 Joris Wauman
Joris Wauman Lennart Zabeau
Lennart Zabeau Jan Tavernier
Jan Tavernier- 1Cytokine Receptor Laboratory, Faculty of Medicine and Health Sciences, Department of Biochemistry, Ghent University, Ghent, Belgium
- 2VIB Medical Biotechnology Center, VIB, Ghent, Belgium
Under normal physiological conditions, leptin and the leptin receptor (ObR) regulate the body weight by balancing food intake and energy expenditure. However, this adipocyte-derived hormone also directs peripheral processes, including immunity, reproduction, and bone metabolism. Leptin, therefore, can act as a metabolic switch connecting the body’s nutritional status to high energy consuming processes. We provide an extensive overview of current structural insights on the leptin–ObR interface and ObR activation, coupling to signaling pathways and their negative regulation, and leptin functioning under normal and pathophysiological conditions (obesity, autoimmunity, cancer, …). We also discuss possible cross-talk with other receptor systems on the receptor (extracellular) and signaling cascade (intracellular) levels.
Introduction
The identification of two spontaneous obese mouse strains at the Jackson Laboratory, ob/ob mice in 1950 and db/db mice in 1965, were the first steps toward the discovery of the leptin–ObR system. A series of elegant parabiosis experiments illustrated that db/db mice overexpressed a strong circulating satiety factor to which they cannot respond themselves. On the other hand, ob/ob animals do not produce this factor but lose weight when parabiotically paired to wild-type or db/db mice (1). This factor was cloned 40 years later by Friedman and colleagues as the product of the ob gene and called leptin after the Greek “leptos” meaning thin (2). The product of the db gene was identified as the ObR using an expression-cloning strategy based on the ability to bind leptin (3).
Leptin
Leptin, a hormone with cytokine-like characteristics, is mainly but not exclusively produced by adipose tissue in a way that its levels positively correlate with the energy stored in the body (4–6). Other sources of (lower) leptin expression include placenta, stomach, mammary epithelium, and skeletal muscle (7–9). Mature leptin is a non-glycosylated 16 kDa protein of 146 amino acids. The crystal structure at 2.4 Å resolution of leptin W110E, a mutation that dramatically increases solubility of the protein without affecting biological activity, shows a typical four-helical bundle structure. Four anti-parallel α-helices (A, B, C, and D) in an up-up-down-down arrangement are connected by one short (BC) and two long (AB and CD) loops. Leptin has two conserved cysteine residues (one in the CD loop and the C-terminal residue) that form a solvent-exposed disulfide bridge that tethers the CD loop to the C-terminal part of helix D. This disulfide bridge is crucial for structural stability, secretion, and biological activity (10, 11). These structural characteristics strongly resemble those found in granulocyte-colony stimulating factor (G-CSF) and interleukin-6 (IL-6) cytokines, and leptin is, therefore, classified as a long-chain cytokine.
Leptin Receptor
ObR is a single membrane-spanning receptor belonging to the class I cytokine receptor family (3). Up to now, six ObR isoforms are produced by alternative splicing or ectodomain shedding: ObRa–ObRf. These include one long form (ObRb; with an intracellular domain of 302 Aa); four short forms (ObRa, ObRc, ObRd, and ObRf, with cytoplasmic tails of 30–40 Aa with unique C-termini); and one soluble form (ObRe). ObRb contains three highly conserved tyrosine residues (Y985, Y1077, Y1138) required for efficient leptin signaling. ObRb is highly expressed in specific nuclei of the hypothalamus, a region of the brain involved in the regulation of body weight (12–14). Expression at functional levels can also be detected in a broad range of other cell types, in line with the pleiotropic effects of leptin (see further). Their expression pattern suggests that the short ObR’s play a role in transport of leptin over the blood–brain barrier (BBB) (15) and/or renal clearance (3). The soluble ObRe isoform, directly secreted in mice while in humans generated by ectodomain shedding (16, 17), modulates bio-availability of the hormone (18). All isoforms have an identical extracellular part consisting of six domains: an N-terminal domain (NTD), two CRH domains (CRH1 and CRH2), an immunoglobulin-like domain (IGD), and two additional membrane-proximal fibronectin type III (FN III) domains. This overall architecture of the extracellular domain and the sequence similarity resemble that of the G-CSF and gp130-related receptors. The ObR is heavily glycosylated causing an increase of 30–70 kDa in molecular weight (19, 20). N-glycosylation is predominant with 18 of 20 NXS motifs in the human receptor glycosylated, although some O-glycosylation is also present (19). N-glycanase F treatment reduces leptin binding of recombinant ObR extracellular domain by 80%, illustrating the importance of this type of modification in this process (20).
Leptin Biology and Disease
As mentioned above, loss-of-function mutations in the leptin or ObR genes (2, 21–23) or genetic ablation of leptin’s central signaling (16, 24) results in severe, early-onset obesity (25). To date, leptin and its receptor are the most crucial of factors identified, which control body weight by balancing food intake and energy expenditure in the adaptive response to altered energy states like fasting or starvation. As a product of adipose tissue, it signals the energy stored in the body and thereby functions as a negative feedback adipostat, an efferent satiety signal and an anti-obesity hormone.
In 2014, more than 1.9 billion adults were overweight (BMI levels 25–29 kg/m2), and of these, over 600 million were obese (BMI levels 30 kg/m2 and greater), making obesity and the associated metabolic syndrome a major health problem worldwide. It is a complex medical condition caused by an accumulation of excess body fat (calorie intake exceeds calorie expenditure) leading to negative effects on health including type 2 diabetes, heart diseases, obstructive sleep apnea, cancer, and joint disease. Obesity is caused by the interplay between environmental, genetic, and epigenetic factors. To date, not less than 50 genes (including leptin, ObR, and mediators of leptin signaling) are related to an increased obesity risk in humans and rodents (26). Paradoxically, levels of biologically active leptin are elevated in most obese subjects, and these patients do not respond to leptin treatment. Mechanisms underlying this “leptin resistance” are discussed later.
Leptin or ObR deficiencies not only cause severe obesity but also abnormalities in hematopoiesis (27), immunity (28), reproduction (29), angiogenesis (30), bone formation (31), and blood pressure (BP) (32). This lead to the concept that leptin can act as a “metabolic switch” that links the body’s energy stores to these high-energy demanding processes (33).
Ozata and colleagues were the first to report that seven members of a Turkish family with congenital leptin deficiency died during childhood due to infections pointing to a role for leptin in human immunity (34). Along the same line, leptin treatment of two children with congenital leptin deficiency (35) and in females with acquired hypoleptinemia (36) normalized absolute T cell numbers and nearly restored T cell proliferation responses and cytokine release profiles. Over the past decade, leptin emerged as a regulator of multiple cell types of both innate and adaptive immune responses (37). In innate immunity, leptin controls the activation of macrophages, neutrophils, monocytes, dendritic, and natural killer cells and promotes the production of pro-inflammatory cytokines. Thymic and splenic homeostasis, naïve CD4+ cell proliferation, promotion of TH1 responses, suppression of regulatory T cells, and activation of TH17 cells are the most important functions in adaptive immunity (37). Its pro-inflammatory characteristics link leptin to the onset and progression of several autoimmune diseases, including multiple sclerosis (38), antigen-induced arthritis (39), hepatitis (40, 41), colitis (41), and glomerulonephritis (42). Leptin-deficient rodents are often protected in experimental models for these diseases, while leptin administration restores sensitivity.
The observations that humans and rodents with congenital leptin deficiencies are sterile and that anorexia and obesity delay and accelerate the onset of puberty, respectively, led to the idea that leptin is an important player in reproduction (43). The hormone functions both directly on the ovaries and indirectly via gonadotrophin-releasing hormone, luteinizing hormone, or kisspeptin release (44).
Ob/ob and db/db mice display significant longer vertebral length and have higher bone mass (31). Leptin influences bone metabolism via central and peripheral pathways [reviewed in Ref. (45)]: it suppresses osteoblast proliferation and promotes osteoclast resorption through activation of neurons in the ventromedial hypothalamus. More direct effects include proliferation, survival, differentiation, or suppression of bone marrow mesenchymal stem cells, osteoblasts, osteoclasts, and chondrocytes, and the synthesis of collagen and extracellular matrix proteins.
Leptin affects BP in an apparently opposite way: on the one hand it causes chronic increase in BP and may contribute to obesity related hypertension, while on the other hand its metabolic actions (lowering appetite and increasing energy expenditure) tend to reduce BP (46).
Excess body fat in obesity can contribute to the development of cancer. Etiological causes not only include elevation of estrogen, insulin, insulin-like growth factors, leptin but also local inflammation and depressed immune function seen with excess adiposity. Elevated leptin levels are linked to an increased risk of myeloma (47) and of prostate (48), breast (49), colorectal (50), and renal (51) cancers. Leptin not only promotes survival and proliferation of several cancer cell lines directly but also promotes adhesion, invasion, metastasis (via upregulation of metalloproteinases, E-cadherin, and extracellular matrix proteins), and angiogenesis (via vascular endothelial growth factor and its receptor) in the tumor environment (52).
ObR Activation
Only a minor fraction of the ObR (10–20%) is expressed at the cellular surface, while the majority is found in intracellular compartments, including endoplasmic reticulum (ER), trans-Golgi, and endosomes (53). This subcellular distribution is the result of the partial retention of the receptor in the biosynthesis pathway and the constitutive, ligand-independent endocytosis of the receptor (see also further). Furthermore, co-immunoprecipitation of differentially tagged ObR’s (54, 55) or the high basal signal in the absence of leptin in bioluminescence resonance energy transfer (BRET) (56) and fluorescence resonance energy transfer (FRET) (57) suggests that the ObR forms pre-formed dimers (or oligomers) on the cellular surface. Numerous cytokine receptors can form these inactive, pre-formed receptor complexes, including the erythropoietin receptor (58–60), the growth hormone receptor (61), and the IL-6 receptor (62). Earlier studies suggested this oligomerization also in solution (63, 64), but more recent multi-angle laser light scattering and small-angle X-ray scattering (SAXS) show that the soluble ObRe is a monomer in the absence of leptin (65, 66).
ObR activation depends on the CRH2, IGD, and FN III domains (Figure 1). The ObR CRH2 domain is the major leptin-binding determinant in the receptor (67, 68). Depending on the technique and proteins used, CRH2 binds leptin with a KD of 0.2–15 nM in solution or 0.2–1.5 nM on the cellular surface [reviewed in Ref. (69)]. The binding affinity to the isolated CRH2 domain or to the complete extracellular domain is comparable, illustrating that this domain is indeed strictly required and sufficient (67, 70). A region of four consecutive hydrophobic residues has been identified as the leptin-binding site in the CRH2 domain (71, 72). The crystal structure of the CRH2 domain, in complex with a Fab fragment of a neutralizing Ab, was determined at 1.95 Å resolution (73). This is the first and, hitherto, the only high-resolution structure for (a part of) the ObR. The IGD and membrane-proximal domains have no detectable affinity for the ligand, but are nonetheless indispensable for receptor activation. Deletion of the IGD results in a receptor with wild-type affinity for leptin, but completely devoid of biological activity (67, 68). A conserved surface patch in the β-sheet formed by β-strands 3, 6, and 7 was identified as the leptin-binding site in this domain (74). Leptin contains a binding site (III) which allows contact with the IGD of a second ObR, thereby inducing dimerization and potential higher-order clustering with evidence for a 2:4 leptin:ObR hexameric structure (74). In the FN III domains, two conserved cysteine are crucial for receptor activation since combined mutation completely blocks activation of the receptor (64). Finally, deletion of the NTD and CRH1 domains hardly affects ObR functionality (67, 68). This seems in contrast to the obese phenotype of fatty Zucker rats resulting from the Q269P mutation in the CRH1 domain (75). Likewise, the naturally occurring single-nucleotide polymorphism Q223R causes obesity in Brazilian multiethnic subjects (76) and the increased susceptibility toward protozoan infections in children (77).
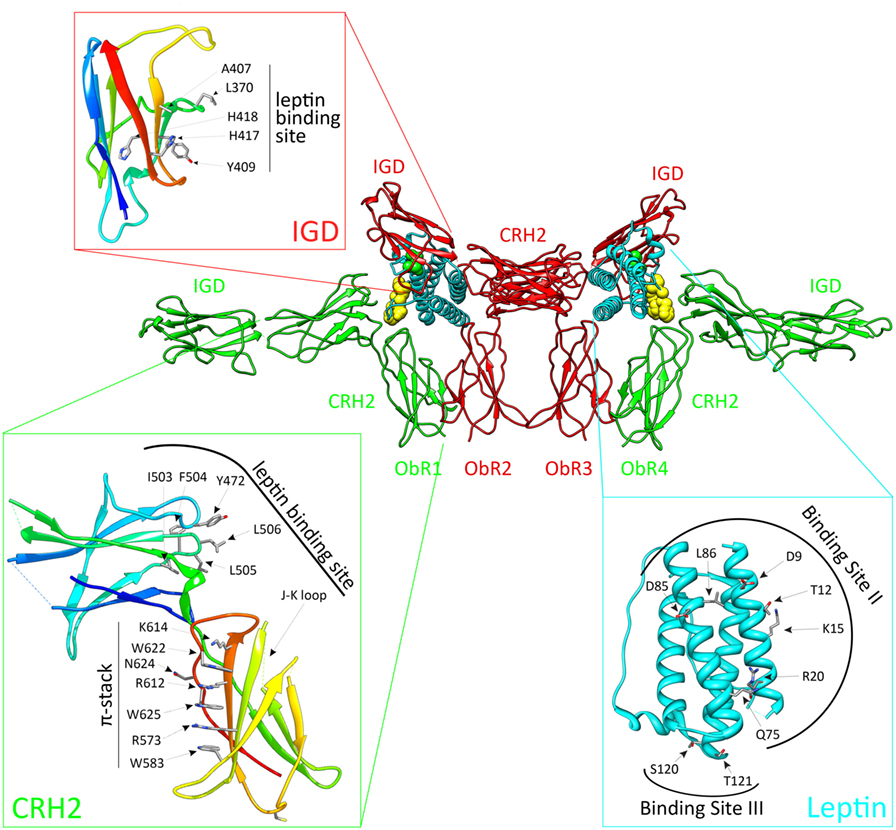
Figure 1. The activated leptin:ObR complex. Leptin clusters two pre-formed ObR dimers to form an activated 2:4 leptin:ObR complex. In this model, the ObR’s are colored green and red, leptin molecules cyan. The hormone binds with its binding site II to the CRH2 domain of the receptor, while site III residues interact with the immunoglobulin-like domain (IGD) of a second receptor. These residues are colored yellow and green, and defined in the cyan panel. Receptor residues involved in these interactions are shown in the red and green panels. For reasons of clarity, only the CRH2 and IGD domains of each receptor are shown.
Based on the structural and evolutionary relationship of leptin and its receptor with the G-CSF and IL-6 receptor systems, we selected and tested a panel of leptin mutants and thereby identified three putative receptor-binding sites (78). Mutations in binding site I (located at the C terminus of helix D) moderately affect binding and signaling. Binding site II residues (at the surface of helices A and C) are crucial for binding to the CRH2 domain, but mutations in this region have only limited effect on signaling (78). Two regions were independently proposed as the binding-site III, which interacts with the ObR IGD: the area around residues S120 and T121 at the N terminus of helix D (78) and the 39-LDFL-42 stretch in the AB loop (79). Mutation of either area is sufficient to create leptin antagonists in vitro and in vivo (78, 79).
