 Veronica Vella1
Veronica Vella1 Maria Luisa Nicolosi2
Maria Luisa Nicolosi2 Stefania Giuliano2
Stefania Giuliano2 Maria Bellomo1
Maria Bellomo1 Antonino Belfiore2*
Antonino Belfiore2* Roberta Malaguarnera2
Roberta Malaguarnera2
- 1Scienze delle Attività Motorie e Sportive, University Kore, Enna, Italy
- 2Endocrinology, Department of Health Sciences, University Magna Graecia of Catanzaro, Catanzaro, Italy
It is now widely accepted that insulin resistance and compensatory hyperinsulinemia are associated to increased cancer incidence and mortality. Moreover, cancer development and progression as well as cancer resistance to traditional anticancer therapies are often linked to a deregulation/overactivation of the insulin-like growth factor (IGF) axis, which involves the autocrine/paracrine production of IGFs (IGF-I and IGF-II) and overexpression of their cognate receptors [IGF-I receptor, IGF-insulin receptor (IR), and IR]. Recently, new drugs targeting various IGF axis components have been developed. However, these drugs have several limitations including the occurrence of insulin resistance and compensatory hyperinsulinemia, which, in turn, may affect cancer cell growth and survival. Therefore, new therapeutic approaches are needed. In this regard, the pleiotropic effects of peroxisome proliferator activated receptor (PPAR)-γ agonists may have promising applications in cancer prevention and therapy. Indeed, activation of PPAR-γ by thiazolidinediones (TZDs) or other agonists may inhibit cell growth and proliferation by lowering circulating insulin and affecting key pathways of the Insulin/IGF axis, such as PI3K/mTOR, MAPK, and GSK3-β/Wnt/β-catenin cascades, which regulate cancer cell survival, cell reprogramming, and differentiation. In light of these evidences, TZDs and other PPAR-γ agonists may be exploited as potential preventive and therapeutic agents in tumors addicted to the activation of IGF axis or occurring in hyperinsulinemic patients. Unfortunately, clinical trials using PPAR-γ agonists as antineoplastic agents have reached conflicting results, possibly because they have not selected tumors with overactivated insulin/IGF-I axis or occurring in hyperinsulinemic patients. In conclusion, the use of PPAR-γ agonists in combined therapies of IGF-driven malignancies looks promising but requires future developments.
Introduction
Peroxisome proliferator activated receptors (PPARs) are transcription factors that regulate gene expression and repression upon binding to natural or synthetic ligands (1). PPARs belong to the nuclear hormone receptor superfamily that includes receptors for steroids, thyroid hormones, vitamin D, and retinoic acid. Different subtypes of PPARs called PPAR-α, PPAR-β, PPAR-γ, and PPAR-δ have been identified. Each of them displays differential tissue distribution and mediates specific functions in early development, cell proliferation, differentiation, apoptosis, and metabolic homeostasis (1). PPAR-γ is expressed at high levels in adipose tissue and at lower levels in several other tissues, such as breast, colon, lung, ovary, prostate, and thyroid (1, 2). Many synthetic PPAR-γ ligands have been developed. The most widely used synthetic agents belong to the thiazolidinedione (TZD) class of antidiabetic drugs (also referred to as glitazones or TZDs) that includes ciglitazone, troglitazone, pioglitazone (PIO), and rosiglitazone (RGZ). Some glitazones are already in the clinical use as insulin sensitizers in patients with type 2 diabetes mellitus (T2DM) (3). Activation of PPAR-γ plays an inhibitory role in cell growth and proliferation by favoring cell differentiation (4). These properties make PPAR-γ activation by natural and synthetic ligands an attractive option in cancer prevention and treatment. However, PPAR-γ ligands exert their effects through both PPAR-γ dependent and independent pathways, often triggering cross talks with other signaling pathways, including the insulin-like growth factor (IGF) system signaling.
Dysregulated activation of IGF axis has recently emerged as a relevant factor in development and progression of a variety of human malignancies (5–8). For instance, cancer cells are frequently characterized by altered expression of various components of the IGF including autocrine and/or paracrine secretion of IGFs (IGF-I and IGF-II) and overexpression of their cognate receptors [the IGF-I receptor, IGF-insulin receptor (IR), and the closely related IR]. In particular, IR overexpression may explain the increased sensitivity of cancer cells to hyperinsulinemia. Notably, in cancer cells, IR is often predominantly expressed as the “fetal” insulin receptor isoform A (IR-A), which binds both insulin and IGF-II (9). IR overexpression also contributes to enhanced signaling of IGF-II and IGF-I through the formation of IR/IGF-IR hybrid receptors (5). Not surprisingly, a number of epidemiological studies have consistently demonstrated that insulin resistance and hyperinsulinemia, common features of obesity and T2DM, are often associated with increased risk for several types of cancer (including cancers of the breast, colorectum, liver, and pancreas) (10–12) (Figure 1). One viable anticancer strategy is, therefore, to reduce insulin resistance and/or target the various IGF system components that are deregulated and that sustain the constitutive overactivation of IGF axis in cancer cells.
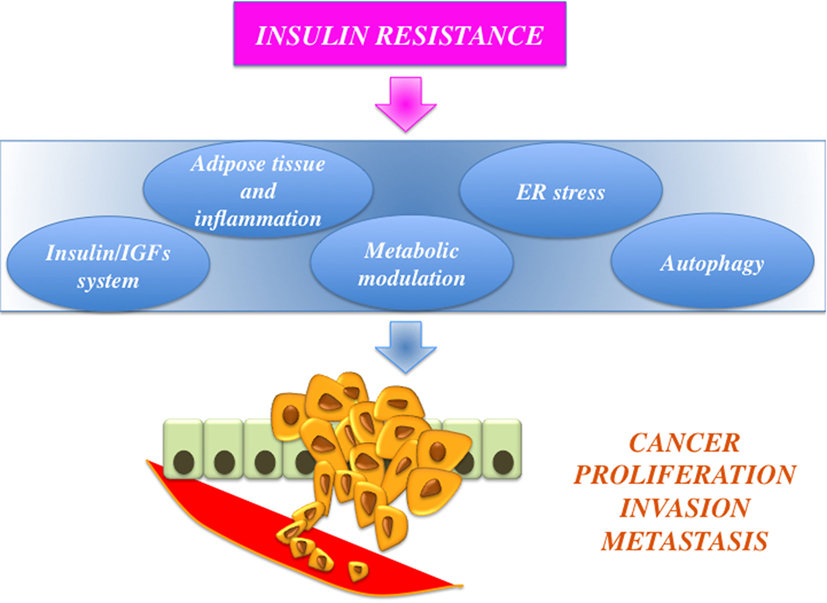
Figure 1. Schematic representation of the links between obesity, insulin resistance, and cancer. Insulin resistance is associated with protumoral actions, such as insulin-like growth factors (IGFs) axis hyperactivation, adiposity-related low-grade inflammation, and modulation of cell metabolism, endoplasmic reticulum (ER) stress, and altered autophagy.
This review will focus on the mechanisms that link insulin resistance to cancer risk and will discuss the possible clinical implications of PPAR-γ agonists in cancers with dysregulated IGF axis.
The Link Between Obesity, Insulin Resistance, and Cancer
Several epidemiological studies have consistently shown that metabolic disorders characterized by hyperinsulinemia and insulin resistance, such as obesity and T2DM, are associated with a significantly increased risk of cancer and cancer-specific mortality (13). The links underlying this association are not entirely clear and appear to involve a number of complex mechanisms.
Mechanisms Involving the Insulin/IGFs System and Estrogens
Insulin is a major mediator of important metabolic functions. However, it is widely accepted that it may exert mitogenic functions through the activation of different signaling pathways. Notably, cancer cells usually overexpress molecules that are typical of fetal life, as they confer them a survival advantage compared to normal cells (14). Specifically, the IR-A, which is predominantly expressed by fetal and cancer cells, exerts proliferative and protumoral effects in response to insulin, proinsulin, and IGF-II. In contrast, the second isoform, IR-B, is mainly involved in metabolic functions (9, 15, 16). IR-A also binds IGF-I with a lower affinity than IGF-II. Both IGFs circulate in the bloodstream but they are also secreted at high levels by cancer cells and/or tumor stroma (17) thus activating IR-A in cancer cells (5, 18). Moreover, while in normoinsulinemic individuals the principal pathway activated by insulin to control whole-body metabolism is the PI3K cascade, in insulin resistant, hyperinsulinemic subjects this metabolic branch is blunted while the more mitogenic MAPK/mTOR cascade is overactivated. This unbalance between the MAPK and the PI3K cascades is associated with impaired glucose/lipid homeostasis in typical target tissues like liver, muscle, and adipose tissue and with increased cell proliferation in other tissues (12, 19). In brief, insulin resistance and hyperinsulinemia are considered the main determinants of cancer initiation/progression in diabetic/obese patients, and therefore the major preventable cancer risk factor in these subjects.
Moreover, hyperinsulinemia may increase the bioavailability of IGF-I and IGF-II through two mechanisms: (1) by inhibiting the synthesis of certain IGF-binding proteins, such as IGF-binding protein 1 (IGF-BP1) and IGF-binding protein 2 (IGF-BP2), (2) by increasing IGF-I hepatic production.
The increased bioavailability of IGFs may contribute to tumor progression through the stimulation of IGF-IR, IR/IGF-IR hybrids, and IR-A itself (8). Partner molecules can be further recruited by these activated receptors contributing to increase cancer proliferation and migration (20, 21). Finally, a cross talk between the insulin/IGF axis and estrogens has been extensively reported in tumors where both signaling pathways are involved in controlling cell survival and proliferation. The functional interactions between these signaling cascades may occur at multiple levels. For instance, increased circulating insulin reduces the sex hormone-binding globulin thus increasing bioavailable sex steroids (22), which are associated with a higher risk for estrogen-dependent cancers such as breast and endometrial cancers (23). Moreover, ligand-dependent and independent activation of estrogen receptors increases insulin/IGF-mediated growth effects in several tumors, such as neuroblastoma, pituitary adenomas, and cancers from breast and prostate (24–26). Additionally, in prostate cancer cells, activation of classical ERs (and of androgen receptors) located at the level of cell membrane induces IGF-IR upregulation via membrane-initiated steroid signaling and enhances IGF-mediated biological effects (27, 28). Yet, in breast cancer cells, ligand-activated IGF-IR and IR upregulate the non-classical estrogen receptor (GPER), which potentiates the protumoral actions of insulin/IGFs and estrogens (29). Overall, all these functional interactions between insulin/IGFs and estrogens may concur to cancer growth and progression (30, 31).
Mechanisms Involving Adipose Tissue and Chronic Low-Grade Inflammation
A second mechanism by which obesity is associated with cancer (32) is related to the adipose tissue expansion. As individuals become obese and their adipocytes enlarge, adipose tissue undergoes molecular and cellular alterations affecting the local and systemic metabolism. Adipocyte function dysregulation and the associated chronic inflammation may also contribute to adiposity-induced tumorigenesis (33, 34) (Figure 1). Yet, the insufficient vascularization of the enlarged adipose tissue results in hypoxia, and infiltration by macrophages, T cells, and natural killer cells. These cells generate large amounts of pro-inflammatory cytokines, including tumor necrosis factor α and interleukin-6, which act as paracrine signaling molecules. Each of these factors might play an etiologic role in regulating malignant transformation and/or cancer progression. Moreover, adipose tissue within the tumor microenvironment actively contributes to tumor growth and metastasis by secreting leptin, adiponectin, free fatty acid (FFA), pro-angiogenic factors, and extracellular matrix constituents (35). Indeed, cancer-associated adipocytes (CAAs), in concert with cancer-associated fibroblasts and tumor-associated macrophages, may influence cancer cell survival (35).
