4 variants. The time-scaled phylogenetic tree constructed using the Bayesian Markov chain Monte Carlo (MCMC) method showed that the common ancestor of the GII.4 VP1 gene diverged from GII.20 in 1840. The GII.4 genotype emerged in 1932, and then formed seven clusters including 14 known variants after 1980. The evolutionary rate of GII.4 strains was estimated to be 7.68 × 10−3 substitutions/site/year. The evolutionary rates probably differed among variants as well as domains [protruding 1 (P1), shell, and P2 domains]. The Osaka 2007 variant strains probably contained more nucleotide substitutions than any other variant. Few conformational epitopes were located in the shell and P1 domains, although most were contained in the P2 domain, which, as previously established, is associated with attachment to host factors and antigenicity. We found that positive selection sites for the whole GII.4 genotype existed in the shell and P1 domains, while Den Haag 2006b, New Orleans 2009, and Sydney 2012 variants were under positive selection in the P2 domain. Amino acid substitutions overlapped with putative epitopes or were located around the epitopes in the P2 domain. The effective population sizes of the present strains increased stepwise for Den Haag 2006b, New Orleans 2009, and Sydney 2012 variants. These results suggest that HuNoV GII.4 rapidly evolved in a few decades, created various variants, and altered its evolutionary rate and antigenicity.
Introduction
Human norovirus (HuNoV) belongs to the genus Norovirus in the family Caliciviridae and is a major causative agent of acute gastroenteritis in humans worldwide (Green, 2013; Robilotti et al., 2015). The large genetic divergence of HuNoV results in two genogroups and many genotypes (Green, 2013; Robilotti et al., 2015; Vinjé, 2015). Previous epidemiological studies revealed that GI.3, GII.2, GII.3, GII.4, GII.6, GII.12, and GII.17 were prevalent among HuNoV genotypes (Mathijs et al., 2011; Hoa-Tran et al., 2013; Vega et al., 2014a; Kumazaki and Usuku, 2015; Thongprachum et al., 2016). Importantly, GII.4 has caused pandemics of acute gastroenteritis in people of all ages in various countries during the past 10 years (Bull et al., 2006; Siebenga et al., 2009; Robilotti et al., 2015). Notably, multiple GII.4 variants strains having different antigenicity suddenly emerged and caused pandemics of gastroenteritis in several countries (Debbink et al., 2012a,b, 2013; Lindesmith et al., 2012a; Kumazaki and Usuku, 2015; Allen et al., 2016; Qiao et al., 2016; Parra et al., 2017). For example, in the 2006/07 season, the GII.4 variant emerged and spread rapidly in a large number of areas, including Japan, leading to pandemics in people of all ages (Tu et al., 2007; Motomura et al., 2008; Siebenga et al., 2009; Lam et al., 2012). The GII.4 variant 2006b, a representative strain (Den Haag 2006b strain), caused pandemics in five seasons (Hoa-Tran et al., 2013; Kumazaki and Usuku, 2015; Sato et al., 2017). In the 2006/07 season in Japan, it was estimated that over 3 million children and adults presented with acute gastroenteritis due to infections with the GII.4 2006b (Umeda, 2007). Furthermore, in the 2012/13 season, the GII.4 variant 2012, a representative strain (Sydney 2012 strain), also suddenly emerged and caused pandemics of gastroenteritis similar to that caused by the GII.4 2006b strain in the USA, Europe, and Japan (Siebenga et al., 2009; Allen et al., 2014; Vega et al., 2014a; Kumazaki and Usuku, 2015; Vinjé, 2015; Parra et al., 2017).
The HuNoV major capsid (VP1) protein is considered to be closely associated with the infectivity and antigenicity of these strains (Debbink et al., 2012a). Many previous reports suggested that the HuNoV VP1 gene rapidly evolved, resulting in a large divergence of antigenicity (Allen et al., 2008; Bok et al., 2009; Yang et al., 2010; Debbink et al., 2012a; Zakikhany et al., 2012). Earlier findings also indicated that the rapid evolution of the VP1 gene in HuNoV is strongly associated with gastroenteritis pandemics caused by HuNoV (Robilotti et al., 2015).
Various bioinformatic techniques for investigating viral genes may contribute to a better understanding of the evolution of the viruses (Andernach et al., 2013; Añez et al., 2013; Sun et al., 2017; Wei and Li, 2017). Of these, phylogenetic analyses using the Bayesian Markov chain Monte Carlo (MCMC) method can characterize evolutionary processes by estimating the date of divergence and evolutionary rates using sequences with a known sample-collection date for various organisms (Drummond and Bouckaert, 2015). Additionally, the Bayesian skyline plot (BSP) allows prediction of the transition of the infectious population size for pathogens (Hall et al., 2016). Conformational epitope analyses using in silico approaches also enable us to identify and map binding sites of host antibodies on three-dimensional structures of proteins (Potocnakova et al., 2016). For HuNoV, a large number of studies on viral evolution have been performed using bioinformatics (Giammanco et al., 2014; Kimura et al., 2015; Kobayashi et al., 2015, 2016; Lu et al., 2016; Parra et al., 2017; Siqueira et al., 2017; Tohma et al., 2017). Previous reports described the evolutionary rates of some HuNoV genotypes using the Bayesian MCMC method and linear regression analyses, which suggested differences in such rates among the genotypes (Parra et al., 2017). In addition, the blockade epitopes on the VP1 protein in some HuNoV genotypes were identified by amino acid variation analyses and molecular biological techniques using the virus-like particle (VLP) (Lindesmith et al., 2014). Previous phylogenetic approaches also showed that GII.4 continuously created genetically different clusters, which resulted in the emergence of many variants (Fioretti et al., 2014; Qiao et al., 2016). However, the differences of evolutionary features among all GII.4 variants, including recently discovered strains, have not been comprehensively elucidated (Bok et al., 2009; Siebenga et al., 2010; Qiao et al., 2016). It may be important to understand why GII.4 has frequently produced escape from previously acquired immunity and repeatedly caused large outbreaks. Moreover, a recent study constructed a system for predicting the HuNoV genotype composition in the peak season using a unique algorithm, namely, NOROCAST (Suzuki et al., 2016). Thus, these bioinformatics technologies may contribute to our understanding of the evolution of the HuNoV GII.4 capsid gene, including the changes of antigenicity, for advancing the prevalent prediction systems for HuNoV. In the present study, using various bioinformatic techniques, we compared the molecular evolution of all GII.4 variants strains using globally collected full-length HuNoV GII.4 VP1 gene sequences.
Materials and Methods
Strains Used in this Study
We obtained the full-length nucleotide sequences of HuNoV VP1 gene from GenBank1 in December 2015. The strains employed were classified using norovirus genotyping tool (Kroneman et al., 2011), and sequences of GII.4 genotype were selected. Of these, strains with ambiguous sequences and unknown year of collection were excluded from the dataset. At this point, complete GII.4 VP1 sequences of approximately 2,000 strains were included in the dataset. However, due to limitation in computing capacity, positive and negative selection analyses could not include sequences with greater than 99.0% identity, as it resulted in over 500 sequences. Thus, the VP1 sequences with >98.9% identity were omitted from the dataset. Moreover, the dataset was analyzed to detect recombination within the VP1 gene using RDP4.95 with seven primary exploratory recombination signal detection methods (RDP, GENECONV, BOOTSCAN/RECSCAN, MAXCHI, CHIMAERA, SISCAN, 3SEQ) (Martin et al., 2015). The p-value threshold for significance was set as 0.001. Recombinant regions was determined when it was detected by more than four of these methods. However, this dataset contained no recombinant sequences. Finally, 466 strains were included in this study (Figure S1, Table S1). The sequences of the dataset were aligned using MAFFT software (Katoh and Standley, 2013) and were then subjected to bioinformatic analysis as described below.
Time-Scaled Phylogenetic Tree Constructed Using the Bayesian Markov Chain Monte Carlo Method
Molecular clock evolutionary dynamics was performed using the Bayesian MCMC method in the BEAST package v2.4.6 (Suchard et al., 2001; Drummond and Rambaut, 2007; Bouckaert et al., 2014). To construct the correct phylogenetic tree (Choudhuri, 2014), we added the nucleotide sequences of all GII genotypes, including porcine NoV GII (GII.11, GII.18, and GII.19) and other HuNoV GII genotypes (18 strains), and an outgroup strain, HuNoV GI genotype (GI.1) into the dataset mentioned above (a total of 488 strains). First, we selected the appropriate substitution model using the jModelTest2 program (Guindon and Gascuel, 2003; Darriba et al., 2012). Next, the best of four clock models (Strict clock, Exponential relaxed clock, Relaxed clock log normal, Random local clock) and two tree prior models (Coalescent constant population, Coalescent exponential population) were determined by the method of path-sampling/stepping stone-sampling marginal-likelihood estimation. Finally, the dataset was analyzed using the strict clock and constant tree prior models. The MCMC chain lengths were 300,000,000 steps with sampling every 20,000 steps using high-performance computers [system: x3850X6, CPU: Intel(R) Xeon (R) CPU E7-8890 v3 @ 2.50GHz, memory: 1TB] with GPU [Tesla K20c; global memory: 5,061 MB, clock speed; 0.71 GHz, number of cores: 2,496]. The analyzed data were evaluated by the effective sample size using Tracer2, and values >200 were accepted. The maximum clade credibility tree was constructed after we discarded the first 10 or 15% (“burn-in”) of the trees using the TreeAnnotator v2.4.6 in the BEAST 2 package. The phylogenetic tree was visualized by the FigTree v1.4.0 program. The reliability of branches was supported by 95% highest posterior densities (HPDs). Furthermore, the evolutionary rates of HuNoV GII.4, the variants including more than 10 strains (US95_96, Farmington Hills 2002, Asia 2003, Hunter 2004, Yerseke 2006a, Den Haag 2006b, Osaka 2007, Apeldoorn 2007, New Orleans 2009, and Sydney 2012 variants) and the domains were also estimated using appropriate models selected for each dataset as described above.
