 Elżbieta Supruniuk
Elżbieta Supruniuk Agnieszka Mikłosz
Agnieszka Mikłosz Adrian Chabowski
Adrian Chabowski
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 15 November 2017
Sec. Lipid and Fatty Acid Research
Volume 8 - 2017 | https://doi.org/10.3389/fphys.2017.00923
PGC-1α coactivator plays a decisive role in the maintenance of lipid balance via engagement in numerous metabolic processes (i.e., Krebs cycle, β-oxidation, oxidative phosphorylation and electron transport chain). It constitutes a link between fatty acids import and their complete oxidation or conversion into bioactive fractions through the coordination of both the expression and subcellular relocation of the proteins involved in fatty acid transmembrane movement. Studies on cell lines and/or animal models highlighted the existence of an upregulation of the total and mitochondrial FAT/CD36, FABPpm and FATPs content in skeletal muscle in response to PGC-1α stimulation. On the other hand, the association between PGC-1α level or activity and the fatty acids transport in the heart and adipocytes is still elusive. So far, the effects of PGC-1α on the total and sarcolemmal expression of FAT/CD36, FATP1, and FABPpm in cardiomyocytes have been shown to vary in relation to the type of PPAR that was coactivated. In brown adipose tissue (BAT) PGC-1α knockdown was linked with a decreased level of lipid metabolizing enzymes and fatty acid transporters (FAT/CD36, FABP3), whereas the results obtained for white adipose tissue (WAT) remain contradictory. Furthermore, dysregulation in lipid turnover is often associated with insulin intolerance, which suggests the coactivator's potential role as a therapeutic target.
A modern lifestyle associated with physical inactivity and over-nutrition inevitably leads to overweight or obesity and has a critical importance in the development of insulin resistance (IR). Such a deleterious alterations in lifestyle result in tremendous expanse of type 2 diabetes (T2DM). According to the International Diabetes Federation (IDF) in 2015 there were 415 million people suffering from diabetes and IDF predicts that by 2,040 this number will grow to 642 million (Nam et al., 2015). For these reasons, researchers focus on the possible mechanisms contributing to IR and its associated comorbidities as well as on the elaboration of effective treatments.
So far, most of the studies suggest that a prolonged increase in plasma non-esterified fatty acid (NEFA) content and subsequent ectopic lipid accumulation interferes with insulin signal transduction and is responsible for the development of IR. Long chain fatty acids (LCFA) plasma concentration has been linked with an enhanced intracellular fatty acid (FA) transport, which exceeds the rate of β-oxidation in a tissue (Serra et al., 2013). As a result, lipid peroxidation and generation of reactive oxygen species (ROS) are induced (Ayala et al., 2014). Nowadays, a lot of data connect lipid-dependent oxidative stress with IR etiology (Schrauwen et al., 2006), for instance 4-hydroxynonenal (4-HNE) reduces protein kinase B (PKB/Akt) and Akt substrate of 160 kDa (AS160) phosphorylation as well as insulin receptor substrates (IRS-1 and IRS-2) expression (Prasannarong et al., 2014). Furthermore, the impaired lipid utilization, characterized by increased triacylglycerols (TAG) and other bioactive lipid fractions levels, such as fatty acyl CoA, diacylglycerols (DAG) and ceramides (CER), admittedly contributes to adverse alternations in insulin signaling pathway (Erion et al., 2016). Briefly, DAG stimulate protein kinase C (PKC-θ, PKC-βII, and PKC-δ isoforms) that in turn dephosphorylates IRS-1, thereby blocking phosphatidylinositol-3 kinase (PI3K) activation (Szendroedi et al., 2014; Łukaszuk et al., 2015a). Excessive accumulation of CER induces phosphatidyl phosphatase 2A (PP2A) and protein kinase C λ/ζ (PKCλ/ζ), thus inhibiting the phosphorylation of PKB/Akt in the position of Thr308 and Ser473 (Hage Hassan et al., 2014; Kurek et al., 2015). Additionally, CER and DAG can upregulate nuclear factor κB (NF-κB) level (Figure 1) (Coll et al., 2008; Hommelberg et al., 2011), which is involved in the activation of inflammatory processes, including IL-6 secretion that impairs insulin action at the level of IRS-1 (Nieto-Vazquez et al., 2008). The CER-mediated induction of signaling pathway of the protein kinase inhibitor complex of IκB and protein kinase c-Jun leads to an inhibition of tyrosine phosphorylation of IRS1 (Boon et al., 2013). Generally, high lipid availability, mitochondrial dysfunction, ROS and lipid intermediates accumulation may induce IR. In the light of the above mentioned statements, understanding of the mechanisms responsible for the control of FA uptake and metabolism is essential for the discovery of potential therapeutic options.
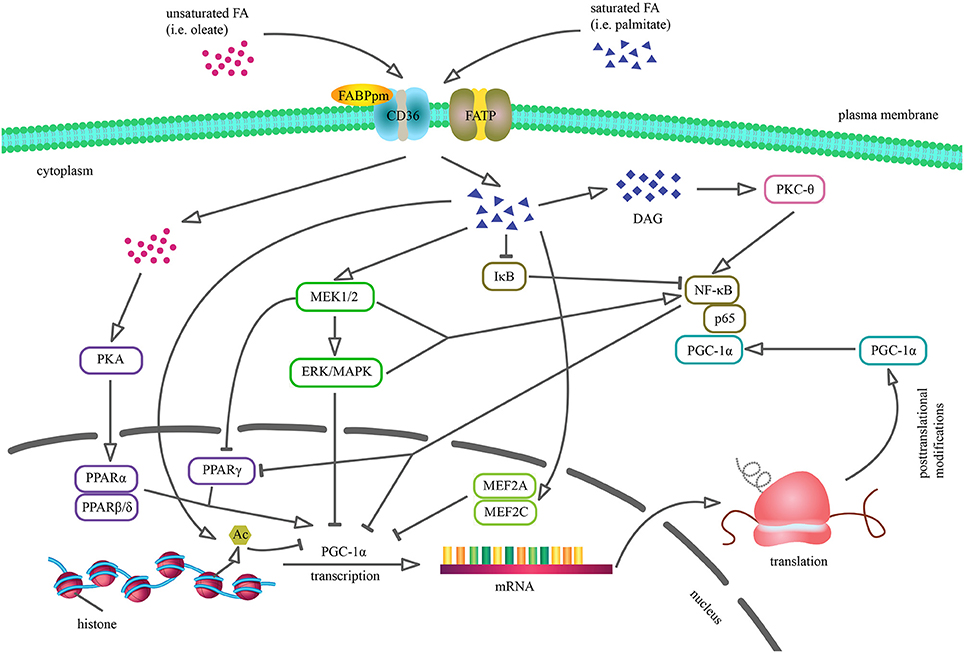
Figure 1. Cellular mechanisms by which saturated and unsaturated fatty acids may alter PGC-1α expression and activity. Palmitic acid incorporation reduces PGC-1α transcription through the activation of ERK/MAPK cascade, reduction of MEF2 transcription factor DNA binding and stimulation of IκB proteasomal degradation. Moreover, saturated FA contribute to MEK1/2-dependent NF-κB stimulation and DAG accumulation that results in PKCθ-mediated NF-κB induction. PGC-1α downregulation via NF-κB may involve several processes, including direct repression as p65 subunit of NF-κB is constitutively physically bound to PGC-1α as well as reduced PPARγ activity, because PGC-1α promoter sequence contains PPRE. Palmitate-induced histone deacetylation results in reduced PGC-1α promoter activity and inhibits PGC-1α expression. Conversely, oleate enhances PGC-1α mRNA level via PKA-PPARα pathway, thus reversing the coactivator's previous level (Coll et al., 2006, 2008; Crunkhorn et al., 2007; Alvarez-Guardia et al., 2010).
The peroxisome proliferator-activated receptor coactivator-1 (PGC-1) family serves as a major regulator of cellular metabolism by activation of a wide range of nuclear receptor (NR) and non-NR transcription factors. The first discovered member of the PGC-1 family was a 91 kDa nuclear protein (Besseiche et al., 2015) identified in brown adipose tissue (BAT) in cold-induced thermogenesis studies and termed PPARγ coactivator 1α (PGC-1α) (Puigserver et al., 1998). A sequence analysis revealed that PGC-1α (also known as PPARGC1A) is composed of several distinct domains, including an amino-terminal activation domain covering nuclear receptor docking region, a central regulatory sequence and a carboxy-terminal RNA recognition motif (Martínez-Redondo et al., 2015). The biological activity of PGC-1α is tightly controlled by transcriptional (multiple promoter regions, alternative splicing) and post-translational (phosphorylation, acetylation or methylation) modifications, which give rise to PGC-1α-a, PGC-1α-b, PGC-1α-c, and NT-PGC-1α mRNA isoforms involved in the cellular adaptation to the environmental conditions (Popov et al., 2015). The dynamic changes in PGC-1α activity also depend on its interactions with other coregulators/coactivators functioning as sensors of cellular energy status. A set of genes involved in energy balance remains under the antagonistic control of PGC-1α and receptor-interacting protein 140 (RIP140) (Kupr and Handschin, 2015). PGC-1α sensitivity to RIP140-mediated repression increases after SUMOylation by small ubiquitin-like modifier (SUMO-1) protein, which reduces transcriptional activity of the coactivator without an impact on its stability and cellular localization (Rytinki and Palvimo, 2009). Additionally, retinoblastoma protein (pRB) promoting white adipose tissue (WAT) development represses PGC-1α promoter activity and diminishes the coactivator's transcription rate (Scimè et al., 2005; Váraljai et al., 2015). In adipocytes PGC-1α is also controlled by transcriptional coregulator PRDM16 involved in attenuation of white fat-selective genes expression. PGC-1α competes with C-terminal binding protein (CtBP) for binding to PRDM16 and in such complex strongly stimulates the expression of brown fat genes, including PGC-1α itself (Kajimura et al., 2008). Moreover, CREB-regulated transcription coactivator (CRTC2) was found to positively stimulate PGC-1α transcription in skeletal muscle cells, whereas dysregulation of CRTC2/CREB pathway in diabetic rats may contribute to a reduced PGC-1α level (Rahnert et al., 2016). The half-life time of the protein is about 20 min and afterwards it undergoes ubiquitylation and proteasomal degradation (Sano et al., 2007). PPARβ can limit PGC-1α degradation via binding to the coactivaor and reducing its ubiquitination in skeletal muscle challenged by exercise (Koh et al., 2017). Recent studies performed in vitro on muscle and liver cells discovered a new regulator of PGC-1α activity, namely flavoprotein NADH quinone oxidoreductase 1 (NQO1). NQO1 binds to PGC-1α and protects the coactivator from ubiquitin-independent proteasomal degradation executed by the 20S proteasome catalytic particle in NADH-dependent manner (Adamovich et al., 2013). What is also important, PGC-1α does not directly bind to DNA, but interacts with a variety of transcription factors and therefore mediates chromatin remodeling (via CBP/p300 and SRC-1) and DNA transcriptional machinery activation (via TRAP/Mediator), eventually regulating gene expression (Wallberg et al., 2003; Sano et al., 2007). Finally, it coordinates such processes as mitochondrial biogenesis and oxidative metabolism (Popov et al., 2015). Therefore, PGC-1α is the most important powerful lipid homeostasis regulator, however efforts for understanding its engagement in LCFA metabolism are still in their infancy.