Mechanisms Involving Modulation of Tumor Metabolism
Emerging data suggest that obesity may stimulate cancer progression by affecting tumor metabolism (36–38). This mechanism has been particularly studied in tumors arising in close proximity to adipose tissue (39). Indeed, Nieman et al. demonstrated that adipocytes act as mediators of ovarian cancer metastases providing fatty acids to the cancer cells (39). This mechanism is not limited to ovarian cancer but can be extended to other cancers. According to a recent view, tumor cells grow in a complex microenvironment characterized by a dynamic exchange of metabolites between stromal cells (fibroblasts and adipocytes) and epithelial cancer cells. Stromal cells provide metabolites (lactate, ketones, glutamines, and fatty acids) that are used by cancer cells to generate energy by oxidative phosphorylation and β-oxidation (reverse Warburg effect) (40). Adipocytes localized in proximity to cancer cells undergo dedifferentiation into pre-adipocytes, and some of them are reprogrammed into CAAs. Fatty acids derived from lipolysis are released by CAAs and utilized by cancer cells to obtain energy from mitochondrial β-oxidation. This availability of energetic substrates in the tumor microenvironment promotes uncontrolled cancer cell growth and tumor progression (35) (Figure 1). Lipid metabolism is, therefore, a new target for the treatment of cancers where adipocytes are a major component of the microenvironment.
Mechanisms Involving Endoplasmic Reticulum (ER) Stress
In obesity, ER stress is due to the increased protein synthesis caused by nutrient excess and elevated levels of saturated FFA.
Cancer cells have developed a capacity to survive under these extreme conditions through the modulation of the unfolded protein response (UPR) pathway. The components of the UPR pathway have also been implicated in cancer (41) and appear to be affected by glucose homeostasis (42, 43). In fact, in many cancers, glucose-regulated protein 78 (GPR78, an ER protein chaperone involved in adaptive response to ER stress) is overexpressed and correlates with cancer recurrence, therapeutic resistance, and stemness phenotype (44–48). Glucose and leptin can induce expression of GPR78 (49, 50), suggesting a link between diabetes and ER stress-related cancer features (Figure 1).
Mechanisms Involving Autophagy
Autophagy is a natural and regulated, destructive process that is activated upon starvation in order to disassemble, unnecessary or dysfunctional cellular components (51) that are broken down and recycled through lysosomes (52). This process is regulated in a biphasic way through short- and long-term responses. Posttranslational protein modifications and protein–protein interactions mediate the rapid response, while nuclear transcriptional mechanisms are induced after a sustained stimulus (53). Autophagy was initially considered a survival strategy during starvation, but it has been recently demonstrated that it can lead to apoptosis if prolonged (autophagic cell death) (54, 55). Autophagy has opposite roles in tumorigenesis and tumor progression. Decreased baseline levels of autophagy are observed in many cancer cells compared to non-cancerous cells from the same tissue. Indeed, inhibition of autophagy can promote carcinogenesis by decreasing protein degradation thus increasing unrepaired and accumulated mutations (Figure 1). On the contrary, in response to hypoxia, acidosis, or nutrient deprivation, autophagic process is enhanced in cancer cells in the later stages of tumor progression. The cells in the inner part of the tumor increase autophagy in order to survive to the low nutrient and hypoxic microenvironment (56). For this reason, in the last years many cancer therapies have been aimed to modulate autophagy.
Multiple signaling pathways, including mTOR, AMPK, B-cell lymphoma 2 (Bcl-2)/Beclin 1 complex, and p53, play important roles in regulating autophagy (57). Insulin inhibits autophagy in several ways: first by activating mTOR in synergy with amino acids (58), which results in the phosphorylation and inhibition of unc-51-like autophagy activating kinase 1 (59); second by inducing Akt-mediated phosphorylation and inhibition of the transcription factor FoxO3, which controls the transcription of autophagy-related genes, including LC3 and Bnip3 (60); third by inhibiting the expression of autophagy-related genes, such as VPS34 and Atg12 in a FoxO1-dependent manner (61).
During insulin resistance, various dysfunctional/damaged components are retained leading to cellular stresses and/or inflammation. At the same time, hyperinsulinemia may inhibit autophagy (60) and favor tumorigenesis and tumor progression. In insulin-resistant mice, autophagy is suppressed in many tissues (61).
PPAR-γ Agonists: History, Mechanisms of Action, and Clinical Use
History
Peroxisome proliferator activated receptor-γ is a nuclear hormone receptor that is activated by multiple agonists. The name derives from the first identified member that belongs to a group of hepatocarcinogens that upregulate the proliferation of peroxisomes (62). The γ variant was first cloned from a Xenopus cDNA library, along with the α and β variants (63). In the same study, all three receptors were observed to have a role in the regulation of β-oxidation. PPARs perform many activities, mainly via endogenous ligands generated from fatty acids. For this reason, they are called lipid sensors. PPAR agonists have different affinities for PPARs, various pharmacokinetic profiles, and specific gene expression profiles (64, 65). Shortly after the role of PPAR-γ in adipocyte differentiation was characterized, potent synthetic ligands of PPAR-γ (TZDs) were discovered (66) and subsequently used in the therapy of T2DM patients to improve insulin sensitivity (67).
Mechanisms of Action
Peroxisome proliferator activated receptor-γ forms a heterodimer with retinoid X receptor (RXR) and then binds to specific DNA sequences, named PPAR-responsive elements, in the promoter region of target genes. Binding of agonist ligands to the PPAR:RXR heterodimer activates the complex and initiates gene transcription. Alternatively, PPAR-γ can recruit transcriptional corepressor proteins to silence gene expression. PPAR-γ is activated by natural ligands such as polyunsaturated fatty acids, eicosanoids, oxidized low-density lipoproteins, J2 type prostaglandins, and by a panel of potent drug agonists that include the TZDs (68). Although PPAR-γ acts mainly as transcription factor (genomic action), it may also exert its function via the activation of cytosolic non-genomic signaling pathways (Figure 2). The latter comprises activation of transmembrane proteinases, EGF-R transactivation, calcium influx, changes in protein biosynthesis, modulation of mitochondrial functions, stress response, interaction with Wnt/β-catenin, induction of signaling pathways involved in proliferation and survival, such as IGF-I/PI3K/AKT/mTOR and MAPK (69). The cross talk with these cascades plays an important role in the regulation and signal transmission of PPAR-γ and its ligands (see Role of PPAR-γ Agonists in Cell Proliferation and Growth: In Vitro Evidences). Overall, the PPAR-γ non-genomic actions, the PPAR-γ genomic functions, and the modulation of the cross talk between PPAR-γ and other key survival pathways support the pleiotropic functions of PPAR-γ that include fat cell formation and differentiation (4, 70–73), glucose and lipid homeostasis (74–77), atherosclerosis regulation (78), and anti-inflammatory effects (79, 80). In addition, PPAR-γ and its ligands exert antiproliferative and anti-tumorigenic functions or induce pro-tumorigenic and antiapoptotic responses on the basis of the cell context (69).
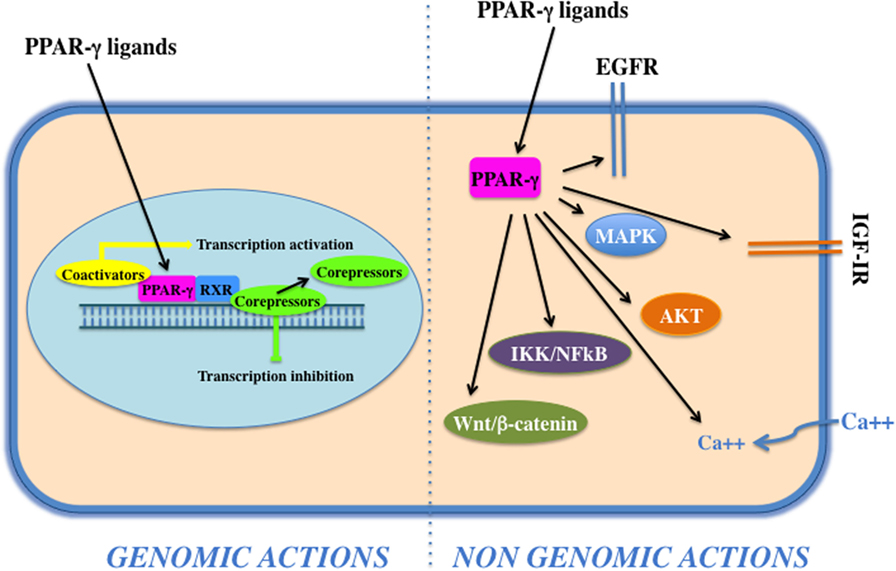
Figure 2. Genomic and non-genomics actions of peroxisome proliferator activated receptor (PPAR)-γ. PPAR-γ belongs to the class of nuclear receptors, containing a transactivation domain and a DNA-binding domain. Upon ligand binding, at the nuclear level, a conformational change leads to the release of corepressors, recruitment of coactivators, heterodimerization, and transactivation of PPAR-responsive element-related promoters (genomic actions). Cytoplasmic PPAR-γ, by interacting with proteins and activating transmembrane proteinases, elicits rapid and transient non-genomic effects that modulate EGF-R/insulin-like growth factor (IGF)-insulin receptor (IR) transactivation, calcium influx, PI3K/Akt, IKK/NFKB, and MAPKs signaling pathways.
Clinical Use
Thiazolidinediones are potent insulin sensitizers that efficiently and sustainably improve glycemic control in patients with T2DM (81). Although both metformin and TZDs decrease hepatic glucose production (82, 83), only TZDs reduce liver fat content (82, 84). They also diminish fasting FFA concentrations (85). This effect probably accounts for the indirect improvement in skeletal-muscle insulin sensitivity and the reduction in liver steatosis.
However, the use of TZDs in clinical practice is currently limited because of side effects, such as weight gain and fluid retention that can precipitate cardiac failure and bone fractures. Troglitazone and RGZ were withdrawn because of hepatotoxicity (86) and suspected to increase cardiovascular risk (87), respectively. In addition, the benefit–risk ratio of PIO has been reassessed recently in light of a putatively increased risk of bladder cancer (see Role of PPAR-γ Agonists in Chemoprevention). A novel synthetic third-generation TZD highly selective for PPAR-γ, i.e., efatutazone (RS5444 and CS-7017) has been recently synthesized. So far, efatutazone is the most potent TZDs in terms of transcriptional response and cell proliferation inhibition (88).
PPAR-γ Agonists as Antitumor Drugs: Cross Talk with the IGF System
As mentioned earlier, PPAR-γ and its ligands may exert pleiotropic effects (pro- vs. antineoplastic functions). However, PPAR-γ is often considered a tumor suppressor by the virtue of promoting growth inhibition, apoptosis, cell cycle arrest, and redifferentiation in several malignancies (Figure 3). In vitro and in vivo evidences suggest that many of these antineoplastic functions are explained by the interference with the IGF system activity at various levels and by the well-established metabolic actions, i.e., reduction of circulating insulin levels and improvement of tissue insulin sensitivity. These findings provide new insights for the potential clinical use of PPAR-γ agonists as anticancer agents especially in tumors characterized by IGFs’ overactivation and/or occurring in patients with insulin resistance and hyperinsulinemia.