Similarity Analysis
Similarities between the aligned nucleotide sequences and the Bristol 1993 variant were visualized using the SimPlot program (Lole et al., 1999). Two strains of Bristol 1993 variant (X76716 and FJ537137) were used as query sequences. The similarity was examined using a window size of 200 nucleotides in length (nt) and a step size of 20 nt in the full-length VP1 genes.
Calculation of the Phylogenetic Distance
Phylogenetic trees were generated from the datasets of all GII.4 strains and each GII.4 variant, including more than three strains, based on the maximum likelihood method in the MEGA6 software. The best substitution models were determined by the jModelTest2. The phylogenetic distances between GII.4 strains were calculated from the constructed phylogenetic trees by the Patristic program (Fourment and Gibbs, 2006).
Selective Pressure Analysis
Non-synonymous (dN) and synonymous (dS) substitutions rates at each codon were calculated by the Datamonkey server to estimate the positive and negative selection sites in capsid VP1 genes of HuNoV GII.4 and each GII.4 variant (Pond and Frost, 2005; Delport et al., 2010). Sites under positive (dN > dS) and negative (dN < dS) selection were determined by three methods (SLAC, FEL, and IFEL) based on a significance level of p < 0.05. For SLAC, the two-tail extended binomial distribution was used to culculate the p-value. The FEL and IFEL were based on the single degree of freedom likelihood ratio test (chi-squared asymptotic distribution was used) to classify a site as positively or negatively selected.
Conformational B-Cell Epitope Prediction of HuNoV GII.4
Conformational epitopes on the capsid VP1 protein of each of the GII.4 variants strains (Bristol 1993: X76716, Camberwell 1994: AF145896, US95_96: AF080558, Kaiso 2003: AB303929, Farmington Hills 2002: AY485642, Lanzou 2002: EU310927, Asia 2003: AB220921, Hunter 2004: AY883096, Yerseke 2006a: EF126963, Den Haag 2006b: EF126965, Osaka 2007: AB434770, Apeldoorn 2007: GU270580, New Orleans 2009: GU445325, Sydney 2012: JX459908) were predicted using the following four tools: DiscoTope 2.0 (Kringelum et al., 2012), BEPro (Sweredoski and Baldi, 2008), EPCES (Liang et al., 2007), and EPSVR (Liang et al., 2010). Cut-off values of epitopes were set at −3.7 (DiscoTope 2.0), 1.3 (BEPro), and 70 (EPCES, EPSVR). Consensus sites in all of the four tools and regions with close residues over two of the sites on the VP1 dimer structures were determined as conformational epitopes.
Mapping of Positive Selection Sites, Predicted Epitopes, and Amino-Acid Substitutions on the Three-Dimensional Structure of HuNoV GII.4 VP1 Proteins
VP1 dimer structural models of each GII.4 variant were constructed using MODELLER v9.15 (Webb and Sali, 2014). The templates for homology modeling were based on the crystal structures of four strains (PDB ID: 1IHM, 4X07, 4OP7, and 4OPS). The capsid structure of GI (PDB ID: 1IHM) was used as a template for the construction of a shell domain in whole VP1 structures. Amino acid sequences of five templates and the targeted strains were aligned by MAFFTash (Katoh et al., 2002; Standley et al., 2007). The constructed models were minimized based on GROMOS96 (van Gunsteren et al., 1996), implemented by Swiss PDB Viewer v4.1 (Guex and Peitsch, 1997), and evaluated by Ramachandran plots through RAMPAGE server (Lovell et al., 2003). The final models were modified and colored by Chimera v1.11.2 (Pettersen et al., 2004). The positive selection sites, conformational epitopes of each variant, and substitution sites of the other variants to the Bristol 1993 strain were mapped on the structure.
Bayesian Skyline Plot Analysis
The genealogical population size of HuNoV GII.4 strains was estimated by BSP inference using BEAST v2.4.6. Appropriate substitution and clock models were selected as described above. Yerseke 2006a strains were analyzed in a short period for 1 year since the strains with >98.9% sequence identity were omitted. The analyzed plots were visualized with the 95% HPDs using Tracer.
Statistical Analyses
Statistical analyses were performed by EZR statistical software based on the Kruskal–Wallis test with multiple comparisons using the Holm test for the evolutionary rates and phylogenetic distance, and Fisher's exact test for the distribution of negative selection sites (Kanda, 2013). We obtained sample sizes of 18,000–135,000 for the statistical analyses of the evolutionary rates. The values obtained by dividing the numbers of chain lengths by that of the log parameters were used as sample sizes. For the phylogenetic distance analyses, we acquired 3–11,440 data that exhibited the number of all combinations between GII.4 strains included in the dataset (Mizukoshi et al., 2017). Detailed statistical data are shown in Tables S2–S4. Sample sizes of each group are also presented in Figures 2, 4.
Results
Time-Scale Evolution of the Globally Collected GII.4 Strains
We constructed a time-scale evolutionary phylogenetic tree by the MCMC method using numerous sequences for all GII.4 variants including recent strains. First, a common ancestor of GII.4 strains diverged from GII.20 in 1840 (95% HPDs, 1820–1858). The GII.4 genotype emerged in 1932 (95% HPDs, 1926–1938) and formed seven clusters (seven sublineages) after 1980 (95% HPDs, 1979–1981). Notably, an ancestor of Den Haag 2006b and Sydney 2012 strains, two pandemic GII.4 variants, diverged in 1994 (95% HPDs, 1993–1995) from Asia 2003 strains as a common ancestor. A common ancestor of Apeldoorn 2007, New Orleans 2009, and Sydney 2012 diverged in 2001 (95% HPDs, 2000–2002). Moreover, both pandemic GII.4 variants strains (Den Haag 2006b and Sydney 2012) formed distinct and unique clusters (Figure 1). All divergence years of the clusters are shown in Figure 1B. These results suggest that GII.4 strains rapidly evolved during the past 35–40 years and formed multiple clusters in the phylogenetic tree.
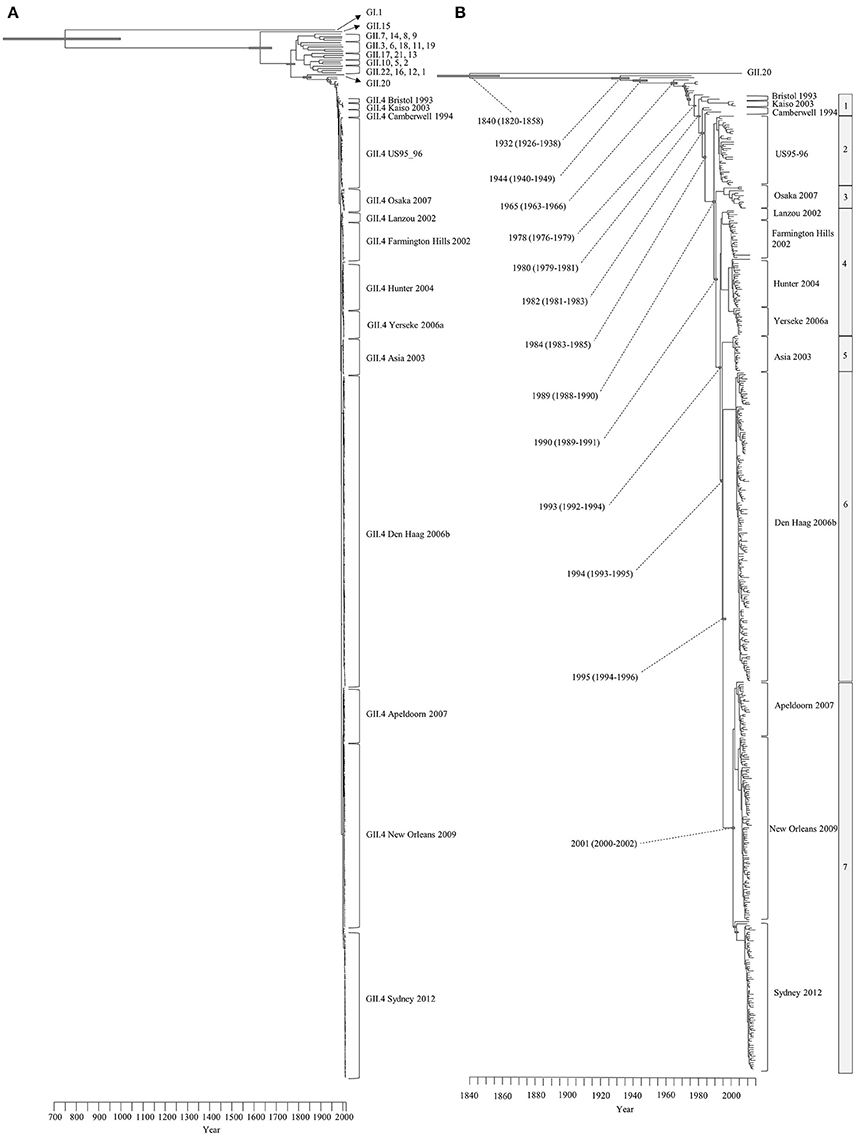
Figure 1. Time-scaled phylogenetic trees of the complete HuNoV capsid VP1 gene constructed by the Bayesian MCMC method. (A) The maximum clade credibility tree with the dataset including HuNoV GI.1 and all GII genotypes; (B) Enlarged tree focused on GII.4. Gray bars indicate the 95% highest probability densities for each branch year.
