
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Pharmacol., 08 February 2018
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 9 - 2018 | https://doi.org/10.3389/fphar.2018.00083
 Won-Hyong Chung1†
Won-Hyong Chung1† Jisu Kang1,2†
Jisu Kang1,2† Mi Young Lim1
Mi Young Lim1 Tae-joong Lim3
Tae-joong Lim3 Sanghyun Lim3
Sanghyun Lim3 Seong Woon Roh4*
Seong Woon Roh4* Young-Do Nam1,2*
Young-Do Nam1,2*Our body has natural defense systems to protect against potentially harmful microbes, including the physical and chemical barriers of the intestinal epithelium (Corfield et al., 2000). The physical barrier of the intestinal epithelium protects the host against pathogenic microbes (Anderson et al., 1993), and the intestinal mucosa coated with mucus excretes pathogens from the intestinal tract (Corfield et al., 2000).
The gut microbiota also controls the number of enteric pathogens by producing anti-microbial molecules, such as proteinaceous bacteriocins (Kamada et al., 2013), and by inhibiting the proliferation of pathogens by generating organic acids-short chain fatty acids (SCFAs) that lower the local pH (Shin et al., 2002), and by regulating the expression of virulence genes in certain pathogenic bacteria, such as Salmonella (Gantois et al., 2006). Moreover, the gut microbiota indirectly prevents pathogenic infection by enhancing the functions of the host mucosal barrier and innate immune system (Kobayashi et al., 2005). Therefore, in a healthy gastrointestinal tract, host cells and the gut microbiota synergistically protect the host from pathogenic infections.
The human intestinal tract harbors a dense population of resident microbiota, consisting of bacteria, archaea, viruses, and fungi. Host genetics, diet, and environmental insults can affect the gut microbial composition (Human Microbiome Project Consortium, 2012), and disruption of the gut microbiota can lead to invasion and overgrowth of certain pathogenic bacteria, such as virulent Escherichia coli, Salmonella enterica serovar Typhimurium, and Clostridium difficile (Bohnhoff et al., 1954; Rupnik et al., 2009; Ayres et al., 2012). In particular, the use of antibiotics can lead to a temporary or long-term reduction of bio-diversity, and this change in the gut microbiota, called dysbiosis, increases susceptibility to microbial infection and the proliferation of antibiotic-resistant strains (Vangay et al., 2015).
On the other hand, probiotics have been also considered for the treatment or prevention of diverse infectious diseases, and previous studies have reported the successful treatment of infection by pathogenic bacteria, including Escherichia, Klebsiella, Shigella, Enterobacter, Pseudomonas, Clostridium, Helicobacter, etc., (Kabir et al., 1997; Brashears et al., 1998; Forestier et al., 2001; Ogawa et al., 2001). Therefore, the use of probiotics could be an alternative strategy for the treatment or prevention of infectious disease that avoids the gut microbiota dysbiosis associated with antibiotic treatment.
In our previous study, we reported that Lactobacillus acidophilus strain LA1 (11869BP), an isolate from a dairy product, had preventive effects against Salmonella infection (Kim et al., 2013). While the anti-pathogenic effects of various probiotics have been widely demonstrated, the exact mechanism of action is not well understood. In addition, our genomic knowledge of L. acidophilus strains is insufficient because only five complete L. acidophilus genomes including LA1 are currently available in NCBI database (https://www.ncbi.nlm.nih.gov/genome/). Therefore, to gain better insight into its probiotic and Salmonella infection-preventing effects, we sequenced and analyzed the genome of L. acidophilus LA1. The availability of this genomic information will allow for further in-depth analysis and a better understanding of the probiotic functions of L. acidophilus strains for the prevention of pathogenic infections.