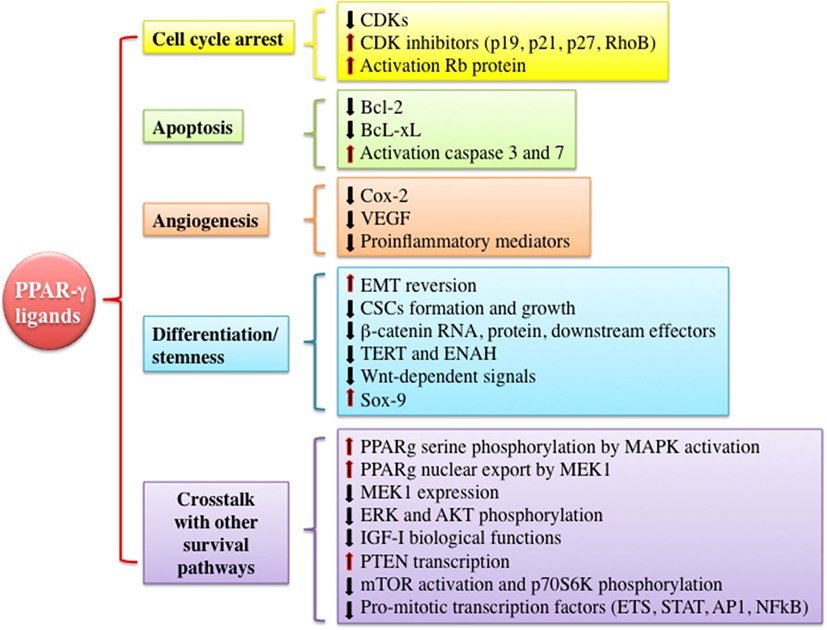
Figure 3. Mechanisms underlying anti-tumorigenic actions of peroxisome proliferator activated receptor (PPAR)-γ agonists. PPAR-γ activation by thiazolidinediones or other ligands may trigger anti-tumorigenic effects through different mechanisms including cell cycle arrest, induction of apoptosis, inhibition of angiogenesis, modulation of differentiation and stemness processes, and cross talk with other signaling pathways involved in proliferation and survival. All of these events may be exerted by PPAR-γ genomic or non-genomic actions. Abbreviations: CDKs, cyclin-dependent kinases; RhoB, rho-related GTP-binding protein; Rb protein, retinoblastoma-associated protein; Bcl-2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma-extra large; Cox-2, cyclooxygenase-2; VEGF, vascular endothelial growth factor; EMT, epithelial to mesenchymal transition; CSCs, cancer stem cells; TERT, telomerase reverse transcriptase; ENAH, enabled homolog; ETS, E26-transformation specific family; STAT, signal transducer and activator of transcription; AP1, activator protein-1.
Role of PPAR-γ Agonists in Cell Proliferation and Growth: In Vitro Evidences
In vitro studies conducted in cancer cell lines derived from many tumors including breast cancer (89), lung cancer (90), colon (91), liposarcoma (92), hematopoietic cancer (93), glioma (94), hepatocarcinoma (95, 96), pancreatic cancer (97), thyroid cancer (98), and prostate cancer (99) have shown that PPAR-γ is often expressed in these cancer cells and that PPAR-γ activation exerts a growth inhibitory effect. Well-known molecular mechanisms implicated in the suppression of tumor growth and progression after PPAR-γ activation by TZDs or other PPAR-γ agonists involve cell cycle arrest, apoptosis, inhibition of angiogenesis, and redifferentiation. For instance, in cancer cells from different organs and tissues including the thyroid gland (98, 100, 101), lung (102, 103), esophagus (104), prostate (105), breast (106), kidney, and urothelium (107, 108), TZDs decrease cyclin-dependent kinase levels (CDKs) such as Cdk4, increase CDKs inhibitors such as p19, p21, p27, and rho-related GTP-binding protein, and activate retinoblastoma-associated protein. In some tumors, TZDs modulate proteins involved in apoptosis process. For example, in lung cancer cells, TGZ reduces the antiapoptotic protein Bcl-2; in anaplastic thyroid cancer, RGZ-induced apoptosis is associated with decrease of B-cell lymphoma-extra large expression and caspase-3 and caspase-7 activation; in many hepatoma cell lines, RGZ induces apoptosis by promoting the expression of PTEN, caspase-3, and caspase-9 (95, 96). In addition to apoptosis, PPAR-γ activation inhibits angiogenesis-associated proteins expression such as cyclooxygenase-2 and vascular endothelial growth factor (VEGF) as well as tumor microenvironment inflammatory mediators (79, 80, 97). Another mechanism by which PPAR-γ activation may act as tumor suppressor is the promotion of cellular differentiation (see next paragraph). Many of these PPAR-γ-mediated anticancer effects may be linked to a direct or indirect cross talk with the IGF axis. In particular, in vitro evidences indicate that PPAR-γ cooperates with the IGF axis downstream signaling pathways such as MAPK, PI3K, and mTOR. These interactions may occur in a synergistic or antagonistic way and in a context-specific manner, supporting the multifacet features of PPAR-γ effects. In accordance with the antitumor role of PPAR-γ and the antiapoptotic role of the IGF system and its downstream signaling pathways, MAPK activation by different growth stimuli (i.e., insulin, IGF-I, EGF, and PDGF) may induce PPAR-γ serine phosphorylation and consequently inactivation of its genomic activity (109–112). Indeed, poorly differentiated cancers typically show constitutive activation of mitogenic pathways downstream tyrosine-kinase receptors that are involved in chemoresistance (113). Yet, MEK1 interacting with PPAR-γ induces its nuclear export and inhibition of its genomic functions (114). On the other hand, PPAR-γ activation reduces MEK1 protein expression and ERK phosphorylation, causing cell growth arrest and apoptosis. Beyond the inhibition of MAPK pathway, PPAR-γ agonists may also inhibit other cascades downstream the insulin/IGF signaling, such as the PI3K/mTOR pathway. In particular, in several cancer cells from thyroid (98), liver (95, 111), colon (115), breast (115), lung (116, 117), and pancreas (118), PPAR-γ agonists inhibit IGF-I mediated biological effects through the reduction in Akt phosphorylation, increased PTEN expression levels (115), and inhibition of mTOR and p70S6K activity (116–120). Taken together, these data support the possibility that PPAR-γ agonists may have a potential role as antineoplastic agents in tumors characterized by overactivation of the insulin/IGF-I axis (121, 122).
At variance with these data, in some tumor cell models, a positive cross talk between PPAR-γ and MAPK/PI3K/mTOR signaling components may also occur. For example, in breast and colon cancer cells, TZDs have been described to induce rapid MAPK and PI3K/mTOR activation that may affect the antitumor PPAR-γ genomic action (115, 123, 124). These latter mechanisms add complexity to the actions of PPAR-γ agonists and may contribute to the absence of therapeutic efficacy of TZDs in some cancer patients.
PPAR-γ Agonists and Autophagy
Peroxisome proliferator activated receptor-γ activation may revert the effect of hyperinsulinemia by favoring the autophagic process at multiple levels. For example, in breast cancer cells, PPAR-γ agonists induced autophagy through HIF-1α and BNIP3 upregulation (125), while in adrenocortical cancer cells, RGZ triggered autophagy by increasing the expression of AMPKα and beclin 1, through both PPAR-γ-dependent and PPAR-γ-independent mechanisms (126). In bladder cancer cells, TGZ treatment enhanced autophagy, and then apoptosis (127). Moreover, RGZ-induced autophagy depends on the cellular context since this effect was not observed in all cell lines. In some in vitro models, PPAR-γ ligands induce autophagy by increased ROS production in mitochondria thus altering the mitochondrial membrane potential (126).
Role of PPAR-γ Agonists in Cell Differentiation and Stemness: In Vitro Evidences
As previously mentioned, PPAR-γ agonists may favor cancer cell differentiation (128). This notion is supported by experiments conducted in lung, breast, and thyroid cancer cells showing a change in epithelial expression profile and a reversion of epithelial–mesenchymal transition (EMT) process after TZD treatment (4, 98, 101, 111). Furthermore, several studies have demonstrated the efficacy of PPAR-γ agonists in inhibiting the survival of cancer stem cells (CSCs) derived from human cell lines or specimens from breast, prostate, colon, bladder, and blood tissues (129–134) supporting a role of PPAR-γ in regulating CSC biology. For example, PIO, in combination with a RXR ligand, was able to reduce the formation of mammospheres from human breast tumors and MCF7 cells (132). Yet, in bladder cancer cells, the combination of a natural ligand of PPAR-γ (i.e., 15d-PGJ2) together with a survivin inhibitor was associated with downregulation of stemness-related genes and reduction of spheres formation (134). The molecular mechanisms through which TZDs regulate differentiation and stemness programs have been studied in adipocytes and normal cells, while in cancer cells and in CSC they remain still incompletely elucidated. In adipocytes, PPAR-γ amplifies differentiation signals and inhibits proliferation by affecting the Wnt/GSK3-β/β-catenin pathway. In particular, PPAR-γ interacts with GSK3-β inducing the differentiation factor C/EBPα and leading to the production of adiponectin (70, 71); yet, PPAR-γ activation reduces β-catenin at both mRNA and protein levels promoting differentiation (135). Similar mechanisms involving the Wnt/β-catenin cascade may also occur in cancer cells and in CSC, as this pathway has also emerged to be essential not only for the differentiation process but also for self-renewal and stemness programs. In human kidney embryonic HEK293 cells and in human metastatic prostate cancer LnCaP cells, PPAR-γ suppresses Wnt signaling by targeting phosphorylated β-catenin to proteasome (136–138). In gastric and colon cancer cells, PPAR-γ inhibits β-catenin expression, subcellular localization, and downstream effectors. All of these events lead to the modulation of a subset of genes, such as telomerase reverse transcriptase, enabled homolog, and Sox9, involved in cell development, differentiation, and survival processes (139–141). Another mechanism through which PPAR-γ could modulate CSC biology is the cross talk with IGF signaling. Indeed, recent studies have provided increasing evidence that the IGF pathway is essential for the growth/expansion of cancer stem-like cells by contributing to regulate pluripotency, EMT, and self-renewal. We have recently found that IR and IGF-IR are overexpressed in human thyroid progenitor/stem cells where they regulate self-renewal ability and stem cell expansion (142–144). Similar findings have been demonstrated in cancer progenitor/stem cells from solid and hematopoietic cancers reinforcing the important role of the IGF system in regulating stem cell biology and the early steps of the carcinogenesis process (145, 146). Furthermore, to control cell reprogramming, the IGF system, in turn, interacts with effectors present in the stem cell niche belonging to GSK3-β/Wnt/β-catenin, Notch, and Shh pathways.