We also estimated the evolutionary rates of the present GII.4 strains. The evolutionary rate of complete VP1 gene in the GII.4 strains was estimated to be 7.68 × 10−3 substitutions/site/year (95% HPDs, 6.69–8.59 × 10−3) (Figure 2A). Moreover, the nucleotide substitution rates were significantly different between the GII.4 variant (Kruskal–Wallis test; p < 0.001). The rate of Osaka 2007 variant was higher than among those of other variants (Figure 2B, Table S3). Furthermore, the mean evolutionary rate of the P2 domain (9.15 × 10−3 substitutions/site/year) was higher than those of the other domains (shell: 6.97 × 10−3, P1 domain: 5.79 × 10−3 substitutions/site/year) (Kruskal–Wallis test; p < 0.001) (Figure 2A, Table S2).
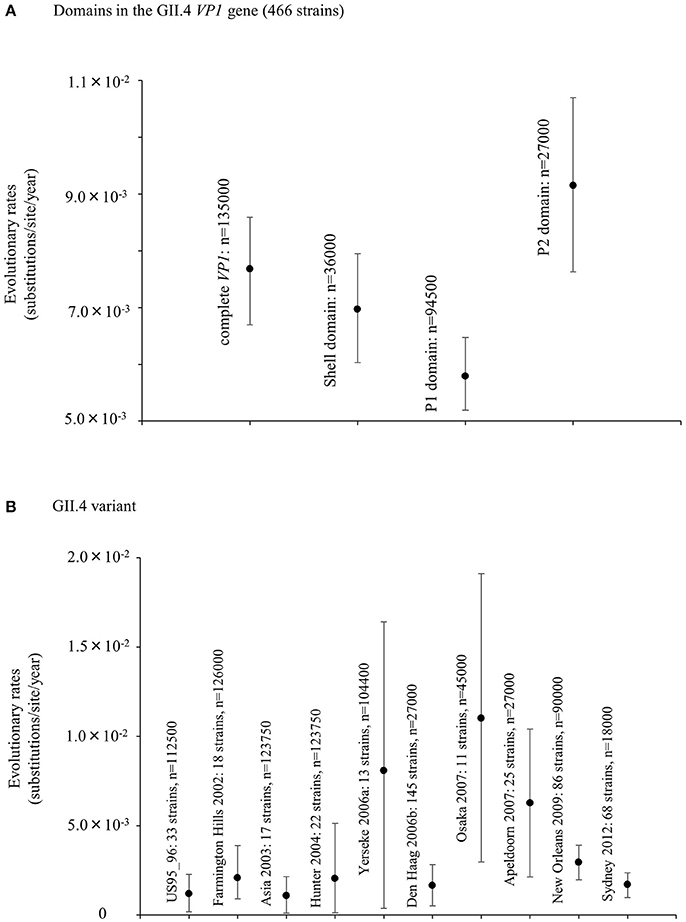
Figure 2. Evolutionary rates of nucleotide sequences in the full-length GII.4 VP1 gene. (A) Evolutionary rates for domains within GII.4 VP1 gene; (B) Evolutionary rates for each GII.4 variant. The y-axis represents the evolutionary rate (substitutions/site/year). Statistical results for multiple comparisons in the domains and the GII.4 variants were shown in Tables S2, S3, respectively.
Simplot Data Analyses of the Capsid VP1 Gene in the Present GII.4 Strains
We performed the SimPlot analysis based on the full-length capsid gene sequences in the present strains. As illustrated in Figure 3, similarities of 85–100% were found in the shell domain, whereas the P1 and P2 domains were characterized by lower similarities (75–95%). These results indicated that the genetic divergence of the VP1 gene in the GII.4 strains differed among domains.
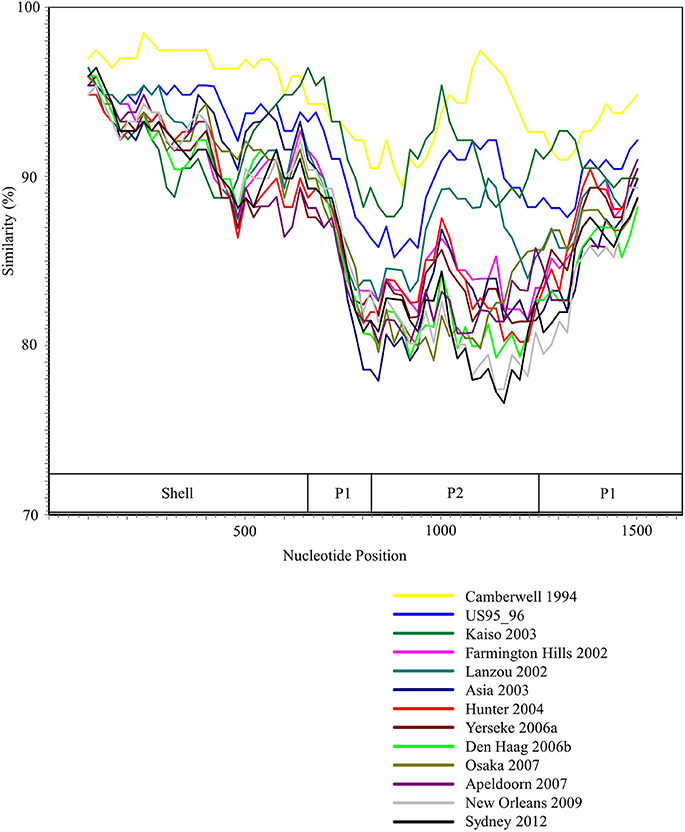
Figure 3. SimPlot analysis of the representative HuNoV GII.4 strains. Each variant's similarity to the Bristol 1993 variant is represented. The positions of the shell, P1, and P2 domains in VP1 gene are shown below the graph.
Phylogenetic Distances of the Capsid VP1 Gene in the GII.4 Strains
We calculated the phylogenetic distance of the VP1 gene in the GII.4 strains studied here. The phylogenetic distance values of all GII.4 strains were 0.210 ± 0.105 (mean ± standard deviation) (Figure 4A). The mean values of each variant strain were within the range 0.0163–0.0698, and the phylogenetic distances were significantly distinct between GII.4 variants. In particular, the Osaka 2007 variant strains probably included the highest number of nucleotide substitutions, whereas the branches of the strains of the Asia 2003 variant tended to be shorter than those of the other variants (Kruskal–Wallis test; p < 0.001) (Figure 4B, Table S4).
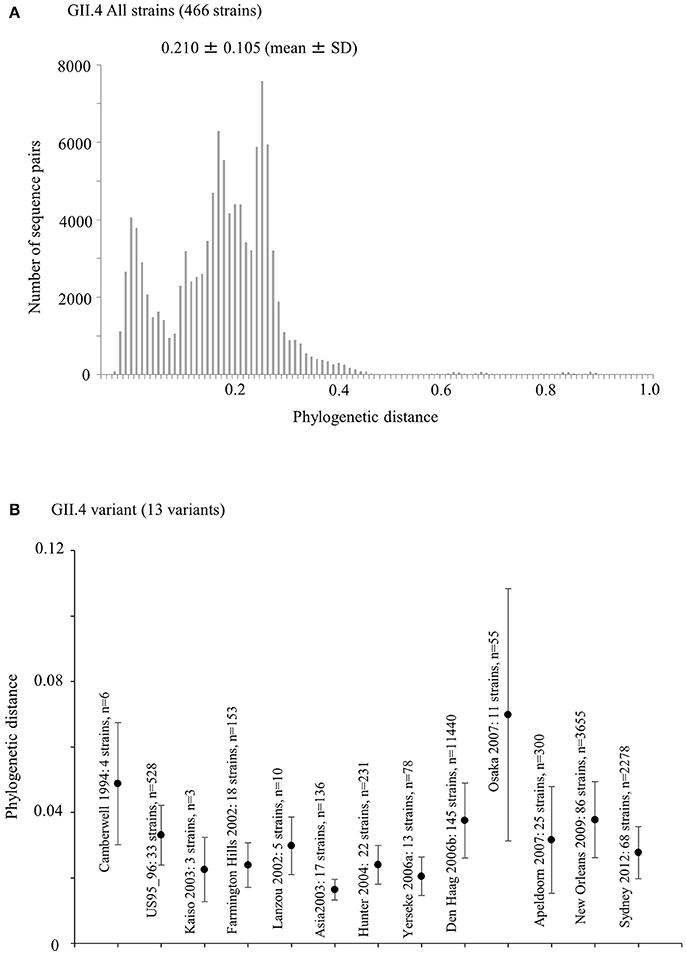
Figure 4. Phylogenetic distance between the nucleotide GII.4 sequences of the full-length VP1 gene. (A) Phylogenetic distance of intra-genotype in HuNoV GII.4 strains. The y-axis represents the number of sequence pairs corresponding to each distance. The x-axis shows phylogenetic distances; (B) Phylogenetic distance of intra-variant in HuNoV GII.4 strains. The y-axis indicates phylogenetic distances. The x-axis represents each variant. Data are expressed as mean ± standard deviation. Statistical results for multiple comparisons were shown in Table S4.