PGC-1α is abundantly expressed in tissues with high energy requirements, such as heart, skeletal muscle and BAT (Liang and Ward, 2006; Vega et al., 2015). Moreover, its expression in subcutaneous and visceral adipose tissue is significantly lower than in skeletal muscle (Heilbronn et al., 2012). Multiple factors enhance muscular PGC-1α content, most notably an increased contractile activity (Irrcher et al., 2003; Mathai et al., 2008), which also stimulates translocation of PGC-1α specifically to subsarcolemmal (SS) mitochondrial subpopulation in a process mediated by AMP-activated protein kinase (AMPK) (Smith et al., 2013). Furthermore, exercise and β3-adrenergic signaling were shown to strongly induce PGC-1α expression in BAT (Ruschke et al., 2010; Kim et al., 2016).
Plasma free fatty acids (FFA) level is responsible for the modulation of tissue PGC-1α expression as evidenced by inverse correlation between PGC-1α mRNA content in human skeletal muscle and FFA concentration (Richardson et al., 2005). Consistent with this notion, the reduced PGC-1α transcription has been observed in skeletal muscle (Crunkhorn et al., 2007) and adipose cells (Semple et al., 2004) of obese subjects. However, transcriptional control is often not readily evident as there are numerous observations showing a poor relationship between PGC-1α mRNA and protein expression (Watt et al., 2004; Sparks et al., 2005). The in vitro studies on myocytes revealed the opposite impact of saturated and unsaturated FA on PGC-1α expression and activity. Palmitate reduced PGC-1α level in a process mediated by mitogen-activated protein kinase (MAPK) and NF-κB, whereas oleate administration prevented the palmitate-induced PGC-1α downregulation (Figure 1) (Coll et al., 2006, 2008; Crunkhorn et al., 2007; Alvarez-Guardia et al., 2010). It is worth to emphasize that these observations may not reflect the physiological or pathophysiological in vivo effects of FA, since palmitic and oleic acids constitute only a part of FFA conglomerate present in the blood plasma. Contradictory results were observed in Huh7 hepatocytes as exposure to palmitic acid resulted in a dose and time dependent increase in PGC-1α expression. Additionally, the upregulation in hepatic PGC-1α level was demonstrated in mice with non-alcoholic fatty liver disease, suggesting different role of the coactivator in response to FA treatment in skeletal muscle and the liver (Maruyama et al., 2016). Understanding the tissue specific functions of the coactivator has been made possible thanks to both gain- and loss-of-function models. Therefore, our review focuses on the comparison and assessment of the lipid metabolism features in both the model types (Tables 1, 2).
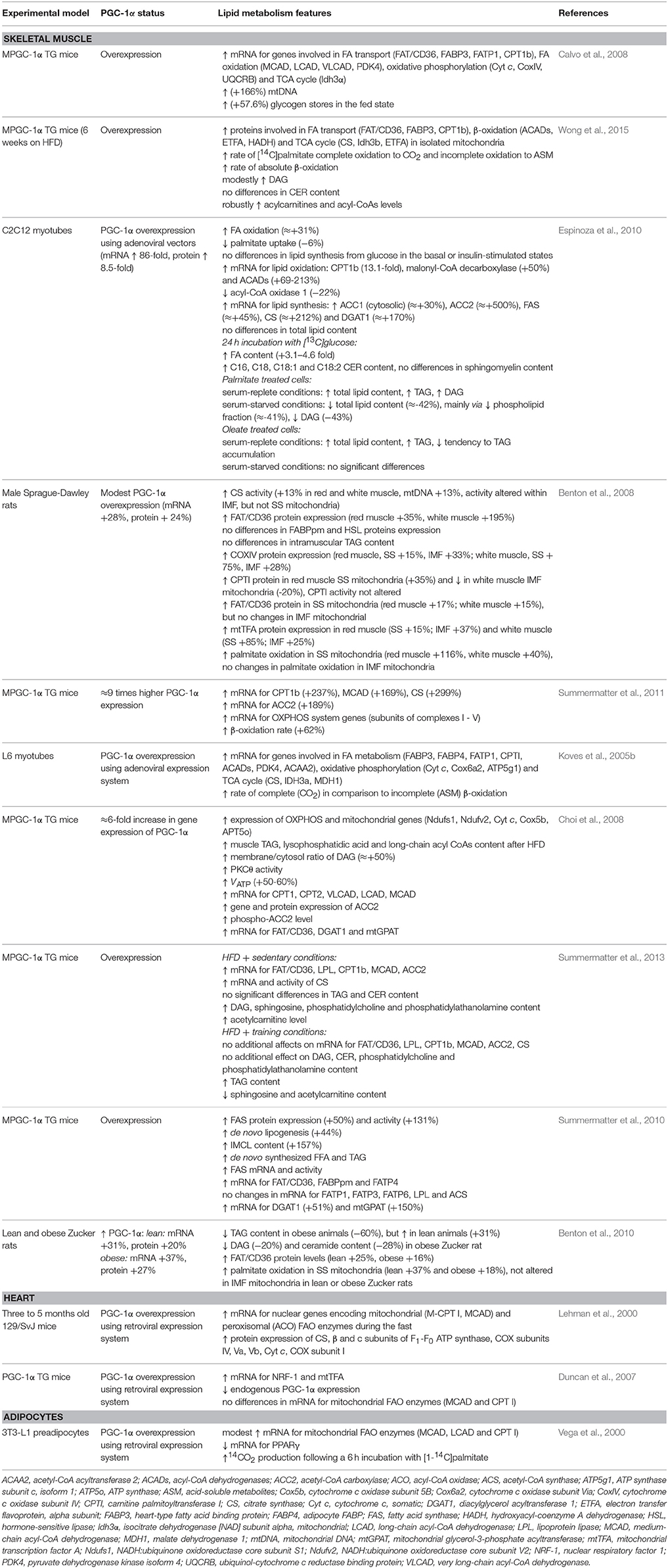
Table 1. The effects of PGC-1α overexpression on the lipids metabolism.
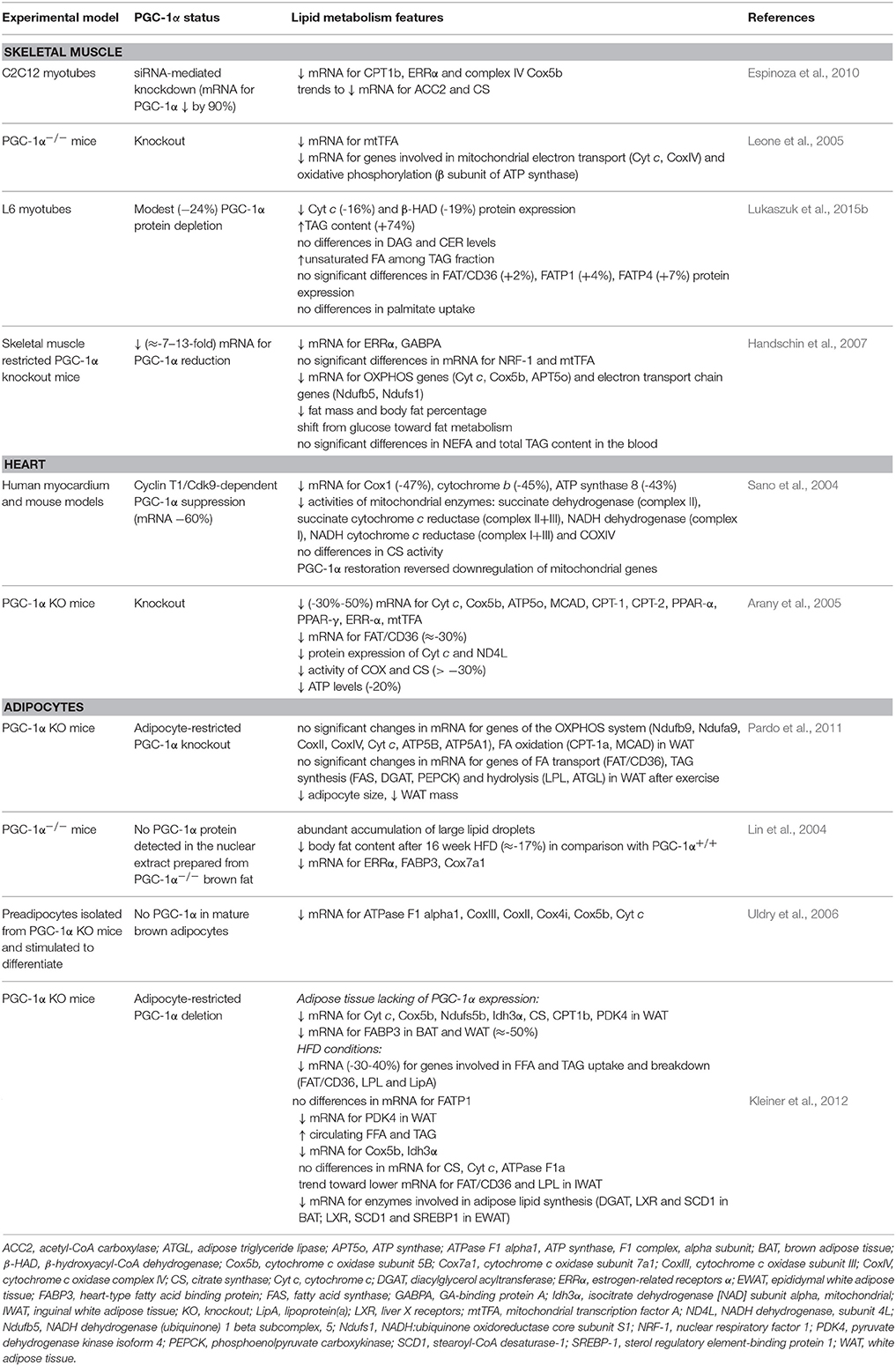
Table 2. The effects of PGC-1α downregulation on the lipids metabolism.
So far, most of the studies indicate that PGC-1α regulates the expression of genes controlling both lipid oxidation and synthesis. Such pleiotropic effects of the coactivator result from its interaction with multiple agents in diverse tissues. Among transcription factor targets of PGC-1α are PPARα, PPARβ/δ and PPARγ, which coordinate the expression of mitochondrial genes as well as indirectly participate in FA transport and utilization (Lin et al., 2005). Furthermore, PGC-1α upregulates the expression of several genes of the tricarboxylic acid (TCA) cycle (Hatazawa et al., 2015) and mitochondrial FA oxidation (FAO) pathway (Calvo et al., 2008). Accordingly, improvement in both FAO and TCA cycle flux in PGC-1α overexpressing cells coincides with a significant increase in the oxidation of palmitate (Espinoza et al., 2010; Wong et al., 2015). Benton et al. demonstrated that this effect was connected with an increase in FAO in SS, but not in intramyofibrillar (IMF) mitochondria, in both insulin sensitive and IR groups (Benton et al., 2008). Importantly, PGC-1α regulates the expression of nuclear and mitochondrial genes that encode the components of electron transport system and oxidative phosphorylation (OXPHOS) via NRF-1 and NRF-2 (nuclear respiratory factor 1 and 2), and estrogen-related receptor α (ERRα) coactivation. This results in increased mitochondrial transcription factor A (mtTFA) expression, which is known to control mtDNA replication and transcription and therefore regulates cellular oxidative metabolism (Dillon et al., 2012). Consequently, the augmented expression of cytochrome c, cytochrome c oxidase (COX) subunits II and IV, and ATP synthase is also a result of PGC-1α action (Table 1) (Lehman et al., 2000; Choi et al., 2008; Espinoza et al., 2010; Smith et al., 2013). Another noteworthy fact is that PGC-1α can stimulate peroxisomal activity and concurrent long-chain and very-long-chain fatty acid oxidation (Huang et al., 2017). Briefly, PGC-1α expression is positively correlated with cellular capacity to completely oxidize FA that in turn reduces intramuscular lipids deposition and improves insulin sensitivity of tissues.