In this paragraph, we will focus only on Wnt/β-catenin signaling cascade, because there are more data regarding possible interactions between this pathway and both the IGF- and PPAR-γ-dependent signals. In particular, in human colon and hepatocellular cancer cells (147–149), IGF-I stimulates tyrosine phosphorylation of β-catenin, IRS-1, and E-cadherin as well as cellular relocation and stabilization of β-catenin. These events result in the disruption of β-catenin/E-cadherin interaction and inhibition of GSK3-β activity (148, 149). Furthermore, IRS-1, in turn, contributes to β-catenin stability, GSK3-β inactivation, IGF-IR/PI3K signaling pathway amplification, and T cell factor/lymphoid enhancer-binding factor-dependent transcription of genes controlling stem cell fate, long-term renewal, and differentiation programs (147). On the other hand, IRS-1 is a downstream target of β-catenin, which regulates IRS-1 expression and localization controlling cancer initiation, self-renewal, and differentiation processes (150–152). Overall, these interactions between GSK3-β/Wnt/β-catenin and the IGF system may contribute to enhance mitogenesis and stemness characteristics. In light of these considerations, PPAR-γ agonists, by inhibiting the activation of IGF axis as well as the GSK3-β/Wnt/β-catenin pathway, could be used in combination with other drugs such as inhibitors of tyrosine kinases (133), PI3K/AKT (153), and MAPK cascades to reach the maximum antitumor and pro-differentiating effect.
Role of PPAR-γ Agonists As Antitumor Drugs and in Chemoprevention: Animal Models
Several in vivo studies conducted in mice and rats support the anticancer PPAR-γ properties (89, 99, 105, 154–159). In general, TZDs have shown univocal effects in impairing progression and metastatic spread in animals injected with human cancer cells. Less clear-cut results have been obtained when looking at the effect of PPAR-γ agonists in cancer prevention. The discrepancy between the anticancer and the tumor-promoting effects of PPAR-γ agonists shown in the different studies reflects, as seen in vitro, the complexity of signaling interactions involved in tumor formation in vivo. In some of these studies, the molecular mechanisms responsible for the antitumor vs. protumour PPAR-γ actions have been identified in the regulation of cell cycle, apoptosis, and signals involved in differentiation and cell fate such as the Wnt/β-catenin cascade and the Notch/Hes1 and NFKB pathways (105, 157, 159). However, so far, no studies in animals have explored the interactions between PPAR-γ and the IGF system or the correlation between PPAR-γ actions and the presence of insulin resistance in animals.
PPAR-γ Agonists As Antitumor Drugs: Clinical Trials
Taken together, data from in vitro studies and from animal models strongly support the concept that PPAR-γ has antineoplastic effects. However, clinical trials, using TZDs as antineoplastic agents, are few and have reached conflicting results. Overall, the ambivalent findings in clinical trials may be due to the inclusion of pretreated refractory cancers or cancers in far advanced stages, or to the activation of PPAR-γ independent pathways.
Furthermore, most of these studies lack clinical information regarding the presence of insulin resistance (diabetes mellitus condition was a criterion of exclusion) as well as data regarding the alterations of IGF axis in the tumors studied. Few studies using PPAR-γ agonists and in combination with other drugs have been conducted. Encouraging results have been obtained in patients affected by chronic myeloid leukemia treated with TZDs and imatinib. Leukemia quiescent cells have been eliminated through a mechanism involving TZD-related inhibition of STAT5 expression responsible of stemness maintenance (129–131, 133). One relevant ongoing clinical phase II trial using PIO (15 mg) as add-on therapy to imatinib has started in July 2009 in patients affected by chronic myelogenous leukemia with residual molecular disease after imatinib monotherapy for more than 2 years (ACTIM EudraCT 2009-011675-79). Although the interim results from this trial are promising, the study was non-randomized. Yet, in hepatocellular carcinoma (HCC) in vitro and in vivo models, the use of RGZ in combination with either AKT pharmacological inhibitors or AKT siRNA significantly enhanced PPAR-γ agonist-mediated inhibition of cell proliferation, stem cell-like properties, and tumor growth (153). Although promising, further investigations regarding the antitumor effects of PPAR-γ agonists in the clinical setting need to be conducted. More data have been collected in diabetic populations with regard to the role of PPAR-γ agonists in chemoprevention (see next paragraph).
Role of PPAR-γ Agonists in Chemoprevention: Lessons from Diabetic Patients Treated with TZDs
Thiazolidinediones are widely used as antidiabetic agents in T2DM patients. According to in vivo studies in diabetic hyperinsulinemic mice showing that TGZ has potent glucose and insulin-reducing effects (160), several reports have demonstrated that both TGZ and PIO reduce hyperglycemia, hyperinsulinemia, and hypertriglyceridemia and improve insulin sensitivity in T2DM patients (161–163). However, studies demonstrating efficacy of TZDs in cancer chemoprevention are difficult to perform because they should ideally be prospective and involving large cohorts of T2DM patients followed up for several years. Moreover, many confounders may affect such studies because T2DM patients are heterogeneous and often subjected to multiple therapies. Therefore, it is not surprising that only few retrospective studies are available and that their results are largely inconclusive.
A recent retrospective study has assessed the influence of TZDs on the risk of lung, prostate, and colon cancers in patients with diabetes. The study population was derived from the Veterans Integrated services Network 16 data warehouse (164). A total of 87,678 male patients met the study inclusion criteria, and 11,289 were treated with TZDs for a median duration of 1 year. After adjusting for covariates (age, ethnicity, body mass index, HbA1c, use of insulin, or other agents), the use of RGZ or PIO was found to significantly (P = 0.0033) reduce the incidence of lung cancer by 33%, as compared with TZDs non-users. A trend for a reduced risk for prostate and colorectal cancer was also observed when the population was analyzed as a whole. However, when results were subjected to subgroup analysis by ethnicity, prostate cancer resulted significantly increased by 15% in white patients while colorectal cancer was significantly reduced in African-Americans. These differences may be explained by possible differences in the metabolism of TZDs between white and African-American populations as well as to the low statistical power of the study (164). Although this study included a large number of patients and had several strengths, it showed several limitations. First, this is a retrospective study, thus some relevant information, such as smoking history and duration of TZD exposure were missing, and other information, such as ethnicity, were not available for all patients.
A different conclusion was reached by a similar retrospective study performed in 1,003 subjects enrolled in the Vermont Diabetes Information System (165). In this community-based diabetic population, the use of TZDs was found significantly and positively associated with the diagnosis of cancer (OR = 1.59, P = 0.04). This association was stronger in women (OR = 2.07, P = 0.01) and in RGZ users than in PIO users. This study has also several limitations, including the relatively small number of patients, the uncertain duration of TZDs treatment, the lack of diagnosis and tumor stage confirmation, and of rigorous controls for genetic and environmental confounders (165).
A recent meta-analysis has been performed in diabetic patients using RGZ with a focus on cancer risk. This meta-analysis includes all trials that can be retrieved from the GSK (GlaxoSmithKline) web site or from Medline with results published up to February 2008. The authors analyzed 80 trials involving 16,322 RGZ users and 12,522 patients using a different antidiabetic treatment. They found no evidence that RGZ was either positively or negatively associated with cancer risk (OR = 0.91, P = 0.44) (166). Case–control studies for specific cancers have also reached discrepant results. In a hospital-based case–control study, carried out in 420 patients (140 diabetics) with HCC and 1,104 controls (115 diabetics), diabetes was found a significant risk factor for HCC, and the treatment with TZDs or biguanides was associated with 70% HCC risk reduction as compared to other treatments. In a similar case–control study involving 973 patients (259 diabetics) with pancreatic adenocarcinoma and 863 controls (109 diabetics), it was found that metformin users, but not TZDs users, were protected from pancreatic cancer as compared to diabetic patients subjected to other treatments (167). Moreover, the risk of breast, colon, and prostate cancers in TZDs users was evaluated in three-nested case–control studies based on 26,971 diabetic patients included in the US Integrated Healthcare Information Services database (168). Data revealed no association between the risk of breast, colon, and prostate cancers and the use of TZDs. The latter study is in agreement with a case–control study performed in 195 diabetic patients and 195 controls, taking into account cancer risk in relation to the antidiabetic treatment used. Also in this study, no association was found between cancer risk and TZD use while a significantly reduced cancer risk was observed in metformin users (166). Yet, a large randomized prospective clinical trial focused on secondary cardiovascular disease prevention in T2DM by PIO (169) has shown that the use of PIO was not associated with significant changes in the incidence of various cancers. The clinical significance of the small increase in bladder cancer (14 vs. 5) and the small decrease in breast cancer (3 vs. 11) remains uncertain. Regarding bladder cancer risk, in another study including a cohort of 193,099 patients with diabetes, the short-term use of PIO was not associated with an increase incidence of bladder cancer, but the use for more than 2 years was weakly associated with increased risk (170). To date, a retrospective cohort study to evaluate the bladder cancer rate in male T2DM subjects aged more than 50 years who are on PIO (7.5–30 mg) therapy for 1 year or more as compared to never users of PIO (http://ClinicalTrials.gov Identifier: NCT01935466, PROBE-PIO) is still ongoing. Similarly, another randomized, double-blind placebo controlled clinical trial evaluating the use of PIO in the chemoprevention of lung cancer (NCT00780234) is also ongoing. In this study, non-diabetic subjects at risk for lung cancer (based on smoking history, lung function testing, and atypical cells in a sputum sample) are randomized to receive either placebo or PIO (30 mg/day). The primary outcome is endobronchial histology and evaluation of cancer progression. Secondary endpoints will include the activation of PPAR-γ dependent signaling pathways.
Taken together, these studies do not provide a definite answer to the question whether the use of TDZs has any association with cancer risk in diabetic population. However, they suggest that, overall, their use is neutral. These results are at odd with those obtained with metformin, which shares with TZDs’ insulin-sensitizing activity, suggesting that the observed effects of these drugs on cancer risk are only partially dependent on their insulin sensitizing effect. It is also possible, that genetic factors, cancer histotype and/or molecular alterations could significantly modulate the clinical effects of TDZs.
PPAR-α Agonists as Antitumor Drugs: Cross Talk with the IGF System
Peroxisome proliferator activated receptor-α is the first identified PPAR, and it has been found widely expressed in several tissues including skeletal muscle, liver, hearth, kidney, and intestine (171, 172). PPAR-α can be activated by endogenous ligands such as fatty acids and derivatives (arachidonic acid, leukotriene B4, and non-esterified fatty acids) or by synthetic compounds such as fenofibrate, clofibrate, bezafibrate, and Wy-14,63.
Activated PPAR-α forms heterodimer with RXRα (PPARs/RXRα) and binds to the consensus sequence in the promoter region of target genes (173). As PPAR-α agonists modulate the expression of genes regulating glucose and fatty acid metabolism, they have been widely sought after as therapeutic agents for several metabolic disorders such as obesity, T2DM, hyperlipidemia, and cardiovascular diseases (174). However, PPAR-α has also a critical role in the regulation of cell proliferation, survival, motility, and metabolism in several cancer cells (175, 176). Activated PPAR-α may decrease or induce tumor progression depending on the specific tissue or the PPAR-α ligand. While few in vivo studies regarding the effects of PPAR-α agonists in cancer are currently available, several in vitro studies indicate that PPAR-α has a remarkable antineoplastic potential. For instance, fenofibrate regulates colon inflammation and proliferation by inhibiting the production of pro-inflammatory cytokines such as IL-17, IFN-g, CXCL10, CCL2, and CCL20 (177). In breast and ovarian cancer cells, clofibrate induces HIF-1α degradation leading to decreased tumor-associated VEGF gene expression (178). Yet, in non-small cell lung cancer, activated PPAR-α increases p53 expression and inhibits PDK1 and NFKB/p65 dependent signaling, leading to inhibition of cell growth (179).