Mapping of Conformational Epitopes, Positive Selection Sites, and Amino-Acid Substitutions on the VP1 Structures of GII.4
We also comprehensively predicted the conformational epitope on the whole deduced GII.4 variants VP1 protein and mapped the epitopes, positive selection sites, and amino acid substitutions on the structures of the GII.4 strains to assess their relationships using in silico methods (Borley et al., 2013; Yao et al., 2013). Five regions were recognized as conformational epitopes, four of which were located in the exterior surface of the P2 domain and overlapped with the epitope regions identified by previous in vitro studies. Moreover, a number of amino-acid substitutions were found on/around the putative epitopes (Table 2, Figure 5, Figure S2). Positive selection sites for the dataset including all GII.4 strains were predicted to be amino acid (aa) 6 (Asn6Ser and Ser6Asn), aa9 (Asn9Ser, Thr, His, Lys, and Ser9Asn), and aa534 (Thr534Ala, Ser, and Ala534Val, Thr) positions, whereas no positive selection site was found in the P2 domain (Table 3). Moreover, a total of 369 residues were under negative selection, whose rates were significantly higher in the P1 domain than in the shell and P2 domains (p < 0.001) (Table 5). The positive selection sites for the dataset separating each GII.4 variant was estimated to be aa393 (Ser393Gly, Asn, and Gly393Ser) and aa412 (Asn412Asp, Ser, and Asp412Gly) in Den Haag 2006b; aa294 (Pro294Ser, Thr, Ser294Ala, Pro, and Ala294Thr) and aa376 (Glu376Asp, Val, Gln, Asp376Val, Glu, and Val376Asp, Glu, Ile) in New Orleans 2009; and aa393 (Ser393Gly, Asn, Thr, and Gly393Ser) in Sydney 2012 (Table 4). The Den Haag 2006b variant contained the most negative selection sites (59 sites), whereas seven GII.4 variants (Camberwell 1994, Kaiso 2003, Farmington Hills 2002, Lanzou 2002, Asia 2003, Hunter 2004, and Yerseke 2006a) were not identified as negative selection sites (Table 5).
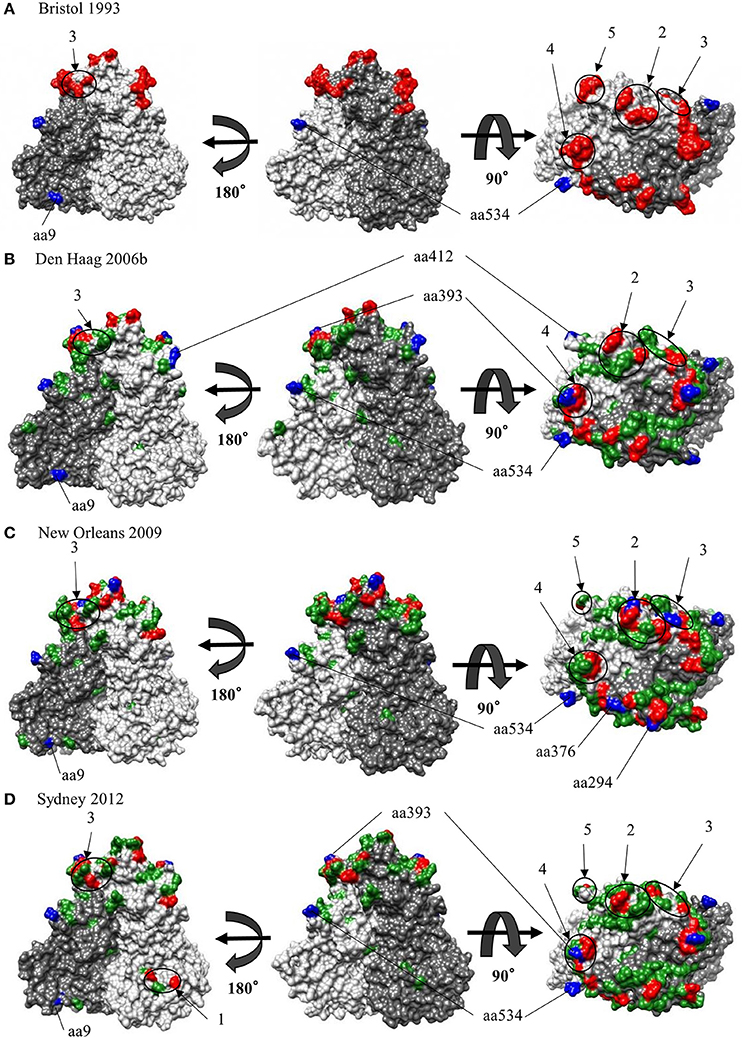
Figure 5. Structural models for the capsid VP1 protein of each HuNoV GII.4 variant. Three-dimensional VP1 dimer structures for the Bristol 1993 (A), the Den Haag 2006b (B), the New Orleans 2009 (C) and the Sydney 2012 (D) variants are shown. Chains that are composed of the dimer structures are colored in gray (chain A) and dim gray (chain B). Predicted epitopes of each variant are colored in red and circled for regions. Positive selection sites are colored in blue for aa9, aa294, aa376, aa393, aa412, and aa534. Of note, aa6 could not be specified due to lack of structure modeling in N terminus. Amino-acid substitutions of the other variants to a GII.4 Bristol 1993 strain are colored in green.
Phylodynamics of the Capsid VP1 Gene in the Present GII.4 Strains
We examined the phylodynamics of human NoV GII.4 strains in the VP1 gene using the BSP analyses with the parameters shown in Table 1. In the present HuNoV GII.4 strains, the mean effective population size remained constant until approximately 1995 and gradually increased around 1994–1996 and 2005–2006 (Figure 6A). In each GII.4 variant, the mean effective population sizes of Den Haag 2006b, New Orleans 2009, and Sydney 2012 grew in 2005–2007, 2004–2010, and 2006–2011, respectively, whereas no changes of the sizes of the other variants were observed (Figures 6B–K). Moreover, the effective population sizes of all NoV GII.4 strains were estimated to be 102 for approximately 10 years (Figure 6A). These results suggested that restricted sublineages of GII.4 increased the effective population sizes, and the GII.4 strains have adapted to humans for approximately 10 years.
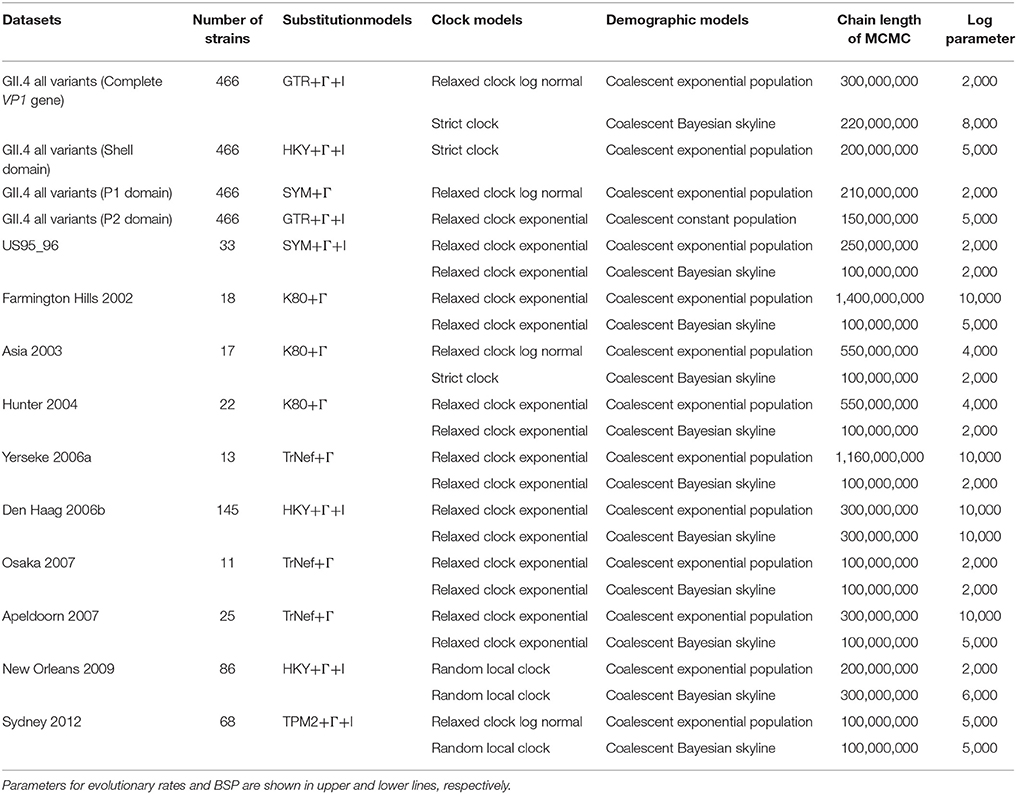
Table 1. Parameters for evolutionary rates and Bayesian skyline plot analyses in HuNoV GII.4 and the variants.