On the other hand, real-time PCR and photometric methods revealed that PGC-1α promotes also lipogenesis in skeletal muscle by increasing the expression and activity of fatty acid synthase (FAS), a multifunctional enzyme engaged in de novo lipid synthesis (Espinoza et al., 2010; Summermatter et al., 2013). Chromatin immunoprecipitation assay showed that the mechanism for this effect involves liver X receptor α (LXRα) coactivation that stimulates PGC-1α binding to the liver X receptor-responsive element (LXRE) in FAS promoter. Additionally, muscle specific PGC-1α transgenic (MPGC-1α TG) mice demonstrated exacerbated rates of de novo synthesized FFA as well as FA esterification and TAG accumulation (Summermatter et al., 2010). Nevertheless, PGC-1α overexpression does not change the level of acetyl-CoA synthase (ACS), an enzyme implicated in linking the imported lipids to coenzyme A. Concomitantly, despite the enhanced expression of acetyl coenzyme A carboxylase 2 (ACC2), there is no difference in AMPK activity between wild type and transgenic animals (Espinoza et al., 2010). Moreover, the coactivator-dependent intramyocellular lipid (IMCL) accumulation (Summermatter et al., 2010) is associated with impaired glucose homeostasis and regarded as a risk factor for the development of IR and/or T2DM (Mikłosz et al., 2017). However, endurance-trained athletes also display elevated IMCL level combined with high insulin sensitivity, a phenomenon known as “an athletes” metabolic “paradox” (Bergman et al., 2010). In particular, the elevated total DAG level found in trained individuals implies a great role of subcellular localization and composition of lipid fractions in IR development (Amati et al., 2011). Since lipotoxic intermediates are crucial for insulin action deterioration, it is also important to notice that PGC-1α overexpression causes a reduction in intramuscular TAG (−60%), DAG (−20%), and CER (−28%) concentration in obese Zucker rats. On the other hand, an increase in TAG content (+31%), but no change in DAG and CER levels has been observed in lean animals (Benton et al., 2010). Paradoxically, robustly overexpressed PGC-1α promoted the development of IR in the skeletal muscle of the sedentary MPGC-1α TG mice, presumably as a consequence of imbalanced lipid uptake and oxidation (Summermatter et al., 2013). Excessive acyl-CoA incorporation into TCA cycle and OXPHOS was reflected in an enhanced acylcarnitine level. Moreover, the animals overexpressing PGC-1α that were fed with a high fat diet (HFD) exhibited partly different features of a lipid profile from the above mentioned, revealing no significant changes in TAG and CER content, but elevated DAG level. In addition, an increased sphingosine concentration in MPGC-1α mice resulted in a diminished glucose influx and aggravated insulin signal transduction (Summermatter et al., 2013) via PKB/Akt inactivation (Taha et al., 2006). Importantly, insulin sensitivity was improved when elevated level of PGC-1α was combined with exercise (Summermatter et al., 2013). On the other hand, Choi et al. observed that after PGC-1α stimulation (mRNA ≈+6-fold) HFD-induced IR occurred due to an increased membrane/cytosol DAG ratio as well as diminished Akt2 and PKCθ activity along with a reduced (−60%) insulin-stimulated glucose influx. The paralleled unequal growth in mitochondria number (+2-fold) and activity (+60%) implies the reduced activity per unit of mitochondrial mass, inevitably linking increased FA uptake with IMCL accumulation and impaired insulin tolerance (Choi et al., 2008). Some studies point out that this may be a consequence of PGC-1α/PPAR-α dependent stimulation of mammalian tribbles homolog 3 (TRB-3), which can bind to Akt1 and Akt2 and inhibit their activation (Mortensen et al., 2006). Furthermore, the unexpected exacerbation of IR in the case of muscle-specific PGC-1α upregulation (10–13-fold) has been also linked with downregulated GLUT4 mRNA level and glucose uptake. This was also supported by the observed lack of changes in GLUT-4 level in tisssues (heart muscle, white and brown adipose tissue), where PGC-1α content was not altered (Miura et al., 2003). Collectively, the alterations in lipid content and composition occur as a consequence of PGC-1α-mediated increase in gene expression of the proteins involved in both lipid oxidation and synthesis. Modestly elevated PGC-1α level accelerates the positive impact of exercise on glucose homeostasis, increases activity of enzymes involved in Krebs cycle and favors the shift from incomplete to complete β-oxidation. However, the oversized expression of the coactivator has been found to trigger mostly unfavorable metabolic changes.
The rate of LCFA deposition/oxidation in tissues is strictly related to the accompanying changes in their movement across both plasma and mitochondrial membranes. For many years, it was believed that the transport of FA across the plasma membrane occurs only via passive diffusion down their electrochemical gradient. However, currently it is well-established that FA are transported into the cells also by protein-mediated mechanism. In metabolically active tissues, such as the striated muscles and adipocytes, fatty acid translocase (FAT/CD36), fatty acid transport proteins (FATP1-6) and plasma membrane fatty acid binding protein (FABPpm) have been identified as putative FA transporters. Furthermore, protein-mediated LCFA uptake is also involved in mitochondrial lipid flow and their subsequent β-oxidation, besides the well-known carnitine-palmitoyltransferase (CPT) system (Schwenk et al., 2010). Importantly, the regulation of FA transporters expression and distribution within the cell compartments is another level of PGC-1α control over lipid homeostasis. In the current paper we summarize the role of PGC-1α in the regulation of the total, plasma membrane and mitochondrial expression of FA transporters in the insulin sensitive tissues.
Skeletal muscle constitute ~40% of the human body mass and serve as a substantial tissue for FA uptake (Egan and Zierath, 2013). It has been proven that facilitated LCFA transport involves a number of transporters, such as FAT/CD36, FABPpm, FATP1, FATP4, and FATP6. However, the expression of FAT/CD36 and FABPpm, but not FATP1, corresponds with the oxidative capacity of muscle cells. Therefore, higher LCFA uptake rate is found in red oxidative muscle fibers in comparison to white glycolytic fibers (Luiken et al., 1999). For this reason, it is worth pointing out that the protein amount of FABPpm, FAT/CD36 and FATP4 highly correlates with PGC-1α expression (Nickerson et al., 2009; Benton et al., 2010), which is consistent with the fact that the protein level of PGC-1α is positively associated with the oxidative fibers ratio in metabolically heterogeneous rat hindlimb muscle (Benton et al., 2008).
Summermater et al. noticed the relationship between PGC-1α mRNA level and the FA transporters' gene expression. The authors revealed that the upregulation of PGC-1α expression in mice results in the elevation of FAT/CD36, FABPpm and FATP4 transcripts abundance, but not the mRNA amount for FATP1, FATP3, and FATP6. The subsequent increase in IMCL content was observed in MPGC-1α TG mice, although a direct analysis proved that the IMCL accumulation is caused rather by de novo lipogenesis than cellular FA import (Summermatter et al., 2010). Interestingly, the stimulatory effect on FAT/CD36 level was significantly greater in red fibers (195%) in comparison with white muscle fibers (35%) (Table 1) (Benton et al., 2008). The concomitant growth in cellular PGC-1α and FAT/CD36 amount was also confirmed in other studies with skeletal muscle-restricted murine PGC-1α transgenic models (Wende et al., 2007; Choi et al., 2008). However, in some studies higher transporter level (FAT/CD36) did not affect TAG muscular concentration (Benton et al., 2008), while in others an enhanced TAG content was present (Summermatter et al., 2010). On the contrary, PGC-1α overexpression in cultured human skeletal muscle cells did not increase FAT/CD36 content, although elevated FAO and expression of other genes connected with lipid metabolism were observed (Mormeneo et al., 2012; Nikolić et al., 2012). Currently, it remains unresolved if PGC-1α may directly control FABPpm expression, because there are some discrepancies in the literature as to whether the PPARγ activator rosiglitazone stimulates the increase in FABPpm content (Coort et al., 2005; Benton et al., 2008). Surprisingly, increased (≈+31%) palmitate β-oxidation may not be a consequence of lipid transport as a trend for a reduction (−6%) in the intracellular palmitate influx was observed in PGC-1α overexpressing C2C12 myotubes (Espinoza et al., 2010).
Interestingly, no significant differences in the whole muscle FAT/CD36, FABPpm and FATP4 expression has been detected between lean and obese individuals (Holloway et al., 2007), although HFD downregulates both PGC-1α and OXPHOS genes (Sparks et al., 2005). Moreover, higher TAG content in obese Zucker rats lowered PGC-1α protein expression. Conversely, mice lacking FAT/CD36 activity a parallel reduction in the number of intramuscular triacylglycerol (IMTG) depots exhibited increased PGC-1α expression. These unexpected changes suggest that the intramuscular lipid milieu may evoke alterations in the coactivator's amount independently of plasma FFA concentration (Benton et al., 2006). Additionally, the effectiveness of training-induced PGC-1α ability to stimulate the downstream genes involved in lipid utilization was impaired in FAT/CD36-knockout mice, implying an important role of FA availability in PPAR targets transcription (Manio et al., 2017). Nevertheless, the suppression of plasma FFA level via nicotinic acid (NA) corresponded with an increased muscle PGC-1α content without any changes after exercise. Simultaneously, the response of its target gene, FAT/CD36, did not differ between NA treated subjects and the control ones (Watt et al., 2004).
Additionally to the previous statements, Choi et al. demonstrated a paradoxical increase in IR observed in MPGC-1α TG mice (a 6-fold increase in mRNA for PGC-1α) fed with HFD, which was linked with a strong concurrent upregulation of FAT/CD36 expression (a 3-fold increase in mRNA) and FA oversupply. Moreover, the authors noticed enhanced transcription of acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) and mitochondrial glycerol-3-phosphate acyltransferase (mtGPAT), enzymes that catalyze TAG reestrification, as well as subsequent DAG and long-chain fatty acyl-CoAs accumulation (Choi et al., 2008). Furthermore, exceessive lipids provision combined with insufficient citrate synthase (CS) activity are attributed to an increased acetylcarnitine level. This in turn may deteriorate glucose metabolism via activation of NF-κB, which enhances insulin-stimulated phosphorylation of IRS and diminishes insulin-stimulated phosphorylatation of PKB/Akt (Zhang et al., 2010). Adverse effects of PGC-1α on insulin sensitivity have been connected with vascular endothelial growth factor β (VEGF-β) dependent stimulation of FATP3 and FATP4 expression and downstream lipid accumulation in HFD-fed MPGC-1α TG mice (Jang et al., 2016; Mehlem et al., 2016). PGC-1α also induces branched-chain amino acids (BCAA) catabolism and subsequent 3-hydroxyisobutyrate (3-HIB) secretion from muscle, resulting in excessive trans-endothelial FA transport into the tissue, deposition of lipotoxic intermediates and impaired insulin signal transmission (Jang et al., 2016). In contrast to the aforementioned findings, a limited PGC-1α-mediated FAT/CD36 upregulation (~25%) elevates insulin sensitivity (Benton et al., 2008, 2010) by stimulating FAO (Nickerson et al., 2009). It is also presumed that the mechanisms responsible for IMCL accumulation in endurance trained athletes differ from those in the individuals with T2DM. Interestingly, in the first case IMTG de novo synthesis prevails, whereas in the latter situation an increased FA uptake is observed (Bergman et al., 2010). Moreover, given the enhanced lipogenesis, lipid turnover seems to be insufficient in sedentary state after ectopic PGC-1α overexpression, despite enhanced oxidative capacity. This is in contrast to an active animal, where energy requirements and provision are tightly coordinated (Summermatter et al., 2010). However, the usage of PGC-1α activator, pioglitazone (thiazolidinedione), reverses disruption of insulin signaling and restores mitochondrial bioenergetics (Pagel-Langenickel et al., 2008). Thereby, a modest overexpression of PGC-1α, which restricts FAT/CD36 upregulation, seems to be a key factor for improvement of insulin sensitivity (Benton et al., 2010). On the basis of these observations, PGC-1α is regarded as one of the factors eliciting FA uptake, which rises a possibility to control their oxidation and esterification in skeletal muscle. However, its therapeutic potential may be questioned, since physiological and supraphysiological PGC-1α doses did not reverse a decreased FAO and mtDNA content in myocytes from extremely obese subjects. Therefore, skeletal muscle oxidative capacity in obesity may be partially independent of PGC-1α (Consitt et al., 2010). Currently available literature data unfortunately do not provide information about the relationship between PGC-1α expression and sarcolemmal content of FA binding and transporting proteins. To date, metabolic challenges with insulin and muscle contraction have been shown to induce the translocation of FAT/CD36 and FABPpm to the plasma membrane, thus enhancing FA delivery into muscle cells (Han et al., 2007; Bradley et al., 2012). Moreover, researchers indicate that FAT/CD36, but not FABPpm, can be constantly relocated to the plasmalemma in IR (T2DM and obesity) human skeletal muscle, whereas its total expression seems to remain unaltered (Aguer et al., 2011).