In our previous study, we isolated L. acidophilus LA1 from a fermented dairy product in Korea (Kim et al., 2013). To analyze the genomic content of strain LA1, we cultivated the bacteria in MRS medium (Difco, USA) at 37°C for 18 h. Genomic DNA was extracted and purified using the QIAamp DNA Mini Kit (Qiagen, Germany). The extracted genomic DNA was quantified with a NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific, USA) and Qubit 2.0 fluorometer (Life Technologies, USA). The genome of L. acidophilus LA1 was sequenced with the PacBio RS II (Menlo Park, USA) sequencing platform.
Genome assembly was performed using HGAP 3.0 (Chin et al., 2013), with default options. The start position of the chromosome was determined by the location of the gene encoding the chromosomal replication initiation protein, dnaA, as well as the GC skew pattern. Annotation of this genome was carried out with the NCBI Prokaryotic Genome Annotation Pipeline (PGAAP) (Tatusova et al., 2016). From the ASN.1-formatted annotation file, the protein-coding genes and rRNA genes were extracted using the NCBI toolbox (http://www.ncbi.nlm.nih.gov/IEB/ToolBox). Coding genes were assigned to COG categories using BLASTP and the COG database (Galperin et al., 2015), with an e-value cutoff of 1e-3.
Nine complete genome sequences that belong to L. acidophilus group were selected for phylogenetic and comparative analysis: L. acidophilus NCFM (assembly accession: GCF_000011985.1), L. acidophilus La-14 (GCF_000389675.2), L. acidophilus FSI4 (GCF_000934625.1), L. gallinarum HFD4 (GCF_001314245.2), L. helveticus CNRZ32 (GCF_000422165.1), L. crispatus ST1 (GCF_000091765.1), L. kefiranofaciens ZW3 (GCF_000214785.1), L. amylovorus GRL1118 (GCF_000194115.1), and L. acetotolerans NBRC 13120 (GCF_001042405.1). L. acetotolerans NBRC 13120 was used as the out-group in the phylogenetic analysis. The three L. acidophilus genomes (NCFM, La-14, and FSI4) and the present sequenced genome, L. acidophilus LA1, were used for the comparative analysis.
A phylogenetic tree based on the 16S gene sequences was constructed by the maximum likelihood method based on the Tamura-Nei model (Tamura and Nei, 1993). Orthologous average nucleotide identity (OrthoANI) between genome sequences was computed (Lee et al., 2016). To obtain genomic distance, the OrthoANI values were converted to distance values with the following formula: distance = 1 – (OrthoANI/100). Evolutionary distance was computed from the genome-distance matrix using the neighbor-joining method (Saitou and Nei, 1987). The tree is drawn to scale, with branch lengths presented in the same units as those for the evolutionary distances used to infer the phylogenetic tree. The phylogenetic tree was generated using MEGA6 (Tamura et al., 2013).
A pan-genomic study was performed to investigate the functional conservation in the sequenced genome by using GET_HOMOLOGOUS (Contreras-Moreira and Vinuesa, 2013). COG triangles was used as the clustering method, and a minimum of 50% amino acid identity and 50% coverage were used for the clustering threshold.
Carbohydrate active enzymes (CAZymes) were searched by using the CAZymes analysis tool kit (Park et al., 2010; Lombard et al., 2014). Different CAZy families were used to identify the key enzymes related to polysaccharide degradation: glycoside hydrolases (GHs), glycosyl transferases (GTs), carbohydrate esterases (CEs), auxiliary activities (AAs), and carbohydrate-binding modules (CBMs).
Bacteriocins were predicted with BAGEL 3 which is a web service for bacteriocin searches that uses a bacteriocin mining tool (Van Heel et al., 2013). BAGEL 3 is based on three types of databases: modified bacteriocins, unmodified bacteriocins, and post-translationally modified peptides.
The CRISPR regions were identified with a CRISPR on-line detection tool, CRISPR finder (Grissa et al., 2007). The sequences of dicers and spacers were downloaded from the analysis server after the finding computation. Aligned pairs of spacers in L. acidophilus NCFM and LA1 were obtained by running BLASTN without the dust masking option. The numerical order of spacers in LA1 followed the order in NCFM. To find counterparts for the spacers, a BLASTN search was performed against the NCBI NT and NR database (downloaded on 16 May, 2017), with no dust option and an e-value of 0.1. The search result was filtered with 90% identity and 80% coverage.