Figure 6. Bayesian skyline plot for VP1 sequences of HuNoV GII.4. Plots for all GII.4 strains (A), US95_96 variant strains (B), Farmington Hills 2002 variant strains (C), Asia 2003 variant strains (D), Hunter 2004 variant strains (E), Yerseke 2006a variant strains (F), Den Haag 2006 variant strains (G), Osaka 2007 variant strains (H), Apeldoorn 2007 variant strains (I), New Orleans 2009 variant strains (J), and Sydney 2012 variant strains (K) are shown. The y-axis represents the effective population size on logarithmic scale, whereas the x-axis denotes the time in years. The solid black line indicates the median posterior value. The intervals with the highest probability densities (95%) are shown by blue lines.
Discussion
In the present study, we show the molecular evolution of the capsid VP1 gene in globally collected HuNoV GII.4, including recent strains, during a period of approximately 40 years. Our new findings can be summarized as follows: (1) The evolutionary pattern, including evolutionary rates, phylogenetic distance, and infectious population size, may differ between GII.4 variants. (2) The evolutionary rates and constraints on amino acid mutations are likely distinct between domains within GII.4 VP1. (3) There may be few epitopes on the exterior surface of the VP1 structure other than those positions on the P2 domain, as previously identified. (4) Prevalent GII.4 variants specifically undergo selective pressure that leads to new variant that can escape the immune system of the host.
The Bayesian MCMC method is used to characterize evolutionary processes in terms of date of divergence and the rates of evolution for organism genes with high evolutionary rates, such as RNA viruses (Drummond and Bouckaert, 2015). This method has already been used to study molecular evolution not only for noroviruses but also for various other RNA viruses (Añez et al., 2013; Vijaykrishna et al., 2015). In particular, it is important to estimate nucleotide evolutionary rates for HuNoV since previous reports showed that the viruses could evolve with the distinct rates among the genotypes depending on difference of polymerase fidelity and immune pressure for each genotype (Bull et al., 2010). Estimation of evolutionary rates using the method may contribute to predict the frequency of emergence of variants that can escape from previously acquired host immunity in the HuNoV genotypes. However, the results of the method are affected by the sequences included in the dataset. In this study, the time-scaled evolutionary phylogenetic tree showed that the common ancestor of GII.4 and GII.20 dated back to about 175 years ago. Furthermore, during the past 40 years, the GII.4 strains evolved and formed seven clusters including 14 known variants (Figure 1). Next, we estimated the HuNoV GII.4 VP1 gene evolutionary rate as 7.68 × 10−3 substitutions/site/year. Our observations are generally consistent with previous observations overall, with GII.4 having a higher evolutionary rate than other genotypes such as GII.7 (2.3 × 10−3 substitutions/site/year), GII.3 (4.16 × 10−3 substitutions/site/year), and GII.2 (1.31 × 10−3 substitutions/site/year) (Bok et al., 2009; Bull et al., 2010; Siebenga et al., 2010; Boon et al., 2011; Fioretti et al., 2014; Qiao et al., 2016; Mizukoshi et al., 2017; Parra et al., 2017). In addition, we estimated the different evolutionary rates among GII.4 variants. The rates among domains (shell, P1, and P2 domains) in the VP1 gene also tended to be distinct, with that of the P2 domain being the highest (Figure 2). A previous report showed that GII.4 had replication rates of polymerase that differed among the variants (Bull et al., 2010). In addition, the prevalence of GII.4 differed among the variants (Vinjé, 2015), which may produce differences of herd immunity pressure for each GII.4 variant. We also performed similarity plot analyses in the GII.4 variant strains. Our data showed that the similarities of the shell domains were relatively high (85–100%), followed by a lower level in the P1 domains, and the lowest level in the P2 domains (75–95%), which is consistent with previous reports that showed similarities in the shell, P1, and P2 domains of around >85, >80, and >75%, respectively (Eden et al., 2013; Doerflinger et al., 2017). Next, we calculated the phylogenetic distance among the GII.4 strains. Notably, the values of the intra-variant distance were different. Of them, Osaka 2007 variant strains tended to have high nucleotide variability, while Asia 2003 probably contained fewer nucleotide substitutions than the other variants (Figure 4). These results suggest that GII.4 has uniquely evolved among these variants having different evolutionary rates.
Earlier molecular biological and bioinformatic studies identified or estimated the blocking epitopes in the P2 domain within the GII.4 VP1 structure (Debbink et al., 2012b, 2013; Lindesmith et al., 2012a,b; Parra et al., 2012; Giammanco et al., 2014). These studies were mainly focused on the P2 domain and based on the amino acid variations on the exterior surface in the domain that arose over time. However, other conformational epitopes may also be contained in the shell and P1 domains. Since amino acid sequences of the shell and P1 domains are conserved than those of the P2 domain, it may be possible to use the epitopes of the shell and P1 domains for the development of diagnostic tools for HuNoV, such as immunochromatography kits. Additionally, antibodies associated with antigenicity in positions different from previously identified epitopes may be produced by the B-cell response in the host. To the best of our knowledge, conformational epitope analyses of the whole VP1 structure, including the shell and P1 domains, of various GII.4 variants using mathematical methods have not yet been conducted. The epitopes predicted in our data were close to or overlapped with those positions identified by previous in vitro experiments using VLP and monoclonal antibodies (Lindesmith et al., 2012a). Furthermore, our prediction identified few epitopes, including in shell and P1 domains, other than the previously reported positions. This result may suggest difficulty in acquiring antibodies broadly responding to various GII.4 variants that can escape from previously acquired herd immunity. To the best of our knowledge, this is the first observation of this kind in this field. This study suggests the need for additional clarification of the immunology of the GII.4 VP1 protein. However, the present study was performed only in silico. To obtain a better understanding of norovirus biology, further studies including in vitro epitope-mapping experiments for all antibodies produced by the host should be performed. Next, our analyses revealed that many common epitopes among the GII.4 variants strains were present in the P2 domain. Moreover, a number of substitutions of amino acids were detected in the conformational epitopes (Table 2). Other in vitro studies using the HuNoV GII.4 VLP established that the antigenicity changes of the GII.4 were associated with the amino acid substitutions in the P2 domains of GII.4 VP1 proteins (White, 2014). Additionally, previous in vitro and bioinformatic studies predicted contact residues specific to antibodies in epitopes A (aa294-298, aa362, 368), B (aa333, aa382), C (aa340, aa376), D (aa393–395), and E (aa407, aa412–413). It has also been confirmed that epitope A is an antigenic region, whereas epitope D is associated with HBGA binding sites. Moreover, epitope C has been predicted to be an evolving epitope and was confirmed using a monoclonal antibody. Epitope E is associated with the potential blockade of Farmington Hills 2002 variant. However, there are no reports on epitope B based on in vitro experiments. Previous bioinformatic studies suggested that the five epitopes are associated with the production of antibodies against various GII.4 strains on the exterior surface in the P2 domain (Debbink et al., 2012b, 2013; Lindesmith et al., 2012a,b; Parra et al., 2012; Giammanco et al., 2014; Garaicoechea et al., 2015). Thus, based on our results and those of other researchers, we propose that previously identified epitopes are robustly associated with the antigenicity of HuNoV GII.4.

Table 2. Putative conformational epitopes for the capsid VP1 proteins of HuNoV GII.4 variants.
We also estimated the selective pressures in the present GII.4 VP1 gene. The results obtained by the SLAC, FEL, and IFEL methods revealed positive selection for the whole GII.4 genotype at aa6, aa9, and aa534, which are located in the shell and P1 domains, whereas no positive selection sites were identified in the P2 domain (Table 3). However, separating for each GII.4 variant, positive selection sites in three prevalent GII.4 variants strains were predicted in the P2 domain (aa393 and aa412 in Den Haag 2006b, aa294, and aa376 in New Orleans 2009, and aa393 in Sydney 2012) (Table 4). These sites mostly overlapped with the epitopes that were identified by our prediction and previous in vitro studies (Table 2). Positive selection implies that non-synonymous substitutions facilitate the survival of organisms, such as escape from the immune response of the host (Kryazhimskiy and Plotkin, 2008). Moreover, a previous report predicted an association with positive selection of efficient RNA translation via secondary RNA structures (Siebenga et al., 2010). These results indicate that the prevalent GII.4 variants specifically undergoes strong selective pressure that may produce immune escape variants due to the numerous infectious populations with herd immunity. However, the importance of the positive selection in the shell and P1 domains requires further investigation by in vitro studies. Previous studies also estimated the positive selection sites in the GII.4 VP1 gene (Lindesmith et al., 2008; Bok et al., 2009; Siebenga et al., 2010; Vega et al., 2014b). For example, using the SLAC method, Bok et al. (2009) predicted six sites of positive selection (aa6, aa9, aa15, aa47, aa395, and aa534). However, the threshold for statistical significance in that study was higher than that in our study (p < 0.25). Siebenga et al. (2010) identified eight positive selection sites using FEL and IFEL (aa6, aa9, aa47, aa352, aa372, aa395, aa407, and aa534), which is partially consistent with our results. The partial discrepancy between our data and those in previous reports might have resulted from the difference in the datasets between studies. Moreover, our results revealed many sites under negative selection. In each domain, negative selection sites accounted for approximately 60–80%. The proportion of negative selection sites tended to be distinct between the domains in the VP1 gene (Table 5). However, low numbers of strains may result in no or few negative selection sites in some GII.4 variants. Negative selection exhibits non-synonymous substitutions reduce the survival of organisms. Namely, the sites should preserve synonymous substitutions to maintain the survival (Suzuki, 2004). These results suggest that a number of mutations in the VP1 gene may be under negative and neutral selection.