Notably, similar to PPAR-γ, PPAR-α may be involved in tumorigenesis by cross talking with the IGF system at different levels. For instance, in brain tumors, fenofibrate exerts anticancer effect by inhibiting IGF-I-mediated signaling and biological responses (180, 181). In medulloblastoma cell lines, fenofibrate strongly inhibited IGF-I-mediated activation of IRS-1, AKT, ERK, and GSK3b phosphorylation (181) as well as IGF-I-mediated cell clonogenic growth, migration, and colony formation in soft agar. Accordingly, the combined treatment with fenofibrate and the IGF-IR inhibitor (NVP-AEW541) resulted in a complete suppression of growth responses to IGF-I, cell cycle arrest, and apoptosis. Similarly, fenofibrate inhibited IGF-I and serum-induced motility of glioma cells and induced ROS accumulation, loss of mitochondrial membrane potential, and reduction in adenosine 5′-triphosphate (ATP) production (180).
In mice bearing 4-nitroquinoline 1-oxide-induced lung hyperplasia, adenoma, and adenocarcinoma (182), fenofibrate downregulated the IGF axis by significantly reducing insulin and IGF-1 serum levels and the immunohistochemical expression of IGF-IR, pAkt, and pERK1/2, indicating a potential as chemoprevention agent. Fenofibrate-mediated inhibition of constitutively activated PI3K/AKT signaling has been reported in melanoma cell lines where this drug decreased both cell invasiveness and clonogenic cell growth (183).
So far, the molecular mechanisms involved in IGF-IR signaling inhibition by fenofibrate are still under investigation and may be at least partially PPAR-α independent, involving changes in the fluidity of plasma membrane, which, in turn, affect ligand-mediated IGF-IR partitioning and consequent initiation of growth-promoting downstream signaling. Indeed, fenofibrate may affect the activities of integral membrane proteins, leading to plasma membrane rigidity and alteration in the activity of membrane-spanning proteins such as IGF-IR (184).
In addition to the attenuation of IGF-IR signaling responses, another mechanism through which fenofibrate exerts antitumor role is the modulation of the energy metabolism of cancer cells, which strongly depend on glycolysis (185). As fenofibrate can switch energy metabolism from glucose to fatty acid oxidation and ketogenesis, as a main source of energy (186, 187), it can favor aberrant mitochondrial oxidative phosphorylation leading to ROS accumulation, oxidative damage, and reduction in ATP production with severe cell energy depletion. As seen in glial neoplasms (180), all of these events may impair growth and survival of cancer cells with defective mitochondrial function. The ability of fenofibrate to force mitochondrial oxidative respiration in tumor cells, without systemic toxicity, suggests that this drug may have clinical benefits in preventing tumorigenesis.
Conclusion
It is now widely accepted that systemic diseases, such as obesity, T2DM, and metabolic syndrome, are not only cardiovascular risk factors but also cancer risk factors. Insulin resistance and hyperinsulinemia, common features of all these metabolic disorders, contribute to the deregulation of the insulin/IGF axis that plays an important role in cancer progression as well as in CSCs. In light of these considerations, the pleiotropic effects of PPAR-γ agonists may have potential applications in cancer prevention and therapy. Indeed, PPAR-γ activation, by TZD or other agonists, improves insulin resistance and reduces circulating levels of insulin and free IGF-I. Furthermore, PPAR-γ agonists downregulate key pathways of the insulin/IGF axis, such as PI3K/mTOR, MAPK, and GSK3β/Wnt/β-catenin cascades, which regulate cancer cell growth, proliferation, cell reprogramming, and differentiation. Thus, PPAR-γ agonists have a great potential to be used as antineoplastic agents in combination therapies with a variety of other compounds. Although extensive in vitro evidence supports the concept of pleiotropic antineoplastic actions of PPAR-γ agonists, unfortunately, the available clinical trials have reached conflicting results, possibly because they have not selected tumors characterized by overactivation of the insulin/IGF-I axis or occurring in hyperinsulinemic patients. Hopefully, future clinical trials with PPAR-γ agonists will include patients with these characteristics. Similarly, studies aiming to show a role of PPAR-γ agonists in cancer prevention should ideally be prospective, randomized, and should involve large cohorts of patients followed up for several years.
Besides PPAR-γ, agonists, also PPAR-α agonists, such as fenofibrate can also inhibit IR/IGF-IR signaling responses and exert complex antineoplastic actions.
In conclusion, the use of PPAR-γ agonists alone or in combination with other agents in cancer prevention and therapy remains promising but still awaits future developments.
Author Contributions
The authors listed below gave the following contributions. VV, MN, SG, MB, AB, and RM: substantial contributions to the conception and design of the paper; final approval of the version to be published; and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; VV, MN, SG, and MB: drafting the work; VV, RM, and AB: revising it critically for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported in part by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) (grant IG 14066/2013, to AB), Ministero della Salute, Italy (grant 67/GR-2010-2319511, to RM), and Ministero dell’Istruzione, dell’Università e della Ricerca, Italy (PON01_01078 to AB).
Abbreviations
PPARs, peroxisome proliferator activated receptors; TZDs, thiazolidinediones; IGF, insulin-like growth factor; T2DM, type 2 diabetes mellitus; IR-A, insulin receptor isoform A; BMI, body mass index; FFA, free fatty acids; IGF-BP1, IGF-binding protein 1; IGF-BP2, IGF-binding protein 2; NK, natural killer; TNF-α, tumor necrosis factor α; IL-6, interleukin-6; ER, endoplasmic reticulum; CAAs, cancer-associated adipocytes; CAFs, cancer-associated fibroblasts; TAMs, tumor-associated macrophages; UPR, unfolded protein response; ATP, adenosine 5′-triphosphate; GPR78, glucose-regulated protein 78; ULK1, unc-51 like autophagy activating kinase 1; RXR, retinoid X receptor; PPRE, PPAR-responsive elements; RGZ, rosiglitazone; PIO, pioglitazone; CSCs, cancer stem cells; TCF/LEF, T cell factor/lymphoid enhancer-binding factor; TKIs, tyrosine-kinase inhibitors; CML, chronic myelogenous leukemia; HCC, hepatocellular carcinoma; DBD, DNA-binding domain; CDKs, cyclin-dependent kinases; RhoB, rho-related GTP-binding protein; Rb protein, retinoblastoma-associated protein; Bcl-2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma-extra large; Cox-2, cyclooxygenase-2; VEGF, vascular endothelial growth factor; EMT, epithelial to mesenchymal transition; TERT, telomerase reverse transcriptase; ENAH, enabled homolog; ETS, E26-transformation specific family; STAT, signal transducer and activator of transcription; AP1, activator protein-1.
References
2. Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer (2004) 4:61–70. doi:10.1038/nrc1254
3. Aleman-Gonzalez-Duhart D, Tamay-Cach F, Alvarez-Almazan S, Mendieta-Wejebe JE. Current advances in the biochemical and physiological aspects of the treatment of type 2 diabetes mellitus with thiazolidinediones. PPAR Res (2016) 2016:7614270. doi:10.1155/2016/7614270
4. Mueller E, Smith M, Sarraf P, Kroll T, Aiyer A, Kaufman DS, et al. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc Natl Acad Sci U S A (2000) 97:10990–5. doi:10.1073/pnas.180329197
5. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev (2009) 30:586–623. doi:10.1210/er.2008-0047
6. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8:915–28. doi:10.1038/nrc2536
7. Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer (2004) 4:505–18. doi:10.1038/nrc1387
8. Belfiore A, Malaguarnera R. Insulin receptor and cancer. Endocr Relat Cancer (2011) 18:R125–47. doi:10.1530/ERC-11-0074
9. Frasca F, Pandini C, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol (1999) 19:3278–88.
10. Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol (2004) 159:1160–7. doi:10.1093/aje/kwh161
11. Strickler HD, Wylie-Rosett J, Rohan T, Hoover DR, Smoller S, Burk RD, et al. The relation of type 2 diabetes and cancer. Diabetes Technol Ther (2001) 3:263–74. doi:10.1089/152091501300209633
12. Vigneri P, Frasca F, Sciacca L, Frittitta L, Vigneri R. Obesity and cancer. Nutr Metab Cardiovasc Dis (2006) 16:1–7. doi:10.1016/j.numecd.2005.10.013
13. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer (2004) 4:579–91. doi:10.1038/nrc1408
14. Ciavardelli D, Bellomo M, Crescimanno C, Vella V. Type 3 deiodinase: role in cancer growth, sternness, and metabolism. Front Endocrinol (2014) 5:215. doi:10.3389/fendo.2014.00215
15. Louvi A, Accili D, Efstratiadis A. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev Biol (1997) 189:33–48. doi:10.1006/dbio.1997.8666
16. Sciacca L, Mineo R, Pandini G, Murabito A, Vigneri R, Belfiore A. In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene (2002) 21:8240–50. doi:10.1038/sj.onc.1206058
17. Vella V, Pandini G, Sciacca L, Mineo R, Vigneri R, Pezzino V, et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metab (2002) 87:245–54. doi:10.1210/jcem.87.1.8142
18. Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr Pharm Des (2007) 13:671–86. doi:10.2174/138161207780249173
19. Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer (2009) 16:1103–23. doi:10.1677/ERC-09-0087
20. Malaguarnera R, Nicolosi ML, Sacco A, Morcavallo A, Vella V, Voci C, et al. Novel cross-talk between IGF-IR and DDR1 regulates IGF-IR trafficking, signaling and biological responses. Oncotarget (2015) 6:16084–105. doi:10.18632/oncotarget.3177
21. Mata R, Palladino C, Nicolosi ML, Lo Presti AR, Malaguarnera R, Ragusa M, et al. IGF-I induces upregulation of DDR1 collagen receptor in breast cancer cells by suppressing MIR-199a-5p through the PI3K/AKT pathway. Oncotarget (2016) 7:7683–700. doi:10.18632/oncotarget.6524
22. Arcidiacono B, Iiritano S, Nocera A, Possidente K, Nevolo MT, Ventura V, et al. Insulin resistance and cancer risk: an overview of the pathogenetic mechanisms. Exp Diabetes Res (2012) 2012:789174. doi:10.1155/2012/789174
23. Pothiwala P, Jain SK, Yaturu S. Metabolic syndrome and cancer. Metab Syndr Relat Disord (2009) 7:279–88. doi:10.1089/met.2008.0065
24. Lanzino M, Morelli C, Garofalo C, Panno ML, Mauro L, Ando S, et al. Interaction between estrogen receptor alpha and insulin/IGF signaling in breast cancer. Curr Cancer Drug Targets (2008) 8:597–610. doi:10.2174/156800908786241104
25. Ma ZQ, Santagati S, Patrone C, Pollio G, Vegeto E, Maggi A. Insulin-like growth-factors activate estrogen-receptor to control the growth and differentiation of the human neuroblastoma cell-line Sk-Er3. Mol Endocrinol (1994) 8:910–8.
26. Newton CJ, Buric R, Trapp T, Brockmeier S, Pagotto U, Stalla GK. The unliganded estrogen-receptor (ER) transduces growth-factor signals. J Steroid Biochem (1994) 48:481–6.