Both 3D reconstructions of 2D negative-stain EM images by Mancour et al. (65) and the SAXS experiments by Moharana et al. (66) point to an analog 2:2 quaternary core leptin:ObR complex. In this model, leptin binds with its site II to the CRH2 domain of a first ObR and engages a second receptor via the site III–IGD interaction. Additional receptor–receptor interactions (e.g., between FN III domains) that occur at the cell surface, but not in solution, could lead to 2:4 or 4:4 leptin:ObR complexes. This higher-order clustering would be in line with our signaling-complementation assay (68). Finally, leptin stimulation increases BRET and FRET signals (see above), suggesting that reorganization within the pre-formed complexes and/or de novo oligomerization likely occur (56, 57).
ObR Signaling
The ObRb is mainly expressed by two distinct neuronal populations in the hypothalamic arcuate nucleus (ARC): the anorexigenic (appetite-depressing) POMC (pro-opiomelanocortin) neurons and the orexigenic (appetite-promoting) NPY/AgRP (neuropeptide Y/agouti-related peptide) neurons (80, 81) (Figure 2). ObRb activation in POMC neurons triggers POMC expression, a precursor peptide, that is further converted by prohormone convertases to α-melanocyte stimulating hormone (αMSH), which is secreted and signals by activating melanocortin receptors, MC3R, and MC4R. Deletion of MC3R and/or MC4R results in leptin resistance and obesity (82, 83). Specific reconstitution of ObRb expression in POMC neurons of ObRb-deficient mice not only modestly reduces body weight but also completely normalizes blood glucose levels, insulin sensitivity, and locomotor activity, indicating that leptin signaling in POMC neurons has a key role in regulating glucose homeostasis (84, 85). In contrast to POMC neurons, leptin suppresses the activity of NPY/AgRP neurons and the associated secretion of its orexigenic neuropeptides. AgRP is a potent antagonist of αMSH. Moreover, NPY/AgRP neurons innervate POMC neurons and inhibit POMC neuronal firing by releasing GABA (γ-aminobutyric acid) (86, 87). Selective deletion of ObRb in POMC or NPY/AgRP neurons only gives rise to a mild obese phenotype indicating that also other regions in the brain are involved in the control of energy homeostasis by leptin (88–90).
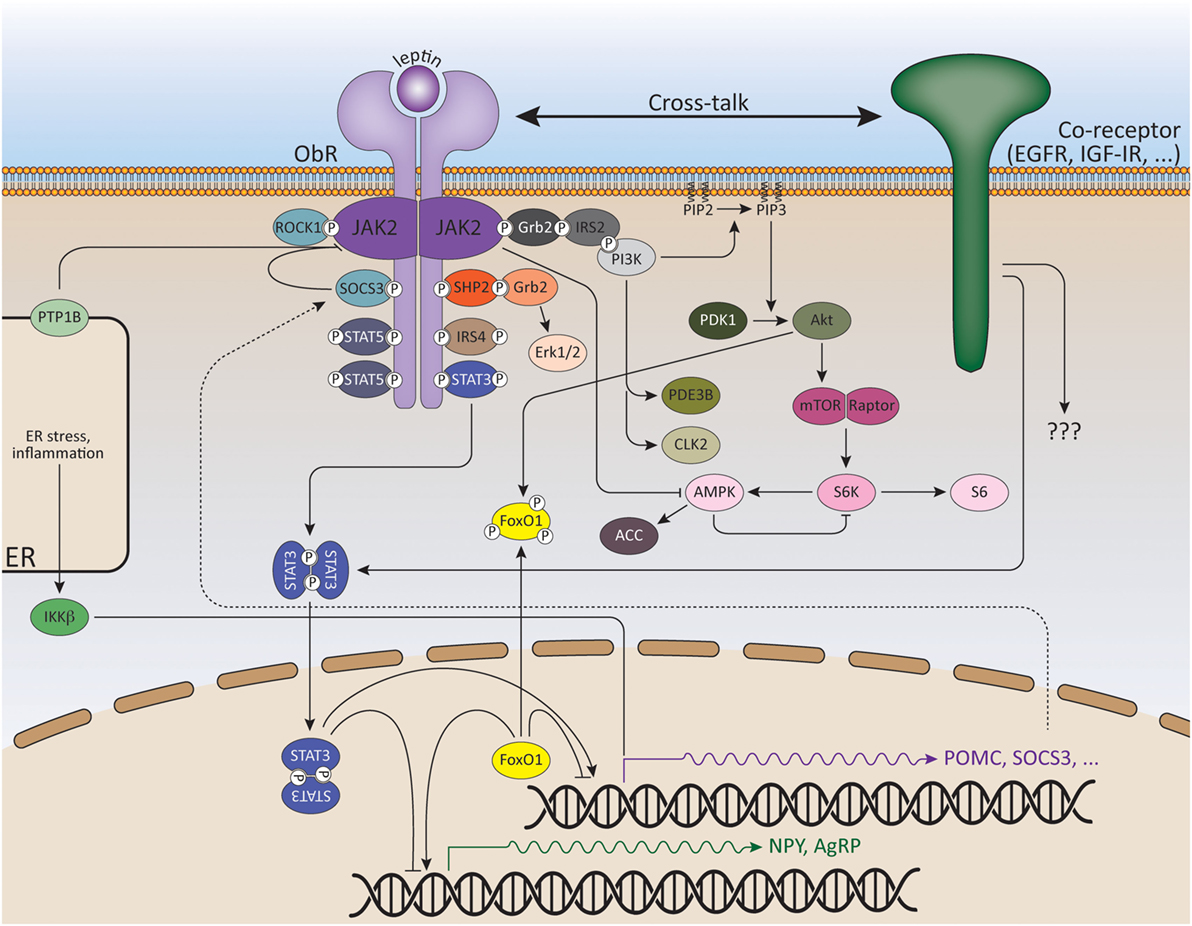
Figure 2. Signaling pathways of leptin and its downstream effectors. ObR oligomerization (here only dimerization shown for reasons of clarity) results in phosphorylation and activation of cytoplasmic associated JAK2 kinases. These activated JAKs phosphorylate tyrosine residues in the cytoplasmic tail of the receptor. Recruitment and activation of secondary signaling molecules allow ObR signaling via the JAK/STAT, MAPK, PI3K, AMPK, and mTOR pathways. See text for more details.
The JAK/STAT Pathway
The JAK/STAT (Janus kinase/Signal transducers and activators of transcription) pathway is probably the best explored pathway activated by leptin. Upon ligand binding and JAK2 activation, the conserved ObRb phosphotyrosine 1138 motif serves as a binding site for the SH2 domain of STAT3. STAT3 itself will subsequently become phosphorylated by the JAKs on Y705 and translocate as dimers to the nucleus, where it will modulate expression of several STAT3-responsive target genes, such as Suppressor of cytokine signaling 3 (SOCS3) (91, 92). SOCS3 acts as a potent negative regulator of the JAK/STAT pathway, thereby forming a negative feedback loop (93). The negative regulation of ObR signaling will be further discussed in the following section.
Experiments with neuron-specific STAT3−/− mice have pinpointed the essential role of STAT3 for the acute anorectic actions of leptin (94–96). Mice in which STAT3 is specifically deleted in ObRb neurons similary develop hyperphagic obesity with some preservation of glucose homeostasis (97). However, despite the fact that the POMC promotor has a STAT3-responsive element and that deletion of STAT3 in POMC neurons completely diminishes POMC induction, POMC-specific STAT3 knock-down did not completely abrogate the anorexigenic effect of leptin and only leads to a mild obese phenotype (98, 99). This indicates that also other signaling pathways in POMC neurons are involved in the energy homeostatic response to leptin and/or STAT3-dependent effects in other, non-ARC cells are involved. Likewise, genetic inactivation of STAT3 in NPY/AgRP neurons leads to mild hyperphagia, a decreased response to leptin and increased levels of basal NPY expression, while AgRP expression remains unaltered (100, 101).
Substitution of tyrosine 1138 with serine (s/s mice) disrupts STAT3 recruitment to the ObRb. S/s mice are, therefore, hyperphagic and obese, similar to db/db mice and show a suppression of melanocortin activity. However, s/s mice are less hyperglycemic and show normal fertility, indicating that STAT3-independent pathways control leptins’ effects on reproduction and glucose metabolism (102–104).
In addition to phosphorylation, leptin-induced STAT3 methylation by protein arginine N-methyltransferase 2 (PRMT2) was also shown to occur, and PRMT2−/− mice are lean, hypophagic, have reduced serum leptin levels and are more resistant to diet-induced obesity (DIO) compared to wild-type littermates (105). More recently, it was demonstrated that the nuclear receptor Nur77 (or TR3) facilitates STAT3 acetylation by recruiting acetylase p300 to and dissociating deacetylase HDAC1 (histone deacetylase 1) from STAT3, thereby enhancing the transcriptional activity of STAT3 and the expression of POMC (106).
Next to STAT3, leptin also activates STAT1, STAT5, and STAT6 in cell culture (107, 108). However, only leptin-dependent hypothalamic STAT5 phosphorylation was observed in vivo (109, 110). Activated STAT5 binds to pY1077, and to lesser extent pY1138 of the ObRb (111, 112). ObRb Y1077 mutants show only mildly increased food intake and adiposity. Furthermore, female mice display impairments in estrous cycling, suggesting that signaling by ObRb Y1077 plays only a modest role in the control of metabolism by leptin, while it may link body adiposity to the reproductive axis (113). However, deletion of STAT5 in ObRb expressing cells has no effect on body weight and fertility (114), indicating that neither STAT3 nor STAT5 are required for the regulation of fertility by leptin and that other signaling pathways depending on Y1077 might be involved in the leptin’s reproductive functions.
The MAPK Pathway
As mentioned before, the ObRb has three conserved tyrosine residues of which the pY985 motif serves as a docking site for the C-terminal SH2 domain of SHP2 (SH2-containing protein tyrosine phosphatase 2) (115–117). On its turn, SHP2 will become phosphorylated by the JAKs and recruit the adaptor protein Grb2 (growth factor receptor-bound protein 2), which ultimately leads to the activation of the mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase 1/2 (ERK1/2) (115, 118, 119). The phosphatase activity of SHP2 is necessary for leptin-mediated ERK activation (115). Weaker ERK activation by the short isoform ObRa (independent of Y985) was also shown, most likely via direct binding of Grb2 to JAK2 (120).
Neuron-specific deletion of SHP2 or pharmacological inhibition of ERK1/2 in the hypothalamus blocks the anorectic effects of leptin (118, 119). Furthermore, POMC-specific deletion of SHP2 results in mild obesity and increased susceptibility to DIO (121, 122), while female mice expressing a constitutively active form of SHP2 in the brain are resistant to DIO (123), supporting the role for SHP2 in the control of energy homeostasis. However, since SHP2 participates in many signaling pathways, the specific contribution of SHP2 downstream the ObRb remains difficult to assess. Mice with a mutation in Y985 are neuroendocrinologically normal and fertile, but especially female mice demonstrate decreased hypothalamic AgRP expression, increased pSTAT3 levels and leptin sensitivity, and resistance to DIO (124). Young homozygous Y985F mice were shown to be slightly leaner, although they exhibit adult-onset obesity (125). These data most likely do not reflect the role of Y985 in leptin-dependent ERK activation, but are consistent with increased ObRb signaling due to decreased feedback inhibition via disruption of SOCS3 binding (see further).
The PI3K–Akt–Foxo1 Pathway
A crucial role for the PI3K (phosphatidylinositol 3-OH kinase) pathway in leptin signaling was first demonstrated when intracerebroventricular (ICV) injection with PI3K inhibitors inhibited leptin’s anorexigenic effects (126). However, the activation mechanism is difficult to dissect since no ObRb phoshotyrosine site has been identified that mediates PI3K activation. Also, the PI3K pathway is shared with other receptors, especially the insulin receptor (IR). This hinders the evaluation of the ObRb-specific contribution of PI3K in the control of energy homeostasis. Although there is evidence that PI3K activation mediates the acute effects of leptin on the neuroelectrical activity of POMC cells and that the acute responses to leptin and insulin are largely segregated in distinct POMC subpopulations (127, 128). Nevertheless, mice with impaired PI3K signaling in POMC neurons have normal body weight, indicating that the PI3K pathway in these neurons is not critical for the leptin-mediated regulation of energy homeostasis (128).
Insulin receptor substrate 1, and particularly IRS2 are recruited to the ObRb via SH2B1, which interacts and upregulates the kinase activity of JAK2 (129, 130). SH2B1−/− mice display hyperphagia and severe early-onset obesity (131). Neuron-specific restauration of SH2B1 rescues this phenotype and improves leptin-dependent signaling and neuropeptide expression in the hypothalamus (132, 133). Alternatively, IRS4 was also shown to recruit the PI3K regulatory p85 subunit via the Y1077 motif of the ObRb (134).
IRS proteins in turn bind the PI3K p85 subunit, leading to PI3K activation and the accumulation of PIP3 (phosphatidylinositol 3,4,5-triphosphate). This leads to the sequential activation of PDK1 (3-phosphoinositide-dependent protein kinase 1) and Akt, resulting in the inhibition of the transcription factor Foxo1 (Forkhead Box O1). Foxo1 mediates the anorectic effects of leptin by regulating the expression of POMC, AgRP, and NPY (135–138). Foxo1 (when activated) stimulates the transcription of AgRP and NPY, but suppresses the transcription of POMC; thereby antagonizing the transcriptional action of STAT3 in these hypothalamic subpopulations. Recently, it was shown that direct interaction of Foxo1 to STAT3 was responsible for the inhibition of STAT3-mediated leptin signaling (137, 139). Therefore, mice with POMC-specific PDK1 or Foxo1 depletion are less or more sensitive, respectively, to the anorectic effects of leptin (140, 141). Likewise, Foxo1 ablation in AgRP neurons results in reduced food intake, improved glucose homeostasis, and increased sensitivity to leptin (142).
As mentioned above, PI3K activity leads to PIP3 accumulation. PTEN (tumor suppressor phosphatase and tensin homology) promotes the opposite reaction via its lipid phosphatase activity. PTEN ablation in ObRb-expressing neurons induces enhanced PI3K activity and reduced body fat (143). On the other hand, POMC-specific PTEN−/− mice develop leptin resistance and obesity, suggesting that chronic elevation of PIP3 in POMC neurons may interfere with hypothalamic leptin activity (144).
Another target activated downstream of PI3K/Akt is mTOR (mammalian target of rapamycin), an evolutionally conserved serine/threonine kinase, which senses nutrient availability and stimulates protein synthesis, cell growth, and proliferation. Leptin stimulates the phosphorylation of p70 S6 kinase (S6K) via mTOR. Selective inhibition/deletion of mTOR or S6K in the hypothalamus attenuates leptin’s anorexigenic effects (145–147). Additionally, PDE3B (phosphodiesterase 3B) is expressed in ObRb-expressing neurons in the hypothalamus (148). Leptin induces PDE3B activity, which results in a decrease in cAMP levels via the PI3K pathway. Inhibition of PDE3B activity by cilostamide reverses leptin’s effects on food intake and body weight, as well as the leptin-induced increase in POMC expression in the hypothalamus (149, 150).