Prophage insert regions were detected with an on-line phage search tool, PHASTER (Arndt et al., 2016). The genomic structure of the inserted prophage and the associated genes were obtained from the computation result. The detected ORFs were annotated based on the highest hit in a BLAST search against the annotated bacterial genome database in the PHASTER system.
We obtained the complete genome sequence of L. acidophilus LA1 using SMRT sequencing, which showed that the genome is composed of a 1.99-Mbp circular chromosome with 34.7% G+C content (Table 1). A total of 1,953 genes were identified in the LA1 genome, including 1,844 protein-coding genes, 76 RNA genes, and 33 pseudo genes. Four sets of ribosomal RNA genes, including 5S, 16S, and 23S genes, were also found. Other RNA genes, including 61 tRNA genes and three non-coding RNA (or ncRNA) genes were found. We also found ncRNA-coding sequences in L. acidophilus NCFM, even though they were not reported in the genome annotation. We hypothesized that these ncRNAs were absent from the annotation because of the difference in the annotation methods, since these types of ncRNA genes were recently introduced in the annotation system (PGAAP 3.1 and above). These genomic features of LA1 are shown in Figure 1A.
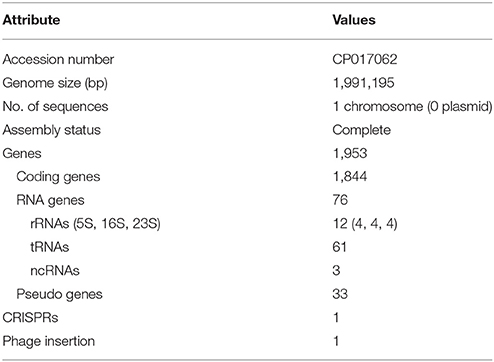
Table 1. Genome features of Lactobacillus acidophilus LA1.
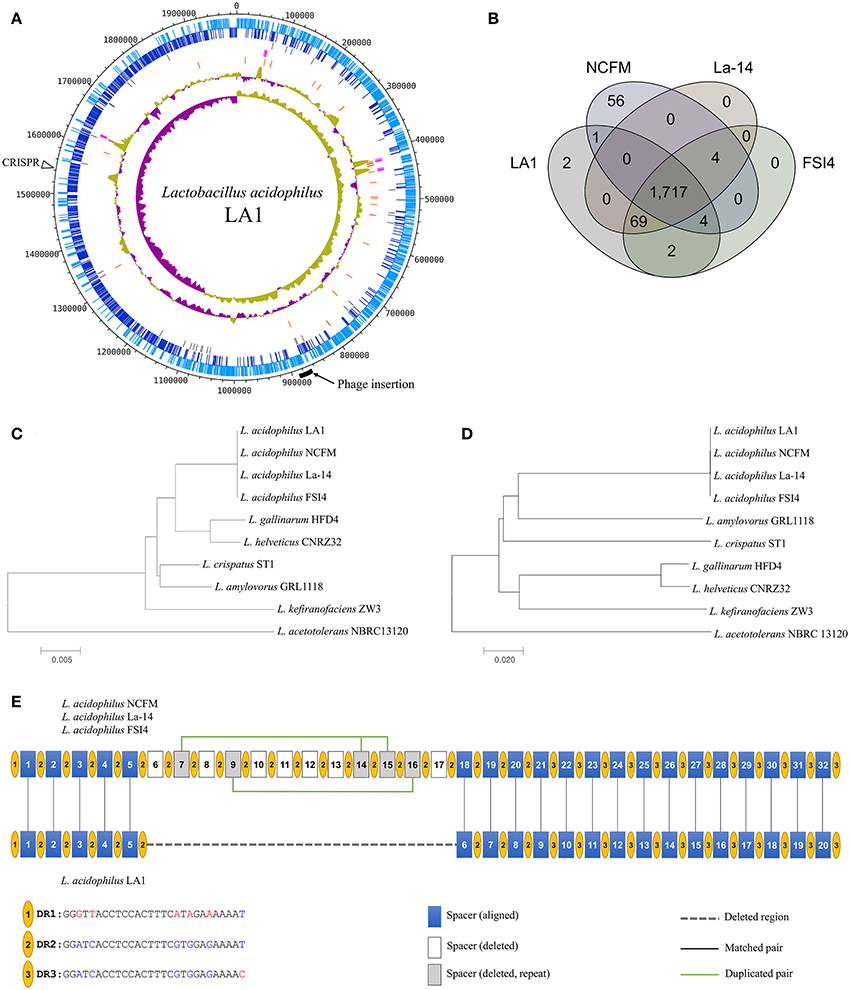
Figure 1. (A) Circular map of the Lactobacillus acidophilus LA1 genome. Tracks (from outset): forward-strand coding CDS, reverse-strand coding CDS, pseudogenes, rRNAs, tRNAs, G+C content, and GC skew. (B) Venn diagram of the shared gene clusters among the four L. acidophilus genomes. (C) Phylogenetic tree of the L. acidophilus groups based on 16S rRNA genes and (D) phylogenetic tree based on genome-wide identities (ANI). (E) Comparison of the CRISPR structure in LA1 and the other L. acidophilus genomes. Yellow circles indicate the dicers. The blue, white, and gray boxes represent the spacers that are aligned in LA1, the spacers deleted in LA1, and the deleted repeat spacers in LA1, respectively. A dashed line fills the deleted region in LA1. Vertical lines indicate aligned pairs of spacers. Green lines link the repeat spacers.