27. Genua M, Pandini G, Sisci D, Castoria G, Maggiolini M, Vigneri R, et al. Role of cyclic amp response element-binding protein in insulin-like growth factor-I receptor up-regulation by sex steroids in prostate cancer cells. Cancer Res (2009) 69:7270–7. doi:10.1158/0008-5472.CAN-09-0088
28. Malaguarnera R, Sacco A, Morcavallo A, Squatrito S, Migliaccio A, Morrione A, et al. Metformin inhibits androgen-induced IGF-IR up-regulation in prostate cancer cells by disrupting membrane-initiated androgen signaling. Endocrinology (2014) 155:1207–21. doi:10.1210/en.2013-1925
29. De Marco P, Romeo E, Vivacqua A, Malaguarnera R, Abonante S, Romeo F, et al. GPER1 is regulated by insulin in cancer cells and cancer-associated fibroblasts. Endocr Relat Cancer (2014) 21:739–53. doi:10.1530/ERC-14-0245
30. De Marco P, Cirillo F, Vivacqua A, Malaguarnera R, Belfiore A, Maggiolini M. Novel aspects concerning the functional cross-talk between the insulin/IGF-I system and estrogen signaling in cancer cells. Front Endocrinol (2015) 6:30. doi:10.3389/fendo.2015.00030
31. Bartella V, De Marco P, Malaguarnera R, Belfiore A, Maggiolini M. New advances on the functional cross-talk between insulin-like growth factor-I and estrogen signaling in cancer. Cell Signal (2012) 24:1515–21. doi:10.1016/j.cellsig.2012.03.012
32. Cowey S, Hardy RW. The metabolic syndrome: a high-risk state for cancer? Am J Pathol (2006) 169:1505–22. doi:10.2353/ajpath.2006.051090
34. Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr (2006) 83:461S–5S.
35. Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta (2013) 1831:1533–41. doi:10.1016/j.bbalip.2013.02.010
36. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res (2011) 71:2455–65. doi:10.1158/0008-5472.CAN-10-3323
37. Manabe Y, Toda S, Miyazaki K, Sugihara H. Mature adipocytes, but not preadipocytes, promote the growth of breast carcinoma cells in collagen gel matrix culture through cancer-stromal cell interactions. J Pathol (2003) 201:221–8. doi:10.1002/path.1430
38. Tokuda Y, Satoh Y, Fujiyama C, Toda S, Sugihara H, Masaki Z. Prostate cancer cell growth is modulated by adipocyte-cancer cell interaction. BJU Int (2003) 91:716–20 doi:10.1046/j.1464-410X.2003.04218.x
39. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med (2011) 17:1498–U207. doi:10.1038/nm.2492
40. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8:3984–4001. doi:10.4161/cc.8.23.10238
41. Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov (2015) 5:586–97. doi:10.1158/2159-8290.CD-14-1490
42. Gao Y, Sartori DJ, Li C, Yu QC, Kushner JA, Simon MC, et al. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol Cell Biol (2012) 32:5129–39. doi:10.1128/MCB.01009-12
43. Hassler JR, Scheuner DL, Wang S, Han J, Kodali VK, Li P, et al. The IRE1alpha/XBP1s pathway is essential for the glucose response and protection of beta cells. PLoS Biol (2015) 13:e1002277. doi:10.1371/journal.pbio.1002277
44. Chiu CC, Lee LY, Li YC, Chen YJ, Lu YC, Li YL, et al. Grp78 as a therapeutic target for refractory head-neck cancer with CD24(-)CD44(+) stemness phenotype. Cancer Gene Ther (2013) 20:606–15. doi:10.1038/cgt.2013.64
45. Cook KL, Clarke R. Role of GRP78 in promoting therapeutic-resistant breast cancer. Future Med Chem (2015) 7:1529–34. doi:10.4155/FMC.15.80
46. Roller C, Maddalo D. The molecular chaperone GRP78/BiP in the development of chemoresistance: mechanism and possible treatment. Front Pharmacol (2013) 4:10. doi:10.3389/fphar.2013.00010
47. Xing X, Li Y, Liu H, Wang L, Sun L. Glucose regulated protein 78 (GRP78) is overexpressed in colorectal carcinoma and regulates colorectal carcinoma cell growth and apoptosis. Acta Histochem (2011) 113:777–82. doi:10.1016/j.acthis.2010.11.006
48. Zheng HC, Takahashi H, Li XH, Hara T, Masuda S, Guan YF, et al. Overexpression of GRP78 and GRP94 are markers for aggressive behavior and poor prognosis in gastric carcinomas. Hum Pathol (2008) 39:1042–9. doi:10.1016/j.humpath.2007.11.009
49. Mote PL, Tillman JB, Spindler SR. Glucose regulation of GRP78 gene expression. Mech Ageing Dev (1998) 104:149–58.
50. Thon M, Hosoi T, Yoshii M, Ozawa K. Leptin induced GRP78 expression through the PI3K-mTOR pathway in neuronal cells. Sci Rep (2014) 4:7096. doi:10.1038/srep07096
51. Kobayashi S. Choose delicately and reuse adequately: the newly revealed process of autophagy. Biol Pharm Bull (2015) 38:1098–103. doi:10.1248/bpb.b15-00096
52. Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol (2008) 3:427–55. doi:10.1146/annurev.pathmechdis.2.010506.091842
53. Settembre C, Ballabio A. Cell metabolism: autophagy transcribed. Nature (2014) 516:40–1. doi:10.1038/nature13939
54. Alva AS, Gultekin SH, Baehrecke EH. Autophagy in human tumors: cell survival or death? Cell Death Differ (2004) 11:1046–8. doi:10.1038/sj.cdd.4401445
55. Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol (2005) 6:505–10. doi:10.1038/nrm1666
56. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell (2008) 132:27–42. doi:10.1016/j.cell.2007.12.018
57. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol (2010) 12:814–22. doi:10.1038/ncb0910-814
58. Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ (2005) 12(Suppl 2):1509–18. doi:10.1038/sj.cdd.4401751
59. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi:10.1038/ncb2152
60. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab (2007) 6:458–71. doi:10.1016/j.cmet.2007.11.001
61. Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem (2009) 284:31484–92. doi:10.1074/jbc.M109.033936
62. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature (1990) 347:645–50.
63. Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell (1992) 68:879–87.
64. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med (2002) 53:409–35. doi:10.1146/annurev.med.53.082901.104018
65. Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem (2000) 43:527–50. doi:10.1021/jm990554g
66. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem (1995) 270:12953–6.
67. Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes (1996) 45:1661–9.
68. Ray DM, Akbiyik F, Phipps RP. The peroxisome proliferator-activated receptor gamma (PPARgamma) ligands 15-deoxy-Delta12,14-prostaglandin J2 and ciglitazone induce human B lymphocyte and B cell lymphoma apoptosis by PPARgamma-independent mechanisms. J Immunol (2006)177:5068–76. doi:10.4049/jimmunol.177.8.5068
69. Belfiore A, Genua M, Malaguarnera R. PPAR-gamma agonists and their effects on IGF-I receptor signaling: implications for cancer. PPAR Res (2009) 2009:830501. doi:10.1155/2009/830501
70. Ohta T, Elnemr A, Yamamoto M, Ninomiya I, Fushida S, Nishimura G, et al. Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, modulates the E-cadherin/beta-catenin system in a human pancreatic cancer cell line, BxPC-3. Int J Oncol (2002) 21:37–42. doi:10.3892/ijo.21.1.37
71. Piwien-Pilipuk G, Van Mater D, Ross SE, MacDougald OA, Schwartz J. Growth hormone regulates phosphorylation and function of CCAAT/enhancer-binding protein beta by modulating Akt and glycogen synthase kinase-3. J Biol Chem (2001) 276:19664–71. doi:10.1074/jbc.M010193200
72. Zhang W, Wu N, Li Z, Wang L, Jin J, Zha XL. PPARgamma activator rosiglitazone inhibits cell migration via upregulation of PTEN in human hepatocarcinoma cell line BEL-7404. Cancer Biol Ther (2006) 5:1008–14.
73. Okuno A, Tamemoto H, Tobe K, Ueki K, Mori Y, Iwamoto K, et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J Clin Invest (1998) 101:1354–61.
74. Berger JP. Role of PPARgamma, transcriptional cofactors, and adiponectin in the regulation of nutrient metabolism, adipogenesis and insulin action: view from the chair. Int J Obes (2005) 29(Suppl 1):S3–4. doi:10.1038/sj.ijo.0802904
75. Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci (2005) 26:244–51. doi:10.1016/j.tips.2005.03.003
76. Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem (2003) 278:34268–76. doi:10.1074/jbc.M300043200
77. Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G, Olson P, et al. Muscle-specific Pparg deletion causes insulin resistance. Nat Med (2003) 9:1491–7. doi:10.1038/nm956
78. Duan SZ, Ivashchenko CY, Usher MG, Mortensen RM. PPAR-gamma in the cardiovascular system. PPAR Res (2008) 2008:745804. doi:10.1155/2008/745804
79. Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest (2002) 110:923–32. doi:10.1172/JCI15634
80. Xin X, Yang S, Kowalski J, Gerritsen ME. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J Biol Chem (1999) 274:9116–21.
81. Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med (2006) 355:2427–43. doi:10.1056/NEJMoa066224
82. Juurinen L, Kotronen A, Graner M, Yki-Jarvinen H. Rosiglitazone reduces liver fat and insulin requirements and improves hepatic insulin sensitivity and glycemic control in patients with type 2 diabetes requiring high insulin doses. J Clin Endocrinol Metab (2008) 93:118–24. doi:10.1210/jc.2007-1825
83. Phielix E, Szendroedi J, Roden M. The role of metformin and thiazolidinediones in the regulation of hepatic glucose metabolism and its clinical impact. Trends Pharmacol Sci (2011) 32:607–16. doi:10.1016/j.tips.2011.06.006
84. Tiikkainen M, Hakkinen AM, Korsheninnikova E, Nyman T, Makimattila S, Yki-Jarvinen H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes (2004) 53:2169–76. doi:10.2337/diabetes.53.8.2169
85. Hevener AL, Reichart D, Janez A, Olefsky J. Thiazolidinedione treatment prevents free fatty acid-induced insulin resistance in male wistar rats. Diabetes (2001) 50:2316–22. doi:10.2337/diabetes.50.10.2316
86. Shibuya A, Watanabe M, Fujita Y, Saigenji K, Kuwao S, Takahashi H, et al. An autopsy case of troglitazone-induced fulminant hepatitis. Diabetes Care (1998) 21:2140–3.
87. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med (2007) 356:2457–71. doi:10.1056/NEJMoa072761
88. Pishvaian MJ, Marshall JL, Wagner AJ, Hwang JJ, Malik S, Cotarla I, et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer (2012) 118:5403–13. doi:10.1002/cncr.27526
89. Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, et al. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A (1998) 95:8806–11.
90. Tsubouchi Y, Sano H, Kawahito Y, Mukai S, Yamada R, Kohno M, et al. Inhibition of human lung cancer cell growth by the peroxisome proliferator- activated receptor-gamma agonists through induction of apoptosis. Biochem Biophys Res Commun (2000) 270:400–5. doi:10.1006/bbrc.2000.2436
91. Kitamura S, Miyazaki Y, Shinomura Y, Kondo S, Kanayama S, Matsuzawa Y. Peroxisome proliferator-activated receptor gamma induces growth arrest and differentiation markers of human colon cancer cells. Jpn J Cancer Res (1999) 90:75–80.
92. Wagner KD, Benchetrit M, Bianchini L, Michiels JF, Wagner N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) is highly expressed in liposarcoma and promotes migration and proliferation. J Pathol (2011) 224:575–88. doi:10.1002/path.2910
93. Fujimura S, Suzumiya J, Nakamura K, Ono J. Effects of troglitazone on the growth and differentiation of hematopoietic cell lines. Int J Oncol (1998) 13:1263–7.