Due to the prominent impact of PGC-1α on mitochondrial expression of the key genes involved in lipid metabolism, it is also relevant to answer whether this coactivator may exhibit a pronounced effect on protein mediated FA delivery to this organellum. So far, most of the studies have demonstrated that PGC-1α can increase the expression of enzymes like carnitine palmitoyltransferase 1b (CPT1b), carnitine-acylcarnitine translocase and malonyl-CoA decarboxylase that are implicated in the long-chain fatty acyl-CoA delivery for β-oxidation (Espinoza et al., 2010; Gacias et al., 2012). In addition, modest overexpression of PGC-1α was associated with elevated FAT/CD36 content in the mitochondrial skeletal muscle fraction from transfected mice. However, such an effect was observed solely within the SS mitochondria (+15–17%), but not in the IMF (Benton et al., 2008). On the contrary, pharmacological PGC-1α downregulation (i.e., clonbuterol administration) and the concomitant reciprocal increase in RIP140 expression evoked a reduction in FAT/CD36 total and, specifically, the IMF mitochondrial protein content in both red (−36 and −36%, respectively) and white (−35 and −58%, respectively) skeletal muscle. Accordingly, the alterations in mitochondrial content and structure together with a reduced expression of the enzymes responsible for FAO were also attributed to PGC-1α decrease (Hoshino et al., 2012). In regard to PGC-1α ability for enhancing cellular oxidative capacity, palmitate utilization was stimulated selectively in the SS mitochondria of both red (+116%) and white (+40%) muscles (Benton et al., 2008). These observations may reflect the fact that palmitate oxidation in both the mitochondrial subpopulations was higher in red muscle fibers (Koves et al., 2005a), although the oxidative capacity of the IMF type was elevated in comparison with the SS mitochondria (Ferreira et al., 2010). Moreover, the rate of palmitate oxidation in the IMF mitochondria did not differ between lean and obese Zucker rats, but the oxidation in the SS subpopulation was higher in the case of obese animals (Holloway et al., 2009). Novel findings revealed that the absence of fat overload in muscles of T2DM rats ensures normal function of the SS and IMF mitochondria (Lai et al., 2017). A comprehensive proteomic analysis confirmed an upregulation of FAT/CD36 and FABP3 (heart-type FABP, H-FABP) protein abundance as well as intensified the rate of β-oxidation in the mitochondria isolated from skeletal muscle of transgenic PGC-1α mice on a HFD. Moreover, exercise and caloric restriction declined the acylcarnitines level in the muscle of these animals, although the opposite response was observed in the case of 18:1 and 18:2 DAG species (Wong et al., 2015). Importantly, the transgenic mitochondria rich in PGC-1α can tolerate greater level of FA supply as well as exhibit an enhanced level of maximal uncoupled respiration (Hoeks et al., 2012). The presented findings support the hypothesis that PGC-1α induces mitochondrial adjustments aimed to cope with a disproportionate lipid delivery.
In summary, the proposed mechanisms of PGC-1α involvement in the rate of lipid movement into skeletal muscle include an upregulation of FAT/CD36, FABPpm and FATP1 gene expression as well as protein content in mitochondrial fraction. The latter effect has been confirmed solely in the case of murine FAT/CD36 and FABP3. However, it should be emphasized that the coactivator's insulin-sensitizing role is connected specifically with limited, i.e., within physiological range, PGC-1α overexpression. The used model also plays a vital role in the evaluation of PGC-1α effects, since the lack of stimulatory effect on FA transporters content was observed in human cultured myocytes. Moreover, insufficient information gives rise to the doubts, whether PGC-1α possesses the ability to relocate FA transporters to the sarcolemma or what are the direct molecular mechanisms responsible for the aforementioned changes.
It is well-documented that under physiological conditions 70–80% of myocardial energy expenditure are covered by β-oxidation of FA, however the rate of LCFA influx depends mainly on their plasma amount (Chabowski et al., 2008). Additionally, the FA transport into isolated cardiomyocytes can occur against the concentration gradient, which was measured by acrylodan labeled intestinal fatty acid binding protein (ADIFAB) (Carley and Kleinfeld, 2011). Importantly, the high PGC-1α content in the heart is connected with some specific features in comparison with other insulin sensitive tissues. For instance, a significant increase in PGC-1 expression is observed at birth in conjunction with an increase in cardiac oxidative capacity and a shift from glucose toward FA as a fundamental energy source during postnatal development (Buroker et al., 2008). Moreover, in the heart muscle the coactivator's mRNA level rises after fasting with no changes observed in BAT (Russell et al., 2004).
A causal relationship between PGC-1α/PPARα activation and an enhanced expression of multiple mitochondrial ATP-generating pathways was supported by animal model study (Duncan et al., 2007). PGC-1α increases the levels of some critical enzymes involved in β-oxidation, including the components of the OXPHOS complex and TCA cycle (Table 1) (Rowe et al., 2010). The expression of FA transporters is also controlled by myocardial PPARs, since the presence of peroxisome proliferator-responsive elements (PPREs) has been identified in the FAT/CD36, FABPpm, and FATP promoters sequence (Frohnert et al., 1999; Teboul et al., 2001). In accordance with this notion, the mRNA levels for FATP1 and FAT/CD36 were significantly blunted in PPARα-null mice as compared to wild type animals. Simultaneously, radioactively labeled palmitic acid uptake and oxidation were decreased due to lower expression of FA transporters (Watanabe et al., 2000). On the other hand, cardiac-restricted overexpression of PPARα in mice did not alter the transcription of FATP1 and FAT/CD36 in basal conditions, although PPARα agonist treatment (Wy-14,643) induced cardiac expression of these FA transporters (Finck et al., 2002). Moreover, the exposure to high concentration of FA coordinately enhanced mRNA content for FAT/CD36 and FABPpm as well as increased cellular oxidative capacity (van der Lee et al., 2000). PGC-1α interaction with ERRα also contributes to the regulation of cardiac lipid metabolism and upregulation of FAT/CD36, FABP3, and lipoprotein lipase (LPL) genes (Huss and Kelly, 2004; Huss et al., 2004). Huss et al. additionally reported a 92% increase in palmitate oxidation in PGC-1a overexpressing cells. Moreover, the compensatory acceleration in PGC-1α content in ERRα null mice, prevented FAT/CD36 and CPT I reduction (Huss et al., 2004).
Some authors speculate that the upregulation of the nuclear receptor transcription factor PPARα and its coactivators PGC-1α/β is one of the preliminary requirements for metabolic switch from glucose to FA consumption in IR and diabetic hearts (Schilling, 2015; Lee et al., 2017). This agrees with an increased level of PPARα as well as the total (+38%) and sarcolemmal (+26%) FAT/CD36 content in the diabetic animal hearts (Mansor et al., 2017). On the contrary, lowered PGC-1α expression was observed in diabetic human blood and myocardium (Fabregat-Andres et al., 2016) as well as in the animal models of T2DM (ob/ob mice) and diabetic dyslipidemia (db/db mice) (Buchanan et al., 2005). PPARα determines the expression of enzymes controlling both the catabolic and anabolic pathways of TAG metabolism (Banke et al., 2010). Cardiac-specific PPARα overexpression induced the occurrence of the hallmarks of diabetes, such as diminished glucose oxidation and increased cardiomyocyte TAG storage (Yang et al., 2007). Simultaneously, an elevated turnover of palmitoyl-CoA between cytosolic and TAG pool prior to β-oxidation and augmented recruitment of TAG-derived LCFA for utilization were observed in MHC-PPARα mice (Banke et al., 2010). Interestingly, PPARα and FAT/CD36 deficient mice [α-myosin heavy chain (MHC)-PPARα/CD36−/−] were not only protected from lipid imbalance in the myocardium, but also demonstrated an enhanced glucose uptake and FATP1 gene expression (Yang et al., 2007). Nevertheless, direct inhibition of plasma membrane FAT/CD36 by sulfo-N-succinimidyl oleate was sufficient to restore normal substrate metabolism and prevent lipotoxicity (Mansor et al., 2017). PPARγ overexpression in transgenic mice contributed to cardiac dysfunction encompassing an increase in FAT/CD36 level, FA uptake and intracellular lipid accumulation with rosiglitazone treatment even aggravating the lipotoxic effects. On the contrary, the action of rosiglitazone in the heart of wild type mice has been connected with a decreased expression of PPARγ target genes and a redistribution of PPAR ligands (i.e., plasma lipids) mainly to adipose tissue (Son et al., 2007). The consequences of PPARβ/δ knockout included a decreased PGC-1α protein content together with diminished FABP3, FATP1 and CPTIb genes expression in the adult mouse heart (Wang et al., 2010). The cardiomyocytes of MHC-PPARβ/δ mice were characterized by increased glucose uptake and utilization, induced FAO genes expression, lack of a stimulating effect on FA uptake and lipogenesis (with no signs of abnormal lipid accumulation) and, as a result, lipotoxic cardiomyopathy did not develop (Burkart et al., 2007).
Most of the studies corroborate the presence of FA transporters in both plasma membrane and the low-density microsome compartment of cardiomyocytes (Chabowski et al., 2005). Chronic translocation of FAT/CD36 and FABPpm to the plasma membrane, as a mechanism underlying TAG accumulation, was shown in the hearts from obese Zucker rats (Luiken et al., 2001; Coort et al., 2004) as well as in moderate and severe streptozotocin-induced diabetes of Sprague-Dawley rats (Luiken et al., 2002). Experiments in our laboratory revealed that chronic activation of PPARα induces the redistribution of FAT/CD36 from low density microsomes to the plasma membrane along with the increased palmitate incorporation into DAG fraction. PPARβ/δ stimulation is responsible for the relocation of both FAT/CD36 and FABPpm to the plasmalemma. Moreover, an enhanced level of myocardial TAG synthesis and palmitate oxidation was observed after PPARβ/δ agonist treatment. On the contrary, the usage of PPARγ activator did not exert any influence on the intracellular localization of these transporters as well as intracellular lipid pools content, although a decreased level of palmitate oxidation was noticed (Kalinowska et al., 2009).