The AMPK Pathway
Like mTOR, adenosine monophosphate-activated protein kinase (AMPK) serves as an intracellular fuel sensor and is activated by elevated AMP/ATP ratios. AMPK is a heterotrimeric serine/threonine kinase consisting of a catalytic α subunit and regulatory β and γ subunits. Leptin regulates AMPK activity in a tissue-specific way: leptin activates AMPK in hepatocytes and muscle tissue (151, 152), while in the hypothalamus, leptin inhibits AMPK, hence reducing food intake and body weight (153, 154). A reduction in AMPK phosphorylation (at T172), observed upon leptin treatment, is followed by decreased phosphorylation and increased activation of the AMPK target, acetyl-CoA carboxylase (ACC), a key enzyme in fatty acid biosynthesis (155, 156). Dominant negative AMPK expression in the hypothalamus is sufficient to reduce food intake and body weight, while constitutively active AMPK attenuates leptin’s anorexigenic effects (153, 155).
The precise mechanism of leptin-mediated AMPK activation is still unclear but requires JAK2 activity and does not seem to depend on intracellular phosphotyrosine motifs in the ObRb (152). Of the two catalytic subunits, α1 and α2, leptin predominantly appears to modulate AMPK activity via alternative phosphorylation sites in the α2 subunit (153, 157). In the hypothalamus, leptin administration increases α2-AMPK phosphorylation at S491, which decreases AMPK activity. Mice lacking α2-AMPK in POMC neurons become obese due to dysregulated food intake and decreased energy expenditure (most likely via altered glucose sensing), while deletion of α2-AMPK in NPY/AgRP neurons results in an age-dependent lean phenotype (154). S6K was shown to form a complex with α2-AMPK, resulting in phosphorylation on S491 (158). Blocking α2-AMPK S491 phosphorylation increases hypothalamic AMPK activity, food intake, and body weight (158). Thus, mTOR-S6K signaling serves as an important signaling pathway upstream of AMPK in hypothalamic leptin signaling.
Other ObRb Signaling Pathways
Mice with replacement of all three ObRb tyrosines with phenylalanines (ObRb3F) are slightly less obese than db/db mice and show significantly ameliorated glycemic control and fertility, illustrating that ObRb exerts crucial metabolic actions not only through ObR tyrosine-dependent but also ObR tyrosine-independent mechanisms (159). Interestingly, mice expressing a truncated ObRb mutant (ObRb∆65c), that retains JAK2 activity but lacks all ObRb tyrosines, have a similar phenotype as db/db mice showing obesity, diabetes, and infertility (160). Thus, the improved phenotype of ObRb3F mice compared to ObR∆65c mice reveals that JAK2-autonomous signaling is not sufficient to mediate these improvements. These data suggest the existence of non-canonical signaling pathways that may emanate from an uncharacterized ObRb site, independent of phosphorylation.
During the last few years, a role for several kinases in hypothalamic leptin signaling was proposed. Rho-kinase 1 (ROCK1) regulates leptin’s effect on body weight homeostasis by binding and activating JAK2. ROCK1 increases JAK2 phosphorylation and downstream activation of STAT3 and Foxo1. Mice lacking ROCK1 in either POMC or NPY/AgRP neurons, display impaired leptin sensitivity and obesity (161). A role for the RIIβ regulatory subunit of PKA (cyclic AMP-dependent protein kinase A) in modulating the magnitude and duration of ObRb signaling was also demonstrated (162). Mice lacking this PKA subunit display reduced adiposity and resistance to DIO. Recently, Cdc2-like kinase 2 (CLK2) activity was proven to be regulated by leptin in a PI3K-dependent manner and reduction of CLK2 expression in the hypothalamus was sufficient to abolish the anorexigenic effect of leptin (163).
Leptin Resistance: Possible Mechanisms
The discovery of leptin in 1994 generated high expectations for its potential use as a therapeutic to combat obesity. However, obese individuals were found to be refractory to leptin therapy (164, 165). In fact, obesity is commonly accompanied with elevated circulating leptin levels (hyperleptinemia) in proportion to the increased body fat mass. This paradox is referred to as leptin resistance and the underlying mechanism is multifactorial: impairment in ObRb signaling, hypothalamic neuronal wiring, leptin transport into the brain and ObR trafficking, ER stress and inflammation (16).
Negative Regulation of ObR Signaling
Leptin signaling is negatively regulated by SOCS3 and phosphatases, such as protein tyrosine phosphatase 1B (PTP1B), T-cell PTP (TCPTP), and RPTPϵ. Their hypothalamic expression levels are elevated in obesity and thus might contribute to the development of leptin resistance (166–168).
Members of the SOCS family are negative regulators of the JAK/STAT pathway. Leptin signaling via ObRb Y1138 and STAT3 rapidly induces SOCS3 expression (92, 166, 169). SOCS3 in turn will initiate a negative feedback loop by binding ObRb Y985, inhibiting further phosphorylation/activation of JAK2 (166, 170, 171). Immunohistochemical studies suggest that the ARC is selectively leptin resistant in DIO mice and that this may be caused by elevated SOCS3 expression (172). Neuron-wide deletion of SOCS3 leads to enhanced leptin-induced hypothalamic STAT3 phosphorylation and POMC induction, resulting in a more pronounced suppression of food intake, body weight loss, and an attenuation of diet-induced leptin resistance compared to wild-type mice (173). Specific deletion of SOCS3 in POMC neurons improves leptin sensitivity and glucose homeostasis (174). Consistently, increased SOCS3 expression in POMC neurons results in impaired STAT3 signaling with subsequent leptin resistance and obesity (175). Likewise, mice overexpressing a constitutively active version of STAT3 in POMC neurons show elevated SOCS3 expression and develop obesity as a result of hyperphagia and decreased POMC expression accompanied by central leptin resistance (176). A temporal and spatial pattern in leptin responsiveness was demonstrated before (177), and more recently, it was shown that SOCS3 activation in AgRP neurons precedes that of POMC and other hypothalamic neurons in the development of DIO (178).
Unexpectedly, SOCS3 upregulation in ObRb-expressing neurons does not lead to obesity, but rather a more lean phenotype (175). This may result from a compensatory increase in basal STAT3 expression and a corresponding increase in pSTAT3 levels after leptin treatment (175). However, inactivation of SOCS3 in ObRb-expressing cells protects mice from diet-induced insulin resistance, indicating that the regulation of leptin signaling by SOCS3 orchestrates diet-induced changes on glycemic control (179). Recently, ObRb-specific SOCS3−/− mice were studied in fasting and refeeding conditions. These mice exhibit increased leptin sensitivity in the hypothalamus and show attenuated food intake and weight regain after 48 h of fasting by a lower transcription of orexigenic neuropeptides (180).
Protein tyrosine phosphatase 1B is localized to the cytoplasmic face of the ER and inhibits leptin signaling by binding and dephosphorylating JAK2 (181, 182). Mice with PTP1B-deficiency in the whole brain, ObRb expressing cells, or POMC neurons are lean, leptin hypersensitive, and display improved glucose homeostasis, supporting an antagonistic role for PTP1B in hypothalamic leptin signaling (121, 183, 184). Moreover, the impact of PTP1B on energy homeostasis seems to rely on hypothalamic ObRb signaling as the reduced adiposity seen after hypothalamic PTP1B depletion is reversed by the concomitant hypothalamic deletion of ObRb (185).
Tyrosine phosphatases (PTPs) such as TCPTP and RPTPϵ were demonstrated to modulate ObR signaling (186), since genetic ablation of neuronal TCPTP or whole-body RPTPϵ enhances leptin sensitivity (167, 186, 187). Other PTPs including SHP2 and PTEN may also have regulatory functions in ObRb signaling and were briefly discussed above.
Hypothalamic Circuitry
Beyond leptin’s regulation of POMC and NPY/AgRP neurons on a transcriptional level, leptin can have a direct effect on these cells by altering neuronal firing (87). In addition, several lines of evidence have shown that the neuronal connectivity between specific subpopulations of hypothalamic and extra-hypothalamic neurons implicated in the regulation of energy balance is changing in response to high-fat diet (HFD) or metabolic hormones, such as leptin (188). For example, leptin treatment of ob/ob mice induces synaptic changes that precede the reduction in food intake and subsequent decrease in body weight (189). Although POMC and NPY/AgRP neurons in the ARC are considered to be the main targets of leptin, the majority of ObRb-expressing neurons lie outside the ARC in other central nervous system (CNS) regions known to modulate energy balance (88). This is underscored by the obese phenotype observed after deletion of ObRb in neurons, such as GABAergic neurons and NOS-1 (nitric oxide synthase-1) expressing neurons (190, 191). Growing evidence points to a critical role of astrocytes in orchestrating the hypothalamic response to metabolic cues by participating in processes of synaptic transmission and plasticity (192). Impaired ObR signaling in astrocytes leads to an altered glial morphology, increases the number of synapses onto POMC, and NPY/AgRP neurons and blunts leptin-induced anorexia (193).
Defective Leptin Transport
To enter the brain, circulating leptin has to cross the BBB. During obesity, the cerebrospinal fluid/serum leptin ratio is decreased, indicating impairment of leptin transport (194, 195). Leptin is actively transported across the BBB in a saturable manner (196). The short ObR isoform, ObRa is believed to be implicated in this process (197, 198). However, more recent data contradict this theory. ObRa−/− mice show only a small decrease in leptin responsiveness, suggesting that ObRa binding is not the only way by which leptin accesses the CNS (199). Moreover, pre-treatment with an ObR-neutralizing antibody, to directly examine the involvement of endothelial ObR in leptin transport, did not alter leptin transport in an in vitro BBB model (200). A role for megalin (or LRP2, low-density lipoprotein receptor-related protein-2) as a leptin transporter in the choroid plexus has been suggested (see also further) (200, 201).
The contribution of defective leptin transport to central leptin resistance remains unclear. Recently, it was demonstrated that the median eminence serves as the route through which leptin is transported into the hypothalamus, and that tanycytes act as a transport checkpoint. Peripherally administered leptin sequentially activates ObRb in median eminence tanycytes followed by neurons in the medio-basal hypothalamus (MBH) in a process that requires tanycytic ERK signaling (202). In obese mice (db/db or DIO) leptin accumulates in the median eminence and fails to reach the MBH, while triggering ERK signaling in tanycytes with epidermal growth factor (EGF) reestablishes leptin transport and its activation of MBH neurons (202).
Leptin Receptor Trafficking
The amount of signaling-competent ObRb on the cell surface is determined by the balance between receptor synthesis, transport to the plasma membrane, internalization, recycling, degradation, and ectodomain shedding. At steady state, the ObRb is mainly retained in the Golgi complex or in a post-Golgi intracellular compartment, resulting in low levels at the cell surface, from where it undergoes constitutive removal via ligand-independent endocytosis leading to lysosomal degradation with no evidence of recycling (53). Feeding seems to control ObRb expression since ObRb levels in the ARC are increased after fasting and decreased by refeeding. Leptin increases ObRb expression in the ARC, but not after high-fat feeding (203). However, overexpression of ObRb in POMC neurons renders mice more susceptible to DIO, further underlining the importance of correct ObRb expression (204).
Endospanin 1 [also known as Ob-RGRP (ObR gene-related protein) or LEPROT (leptin receptor overlapping transcript)], whose expression is genetically linked to the ObRb transcript, negatively controls ObRb cell surface expression (205). Endospanin 1 interacts with ObRb and targets ObRb from endosomes to lysosomes, thereby increasing its degradation (206). Hence, endospanin 1 silencing in the ARC is sufficient to prevent or reverse the development of obesity after high-fat diet in lean or fully obese mice, respectively (205, 207).
Our group identified the E3 ubiquitin ligase RNF41 (Ring Finger Protein 41) as an interaction partner of the ObRb complex (17). RNF41 acts as a key regulator of basal cytokine receptor trafficking, proteolytic processing, and signaling. RNF41 controls the constitutive intracellular trafficking of the ObRb, by preventing its lysosomal receptor degradation, and concomitantly enhancing receptor ectodomain shedding by the metalloprotease ADAM10 (17). We further demonstrated that this results from RNF41-dependent ubiquitination and suppression of the deubiquitinating enzyme USP8, which abrogates ESCRT-0 functionality and accounts for the rerouting of cytokine receptors (208).
Accumulating evidence suggests that the neuronal cilia basal body complex acts as a platform for ObRb signaling. Neuronal cilia lengths were selectively reduced in the hypothalamus of obese mice with leptin deficiency and leptin resistance, while treatment of hypothalamic neurons with leptin-stimulated cilia assembly via inhibition of PTEN and glycogen synthase kinase 3β (GSK3β) (209, 210). Moreover, mice with short hypothalamic cilia exhibit increased food intake, decreased energy expenditure, and attenuated anorectic responses to leptin, which indicates that leptin-induced cilia assembly is essential for sensing leptin by hypothalamic neurons (209). Bardet–Biedl syndrome (BBS) proteins form the stable BBSome complex, which mediates protein trafficking to the ciliary membrane (211). The BBSome influences energy homeostasis through the control of ObRb transport to the cell surface expression as targeted disruption of the BBSome by deleting BBS1 in ObRb-expressing cells causes obesity in mice (212, 213). Suppression of another ciliary gene, retina pigmentosa GTPase regulator-interacting protein-1 (RPGRIP1L), in neuronal cultures decreases localization of ObRb near the cilium and activation of the downstream signaling cascade (214). Like BBS proteins, RPGRIP1L interacts with the ObRb and mediates its trafficking to the periciliary area (215). Mice hypomorphic for RPGRP1L exhibit hyperphagic obesity as the result of diminished leptin sensitivity in ObRb-expressing neurons (216).
ER Stress and Inflammation
Several studies have provided evidence that ER stress and the activated adaptive unfolded protein response (UPR) impair leptin signaling and are highly increased in hypothalamic neurons in the context of obesity (217, 218). Reducing hypothalamic ER stress by chemical and natural chaperones can re-establish leptin sensitivity (217, 219, 220). Xbp1s (spliced X-box binding protein 1) is one of the ER-stress-induced genes and neuron-specific Xbp1−/− mice have ER stress, severe hyperleptinemia, leptin resistance, and obesity (217). In contrast, induction of Xbp1s in POMC neurons alone is sufficient to protect against DIO and to improve leptin sensitivity by suppressing SOCS3 and PTP1B, even in the presence of strong ER stress activators (221). Several mechanisms are suggested to bridge HFD-induced ER stress to impaired ObR signaling (222). Overnutrition atypically activates IKKβ-NFκB signaling in the hypothalamus through ER stress responses, which implies a connection between ER stress and hypothalamic inflammation (218). Indeed, obesity seems to be associated with low-grade chronic inflammation (223). Saturated fatty acids, which are elevated in obesity, are able to bind and activate toll-like receptor 4 (TLR4) and inhibition of TLR4 or neuronal deletion of the TLR adaptor molecule MyD88 protects from HFD-induced leptin resistance and obesity (224–226). Inhibition of IKKϵ, a downstream molecule of NFκB signaling, reduces leptin resistance by restoring JAK2-STAT3 and PI3K signaling in the hypothalamus of HFD-fed mice (227).
It was recently shown that disruption of mitochondria–ER contacts may also contribute to leptin resistance development in POMC neurons. Mitochondrial-ER contacts are decreased in POMC neurons of mice receiving HFD (228). Furthermore, deletion of PPARγ in POMC neurons enhances mitochondrial-ER interactions and sensitizes POMC neurons to leptin during HFD (229). On the other hand, mice with a POMC-specific deletion of MFN2 (mitofusin 2), a key protein for mitochondrial fusion and the formation of ER–mitochondria contacts, display a loss of these interactions, defective POMC processing, ER stress-induced leptin resistance, and obesity (228). In contrast, ablation of mitofusins in NPY/AgRP neurons disrupts mitochondrial fusion without inducing ER stress and alleviates HFD-induced obesity (230). These findings indicate the importance of mitochondrial dynamics in hypothalamic neurons during the establishment of DIO.