94. Ellis HP, Kurian KM. Biological rationale for the use of PPARgamma agonists in glioblastoma. Front Oncol (2014) 4:52. doi:10.3389/fonc.2014.00052
95. Cao LQ, Chen XL, Wang Q, Huang XH, Zhen MC, Zhang LJ, et al. Upregulation of PTEN involved in rosiglitazone-induced apoptosis in human hepatocellular carcinoma cells. Acta Pharmacol Sin (2007) 28:879–87. doi:10.1111/j.1745-7254.2007.00571.x
96. Cao LQ, Wang XL, Wang Q, Xue P, Jiao XY, Peng HP, et al. Rosiglitazone sensitizes hepatocellular carcinoma cell lines to 5-fluorouracil antitumor activity through activation of the PPARgamma signaling pathway. Acta Pharmacol Sin (2009) 30:1316–22. doi:10.1038/aps.2009.119
97. Ninomiya I, Yamazaki K, Oyama K, Hayashi H, Tajima H, Kitagawa H, et al. Pioglitazone inhibits the proliferation and metastasis of human pancreatic cancer cells. Oncol Lett (2014) 8:2709–14. doi:10.3892/ol.2014.2553
98. Aiello A, Pandini G, Frasca F, Conte E, Murabito A, Sacco A, et al. Peroxisomal proliferator-activated receptor-gamma agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology (2006) 147:4463–75. doi:10.1210/en.2005-1610
99. Kubota T, Koshizuka K, Williamson EA, Asou H, Said JW, Holden S, et al. Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res (1998) 58:3344–52.
100. Copland JA, Marlow LA, Kurakata S, Fujiwara K, Wong AK, Kreinest PA, et al. Novel high-affinity PPARgamma agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene (2006) 25:2304–17. doi:10.1038/sj.onc.1209267
101. Wood WM, Sharma V, Bauerle KT, Pike LA, Zhou Q, Fretwell DL, et al. PPARgamma promotes growth and invasion of thyroid cancer cells. PPAR Res (2011) 2011:171765. doi:10.1155/2011/171765
102. Keshamouni VG, Reddy RC, Arenberg DA, Joel B, Thannickal VJ, Kalemkerian GP, et al. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor progression in non-small-cell lung cancer. Oncogene (2004) 23:100–8. doi:10.1038/sj.onc.1206885
103. Reddy RC, Srirangam A, Reddy K, Chen J, Gangireddy S, Kalemkerian GP, et al. Chemotherapeutic drugs induce PPAR-gamma expression and show sequence-specific synergy with PPAR-gamma ligands in inhibition of nonsmall cell lung cancer. Neoplasia (2008) 10:597–603. doi:10.1593/neo.08134
104. Sawayama H, Ishimoto T, Watanabe M, Yoshida N, Sugihara H, Kurashige J, et al. Small molecule agonists of PPAR-gamma exert therapeutic effects in esophageal cancer. Cancer Res (2014) 74:575–85. doi:10.1158/0008-5472.CAN-13-1836
105. Annicotte JS, Iankova I, Miard S, Fritz V, Sarruf D, Abella A, et al. Peroxisome proliferator-activated receptor gamma regulates E-cadherin expression and inhibits growth and invasion of prostate cancer. Mol Cell Biol (2006) 26:7561–74. doi:10.1128/MCB.00605-06
106. Musgrove EA, Lee CS, Buckley MF, Sutherland RL. Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc Natl Acad Sci U S A (1994) 91:8022–6.
107. Kawakami S, Arai G, Hayashi T, Fujii Y, Xia G, Kageyama Y, et al. PPARgamma ligands suppress proliferation of human urothelial basal cells in vitro. J Cell Physiol (2002) 191:310–9. doi:10.1002/jcp.10099
108. Yang FG, Zhang ZW, Xin DQ, Shi CJ, Wu JP, Guo YL, et al. Peroxisome proliferator-activated receptor gamma ligands induce cell cycle arrest and apoptosis in human renal carcinoma cell lines. Acta Pharmacol Sin (2005) 26:753–61. doi:10.1111/j.1745-7254.2005.00753.x
109. Camp HS, Tafuri SR. Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J Biol Chem (1997) 272:10811–6.
110. Chang AJ, Song DH, Wolfe MM. Attenuation of peroxisome proliferator-activated receptor gamma (PPARgamma) mediates gastrin-stimulated colorectal cancer cell proliferation. J Biol Chem (2006) 281:14700–10. doi:10.1074/jbc.M602623200
111. Zhang B, Berger J, Zhou G, Elbrecht A, Biswas S, White-Carrington S, et al. Insulin- and mitogen-activated protein kinase-mediated phosphorylation and activation of peroxisome proliferator-activated receptor gamma. J Biol Chem (1996) 271:31771–4.
112. Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem (1997) 272:5128–32.
113. Frasca F, Vella V, Nicolosi ML, Messina RL, Giani F, Lotta S, et al. Thyroid cancer cell resistance to gefitinib depends on the constitutive oncogenic activation of the ERK pathway. J Clin Endocrinol Metab (2013) 98:2502–12. doi:10.1210/jc.2012-3623
114. Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor gamma. Mol Cell Biol (2007) 27:803–17. doi:10.1128/MCB.00601-06
115. Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol (2001) 11:764–8. doi:10.1016/S0960-9822(01)00225-1
116. Han S, Roman J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol Cancer Ther (2006) 5:430–7. doi:10.1158/1535-7163.MCT-05-0347
117. Lee SY, Hur GY, Jung KH, Jung HC, Lee SY, Kim JH, et al. PPAR-gamma agonist increase gefitinib’s antitumor activity through PTEN expression. Lung Cancer (2006) 51:297–301. doi:10.1016/j.lungcan.2005.10.010
118. Farrow B, Evers BM. Activation of PPARgamma increases PTEN expression in pancreatic cancer cells. Biochem Biophys Res Commun (2003) 301:50–3. doi:10.1016/S0006-291X(02)02983-2
119. Cho DH, Choi YJ, Jo SA, Ryou J, Kim JY, Chung J, et al. Troglitazone acutely inhibits protein synthesis in endothelial cells via a novel mechanism involving protein phosphatase 2A-dependent p70 S6 kinase inhibition. Am J Physiol Cell Physiol (2006) 291:C317–26. doi:10.1152/ajpcell.00491.2005
120. He G, Sung YM, Digiovanni J, Fischer SM. Thiazolidinediones inhibit insulin-like growth factor-I-induced activation of p70S6 kinase and suppress insulin-like growth factor-I tumor-promoting activity. Cancer Res (2006) 66:1873–8. doi:10.1158/0008-5472.CAN-05-3111
121. Hashimoto K, Farrow BJ, Evers BM. Activation and role of MAP kinases in 15d-PGJ2 induced apoptosis in the human pancreatic cancer cell line MIA PaCa-2. Pancreas (2004) 28:153–9. doi:10.1097/00006676-200403000-00006
122. Motomura W, Okumura T, Takahashi N, Obara T, Kohgo Y. Activation of peroxisome proliferator-activated receptor gamma by troglitazone inhibits cell growth through the increase of p27KiP1 in human. Pancreatic carcinoma cells. Cancer Res (2000) 60:5558–4.
123. Kim EH, Na HK, Surh YJ. Upregulation of VEGF by 15-deoxy-Delta12,14- prostaglandin J2 via heme oxygenase-1 and ERK1/2 signaling in MCF-7 cells. Ann N Y Acad Sci (2006)1090:375–84. doi:10.1196/annals.1378.041
124. Kim KH, Cho YS, Park JM, Yoon SO, Kim KW, Chung AS. Pro-MMP-2 activation by the PPARgamma agonist, ciglitazone, induces cell invasion through the generation of ROS and the activation of ERK. FEBS Lett (2007) 581:3303–10. doi:10.1016/j.febslet.2007.06.012
125. Zhou J, Zhang W, Liang B, Casimiro MC, Whitaker-Menezes D, Wang M, et al. PPARgamma activation induces autophagy in breast cancer cells. Int J Biochem Cell Biol (2009) 41:2334–42. doi:10.1016/j.biocel.2009.06.007
126. Cerquetti L, Sampaoli C, Amendola D, Bucci B, Masuelli L, Marchese R, et al. Rosiglitazone induces autophagy in H295R and cell cycle deregulation in SW13 adrenocortical cancer cells. Exp Cell Res (2011) 317:1397–410. doi:10.1016/j.yexcr.2011.02.014
127. Yan S, Yang X, Chen T, Xi Z, Jiang X. The PPARgamma agonist troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther (2014) 21:188–93. doi:10.1038/cgt.2014.16
128. Kotta-Loizou I, Giaginis C, Theocharis S. The role of peroxisome proliferator-activated receptor-gamma in breast cancer. Anticancer Agents Med Chem (2012) 12:1025–44. doi:10.2174/187152012803529664
129. Apsel Winger B, Shah NP. PPARgamma: welcoming the new kid on the CML stem cell block. Cancer Cell (2015) 28:409–11. doi:10.1016/j.ccell.2015.09.017
130. Egan JM. Targeting stem cells in chronic myeloid leukemia with a PPARgamma agonist. N Engl J Med (2015) 373:1973–5. doi:10.1056/NEJMcibr1511276
131. Holyoake T, Vetrie D. Cancer: repositioned to kill stem cells. Nature (2015) 525:328–9. doi:10.1038/nature15213
132. Papi A, De Carolis S, Bertoni S, Storci G, Sceberras V, Santini D, et al. PPARgamma and RXR ligands disrupt the inflammatory cross-talk in the hypoxic breast cancer stem cells niche. J Cell Physiol (2014) 229:1595–1606. doi:10.1002/jcp.24601
133. Prost S, Relouzat F, Spentchian M, Ouzegdouh Y, Saliba J, Massonnet G, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature (2015) 525:380–3. doi:10.1038/nature15248
134. Wang Y, Tan H, Xu D, Ma A, Zhang L, Sun J, et al. The combinatory effects of PPAR-gamma agonist and survivin inhibition on the cancer stem-like phenotype and cell proliferation in bladder cancer cells. Int J Mol Med (2014) 34:262–8. doi:10.3892/ijmm.2014.1774
135. Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science (2000) 289:950–3. doi:10.1126/science.289.5481.950
136. Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol (2006) 26:5827–37. doi:10.1128/MCB.00441-06
137. Liu JJ, Dai XJ, Xu Y, Liu PQ, Zhang Y, Liu XD, et al. Inhibition of lymphoma cell proliferation by peroxisomal proliferator-activated receptor-gamma ligands via Wnt signaling pathway. Cell Biochem Biophys (2012) 62:19–27. doi:10.1007/s12013-011-9253-x
138. Lu D, Cottam HB, Corr M, Carson DA. Repression of beta-catenin function in malignant cells by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A (2005) 102:18567–71. doi:10.1073/pnas.0509316102
139. Guo F, Ren X, Dong Y, Hu X, Xu D, Zhou H, et al. Constitutive expression of PPARgamma inhibits proliferation and migration of gastric cancer cells and down-regulates Wnt/beta-catenin signaling pathway downstream target genes TERT and ENAH. Gene (2016) 584:31–7. doi:10.1016/j.gene.2016.03.003
140. Panza A, Pazienza V, Ripoli M, Benegiamo G, Gentile A, Valvano MR, et al. Interplay between SOX9, beta-catenin and PPARgamma activation in colorectal cancer. Biochim Biophys Acta (2013) 1833:1853–65. doi:10.1016/j.bbamcr.2013.04.004
141. Ren X, Zheng D, Guo F, Liu J, Zhang B, Li H, et al. PPARgamma suppressed Wnt/beta-catenin signaling pathway and its downstream effector SOX9 expression in gastric cancer cells. Med Oncol (2015) 32:91. doi:10.1007/s12032-015-0536-8
142. Giani F, Vella V, Nicolosi ML, Fierabracci A, Lotta S, Malaguarnera R, et al. Thyrospheres from normal or malignant thyroid tissue have different biological, functional, and genetic features. J Clin Endocrinol Metab (2015) 100:E1168–78. doi:10.1210/JC.2014-4163
143. Malaguarnera R, Frasca F, Garozzo A, Giani F, Pandini G, Vella V, et al. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab (2011) 96:766–74. doi:10.1210/jc.2010-1255
144. Malaguarnera R, Morcavallo A, Giuliano S, Belfiore A. Thyroid cancer development and progression: emerging role of cancer stem cells. Minerva Endocrinol (2012) 37:103–15.