Functional classifications of the protein-coding genes of LA1 were categorized into 26 COG functional codes (Supplementary Figure S1, Supplementary Table S2). Of the 1,844 protein-coding genes in LA1, 1,553 were assigned to COG, and 291 genes were not assigned. Because 135 genes were assigned multiple codes, the number of COG codes was 1,705. The percentage of proteins with unknown function, including “General function prediction only (R),” “Function unknown (S),” and “Not assigned (–),” was 29.3%. The four analyzed L. acidophilus strains, including LA1, have nearly identical numbers of functional genes, and they showed >99% genomic identity (Supplementary Table S3).
Lactobacillus acidophilus strains including LA1 showed high conservation each other by comparison of the L. acidophilus group (Bull et al., 2013, 2014), a subgroup of Lactobacillus. A comparative study of the 10 complete genomes in the L. acidophilus group was performed to confirm the genomic distance based on the 16S rRNA genes and average nucleotide identity (ANI). The genomic similarities ranged from 75 to 99.9%, and the similarities based on the 16S rRNA genes ranged from 94 to 100% (Supplementary Table S3). Four L. acidophilus strains were located on the same node of the both 16S rRNA phylogenetic and ANI trees (Figures 1C,D). Those genomes had identical 16S rRNA sequences and very similar ANI values (≥99%); therefore, the L. acidophilus genomes were difficult to distinguish by sequence similarity. We found different phylogenetic relationships between the 16S rRNA sequence-based and ANI value-based phylogenetic trees. L. gallinarum and L. helveticus were the closest to L. acidophilus on the 16S rRNA phylogenetic tree (Figure 1C). However, L. amylovorus was the closet to L. acidophilus on the ANI phylogenetic tree (Figure 1D). Phylogenetic studies using ANI better reflect the functional relationship between strains than studies based on 16S rRNA sequences, as shown by the statistics for COG categories. L. gallinarum and L. helveticus (marked as triangles in Supplementary Figure S1) showed distinguishable profiles when compared to the L. acidophilus genomes, and there was a greater than five-fold difference in “Mobilome: prophages, transposons (X).”