Redefine the View on “Leptin Resistance”?
Recently, Ottaway et al. reported that central or peripheral administration of an ObR antagonist induces comparable changes in food intake, body weight, and hypothalamic POMC and SOCS3 expression in lean and DIO mice, illustrating that endogenous ObR signaling may not be reduced in the context of DIO, thus challenging the established concept of leptin resistance under dietary-induced conditions (231). More efforts are required to further comprehend the link between the different mechanisms mentioned above, and the precise sequence of appearance of the alterations, discriminating between causes and consequences (232). Moreover, many other nutrient signals, such as insulin and ghrelin, contribute to the control of energy homeostasis. Cross-talk and redundancy between these signals complicate the precise assessment of the contribution of leptin-mediated signaling in the context of leptin resistance and obesity.
Leptin Signaling Beyond the ObR: Cross-Talk at the Cellular Surface
Several independent lines of evidence suggest that leptin signaling in the brain or the periphery can be different. In a first example, Nizard and colleagues reported the pregnancy of a morbidly obese patient with a rare ObR mutation (233). Normally, such loss-of-function mutations have been linked to infertility in humans and rodents (29, 234). Despite neonatal hypoglycemia, the child’s growth and development have been normal (233). Second, a serine to leucine mutation on position 72 in leptin hampers the secretion, but not the expression of leptin in adipose tissue in a 14-year-old child of non-obese Austrian parents (235). The child showed signs of a hypogonadotropic hypogonadism, but in contrast to previous studies only mild obesity and a normal T cell responsiveness. Third, a spontaneous splice-mutation causes the deletion of the complete IGD domain in fatt/fatt mice (236). These animals are hyperphagic and obese, but show minimal changes in size and cellularity of the thymus and respond comparable to wild-type animals to concavalin A in a model for autoimmune hepatitis. Finally, treatment of healthy mice with a IGD-specific neutralizing nanobody induced clear weight gain and hyperinsulinemia, but failed to block development of experimentally induced autoimmune multiple sclerosis, arthritis, and hepatitis (236).
Cross-talk with other (cytokine) receptors could explain this observed uncoupling of leptin’s central and peripheral functions. In a simplistic view, a cytokine triggers intracellular signaling by binding and activation of a cognate homo- or heteromeric receptor pair. However, when combined, cytokines, hormones, and other stimuli might have additive, synergistic, or antagonistic effects. This so-called “cross-talk” does not only occur at the level of signaling pathways but also at the cell surface between cytokines, hormones, and their receptors. The process that a certain cytokine activates another receptor complex is called cross-activation. At the moment, it is mostly unclear how a cytokine discriminates between different options, but cell-specific co-expression is likely a determining factor.
At the cell surface, the leptin:ObR system can interact with the epidermal growth factor receptor (EGFR), estrogen receptor alpha (ERα), insulin-like growth factor I receptor (IGF-IR), lipoprotein receptor-related protein 1 and 2 (LRP1 and LRP2), and vascular endothelial growth factor receptor (VEGFR). Studies describing ObR cross-talk and cross-activation are summarized in Table 1.
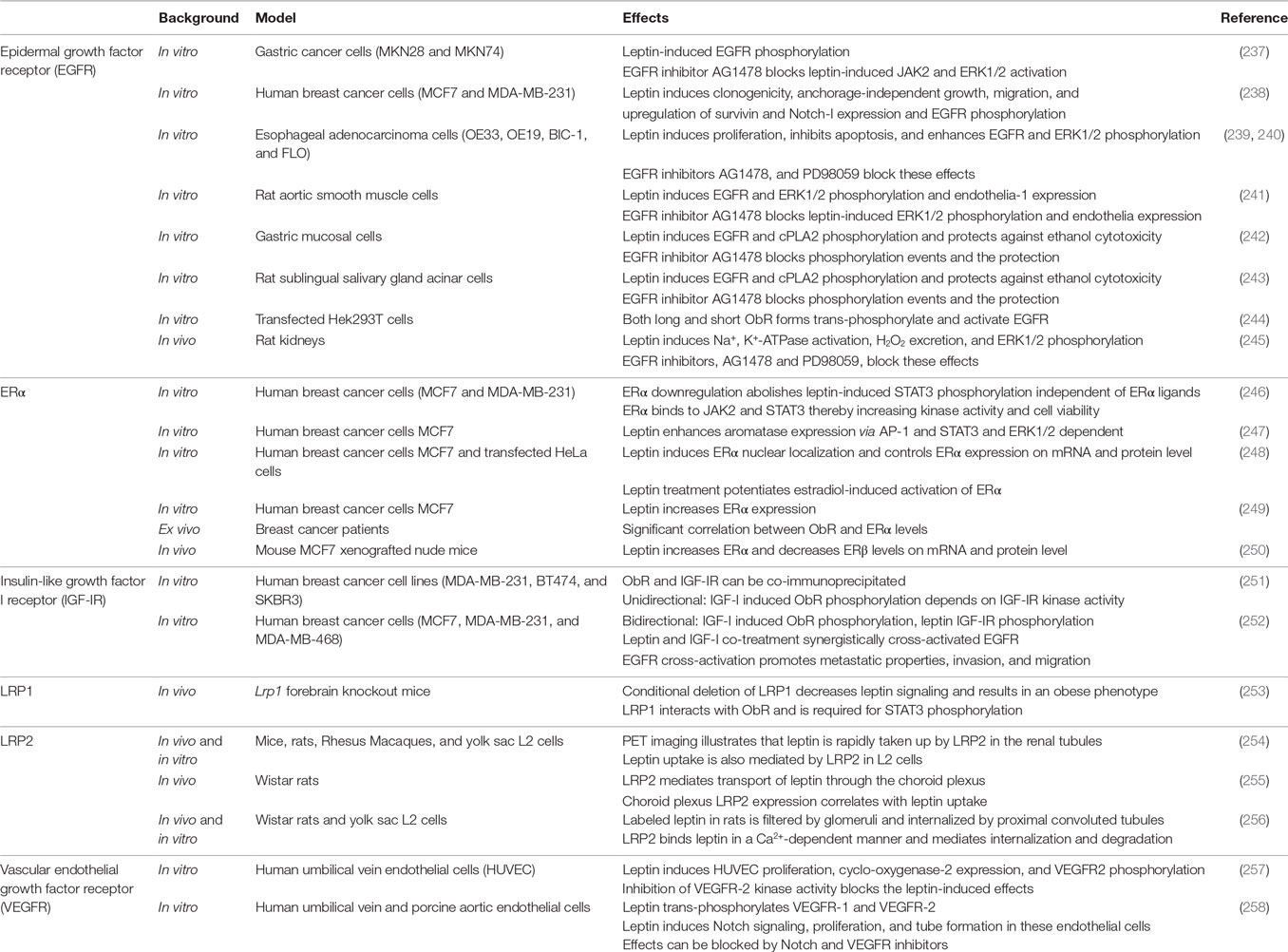
Table 1. Overview of studies describing ObR cross-talk and/or cross-activation (see text for more details).
Epidermal Growth Factor Receptor
The EGFR is a single membrane-spanning receptor with a cytoplasmic tyrosine kinase domain. This receptor can be activated not only by its “natural” ligands, including EGF, transforming growth factor-α (TGF-α), heparin-binding EGF-like growth factor, amphiregulin, and epiregulin but also by cross-activation by G-protein-coupled receptors, the tumor necrosis factor receptor, or IGF-IR [reviewed in Ref. (259)]. ObR:EGFR cross-talk and cross-activation is not restricted to cancer cell lines, but could also be demonstrated in muscle, salivary gland, and mucosal cells and in rat kidneys (see Table 1). The cross-talk often involves the activity of ERK and Src kinases and can be blocked by EGFR kinase inhibitors. Both ObR long and short forms are able to phosphorylate the EGFR, suggesting that this cross-activation is independent of downstream ObR signaling (244). It was suggested that this cross-talk depends on the proteolytic release of EGFR ligands as broad-spectrum matrix metalloproteinase inhibitor (e.g., GM6001) blocks the leptin-induced effects. However, this pathway likely does not explain the rapid (within 5 min) and transient increase in EGFR tyrosine phosphorylation by leptin in, for example, rat aortic smooth muscle cells (241).
ERα
A significant co-expression between ObR and ERα and a functional cross-talk between the leptin and estrogen signaling networks are associated with breast tumor progression [reviewed in Ref. (260)]. Leptin not only controls ERα and aromatase mRNA expression both in vitro and in vivo and in an ERK and STAT3-dependent manner (247, 249, 250), one report shows that it can directly cross-activate ERα in the absence of cognate ligand (248).
Insulin-Like Growth Factor I Receptor
Insulin-like growth factor I receptor is an α2: β2 disulfide-linked receptor that shares more than 50% of overall sequence homology with the IR. It is overexpressed in 50% of primary breast tumors compared with normal tissue (261), and IGF-IR inhibition reduces cancer growth and metastasis in vivo (262). Saxena et al. and Ozbay et al. independently demonstrated the ObR:IGF-IR cross-talk in breast cancer cells: both IGF-I and leptin phosphorylate the reciprocal receptor, ObR and IGF-IR can be co-immunoprecipitated, and combined treatment increased proliferation, invasion, and migration of breast cancer cells (251, 252).
LRP1/LRP2
LRP1 is highly expressed in neurons of the CNS and plays a role in lipoprotein metabolism, neurotransmission, synaptic plasticity, and cell survival (263). Liu et al. showed that conditional deletion of LRP1 results in an obese phenotype, characterized by increased food intake, decreased energy expenditure, and hampered leptin signaling. LRP1 directly interacts with ObR and is necessary for STAT3 activation (253).
LRP2 or megalin is a large glycoprotein abundantly expressed at the apical membranes of proximal tubule cells that reabsorb and metabolize proteins filtered by glomeruli in the kidney (264). A role of LRP2 in leptin clearance (254, 256), and/or transport over the BBB has been suggested (see above) (255).
Vascular Endothelial Growth Factor Receptor
Vascular endothelial growth factor receptor and its receptor play a crucial role in the angiogenic process in physiologic and pathological scenarios (265). In human umbilical vein endothelial cells and porcine endothelial cells, leptin trans-phosphorylates VEGFR-1 and VEGFR-2, induces cyclo-oxygenase-2 and Notch signaling, proliferation and tube formation (257, 258).
Leptin Signaling Beyond the ObR: Cross-Talk Between Downstream Signal Cascades
Cross-talk does not only occur at the receptor level (receptor co-complexes, cross-activation) but also more downstream at the level of activated kinases and transcription factors or due to leptin-dependent expression of other cytokines.
Leptin–Insulin
Both leptin and insulin elicit strong anorectic responses within the ARC and their central administration, which mimics a state of energy surplus, inhibits food intake, and decreases body weight (266). One prevailing view is that different POMC neurons exist and that leptin and insulin may act on distinct POMC neuronal subsets (127). However, many studies demonstrate cross-talk between leptin and insulin signaling at many levels [reviewed in Ref. (267)]. The insulin pathway, after insulin binding to the IR and activation of the IRS proteins, converges with the leptin pathway at the same point, the activation of PI3K, to modulate body weight and glucose homeostasis (268). Also, both leptin and insulin regulate the AMPK pathway in the hypothalamus and inhibit AMPK activation and its downstream targets (153, 154). Recently, it was shown that insulin can potentiate leptin-induced STAT3 activation by the induction of GRP78 (glucose-regulated protein 78), a UPR chaperone that is required to maintain ER capacity and protect against ER stress (269). Strikingly, ICV administration of leptin in mice devoid of insulin and lacking ObRb demonstrates that concomitant re-expression of ObRb only in hypothalamic GABAergic and POMC neurons is sufficient to fully mediate the anti-diabetic actions of leptin in insulin deficiency (270). Likewise, cellular insulin resistance disrupts leptin-mediated control of neuronal signaling and transcription (271).
NILCO (Notch, IL-1, and Leptin Cross-talk Outcome)
Functional cross-talk between leptin, IL-1, and Notch signaling (NILCO) is found in breast cancer cells and could represent the integration of developmental, pro-inflammatory, and pro-angiogenic signals which are critical for leptin-induced breast cancer cell proliferation/migration and tumor angiogenesis. Inhibition of leptin signaling significantly reduces the establishment and growth of breast cancer and simultaneously decreases the levels of VEGF/VEGFR2, IL-1 and Notch (272–274). Therefore, inhibition of leptin–cytokine cross-talk might serve as a preventative or adjuvant measure to target breast cancer, particularly in obese women.
Cross-talk with Pro-inflammatory Cytokines
Pro-inflammatory cytokines may have differential roles in hypothalamic leptin signaling. As mentioned before, overnutrition leads to increased expression of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), IL-1β, and IL-6 (223, 275). Lack of TNFR1 mitigates leptin resistance under HFD conditions, while ICV co-injection of TNFα partially blocks leptin’s anorexigenic effect through the inhibition of PI3K–Foxo1 signaling (276, 277). Conversely, central injection of an IL-1R antagonist or IL-1R1 knock-down blunts the suppression of food intake in response to leptin (278). Likewise, central infusion of IL-6 enhances hypothalamic STAT3 phosphorylation and suppresses hypothalamic IKKβ activation and hyperphagia in DIO (279).
Toll-Like Receptor 4
Toll-like receptor 4 and ObRb activation seem to converge at a common signaling point in the hypothalamus. LPS, a ligand of TLR4, stimulates PI3K and STAT3 signaling pathways in cells expressing ObRb. Genetic deletion of the PI3K p110β catalytic subunit in ObRb-expressing cells leads to blunted suppression of food intake by LPS which demonstrates that lowered food intake during an inflammatory challenge depends on the PI3K pathway activated by cytokines and leptin in hypothalamic neurons (280). Moreover, NFκB signaling in POMC neurons is activated by leptin and mediates leptin-stimulated POMC transcription, indicating that hypothalamic NFκB also serves as a downstream transcription factor of the ObRb (281). Increased hypothalamic POMC promoter methylation in mice with DIO limits NFκB binding, which limits the ability of leptin to increase POMC expression (282).
Future Perspectives
It is more than 20 years ago that leptin and its receptor have been identified as key regulators of body weight and energy homeostasis. However, the hormone mostly failed in the clinic to treat obesity due to the fact that obese people are almost always hyperleptinemic and resistant to leptin. The observation that leptin has functions in immunity, hematopoiesis, angiogenesis, reproduction, and BP, and is involved in the pathology of, e.g., autoimmune diseases and cancers, reopened the interest in leptin and ObR-based therapeutics. Current strategies, including leptin mutants, leptin peptide antagonists, neutralizing antibodies, and soluble receptors, were shown to be effective in the treatment of several autoimmune diseases and in some cancer models [reviewed in Ref. (283)]. Their clinical application is, however, hampered by the unwanted weight-gain (10–15% per week in rodent models) upon treatment.
The ability of the ObR to interact with other receptor systems at the receptor level or intracellulary illustrates that the leptin receptor complex may be “heavier” than expected. This creates new exciting opportunities including the possibility to uncouple leptin’s central role in weight regulation and its peripheral functions. More in-depth insights in these complex leptin/ObR activation and downstream signaling mechanisms may ultimately allow the design of selective antagonists.
Author Note
The authors apologize to their colleagues that space limitations did not allow us to cite all the relevant literature.
Author Contributions
JW and LZ wrote the review, while JT was responsible for revising.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was funded by IUAP (P6/36). JT is a recipient of an ERC Advanced Grant (No. 340941-CYRE).