Mitochondria constitute 20–30% of a cardiomyocyte volume, therefore they are substantial for the maintenance of an optimal FAO rate (Dillon et al., 2012). However, little is known regarding the possibility of FA transporters relocation into an out of cardiac mitochondrial membranes. Currently, solely the presence of FAT/CD36 has been confirmed. Interestingly, the reported mitochondrial expression of FAT/CD36 in the heart was ~10-fold greater than the one observed in red and white skeletal muscle (Campbell et al., 2004; King et al., 2007). Moreover, in the hearts of mice FAT/CD36 depletion did not affect the rate of palmitate, palmitoyl-CoA and palmitoylcarnitine mitochondrial β-oxidation, indicating considerably unessential role of this carrier in the mitochondrial lipid utilization in that tissue (King et al., 2007).
In conclusion, PGC-1α may play a key role in orchestrating FA supply to the heart via PPARs coactivation. However, potentially beneficial therapeutic window for PGC-1α in the cardiomyocytes is relatively narrow, since its overexpression leads to uncontrolled mitochondrial proliferation, abnormal sarcomeric structure and dilated cardiomyopathy (Lehman et al., 2000; Lehman and Kelly, 2002). Additionally, PPARα stimulation is thought to be a main culprit responsible for the excessive TAG, CER and DAG accumulation through incorporation of FAT/CD36 to the membrane structure in IR cardiomyocytes.
Adipose tissue serves as a primary tissue involved in lipolysis and lipogenesis, thereby contributing to the maintenance of FA uptake and release from/to the blood. In contrast to striated muscle, the driving gradient for FA diffusion between circulation and adipocytes may not occur, what suggests differences in the regulation of protein-mediated LCFA transport between the both tissues. Indeed, the facilitated FA transfer across plasma membrane constitutes more than 90% of their total flux in fat tissue (Stump et al., 2001). During their entry to the cell FA are accompanied by adipocyte cytoplasmic FABP (A-FABPc) that traffics them to a specific compartment and thereby decides about their further destination (Furuhashi and Hotamisligil, 2008). Pivotal transporters governing LCFA uptake into adipocytes are FAT/CD36, FABPpm, FATP1, and FATP4. Moreover, continuous recycling of these proteins from intracellular pool to the plasma membrane guarantees an appropriate plasma lipid concentration. In view of a significant role of FA in energy homeostasis, disturbances in the incorporation of LCFA into adipocytes may lead to IR in obesity and T2DM.
PGC-1α controls multiple morphological and molecular features required for the differentiation and proper function of brown adipocytes. It has been demonstrated that PGC-1α content is greater in subcutaneous than in omental adipose tissue regardless of obesity and fat distribution. The mRNA level for the coactivator inversely correlates with augmented body fat mass, visceral fat accumulation, as well as impaired glucose and insulin tolerance (Ruschke et al., 2010). Additionally, PGC-1α expression is significantly decreased in the subcutaneous adipose tissue of non-obese, non-diabetic IR individuals (Hammarstedt et al., 2003) and morbidly obese subjects (Semple et al., 2004).
Mice with adipose-restricted PGC-1α deficiency exhibited multiple alterations, including decreased lipid clearance arising as a result of FA transporter (FABP3), OXPHOS, TCA cycle and FAO genes downregulation. However, apart from FABP3 these observations affected solely WAT, while in BAT no significant changes occurred. Similar reduction (−20–40%) in mitochondrial genes expression was demonstrated in the cultured adipocytes derived from the inguinal stromal-vascular fraction (Kleiner et al., 2012). HFD fed mice lacking PGC-1α are characterized by increased FFA and TAG plasma levels along with decreased transcription of the genes involved in FAO as well as lipogenesis. Moreover, their FA uptake and breakdown were impaired as a consequence of reduced gene expression of FABP3 by 50%, FAT/CD36 by 30–40%, LPL and lipoprotein(a) in BAT (Kleiner et al., 2012). In WAT only a downward trend for FAT/CD36 and LPL transcripts content has been revealed. In both types of adipose tissue no changes in FATP1 mRNA level were noticed in response to PGC-1α ablation (Kleiner et al., 2012). As observed, dysregulation of genes involved in FA uptake, oxidation and synthesis strongly affected whole-body insulin sensitivity, mainly by increased hepatic glucose output (Kleiner et al., 2012). Interestingly, the knockout of adipocyte FABP (FABP4) reduced the potency of HFD for the increase in PGC-1α in BAT, while FAT/CD36 and FATP1 gene expression was upregulated to a similar extent as in a wild type mice (Shu et al., 2017). The role of PGC-1α in WAT has been questioned by Pardo et al. since the authors had not found significant differences in the expression of the genes involved in FA transport (FAT/CD36) and metabolism in adipocyte-restricted PGC-1α knockout mice (Pardo et al., 2011). Additionally, rosiglitazone treatment of these animals improved peripheral insulin sensitivity indicating that PGC-1α activity in WAT is not crucial for the whole body glucose and lipid balance. Furthermore, rosiglitazone effectively stimulated the expression of OXPHOS, TCA, and FAO genes, implying non-essential PGC-1α function in the mitochondrial biogenesis in WAT. Therefore, Pardo et al. performed studies on 3T3-L1 adipocytes and demonstrated that PGC-1β, instead of PGC-1α, acts as a primary modulator of the mitochondrial function in WAT, whereas PGC-1α is more important in the induction of brown adipocyte markers in white fat cells (Pardo et al., 2011). Surprisingly, Lin et al. showed that mice with whole-body PGC-1α ablation are resistant to HFD-induced obesity as well as exhibit increased insulin sensitivity and ameliorated glycemic control as indicated by glucose tolerance test (Lin et al., 2004). These findings are similar to the observations obtained by Leone et al. in PGC-1α−/− mice, which were less susceptible to HFD-induced insulin resistance, despite an increased body fat mass (Leone et al., 2005). Additionally, accumulating evidence suggest that the hypoglycemic effect exerted by the activation of AMPK/PGC-1α axis (metformin) may be of secondary importance, since AMPK deletion did not abolish metformin's action (Viollet et al., 2012). Nevertheless, high basal PGC-1α and FABP4 expression in adipose tissue are the predictors of good treatment response to pioglitazone, independently of the changes in PGC-1α mRNA level (Hammarstedt et al., 2008).
To summarize, the collected data corroborate the fact that FA transporters translocate to the cell membranes in the adipose tissue of obese Zucker rats. However, PGC-1α contribution to the regulation of their expression and cellular localization remains undermined or described in limited extent. Nonetheless, the reduction of protein transporters content (FAT/CD36, FABP3) in BAT of PGC-1α knockout mice indicates the connection between intracellular FA transport and PGC-1α activity.
Inefficient utilization and excessive lipids accumulation in tissues consequently result in a disruption of the insulin signaling pathway. The complexity of interactions between FA delivery to the insulin-sensitive tissues and their subsequent downstream metabolism makes it difficult to find one obvious target that may enable to control the rate of these processes. Therefore, the role of PGC-1α in modulating the activity and cellular localization of FA transport proteins (FAT/CD36, FABPpm, FATP1-6) has attracted considerable attention. This coactivator is widely known to increase the expression of critical genes involved in OXPHOS and TCA cycle, but also in lipid synthesis (i.e., FA, DGAT1, CS). So far, there is a limited number of studies linking PGC-1α protein expression (down- and upregulation models) with the total, membrane and mitochondrial content of FA transport proteins in insulin sensitive tissues. As previously shown, PGC-1α upregulates both protein expression and mitochondrial content of LCFA transporters in skeletal muscle, thereby modulating the rate of lipid oxidation. Thus, PGC-1α-stimulated FA targeting for mitochondrial β-oxidation may diminish the rate of FA storage and the synthesis of lipotoxic derivatives. On the other hand, despite much progress in describing cardiac- and adipocyte-specific coactivator's effects, its influence on FA protein transporters has yet to be elucidated. In conclusion, regulation of the expression of FA transporters may indicate PGC-1α as a prospective target for the prevention and/or treatment of IR and, as a consequence, of T2DM.
ES participated in the design of the work, drafted the manuscript and approved the final version submitted. AM provided substantial contributions to the conception and design of the work, helped to draft the manuscript and approved the final version submitted. AC participated in the design of the study, revised it critically for important intellectual content and approved the final version submitted. All authors agree to be accountable for all aspects of the work.
The project was funded by the Medical University of Bialystok, grant number N/ST/MN/17/004/1118.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DV and handling Editor declared their shared affiliation.