To obtain better insight into the specific features of LA1, we compared LA1 to nine complete genomes in the L. acidophilus group (Supplementary Table S1). A distinguishing feature of the four L. acidophilus genomes (LA1, NCFM, La-14, and FSI4) was the lower G+C content (2–3%) compared to the other species. Gene clustering was performed to determine the differences between these 10 genomes based on COG functional annotation (Supplementary Table S4). The genes were categorized into 3,810 functional clusters using the pan-genome analysis program, GET_HOMOLOGOUS. The four L. acidophilus genomes were compared by orthologous protein clustering to determine the LA1-specific genes (Figure 1B). Among the total 1,955 gene clusters, we found 1,717 core gene clusters. Only two gene clusters were identified as LA1 strain specific. Even though there were high similarities between the L. acidophilus genomes, four gene clusters were absent in the LA1 genome. Among the other L. acidophilus strains, 125 gene clusters were unique; 56 clusters belonged to NCFM, and the 69 clusters belonged to the other strains.
Carbohydrate active enzymes (CAZymes) analysis showed that LA1 contains 344 genes in the five CAZymes gene families (Supplementary Table S5); 148 glycoside hydrolase (GH) genes, 130 glycosyl transferase (GT) genes, 25 carbohydrate esterase (CE) genes, 11 auxiliary activity (AA) genes, and 30 CBMs. These numbers of carbohydrate-active enzymes were relatively larger than those in Lactobacillus plantarum, which is an important probiotic species. For example, L. plantarum KLDS1.0391 contains only 34 GHs, 23 GTs, 14 CEs, 2 AAs, and 21 CPMs (Jia et al., 2017). GTs that catalyze the transfer of sugars from activated donor molecules to specific acceptors are important for the formation of surface structures recognized by host immune systems (Mazmanian et al., 2008). Therefore, the six-fold larger number of GT genes in L. acidophilus LA1 compared to L. plantarum KLDS1.0391 suggests the probiotic potential of LA1, especially for immune stimulation and pathogen defense.
Lactobacillus acidophilus genomes have three area(s) of interest (AOI) that include one class II bacteriocin and two class III bacteriocins in common: AOI_1 (bacteriocin III, Enterolysin A), AOI_2 (bacteriocin III, Helventicin J), and AOI_3 (bacteriocin II, Acidocin J) (Supplementary Table S6). Enterolysin A is a cell wall-degrading bacteriocin (Nilsen et al., 2003) with broad-spectrum antibiotic activity that acts by cleaving stem peptide bonds and lysing peptidoglycan in cell walls (Riley and Chavan, 2007; Khan et al., 2013). Helveticin J is antimicrobial protein that was first characterized in L. helveticus 481 (Joerger and Klaenhammer, 1986). However, a recent report revealed that helveticin is also found in other Lactobacillus species (Collins et al., 2017). Acidocin J is a heat-stable class II bacteriocin, which is mainly found in L. acidophilus (Riley and Chavan, 2007; Yang et al., 2014). In this study, Enterolysin A and Helventicin J were found in all Lactobacillus spp. whereas Acidocin J was only encoded in L. acidophilus genomes. In addition, all L. acidophilus genomes, except for L. acidophilus NCFM, have the same start position for each AOI.