Abbreviations
ACC, acetyl-CoA carboxylase; AgRP, agouti-related peptide; AMPK, adenosine monophosphate-activated protein kinase; α-MSH, α-melanocyte stimulating hormone; AR, amphiregulin; ARC, arcuate nucleus; BBB, blood–brain barrier; BBS, Bardet–Biedl syndrome; BP, blood pressure; BRET, bioluminescence resonance energy transfer; CNS, central nervous system; CRH, cytokine receptor homology domain; EGF, epidermal growth factor; EGFR, EGF receptor; EPR, epiregulin; ER, endoplasmic reticulum; ERK, extracellular signal-related kinase; FN III, fibronectin type III; Foxo1, forkhead box O1; FRET, fluorescence resonance energy transfer; GABA, γ-aminobutyric acid; G-CSF, granulocyte-colony stimulating factor; Grb2, growth factor receptor-bound protein 2; GRP78, glucose-regulated protein 78; GSK3β, glycogen synthase kinase 3β; HB-EGF, heparin-binding EGF-like growth factor; HFD, high-fat diet; HUVEC, human umbilical vein endothelial cell; ICV, intracerebroventricular; IGD, immunoglobulin-like domain; IGF-IR, insulin-like growth factor I receptor; IL, interleukin; IR, insulin receptor; IRS, insulin receptor substrate; JAK, Janus kinase; LEPROT, leptin receptor overlapping transcript; LRP, lipoprotein receptor-related protein; MALLS, multi-angle laser light scattering; MAPK, mitogen-activated protein kinase; MBH, medio-basal hypothalamus; MFN2, mitofusin 2; mTOR, mammalian target of rapamycin; NOS-1, nitric oxide synthase-1; NPY, neuropeptide Y; NTD, N-terminal domain; ObR, leptin receptor; PDE3B, phosphodiesterase 3B; PDK1, 3-phosphoinositide-dependent protein kinase; PI3K, phosphatidylinositol 3 kinase; PIP3, phosphatidylinositol 3,4,5-triphosphate; POMC, pro-opiomelanocortin; PRMT2, protein arginine N-methyltransferase 2; PTEN, tumor suppressor phosphatase and tensin homology; PTP, tyrosine phosphatase; PTP1B, protein tyrosine phosphatase 1B; RNF41, ring finger protein 41; RPGRIP1L, retina pigmentosa GTPase regulator-interacting protein-1; S6K, S6 kinase; SAXS, small-angle X-ray scattering; SHP2, SH2-containing protein tyrosine phosphatase 2; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; TGF-α, transforming growth factor-α; TLR, toll-like receptor; TNF-α, tumor necrosis factor-α; UPR, unfolded protein response; VEGF, vascular endothelial growth factor receptor; Xbp1s, spliced X-box binding protein 1.
References
1. Coleman DL. A historical perspective on leptin. Nat Med (2010) 16:1097–9. doi:10.1038/nm1010-1097
2. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi:10.1038/372425a0
3. Tartaglia LA, Dembski M, Weng X, Deng NH, Culpepper J, Devos R, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell (1995) 83:1263–71.
4. Frederich RC, Lollmann B, Hamann A, Napolitano-Rosen A, Kahn BB, Lowell BB, et al. Expression of ob mRNA and its encoded protein in rodents. Impact of nutrition and obesity. J Clin Invest (1995) 96:1658–63. doi:10.1172/JCI118206
5. Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science (1995) 269:543–6.
6. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med (1996) 334:292–5.
7. Señarís R, Garcia-Caballero T, Casabiell X, Gallego R, Castro R, Considine RV, et al. Synthesis of leptin in human placenta. Endocrinology (1997) 138:4501–4.
8. Bado A, Levasseur S, Attoub S, Kermorgant S, Laigneau JP, Bortoluzzi MN, et al. The stomach is a source of leptin. Nature (1998) 394:790–3. doi:10.1038/29547
9. Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature (1998) 393:684–8. doi:10.1038/31474
10. Rock FL, Altmann SW, van Heek M, Kastelein RA, Bazan JF. The leptin haemopoietic cytokine fold is stabilized by an intrachain disulfide bond. Horm Metab Res (1996) 28:649–52. doi:10.1055/s-2007-979871
11. Haglund E, Sułkowska JI, He Z, Feng G-S, Jennings PA, Onuchic JN. The unique cysteine knot regulates the pleotropic hormone leptin. PLoS One (2012) 7:e45654. doi:10.1371/journal.pone.0045654
12. Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett (1996) 387:113–6.
13. Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest (1996) 98:1101–6.
14. Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, et al. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A (1997) 94:7001–5.
15. Hileman SM, Tornoe J, Flier JS, Bjorbaek C. Transcellular transport of leptin by the short leptin receptor isoform ObRa in Madin-Darby Canine Kidney cells. Endocrinology (2000) 141:1955–61. doi:10.1210/endo.141.6.7450
16. Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci (2011) 16:2771–93. doi:10.2741/3885
17. Wauman J, De Ceuninck L, Vanderroost N, Lievens S, Tavernier J. RNF41 (Nrdp1) controls type 1 cytokine receptor degradation and ectodomain shedding. J Cell Sci (2011) 124:921–32. doi:10.1242/jcs.078055
18. Ge HF, Huang L, Pourbahrami T, Li C. Generation of soluble leptin receptor by ectodomain shedding of membrane-spanning receptors in vitro and in vivo. J Biol Chem (2002) 277:45898–903. doi:10.1074/jbc.M205825200
19. Haniu M, Arakawa T, Bures EJ, Young Y, Hui JO, Rohde MF, et al. Human leptin receptor. Determination of disulfide structure and N-glycosylation sites of the extracellular domain. J Biol Chem (1998) 273:28691–9. doi:10.1074/jbc.273.44.28691
20. Kamikubo Y, Dellas C, Loskutoff DJ, Quigley JP, Ruggeri ZM. Contribution of leptin receptor N-linked glycans to leptin binding. Biochem J (2008) 410:595–604. doi:10.1042/BJ20071137
21. Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell (1996) 84:491–5.
22. Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature (1998) 392:398–401. doi:10.1038/32911
23. Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature (1997) 387:903–8.
24. Xu X, Zeng H, Xiao D, Zhou H, Liu Z. Genome wide association study of obesity. Zhong Nan Da Xue Xue Bao Yi Xue Ban (2013) 38:95–100. doi:10.3969/j.issn.1672-7347.2013.01.018
26. Farooqi IS, O’Rahilly S. 20 years of leptin: human disorders of leptin action. J Endocrinol (2014) 223:T63–70. doi:10.1530/JOE-14-0480
27. Bennett BD, Solar GP, Yuan JQ, Mathias J, Thomas GR, Matthews W. A role for leptin and its cognate receptor in hematopoiesis. Curr Biol (1996) 6:1170–80.
28. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature (1998) 394:897–901. doi:10.1038/29795
29. Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet (1996) 12:318–20. doi:10.1038/ng0396-318
30. Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, Sessa WC, et al. Biological action of leptin as an angiogenic factor. Science (1998) 281:1683–6.
31. Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell (2000) 100:197–207. doi:10.1016/S0092-8674(00)81558-5
32. Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens (1999) 17:1949–53.
33. Matarese G, La Cava A, Sanna V, Lord GM, Lechler RI, Fontana S, et al. Balancing susceptibility to infection and autoimmunity: a role for leptin? Trends Immunol (2002) 23:182–7. doi:10.1016/S1471-4906(02)02188-9
34. Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab (1999) 84:3686–95. doi:10.1210/jcem.84.10.5999
35. Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest (2002) 110:1093–103. doi:10.1172/JCI15693.another
36. Matarese G, La Rocca C, Moon H-S, Huh JY, Brinkoetter MT, Chou S, et al. Selective capacity of metreleptin administration to reconstitute CD4+ T-cell number in females with acquired hypoleptinemia. Proc Natl Acad Sci U S A (2013) 110:E818–27. doi:10.1073/pnas.1214554110
37. Naylor C, Petri WA. Leptin regulation of immune responses. Trends Mol Med (2016) 22:88–98. doi:10.1016/j.molmed.2015.12.001
38. Matarese G, Di Giacomo A, Sanna V, Lord GM, Howard JK, Di Tuoro A, et al. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol (2001) 166:5909–16. doi:10.4049/jimmunol.166.10.5909
39. Busso N, So A, Chobaz-Peclat V, Morard C, Martinez-Soria E, Talabot-Ayer D, et al. Leptin signaling deficiency impairs humoral and cellular immune responses and attenuates experimental arthritis. J Immunol (2002) 168:875–82. doi:10.4049/jimmunol.168.2.875
40. Faggioni R, Jones-Carson J, Reed DA, Dinarello CA, Feingold KR, Grunfeld C, et al. Leptin-deficient (ob/ob) mice are protected from T cell-mediated hepatotoxicity: role of tumor necrosis factor alpha and IL-18. Proc Natl Acad Sci U S A (2000) 97:2367–72. doi:10.1073/pnas.040561297
41. Siegmund B, Lear-Kaul KC, Faggioni R, Fantuzzi G. Leptin deficiency, not obesity, protects mice from Con-A induced hepatitis. Eur J Immunol (2002) 32:552–60. doi:10.1002/1521-4141(200202)32:2<552::AID-IMMU552>3.0.CO;2-H
42. Tarzi RM, Cook HT, Jackson I, Pusey CD, Lord GM. Leptin-deficient mice are protected from accelerated nephrotoxic nephritis. Am J Pathol (2004) 164:385–90. doi:10.1016/S0002-9440(10)63128-8
43. Chehab FF. 20 years of leptin: leptin and reproduction: past milestones, present undertakings, and future endeavors. J Endocrinol (2014) 223:T37–48. doi:10.1530/JOE-14-0413
44. Catteau A, Caillon H, Barrière P, Denis MG, Masson D, Fréour T. Leptin and its potential interest in assisted reproduction cycles. Hum Reprod Update (2015) 22:dmv057. doi:10.1093/humupd/dmv057
45. Chen XX, Yang T. Roles of leptin in bone metabolism and bone diseases. J Bone Miner Metab (2015) 33:474–85. doi:10.1007/s00774-014-0569-7
46. Abel ED, Sweeney G. Modulation of the cardiovascular system by leptin. Biochimie (2012) 94:2097–103. doi:10.1016/j.biochi.2012.03.019
47. Gogas H, Trakatelli M, Dessypris N, Terzidis A, Katsambas A, Chrousos GP, et al. Melanoma risk in association with serum leptin levels and lifestyle parameters: a case-control study. Ann Oncol (2008) 19:384–9. doi:10.1093/annonc/mdm464
48. Garofalo C, Surmacz E. Leptin and cancer. J Cell Physiol (2006) 207:12–22. doi:10.1002/jcp.20472
49. Cirillo D, Rachiglio AM, la Montagna R, Giordano A, Normanno N. Leptin signaling in breast cancer: an overview. J Cell Biochem (2008) 105:956–64. doi:10.1002/jcb.21911
50. Pais R, Silaghi H, Silaghi A-C, Rusu M-L, Dumitrascu D-L. Metabolic syndrome and risk of subsequent colorectal cancer. World J Gastroenterol (2009) 15:5141–8. doi:10.3748/wjg.15.5141
51. Liao LM, Schwartz K, Pollak M, Graubard BI, Li Z, Ruterbusch J, et al. Serum leptin and adiponectin levels and risk of renal cell carcinoma. Obesity (Silver Spring) (2013) 21:1478–85. doi:10.1002/oby.20138
52. García-Robles MJ, Segura-Ortega JE, Fafutis-Morris M. The biology of leptin and its implications in breast cancer: a general view. J Interferon Cytokine Res (2013) 33:717–27. doi:10.1089/jir.2012.0168
53. Belouzard S, Delcroix D, Rouille Y. Low levels of expression of leptin receptor at the cell surface result from constitutive endocytosis and intracellular retention in the biosynthetic pathway. J Biol Chem (2004) 279:28499–508. doi:10.1074/jbc.M400508200
54. Nakashima K, Narazaki M, Taga T. Leptin receptor (OB-R) oligomerizes with itself but not with its closely related cytokine signal transducer gp130. FEBS Lett (1997) 403:79–82.
55. White DW, Tartaglia LA. Evidence for ligand-independent homo-oligomerization of leptin receptor (OB-R) isoforms: a proposed mechanism permitting productive long-form signaling in the presence of excess short-form expression. J Cell Biochem (1999) 73:278–88.
56. Couturier C, Jockers R. Activation of the leptin receptor by a ligand-induced conformational change of constitutive receptor dimers. J Biol Chem (2003) 278:26604–11. doi:10.1074/jbc.M302002200
57. Biener E, Charlier M, Ramanujan VK, Daniel N, Eisenberg A, Bjorbaek C, et al. Quantitative FRET imaging of leptin receptor oligomerization kinetics in single cells. Biol Cell (2005) 97:905–19. doi:10.1042/BC20040511
58. Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK, Wilson IA. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science (1999) 283:987–90.
59. Remy I, Wilson IA, Michnick SW. Erythropoietin receptor activation by a ligand-induced conformation change. Science (1999) 283:990–3.
60. Constantinescu SN, Keren T, Socolovsky M, Nam H, Henis YI, Lodish HF. Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain. Proc Natl Acad Sci U S A (2001) 98:4379–84. doi:10.1073/pnas.081069198
61. Gent J, van Kerkhof P, Roza M, Bu G, Strous GJ. Ligand-independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system-dependent endocytosis. Proc Natl Acad Sci U S A (2002) 99:9858–63. doi:10.1073/pnas.152294299
62. Schuster B, Meinert W, Rose-John S, Kallen K-J. The human interleukin-6 (IL-6) receptor exists as a preformed dimer in the plasma membrane. FEBS Lett (2003) 538:113–6. doi:10.1016/S0014-5793(03)00154-6
63. Devos R, Guisez Y, VanderHeyden J, White DW, Kalai M, Fountoulakis M, et al. Ligand-independent dimerization of the extracellular domain of the leptin receptor and determination of the stoichiometry of leptin binding. J Biol Chem (1997) 272:18304–10.
64. Zabeau L, Defeau D, Iserentant H, Vandekerckhove J, Peelman F, Tavernier J. Leptin receptor activation depends on critical cysteine residues in its fibronectin type III subdomains. J Biol Chem (2005) 280:22632–40. doi:10.1074/jbc.M413308200
65. Mancour LV, Daghestani HN, Dutta S, Westfield GH, Schilling J, Oleskie AN, et al. Ligand-induced architecture of the leptin receptor signaling complex. Mol Cell (2012) 48:655–61. doi:10.1016/j.molcel.2012.09.003
66. Moharana K, Zabeau L, Peelman F, Ringler P, Stahlberg H, Tavernier J, et al. Structural and mechanistic paradigm of leptin receptor activation revealed by complexes with wild-type and antagonist leptins. Structure (2014) 22:866–77. doi:10.1016/j.str.2014.04.012
67. Fong TM, Huang RRC, Tota MR, Mao C, Smith T, Varnerin J, et al. Localization of leptin binding domain in the leptin receptor. Mol Pharmacol (1998) 53:234–40.