Table 3. Positive selection sites in the present strains of HuNoV GII.4.
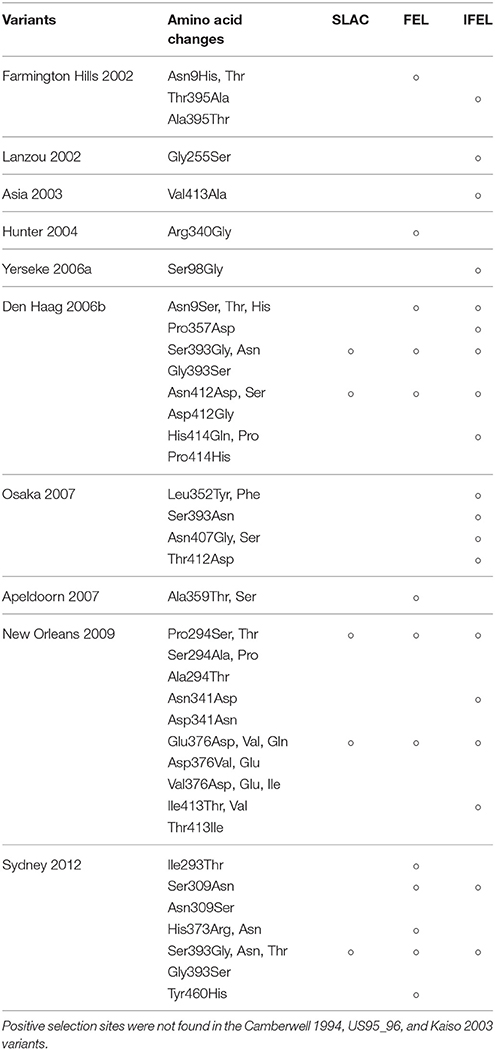
Table 4. Positive selection sites in HuNoV GII.4 variants.
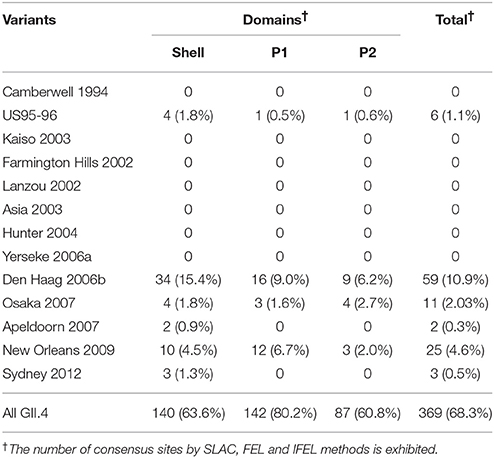
Table 5. Number of negative selection sites in the present strains of HuNoV GII.4.
Next, we mapped the conformational epitopes, positive selection, and amino acid substitutions on the VP1 structures in GII.4 variants. Most putative epitopes and substitutions were located on the exterior surface of the P2 domain in all variants. The epitopes overlapped with or were proximal to the substitution sites. Moreover, positive selection sites for shell and P1 domains existed at the exterior surface of the structure that was distal to the predicted epitopes. However, the positive selection sites for Den Haag 2006b, New Orleans 2009, and Sydney 2012 variants were located on the exterior surface of the epitopes (Figure 5). These results indicate that HuNoV GII.4 has evolved with frequent alteration of antigenicity. In addition, the circulation of GII.4 may also be associated with mechanisms other than immune escape.
In addition, we estimated the fluctuation in the size of infectious populations in HuNoV GII.4. The population size increased around 1994–1996 and 2005–2006. Moreover, Den Haag 2006b, New Orleans 2009, and Sydney 2012 variants accumulated infectious populations in 2005–2007, 2004–2010, and 2006–2011, respectively (Figure 6). These results suggest that GII.4 has a capacity to repeatedly produce highly epidemic variants. A previous report showed evidence of higher affinity to a broad range of HBGA types in the Den Haag 2006b strains compared with that in other variants (de Rougemont et al., 2011). Thus, the prevalence of HuNoV GII.4 may be associated with multiple factors, such as the capacity to bind to host receptors and immune evasion.
Overall, this study utilized limited numbers of GII.4 VP1 sequences. For example, there were some biases among GII.4 variants in the amount of original data deposited in GenBank. Additionally, sequences with >98.9% identity were omitted. These biases may have affected the results of the bioinformatic analyses.
In conclusion, our findings show that the evolutionary pattern of GII.4 may be distinct among various variants. Moreover, our data may contribute to developing candidates of an efficient vaccine and prevalent prediction systems for HuNoV. In the near future, the accumulation of data from molecular epidemiological studies with continuous surveillance will be needed.
Author Contributions
TM, KK, and HK designed this study. TM, KN, YMa, NN, AR, TS, AY, MK, YMo, YS, and NS analyzed the data. TM, KN, YMa, and HK wrote the manuscript. KK and HK supervised this study. All authors read and approved the manuscript.
Funding
This work was partly supported by a commissioned project for Research on Emerging and Re-emerging Infectious Diseases from Japan Agency for Medical Research and Development, AMED.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles//10.3389/fmicb.2017.02399/full#supplementary-material
Footnotes
References
Añez, G., Grinev, A., Chancey, C., Ball, C., Akolkar, N., Land, K. J., et al. (2013). Evolutionary dynamics of West Nile virus in the United States, 1999-2011: phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl. Trop. Dis. 7:e2245. doi: 10.1371/journal.pntd.0002245
Allen, D. J., Adams, N. L., Aladin, F., Harris, J. P., and Brown, D. W. (2014). Emergence of the GII-4 Norovirus Sydney2012 Strain in England, Winter 2012-2013. PLoS ONE 9:e88978. doi: 10.1371/journal.pone.0088978
Allen, D. J., Gray, J. J., Gallimore, C. I., Xerry, J., and Iturriza-Gómara, M. (2008). Analysis of amino acid variation in the P2 domain of the GII-4 norovirus VP1 protein reveals putative variant-specific epitopes. PLoS ONE 3:e1485. doi: 10.1371/journal.pone.0001485
Allen, D. J., Trainor, E., Callaghan, A., O'Brien, S. J., Cunliffe, N. A., and Iturriza-Gómara, M. (2016). Early detection of epidemic GII-4 norovirus strains in UK and Malawi: role of surveillance of sporadic acute gastroenteritis in anticipating global epidemics. PLoS ONE 11:e0146972. doi: 10.1371/journal.pone.0146972
Andernach, I. E., Hunewald, O. E., and Muller, C. P. (2013). Bayesian inference of the evolution of HBV/E. PLoS ONE 8:e81690. doi: 10.1371/journal.pone.0081690
Bok, K., Abente, E. J., Realpe-Quintero, M., Mitra, T., Sosnovtsev, S. V., Kapikian, A. Z., et al. (2009). Evolutionary dynamics of GII.4 noroviruses over a 34-year period. J. Virol. 83, 11890–11901. doi: 10.1128/JVI.00864-09
Boon, D., Mahar, J. E., Abente, E. J., Kirkwood, C. D., Purcell, R. H., Kapikian, A. Z., et al. (2011). Comparative evolution of GII.3 and GII.4 norovirus over a 31-year period. J. Virol. 85, 8656–8666. doi: 10.1128/JVI.00472-11
Borley, D. W., Mahapatra, M., Paton, D. J., Esnouf, R. M., Stuart, D. I., and Fry, E. E. (2013). Evaluation and use of in-silico structure-based epitope prediction with foot-and-mouth disease virus. PLoS ONE 8:e61122. doi: 10.1371/journal.pone.0061122
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Bull, R. A., Eden, J. S., Rawlinson, W. D., and White, P. A. (2010). Rapid evolution of pandemic noroviruses of the GII.4 lineage. PLoS Pathog. 6:e1000831. doi: 10.1371/journal.ppat.1000831
Bull, R. A., Tu, E. T., McIver, C. J., Rawlinson, W. D., and White, P. A. (2006). Emergence of a new norovirus genotype II.4 variant associated with global outbreaks of gastroenteritis. J. Clin. Microbiol. 44, 327–333. doi: 10.1128/JCM.44.2.327-333.2006
Choudhuri, S. (2014). Bioinformatics for Beginners: Genes, Genomes, Molecular Evolution, Databases and Analytical Tools. Amsterdam: Elsevier/Academic Press.