We found a large deletion in the CRISPR region in LA1 when compared to NCFM, La-14, and FSI4. All the genomes, except LA1, have the same structure in the CRISPR region, with three types of 28-bp dicer sequences and 32 spacer sequences, whereas LA1 has only 20 spacer sequences (Figure 1E). The clean-cut of 12 spacers (6–17) was observed in this region. The CRISPR region has three types of dicers, and the deletion in LA1 was located in the middle of the array of type 2 dicers (DR2). The deleted spacers in LA1 include five duplicated spacers of two types, one type consists of spacers 7, 14, and 15, and the other consists of spacers 9 and 16. Considering the high conservation of the CRISPR region, this deletion may be a very recent event. Because one of the major roles of the CRISPR region is defense against foreign DNA (Yin et al., 2013), this shortage of spacers to detect unwanted nucleic acids in LA1 may lower its immune power. The structural variation in the CRISPR region can be found even within the same species, and could be used to interpret the evolutional history of a strain because the CRISPR region may reflect the phages that have tried to invade the cell. Therefore, the CRISPR region has been recently proposed for the identification of industrially important microorganisms (Barrangou and Horvath, 2012). Therefore, this difference in the CRISPR region can be used as a marker for distinguishing LA1 from other L. acidophilus strains.
A 26-kbp prophage insert was identified in the LA1 genome at chromosomal position 863,940–890,001 (Supplementary Table S7). The position and size of the insert region were similar to that in other L. acidophilus genomes, except for the number of proteins encoded in that region. Despite their high similarity, the number of genes varies from 10 to 15 (Supplementary Table S8). LA1 contains the smallest number of genes, FSI4 has the largest number of genes, and NCFM and La-14 have the same number of genes (12). Genes required for phage invasion, including attL (ORF1), phage integrase (ORF6), and attR (ORF15), were found in the inserted region. LA1 showed the highest rate of decay in the inserted phage genome, and five genes, ORF2–5 and ORF9, were degraded when compared to FSI4.
Here, we sequenced and analyzed the complete genome of a probiotic strain with the potential to prevent Salmonella infection, L. acidophilus LA1. In the current study, we demonstrated that the LA1 genome contains the genes required for the biosynthesis of the three bacteriocins, Enterolysin A, Helveticin J, and Acidocin J. In addition, the six-fold larger number of GT genes in LA1 compared to the number in one of the most well-described probiotic strains, L. plantarum KLDS1.0391, suggests the probiotic potential of LA1, especially in terms of immune stimulation and fortification of pathogen defense. Interestingly, one of the most unique features of LA1 when compared to the other currently available L. acidophilus genome sequences is the large, clean-cut of 12 spacers from the CRISPR region and the difference in the historical record of phage infection, which can be used as a genetic marker for identification of this industrially and medically important probiotic strain to distinguish it from genetically related L. acidophilus strains. Considering the possible use of L. acidophilus LA1 as beneficial probiotic, the availability of the LA1 genome is an important step for understanding its evolution and probiotic function against pathogenic bacteria.
The Lactobacillus acidophilus LA1 genome sequencing project has been deposited into GenBank under accession number CP017062. This strain has been deposited in the Korean Collection for Type Cultures (deposit ID: KCTC 11906BP).
Y-DN and SR: Designed and coordinated all the experiments; T-jL and SL: Performed the bacterial cultivation, and DNA extraction and purification; W-HC, JK, and ML: Performed the genome analysis; W-HC, JK, and Y-DN: prepared the manuscript; All authors have read and approved the manuscript.