68. Zabeau L, Defeau D, Van der Heyden J, Iserentant H, Vandekerckhove J, Tavernier J. Functional analysis of leptin receptor activation using a Janus kinase/signal transducer and activator of transcription complementation assay. Mol Endocrinol (2004) 18:150–61. doi:10.1210/me.2003-0078
69. Peelman F, Zabeau L, Moharana K, Savvides SN, Tavernier J. 20 years of leptin: insights into signaling assemblies of the leptin receptor. J Endocrinol (2014) 223:T9–23. doi:10.1530/JOE-14-0264
70. Sandowski Y, Raver N, Gussakovsky EE, Shochat S, Dym O, Livnah O, et al. Subcloning, expression, purification, and characterization of recombinant human leptin-binding domain. J Biol Chem (2002) 277:46304–9. doi:10.1074/jbc.M207556200
71. Iserentant H, Peelman F, Defeau D, Vandekerckhove J, Zabeau L, Tavernier J. Mapping of the interface between leptin and the leptin receptor CRH2 domain. J Cell Sci (2005) 118:2519–27. doi:10.1242/jcs.02386
72. Niv-Spector L, Raver N, Friedman-Einat M, Grosclaude J, Gussakovsky EE, Livnah O, et al. Mapping leptin-interacting sites in recombinant leptin-binding domain (LBD) subcloned from chicken leptin receptor. Biochem J (2005) 390:475–84. doi:10.1042/bj20050233
73. Carpenter B, Hemsworth GR, Wu Z, Maamra M, Strasburger CJ, Ross RJ, et al. Structure of the human obesity receptor leptin-binding domain reveals the mechanism of leptin antagonism by a monoclonal antibody. Structure (2012) 20:487–97. doi:10.1016/j.str.2012.01.019
74. Peelman F, Iserentant H, De Smet AS, Vandekerckhove J, Zabeau L, Tavernier J. Mapping of binding site III in the leptin receptor and modeling of a hexameric leptin:leptin receptor complex. J Biol Chem (2006) 281:15496–504. doi:10.1074/jbc.M512622200
75. Takaya K, Ogawa Y, Isse N, Okazaki T, Satoh N, Masuzaki H, et al. Molecular cloning of rat leptin receptor isoform complementary DNAs – identification of a missense mutation in Zucker fatty (fa/fa) rats. Biochem Biophys Res Commun (1996) 225:75–83. doi:10.1006/bbrc.1996.1133
76. Duarte SFP, Francischetti EA, Genelhu-Abreu V, Barroso SG, Braga JU, Cabello PH, et al. Q223R leptin receptor polymorphism associated with obesity in Brazilian multiethnic subjects. Am J Hum Biol (2006) 18:448–53. doi:10.1002/ajhb.20519
77. Duggal P, Guo X, Haque R, Peterson KM, Ricklefs S, Mondal D, et al. A mutation in the leptin receptor is associated with Entamoeba histolytica infection in children. J Clin Invest (2011) 121:1191–8. doi:10.1172/JCI45294
78. Peelman F, Van Beneden K, Zabeau L, Iserentant H, Ulrichts P, Defeau D, et al. Mapping of the leptin binding sites and design of a leptin antagonist. J Biol Chem (2004) 279:41038–46. doi:10.1074/jbc.M404962200
79. Niv-Spector L, Gonen-Berger D, Gourdou I, Biener E, Gussakovsky EE, Benomar Y, et al. Identification of the hydrophobic strand in the A-B loop of leptin as major binding site III: implications for large-scale preparation of potent recombinant human and ovine leptin antagonists. Biochem J (2005) 391:221–30. doi:10.1042/bj20050457
80. Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci (1998) 1:271–2.
81. Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology (1997) 138:4489–92.
82. Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet (2000) 26:97–102. doi:10.1038/79254
83. Huszar D, Lynch CA, FairchildHuntress V, Dunmore JH, Fang Q, Berkemeier LR, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell (1997) 88:131–41.
84. Huo L, Gamber K, Greeley S, Silva J, Huntoon N, Leng XH, et al. Leptin-dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab (2009) 9:537–47. doi:10.1016/j.cmet.2009.05.003
85. Berglund ED, Vianna CR, Donato J, Kim MH, Chuang J-C, Lee CE, et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest (2012) 122:1000–9. doi:10.1172/JCI59816
86. Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen YR, Gantz I, et al. Antagonism of central melanocortin receptors in vitro and in vivo by Agouti-related protein. Science (1997) 278:135–8.
87. Cowley MA, Smart JL, Rubinstein M, Cordan MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature (2001) 411:480–4. doi:10.1038/35078085
88. Myers MG, Munzberg H, Leinninger GM, Leshan RL. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab (2009) 9:117–23. doi:10.1016/j.cmet.2008.12.001
89. Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron (2004) 42:983–91. doi:10.1016/j.neuron.2004.06.004
90. de Wall EV, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, et al. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology (2008) 149:1773–85. doi:10.1210/en.2007-1132
91. Vaisse C, Halaas JL, Horvath CM, Darnell JE, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wildtype and ob/ob mice but not db/db mice. Nat Genet (1996) 14:95–7.
92. Waelput W, Verhee A, Broekaert D, Eyckerman S, Vandekerckhove J, Beattie JH, et al. Identification and expression analysis of leptin-regulated immediate early response and late target genes. Biochem J (2000) 348(Pt 1):55–61. doi:10.1042/0264-6021:3480055
93. Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem (1999) 274:30059–65.
94. Cui YX, Huang L, Elefteriou F, Yang GQ, Shelton JM, Giles JE, et al. Essential role of STAT3 in body weight and glucose homeostasis. Mol Cell Biol (2004) 24:258–69. doi:10.1128/MCB.24.1.258-269.2004
95. Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, et al. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A (2004) 101:4661–6. doi:10.1073/pnas.0303992101
96. Buettner C, Pocai A, Muse ED, Etgen AM, Myers MG Jr, Rossetti L. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab (2006) 4:49–60. doi:10.1016/j.cmet.2006.04.014
97. Piper ML, Unger EK, Myers MG, Xu AW. Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol Endocrinol (2008) 22:751–9. doi:10.1210/me.2007-0389
98. Munzberg H, Huo LH, Nillni EA, Hollenberg AN, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology (2003) 144:2121–31. doi:10.1210/en.2002-221037
99. Xu AW, Ste-Marie L, Kaelin CB, Barsh GS. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased Pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology (2007) 148:72–80. doi:10.1210/En.2006-1119
100. Kaelin CB, Gong LJ, Xu AW, Yao FY, Hockman K, Morton GJ, et al. Signal transducer and activator of transcription (Stat) binding sites but not Stat3 are required for fasting-induced transcription of Agouti-related protein messenger ribonucleic acid. Mol Endocrinol (2006) 20:2591–602. doi:10.1210/me.2006-0107
101. Gong L, Yao F, Hockman K, Heng HH, Morton GJ, Takeda K, et al. Signal transducer and activator of transcription-3 is required in hypothalamic agouti-related protein/neuropeptide Y neurons for normal energy homeostasis. Endocrinology (2008) 149:3346–54. doi:10.1210/En.2007-0945
102. Bates SH, Kulkarni RN, Seifert M, Myers MG. Roles for leptin receptor/STAT3-dependent and -independent signals in the regulation of glucose homeostasis. Cell Metab (2005) 1:169–78. doi:10.1016/j.cmet.2005.02.001
103. Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AWK, Wang YP, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature (2003) 421:856–9. doi:10.1038/Nature01388
104. Bates SH, Dundon TA, Seifert M, Carlson M, Maratos-Filer E, Myers MG. LRb-STAT3 signaling is required for the neuroendocrine regulation of energy expenditure by leptin. Diabetes (2004) 53:3067–73. doi:10.2337/diabetes.53.12.3067
105. Iwasaki H, Kovacic JC, Olive M, Beers JK, Yoshimoto T, Crook MF, et al. Disruption of protein arginine N-methyltransferase 2 regulates leptin signaling and produces leanness in vivo through loss of STAT3 methylation. Circ Res (2010) 107:992–1001. doi:10.1161/circresaha.110.225326
106. Chen Y, Wu R, Chen H, Xiao Q, Wang W, He J, et al. Enhancement of hypothalamic STAT3 acetylation by nuclear receptor Nur77 dictates leptin sensitivity. Diabetes (2015) 64:2069–81. doi:10.2337/db14-1206
107. Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC. Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci U S A (1996) 93:6231–5.
108. Rosenblum CI, Tota M, Cully D, Smith T, Collum R, Qureshi S, et al. Functional STAT 1 and 3 signaling by the leptin receptor (OB-R); reduced expression of the rat fatty leptin receptor in transfected cells. Endocrinology (1996) 137:5178–81.
109. Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Muenzberg H, Myers MG. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J Biol Chem (2007) 282:31019–27. doi:10.1074/jbc.M702838200
110. Mutze J, Roth J, Gerstberger R, Hubschle T. Nuclear translocation of the transcription factor STAT5 in the rat brain after systemic leptin administration. Neurosci Lett (2007) 417:286–91. doi:10.1016/j.neulet.2007.02.074
111. Hekerman P, Zeidler J, Bamberg-Lemper S, Knobelspies H, Lavens D, Tavernier J, et al. Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS J (2005) 272:109–19. doi:10.1111/j.1432-1033.2004.04391.x
112. Montoye T, Piessevaux J, Lavens D, Wauman J, Catteeuw D, Vandekerckhove J, et al. Analysis of leptin signalling in hematopoietic cells using an adapted MAPPIT strategy. FEBS Lett (2006) 580:3301–7. doi:10.1016/j.febslet.2006.04.094
113. Patterson CM, Villanueva EC, Greenwald-Yarnell M, Rajala M, Gonzalez IE, Saini N, et al. Leptin action via LepR-b Tyr(1077) contributes to the control of energy balance and female reproduction. Mol Metab (2012) 1:61–9. doi:10.1016/j.molmet.2012.05.001
114. Singireddy AV, Inglis MA, Zuure WA, Kim JS, Anderson GM. Neither signal transducer and activator of transcription 3 (STAT3) or STAT5 signaling pathways are required for Leptin’s effects on fertility in mice. Endocrinology (2013) 154:2434–45. doi:10.1210/en.2013-1109
115. Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, et al. Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem (2001) 276:4747–55. doi:10.1074/jbc.M007439200
116. Carpenter LR, Farruggella TJ, Symes A, Karow ML, Yancopoulos GD, Stahl N. Enhancing leptin response by preventing SH2-containing phosphatase 2 interaction with Ob receptor. Proc Natl Acad Sci U S A (1998) 95:6061–6.
117. Li C, Friedman JM. Leptin receptor activation of SH2 domain containing protein tyrosine phosphatase 2 modulates Ob receptor signal transduction. Proc Natl Acad Sci U S A (1999) 96:9677–82.
118. Rahmouni K, Sigmund CD, Haynes WG, Mark AL. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes (2009) 58:536–42. doi:10.2337/db08-0822
119. Zhang EE, Chapeau E, Hagihara K, Feng GS. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A (2004) 101:16064–9. doi:10.1073/pnas.0405041101
120. Bjorbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem (1997) 272:32686–95.
121. Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, et al. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest (2010) 120:720–34. doi:10.1172/JCI39620
122. do Carmo JM, da Silva AA, Ebaady SE, Sessums PO, Abraham RS, Elmquist JK, et al. Shp2 signaling in POMC neurons is important for leptin’s actions on blood pressure, energy balance, and glucose regulation. Am J Physiol Regul Integr Comp Physiol (2014) 307:R1438–47. doi:10.1152/ajpregu.00131.2014
123. He Z, Zhang SS, Meng Q, Li S, Zhu HH, Raquil M-A, et al. Shp2 controls female body weight and energy balance by integrating leptin and estrogen signals. Mol Cell Biol (2012) 32:1867–78. doi:10.1128/MCB.06712-11
124. Bjornholm M, Munzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, et al. Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest (2007) 117:1354–60. doi:10.1172/JCI30688
125. You J, Yu Y, Jiang L, Li WX, Yu XX, Gonzalez L, et al. Signaling through Tyr(985) of leptin receptor as an age/diet-dependent switch in the regulation of energy balance. Mol Cell Biol (2010) 30:1650–9. doi:10.1128/MCB.01307-09
126. Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Schwartz MW. Intracellular signalling – key enzyme in leptin-induced anorexia. Nature (2001) 413:794–5. doi:10.1038/35101657
127. Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, et al. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci (2010) 30:2472–9. doi:10.1523/JNEUROSCI.3118-09.2010
128. Hill JW, Williams KW, Ye CP, Luo J, Balthasar N, Coppari R, et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest (2008) 118:1796–805. doi:10.1172/JCI32964
129. Duan CJ, Li MH, Rui LY. SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J Biol Chem (2004) 279:43684–91. doi:10.1074/jbc.M408495200
130. Li ZQ, Zhou YJ, Carter-Su C, Myers MG, Rui L. SH2B1 enhances leptin signaling by both janus kinase 2 Tyr(813) phosphorylation-dependent and -independent mechanisms. Mol Endocrinol (2007) 21:2270–81. doi:10.1210/me.2007-0111
131. Ren DC, Li MH, Duan CJ, Rui LY. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab (2005) 2:95–104. doi:10.1016/j.cmet.2005.07.004
132. Ren DC, Zhou YJ, Morris D, Li MH, Li ZQ, Rui LY. Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J Clin Invest (2007) 117:397–406. doi:10.1172/jci29417
133. Morris DL, Cho KW, Rui L. Critical role of the Src homology 2 (SH2) domain of neuronal SH2B1 in the regulation of body weight and glucose homeostasis in mice. Endocrinology (2010) 151:3643–51. doi:10.1210/en.2010-0254
134. Wauman J, De Smet AS, Catteeuw D, Belsham D, Tavernier J. Insulin receptor substrate 4 couples the leptin receptor to multiple signaling pathways. Mol Endocrinol (2008) 22:965–77. doi:10.1210/me.2007-0414
135. Kitamura T, Feng Y, Kitamura YI, Chua SC Jr, Xu AW, Barsh GS, et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med (2006) 12:534–40. doi:10.1038/nm1392
136. Kim M-S, Pak YK, Jang P-G, Namkoong C, Choi Y-S, Won J-C, et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci (2006) 9:901–6. doi:10.1038/nn1731
137. Yang G, Lim CY, Li C, Xiao X, Radda GK, Li C, et al. FoxO1 inhibits leptin regulation of pro-opiomelanocortin promoter activity by blocking STAT3 interaction with specificity protein 1. J Biol Chem (2009) 284:3719–27. doi:10.1074/jbc.M804965200
138. Cao Y, Nakata M, Okamoto S, Takano E, Yada T, Minokoshi Y, et al. PDK1-Foxo1 in agouti-related peptide neurons regulates energy homeostasis by modulating food intake and energy expenditure. PLoS One (2011) 6:e18324. doi:10.1371/journal.pone.0018324
139. Ma W, Fuentes G, Shi X, Verma C, Radda GK, Han W. FoxO1 negatively regulates leptin-induced POMC transcription through its direct interaction with STAT3. Biochem J (2015) 466:291–8. doi:10.1042/BJ20141109
140. Belgardt BF, Husch A, Rother E, Ernst MB, Wunderlich FT, Hampel B, et al. PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab (2008) 7:291–301. doi:10.1016/j.cmet.2008.01.006
141. Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, et al. The obesity susceptibility gene carboxypeptidase E links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat Med (2009) 15:1195–201. doi:10.1038/nm.2026
142. Ren H, Orozco IJ, Su Y, Suyama S, Gutiérrez-Juárez R, Horvath TL, et al. G protein-coupled purinergic receptor GPR17 mediates orexigenic effects of FoxO1 in AgRP neurons. Cell (2012) 149:1314–26. doi:10.1016/j.cell.2012.04.032
143. Plum L, Rother E, Munzberg H, Wunderlich FT, Morgan DA, Hampel B, et al. Enhanced leptin-stimulated PI3K activation in the CNS promotes white adipose tissue transdifferentiation. Cell Metab (2007) 6:431–45. doi:10.1016/j.cmet.2007.10.012
144. Plum L, Ma XS, Hampel B, Balthasar N, Coppari R, Munzberg H, et al. Enhanced PIP3 signaling in POMC neurons causes K-ATP channel activation and leads to diet-sensitive obesity. J Clin Invest (2006) 116:1886–901. doi:10.1172/jci27123
145. Cota D, Proulx K, Smith KAB, Kozma SC, Thomas G, Woods SC, et al. Hypothalamic mTOR signaling regulates food intake. Science (2006) 312:927–30. doi:10.1126/science.1124147
146. Cota D, Matter EK, Woods SC, Seeley RJ. The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J Neurosci (2008) 28:7202–8. doi:10.1523/jneurosci.1389-08.2008
147. Blouet C, Ono H, Schwartz GJ. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab (2008) 8:459–67. doi:10.1016/j.cmet.2008.10.004
148. Sahu M, Sahu A. Leptin receptor expressing neurons express phosphodiesterase-3B (PDE3B) and leptin induces STAT3 activation in PDE3B neurons in the mouse hypothalamus. Peptides (2015) 73:35–42. doi:10.1016/j.peptides.2015.08.011
149. Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci (2002) 5:727–8. doi:10.1038/nn885nn885
150. Sahu A. A role of phosphodiesterase-3B pathway in mediating leptin action on proopiomelanocortin and neurotensin neurons in the hypothalamus. Neurosci Lett (2010) 479:18–21. doi:10.1016/j.neulet.2010.05.018
151. Minokoshi Y, Kim YB, Peroni OD, Fryer LGD, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature (2002) 415:339–43. doi:10.1038/415339a
152. Uotani S, Abe T, Yamaguchi Y. Leptin activates AMP-activated protein kinase in hepatic cells via a JAK2-dependent pathway. Biochem Biophys Res Commun (2006) 351:171–5. doi:10.1016/j.bbrc.2006.10.015
153. Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature (2004) 428:569–74. doi:10.1038/nature02440
154. Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LGD, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest (2007) 117:2325–36. doi:10.1172/jci31516
155. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem (2004) 279:12005–8. doi:10.1074/jbc.C300557200C300557200
156. Gao S, Kinzig KP, Aja S, Scott KA, Keung W, Kelly S, et al. Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc Natl Acad Sci U S A (2007) 104:17358–63. doi:10.1073/pnas.0708385104
157. Tanida M, Yamamoto N, Shibamoto T, Rahmouni K. Involvement of hypothalamic AMP-activated protein kinase in leptin-induced sympathetic nerve activation. PLoS One (2013) 8:e56660. doi:10.1371/journal.pone.0056660
158. Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB. p70S6 kinase phosphorylates AMPK on serine 491 to mediate Leptin’s effect on food intake. Cell Metab (2012) 16:104–12. doi:10.1016/j.cmet.2012.05.010
159. Jiang L, You J, Yu XX, Gonzalez L, Yu Y, Wang Q, et al. Tyrosine-dependent and -independent actions of leptin receptor in control of energy balance and glucose homeostasis. Proc Natl Acad Sci U S A (2008) 105:18619–24. doi:10.1073/pnas.0804589105
160. Robertson S, Ishida-Takahashi R, Tawara I, Hu J, Patterson CM, Jones JC, et al. Insufficiency of Janus kinase 2-autonomous leptin receptor signals for most physiologic leptin actions. Diabetes (2010) 59:782–90. doi:10.2337/db09-1556
161. Huang H, Kong D, Byun KH, Ye C, Koda S, Lee DH, et al. Rho-kinase regulates energy balance by targeting hypothalamic leptin receptor signaling. Nat Neurosci (2012) 15:1391–8. doi:10.1038/nn.3207
162. Yang L, McKnight GS. Hypothalamic PKA regulates leptin sensitivity and adiposity. Nat Commun (2015) 6:8237. doi:10.1038/ncomms9237
163. Quaresma PGF, Weissmann L, Zanotto TM, Santos AC, de Matos AHB, Furigo IC, et al. Cdc2-like kinase 2 (CLK2) in the hypothalamus is necessary to maintain energy homeostasis. Int J Obes (2017) 41:268–78. doi:10.1038/ijo.2016.174
164. Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA (1999) 282:1568–75.