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
de Rougemont, A., Ruvoen-Clouet, N., Simon, B., Estienney, M., Elie-Caille, C., Aho, S., et al. (2011). Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J. Virol. 85, 4057–4070. doi: 10.1128/JVI.02077-10
Debbink, K., Donaldson, E. F., Lindesmith, L. C., and Baric, R. S. (2012b). Genetic mapping of a highly variable norovirus GII.4 blockade epitope: potential role in escape from human herd immunity. J. Virol. 86, 1214–1226. doi: 10.1128/JVI.06189-11
Debbink, K., Lindesmith, L. C., Donaldson, E. F., and Baric, R. S. (2012a). Norovirus immunity and the great escape. PLoS Pathog. 8:e1002921. doi: 10.1371/journal.ppat.1002921
Debbink, K., Lindesmith, L. C., Donaldson, E. F., Costantini, V., Beltramello, M., Corti, D., et al. (2013). Emergence of new pandemic GII.4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 208, 1877–1887. doi: 10.1093/infdis/jit370
Delport, W., Poon, A. F., Frost, S. D., and Kosakovsky Pond, S. L. (2010). Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457. doi: 10.1093/bioinformatics/btq429
Doerflinger, S. Y., Weichert, S., Koromyslova, A., Chan, M., Schwerk, C., Adam, R., et al. (2017). Human norovirus evolution in a chronically infected host. mSphere 2, e00352-16. doi: 10.1128/mSphere.00352-16
Drummond, A. J., and Bouckaert, R. R. (2015). Bayesian Evolutionary Analysis with BEAST. Cambridge: Cambridge University.
Drummond, A. J., and Rambaut, A. (2007). BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Eden, J. S., Tanaka, M. M., Boni, M. F., Rawlinson, W. D., and White, P. A. (2013). Recombination within the pandemic norovirus GII.4 lineage. J. Virol. 87, 6270–6282. doi: 10.1128/JVI.03464-12
Fioretti, J. M., Bello, G., Rocha, M. S., Victoria, M., Leite, J. P., and Miagostovich, M. P. (2014). Temporal dynamics of norovirus GII.4 variants in Brazil between 2004 and 2012. PLoS ONE 9:e92988. doi: 10.1371/journal.pone.0092988
Fourment, M., and Gibbs, M. J. (2006). PATRISTIC: a program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol. Biol. 6:1. doi: 10.1186/1471-2148-6-1
Garaicoechea, L., Aguilar, A., Parra, G. I., Bok, M., Sosnovtsev, S. V., Canziani, G., et al. (2015). Llama nanoantibodies with therapeutic potential against human norovirus diarrhea. PLoS ONE 10:e0133665. doi: 10.1371/journal.pone.0133665
Giammanco, G. M., De Grazia, S., Terio, V., Lanave, G., Catella, C., Bonura, F., et al. (2014). Analysis of early strains of the norovirus pandemic variant GII.4 Sydney 2012 identifies mutations in adaptive sites of the capsid protein. Virology 450–451, 355–358. doi: 10.1016/j.virol.2013.12.007
Green, K. Y. (2013). “Caliciviridae: the noroviruses,” in Fields Virology, 6th Edn., eds D. M. Knipe and P. M. Howley (Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins), 582–608.
Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723. doi: 10.1002/elps.1150181505
Guindon, S., and Gascuel, O. (2003). A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Hall, M. D., Woolhouse, M. E., and Rambaut, A. (2016). The effects of sampling strategy on the quality of reconstruction of viral population dynamics using Bayesian skyline family coalescent methods: a simulation study. Virus Evol. 2:vew003. doi: 10.1093/ve/vew003
Hoa-Tran, T. N., Trainor, E., Nakagomi, T., Cunliffe, N. A., and Nakagomi, O. (2013). Molecular epidemiology of noroviruses associated with acute sporadic gastroenteritis in children: global distribution of genogroups, genotypes and GII.4 variants. J. Clin. Virol. 56, 185–193. doi: 10.1016/j.jcv.2012.11.011
Kanda, Y. (2013). Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 48, 452–458. doi: 10.1038/bmt.2012.244
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kimura, H., Saitoh, M., Kobayashi, M., Ishii, H., Saraya, T., Kurai, D., et al. (2015). Molecular evolution of haemagglutinin (H) gene in measles virus. Sci. Rep. 5:11648. doi: 10.1038/srep11648
Kobayashi, M., Matsushima, Y., Motoya, T., Sakon, N., Shigemoto, N., Okamoto-Nakagawa, R., et al. (2016). Molecular ecolution of the capsid gene in human norovirus genogroupII. Sci. Rep. 6:29400. doi: 10.1038/srep29400
Kobayashi, M., Yoshizumi, S., Kogawa, S., Takahashi, T., Ueki, Y., Shinohara, M., et al. (2015). Molecular evolution of the capsid gene in norovirus genogroup I. Sci. Rep. 5:13806. doi: 10.1038/srep13806
Kringelum, J. V., Lundegaard, C., Lund, O., and Nielsen, M. (2012). Reliable B cell epitope predictions: Impacts of method development and improved benchmarking. PLoS Comput. Biol. 8:e1002829. doi: 10.1371/journal.pcbi.1002829
Kroneman, A., Vennema, H., Deforche, K., v d Avoort, H., Pe-aranda, S., Oberste, M. S., et al. (2011). An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 51, 121–125. doi: 10.1016/j.jcv.2011.03.006
Kryazhimskiy, S., and Plotkin, J. B. (2008). The population genetics of dN/dS. PLoS Genet. 4:e1000304. doi: 10.1371/journal.pgen.1000304
Kumazaki, M., and Usuku, S. (2015). Genetic analysis of norovirus GII.4 variant strains detected in outbreaks of gastroenteritis in Yokohama, Japan, from the 2006-2007 to the 2013-2014 seasons. PLoS ONE 10:e0142568. doi: 10.1371/journal.pone.0142568
Lam, T. T., Zhu, H., Smith, D. K., Guan, Y., Holmes, E. C., and Pybus, O. G. (2012). The recombinant origin of emerging human norovirus GII.4/2008: intra-genotypic exchange of the capsid P2 domain. J. Gen. Virol. 93, 817–822. doi: 10.1099/vir.0.039057-0
Liang, S., Liu, S., Zhang, C., and Zhou, Y. (2007). A simple reference state makes a significant improvement in near-native selections from structurally refined docking decoys. Proteins 69, 244–253. doi: 10.1002/prot.21498
Liang, S., Zheng, D., Standley, D. M., Yao, B., Zacharias, M., and Zhang, C. (2010). EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results. BMC Bioinformatics 11:381. doi: 10.1186/1471-2105-11-381
Lindesmith, L. C., Beltramello, M., Donaldson, E. F., Corti, D., Swanstrom, J., Debbink, K., et al. (2012a). Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 8:e1002705. doi: 10.1371/journal.ppat.1002705
Lindesmith, L. C., Debbink, K., Swanstrom, J., Vinjé, J., Costantini, V., Baric, R. S., et al. (2012b). Monoclonal antibody-based antigenic mapping of norovirus GII.4-2002. J. Virol. 86, 873–883. doi: 10.1128/JVI.06200-11
Lindesmith, L. C., Donaldson, E. F., Beltramello, M., Pintus, S., Corti, D., Swanstrom, J., et al. (2014). Particle conformation regulates antibody access to a conserved, G. I. I.4 norovirus blockade epitope. J Virol. 88, 8826–8842. doi: 10.1128/JVI.01192-14
Lindesmith, L. C., Donaldson, E. F., Lobue, A. D., Cannon, J. L., Zheng, D. P., Vinje, J., et al. (2008). Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 5:e31. doi: 10.1371/journal.pmed.0050031
Lole, K. S., Bollinger, R. C., Paranjape, R. S., Gadkari, D., Kulkarni, S. S., Novak, N. G., et al. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160.