This research was supported by the Main Research Program (E0170602-01) of the Korea Food Research Institute (KFRI) and the World Institute of Kimchi (KE1702-2) funded by the Ministry of Science, ICT, & Future Planning.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00083/full#supplementary-material
Anderson, J. M., Balda, M. S., and Fanning, A. S. (1993). The structure and regulation of tight junctions. Curr. Opin. Cell Biol. 5, 772–778. doi: 10.1016/0955-0674(93)90024-K
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Ayres, J. S., Trinidad, N. J., and Vance, R. E. (2012). Lethal inflammasome activation by a multi-drug resistant pathobiont upon antibiotic disruption of the microbiota. Nat. Med. 18, 799–806. doi: 10.1038/nm.2729
Barrangou, R., and Horvath, P. (2012). CRISPR: new horizons in phage resistance and strain identification. Annu. Rev. Food Sci. Technol. 3, 143–162. doi: 10.1146/annurev-food-022811-101134
Bohnhoff, M., Drake, B. L., and Miller, C. P. (1954). Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc. Soc. Exp. Biol. Med. 86, 132–137. doi: 10.3181/00379727-86-21030
Brashears, M. M., Reilly, S. S., and Gilliland, S. E. (1998). Antagonistic action of cells of Lactobacillus lactis toward Escherichia coli O157:H7 on refrigerated raw chicken meat. J. Food Prot. 61, 166–170. doi: 10.4315/0362-028X-61.2.166
Bull, M., Plummer, S., Marchesi, J., and Mahenthiralingam, E. (2013). The life history of Lactobacillus acidophilus as a probiotic: a tale of revisionary taxonomy, misidentification and commercial success. FEMS Microbiol. Lett. 349, 77–87. doi: 10.1111/1574-6968.12293
Bull, M. J., Jolley, K. A., Bray, J. E., Aerts, M., Vandamme, P., Maiden, M. C., et al. (2014). The domestication of the probiotic bacterium Lactobacillus acidophilus. Sci. Rep. 4:7202. doi: 10.1038/srep07202
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Collins, F. W., O'connor, P. M., O'sullivan, O., Gómez-Sala, B., Rea, M. C., Hill, C., et al. (2017). Bacteriocin Gene-Trait matching across the complete Lactobacillus Pan-genome. Sci. Rep. 7:3481. doi: 10.1038/s41598-017-03339-y
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Corfield, A., Myerscough, N., Longman, R., Sylvester, P., Arul, S., and Pignatelli, M. (2000). Mucins and mucosal protection in the gastrointestinal tract: new prospects for mucins in the pathology of gastrointestinal disease. Gut 47, 589–594. doi: 10.1136/gut.47.4.589
Forestier, C., De Champs, C., Vatoux, C., and Joly, B. (2001). Probiotic activities of Lactobacillus casei rhamnosus: in vitro adherence to intestinal cells and antimicrobial properties. Res. Microbiol. 152, 167–173. doi: 10.1016/S0923-2508(01)01188-3
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Gantois, I., Ducatelle, R., Pasmans, F., Haesebrouck, F., Hautefort, I., Thompson, A., et al. (2006). Butyrate specifically down-regulates salmonella pathogenicity island 1 gene expression. Appl. Environ. Microbiol. 72, 946–949. doi: 10.1128/AEM.72.1.946-949.2006
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Jia, F.-F., Zhang, L.-J., Pang, X.-H., Gu, X.-X., Abdelazez, A., Liang, Y., et al. (2017). Complete genome sequence of bacteriocin-producing Lactobacillus plantarum KLDS1. 0391, a probiotic strain with gastrointestinal tract resistance and adhesion to the intestinal epithelial cells. Genomics 109, 432–437. doi: 10.1016/j.ygeno.2017.06.008
Joerger, M. C., and Klaenhammer, T. R. (1986). Characterization and purification of helveticin J and evidence for a chromosomally determined bacteriocin produced by Lactobacillus helveticus 481. J. Bacteriol. 167, 439–446. doi: 10.1128/jb.167.2.439-446.1986
Kabir, A., Aiba, Y., Takagi, A., Kamiya, S., Miwa, T., and Koga, Y. (1997). Prevention of Helicobacter pylori infection by lactobacilli in a gnotobiotic murine model. Gut 41, 49–55. doi: 10.1136/gut.41.1.49