165. Mantzoros CS, Flier JS. Leptin as a therapeutic agent – trials and tribulations. J Clin Endocrinol Metab (2000) 85:4000–2. doi:10.1210/jcem.85.11.7062
166. Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell (1998) 1:619–25.
167. Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab (2011) 14:684–99. doi:10.1016/j.cmet.2011.09.011
168. White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab (2009) 296:E291–9. doi:10.1152/ajpendo.90513.2008
169. Dunn SL, Bjornholm M, Bates SH, Chen ZB, Seifert M, Myers MG. Feedback inhibition of leptin receptor/Jak2 signaling via Tyr(1138) of the leptin receptor and suppressor of cytokine signaling 3. Mol Endocrinol (2005) 19:925–38. doi:10.1210/Me.2004-0353
170. Bjorbaek C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, et al. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr(985). J Biol Chem (2000) 275:40649–57. doi:10.1074/jbc.M007577200
171. Eyckerman S, Broekaert D, Verhee A, Vandekerckhove J, Tavernier J. Identification of the Y985 and Y1077 motifs as SOCS3 recruitment sites in the murine leptin receptor. FEBS Lett (2000) 486:33–7. doi:10.1016/S0014-5793(00)02205-5
172. Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology (2004) 145:4880–9. doi:10.1210/En.2004-0726
173. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med (2004) 10:739–43. doi:10.1038/nm1071
174. Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, et al. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab (2006) 4:123–32. doi:10.1016/j.cmet.2006.06.010
175. Reed AS, Unger EK, Olofsson LE, Piper ML, Myers MG, Xu AW. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes (2010) 59:894–906. doi:10.2337/db09-1024
176. Ernst MB, Wunderlich CM, Hess S, Paehler M, Mesaros A, Koralov SB, et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci (2009) 29:11582–93. doi:10.1523/JNEUROSCI.5712-08.2009
177. Faouzi M, Leshan R, Bjornholm M, Hennessey T, Jones J, Munzberg H. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology (2007) 148:5414–23. doi:10.1210/en.2007-0655
178. Olofsson LE, Unger EK, Cheung CC, Xu AW. Modulation of AgRP-neuronal function by SOCS3 as an initiating event in diet-induced hypothalamic leptin resistance. Proc Natl Acad Sci U S A (2013) 110:E697–706. doi:10.1073/pnas.1218284110
179. Pedroso JAB, Buonfiglio DC, Cardinali LI, Furigo IC, Ramos-Lobo AM, Tirapegui J, et al. Inactivation of SOCS3 in leptin receptor-expressing cells protects mice from diet-induced insulin resistance but does not prevent obesity. Mol Metab (2014) 3:608–18. doi:10.1016/j.molmet.2014.06.001
180. Pedroso JAB, Silveira MA, Lima LB, Furigo IC, Zampieri TT, Ramos-Lobo AM, et al. Changes in leptin signaling by SOCS3 modulate fasting-induced hyperphagia and weight regain in mice. Endocrinology (2016) 157(10):3901–14. doi:10.1210/en.2016-1038
181. Kaszubska W, Falls HD, Schaefer VG, Haasch D, Frost L, Hessler P, et al. Protein tyrosine phosphatase 1B negatively regulates leptin signaling in a hypothalamic cell line. Mol Cell Endocrinol (2002) 195:109–18. doi:10.1016/S0303-7207(02)00178-8
182. Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell (2002) 2:489–95. doi:10.1016/S1534-5807(02)00148-X
183. Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med (2006) 12:917–24. doi:10.1038/nm1435
184. Tsou RC, Zimmer DJ, De Jonghe BC, Bence KK. Deficiency of PTP1B in leptin receptor-expressing neurons leads to decreased body weight and adiposity in mice. Endocrinology (2012) 153:4227–37. doi:10.1210/en.2012-1548
185. Tsou RC, Rak KS, Zimmer DJ, Bence KK. Improved metabolic phenotype of hypothalamic PTP1B-deficiency is dependent upon the leptin receptor. Mol Metab (2014) 3:301–12. doi:10.1016/j.molmet.2014.01.008
186. St-Pierre J, Tremblay ML. Modulation of leptin resistance by protein tyrosine phosphatases. Cell Metab (2012) 15:292–7. doi:10.1016/j.cmet.2012.02.004
187. Rousso-Noori L, Knobler H, Levy-Apter E, Kuperman Y, Neufeld-Cohen A, Keshet Y, et al. Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner. Cell Metab (2011) 13:562–72. doi:10.1016/j.cmet.2011.02.017
188. Horvath TL, Sarman B, García-Cáceres C, Enriori PJ, Sotonyi P, Shanabrough M, et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A (2010) 107:14875–80. doi:10.1073/pnas.1004282107
189. Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science (2004) 304:110–5. doi:10.1126/science.1089459
190. Leshan RL, Greenwald-Yarnell M, Patterson CM, Gonzalez IE, Myers MG. Leptin action via hypothalamic nitric oxide synthase-1 neurons controls energy balance. Nat Med (2012) 18:820–3. doi:10.1038/nm.2724
191. Vong L, Ye C, Yang Z, Choi B, Chua S, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron (2011) 71:142–54. doi:10.1016/j.neuron.2011.05.028
192. Chowen JA, Argente-Arizón P, Freire-Regatillo A, Frago LM, Horvath TL, Argente J. The role of astrocytes in the hypothalamic response and adaptation to metabolic signals. Prog Neurobiol (2016) 144:68–87. doi:10.1016/j.pneurobio.2016.03.001
193. Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, et al. Leptin signaling in GFAP-expressing adult glia cells regulates hypothalamic neuronal circuits and feeding. Nat Neurosci (2014) 17:908–10. doi:10.1038/nn.3725
194. Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med (1996) 2:589–93.
195. Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet (1996) 348:159–61.
196. Banks WA, Kastin AJ, Huang WT, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides (1996) 17:305–11.
197. Hileman SM, Pierroz DD, Masuzaki H, Bjorbaek C, El-Haschimi K, Banks WA, et al. Characterizaton of short isoforms of the leptin receptor in rat cerebral microvessels and of brain uptake of leptin in mouse models of obesity. Endocrinology (2002) 143:775–83. doi:10.1210/en.143.3.775
198. Kastin AJ, Pan WH, Maness LM, Koletsky RJ, Ernsberger P. Decreased transport of leptin across the blood-brain barrier in rats lacking the short form of the leptin receptor. Peptides (1999) 20:1449–53.
199. Li Z, Ceccarini G, Eisenstein M, Tan K, Friedman JM. Phenotypic effects of an induced mutation of the ObRa isoform of the leptin receptor. Mol Metab (2013) 2:364–75. doi:10.1016/j.molmet.2013.07.007
200. Gonzalez-Carter D, Goode AE, Fiammengo R, Dunlop IE, Dexter DT, Porter AE. Inhibition of Leptin–ObR interaction does not prevent leptin translocation across a human blood–brain barrier model. J Neuroendocrinol (2016) 28(6). doi:10.1111/jne.12392
201. Dietrich MO, Spuch C, Antequera D, Rodal I, de Yébenes JG, Molina JA, et al. Megalin mediates the transport of leptin across the blood-CSF barrier. Neurobiol Aging (2008) 29:902–12. doi:10.1016/j.neurobiolaging.2007.01.008
202. Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab (2014) 19:293–301. doi:10.1016/j.cmet.2013.12.015
203. Mitchell SE, Nogueiras R, Morris A, Tovar S, Grant C, Cruickshank M, et al. Leptin receptor gene expression and number in the brain are regulated by leptin level and nutritional status. J Physiol (2009) 587:3573–85. doi:10.1113/jphysiol.2009.173328
204. Gamber KM, Huo L, Ha S, Hairston JE, Greeley S, Bjørbaek C. Over-expression of leptin receptors in hypothalamic POMC neurons increases susceptibility to diet-induced obesity. PLoS One (2012) 7:e30485. doi:10.1371/journal.pone.0030485
205. Couturier C, Sarkis C, Séron K, Belouzard S, Chen P, Lenain A, et al. Silencing of OB-RGRP in mouse hypothalamic arcuate nucleus increases leptin receptor signaling and prevents diet-induced obesity. Proc Natl Acad Sci U S A (2007) 104:19476–81. doi:10.1073/pnas.0706671104
206. Séron K, Couturier C, Belouzard S, Bacart J, Monté D, Corset L, et al. Endospanins regulate a postinternalization step of the leptin receptor endocytic pathway. J Biol Chem (2011) 286:17968–81. doi:10.1074/jbc.M111.224857
207. Vauthier V, Swartz TD, Chen P, Roujeau C, Pagnon M, Mallet J, et al. Endospanin 1 silencing in the hypothalamic arcuate nucleus contributes to sustained weight loss of high fat diet obese mice. Gene Ther (2014) 21:638–44. doi:10.1038/gt.2014.36
208. De Ceuninck L, Wauman J, Masschaele D, Peelman F, Tavernier J. Reciprocal cross-regulation between RNF41 and USP8 controls cytokine receptor sorting and processing. J Cell Sci (2013) 126:3770–81. doi:10.1242/jcs.131250
209. Han YM, Kang GM, Byun K, Ko HW, Kim J, Shin M-S, et al. Leptin-promoted cilia assembly is critical for normal energy balance. J Clin Invest (2014) 124:2193–7. doi:10.1172/JCI69395
210. Kang GM, Han YM, Ko HW, Kim J, Oh BC, Kwon I, et al. Leptin elongates hypothalamic neuronal cilia via transcriptional regulation and actin destabilization. J Biol Chem (2015) 290:18146–55. doi:10.1074/jbc.M115.639468
211. Guo D-F, Rahmouni K. Molecular basis of the obesity associated with Bardet-Biedl syndrome. Trends Endocrinol Metab (2011) 22:286–93. doi:10.1016/j.tem.2011.02.009
212. Guo D-F, Cui H, Zhang Q, Morgan DA, Thedens DR, Nishimura D, et al. The BBSome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. PLoS Genet (2016) 12:e1005890. doi:10.1371/journal.pgen.1005890
213. Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet (2009) 18:1323–31. doi:10.1093/hmg/ddp031
214. Stratigopoulos G, LeDuc CA, Cremona ML, Chung WK, Leibel RL. Cut-like homeobox 1 (CUX1) regulates expression of the fat mass and obesity-associated and retinitis pigmentosa GTPase regulator-interacting protein-1-like (RPGRIP1L) genes and coordinates leptin receptor signaling. J Biol Chem (2011) 286:2155–70. doi:10.1074/jbc.M110.188482
215. Stratigopoulos G, Martin Carli JF, O’Day DR, Wang L, LeDuc CA, Lanzano P, et al. Hypomorphism for RPGRIP1L, a ciliary gene vicinal to the FTO locus, causes increased adiposity in mice. Cell Metab (2014) 19:767–79. doi:10.1016/j.cmet.2014.04.009
216. Stratigopoulos G, Burnett LC, Rausch R, Gill R, Penn DB, Skowronski AA, et al. Hypomorphism of Fto and Rpgrip1l causes obesity in mice. J Clin Invest (2016) 126:1897–910. doi:10.1172/JCI85526
217. Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab (2009) 9:35–51. doi:10.1016/j.cmet.2008.12.004
218. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKK[beta]/NF-[kappa]B and ER stress link overnutrition to energy imbalance and obesity. Cell (2008) 135:61–73. doi:10.1016/j.cell.2008.07.043
219. Liu J, Lee J, Hernandez MAS, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell (2015) 161:999–1011. doi:10.1016/j.cell.2015.05.011
220. Hosoi T, Ozawa K. Possible pharmacological approach targeting endoplasmic reticulum stress to ameliorate leptin resistance in obesity. Front Endocrinol (2016) 7:59. doi:10.3389/fendo.2016.00059
221. Williams KW, Liu T, Kong X, Fukuda M, Deng Y, Berglund ED, et al. Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab (2014) 20:471–82. doi:10.1016/j.cmet.2014.06.002
222. Ramírez S, Claret M. Hypothalamic ER stress: a bridge between leptin resistance and obesity. FEBS Lett (2015) 589:1678–87. doi:10.1016/j.febslet.2015.04.025
223. Thaler JP, Yi C-X, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest (2012) 122:153–62. doi:10.1172/JCI59660
224. Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab (2009) 10:249–59. doi:10.1016/j.cmet.2009.08.013
225. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest (2006) 116:3015–25. doi:10.1172/JCI28898
226. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci (2009) 29:359–70. doi:10.1523/jneurosci.2760-08.2009
227. Weissmann L, Quaresma PGF, Santos AC, de Matos AHB, Pascoal VDB, Zanotto TM, et al. IKKε is key to induction of insulin resistance in the hypothalamus, and its inhibition reverses obesity. Diabetes (2014) 63:3334–45. doi:10.2337/db13-1817
228. Schneeberger M, Dietrich MO, Sebastián D, Imbernón M, Castaño C, Garcia A, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell (2013) 155:172–87. doi:10.1016/j.cell.2013.09.003
229. Long L, Toda C, Jeong JK, Horvath TL, Diano S. PPARγ ablation sensitizes proopiomelanocortin neurons to leptin during high-fat feeding. J Clin Invest (2014) 124:4017–27. doi:10.1172/JCI76220
230. Dietrich MO, Liu Z-W, Horvath TL. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell (2013) 155:188–99. doi:10.1016/j.cell.2013.09.004
231. Ottaway N, Mahbod P, Rivero B, Norman LA, Gertler A, D’Alessio D, et al. Diet-induced obese mice retain endogenous leptin action. Cell Metab (2015) 21:877–82. doi:10.1016/j.cmet.2015.04.015
232. Myers MG, Heymsfield SB, Haft C, Kahn BB, Laughlin M, Leibel RL, et al. Challenges and opportunities of defining clinical leptin resistance. Cell Metab (2012) 15:150–6. doi:10.1016/j.cmet.2012.01.002
233. Nizard J, Dommergue M, Clément K. Pregnancy in a woman with a leptin-receptor mutation. N Engl J Med (2012) 366:1064–5. doi:10.1056/NEJMc1200116
234. Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature (1998) 392:398–401.