Adamovich, Y., Shlomai, A., Tsvetkov, P., Umansky, K. B., Reuven, N., Estall, J. L., et al. (2013). The protein level of PGC-1α, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell. Biol. 33, 2603–2613. doi: 10.1128/MCB.01672-12
Aguer, C., Foretz, M., Lantier, L., Hebrard, S., Viollet, B., Mercier, J., et al. (2011). Increased FAT/CD36 cycling and lipid accumulation in myotubes derived from obese type 2 diabetic patients. PLoS ONE 6:e28981. doi: 10.1371/journal.pone.0028981
Alvarez-Guardia, D., Palomer, X., Coll, T., Davidson, M. M., Chan, T. O., Feldman, A. M., et al. (2010). The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res. 87, 449–458. doi: 10.1093/cvr/cvq080
Amati, F., Dubé, J. J., Alvarez-Carnero, E., Edreira, M. M., Chomentowski, P., Coen, P. M., et al. (2011). Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes 60, 2588–2597. doi: 10.2337/db10-1221
Arany, Z., He, H., Lin, J., Hoyer, K., Handschin, C., Toka, O., et al. (2005). Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 1, 259–271. doi: 10.1016/j.cmet.2005.03.002
Ayala, A., Muñoz, M. F., and Argüelles, S. (2014). Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014:360438. doi: 10.1155/2014/360438
Banke, N. H., Wende, A. R., Leone, T. C., O'Donnell, J. M., Abel, E. D., Kelly, D. P., et al. (2010). Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ. Res. 107, 233–241. doi: 10.1161/CIRCRESAHA.110.221713
Benton, C. R., Han, X. X., Febbraio, M., Graham, T. E., and Bonen, A. (2006). Inverse relationship between PGC-1alpha protein expression and triacylglycerol accumulation in rodent skeletal muscle. J. Appl. Physiol. 100, 377–383. doi: 10.1152/japplphysiol.00781.2005
Benton, C. R., Holloway, G. P., Han, X. X., Yoshida, Y., Snook, L. A., Lally, J., et al. (2010). Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1alpha) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia 53, 2008–2019. doi: 10.1007/s00125-010-1773-1
Benton, C. R., Nickerson, J. G., Lally, J., Han, X. X., Holloway, G. P., Glatz, J. F., et al. (2008). Modest PGC-1alpha overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in subsarcolemmal, not intermyofibrillar, mitochondria. J. Biol. Chem. 283, 4228–4240. doi: 10.1074/jbc.M704332200
Bergman, B. C., Perreault, L., Hunerdosse, D. M., Koehler, M. C., Samek, A. M., and Eckel, R. H. (2010). Increased intramuscular lipid synthesis and low saturation relate to insulin sensitivity in endurance-trained athletes. J. Appl. Physiol. 108, 1134–1141. doi: 10.1152/japplphysiol.00684.2009
Besseiche, A., Riveline, J. P., Gautier, J. F., Bréant, B., and Blondeau, B. (2015). Metabolic roles of PGC-1α and its implications for type 2 diabetes. Diabetes Metab. 41, 347–357. doi: 10.1016/j.diabet.2015.02.002
Boon, J., Hoy, A. J., Stark, R., Brown, R. D., Meex, R. C., Henstridge, D. C., et al. (2013). Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes 62, 401–410. doi: 10.2337/db12-0686
Bradley, N. S., Snook, L. A., Jain, S. S., Heigenhauser, G. J., Bonen, A., and Spriet, L. L. (2012). Acute endurance exercise increases plasma membrane fatty acid transport proteins in rat and human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 302, E183–E189. doi: 10.1152/ajpendo.00254.2011
Buchanan, J., Mazumder, P. K., Hu, P., Chakrabarti, G., Roberts, M. W., Yun, U. J., et al. (2005). Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146, 5341–5349. doi: 10.1210/en.2005-0938
Burkart, E. M., Sambandam, N., Han, X., Gross, R. W., Courtois, M., Gierasch, C. M., et al. (2007). Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Invest. 117, 3930–3939. doi: 10.1172/JCI32578
Buroker, N. E., Ning, X. H., and Portman, M. (2008). Cardiac PPARalpha protein expression is constant as alternate nuclear receptors and PGC-1 coordinately increase during the postnatal metabolic transition. PPAR Res. 2008:279531. doi: 10.1155/2008/279531
Calvo, J. A., Daniels, T. G., Wang, X., Paul, A., Lin, J., Spiegelman, B. M., et al. (2008). Muscle-specific expression of PPARgamma coactivator-1alpha improves exercise performance and increases peak oxygen uptake. J. Appl. Physiol. 104, 1304–1312. doi: 10.1152/japplphysiol.01231.2007
Campbell, S. E., Tandon, N. N., Woldegiorgis, G., Luiken, J. J., Glatz, J. F., and Bonen, A. (2004). A novel function for fatty acid translocase (FAT)/CD36: involvement in long chain fatty acid transfer into the mitochondria. J. Biol. Chem. 279, 36235–36241. doi: 10.1074/jbc.M400566200
Carley, A. N., and Kleinfeld, A. M. (2011). Fatty acid (FFA) transport in cardiomyocytes revealed by imaging unbound FFA is mediated by an FFA pump modulated by the CD36 protein. J. Biol. Chem. 286, 4589–4597. doi: 10.1074/jbc.M110.182162
Chabowski, A., Coort, S. L., Calles-Escandon, J., Tandon, N. N., Glatz, J. F., Luiken, J. J., et al. (2005). The subcellular compartmentation of fatty acid transporters is regulated differently by insulin and by AICAR. FEBS Lett. 579, 2428–2432. doi: 10.1016/j.febslet.2004.11.118
Chabowski, A., Górski, J., Glatz, J. F., P Luiken, J. J., and Bonen, A. (2008). Protein-mediated fatty acid uptake in the heart. Curr. Cardiol. Rev. 4, 12–21. doi: 10.2174/157340308783565429
Choi, C. S., Befroy, D. E., Codella, R., Kim, S., Reznick, R. M., Hwang, Y. J., et al. (2008). Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. U.S.A. 105, 19926–19931. doi: 10.1073/pnas.0810339105
Coll, T., Eyre, E., Rodríguez-Calvo, R., Palomer, X., Sánchez, R. M., Merlos, M., et al. (2008). Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J. Biol. Chem. 283, 11107–11116. doi: 10.1074/jbc.M708700200
Coll, T., Jové, M., Rodríguez-Calvo, R., Eyre, E., Palomer, X., Sánchez, R. M., et al. (2006). Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes 55, 2779–2787. doi: 10.2337/db05-1494
Consitt, L. A., Bell, J. A., Koves, T. R., Muoio, D. M., Hulver, M. W., Haynie, K. R., et al. (2010). Peroxisome proliferator-activated receptor-gamma coactivator-1alpha overexpression increases lipid oxidation in myocytes from extremely obese individuals. Diabetes 59, 1407–1415. doi: 10.2337/db09-1704
Coort, S. L., Coumans, W. A., Bonen, A., van der Vusse, G. J., Glatz, J. F., and Luiken, J. J. (2005). Divergent effects of rosiglitazone on protein-mediated fatty acid uptake in adipose and in muscle tissues of Zucker rats. J. Lipid Res. 46, 1295–1302. doi: 10.1194/jlr.M400426-JLR200
Coort, S. L., Hasselbaink, D. M., Koonen, D. P., Willems, J., Coumans, W. A., Chabowski, A., et al. (2004). Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese zucker rats. Diabetes 53, 1655–1663. doi: 10.2337/diabetes.53.7.1655
Crunkhorn, S., Dearie, F., Mantzoros, C., Gami, H., da Silva, W. S., Espinoza, D., et al. (2007). Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 282, 15439–15450. doi: 10.1074/jbc.M611214200
Dillon, L. M., Rebelo, A. P., and Moraes, C. T. (2012). The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 64, 231–241. doi: 10.1002/iub.608
Duncan, J. G., Fong, J. L., Medeiros, D. M., Finck, B. N., and Kelly, D. P. (2007). Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation 115, 909–917. doi: 10.1161/CIRCULATIONAHA.106.662296
Egan, B., and Zierath, J. R. (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 17, 162–184. doi: 10.1016/j.cmet.2012.12.012
Erion, D. M., Park, H. J., and Lee, H. Y. (2016). The role of lipids in the pathogenesis and treatment of type 2 diabetes and associated co-morbidities. BMB Rep. 49, 139–148. doi: 10.5483/BMBRep.2016.49.3.268
Espinoza, D. O., Boros, L. G., Crunkhorn, S., Gami, H., and Patti, M. E. (2010). Dual modulation of both lipid oxidation and synthesis by peroxisome proliferator-activated receptor-gamma coactivator-1alpha and−1beta in cultured myotubes. FASEB J. 24, 1003–1014. doi: 10.1096/fj.09-133728
Fabregat-Andres, O., Paredes, F., Monsalve, M., Milara, J., Ridocci-Soriano, F., Gonzalez-Hervas, S., et al. (2016). mRNA PGC-1α levels in blood samples reliably correlates with its myocardial expression: study in patients undergoing cardiac surgery. Anatol. J. Cardiol. 16, 622–629. doi: 10.5152/AnatolJCardiol.2015.6466
Ferreira, R., Vitorino, R., Alves, R. M., Appell, H. J., Powers, S. K., Duarte, J. A., et al. (2010). Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 10, 3142–3154. doi: 10.1002/pmic.201000173
Finck, B. N., Lehman, J. J., Leone, T. C., Welch, M. J., Bennett, M. J., Kovacs, A., et al. (2002). The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Invest. 109, 121–130. doi: 10.1172/JCI0214080
Frohnert, B. I., Hui, T. Y., and Bernlohr, D. A. (1999). Identification of a functional peroxisome proliferator-responsive element in the murine fatty acid transport protein gene. J. Biol. Chem. 274, 3970–3977. doi: 10.1074/jbc.274.7.3970
Furuhashi, M., and Hotamisligil, G. S. (2008). Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 7, 489–503. doi: 10.1038/nrd2589
Gacias, M., Pérez-Martí, A., Pujol-Vidal, M., Marrero, P. F., Haro, D., and Relat, J. (2012). PGC-1β regulates mouse carnitine-acylcarnitine translocase through estrogen-related receptor α. Biochem. Biophys. Res. Commun. 423, 838–843. doi: 10.1016/j.bbrc.2012.06.051
Hage Hassan, R., Bourron, O., and Hajduch, E. (2014). Defect of insulin signal in peripheral tissues: Important role of ceramide. World J. Diabetes 5, 244–257. doi: 10.4239/wjd.v5.i3.244
Hammarstedt, A., Jansson, P. A., Wesslau, C., Yang, X., and Smith, U. (2003). Reduced expression of PGC-1 and insulin-signaling molecules in adipose tissue is associated with insulin resistance. Biochem. Biophys. Res. Commun. 301, 578–582. doi: 10.1016/S0006-291X(03)00014-7
Hammarstedt, A., Pihlajamäki, J., Graham, T. E., Kainulainen, S., Kahn, B. B., Laakso, M., et al. (2008). High circulating levels of RBP4 and mRNA levels of aP2, PGC-1alpha and UCP-2 predict improvement in insulin sensitivity following pioglitazone treatment of drug-naïve type 2 diabetic subjects. J. Intern. Med. 263, 440–449. doi: 10.1111/j.1365-2796.2007.01914.x
Han, X. X., Chabowski, A., Tandon, N. N., Calles-Escandon, J., Glatz, J. F., Luiken, J. J., et al. (2007). Metabolic challenges reveal impaired fatty acid metabolism and translocation of FAT/CD36 but not FABPpm in obese Zucker rat muscle. Am. J. Physiol. Endocrinol. Metab. 293, E566–E575. doi: 10.1152/ajpendo.00106.2007
Handschin, C., Choi, C. S., Chin, S., Kim, S., Kawamori, D., Kurpad, A. J., et al. (2007). Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J. Clin. Invest. 117, 3463–3474. doi: 10.1172/JCI31785
Hatazawa, Y., Senoo, N., Tadaishi, M., Ogawa, Y., Ezaki, O., Kamei, Y., et al. (2015). Metabolomic analysis of the skeletal muscle of mice overexpressing PGC-1α. PLoS ONE 10:e0129084. doi: 10.1371/journal.pone.0129084
Heilbronn, L., Chen, M., Macpherson, A., Owens, J., and Wittert, G. (2012). Obesity alone or with type 2 diabetes is associated with tissue specific alterations in DNA methylation and gene expression of PPARGC1A and IGF2. J. Diabetes Res. Clin. Metab. 1:16. doi: 10.7243/2050-0866-1-16
Hoeks, J., Arany, Z., Phielix, E., Moonen-Kornips, E., Hesselink, M. K., and Schrauwen, P. (2012). Enhanced lipid-but not carbohydrate-supported mitochondrial respiration in skeletal muscle of PGC-1α overexpressing mice. J. Cell. Physiol. 227, 1026–1033. doi: 10.1002/jcp.22812