Kamada, N., Chen, G. Y., Inohara, N., and Núñez, G. (2013). Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 14, 685–690. doi: 10.1038/ni.2608
Khan, H., Flint, S. H., and Yu, P. L. (2013). Determination of the mode of action of enterolysin A, produced by Enterococcus faecalis B9510. J. Appl. Microbiol. 115, 484–494. doi: 10.1111/jam.12240
Kim, M. S., Yoon, Y. S., Seo, J. G., Lee, H. G., Chung, M. J., and Yum, D. Y. (2013). A study on the prevention of salmonella infection by using the aggregation characteristics of lactic acid bacteria. Toxicol. Res. 29, 129–135. doi: 10.5487/TR.2013.29.2.129
Kobayashi, K. S., Chamaillard, M., Ogura, Y., Henegariu, O., Inohara, N., Nuñez, G., et al. (2005). Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734. doi: 10.1126/science.1104911
Lee, I., Kim, Y. O., Park, S.-C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Mazmanian, S. K., Round, J. L., and Kasper, D. L. (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625. doi: 10.1038/nature07008
Nilsen, T., Nes, I. F., and Holo, H. (2003). Enterolysin A, a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Appl. Environ. Microbiol. 69, 2975–2984. doi: 10.1128/AEM.69.5.2975-2984.2003
Ogawa, M., Shimizu, K., Nomoto, K., Tanaka, R., Hamabata, T., Yamasaki, S., et al. (2001). Inhibition of in vitro growth of Shiga toxin-producing Escherichia coli O157:H7 by probiotic Lactobacillus strains due to production of lactic acid. Int. J. Food Microbiol. 68, 135–140. doi: 10.1016/S0168-1605(01)00465-2
Park, B. H., Karpinets, T. V., Syed, M. H., Leuze, M. R., and Uberbacher, E. C. (2010). CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20, 1574–1584. doi: 10.1093/glycob/cwq106
Rupnik, M., Wilcox, M. H., and Gerding, D. N. (2009). Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7, 526–536. doi: 10.1038/nrmicro2164
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Shin, R., Suzuki, M., and Morishita, Y. (2002). Influence of intestinal anaerobes and organic acids on the growth of enterohaemorrhagic Escherichia coli O157: H7. J. Med. Microbiol. 51, 201–206. doi: 10.1099/0022-1317-51-3-201
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Vangay, P., Ward, T., Gerber, J. S., and Knights, D. (2015). Antibiotics, pediatric dysbiosis, and disease. Cell Host Microbe 17, 553–564. doi: 10.1016/j.chom.2015.04.006
Van Heel, A. J., De Jong, A., Montalbán-López, M., Kok, J., and Kuipers, O. P. (2013). BAGEL3: automated identification of genes encoding bacteriocins and (non-) bactericidal posttranslationally modified peptides. Nucleic Acids Res. 41, W448–W453. doi: 10.1093/nar/gkt391
Yang, S.-C., Lin, C.-H., Sung, C. T., and Fang, J.-Y. (2014). Antibacterial activities of bacteriocins: application in foods and pharmaceuticals. Front. Microbiol. 5:241. doi: 10.3389/fmicb.2014.00241
Yin, S., Jensen, M. A., Bai, J., Debroy, C., Barrangou, R., and Dudley, E. G. (2013). The evolutionary divergence of Shiga toxin-producing Escherichia coli is reflected in clustered regularly interspaced short palindromic repeat (CRISPR) spacer composition. Appl. Environ. Microbiol. 79, 5710–5720. doi: 10.1128/AEM.00950-13
Keywords: probiotics, Lactobacillus acidophilus, Salmonella infection, genome sequence, PacBio
Citation: Chung W-H, Kang J, Lim MY, Lim T-j, Lim S, Roh SW and Nam Y-D (2018) Complete Genome Sequence and Genomic Characterization of Lactobacillus acidophilus LA1 (11869BP). Front. Pharmacol. 9:83. doi: 10.3389/fphar.2018.00083
Received: 24 October 2017; Accepted: 25 January 2018;
Published: 08 February 2018.
Edited by:
Raffaele Capasso, University of Naples Federico II, ItalyReviewed by:
Hosni M. Hassan, North Carolina State University, United StatesCopyright © 2018 Chung, Kang, Lim, Lim, Lim, Roh and Nam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seong Woon Roh, c3dyb2hAd2lraW0ucmUua3I=
Young-Do Nam, eW91bmdkbzk4QGtmcmkucmUua3I=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.