235. Fischer-Posovszky P, von Schnurbein J, Moepps B, Lahr G, Strauss G, Barth TF, et al. A new missense mutation in the leptin gene causes mild obesity and hypogonadism without affecting T cell responsiveness. J Clin Endocrinol Metab (2010) 95:2836–40. doi:10.1210/jc.2009-2466
236. Zabeau L, Jensen CJ, Seeuws S, Venken K, Verhee A, Catteeuw D, et al. Leptin’s metabolic and immune functions can be uncoupled at the ligand/receptor interaction level. Cell Mol Life Sci (2015) 72:629–44. doi:10.1007/s00018-014-1697-x
237. Shida D, Kitayama J, Mori K, Watanabe T, Nagawa H. Transactivation of epidermal growth factor receptor is involved in leptin-induced activation of janus-activated kinase 2 and extracellular signal-regulated kinase 1/2 in human gastric cancer cells. Cancer Res (2005) 65:9159–63. doi:10.1158/0008-5472.CAN-05-0598
238. Knight BB, Oprea-Ilies GM, Nagalingam A, Yang L, Cohen C, Saxena NK, et al. Survivin upregulation, dependent on leptin-EGFR-Notch1 axis, is essential for leptin-induced migration of breast carcinoma cells. Endocr Relat Cancer (2011) 18:413–28. doi:10.1530/ERC-11-0075
239. Beales ILP, Ogunwobi OO. Leptin synergistically enhances the anti-apoptotic and growth-promoting effects of acid in OE33 oesophageal adenocarcinoma cells in culture. Mol Cell Endocrinol (2007) 274:60–8. doi:10.1016/j.mce.2007.05.017
240. Ogunwobi OO, Beales ILP. Leptin stimulates the proliferation of human oesophageal adenocarcinoma cells via HB-EGF and TGF alpha mediated transactivation of the epidermal growth factor receptor. Br J Biomed Sci (2008) 65:121–7. doi:10.1080/09674845.2008.11732814
241. Chao H-H, Hong H-J, Liu J-C, Lin J-W, Chen Y-L, Chiu W-T, et al. Leptin stimulates endothelin-1 expression via extracellular signal-regulated kinase by epidermal growth factor receptor transactivation in rat aortic smooth muscle cells. Eur J Pharmacol (2007) 573:49–54. doi:10.1016/j.ejphar.2007.06.051
242. Slomiany BL, Slomiany A. Leptin-induced cytosolic phospholipase A2 activation in gastric mucosal protection against ethanol cytotoxicity involves epidermal growth factor receptor transactivation. Inflammopharmacology (2009) 17:6–14. doi:10.1007/s10787-008-8037-4
243. Slomiany BL, Slomiany A. Role of epidermal growth factor receptor transactivation in the activation of cytosolic phospholipase A(2) in leptin protection of salivary gland acinar cells against ethanol cytotoxicity. J Physiol Pharmacol (2009) 60:49–55.
244. Eisenberg A, Biener E, Charlier M, Krishnan RV, Djiane J, Herman B, et al. Transactivation of erbB2 by short and long isoforms of leptin receptors. FEBS Lett (2004) 565:139–42. doi:10.1016/j.febslet.2004.03.089
245. Beltowski J. Leptin and atherosclerosis. Atherosclerosis (2006) 189:47–60. doi:10.1016/j.atherosclerosis.2006.03.003
246. Binai NA, Damert A, Carra G, Steckelbroeck S, Löwer J, Löwer R, et al. Expression of estrogen receptor alpha increases leptin-induced STAT3 activity in breast cancer cells. Int J Cancer (2010) 127:55–66. doi:10.1002/ijc.25010
247. Catalano S, Marsico S, Giordano C, Mauro L, Rizza P, Panno ML, et al. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J Biol Chem (2003) 278:28668–76. doi:10.1074/jbc.M301695200
248. Catalano S, Mauro L, Marsico S, Giordano C, Rizza P, Rago V, et al. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J Biol Chem (2004) 279:19908–15. doi:10.1074/jbc.M313191200
249. Fusco R, Galgani M, Procaccini C, Franco R, Pirozzi G, Fucci L, et al. Cellular and molecular crosstalk between leptin receptor and estrogen receptor-alpha in breast cancer: molecular basis for a novel therapeutic setting. Endocr Relat Cancer (2010) 17:373–82. doi:10.1677/ERC-09-0340
250. Yu W, Gu J, Liu J, Wang S, Wang Y, Zhang Z, et al. Regulation of estrogen receptors alpha and beta in human breast carcinoma by exogenous leptin in nude mouse xenograft model. Chin Med J (Engl) (2010) 123:337–43. doi:10.3760/cma.j.issn.0366-6999.2010.03.015
251. Ozbay T, Nahta R. A novel unidirectional cross-talk from the insulin-like growth factor-I receptor to leptin receptor in human breast cancer cells. Mol Cancer Res (2008) 6:1052–8. doi:10.1158/1541-7786.MCR-07-2126
252. Saxena NK, Taliaferro-Smith L, Knight BB, Merlin D, Anania FA, O’Regan RM, et al. Bidirectional crosstalk between leptin and insulin-like growth factor-I signaling promotes invasion and migration of breast cancer cells via transactivation of epidermal growth factor receptor. Cancer Res (2008) 68:9712–22. doi:10.1158/0008-5472.CAN-08-1952
253. Liu Q, Zhang J, Zerbinatti C, Zhan Y, Kolber BJ, Herz J, et al. Lipoprotein receptor LRP1 regulates leptin signaling and energy homeostasis in the adult central nervous system. PLoS Biol (2011) 9:e1000575. doi:10.1371/journal.pbio.1000575
254. Ceccarini G, Flavell RR, Butelman ER, Synan M, Willnow TE, Bar-Dagan M, et al. PET imaging of leptin biodistribution and metabolism in rodents and primates. Cell Metab (2009) 10:148–59. doi:10.1016/j.cmet.2009.07.001
255. Dietrich MO, Spuch C, Antequera D, Rodal I, de Yebenes JG, Molina JA, et al. Megalin mediates the transport of leptin across the blood-CSF barrier. Neurobiol Aging (2008) 29:902–12. doi:10.1016/j.neurobiolaging.2007.01.008
256. Hama H, Saito A, Takeda T, Tanuma A, Xie Y, Sato K, et al. Evidence indicating that renal tubular metabolism of leptin is mediated by megalin but not by the leptin receptors. Endocrinology (2004) 145:3935–40. doi:10.1210/en.2004-0074
257. Garonna E, Botham KM, Birdsey GM, Randi AM, Gonzalez-Perez RR, Wheeler-Jones CPD. Vascular endothelial growth factor receptor-2 couples cyclo-oxygenase-2 with pro-angiogenic actions of leptin on human endothelial cells. PLoS One (2011) 6:e18823. doi:10.1371/journal.pone.0018823
258. Lanier V, Gillespie C, Leffers M, Daley-Brown D, Milner J, Lipsey C, et al. Leptin-induced transphosphorylation of vascular endothelial growth factor receptor increases Notch and stimulates endothelial cell angiogenic transformation. Int J Biochem Cell Biol (2016) 79:139–50. doi:10.1016/j.biocel.2016.08.023
259. Beltowski J, Jazmroz-Wisniewska A. Transactivation of ErbB receptors by leptin in the cardiovascular system: mechanisms, consequences and target for therapy. Curr Pharm Des (2014) 20:616–24.
260. Saxena NK, Sharma D. Multifaceted leptin network: the molecular connection between obesity and breast cancer. J Mammary Gland Biol Neoplasia (2013) 18:309–20. doi:10.1007/s10911-013-9308-2
261. Shimizu C, Hasegawa T, Tani Y, Takahashi F, Takeuchi M, Watanabe T, et al. Expression of insulin-like growth factor 1 receptor in primary breast cancer: immunohistochemical analysis. Hum Pathol (2004) 35:1537–42. doi:10.1016/J.HUMPATH.2004.09.005
262. Sachdev D. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J Biol Chem (2003) 279:5017–24. doi:10.1074/jbc.M305403200
263. Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci (2009) 10:333–44. doi:10.1038/nrn2620
264. Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol (1995) 6:35–47.
265. Fischer C, Schneider M, Carmeliet P. Principles and therapeutic implications of angiogenesis, vasculogenesis and arteriogenesis. Handb Exp Pharmacol (2006) 176:157–212.
266. Brown LM, Clegg DJ, Benoit SC, Woods SC. Intraventricular insulin and leptin reduce food intake and body weight in C57BL/6J mice. Physiol Behav (2006) 89:687–91. doi:10.1016/j.physbeh.2006.08.008
267. Thon M, Hosoi T, Ozawa K. Possible integrative actions of leptin and insulin signaling in the hypothalamus targeting energy homeostasis. Front Endocrinol (2016) 7:138. doi:10.3389/fendo.2016.00138
268. Niswender KD, Schwartz MW. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocrinol (2003) 24:1–10. doi:10.1016/s0091-3022(02)00105-x
269. Thon M, Hosoi T, Ozawa K. Insulin enhanced leptin-induced STAT3 signaling by inducing GRP78. Sci Rep (2016) 6:34312. doi:10.1038/srep34312
270. Fujikawa T, Berglund ED, Patel VR, Ramadori G, Vianna CR, Vong L, et al. Leptin engages a hypothalamic neurocircuitry to permit survival in the absence of insulin. Cell Metab (2013) 18:431–44. doi:10.1016/j.cmet.2013.08.004
271. Nazarians-Armavil A, Menchella JA, Belsham DD. Cellular insulin resistance disrupts leptin-mediated control of neuronal signaling and transcription. Mol Endocrinol (2013) 27:990–1003. doi:10.1210/me.2012-1338
272. Gonzalez-Perez RR, Xu YB, Guo SC, Watters A, Zhou WQ, Leibovich SJ. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NF kappa B/HIF-1 alpha activation. Cell Signal (2010) 22:1350–62. doi:10.1016/j.cellsig.2010.05.003
273. Guo S, Gonzalez-Perez RR. Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS One (2011) 6:e21467. doi:10.1371/journal.pone.0021467
274. Battle M, Gillespie C, Quarshie A, Lanier V, Harmon T, Wilson K, et al. Obesity induced a leptin-notch signaling axis in breast cancer. Int J Cancer (2014) 134:1605–16. doi:10.1002/ijc.28496
275. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology (2005) 146:4192–9. doi:10.1210/en.2004-1520
276. Romanatto T, Roman EA, Arruda AP, Denis RG, Solon C, Milanski M, et al. Deletion of tumor necrosis factor-α receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J Biol Chem (2009) 284:36213–22. doi:10.1074/jbc.M109.030874
277. Romanatto T, Cesquini M, Amaral ME, Roman ÉA, Moraes JC, Torsoni MA, et al. TNF-α acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient – effects on leptin and insulin signaling pathways. Peptides (2007) 28:1050–8. doi:10.1016/j.peptides.2007.03.006
278. Luheshi GN, Gardner JD, Rushforth DA, Loudon AS, Rothwell NJ. Leptin actions on food intake and body temperature are mediated by IL-1. Proc Natl Acad Sci U S A (1999) 96:7047–52.
279. Ropelle ER, Flores MB, Cintra DE, Rocha GZ, Pauli JR, Morari J, et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol (2010) 8:e1000465. doi:10.1371/journal.pbio.1000465
280. Borges BC, Garcia-Galiano D, Rorato R, Elias LLK, Elias CF. PI3K p110β subunit in leptin receptor expressing cells is required for the acute hypophagia induced by endotoxemia. Mol Metab (2016) 5:379–91. doi:10.1016/j.molmet.2016.03.003
281. Jang P-G, Namkoong C, Kang GM, Hur M-W, Kim S-W, Kim GH, et al. NF-κB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. J Biol Chem (2010) 285:9706–15. doi:10.1074/jbc.M109.070706
282. Shi X, Wang X, Li Q, Su M, Chew E, Wong ET, et al. Nuclear factor κB (NF-κB) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia (2013) 56:925–36. doi:10.1007/s00125-013-2831-2
Keywords: leptin, leptin receptor, activation, signaling, cross-talk, leptin resistance
Citation: Wauman J, Zabeau L and Tavernier J (2017) The Leptin Receptor Complex: Heavier Than Expected? Front. Endocrinol. 8:30. doi: 10.3389/fendo.2017.00030
Received: 01 December 2016; Accepted: 01 February 2017;
Published: 21 February 2017
Edited by:
Stefan N. Constantinescu, Ludwig Institute for Cancer Research, Belgium and Université catholique de Louvain, BelgiumReviewed by:
Serge Haan, University of Luxembourg, LuxembourgJulie Dam, Institut national de la santé et de la recherche médicale, France
Copyright: © 2017 Wauman, Zabeau and Tavernier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Tavernier, amFuLnRhdmVybmllciYjeDAwMDQwO3ZpYi11Z2VudC5iZQ==