145. Jenkins CR, Shevchuk OO, Giambra V, Lam SH, Carboni JM, Gottardis MM, et al. IGF signaling contributes to malignant transformation of hematopoietic progenitors by the MLL-AF9 oncoprotein. Exp Hematol (2012) 40:715e–23e. doi:10.1016/j.exphem.2012.05.003
146. Medyouf H, Gusscott S, Wang H, Tseng JC, Wai C, Nemirovsky O, et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J Exp Med (2011) 208:1809–22. doi:10.1084/jem.20110121
147. Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Cherqui G, Perret C, et al. Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene (2001) 20:252–9. doi:10.1038/sj.onc.1204064
148. Playford MP, Bicknell D, Bodmer WF, Macaulay VM. Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of beta-catenin. Proc Natl Acad Sci U S A (2000) 97:12103–8. doi:10.1073/pnas.210394297
149. Vanamala J, Reddivari L, Radhakrishnan S, Tarver C. Resveratrol suppresses IGF-1 induced human colon cancer cell proliferation and elevates apoptosis via suppression of IGF-1R/Wnt and activation of p53 signaling pathways. BMC Cancer (2010) 10:238. doi:10.1186/1471-2407-10-238
150. Bommer GT, Feng Y, Iura A, Giordano TJ, Kuick R, Kadikoy H, et al. IRS1 regulation by Wnt/beta-catenin signaling and varied contribution of IRS1 to the neoplastic phenotype. J Biol Chem (2010) 285:1928–38. doi:10.1074/jbc.M109.060319
151. Chen J, Wu A, Sun H, Drakas R, Garofalo C, Cascio S, et al. Functional significance of type 1 insulin-like growth factor-mediated nuclear translocation of the insulin receptor substrate-1 and beta-catenin. J Biol Chem (2005) 280:29912–20. doi:10.1074/jbc.M504516200
152. Rubin R, Arzumanyan A, Soliera AR, Ross B, Peruzzi F, Prisco M. Insulin receptor substrate (IRS)-1 regulates murine embryonic stem (mES) cells self-renewal. J Cell Physiol (2007) 213:445–53. doi:10.1002/jcp.21185
153. Liu L, Yang Z, Xu Y, Li J, Xu D, Zhang L, et al. Inhibition of oxidative stress-elicited AKT activation facilitates PPARgamma agonist-mediated inhibition of stem cell character and tumor growth of liver cancer cells. PLoS One (2013) 8:e73038. doi:10.1371/journal.pone.0073038
154. Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARgamma agonists: time for a reassessment. Trends Endocrinol Metab (2012) 23:205–15. doi:10.1016/j.tem.2012.03.001
155. Dobson ME, Diallo-Krou E, Grachtchouk V, Yu J, Colby LA, Wilkinson JE, et al. Pioglitazone induces a proadipogenic antitumor response in mice with PAX8-PPARgamma fusion protein thyroid carcinoma. Endocrinology (2011) 152:4455–65. doi:10.1210/en.2011-1178
156. Itami A, Watanabe G, Shimada Y, Hashimoto Y, Kawamura J, Kato M, et al. Ligands for peroxisome proliferator-activated receptor gamma inhibit growth of pancreatic cancers both in vitro and in vivo. Int J Cancer (2001) 94:370–6. doi:10.1002/ijc.1488
157. Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, et al. PPARgamma insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-kappaB signaling pathway. Oncogene (2006) 25:2736–47. doi:10.1038/sj.onc.1209299
158. Ohta K, Endo T, Haraguchi K, Hershman JM, Onaya T. Ligands for peroxisome proliferator-activated receptor gamma inhibit growth and induce apoptosis of human papillary thyroid carcinoma cells. J Clin Endocrinol Metab (2001) 86:2170–7. doi:10.1210/jcem.86.5.7493
159. Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med (1998) 4:1046–52.
160. Fujiwara T, Yoshioka S, Yoshioka T, Ushiyama I, Horikoshi H. Characterization of new oral antidiabetic agent CS-045. Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes (1988) 37:1549–58.
161. Iwamoto Y, Kuzuya T, Matsuda A, Awata T, Kumakura S, Inooka G, et al. Effect of new oral antidiabetic agent CS-045 on glucose tolerance and insulin secretion in patients with NIDDM. Diabetes Care (1991) 14:1083–6.
162. Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med (1994) 331:1188–93.
163. Suter SL, Nolan JJ, Wallace P, Gumbiner B, Olefsky JM. Metabolic effects of new oral hypoglycemic agent CS-045 in NIDDM subjects. Diabetes Care (1992) 15:193–203.
164. Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, Kim L, et al. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncol (2007) 25:1476–81. doi:10.1200/JCO.2006.07.2777
165. Ramos-Nino ME, MacLean CD, Littenberg B. Association between cancer prevalence and use of thiazolidinediones: results from the Vermont Diabetes Information System. BMC Med (2007) 5:17. doi:10.1186/1741-7015-5-17
166. Monami M, Lamanna C, Marchionni N, Mannucci E. Rosiglitazone and risk of cancer: a meta-analysis of randomized clinical trials. Diabetes Care (2008) 31:1455–60. doi:10.2337/dc07-2308
167. Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology (2009) 137:482–8. doi:10.1053/j.gastro.2009.04.013
168. Koro C, Barrett S, Qizilbash N. Cancer risks in thiazolidinedione users compared to other anti-diabetic agents. Pharmacoepidemiol Drug Saf (2007) 16:485–92. doi:10.1002/pds.1352
169. Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet (2005) 366:1279–89. doi:10.1016/S0140-6736(05)67528-9
170. Lewis JD, Ferrara A, Peng T, Hedderson M, Bilker WB, Quesenberry CP Jr, et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care (2011) 34:916–22. doi:10.2337/dc10-1068
171. Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, et al. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol (1995) 15:3012–22.
172. Mukherjee R, Jow L, Noonan D, McDonnell DP. Human and rat peroxisome proliferator activated receptors (PPARs) demonstrate similar tissue distribution but different responsiveness to PPAR activators. J Steroid Biochem Mol Biol (1994) 51:157–66.
173. Palmer CN, Hsu MH, Griffin HJ, Johnson EF. Novel sequence determinants in peroxisome proliferator signaling. J Biol Chem (1995) 270:16114–21.
174. Ahmed W, Ziouzenkova O, Brown J, Devchand P, Francis S, Kadakia M, et al. PPARs and their metabolic modulation: new mechanisms for transcriptional regulation? J Intern Med (2007) 262:184–98. doi:10.1111/j.1365-2796.2007.01825.x
175. Panigrahy D, Kaipainen A, Huang S, Butterfield CE, Barnes CM, Fannon M, et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci U S A (2008) 105:985–90. doi:10.1073/pnas.0711281105
176. Yamasaki D, Kawabe N, Nakamura H, Tachibana K, Ishimoto K, Tanaka T, et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARalpha-independent mechanisms. Eur J Cell Biol (2011) 90:657–64. doi:10.1016/j.ejcb.2011.02.005
177. Lee JW, Bajwa PJ, Carson MJ, Jeske DR, Cong Y, Elson CO, et al. Fenofibrate represses interleukin-17 and interferon-gamma expression and improves colitis in interleukin-10-deficient mice. Gastroenterology (2007) 133:108–23. doi:10.1053/j.gastro.2007.03.113
178. Zhou J, Zhang S, Xue J, Avery J, Wu J, Lind SE, et al. Activation of peroxisome proliferator-activated receptor alpha (PPARalpha) suppresses hypoxia-inducible factor-1alpha (HIF-1alpha) signaling in cancer cells. J Biol Chem (2012) 287:35161–9 doi:10.1074/jbc.M112.367367
179. Hann SS, Zheng F, Zhao S. Targeting 3-phosphoinositide-dependent protein kinase 1 by N-acetyl-cysteine through activation of peroxisome proliferators activated receptor alpha in human lung cancer cells, the role of p53 and p65. J Exp Clin Cancer Res (2013) 32:43. doi:10.1186/1756-9966-32-43
180. Drukala J, Urbanska K, Wilk A, Grabacka M, Wybieralska E, Del Valle L, et al. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARalpha-mediated inhibition of glioma cell motility in vitro. Mol Cancer (2010) 9:159. doi:10.1186/1476-4598-9-159
181. Urbanska K, Pannizzo P, Grabacka M, Croul S, Del Valle L, Khalili K, et al. Activation of PPARalpha inhibits IGF-I-mediated growth and survival responses in medulloblastoma cell lines. Int J Cancer (2008) 123:1015–24. doi:10.1002/ijc.23588
182. Kuno T, Hata K, Takamatsu M, Hara A, Hirose Y, Takahashi S, et al. The peroxisome proliferator-activated receptor (PPAR) alpha agonist fenofibrate suppresses chemically induced lung alveolar proliferative lesions in male obese hyperlipidemic mice. Int J Mol Sci (2014) 15:9160–72. doi:10.3390/ijms15059160
183. Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator- activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res (2006) 12:3028–36. doi:10.1158/1078-0432.CCR-05-2556
184. Gamerdinger M, Clement AB, Behl C. Cholesterol-like effects of selective cyclooxygenase inhibitors and fibrates on cellular membranes and amyloid-beta production. Mol Pharmacol (2007) 72:141–51 doi:10.1124/mol.107.034009
186. Guzman M, Lo Verme J, Fu J, Oveisi F, Blazquez C, Piomelli D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor alpha (PPAR-alpha). J Biol Chem (2004) 279:27849–54. doi:10.1074/jbc.M404087200
Keywords: insulin/IGF signaling pathway, insulin resistance, hyperinsulinemia, PPAR-γ, cancer, thiazolidinediones
Citation: Vella V, Nicolosi ML, Giuliano S, Bellomo M, Belfiore A and Malaguarnera R (2017) PPAR-γ Agonists As Antineoplastic Agents in Cancers with Dysregulated IGF Axis. Front. Endocrinol. 8:31. doi: 10.3389/fendo.2017.00031
Received: 23 November 2016; Accepted: 06 February 2017;
Published: 22 February 2017
Edited by:
Claire Perks, University of Bristol, UKReviewed by:
Krzysztof Reiss, LSU Health Sciences Center New Orleans, USALeonard Girnita, Karolinska Institutet, Sweden
Copyright: © 2017 Vella, Nicolosi, Giuliano, Bellomo, Belfiore and Malaguarnera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonino Belfiore, YmVsZmlvcmVAdW5pY3ouaXQ=