Lovell, S. C., Davis, I. W., Arendall, W. B. III., de Bakker, P. I., Word, J. M., Prisant, M. G., et al. (2003). Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins 50, 437–450. doi: 10.1002/prot.10286
Lu, J., Fang, L., Zheng, H., Lao, J., Yang, F., Sun, L., et al. (2016). The Evolution and Transmission of Epidemic, G. I. I.17 Noroviruses. J. Infect. Dis. 214, 556–564. doi: 10.1093/infdis/jiw208
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003. doi: 10.1093/ve/vev003
Mathijs, E., Denayer, S., Palmeira, L., Botteldoorn, N., Scipioni, A., Vanderplasschen, A., et al. (2011). Novel norovirus recombinants and of GII.4 sub-lineages associated with outbreaks between 2006 and 2010 in Belgium. Virol. J. 8:310. doi: 10.1186/1743-422X-8-310
Mizukoshi, F., Nagasawa, K., Doan, Y. H., Haga, K., Yoshizumi, S., Ueki, Y., et al. (2017). Molecular evolution of the RNA-dependent RNA polymerase and capsid genes of human norovirus genotype GII.2 in Japan during 2004-2015. Front. Microbiol. 8:705. doi: 10.3389/fmicb.2017.00705
Motomura, K., Oka, T., Yokoyama, M., Nakamura, H., Mori, H., Ode, H., et al. (2008). Identification of monomorphic and divergent haplotypes in the 2006-2007 norovirus GII/4 epidemic population by genomewide tracing of evolutionary history. J. Virol. 82, 11247–11262. doi: 10.1128/JVI.00897-08
Parra, G. I., Abente, E. J., Sandoval-Jaime, C., Sosnovtsev, S. V., Bok, K., and Green, K. Y. (2012). Multiple antigenic sites are involved in blocking the interaction of GII.4 norovirus capsid with ABH histo-blood group antigens. J. Virol. 86, 7414–7426. doi: 10.1128/JVI.06729-11
Parra, G. I., Squires, R. B., Karangwa, C. K., Johnson, J. A., Lepore, C. J., Sosnovtsev, S. V., et al. (2017). Static and evolving norovirus genotypes: implications for epidemiology and immunity. PLoS Pathog. 13:e1006136. doi: 10.1371/journal.ppat.1006136
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Pond, S. L., and Frost, S. D. (2005). Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21, 2531–2533. doi: 10.1093/bioinformatics/bti320
Potocnakova, L., Bhide, M., and Pulzova, L. B. (2016). An introduction to B-cell epitope mapping and in silico epitope prediction. J. Immunol. Res. 2016:6760830. doi: 10.1155/2016/6760830
Qiao, N., Wang, X. Y., and Liu, L. (2016). Temporal evolutionary dynamics of norovirus GII.4 variants in China between 2004 and 2015. PLoS ONE11:e0163166. doi: 10.1371/journal.pone.0163166
Robilotti, E., Deresinski, S., and Pinsky, B. A. (2015). Norovirus. Clin. Microbiol. Rev. 28, 134–164. doi: 10.1128/CMR.00075-14
Sato, H., Yokoyama, M., Nakamura, H., Oka, T., Katayama, K., Takeda, N., et al. (2017). Evolutionary constraints on the norovirus pandemic variant GII.4_2006b over the five-year persistence in Japan. Front. Microbiol. 8:410. doi: 10.3389/fmicb.2017.00410
Siebenga, J. J., Lemey, P., Kosakovsky, S. L., Rambaut, A., Vennema, H., and Koopmans, M. (2010). Phylodynamic reconstruction reveals norovirus GII.4 epidemic expansions and their molecular determinants. PLoS Pathog. 6:e1000884. doi: 10.1371/journal.ppat.1000884
Siebenga, J. J., Vennema, H., Zheng, D. P., Vinjé, J., Lee, B. E., Pang, X. L., et al. (2009). Norovirus illness is a global problem: emergence and spread of norovirus GII.4 variants, 2001-2007. J. Infect. Dis. 200, 802–812. doi: 10.1086/605127
Siqueira, J. A. M., Bandeira, R. D. S., Oliveira, D. S., Dos Santos, L. F. P., and Gabbay, Y. B. (2017). Genotype diversity and molecular evolution of noroviruses: a 30-year (1982-2011) comprehensive study with children from Northern Brazil. PLoS ONE 12:e0178909. doi: 10.1371/journal.pone.0178909
Standley, D. M., Toh, H., and Nakamura, H. (2007). ASH structure alignment package: sensitivity and selectivity in domain classification. BMC Bioinformatics 8:116. doi: 10.1186/1471-2105-8-116
Suchard, M. A., Weiss, R. E., and Sinsheimer, J. S. (2001). Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Biol. Evol. 18, 1001–1013. doi: 10.1093/oxfordjournals.molbev.a003872
Sun, J., Wu, D., Zhong, H., Guan, D., Zhang, H., Tan, Q., et al. (2017). Returning ex-patriot Chinese to Guangdong, China, increase the risk for local transmission of Zika virus. J. Infect. S0163–S4453, 30231–30231. doi: 10.1016/j.jinf.2017.07.001
Suzuki, Y. (2004). Negative selection on neutralization epitopes of poliovirus surface proteins: implications for prediction of candidate epitopes for immunization. Gene 328, 127–133. doi: 10.1016/j.gene.2003.11.020
Suzuki, Y., Doan, Y. H., Kimura, H., Shinomiya, H., Shirabe, K., and Katayama, K. (2016). Predicting genotype compositions in norovirus seasons in Japan. Microbiol. Immunol. 60, 418–426. doi: 10.1111/1348-0421.12384
Sweredoski, M. J., and Baldi, P. (2008). PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics 24, 1459–1460. doi: 10.1093/bioinformatics/btn199
Thongprachum, A., Khamrin, P., Maneekarn, N., Hayakawa, S., and Ushijima, H. (2016). Epidemiology of gastroenteritis viruses in Japan: prevalence, seasonality, and outbreak. J. Med. Virol. 88, 551–570. doi: 10.1002/jmv.24387
Tohma, K., Lepore, C. J., Ford-Siltz, L. A., and Parra, G. I. (2017). Phylogenetic analyses suggest that factors other than the capsid protein play a role in the epidemic potential of GII.2 Norovirus. MSphere 2:e0018717. doi: 10.1128/mSphereDirect.00187-17
Tu, E. T., Nguyen, T., Lee, P., Bull, R. A., Musto, J., Hansman, G., et al. (2007). Norovirus, G. I. I.4 strains and outbreaks, Australia. Emerg. Infect. Dis. 13, 1128–1130. doi: 10.3201/eid1307.060999
Umeda, Y. (2007). Norovirus Infection (in Japanese). Jibiinkoukarinsyou 100, 497–504. doi: 10.1097/INF.0b013e3181d742bf
van Gunsteren, W. F., Billeter, S. R., Eising, A. A., Hünenberger, P. H., Krüger, P., Mark, A. E., et al. (1996). Biomolecular Simulation: The GROMOS96 Manual and User Guide. Zürich: Vdf Hochschulverlag, A. G., an der ETH Zürich Press.
Vega, E., Barclay, L., Gregoricus, N., Shirley, S. H., Lee, D., and Vinjé, J. (2014a). Genotypic and epidemiologic trends of norovirus outbreaks in the United States, 2009 to 2013. J. Clin. Microbiol. 52, 147–155. doi: 10.1128/JCM.02680-13
Vega, E., Donaldson, E., Huynh, J., Barclay, L., Lopman, B., Baric, R., et al. (2014b). RNA populations in immunocompromised patients as reservoirs for novel norovirus variants. J. Virol. 88, 14184–14196. doi: 10.1128/JVI.02494-14
Vijaykrishna, D., Holmes, E. C., Joseph, U., Fourment, M., Su, Y. C., Halpin, R., et al. (2015). The contrasting phylodynamics of human influenza B viruses. Elife 4:e05055. doi: 10.7554/eLife.05055
Vinjé, J. (2015). Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 53, 373–381. doi: 10.1128/JCM.01535-14
Webb, B., and Sali, A. (2014). Protein structure modeling with MODELLER. Methods Mol. Biol. 1137, 1–15. doi: 10.1007/978-1-4939-0366-5_1
Wei, K., and Li, Y. (2017). Global evolutionary history and spatio-temporal dynamics of dengue virus type 2. Sci. Rep. 7:45505. doi: 10.1038/srep45505
White, P. A. (2014). Evolution of norovirus. Clin. Microbiol. Infect. 20, 741–745. doi: 10.1111/1469-0691.12746
Yang, Y., Xia, M., Tan, M., Huang, P., Zhong, W., Pang, X. L., et al. (2010). Genetic and phenotypic characterization of GII-4 noroviruses that circulated during 1987 to 2008. J. Virol. 84, 9595–9607. doi: 10.1128/JVI.02614-09
Yao, B., Zheng, D., Liang, S., and Zhang, C. (2013). Conformational B-cell epitope prediction on antigen protein structures: a review of current algorithms and comparison with common binding site prediction methods. PLoS ONE 8:e62249. doi: 10.1371/journal.pone.0062249
Keywords: bioinformatics, GII.4, molecular evolution, Norovirus, VP1
Citation: Motoya T, Nagasawa K, Matsushima Y, Nagata N, Ryo A, Sekizuka T, Yamashita A, Kuroda M, Morita Y, Suzuki Y, Sasaki N, Katayama K and Kimura H (2017) Molecular Evolution of the VP1 Gene in Human Norovirus GII.4 Variants in 1974–2015. Front. Microbiol. 8:2399. doi: 10.3389/fmicb.2017.02399
Received: 18 September 2017; Accepted: 20 November 2017;
Published: 05 December 2017.
Edited by:
Akio Adachi, Tokushima University, JapanReviewed by:
Chang-jun Bao, Jiangsu Provincial Center for Disease Control and Prevention, ChinaMatthew D. Moore, University of Massachusetts Amherst, United States
Copyright © 2017 Motoya, Nagasawa, Matsushima, Nagata, Ryo, Sekizuka, Yamashita, Kuroda, Morita, Suzuki, Sasaki, Katayama and Kimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhiko Katayama, a2F0YXlhbWFAbGlzY2kua2l0YXNhdG8tdS5hYy5qcA==
Hirokazu Kimura, aC1raW11cmFAcGF6LmFjLmpw