Holloway, G. P., Benton, C. R., Mullen, K. L., Yoshida, Y., Snook, L. A., Han, X. X., et al. (2009). In obese rat muscle transport of palmitate is increased and is channeled to triacylglycerol storage despite an increase in mitochondrial palmitate oxidation. Am. J. Physiol. Endocrinol. Metab. 296, E738–E747. doi: 10.1152/ajpendo.90896.2008
Holloway, G. P., Thrush, A. B., Heigenhauser, G. J., Tandon, N. N., Dyck, D. J., Bonen, A., et al. (2007). Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am. J. Physiol. Endocrinol. Metab. 292, E1782–E1789. doi: 10.1152/ajpendo.00639.2006
Hommelberg, P. P., Plat, J., Sparks, L. M., Schols, A. M., van Essen, A. L., Kelders, M. C., et al. (2011). Palmitate-induced skeletal muscle insulin resistance does not require NF-κB activation. Cell. Mol. Life Sci. 68, 1215–1225. doi: 10.1007/s00018-010-0515-3
Hoshino, D., Yoshida, Y., Holloway, G. P., Lally, J., Hatta, H., and Bonen, A. (2012). Clenbuterol, a β2-adrenergic agonist, reciprocally alters PGC-1 alpha and RIP140 and reduces fatty acid and pyruvate oxidation in rat skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R373–R384. doi: 10.1152/ajpregu.00183.2011
Huang, T. Y., Zheng, D., Houmard, J. A., Brault, J. J., Hickner, R. C., and Cortright, R. N. (2017). Overexpression of PGC-1α increases peroxisomal activity and mitochondrial fatty acid oxidation in human primary myotubes. Am. J. Physiol. Endocrinol. Metab. 312, E253–E263. doi: 10.1152/ajpendo.00331.2016
Huss, J. M., and Kelly, D. P. (2004). Nuclear receptor signaling and cardiac energetics. Circ. Res. 95, 568–578. doi: 10.1161/01.RES.0000141774.29937.e3
Huss, J. M., Torra, I. P., Staels, B., Giguère, V., and Kelly, D. P. (2004). Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 24, 9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004
Irrcher, I., Adhihetty, P. J., Sheehan, T., Joseph, A. M., and Hood, D. A. (2003). PPARgamma coactivator-1alpha expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am. J. Physiol. Cell Physiol. 284, C1669–C1677. doi: 10.1152/ajpcell.00409.2002
Jang, C., Oh, S. F., Wada, S., Rowe, G. C., Liu, L., Chan, M. C., et al. (2016). A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 22, 421–426. doi: 10.1038/nm.4057
Kajimura, S., Seale, P., Tomaru, T., Erdjument-Bromage, H., Cooper, M. P., Ruas, J. L., et al. (2008). Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 22, 1397–1409. doi: 10.1101/gad.1666108
Kalinowska, A., Górski, J., Harasim, E., Harasiuk, D., Bonen, A., and Chabowski, A. (2009). Differential effects of chronic, in vivo, PPAR's stimulation on the myocardial subcellular redistribution of FAT/CD36 and FABPpm. FEBS Lett. 583, 2527–2534. doi: 10.1016/j.febslet.2009.07.008
Kim, J., Fernand, V. E., Henagan, T. M., Shin, J., Huypens, P., Newman, S., et al. (2016). Regulation of brown and white adipocyte transcriptome by the transcriptional coactivator NT-PGC-1α. PLoS ONE 11:e0159990. doi: 10.1371/journal.pone.0159990
King, K. L., Stanley, W. C., Rosca, M., Kerner, J., Hoppel, C. L., and Febbraio, M. (2007). Fatty acid oxidation in cardiac and skeletal muscle mitochondria is unaffected by deletion of CD36. Arch. Biochem. Biophys. 467, 234–238. doi: 10.1016/j.abb.2007.08.020
Kleiner, S., Mepani, R. J., Laznik, D., Ye, L., Jurczak, M. J., Jornayvaz, F. R., et al. (2012). Development of insulin resistance in mice lacking PGC-1α in adipose tissues. Proc. Natl. Acad. Sci. U.S.A. 109, 9635–9640. doi: 10.1073/pnas.1207287109
Koh, J. H., Hancock, C. R., Terada, S., Higashida, K., Holloszy, J. O., and Han, D. H. (2017). PPARβ is essential for maintaining normal levels of PGC-1α and mitochondria and for the increase in muscle mitochondria induced by exercise. Cell Metab. 25:e1175. doi: 10.1016/j.cmet.2017.04.029
Koves, T. R., Li, P., An, J., Akimoto, T., Slentz, D., Ilkayeva, O., et al. (2005a). Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 280, 33588–33598. doi: 10.1074/jbc.M507621200
Koves, T. R., Noland, R. C., Bates, A. L., Henes, S. T., Muoio, D. M., and Cortright, R. N. (2005b). Subsarcolemmal and intermyofibrillar mitochondria play distinct roles in regulating skeletal muscle fatty acid metabolism. Am. J. Physiol. Cell Physiol. 288, C1074–C1082. doi: 10.1152/ajpcell.00391.2004
Kupr, B., and Handschin, C. (2015). Complex coordination of cell plasticity by a PGC-1α-controlled transcriptional network in skeletal muscle. Front. Physiol. 6:325. doi: 10.3389/fphys.2015.00325
Kurek, K., Mikłosz, A., Łukaszuk, B., Chabowski, A., Górski, J., and Zendzian-Piotrowska, M. (2015). Inhibition of ceramide de novo synthesis ameliorates diet induced skeletal muscles insulin resistance. J. Diabetes Res. 2015:154762. doi: 10.1155/2015/154762
Lai, N., Kummitha, C., and Hoppel, C. (2017). Defects in skeletal muscle subsarcolemmal mitochondria in a non-obese model of type 2 diabetes mellitus. PLoS ONE 12:e0183978. doi: 10.1371/journal.pone.0183978
Lee, T. W., Bai, K. J., Lee, T. I., Chao, T. F., Kao, Y. H., and Chen, Y. J. (2017). PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J. Biomed. Sci. 24:5. doi: 10.1186/s12929-016-0309-5
Lehman, J. J., and Kelly, D. P. (2002). Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin. Exp. Pharmacol. Physiol. 29, 339–345. doi: 10.1046/j.1440-1681.2002.03655.x
Lehman, J. J., Barger, P. M., Kovacs, A., Saffitz, J. E., Medeiros, D. M., and Kelly, D. P. (2000). Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106, 847–856. doi: 10.1172/JCI10268
Leone, T. C., Lehman, J. J., Finck, B. N., Schaeffer, P. J., Wende, A. R., Boudina, S., et al. (2005). PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3:e101. doi: 10.1371/journal.pbio.0030101
Liang, H., and Ward, W. F. (2006). PGC-1alpha: a key regulator of energy metabolism. Adv. Physiol. Educ. 30, 145–151. doi: 10.1152/advan.00052.2006
Lin, J., Handschin, C., and Spiegelman, B. M. (2005). Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370. doi: 10.1016/j.cmet.2005.05.004
Lin, J., Wu, P. H., Tarr, P. T., Lindenberg, K. S., St-Pierre, J., Zhang, C. Y., et al. (2004). Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119, 121–135. doi: 10.1016/j.cell.2004.09.013
Luiken, J. J., Arumugam, Y., Bell, R. C., Calles-Escandon, J., Tandon, N. N., Glatz, J. F., et al. (2002). Changes in fatty acid transport and transporters are related to the severity of insulin deficiency. Am. J. Physiol. Endocrinol. Metab. 283, E612–E621. doi: 10.1152/ajpendo.00011.2002
Luiken, J. J., Arumugam, Y., Dyck, D. J., Bell, R. C., Pelsers, M. M., Turcotte, L. P., et al. (2001). Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J. Biol. Chem. 276, 40567–40573. doi: 10.1074/jbc.M100052200
Luiken, J. J., Turcotte, L. P., and Bonen, A. (1999). Protein-mediated palmitate uptake and expression of fatty acid transport proteins in heart giant vesicles. J. Lipid Res. 40, 1007–1016.
Łukaszuk, B., Kurek, K., Mikłosz, A., Zendzian-Piotrowska, M., and Chabowski, A. (2015a). The role of PGC-1alpha in the development of insulin resistance in skeletal muscle - revisited. Cell. Physiol. Biochem. 37, 2288–2296. doi: 10.1159/000438584
Lukaszuk, B., Miklosz, A., Chabowski, A., and Górski, J. (2015b). Modest decrease in PGC1α results in TAG accumulation but not in insulin resistance in L6 myotubes. Cell. Physiol. Biochem. 35, 1609–1622. doi: 10.1159/000373975
Manio, M. C. C., Matsumura, S., Masuda, D., and Inoue, K. (2017). CD36 is essential for endurance improvement, changes in whole-body metabolism, and efficient PPAR-related transcriptional responses in the muscle with exercise training. Physiol. Rep. 5:e12382. doi: 10.14814/phy2.13282
Mansor, L. S., Sousa Fialho, M. D. L., Yea, G., Coumans, W. A., West, J. A., Kerr, M., et al. (2017). Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc. Res. 113, 737–748. doi: 10.1093/cvr/cvx045
Martínez-Redondo, V., Pettersson, A. T., and Ruas, J. L. (2015). The hitchhiker's guide to PGC-1α isoform structure and biological functions. Diabetologia 58, 1969–1977. doi: 10.1007/s00125-015-3671-z
Maruyama, H., Kiyono, S., Kondo, T., Sekimoto, T., and Yokosuka, O. (2016). Palmitate-induced regulation of PPARγ via PGC1α: a mechanism for lipid accumulation in the liver in nonalcoholic fatty liver disease. Int. J. Med. Sci. 13, 169–178. doi: 10.7150/ijms.13581
Mathai, A. S., Bonen, A., Benton, C. R., Robinson, D. L., and Graham, T. E. (2008). Rapid exercise-induced changes in PGC-1alpha mRNA and protein in human skeletal muscle. J. Appl. Physiol. 105, 1098–1105. doi: 10.1152/japplphysiol.00847.2007
Mehlem, A., Palombo, I., Wang, X., Hagberg, C. E., Eriksson, U., and Falkevall, A. (2016). PGC-1α Coordinates Mitochondrial Respiratory Capacity and Muscular Fatty Acid Uptake via Regulation of VEGF-B. Diabetes 65, 861–873. doi: 10.2337/db15-1231
Mikłosz, A., Łukaszuk, B., Zendzian-Piotrowska, M., Branska-Januszewska, J., Ostrowska, H., and Chabowski, A. (2017). Challenging of AS160/TBC1D4 alters intracellular lipid milieu in L6 myotubes incubated with palmitate. J. Cell. Physiol. 232, 2373–2386. doi: 10.1002/jcp.25632
Miura, S., Kai, Y., Ono, M., and Ezaki, O. (2003). Overexpression of peroxisome proliferator-activated receptor gamma coactivator-1alpha down-regulates GLUT4 mRNA in skeletal muscles. J. Biol. Chem. 278, 31385–31390. doi: 10.1074/jbc.M304312200
Mormeneo, E., Jimenez-Mallebrera, C., Palomer, X., De Nigris, V., Vázquez-Carrera, M., Orozco, A., et al. (2012). PGC-1α induces mitochondrial and myokine transcriptional programs and lipid droplet and glycogen accumulation in cultured human skeletal muscle cells. PLoS ONE 7:e29985. doi: 10.1371/journal.pone.0029985
Mortensen, O. H., Frandsen, L., Schjerling, P., Nishimura, E., and Grunnet, N. (2006). PGC-1alpha and PGC-1beta have both similar and distinct effects on myofiber switching toward an oxidative phenotype. Am. J. Physiol. Endocrinol. Metab. 291, E807–E816. doi: 10.1152/ajpendo.00591.2005
Nam, H. C., Whiting, D., Forouhi, N., Guariguata, L., Hambleton, I., Li, R., et al. (2015). IDF Diabetes Atlas, 7th Edn. International Diabetes Federation. Available online at: http://www.diabetesatlas.org/
Nickerson, J. G., Alkhateeb, H., Benton, C. R., Lally, J., Nickerson, J., Han, X. X., et al. (2009). Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1 and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J. Biol. Chem. 284, 16522–16530. doi: 10.1074/jbc.M109.004788
Nieto-Vazquez, I., Fernández-Veledo, S., de Alvaro, C., and Lorenzo, M. (2008). Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes 57, 3211–3221. doi: 10.2337/db07-1062
Nikolić, N., Rhedin, M., Rustan, A. C., Storlien, L., Thoresen, G. H., and Strömstedt, M. (2012). Overexpression of PGC-1α increases fatty acid oxidative capacity of human skeletal muscle cells. Biochem. Res. Int. 2012:714074. doi: 10.1155/2012/714074
Pagel-Langenickel, I., Bao, J., Joseph, J. J., Schwartz, D. R., Mantell, B. S., Xu, X., et al. (2008). PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J. Biol. Chem. 283, 22464–22472. doi: 10.1074/jbc.M800842200
Pardo, R., Enguix, N., Lasheras, J., Feliu, J. E., Kralli, A., and Villena, J. A. (2011). Rosiglitazone-induced mitochondrial biogenesis in white adipose tissue is independent of peroxisome proliferator-activated receptor γ coactivator-1α. PLoS ONE6:e26989. doi: 10.1371/journal.pone.0026989
Popov, D. V., Lysenko, E. A., Kuzmin, I. V., Vinogradova, V., and Grigoriev, A. I. (2015). Regulation of PGC-1α isoform expression in skeletal muscles. Acta Nature 7, 48–59.
Prasannarong, M., Santos, F. R., Hooshmand, P., Hooshmand, P., Giovannini, F. J., and Henriksen, E. J. (2014). The lipid peroxidation end-product and oxidant 4-hydroxynonenal induces insulin resistance in rat slow-twitch skeletal muscle. Arch. Physiol. Biochem. 120, 22–28. doi: 10.3109/13813455.2013.834937
Puigserver, P., Wu, Z., Park, C. W., Graves, R., Wright, M., and Spiegelman, B. M. (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839. doi: 10.1016/S0092-8674(00)81410-5
Rahnert, J. A., Zheng, B., Hudson, M. B., Woodworth-Hobbs, M. E., and Price, S. R. (2016). Glucocorticoids alter CRTC-CREB signaling in muscle cells: impact on PGC-1α expression and atrophy markers. PLoS ONE 11:e0159181. doi: 10.1371/journal.pone.0159181
Richardson, D. K., Kashyap, S., Bajaj, M., Cusi, K., Mandarino, S. J., Finlayson, J., et al. (2005). Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J. Biol. Chem. 280, 10290–10297. doi: 10.1074/jbc.M408985200
Rowe, G. C., Jiang, A., and Arany, Z. (2010). PGC-1 coactivators in cardiac development and disease. Circ. Res. 107, 825–838. doi: 10.1161/CIRCRESAHA.110.223818
Ruschke, K., Fishbein, L., Dietrich, A., Klöting, N., Tönjes, A., Oberbach, A., et al. (2010). Gene expression of PPARgamma and PGC-1alpha in human omental and subcutaneous adipose tissues is related to insulin resistance markers and mediates beneficial effects of physical training. Eur. J. Endocrinol. 162, 515–523. doi: 10.1530/EJE-09-0767
Russell, L. K., Mansfield, C. M., Lehman, J. J., Kovacs, A., Courtois, M., Saffitz, J. E., et al. (2004). Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 94, 525–533. doi: 10.1161/01.RES.0000117088.36577.EB
Rytinki, M. M., and Palvimo, J. J. (2009). SUMOylation attenuates the function of PGC-1alpha. J. Biol. Chem. 284, 26184–26193. doi: 10.1074/jbc.M109.038943
Sano, M., Tokudome, S., Shimizu, N., Yoshikawa, N., Ogawa, C., Shirakawa, K., et al. (2007). Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor gamma coactivator 1alpha. J. Biol. Chem. 282, 25970–25980. doi: 10.1074/jbc.M703634200
Sano, M., Wang, S. C., Shirai, M., Scaglia, F., Xie, M., Sakai, S., et al. (2004). Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 23, 3559–3569. doi: 10.1038/sj.emboj.7600351
Schilling, J. D. (2015). The mitochondria in diabetic heart failure: from pathogenesis to therapeutic promise. Antioxid. Redox Signal. 22, 1515–1526. doi: 10.1089/ars.2015.6294
Schrauwen, P., Hoeks, J., and Hesselink, M. K. (2006). Lipid-induced cell stress and insulin resistance. Scand. J. Food Nutr. 50(Suppl. 2), 62–67. doi: 10.1080/17482970601066132
Schwenk, R. W., Holloway, G. P., Luiken, J. J., Bonen, A., and Glatz, J. F. (2010). Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fatty Acids 82, 149–154. doi: 10.1016/j.plefa.2010.02.029
Scimè, A., Grenier, G., Huh, M. S., Gillespie, M. A., Bevilacqua, L., Harper, M. E., et al. (2005). Rb and p107 regulate preadipocyte differentiation into white versus brown fat through repression of PGC-1alpha. Cell Metab. 2, 283–295. doi: 10.1016/j.cmet.2005.10.002
Semple, R. K., Crowley, V. C., Sewter, C. P., Laudes, M., Christodoulides, C., Considine, R. V., et al. (2004). Expression of the thermogenic nuclear hormone receptor coactivator PGC-1alpha is reduced in the adipose tissue of morbidly obese subjects. Int. J. Obes. Relat. Metab. Disord. 28, 176–179. doi: 10.1038/sj.ijo.0802482
Serra, D., Mera, P., Malandrino, M. I., Mir, J. F., and Herrero, L. (2013). Mitochondrial fatty acid oxidation in obesity. Antioxid. Redox Signal. 19, 269–284. doi: 10.1089/ars.2012.4875
Shu, L., Hoo, R. L., Wu, X., Pan, Y., Lee, I. P., Cheong, L. Y., et al. (2017). A-FABP mediates adaptive thermogenesis by promoting intracellular activation of thyroid hormones in brown adipocytes. Nat. Commun. 8:14147. doi: 10.1038/ncomms14147
Smith, B. K., Mukai, K., Lally, J. S., Maher, A. C., Gurd, B. J., Heigenhauser, G. J., et al. (2013). AMP-activated protein kinase is required for exercise-induced peroxisome proliferator-activated receptor co-activator 1 translocation to subsarcolemmal mitochondria in skeletal muscle. J. Physiol. 591, 1551–1561. doi: 10.1113/jphysiol.2012.245944
Son, N. H., Park, T. S., Yamashita, H., Yokoyama, M., Huggins, L. A., Okajima, K., et al. (2007). Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J. Clin. Invest. 117, 2791–2801. doi: 10.1172/JCI30335
Sparks, L. M., Xie, H., Koza, R. A., Mynatt, R., Hulver, M. W., Bray, G. A., et al. (2005). A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 54, 1926–1933. doi: 10.2337/diabetes.54.7.1926
Stump, D. D., Fan, X., and Berk, P. D. (2001). Oleic acid uptake and binding by rat adipocytes define dual pathways for cellular fatty acid uptake. J. Lipid Res. 42, 509–520.
Summermatter, S., Baum, O., Santos, G., Hoppeler, H., and Handschin, C. (2010). Peroxisome proliferator-activated receptor {gamma} coactivator 1{alpha} (PGC-1{alpha}) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J. Biol. Chem. 285, 32793–32800. doi: 10.1074/jbc.M110.145995
Summermatter, S., Shui, G., Maag, D., Santos, G., Wenk, M. R., and Handschin, C. (2013). PGC-1α improves glucose homeostasis in skeletal muscle in an activity-dependent manner. Diabetes 62, 85–95. doi: 10.2337/db12-0291
Summermatter, S., Troxler, H., Santos, G., and Handschin, C. (2011). Coordinated balancing of muscle oxidative metabolism through PGC-1α increases metabolic flexibility and preserves insulin sensitivity. Biochem. Biophys. Res. Commun. 408, 180–185. doi: 10.1016/j.bbrc.2011.04.012
Szendroedi, J., Yoshimura, T., Phielix, E., Koliaki, C., Marcucci, M., Zhang, D., et al. (2014). Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. U.S.A. 111, 9597–9602. doi: 10.1073/pnas.1409229111
Taha, T. A., Mullen, T. D., and Obeid, L. M. (2006). A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta. 1758, 2027–2036. doi: 10.1016/j.bbamem.2006.10.018
Teboul, L., Febbraio, M., Gaillard, D., Amri, E. Z., Silverstein, R., and Grimaldi, P. A. (2001). Structural and functional characterization of the mouse fatty acid translocase promoter: activation during adipose differentiation. Biochem. J. 360 (Pt 2), 305–312. doi: 10.1042/bj3600305
Uldry, M., Yang, W., St-Pierre, J., Lin, J., Seale, P., and Spiegelman, B. M. (2006). Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 3, 333–341. doi: 10.1016/j.cmet.2006.04.002
van der Lee, K. A., Vork, M. M., De Vries, J. E., Willemsen, P. H., Glatz, J. F., Reneman, R. S., et al. (2000). Long-chain fatty acid-induced changes in gene expression in neonatal cardiac myocytes. J. Lipid Res. 41, 41–47.
Váraljai, R., Islam, A. B., Beshiri, M. L., Rehman, J., Lopez-Bigas, N., and Benevolenskaya, E. V. (2015). Increased mitochondrial function downstream from KDM5A histone demethylase rescues differentiation in pRB-deficient cells. Genes Dev. 29, 1817–1834. doi: 10.1101/gad.264036.115
Vega, R. B., Horton, J. L., and Kelly, D. P. (2015). Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circ. Res. 116, 1820–1834. doi: 10.1161/CIRCRESAHA.116.305420
Vega, R. B., Huss, J. M., and Kelly, D. P. (2000). The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 20, 1868–1876. doi: 10.1128/MCB.20.5.1868-1876.2000
Viollet, B., Guigas, B., Sanz Garcia, N., Leclerc, J., Foretz, M., and Andreelli, F. (2012). Cellular and molecular mechanisms of metformin: an overview. Clin. Sci. 122, 253–270. doi: 10.1042/CS20110386
Wallberg, A. E., Yamamura, S., Malik, S., Spiegelman, B. M., and Roeder, R. G. (2003). Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1alpha. Mol. Cell. 12, 1137–1149. doi: 10.1016/S1097-2765(03)00391-5
Wang, P., Liu, J., Li, Y., Wu, S., Luo, J., Yang, H., et al. (2010). Peroxisome proliferator-activated receptor {delta} is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ. Res. 106, 911–919. doi: 10.1161/CIRCRESAHA.109.206185
Watanabe, K., Fujii, H., Takahashi, T., Kodama, M., Aizawa, Y., Ohta, Y., et al. (2000). Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J. Biol. Chem. 275, 22293–22299. doi: 10.1074/jbc.M000248200
Watt, M. J., Southgate, R. J., Holmes, A. G., and Febbraio, M. A. (2004). Suppression of plasma free fatty acids upregulates peroxisome proliferator-activated receptor (PPAR) alpha and delta and PPAR coactivator 1alpha in human skeletal muscle, but not lipid regulatory genes. J. Mol. Endocrinol. 33, 533–544. doi: 10.1677/jme.1.01499
Wende, A. R., Schaeffer, P. J., Parker, G. J., Zechner, C., Han, D. H., Chen, M. M., et al. (2007). A role for the transcriptional coactivator PGC-1alpha in muscle refueling. J. Biol. Chem. 282, 36642–36651. doi: 10.1074/jbc.M707006200
Wong, K. E., Mikus, C. R., Slentz, D. H., Seiler, S. E., DeBalsi, K. L., Ilkayeva, O. R., et al. (2015). Muscle-specific overexpression of PGC-1α does not augment metabolic improvements in response to exercise and caloric restriction. Diabetes 64, 1532–1543. doi: 10.2337/db14-0827
Yang, J., Sambandam, N., Han, X., Gross, R. W., Courtois, M., Kovacs, A., et al. (2007). CD36 deficiency rescues lipotoxic cardiomyopathy. Circ. Res. 100, 1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6
Keywords: PGC-1α, FATPs, FABPpm, FAT/CD36, lipid metabolism, insulin sensitive tissues
Citation: Supruniuk E, Mikłosz A and Chabowski A (2017) The Implication of PGC-1α on Fatty Acid Transport across Plasma and Mitochondrial Membranes in the Insulin Sensitive Tissues. Front. Physiol. 8:923. doi: 10.3389/fphys.2017.00923
Received: 19 July 2017; Accepted: 31 October 2017;
Published: 15 November 2017.
Edited by:
Anna Maria Giudetti, University of Salento, ItalyReviewed by:
Daniele Vergara, University of Salento, ItalyCopyright © 2017 Supruniuk, Mikłosz and Chabowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnieszka Mikłosz, YWduaWVzemthLm1pa2xvc3pAdW1iLmVkdS5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.